Tissue-Resident Memory B Cells: Decoding Their Critical Role in Mucosal vs. Systemic Vaccination
This review synthesizes current knowledge on tissue-resident memory B cells (BRM), a specialized lymphocyte subset residing in mucosal tissues that provides localized, rapid-response defense against pathogens.
Tissue-Resident Memory B Cells: Decoding Their Critical Role in Mucosal vs. Systemic Vaccination
Abstract
This review synthesizes current knowledge on tissue-resident memory B cells (BRM), a specialized lymphocyte subset residing in mucosal tissues that provides localized, rapid-response defense against pathogens. We explore the fundamental biology of BRM, including their generation through CD40-dependent germinal center responses and their role as frontline defenders in the airways and other mucosal surfaces. The article contrasts the potent, localized antibody responses elicited by mucosal vaccination with the predominantly systemic immunity induced by traditional parenteral vaccines. We detail cutting-edge methodological approaches for studying BRM, address key challenges in vaccine design such as durability and adjuvant selection, and present comparative evidence validating the superior ability of mucosal vaccines to establish protective tissue-resident memory. This resource is intended to guide researchers and drug development professionals in the rational design of next-generation vaccines that leverage BRM for enhanced protection against respiratory and other mucosal pathogens.
Frontline Defenders: Unraveling the Biology and Generation of Tissue-Resident Memory B Cells
Within the intricate landscape of adaptive immunity, a specialized sentinel has emerged as a critical mediator of localized protection: the tissue-resident memory B cell (BRM). These cells are a distinct subset of memory B cells that take up long-term residence in tissues without recirculating, providing a first line of defense against pathogen re-exposure at mucosal surfaces and other barrier sites [1] [2]. Unlike their circulating counterparts, BRM cells are strategically positioned at common sites of pathogen entry, enabling them to mount rapid, localized antibody responses during secondary challenges [1]. This guide provides a comprehensive comparison of BRM phenotypic and functional characteristics, situating their role within the broader context of mucosal versus systemic vaccination strategies. We synthesize current experimental data and methodologies to offer researchers a foundational resource for investigating these crucial cellular sentinels.
Phenotypic Profile and Tissue Distribution of BRM
BRM cells are defined by their long-term persistence in tissues and a characteristic molecular profile that distinguishes them from circulating memory B cells. A core set of surface markers facilitates their identification and isolation from tissue samples, though their specific phenotype can exhibit notable heterogeneity across different tissue environments [2].
Table 1: Core Phenotypic Markers of Tissue-Resident Memory B Cells (BRM)
| Marker | Expression Status | Function and Significance |
|---|---|---|
| CD69 | Consistently expressed | A canonical tissue-residency marker; inhibits the sphingosine-1-phosphate receptor 1 (S1P1), thereby preventing egress from tissues [2]. |
| CD49a (VLA-1) | Variably expressed | An integrin that facilitates adhesion to collagen in the extracellular matrix; more commonly expressed in non-mucosal tissues [2]. |
| CD103 (αE integrin) | Variably expressed | Binds to E-cadherin on epithelial cells; predominantly expressed by BRM in mucosal tissues [2]. |
The anatomic distribution of BRM cells is a key determinant of their sentinel function. They have been identified in multiple mucosal tissues, including the lung and airways, where they are derived from CD40-dependent germinal center responses [1]. The inducible bronchus-associated lymphoid tissue (iBALT) is a specific site that contributes to the formation and maintenance of BRM populations in the lung [2]. Their presence has been confirmed through advanced techniques such as parabiosis surgery and intravascular staining, which definitively distinguish tissue-resident from circulating cell populations [2].
Functional Capacities and Role in Protective Immunity
The functional specialization of BRM cells is directly linked to their tissue-resident nature, enabling a unique mode of immune defense.
Primary Functions and Mechanisms
- Rapid Plasma Cell Differentiation: Upon re-encounter with their cognate antigen, BRM cells swiftly differentiate into antibody-secreting plasma cells. This provides a robust and localized source of antibodies directly at the site of pathogen entry, a critical advantage over the slower recall response from circulating memory B cells [1].
- Localized Antibody Production: The antibodies produced by BRM-derived plasma cells can include all major isotypes, such as IgG and IgA. This local production creates a specific immune environment at mucosal surfaces, neutralizing pathogens before they can establish a systemic infection [1] [2].
Comparative Functional Output
The functional profile of BRM cells can be contrasted with other B cell subsets to highlight their unique role. The following diagram illustrates the core functional characteristics and protective mechanisms of BRM cells.
Methodologies for BRM Research: A Toolkit for the Scientist
Studying BRM cells requires a combination of established immunological techniques and advanced technologies to confirm their tissue residency and elucidate their unique biology.
Core Experimental Protocols
Confirming Tissue Residency:
- Parabiosis Surgery: This gold-standard method involves surgically joining two mice to create a shared circulatory system. Circulating cells equilibrate between both partners, while true tissue-resident cells (like BRM) remain in the host tissue and are not found in the partner's tissues [2].
- Intravascular Staining: An intravenous injection of a fluorescently-labeled antibody is administered moments before euthanasia. This antibody labels all cells within the blood vasculature. During subsequent flow cytometry, true tissue-resident cells are identified as the population that is not labeled by the intravascular dye [2].
Profiling and Phenotyping:
- High-Parameter Flow Cytometry: This is essential for identifying the complex surface phenotype of BRM cells (e.g., CD19⁺, CD69⁺, CD49a⁺/CD103⁺) and for isolating pure populations via Fluorescence-Activated Cell Sorting (FACS) for functional assays [2].
- Single-Cell RNA Sequencing (scRNA-seq): This powerful technology reveals the complete transcriptional landscape of BRM cells, uncovering heterogeneity, developmental pathways, and functional states by analyzing gene expression cell-by-cell [2].
Functional Assays:
- B Cell Receptor (BCR) Sequencing: This technique sequences the variable regions of the BCRs of BRM cells, providing insights into clonality, antigenic experience, and affinity maturation [2].
- In Vitro Re-stimulation and Differentiation Assays: Isolated BRM cells can be stimulated with antigens or mitogens in vitro to measure their proliferation, differentiation into plasma cells, and antibody secretion, directly quantifying their functional capacity [1].
Essential Research Reagent Solutions
Table 2: Key Reagents for BRM Cell Research
| Reagent / Tool | Primary Function | Example Application |
|---|---|---|
| Anti-CD69 Antibody | Blocking/Saturation | Used in intravascular staining protocols to discriminate resident vs. circulating cells; critical for flow cytometry panels to identify BRM. |
| Anti-CD49a & Anti-CD103 Antibodies | Phenotypic Identification | Key for defining the BRM population via flow cytometry, especially to distinguish subsets in mucosal vs. non-mucosal sites. |
| Recombinant Cytokines | Cell Culture & Differentiation | IL-4, IL-5, IL-21, and BAFF are used in in vitro cultures to support B cell survival and promote plasma cell differentiation. |
| TaqMan Assays for BCR/Expression | Genotyping & Gene Expression | Custom TaqMan assays (as used in genotyping studies) can be adapted for quantifying BCR clonotypes or BRM-specific gene expression [3]. |
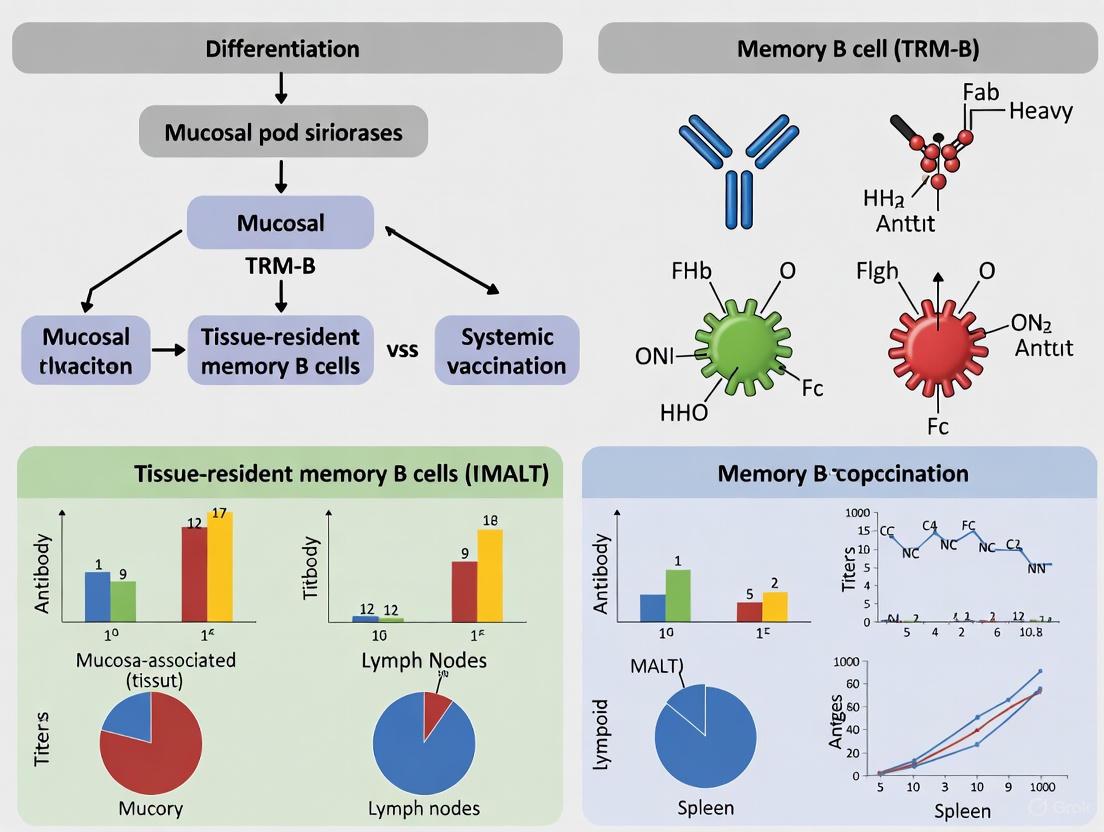
BRM in Mucosal vs. Systemic Vaccination: A Comparative Analysis
The differential induction of BRM cells is a fundamental consideration when comparing mucosal and systemic vaccination routes. The following workflow diagram outlines the key experimental and analytical steps for characterizing BRM cells in vaccination studies.
Table 3: Comparison of BRM Induction by Vaccination Route
| Characteristic | Mucosal Vaccination | Systemic Vaccination |
|---|---|---|
| BRM Induction Efficiency | High; directly targets immune induction at mucosal sites. | Typically low; primarily generates circulating memory and plasma cells that home to bone marrow. |
| Location of Induced BRM | Local mucosal tissues (e.g., lung airways, iBALT) [2]. | Largely absent from mucosal sites; may induce some populations in secondary lymphoid organs. |
| Functional Outcome | Elicits strong local antibody secretion at the portal of entry for many pathogens, providing immediate sterilizing immunity. | Elicits strong systemic antibody titers but poor mucosal immunity, potentially allowing initial infection at mucosae. |
| Protective Efficacy | Superior for preventing initial infection at mucosal surfaces (e.g., against influenza, SARS-CoV-2). | Superior for controlling pathogens after they have entered the systemic circulation. |
BRM cells represent a critical axis of adaptive immunity, defined by their non-recirculating nature, canonical marker expression (CD69, often with CD49a or CD103), and function as rapid local responders. A clear understanding of their phenotypic and functional characteristics, as detailed in this guide, is paramount for the rational design of next-generation vaccines. The current data strongly indicates that mucosal vaccination strategies are the most effective means to generate these sentinel cells at relevant portals of pathogen entry. Future research should focus on optimizing delivery platforms and adjuvants that robustly induce durable BRM populations, thereby bridging the gap between systemic immunity and true mucosal protection.
Tissue-resident memory B cells (BRM cells) represent a specialized subset of memory B cells that reside within mucosal tissues and play a critical role in frontline defense against respiratory pathogens. Unlike their circulating counterparts, BRM cells establish permanent residence in barrier tissues, particularly the airways, where they provide rapid, localized immune protection against reinfection. The strategic positioning of these cells at common sites of pathogen entry enables immediate response capabilities that far exceed those of systemically generated memory responses. This review examines the precise anatomical localization of BRM cells within mucosal tissues and the airways, comparing the effectiveness of mucosal versus systemic vaccination strategies in establishing these sentinel populations. Understanding the factors governing BRM cell development, maintenance, and function provides crucial insights for designing next-generation vaccines capable of eliciting robust tissue-localized immunity against respiratory pathogens.
Biological Basis of BRM Localization
Definition and Key Characteristics
BRM cells are a distinct population of non-recirculating memory B cells that persist long-term in mucosal tissues after antigen exposure. These cells are strategically positioned at barrier surfaces where they interface directly with the external environment [1]. Unlike circulating memory B cells that survey the body through lymphoid organs, BRM cells remain sessile within peripheral tissues, forming a network of antigen-experienced sentinels [4]. Their presence has been most comprehensively characterized in the respiratory tract, though evidence suggests similar populations may exist in other mucosal sites.
Key identifying features of BRM cells include their non-recirculating nature, demonstrated through parabiosis experiments and intravenous antibody labeling techniques [4]. These cells typically display surface markers associated with tissue residency, such as CD69, and often express CXCR3, a chemokine receptor guiding migration to inflammatory sites [4]. Functionally, BRM cells maintain the capacity to rapidly differentiate into antibody-secreting plasma cells upon re-encounter with cognate antigen, providing immediate local antibody production at the infection site [1].
Mechanisms of Tissue Entry and Retention
The establishment of BRM populations in mucosal tissues follows a coordinated sequence of trafficking, retention, and local maintenance. The process begins when antigen-activated B cells are recruited to peripheral tissues through tissue-specific homing signals. In the respiratory tract, this recruitment is driven by inflammatory cytokines and chemokines upregulated during local infection [5].
Once within the tissue, developing BRM cells undergo a program of * tissue residency differentiation, mediated by microenvironmental cues including cytokines and cellular interactions. The *upregulation of CD69 contributes to retention by countering S1P1-mediated egress signals [4]. Local survival factors, including BAFF (B cell-activating factor) and interactions with tissue-resident stromal cells, support the long-term maintenance of BRM populations without requiring continuous replenishment from circulation [4].
Critical to BRM development is the requirement for local antigen encounter. Systemic immunization typically fails to generate substantial BRM populations in mucosal tissues, whereas localized infection or mucosal vaccination efficiently establishes these resident sentinels [5] [6]. This highlights the importance of tissue context in instructing the residency program and ensuring BRM cells are positioned at sites of potential pathogen rechallenge.
Comparative Analysis of BRM in Mucosal vs. Systemic Vaccination
Formation and Maintenance
The differentiation of BRM cells follows distinct pathways depending on the route of antigen encounter, with significant implications for the quality, quantity, and durability of local immune protection.
Table 1: BRM Formation and Maintenance Characteristics
| Aspect | Mucosal Vaccination/Infection | Systemic Vaccination |
|---|---|---|
| Induction Requirement | Local antigen exposure in mucosal tissue | Primarily systemic immune activation |
| Primary Induction Site | Local germinal centers (e.g., iBALT) | Draining lymph nodes, spleen |
| Tissue Homing | Efficient migration to and retention in mucosal sites | Limited mucosal homing capacity |
| Persistence | Long-term residence (≥6 months documented) | Typically transient in peripheral tissues |
| Maintenance Mechanism | Local survival signals, tissue niche support | Continuous replenishment from circulation (limited) |
Mucosal vaccination or natural infection establishes BRM populations through local germinal center reactions occurring in inducible bronchus-associated lymphoid tissue (iBALT) and other mucosal-associated lymphoid structures [5]. These tissue-based germinal centers generate BRM cells specifically imprinted for residence in the local mucosal environment. Once established, BRM cells can persist for extended periods, with studies documenting their presence in lungs for at least six months post-infection [6]. This persistence occurs independently of continuous replenishment from circulation, as demonstrated by FTY720 treatment experiments that block lymphocyte egress from lymphoid organs without depleting lung BRM populations [4].
In contrast, systemic vaccination primarily generates circulating memory B cells with limited capacity to establish resident populations in mucosal tissues [5]. While some circulating cells may be recruited to tissues during subsequent inflammatory events, they typically fail to undergo the full differentiation program required for long-term residence. The few memory B cells that do enter tissues following systemic immunization often display transient persistence and may lack the optimal positioning and functional programming of bona fide BRM cells generated via mucosal routes.
Functional Efficacy in Protection
The strategic positioning of BRM cells within mucosal tissues confers significant functional advantages for rapid pathogen control, particularly against respiratory infections.
Table 2: Functional Characteristics of BRM Cells in Protection
| Functional Aspect | Mucosal-Derived BRM | Systemically Primed Memory B Cells |
|---|---|---|
| Response Kinetics | Rapid differentiation (within days) | Delayed response (≥7 days) |
| Antibody Production Site | Directly in infected alveoli | Primarily in iBALT structures |
| Spatial Organization | Distributed network throughout parenchyma | Confined to lymphoid aggregates |
| Early Protection | Significantly reduces early viral loads | Limited impact on early replication |
| Response to Heterologous Strains | Cross-reactive potential documented | Varies depending on immunization |
During secondary challenge, mucosally-established BRM cells demonstrate superior response kinetics, differentiating into antibody-secreting plasma cells within days of reinfection [1]. This rapid response occurs directly at sites of viral replication in the alveolar space, where BRM cells interface with infected cells [5]. The spatial organization of BRM cells throughout the lung parenchyma creates a network of sentinels that collectively surveil large tissue volumes, enabling detection and response to infection even in areas distant from organized lymphoid structures [5].
The functional superiority of mucosally-primed BRM responses was clearly demonstrated in influenza challenge experiments. Mice primed intranasally developed BRM cells that, upon rechallenge, rapidly differentiated into alveolar plasma cells and accumulated at infection sites within four days [5]. In contrast, systemically primed mice lacked this early response wave, with plasma cell development confined to iBALT structures and delayed until seven days post-challenge [5]. This temporal advantage translates to significantly enhanced early viral control, potentially limiting both disease severity and transmission.
Furthermore, evidence suggests that BRM cells generated through mucosal routes may exhibit broader reactivity against heterologous viral strains compared to systemically induced memory B cells [4]. This cross-reactive potential, likely shaped by local germinal center reactions in mucosal tissues, represents a significant advantage against rapidly evolving respiratory pathogens.
Experimental Models and Methodologies
Key Experimental Models for BRM Research
Investigating BRM cells requires specialized experimental approaches capable of distinguishing tissue-resident from circulating populations and assessing their functional capabilities.
Table 3: Key Experimental Models for BRM Research
| Model Type | Methodology | Key Applications |
|---|---|---|
| Parabiosis | Surgical joining of two mice with shared circulation | Demonstration of non-recirculating nature |
| IV Antibody Labeling | Intravenous antibody administration prior to tissue collection | Distinguishing circulating versus tissue-resident cells |
| Adoptive Transfer | Transfer of labeled cells into recipient animals | Tracking migration and tissue integration |
| FTY720 Treatment | Sphingosine-1-phosphate receptor modulator blocking egress | Assessing independence from circulating precursors |
| Tissue Transplantation | Transfer of infected tissue to naïve recipients | Demonstrating autonomous local memory |
The parabiosis model has been instrumental in establishing the sessile nature of BRM cells. In this approach, two mice are surgically joined, developing a shared circulatory system over several weeks. While circulating lymphocytes reach equilibrium between partners, tissue-resident cells remain confined to their host of origin [4]. Research using this model has demonstrated that lung BRM cells do not equilibrate between parabionts, confirming their non-recirculating nature [4].
Intravenous antibody labeling provides a rapid method for distinguishing tissue-resident from circulating cells. Minutes before tissue collection, mice receive intravenous anti-CD45 or other lymphocyte surface antibodies, which label cells within the circulation but cannot penetrate tissues. During subsequent flow cytometry, unlabeled cells are identified as tissue-resident, while labeled cells are classified as circulating [4]. This technique has revealed that a significant proportion of memory B cells in previously infected lungs are protected from labeling, confirming their tissue-resident status.
For functional studies, adoptive transfer experiments have elucidated the requirements for BRM establishment. When memory B cells from systemically immunized donors are transferred to naïve recipients, they show limited capacity to establish resident populations in mucosal tissues unless the recipients have previously experienced local infection [5]. This highlights the importance of both cellular intrinsic programming and permissive tissue environments in BRM development.
Methodologies for Assessing BRM Localization and Function
Advanced imaging and analytical techniques have provided unprecedented insights into the spatial organization and functional dynamics of BRM populations.
Two-photon microscopy of lung tissues has revealed that BRM cells form a homogeneously distributed network throughout the parenchyma in close association with alveoli [5]. This strategic positioning enables comprehensive tissue surveillance. During rechallenge, BRM cells increase their migration speeds and accumulate within infected foci before differentiating into plasma cells directly at sites of viral replication [5].
For molecular characterization, single-cell RNA sequencing has identified distinct transcriptional profiles distinguishing lung BRM cells from their circulating counterparts or memory B cells in lung-draining lymph nodes [4]. These analyses have revealed upregulated expression of tissue retention genes and chemokine receptors guiding positioning within mucosal environments.
Functional assessments often employ secondary challenge models comparing viral loads and immune responses between animals with and without established BRM populations. Such experiments consistently demonstrate that BRM cells confer superior protection against respiratory pathogens compared to systemic memory alone [5] [4]. Protection correlates with rapid local antibody production and significantly reduced early viral replication.
Signaling Pathways and Molecular Regulation
Diagram 1: Molecular regulation of BRM differentiation and activation. BRM development depends on CD40-mediated germinal center responses following initial antigen recognition. During secondary challenge, chemokine signaling (CXCL9/10-CXCR3 axis) guides BRM migration to infection sites where they differentiate into antibody-producing plasma cells.
The establishment and function of BRM cells are regulated by coordinated molecular signals that program their tissue residency and rapid response capabilities. Key pathways include:
CD40-Mediated Germinal Center Formation: BRM cells originate from CD40-dependent germinal center responses within mucosal tissues [1]. This T cell-dependent pathway ensures the generation of high-affinity, class-switched memory B cells equipped for tissue residence.
Chemokine-Mediated Positioning and Recruitment: The CXCL9/CXCL10-CXCR3 axis plays a critical role in BRM mobilization during secondary challenge. Alveolar macrophages produce these chemokines in response to viral detection, creating gradients that guide CXCR3-expressing BRM cells to sites of infection [4]. This chemotactic response ensures rapid accumulation of BRM cells precisely where their effector functions are required.
Tissue Residency Program: The upregulation of CD69 promotes tissue retention by antagonizing sphingosine-1-phosphate receptor 1 (S1P1), a key mediator of lymphocyte egress from tissues [4]. Additional transcription factors, including BATF and T-bet, contribute to the tissue residency program and functional specialization of BRM populations.
Innate-Like Activation Pathways: BRM cells exhibit a low activation threshold, responding to innate stimuli even in the absence of cognate antigen. Experimental evidence demonstrates that virus-like particles containing ssRNA can trigger BRM cell differentiation through innate pattern recognition, suggesting T cell-independent activation potential under certain conditions [5].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for BRM Investigation
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Mouse Models | BAT reporter mice (BLIMP1mVenus AIDCre/+ Rosa26tdTomato), ChAT-GFP reporter mice, mb-1Cre+/−ChATfl/fl (ChatBKO) | Lineage tracing, cell fate mapping, conditional gene deletion |
| Antibodies for Flow Cytometry | Anti-CD45 (IV labeling), anti-CD69, anti-CXCR3, anti-CD19, anti-B220, anti-IgM, anti-IgD | Cell phenotyping, identification of tissue-resident populations |
| Cell Isolation Kits | Lung dissociation kits, magnetic bead-based B cell isolation | Preparation of single-cell suspensions from tissues |
| Cytokines/Chemokines | Recombinant CXCL9, CXCL10 | Chemotaxis assays, migration studies |
| Viral Strains | Influenza A/PR8, CFP-S-flu reporter strain | Infection models, tracking viral replication foci |
| Activation Reagents | LPS, anti-IgM, CD40L, TLR agonists (CL097, poly(I:C)) | B cell stimulation, in vitro differentiation assays |
The investigation of BRM cells relies on specialized reagents and model systems that enable precise identification, isolation, and functional characterization of these tissue-resident populations. Critical tools include:
Reporter Mouse Models: Genetically engineered mice expressing fluorescent proteins under the control of B cell-specific promoters permit real-time tracking of BRM differentiation and localization. The BAT reporter system (BLIMP1mVenus AIDCre/+ Rosa26tdTomato) has been particularly valuable for tracing memory B cell and plasma cell differentiation during influenza infection [5]. Similarly, ChAT-GFP reporter mice have identified acetylcholine-producing B cell subsets in the respiratory tract, revealing novel immunoregulatory functions [7].
Isolation and Phenotyping Reagents: Comprehensive BRM characterization requires antibodies against surface markers including CD19, CD69, CXCR3, and various immunoglobulin isotypes. Intravenous anti-CD45 antibody labeling is essential for distinguishing tissue-resident from circulating populations during flow cytometric analysis [4]. Specialized tissue dissociation protocols preserve cell surface markers while generating single-cell suspensions from complex organs like lungs.
Challenge Strains and Immunogens: Pathogen-specific BRM responses are often studied using influenza virus strains (e.g., A/PR8) or engineered reporter viruses (e.g., CFP-S-flu) that enable visualization of infection foci [5]. For vaccination studies, mucosal vaccine candidates employing viral vectors, outer membrane vesicles, or minicell technologies provide platforms for comparing mucosal versus systemic immunization strategies [6] [8].
The strategic localization of BRM cells within mucosal tissues and airways represents a critical determinant of protective immunity against respiratory pathogens. Evidence consistently demonstrates that mucosal vaccination or infection establishes resident populations that confer superior protection compared to systemically generated memory responses. The functional advantages of BRM cells include their strategic positioning at potential sites of infection, rapid response kinetics, and capacity for local antibody production directly at replication sites. Future vaccine development should prioritize strategies that effectively engage these tissue-resident mechanisms, particularly through mucosal immunization routes that recapitulate the natural signals guiding BRM development and maintenance. As our understanding of BRM biology continues to evolve, so too will opportunities to design next-generation vaccines that leverage these sentinel cells for enhanced protection against respiratory infections.
The generation of long-lasting, high-affinity antibody responses and memory B cells constitutes a cornerstone of adaptive immunity. This process, essential for effective vaccination and pathogen clearance, occurs primarily within specialized structures called germinal centers (GCs) and is fundamentally regulated by CD40-CD40 ligand (CD40L) interactions. This review compares the cellular and molecular mechanisms governing GC responses, detailing how CD40 signaling orchestrates B cell fate. Furthermore, we frame this molecular machinery within the modern paradigm of mucosal immunity, contrasting how systemic versus mucosal vaccination strategies engage these pathways to generate protective tissue-resident memory B cells ((B_{RM})).
The germinal center reaction represents a critical phase of the T cell-dependent humoral immune response. These transient structures form in secondary lymphoid organs and serve as central factories for the production of high-affinity antibodies, long-lived plasma cells, and memory B cells [9]. The GC response is absolutely dependent on costimulatory signals, among which the interaction between the CD40 receptor (on B cells) and its CD40 ligand (CD40L, primarily on T cells) is one of the best-characterized and most essential [10].
CD40 is a 48-kDa type I transmembrane protein belonging to the tumor necrosis factor receptor (TNFR) superfamily. Its ligand, CD40L (CD154), is a type II transmembrane protein expressed mainly on activated T cells. The engagement of CD40 by CD40L promotes the recruitment of adapter proteins known as TNFR-associated factors (TRAFs) to the cytoplasmic domain of CD40, initiating a complex signaling cascade that is indispensable for an effective adaptive immune response [10].
The Germinal Center: A Structured Microenvironment for B Cell Maturation
The germinal center is a highly organized and dynamic microenvironment, physically partitioned into two distinct zones that facilitate a cyclic process of mutation and selection.
Dark Zone: Proliferation and Somatic Hypermutation
In the dark zone (DZ), B cells, known as centroblasts, undergo rapid proliferation and somatic hypermutation (SHM) [9] [11]. SHM is a process mediated by the activation-induced cytidine deaminase (AID) enzyme, which introduces pseudo-random mutations into the variable regions of the antibody genes. This mutative process generates a diverse repertoire of B cell clones with varying affinities for the antigen [9].
Light Zone: Selection and Differentiation
Following mutation, B cells migrate to the light zone (LZ), where they are called centrocytes. Here, they encounter antigen presented as immune complexes by follicular dendritic cells (FDCs) and compete for survival signals from T follicular helper ((T{FH})) cells [9] [11]. Centrocytes that have gained B cell receptors (BCRs) with higher affinity for the antigen are more effective at internalizing and presenting it to (T{FH}) cells. This interaction, which includes CD40L on (T_{FH}) cells engaging CD40 on B cells, provides a critical survival signal. Positively selected B cells can then re-enter the DZ for further rounds of mutation, differentiate into antibody-secreting plasma cells, or become memory B cells [9].
Table 1: Key Characteristics of Germinal Center Zones
| Feature | Dark Zone (DZ) | Light Zone (LZ) |
|---|---|---|
| Primary B Cell Type | Centroblasts | Centrocytes |
| Proliferative Activity | High | Low |
| Key Processes | Somatic hypermutation, clonal expansion | Antigen presentation, positive selection, differentiation |
| Critical Interacting Cells | - | Follicular Dendritic Cells (FDCs), T Follicular Helper ((T_{FH})) Cells |
| Master Regulator | BCL6 | BCL6 |
The following diagram illustrates the cyclical process of germinal center B cell maturation:
CD40 Signaling: The Molecular Engine of the GC Reaction
CD40 engagement acts as a master regulator of the GC response, triggering multiple downstream signaling pathways that determine B cell fate.
TRAF-Dependent Signaling Pathways
Upon CD40L binding, CD40 clusters and recruits TNFR-associated factor (TRAF) proteins (TRAF1, 2, 3, 5, 6) to its cytoplasmic tail. This recruitment activates several key pathways [10]:
- Canonical NF-κB Pathway: Leads to the degradation of the IκB complex and translocation of NF-κB heterodimers (e.g., p50/p65) to the nucleus, promoting cell survival and proliferation.
- Non-Canonical NF-κB Pathway: Involves the processing of p100 to p52, which complexes with RelB and translocates to the nucleus to regulate specific target genes.
- MAPK Pathways: Including Jnk and p38, which are involved in cellular stress responses and differentiation.
TRAF-Independent and Other Signaling
CD40 can also signal through TRAF-independent mechanisms. For instance, it can bind directly to Janus family kinase 3 (Jak3), leading to the phosphorylation of signal transducer and activator of transcription 5 (STAT5) [10]. Furthermore, CD40 ligation on T cells can stimulate a signaling cascade involving p56lck, Rac1, and the activation of Jun-N-terminal kinase (JNK) and p38-K [12].
The complexity of CD40 signaling is depicted in the following pathway diagram:
Comparative Analysis: Mucosal vs. Systemic Vaccination
The route of vaccine administration significantly influences the engagement of GC and CD40-dependent pathways, ultimately shaping the quality, duration, and anatomical localization of protective immunity. This is particularly relevant for the generation of tissue-resident memory B cells ((B_{RM})), which are crucial for frontline defense at mucosal surfaces.
Table 2: Comparison of Mucosal vs. Systemic Vaccination Strategies
| Parameter | Systemic Vaccination (e.g., Intramuscular) | Mucosal Vaccination (e.g., Intranasal, Intravaginal) |
|---|---|---|
| Primary GC/Immune Induction Site | Draining lymph nodes (e.g., axillary, inguinal) | Mucosa-associated lymphoid tissue (MALT) and regional lymph nodes |
| Strength of Systemic Immunity | Strong systemic antibody and CD8 T cell responses [13] | Variable, often lower systemic antibody titers [13] |
| Strength of Mucosal Immunity | Weaker mucosal T and B cell responses [13] | Stronger local CD4 T cell central memory responses and tissue-resident memory [1] [13] |
| Generation of Mucosal (B_{RM}) | Limited | Robust, derived from CD40-dependent GC responses in mucosal sites [1] [14] |
| Protection against Mucosal Challenge | Partial control of viral load [13] [15] | Enhanced control; lower viral set points in some studies [13] |
| Practical Advantages | Standardized, well-established delivery | Non-invasive, potential for mass administration (e.g., in aquaculture [16]) |
Evidence from Preclinical Models
- Primate SHIV Model: A comparative study in rhesus macaques immunized with helper-dependent adenoviral (HD-Ad) vectors expressing HIV-1 envelope found that intramuscular (i.m.) immunization generated stronger systemic CD8 T cell responses in PBMCs, but weaker mucosal responses. Conversely, intravaginal (ivag.) mucosal immunization generated stronger CD4 T cell central memory responses in the colon. After rectal SHIV challenge, both groups controlled the virus better than controls, but a greater number of animals in the mucosally immunized group had lower viral set points, suggesting improved protection against sexually-transmitted virus [13].
- SIV Challenge Model: Another study comparing aerosol (AE) vaccination with intramuscular (IM) delivery found that both routes controlled acute viremia in an intravenous challenge model. However, after an intrarectal challenge, only peak viremia was reduced by immunization, with no significant effect on the acquisition rate. This underscores the difficulty in achieving sterile immunity against mucosal challenge but also highlights that aerosol delivery induced strong mucosal cellular responses and systemic humoral responses equivalent to the intramuscular route [15].
The Scientist's Toolkit: Key Research Reagents and Models
Table 3: Essential Research Reagents for Studying GC and CD40 Pathways
| Reagent / Model | Function / Application | Key Insight from Usage |
|---|---|---|
| Anti-CD40L Antibody | Blocks CD40-CD40L interaction in vivo | Abrogates GC formation, Ig isotype switching, and affinity maturation, confirming pathway necessity [10]. |
| CD40 or CD40L KO Mice | Genetic models to study loss of function | Demonstrate defective GC formation, humoral immunity, and failure to generate long-lived plasma cells and memory B cells [10]. |
| BCL6 Reporter Mice | Fate-mapping of GC B cells and (T_{FH}) cells | Identified BCL6 as the master transcriptional regulator of GC B and (T_{FH}) cell differentiation [9]. |
| c-Myc Reporter Mice | Tracking positively selected GC B cells | Revealed that CD40 signaling from (T_{FH}) cells induces c-Myc in LZ B cells in proportion to antigen amount, licensing them for DZ re-entry [11]. |
| Metabolic Inhibitors (e.g., 2-DG, DON) | Inhibit glycolysis (2-DG) and glutaminolysis (DON) | Uncovered distinct metabolic dependencies; glutamine is crucial for GC development, while autoreactive GC B cells may be uniquely glucose-dependent [11]. |
| Adenovirus Vectors (HD-Ad) | Vaccine vectors for immunization studies | Used in primate models to compare immunization routes; effective for serotype-switching to overcome pre-existing immunity [13]. |
Detailed Experimental Protocols
To provide context for the data discussed, here are detailed methodologies for key experiments cited.
This protocol outlines the method used to compare intramuscular (i.m.) and intravaginal (ivag.) vaccination.
- Priming: Rhesus macaques are first immunized intranasally with 1x10^11 virus particles (vp) of first-generation adenovirus serotype 5 (FG-Ad5) expressing a marker gene (e.g., β-galactosidase) to establish anti-vector immunity.
- Serotype-Switch Vaccination:
- Group 1 (Systemic): Immunized by i.m. injection with 1x10^11 vp of HD-Ad6 expressing the antigen of interest (e.g., HIV-1 Env gp140).
- Group 2 (Mucosal): Immunized by intravaginal (ivag.) lavage with the same dose of HD-Ad6-Env.
- Boosting: Both groups are boosted at 3-week intervals using the same routes but switching the adenovirus serotype (e.g., to HD-Ad1-Env, then HD-Ad5-Env, then HD-Ad2-Env) to evade neutralizing antibodies.
- Immune Monitoring:
- T cell responses: Measured by IFN-γ ELISPOT on PBMCs stimulated with peptide pools spanning the antigen.
- Mucosal T cells: Isolated from biopsies (e.g., colon) and analyzed by flow cytometry for central memory markers.
- Antibody responses: Quantified by ELISA for antigen-binding antibodies.
- Challenge: At week 32, animals are challenged rectally with a pathogenic virus (e.g., 1000 TCID50 of SHIV-SF162P3) and monitored for viral load (plasma viremia) and disease progression.
- Cell Isolation: Collect PBMCs from vaccinated subjects via density gradient centrifugation.
- Peptide Stimulation: Plate 1x10^5 PBMCs per well in an ELISPOT plate pre-coated with an IFN-γ capture antibody. Stimulate cells with 3 pools of 15-mer peptides (50-70 peptides per pool) overlapping the sequence of the vaccine antigen. Include negative (medium only) and positive (mitogen) control wells.
- Incubation: Incubate plates for 36 hours at 37°C, 5% CO2.
- Detection: After incubation, discard cells and add a biotinylated detection antibody against IFN-γ, followed by enzyme-conjugated streptavidin.
- Development: Add a precipitating substrate to develop spots. Each spot represents a single IFN-γ-secreting T cell.
- Analysis: Enumerate spot-forming units (SFU) using an automated ELISPOT reader. Data are expressed as SFU per 10^6 input cells after subtracting background from negative control wells.
The germinal center, powered by CD40 signaling, is the essential crucible for the birth of protective, high-affinity B cell responses. The CD40-CD40L axis is non-redundant in initiating the signaling cascades that drive B cell proliferation, SHM, and differentiation into plasma cells and memory B cells. The choice between systemic and mucosal vaccination represents a critical strategic decision. While systemic vaccination robustly engages GC pathways in draining lymph nodes, mucosal vaccination uniquely leverages these pathways within mucosal tissues to generate frontline defenders—tissue-resident memory B cells. A deep understanding of these intertwined cellular, molecular, and spatial relationships is fundamental for advancing novel vaccine strategies against pathogens that enter through mucosal surfaces.
In the defense against recurrent pathogens, the speed and location of the immune response are paramount. The "recall advantage" refers to the unique capacity of antigen-experienced memory B cells to rapidly differentiate into antibody-secreting cells (ASCs) upon reinfection, a cornerstone of protective immunity. This process is markedly different from the primary response, which requires days to weeks to generate specific antibodies, whereas memory responses can be mounted within hours to days. Within this paradigm, tissue-resident memory B cells (Brm) situated at mucosal surfaces have emerged as crucial frontline defenders, particularly against respiratory pathogens. Their strategic positioning at sites of pathogen entry enables immediate reaction, bypassing the time required for recruitment from circulation [17] [1]. Understanding the mechanisms governing this rapid differentiation is not merely an academic exercise but a critical endeavor for developing next-generation vaccines, especially mucosal vaccines that can establish these localized sentinels [6].
The broader thesis of modern vaccinology is increasingly shifting toward strategies that leverage these tissue-resident responses. While conventional systemic vaccination excels at generating circulating antibodies and protection against severe disease, it often fails to induce robust mucosal immunity, creating a potential gap in sterilizing immunity that prevents initial infection and transmission [6]. This review objectively compares the performance of mucosal versus systemic vaccination strategies in generating Brm and eliciting rapid ASC differentiation upon reinfection, providing experimental data and methodologies central to this evolving field.
Comparative Performance: Mucosal vs. Systemic Vaccination
The route of vaccine administration fundamentally shapes the quality, location, and kinetics of the resulting B cell memory. The table below synthesizes key performance characteristics based on current research.
Table 1: Comparison of Immune Responses Following Mucosal vs. Systemic Vaccination
| Parameter | Mucosal Vaccination (e.g., Intranasal) | Systemic Vaccination (e.g., Intramuscular) |
|---|---|---|
| Induction of Tissue-Resident Memory B Cells (Brm) | Directly induces Brm in respiratory mucosa [17] [1] | Fails to generate pulmonary Brm; memory B cells are primarily circulating [17] |
| Antibody Secreting Cell (ASC) Kinetics | Brm rapidly differentiate into ASCs upon challenge, providing local antibody [1] | Recall responses involve activation of circulating memory B cells, with a potential delay in local antibody production |
| Key Signaling for Brm Formation | CD40-dependent germinal center responses in local mucosal tissues [1] | Germinal center responses in systemic lymphoid organs (e.g., spleen, lymph nodes) |
| Local Secretory IgA (SIgA) | Robust induction of SIgA at the site of pathogen entry [6] | Ineffective induction of mucosal SIgA; primarily induces serum IgG [6] |
| Protection against Infection & Transmission | Potently blocks initial infection and reduces viral transmission [6] | Primarily protects against severe disease but may be less effective at blocking initial infection [6] |
Quantitative data further underscores this comparison. A study on SARS-CoV-2 breakthrough infections in vaccinated individuals revealed that Spike-specific CD4 T cells and plasmablasts expanded, and CD8 T cells were robustly activated during the first week of infection. In contrast, memory B cell activation and the production of potent neutralizing antibodies were features of the second week, highlighting the coordinated, multi-layered nature of the recall response [18]. Furthermore, intranasal vaccination in mice with a chimpanzee adenoviral-based SARS-CoV-2 vaccine induced CD103+CD69+CD8 T cells in the lungs, a hallmark of tissue-resident memory T cells (Trm), while intramuscular vaccination failed to do so [17]. This principle extends to B cells, where local antigen encounter is a known requirement for establishing a tissue-resident population [17].
Decoding the Experiments: Key Methodologies and Findings
The conclusions drawn in the previous section are supported by rigorous experimental models. Below is a summary of foundational protocols used to investigate Brm and recall responses.
Table 2: Key Experimental Models and Methodologies for Studying Brm and Recall Responses
| Experimental Approach | Key Methodology | Primary Readout / Insight |
|---|---|---|
| Parabiosis / Adoptive Transfer | Surgically connecting two mice or transferring cells from a donor to a recipient mouse, often followed by intravenous labeling to distinguish circulating vs. resident cells [17]. | Confirms the non-circulating, tissue-resident nature of Brm populations in mucosal tissues like the lung [17]. |
| In Vivo Challenge Models | Immunizing mice via different routes (e.g., intranasal vs. intramuscular) and later challenging with a pathogen to assess protection and immune kinetics [17]. | Demonstrates that mucosal vaccination leads to superior local protection and faster ASC responses upon rechallenge compared to systemic vaccination [17]. |
| Longitudinal BCR Repertoire Sequencing | Using high-throughput sequencing to track B cell receptor (BCR) clonotypes in memory B cells and ASCs over time in humans or model systems [19]. | Reveals clonal persistence and reactivation of memory B cells, showing they can undergo new rounds of affinity maturation when differentiating into ASCs upon antigen re-encounter [19]. |
| Single-Cell RNA Sequencing (scRNA-seq) | Profiling the transcriptome of individual B cells during activation and differentiation in vivo [20]. | Identified an early cell fate bifurcation upon B cell activation; ASC-destined cells downregulate CD62L and induce IRF4, MYC-target genes, and oxidative phosphorylation [20]. |
| Flow Cytometry / FACS Panels | Using multicolor antibody panels to identify and isolate specific B cell subsets (e.g., Brm: CD19+ CD69+; Plasmablasts: CD19low CD20- CD27high CD138-) [21] [19]. | Allows for phenotypic characterization and functional assessment of B cell populations, defining ASCs as CD27high CD38high among CD3- CD20- cells [21]. |
A critical study elucidating the requirements for ASC differentiation used an adoptive transfer model where labeled B cells were tracked in vivo. This research found that a minimum of eight cell divisions were required for ASC formation in response to T-cell independent antigens like LPS. The transcription factor BLIMP-1 was essential for this differentiation at division eight, underscoring the link between cellular proliferation and fate determination [20].
Molecular Mechanisms: From Memory B Cell to Antibody Factory
The rapid differentiation of Brm into ASCs is governed by a well-orchestrated molecular program. The following diagram illustrates the key signaling pathways and transcriptional regulators involved in this process.
Diagram Title: Signaling Pathway for Brm to ASC Differentiation
The journey begins when a tissue-resident memory B cell (Brm) re-encounters its cognate antigen. B cell receptor (BCR) engagement, coupled with CD40 signaling (recapitulating T cell help) and cytokine signals (such as IL-21), triggers a pivotal early event: the rapid upregulation of the transcription factor IRF4 [20]. IRF4 initiates a cascade of changes that commit the cell to the ASC fate, including the induction of MYC-target genes to sustain proliferation and a metabolic shift toward oxidative phosphorylation to meet energy demands [20]. A key downstream effector is BLIMP-1 (encoded by Prdm1), a master regulator that extinguishes the B cell gene expression program and drives the expression of XBP1, facilitating the expansion of the endoplasmic reticulum necessary for massive antibody production [20]. This pathway enables the swift conversion of a quiescent Brm into a dedicated antibody factory.
The Scientist's Toolkit: Essential Reagents for Brm and ASC Research
Progress in this field relies on a specific set of research tools and reagents that allow for the identification, isolation, and functional characterization of memory B cells and ASCs.
Table 3: Essential Research Reagent Solutions for Brm and ASC Studies
| Research Reagent / Tool | Primary Function | Key Application in the Field |
|---|---|---|
| Fluorescently-Labeled Antigen Probes | To identify antigen-specific B cells via flow cytometry. | Critical for tracking pathogen-specific B cell responses (e.g., using SARS-CoV-2 Spike probes) without relying on phenotypic markers alone [18]. |
| Multicolor Flow Cytometry Antibody Panels | To phenotype and isolate distinct B cell subsets based on surface and intracellular markers. | Panels including CD19, CD20, CD27, CD38, CD138, CD69, CD103 are used to define human ASCs (CD19low CD20- CD27high CD38high) and tissue-resident cells (CD69+) [21] [19]. |
| ELISpot/Fluorospot Assays | To enumerate and characterize functional antibody-secreting cells (ASCs) at a single-cell level. | Measures the prevalence and antigen-specificity of ASCs from tissues or blood; robust for quantifying recall responses [21]. |
| BCR Sequencing Kits | For high-throughput sequencing of B cell receptor repertoires. | Enables clonal tracking, analysis of somatic hypermutation, and understanding of B cell lineage relationships over time and between subsets [19]. |
| Recombinant Cytokines & Signaling Agonists/Antagonists | To modulate specific signaling pathways in vitro and in vivo. | Used to dissect the role of pathways like CD40 (agonistic antibodies), IL-21, and BCR signaling in Brm reactivation and ASC differentiation. |
The workflow for a typical deep immune profiling experiment might involve using fluorescent antigen probes and flow cytometry to sort specific B cell populations (e.g., Brm, plasmablasts), followed by single-cell RNA sequencing to understand their transcriptional state and BCR sequencing to track their clonal history [18] [21] [19]. This integrated approach provides an unprecedented view of the immune response at the clonal level.
The evidence unequivocally demonstrates that the "recall advantage"—the rapid differentiation of memory B cells into ASCs—is profoundly enhanced when the memory pool is strategically positioned at the site of potential infection. Tissue-resident memory B cells (Brm), induced most effectively by mucosal vaccination or infection, represent a key mediator of this frontline defense [17] [1]. Their ability to swiftly produce local antibody upon rechallenge provides a kinetic and spatial advantage that systemic memory responses cannot match.
The current data underscores a clear performance dichotomy: mucosal vaccines excel at inducing local immunity that can block infection and transmission, while systemic vaccines are highly effective at preventing severe disease but less so at providing sterilizing mucosal immunity [6]. This is not a failure of systemic vaccination but rather a reflection of compartmentalized immunity. The future of vaccine development, particularly against respiratory pathogens, therefore lies in the strategic combination of both approaches or the optimization of mucosal platforms to reliably generate robust and durable Brm populations. Key challenges remain, including the identification of definitive correlates of protection for mucosal immunity and the development of safe and effective adjuvants for human mucosal vaccines [17] [6]. As these hurdles are overcome, leveraging the full potential of the recall advantage offered by tissue-resident memory B cells will be fundamental to achieving next-generation protective immunity.
From Bench to Barrier: Techniques and Strategies for Inducing and Analyzing BRM Responses
While peripheral blood is the most common source for human immunology studies, a growing body of evidence reveals that B cells in tissues are not in homeostasis with circulatory compartments. Advanced profiling technologies now enable direct investigation of tissue-localized B cell repertoires, revealing specialized tissue-resident memory B cells (Brm) with unique phenotypic signatures and functional capabilities. This guide compares methodological approaches for tissue-based B cell repertoire analysis and synthesizes recent findings on tissue-specific B cell compartments, providing researchers with frameworks for selecting appropriate profiling strategies based on research objectives and tissue accessibility.
The conventional reliance on peripheral blood for B cell analysis presents a fundamental limitation in understanding adaptive immunity, as blood B cells are not in homeostasis with tissue compartments [22]. Studies utilizing tissues from organ donors have demonstrated striking differences in B cell subset distribution, isotype usage, and functional capabilities between mucosal tissues, lymphoid organs, and circulatory compartments [22] [23]. This compartmentalization is particularly relevant in vaccine research, where successful mucosal vaccination strategies must induce not only systemic immunity but also tissue-localized Brm cells that provide frontline defense at pathogen entry sites [17] [1].
The development of tissue-resident memory B cells represents a critical adaptation for localized protection. These non-circulating cells persist in tissues long after antigen clearance and mount rapid, robust responses upon re-exposure to pathogens [17]. Their induction requires local antigen encounter and is influenced by vaccination route, with mucosal vaccination strategies demonstrating superior ability to generate these tissue-localized protective populations [17] [6].
Methodological Approaches for Tissue-Based B Cell Repertoire Analysis
Comparative Analysis of Profiling Technologies
| Technology | Key Applications | Sample Requirements | Output Data | Limitations |
|---|---|---|---|---|
| BCR-Seq (Bulk) | Repertoire diversity, clonality, V(D)J usage [24] | Tissue DNA/RNA (120ng for library prep) [24] | Clonotype frequencies, V/D/J gene usage, SHM analysis [24] | Loses cellular resolution, cannot pair heavy/light chains |
| CITE-seq (Single-Cell) | Multimodal analysis of phenotype + BCR [23] | Viable single-cell suspensions from tissues [23] | Paired BCR sequences + surface protein expression (127+ parameters) [23] | Higher cost, complex computational analysis |
| Multicolor Flow Cytometry | B cell subset identification and sorting [22] | Fresh or cryopreserved tissue single-cell suspensions [22] | Surface marker expression (CD19, CD27, CD45RB, CD69, etc.) [22] | Limited to pre-defined marker panels, no sequence information |
| ViCloD Web Server | Clonality and intraclonal diversity analysis [25] | Preprocessed AIRR-seq data (e.g., IMGT/HighV-QUEST) [25] | Clonal abundance, lineage trees, SHM patterns [25] | Dependent on quality of input sequencing data |
Experimental Workflow for Tissue B Cell Repertoire Profiling
The following diagram illustrates a comprehensive workflow for profiling tissue-localized B cell repertoires, integrating both phenotypic and sequencing approaches:
The Scientist's Toolkit: Essential Research Reagents
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Tissue Processing | Collagenase/DNase cocktails, mechanical dissociation systems | Isolation of viable lymphocytes from solid tissues |
| Cell Staining Panels | CD19, CD27, CD45RB, CD69, CD38, IgD, IgM, IgA [22] | Identification of B cell subsets and tissue-resident populations |
| Library Prep Kits | 7genes DNA multiplexing kits (MiLaboratories) [24] | Simultaneous profiling of T and B cell repertoires from same sample |
| Sequencing Platforms | Illumina NovaSeq 6000 (150bp paired-end) [24] | High-throughput immune repertoire sequencing |
| Analysis Tools | MiXCR, ViCloD, Immcantation framework [24] [25] | Clonotype identification, lineage tree reconstruction, diversity analysis |
Tissue-Specific B Cell Signatures: Key Findings from Multi-Tissue Studies
Comparative Distribution of B Cell Subsets Across Tissues
Comprehensive analysis of B lineage cells across multiple tissues from the same donors has revealed striking tissue-specific distributions that are not reflected in peripheral blood [22] [23]. The table below summarizes key quantitative differences in B cell subsets across major tissue compartments:
| Tissue Site | CD27+ Memory B Cells | Naive B Cells | Plasma Cells/Plasmablasts | Tissue-Resident (CD69+) B Cells | Dominant Isotype |
|---|---|---|---|---|---|
| Peripheral Blood | 20.37% [22] | 79.63% [22] | 0.24% [22] | Absent/Low [22] | IgG [23] |
| Spleen | 70.39% [22] | 29.61% [22] | 1.00% [22] | Moderate [22] | IgG [23] |
| Gut (Jejunum) | 86.44% [22] | 13.56% [22] | 1.92% [22] | High (CD69+CD45RB+) [22] | IgA [23] |
| Bone Marrow | 46.00% [22] | 54.00% [22] | 0.51% [22] | Low [22] | IgG [23] |
| Lymph Nodes | Variable (LN-dependent) [23] | Variable [23] | 0.42% [22] | Moderate (CD69+) [23] | IgG [23] |
Identification and Characterization of Tissue-Resident Memory B Cells
Tissue-resident memory B cells (Brm) represent a distinct B cell subset characterized by their non-circulating nature and tissue-localized persistence. The following diagram illustrates the phenotypic markers and differentiation pathways of Brm cells:
The generation of Brm cells occurs through both germinal center (GC)-dependent and GC-independent pathways [26]. GC-independent Brm development is driven by CD40 signaling and occurs with relatively brief T follicular helper (Tfh) cell contact, while GC-dependent development involves sustained Tfh help and cytokine signaling (particularly IL-21), which upregulates BCL-6 and promotes GC formation [26]. These developmental pathways converge on the establishment of CD69+ Brm populations that persist in tissues.
Implications for Mucosal Versus Systemic Vaccination Research
Differential Immune Responses by Vaccination Route
The route of vaccine administration dramatically influences the generation of tissue-localized B cell memory. Studies comparing mucosal versus parenteral vaccination have revealed fundamental differences in the quality, location, and durability of B cell responses [17] [6]. The table below compares key features of B cell responses induced by different vaccination routes:
| Vaccination Parameter | Mucosal Vaccination | Systemic Vaccination |
|---|---|---|
| Tissue Brm Induction | Robust generation of Brm at site of administration [17] [1] | Limited tissue Brm generation [17] |
| Secretory Antibody Production | Induces secretory IgA (SIgA) at mucosal surfaces [6] | Minimal SIgA production [6] |
| Systemic Antibody Response | Variable, often lower systemic IgG [6] | Strong systemic IgG response [6] |
| Germinal Center Reactions | Local GC formation in mucosal tissues [6] | Primarily systemic GC in spleen/LNs [6] |
| Protection Against Infection | Blocks initial infection/colonization [6] | Reduces severe disease but limited blocking of initial infection [6] |
| Durability of Response | May wane faster for mucosal IgA [6] | Often longer-lasting systemic immunity [6] |
Mucosal Vaccination Strategies for Optimal Brm Induction
Successful mucosal vaccine strategies leverage specific mechanisms to induce tissue-resident memory. Intranasal administration of live-attenuated influenza virus induces development of CD4+ and CD8+ Trm cells in lungs, whereas systemic immunization fails to generate similar Trm responses [17]. The "prime and pull" strategy, involving systemic priming followed by local mucosal recruitment through chemokine application, has proven effective for generating tissue-resident memory in vaginal tract and respiratory mucosa [17].
For Brm induction, local antigen encounter is essential [17]. This requirement makes mucosal vaccination particularly advantageous, as it ensures direct antigen presentation to tissue-localized B cells. The local microenvironment, including cytokine signals from innate lymphoid cells and stromal elements, further supports Brm differentiation and maintenance [6].
Technical Considerations and Protocol Details
Detailed Methodology for Simultaneous T and B Cell Repertoire Profiling
The protocol below, adapted from PMC12447526 [24], enables comprehensive profiling of adaptive immune repertoires from limited tissue samples:
Sample Preparation:
- Collect tissues (e.g., colon biopsies) during diagnostic procedures and immerse in Allprotect reagent overnight at 2-8°C before freezing at -80°C
- Extract DNA using automated systems (e.g., Chemagic STAR or QIAcube Connect with DNeasy Blood and Tissue Kit)
- Perform quality control using Agilent TapeStation Systems (acceptable DIN score: 6.5-9.7)
- Quantify DNA using fluorometric methods (e.g., Qubit dsDNA-BR assay)
Library Preparation and Sequencing:
- Use 120ng of DNA as input for library preparation with 7genes DNA multiplexing kits (MiLaboratories)
- Follow manufacturer's instructions for simultaneous T and B cell repertoire amplification
- Pool libraries and clean with magnetic beads (AMPure XP Beads, 1:0.8 sample:beads ratio)
- Dilute to ~2nM final concentration and sequence on Illumina NovaSeq 6000 using paired 150bp x 2 approach
- Include ~10% PhiX control to improve base calling accuracy
Data Processing and Analysis:
- Process raw sequencing data using MiXCR with "milab-human-dna-xcr-7genes-multiplex" preset
- Expect approximately 45-48% of reads to align successfully to immune receptor loci
- Typical distribution of aligned reads: TRA (~10.9%), TRB (~3.43%), TRG (~52.2%), TRD (~11.55%), IGH (~4.93%), IGK (~5.37%), IGL (~2.28%)
- Remove samples with initial low-quality input DNA from downstream analysis
Analysis of Intracional Diversity and Lineage Relationships
Tools like ViCloD provide specialized functionality for analyzing B cell intraclonal diversity and evolutionary relationships [25]. The web server:
- Accepts preprocessed data in Adaptive Immune Receptor Repertoire (AIRR) Community format
- Performs clonal grouping based on shared IGHV, IGHD, IGHJ genes and identical CDR3 amino acid sequences
- Reconstructs intraclonal evolutionary trees to visualize somatic hypermutation patterns
- Enables navigation of repertoire clonality and identification of expanded clones
- Generates downloadable tables and publication-quality plots of lineage relationships
Advanced profiling of tissue-localized B cell repertoires has fundamentally transformed our understanding of adaptive immunity, revealing specialized tissue-resident populations that are not reflected in circulatory compartments. The methodologies and findings summarized in this guide provide researchers with frameworks for investigating these localized immune responses, with significant implications for vaccine development, particularly in the context of mucosal protection. As tissue-based immunology continues to advance, integrating multimodal single-cell technologies with functional assays will further elucidate the unique biology of tissue-resident B cells and their role in protective immunity.
Respiratory infections remain a major global public health threat, as demonstrated by the COVID-19 pandemic [27]. While traditional intramuscular vaccines have proven effective at reducing severe disease and hospitalization, they primarily elicit systemic IgG responses and are limited in their ability to induce robust immunity at mucosal surfaces—the primary site of pathogen entry for many viruses [27] [28]. This immunological gap has accelerated development of mucosal vaccine platforms designed to stimulate localized responses at pathogen entry points. Mucosal vaccines, administered via intranasal, oral, or sublingual routes, aim to induce secretory IgA (SIgA) production and establish tissue-resident memory T cells (TRM) and B cells within mucosal tissues, providing a critical first line of defense against infection and potentially reducing viral transmission [27] [28].
The fundamental advantage of mucosal vaccination lies in its capacity to establish protective immunity at the site of pathogen invasion. Unlike intramuscular vaccines, which rely on circulatory antibodies and cells migrating to infection sites, mucosal vaccines stimulate the common mucosal immune system, enabling activated immune cells to home to distant mucosal tissues [27]. Intranasal vaccination, in particular, induces the formation of CD69⁺CD103⁺ TRM cells, germinal center reactions, and memory B cells that persist locally in the respiratory tract for at least six months, playing a pivotal role in limiting viral replication and transmission [27]. This review comprehensively compares three prominent mucosal delivery platforms—intranasal, oral, and sublingual—focusing on their immunological mechanisms, current developmental status, and relative advantages and challenges within the context of establishing protective tissue-resident memory responses.
Comparative Analysis of Mucosal Vaccine Platforms
The table below summarizes the key characteristics, advantages, and challenges of the three primary mucosal vaccine delivery platforms.
Table 1: Comparison of Mucosal Vaccine Delivery Platforms
| Feature | Intranasal | Oral | Sublingual |
|---|---|---|---|
| Immune Induction Site | Nasal-associated lymphoid tissue (NALT) [27] | Gut-associated lymphoid tissue (GALT) [29] | Mucosal-associated lymphoid tissue [30] |
| Primary Antibody Response | Secretory IgA (SIgA) in upper and lower respiratory tract [27] [28] | Secretory IgA (SIgA) in gut mucosa and other mucosal sites [29] | Secretory IgA (SIgA) in respiratory and other mucosal tissues [30] |
| Cellular Immunity | Strong tissue-resident memory T cell (TRM) formation in respiratory tract [27] | Systemic and mucosal T cell responses [29] | Mucosal and systemic T cell responses [30] |
| Key Advantages | Directly targets respiratory pathogen entry point; potential to block transmission [28] [31] | Non-invasive, high patient compliance, ease of distribution [29] | Avoids gastric degradation; favorable safety profile vs. intranasal [30] |
| Major Challenges | Potential safety concerns regarding brain access via olfactory bulb; mucosal barriers [28] [30] | Degradation in GI tract; variable efficacy influenced by gut microbiota [29] [32] | Mucin barrier and saliva dilution require formulation solutions [30] |
| Authorized Examples | FluMist (USA), COVID-19 vaccines in China, India, Iran, Russia [33] [28] | Oral polio, cholera, rotavirus, and typhoid vaccines [29] | None widely authorized to date (still in development) [30] |
| Clinical Trial Progress | 36 mucosal COVID-19 vaccines in clinical trials as of late 2025 [34] | Vaxart's oral COVID-19 vaccine in Project NextGen-funded trial [33] | Primarily in preclinical and early clinical development stages [30] |
Immunological Mechanisms and Key Research Methodologies
Immune Mechanisms of Mucosal Vaccination
The following diagram illustrates the general immunological mechanisms shared by mucosal vaccines, which lead to the generation of tissue-resident memory and secretory IgA responses.
The diagram above outlines the core adaptive immune mechanism leading to protective mucosal immunity. Upon administration, mucosal vaccines are sampled by specialized microfold (M) cells that transport antigens across the epithelial barrier to underlying antigen-presenting cells, particularly dendritic cells (DCs) [27] [29]. These DCs then migrate to mucosal-associated lymphoid tissues (MALT) such as nasal-associated lymphoid tissue (NALT) for intranasal vaccines or gut-associated lymphoid tissue (GALT) for oral vaccines, where they present antigens to naïve T cells [27] [29].
Activated T follicular helper (Tfh) cells are crucial for supporting B cell maturation and antibody class switching in germinal centers [27]. This process leads to the generation of plasma cells that produce dimeric IgA, which is transported across the epithelium as secretory IgA (SIgA) by the polymeric immunoglobulin receptor (pIgR) [27]. SIgA acts to neutralize and clear pathogens directly at the mucosal surface. Concurrently, antigen-specific tissue-resident memory T cells (TRM), characterized by surface expression of CD69 and CD103, are established in the mucosal tissue [27]. These non-circulating cells provide rapid, localized protection upon subsequent pathogen exposure and are a key correlate of protection for mucosal vaccines [27].
Experimental Workflow for Mucosal Vaccine Evaluation
The diagram below illustrates a standard experimental workflow used in preclinical studies to evaluate the immunogenicity and protective efficacy of mucosal vaccine candidates.
The experimental workflow for evaluating mucosal vaccines involves multiple coordinated steps. Following vaccine administration to animal models (typically mice or non-human primates), researchers collect mucosal samples such as nasal washes, bronchoalveolar lavage (BAL) fluid, and saliva, alongside systemic samples like blood and spleen tissue [27] [30]. These samples are analyzed for antigen-specific SIgA and IgG antibodies using ELISA and neutralization assays, while flow cytometry characterizes cellular immune populations, particularly TRM cells (CD69⁺CD103⁺) and memory B cells [27].
A critical component of mucosal vaccine assessment involves live pathogen challenge studies. Vaccinated and control animals are exposed to the target pathogen (e.g., SARS-CoV-2), and researchers monitor viral loads in respiratory tissues, clinical signs of disease, and tissue histopathology [31]. Some studies also incorporate transmission models where vaccinated animals are co-housed with naïve counterparts to assess the vaccine's ability to prevent viral spread [33] [31]. Additionally, safety evaluations include monitoring inflammatory gene expression in tissues like the olfactory bulb and lungs using RT-qPCR, especially for intranasal vaccines [30].
Research Reagent Solutions and Essential Materials
Table 2: Key Reagents and Materials for Mucosal Vaccine Research
| Reagent/Material | Function/Application | Example Usage |
|---|---|---|
| Poly(I:C) HMW | TLR3 agonist adjuvant that stimulates proinflammatory cytokine production and enhances vaccine immunogenicity [30]. | Used in sublingual and intranasal vaccine formulations to boost immune responses [30]. |
| N-acetyl cysteine (NAC) | Mucolytic agent that disrupts the mucin barrier, improving antigen access to underlying immune cells [30]. | Pre-treatment of sublingual mucosa before vaccine application to enhance delivery [30]. |
| Recombinant Trimeric Proteins | Stabilized antigen forms that mimic native viral structures, improving antibody recognition and immunogenicity [31]. | Key component in two-component intranasal vaccines (e.g., RBDXBB.1.5-HR) [31]. |
| Adenovirus Vectors | Vaccine delivery platform that also functions as a natural adjuvant by activating STING and TLR pathways [31]. | Used in intranasal vaccines (e.g., Ad5XBB.1.5) to deliver antigen genes and enhance immunity [31]. |
| Fluorescence-Labeled Antibodies | Cell population characterization via flow cytometry to identify TRM (CD69, CD103) and other immune cells [27]. | Phenotyping tissue-resident memory T cells in mucosal tissues and BAL fluid [27]. |
| ELISA Kits (SIgA, IgG) | Quantification of antibody responses in mucosal secretions and serum to assess immunogenicity [27] [29]. | Measuring antigen-specific SIgA in nasal washes and saliva post-vaccination [27]. |
Current Status and Future Directions in Mucosal Vaccine Development
Clinical Development Progress
The mucosal vaccine field has expanded rapidly, with 36 mucosal COVID-19 vaccines having reached clinical trials as of late 2025 [34]. Currently, five mucosal COVID-19 vaccines are authorized for use in at least six countries, though none have yet received WHO approval for global use [33]. The most advanced platforms include viral-vectored (adenovirus, Newcastle disease virus), live-attenuated, and protein-subunit formulations [33] [31].
Recent clinical advances include the progression of Vaxart's oral vaccine into a Project NextGen-funded 10,000-participant trial in the US and the initiation of a phase 2a trial for Castlevax's intranasal Newcastle virus-based vaccine in high-risk individuals [33] [34]. Additionally, a novel two-component intranasal vaccine combining an adenovirus vector (Ad5XBB.1.5) with a trimeric protein (RBDXBB.1.5-HR) has demonstrated superior immunogenicity and protective efficacy in preclinical models and early human trials [31].
Key Research Findings and Experimental Data
Recent studies have provided compelling evidence for the potential of mucosal vaccines. The following table summarizes quantitative results from key preclinical and clinical studies, highlighting the immune responses and protective efficacy elicited by different mucosal vaccine platforms.
Table 3: Experimental Data from Mucosal Vaccine Studies
| Vaccine Platform | Immune Response Findings | Protection/Transmission Results | Reference |
|---|---|---|---|
| Two-Component Intranasal (Ad5XBB.1.5 + RBDXBB.1.5-HR) | Superior humoral and cellular immunity vs. single components; induced high levels of neutralizing antibodies in all human participants [31]. | Protected against live XBB.1.16 virus in mice; prevented XBB.1.5 virus transmission in hamster model [31]. | [31] |
| Intranasal DNA Vaccine (GLS-5310) | Phase 1 trial: Reduced rate of COVID-19 after nasal booster (1/17 infected vs. 16/53 in control groups) [33]. | Preclinical study in rabbits showed increased mucosal immunity after intranasal vaccination [33]. | [33] |
| Sublingual vs. Intranasal (Poly(I:C)-adjuvanted influenza) | No adverse effects on olfactory bulb or pons in mice/macaques with sublingual route [30]. | Intranasal vaccination upregulated inflammatory genes (Saa3, Tnf, IL6) in olfactory bulb 1 & 7 days post-vaccination [30]. | [30] |
| Oral Vaccine (Convidecia Air platform) | In mice: Intramuscular version immunity waned, but oral version effects were more durable up to 250 days [34]. | Both versions induced similar peak immune responses [34]. | [34] |
Future Research Directions
Future development of mucosal vaccines faces several key challenges and opportunities. Safety optimization remains paramount, particularly for intranasal vaccines where potential neurological effects must be carefully evaluated [30]. The promising safety profile of sublingual administration suggests it may offer a favorable alternative, though technical challenges like saliva dilution and the mucin barrier require innovative formulation solutions [30].
Durability of protection represents another critical research frontier. While mucosal vaccines can induce TRM cells that persist for at least six months [27], the factors governing their long-term maintenance are not fully understood. Additionally, the impact of prior immunity on mucosal vaccine performance is significant, with studies showing stronger nasal IgA and IgG responses in previously exposed individuals compared to naïve subjects [27].
The emerging strategy of heterologous prime-boost regimens—combining intramuscular priming with mucosal boosting—shows particular promise for enhancing mucosal immunity in previously vaccinated populations [28]. Furthermore, antigen design continues to evolve, with multivalent approaches (e.g., three-component vaccines incorporating antigens from multiple variants) demonstrating enhanced breadth of protection against diverse viral variants [31].
As the field advances, validating reliable correlates of protection for mucosal vaccines will be essential for accelerating their clinical development and regulatory approval [27]. While systemic neutralizing antibodies serve as established correlates for intramuscular vaccines, SIgA levels in upper airways and TRM cells in respiratory tissues are emerging as key potential correlates for mucosal protection [27].
The pursuit of next-generation vaccines has increasingly focused on the mucosal immune system, which serves as the body's first line of defense at barrier surfaces such as the respiratory and gastrointestinal tracts. Comprising a surface area approximately 200 times greater than the skin, mucosal tissues represent a critical interface where most pathogens initially encounter host immunity [35]. While traditional intramuscular vaccines effectively induce systemic IgG responses, they often fail to elicit robust protection at mucosal entry points, limiting their ability to prevent initial infection and transmission [6]. This recognition has catalyzed the development of mucosal vaccination strategies designed to induce localized immune responses, including the production of secretory IgA (SIgA) and the establishment of tissue-resident memory lymphocytes [35] [6].
A particularly promising target for mucosal vaccines is the induction of tissue-resident memory B cells (BRM), which have recently emerged as crucial mediators of rapid, localized protection against respiratory pathogens [1]. These non-circulating cells persist in mucosal tissues after infection or vaccination and can quickly differentiate into antibody-secreting plasma cells upon re-exposure to pathogens [36]. BRM cells localize strategically within mucosal tissues—such as the lung alveoli—where they display confined probing behavior under steady-state conditions but rapidly increase their motility and accumulate at infected sites within 24 hours of challenge [36]. This coordinated response, orchestrated in part by alveolar macrophages through the induction of IFNγ and CXCR3 ligands, enables highly localized antibody production directly at sites of viral replication [36].
The strategic induction of BRM cells through vaccination represents a paradigm shift in prophylactic strategy, moving beyond mere disease mitigation toward preventing initial infection and interrupting community transmission. This comparison guide examines how novel adjuvants and delivery systems can enhance mucosal immunogenicity and BRM recruitment, providing researchers with objective experimental data and methodological frameworks to advance this promising field.
Comparative Analysis of Mucosal Adjuvant Platforms and Delivery Systems
Table 1: Comparison of Novel Adjuvant and Delivery System Platforms for Mucosal Vaccines
| Platform Category | Key Formulations/Examples | Proposed Mechanism of Action | Impact on Mucosal Immunity | BRM Recruitment Evidence |
|---|---|---|---|---|
| Lipid-Based Nanoparticles | Lipid Nanoparticles (LNPs) [37] | Enhance antigen stability and cellular uptake; can act as immune-activating adjuvants [37]. | Improved antigen presentation to mucosal dendritic cells [37]. | Emerging data for mRNA-LNP vaccines; potential to induce tissue-residency signatures [38]. |
| Viral Vectors | Adenoviral vectors [35] [6] | Efficient delivery of genetic material; inherent adjuvant properties through viral recognition [35]. | Strong T and B cell responses in mucosal tissues; induction of SIgA [6]. | Intranasal administration shown to induce CD103+CD69+CD8+ T cells in lungs [38]. |
| Emulsion Systems | Not specified in results | Activation of innate immune pathways; enhanced antigen persistence at mucosal sites. | Induction of local inflammatory responses that recruit immune cells. | Potential via local inflammation and chemokine production [36]. |
| Biomimetic Systems | Bacterial Outer Membrane Vesicles (OMVs), Cell membrane-coated systems [37] | Natural delivery vehicles with inherent adjuvant properties; target specific tissues [37]. | Activation of mucosal innate immunity; enhanced antigen sampling. | Theoretical potential via tissue-targeted delivery; limited direct evidence. |
| Programmable Nanoparticles | pH-, enzyme-, or light-responsive systems [37] | Controlled release of antigens and adjuvants in response to specific biological triggers [37]. | Precise temporal and spatial activation of immune responses in mucosal tissues. | Potential for controlled induction of tissue-residency signals. |
Table 2: Comparative Efficacy of Vaccination Routes in Inducing Mucosal Immunity
| Vaccination Route | Tissue-Resident Memory T Cell (Trm) Induction | Tissue-Resident Memory B Cell (BRM) Induction | Secretory IgA Production | Key Supporting Evidence |
|---|---|---|---|---|
| Intranasal | Strong induction of CD4+ and CD8+ Trm in lungs [38] | Development of lung BRM post-influenza infection [36] | Robust SIgA responses in upper and lower respiratory tract [6] | Intranasal adenoviral-based SARS-CoV-2 vaccine induced pulmonary Trm; intramuscular did not [38] |
| Intramuscular | Limited pulmonary Trm induction [38] | Limited evidence for mucosal BRM induction | Minimal SIgA production [6] | Current COVID-19 mRNA vaccines induce limited polyfunctional Trm in lungs compared to infection [38] |
| Prime and Pull | Establishment of Trm at targeted mucosal sites [38] | Not specifically documented | Not specifically documented | Subcutaneous prime with vaginal chemokine pull generated genital Trm [38] |
| Oral | Induction of gut-homing lymphocytes [35] | Evidence for intestinal BRM subsets [38] | Robust gut SIgA responses [35] | Nine FDA-approved oral mucosal vaccines demonstrate clinical efficacy [35] |
Mucosal Immune Mechanisms and BRM Biology
The Mucosal Immune Landscape
The respiratory mucosal immune system constitutes a sophisticated defensive network incorporating physical, chemical, and immunological barriers. Structural components include epithelial cells interconnected by tight junctions and covered by a mucus layer secreted by goblet cells [6]. This mucus, consisting of mucins, cytokines, antimicrobial peptides, and SIgA, serves as both a physical trap for pathogens and a biochemical filter that can modulate viral entry [6]. Beneath this structural barrier resides a coordinated immune network featuring specialized antigen-sampling mechanisms, including microfold (M) cells that transport antigens to underlying immune cells and dendritic cells that extend dendrites across the epithelial layer to capture antigens directly from the lumen [6].
Upon antigen encounter, dendritic cells migrate to draining lymph nodes where they present antigens to naïve T cells, initiating adaptive immune responses [6]. In mucosal-associated lymphoid tissues (MALT), such as inducible bronchus-associated lymphoid tissue (iBALT), T follicular helper (Tfh) cells support B cell maturation through germinal center reactions where somatic hypermutation and class-switch recombination occur [6]. This process generates plasma cells that produce dimeric IgA (dIgA), which is transported across epithelial cells by the polymeric immunoglobulin receptor (pIgR) to form SIgA at the mucosal surface [6]. SIgA plays a crucial role in neutralization of pathogens at their entry points and prevents their adhesion and dissemination [6].
Tissue-Resident Memory B Cell Development and Function
Tissue-resident memory B cells (BRM) represent a recently characterized subset of memory B cells that persist in mucosal tissues following infection or vaccination. While our understanding of BRM lags behind that of tissue-resident memory T cells (Trm), emerging evidence indicates that BRM development requires local antigen encounter and contributes substantially to early plasmablast responses against viral and bacterial pathogens [38]. These cells express characteristic tissue-residency markers including CD69, with some subsets also expressing CXCR3 and CD44 [38].
The functional capacity of BRM cells has been illuminated through sophisticated intravital imaging studies. In influenza-infected mice, BRM cells reside sparsely throughout lung tissue under steady-state conditions, displaying limited motility [36]. Within 24 hours of rechallenge, these cells significantly increase their migratory capacity, actively localize to infected foci, and subsequently differentiate into antibody-secreting plasma cells [36]. This rapid relocation is orchestrated by alveolar macrophages, which induce expression of chemokines CXCL9 and CXCL10 from infiltrating inflammatory cells, thereby recruiting CXCR3-expressing BRM cells to sites of viral replication [36]. This highly coordinated process results in increased local antibody concentrations precisely where needed to combat infection.
Figure 1: Signaling Pathway for Mucosal Vaccine-Induced BRM Formation and Recall Response. This diagram illustrates the sequential immunological events from intranasal vaccination through the rapid recall response mediated by tissue-resident memory B cells upon pathogen challenge.
Experimental Approaches and Methodologies
Key Experimental Models and Assessment Techniques
Table 3: Essential Methodologies for Evaluating Mucosal Immunity and BRM Responses
| Methodology | Key Applications | Technical Considerations | Representative Findings |
|---|---|---|---|
| Intravital Two-Photon Microscopy | Visualization of BRM cell dynamics in living tissue [36] | Requires reporter mice or labeled cells; technically challenging | BRM cells increase motility and accumulate at infected sites within 24h of rechallenge [36] |
| Parabiosis Models | Distinguishing tissue-resident from circulating lymphocytes [38] | Surgical joining of circulatory systems of two mice | Confirmed tissue-resident nature of memory T and B cell subsets [38] |
| Flow Cytometry with Intravenous Staining | Identifying tissue-resident vs. circulating cells [38] | IV administration of antibody before sacrifice labels circulating but not resident cells | Identification of CD69+ BRM cells in lung tissue [38] |
| ELISpot / Intracellular Cytokine Staining | Quantifying antigen-specific T cell responses [39] | Requires antigen stimulation; measures cytokine production | mRNA vaccination induces polyfunctional CD4+ and CD8+ T cells [39] |
| Bronchoalveolar Lavage (BAL) Analysis | Sampling airway immune components [6] | Human clinical application feasible; measures local antibodies and cells | Identification of tissue-resident memory cells in lower respiratory tract [6] |
Detailed Experimental Protocol: Evaluating BRM Recruitment
For researchers seeking to evaluate adjuvant strategies for BRM recruitment, the following integrated methodology synthesizes approaches from key studies:
Step 1: Immunization and Challenge Model
- Utilize C57BL/6 mice (8-12 weeks old) anesthetized with ketamine/xylazine
- Administer test vaccine formulations via intranasal route (50μL volume) containing candidate antigen plus experimental adjuvant/delivery system
- Include control groups receiving: (1) antigen alone, (2) established mucosal adjuvant, (3) intramuscular delivery
- Challenge with pathogen (e.g., influenza A/PR8/34 strain) at minimum 4 weeks post-immunization
Step 2: Tissue-Resident Cell Identification
- 5 minutes prior to sacrifice, administer 3μg anti-CD45 antibody intravenously to label circulating lymphocytes
- Harvest lung tissue and process to single-cell suspension using collagenase/DNase digestion
- Stain cells with fluorochrome-conjugated antibodies against: CD19, CD69, CD45, CD3, B220, CXCR3, CD73, IgG, IgA
- Analyze by flow cytometry with gating strategy: CD45+ (excluding IV+ cells) → CD3- B220+ CD19+ → CD69+ CXCR3+ for BRM identification [38] [36]
Step 3: Functional Assessment
- For microscopy studies, utilize Cd19-Cre × R26-LSL-tdTomato reporter mice to visualize B cells
- Prepare lung slices (300-400μm) for ex vivo two-photon imaging pre- and post-challenge
- Track cell motility parameters: velocity, meandering index, confinement ratio
- Quantify BRM accumulation at infected foci identified by influenza nucleoprotein staining [36]
Step 4: Local Antibody Measurement
- Collect bronchoalveolar lavage fluid pre- and post-challenge
- Measure antigen-specific IgA and IgG by ELISA
- Perform enzyme-linked immunosorbent spot (ELISpot) on lung homogenates to quantify antibody-secreting cells [36]
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Mucosal Immunity Studies
| Reagent Category | Specific Examples | Research Application | Technical Function |
|---|---|---|---|
| Flow Cytometry Antibodies | Anti-CD69, Anti-CD45 (IV staining), Anti-CXCR3, Anti-CD103 [38] | Identification of tissue-resident lymphocytes | Distinguish resident (CD69+ CD45 IV-) from circulating immune cells |
| Cell Isolation Reagents | Collagenase IV, DNase I [36] | Preparation of single-cell suspensions from tissues | Enzymatic digestion of extracellular matrix while preserving cell surface markers |
| Cytokine/Chemokine Detection | CXCL9/CXCL10 ELISA, IFN-γ ELISpot [39] [36] | Measurement of key signaling molecules | Quantify chemokines driving BRM recruitment and T cell cytokines |
| Animal Models | Cd19-Cre × R26-LSL-tdTomato mice [36], Parabiosis models [38] | Fate mapping and cell tracking | Visualize B cell behavior and distinguish tissue-resident from circulating populations |
| Adjuvant Formulations | Lipid nanoparticles, TLR agonists [37] | Enhancing mucosal immunogenicity | Activate innate immunity and improve antigen presentation |
The strategic induction of tissue-resident memory B cells through mucosal vaccination represents a transformative approach for combating respiratory pathogens. The experimental evidence compiled in this guide demonstrates that novel adjuvant and delivery systems—particularly lipid nanoparticles, viral vectors, and biomimetic systems—show significant promise for enhancing mucosal immunogenicity and BRM recruitment. The route of administration emerges as a critical determinant, with intranasal delivery proving consistently superior to intramuscular immunization for generating tissue-resident memory populations in the respiratory tract.
Future research priorities should include the optimization of antigen design to focus on conserved epitopes that elicit cross-reactive T and B cell responses, the development of safe mucosal adjuvants that specifically promote tissue-residency programming, and the establishment of standardized correlates of protection that include BRM quantification. As these technologies mature, mucosal vaccines capable of inducing robust BRM responses offer the potential to not only reduce disease severity but to prevent infection altogether, representing a crucial advancement in our ongoing battle against respiratory pathogens.
In the evolving landscape of vaccinology, tissue-resident memory B cells (BRM) have emerged as a crucial frontier, particularly in the quest to elicit robust, long-lasting protection at pathogen entry sites. These non-circulating cells take up permanent residence in peripheral tissues, where they stand guard, enabling rapid and localized antibody responses upon pathogen re-encounter [1]. The critical distinction between mucosal and systemic vaccination strategies lies in their capacity to generate these tissue-localized sentinels. While conventional intramuscular vaccines primarily induce systemic IgG responses and circulating memory, they often fail to establish substantial populations of BRM cells at mucosal surfaces, leaving these entry portals more vulnerable to initial infection and transmission [27]. Consequently, identifying specific correlates of protection (CoP) for BRM cells—measurable immune parameters that predict effective defense—has become a central focus for researchers and drug development professionals aiming to design next-generation vaccines that block infection at its inception.
This guide provides a comparative analysis of the leading immune markers and methodologies for quantifying BRM-specific responses, framing the discussion within the broader thesis that mucosal vaccination strategies are superior for generating these critical cellular populations. We present structured experimental data, detailed protocols, and essential research tools to standardize the assessment of BRM correlates of protection across research and development pipelines.
Comparative Analysis of Key BRM Immune Markers
The protective efficacy conferred by BRM cells is mediated through several distinct, measurable mechanisms. The table below summarizes the primary and secondary correlates of protection, their molecular or cellular basis, and their functional significance in mucosal immunity.
Table 1: Key Correlates of Protection for Tissue-Resident Memory B Cells
| Correlate of Protection | Molecular/Cellular Basis | Function in Mucosal Immunity | Detection Method |
|---|---|---|---|
| Local Secretory IgA (SIgA) | Dimeric IgA transported across epithelium by polymeric Ig receptor (pIgR) [27]. | Neutralizes pathogens at mucosal entry points; prevents adhesion and dissemination [27]. | ELISA of mucosal lavage (e.g., BALF); ELISpot [40]. |
| BRM-Derived Plasma Cells | Local, long-lived plasma cells differentiated from resident BRM [1]. | Provides sustained, local antibody production independent of circulating B cells [1]. | Intracellular staining (ICS) for IgA/BLIMP1; flow cytometry [40]. |
| Antigen-Specific BRM Cells | CD38+GL7−IgM−IgD− class-switched B cells resident in tissue [40]. | Rapidly differentiate into antibody-secreting cells (ASCs) upon antigen re-encounter [1]. | MHC class II tetramers; antigen-specific B cell staining [40]. |
| Tissue-Residency Markers | Expression of CD69, CD49a, CD103; downregulation of S1PR1 [41]. | Promotes permanent tissue retention, enabling immediate local response [41]. | Multiplex flow cytometry; transcriptomic analysis [41]. |
Methodologies for Evaluating BRM Correlates of Protection
Quantifying Mucosal Antibody Secreting Cells and Immunoglobulins
Experimental Objective: To measure the functional output of BRM cells and their progeny in mucosal tissues by quantifying the frequency of antibody-secreting cells (ASCs) and the concentration of secreted immunoglobulins.
Detailed Protocol:
- Sample Collection: Collect bronchoalveolar lavage fluid (BALF) and blood serum. Process lung tissue using a gentle MACS Dissociator with collagenase/DNase I to generate single-cell suspensions for intracellular staining [40].
- Enzyme-Linked Immunosport (ELISpot):
- Coat multiscreen IP plates with recombinant antigen (e.g., SARS-CoV-2 RBD or spike protein) or anti-immunoglobulin capture antibody.
- Block plates and seed isolated lymphocyte suspensions from lung or other mucosal tissues.
- Incubate cells for 12-24 hours to allow antibody secretion.
- Detect captured antibodies using biotinylated detection antibodies (anti-IgA or antigen-specific), followed by streptavidin-ALP and BCIP/NBT substrate.
- Quantify spot-forming units (SFUs) representing individual ASCs using an automated ELISpot reader [40].
- Enzyme-Linked Immunosorbent Assay (ELISA):
- Coat high-binding plates with antigen or capture antibody.
- Add BALF or serum samples, followed by detection with horseradish peroxidase (HRP)-conjugated anti-IgA/IgG antibodies.
- Develop with TMB substrate, stop the reaction, and read absorbance to determine antigen-specific immunoglobulin concentrations [40].
- Intracellular Staining (ICS) and Flow Cytometry:
- Stimulate single-cell suspensions ex vivo with PMA/ionomycin in the presence of brefeldin A for 4-6 hours.
- Surface stain for B cell markers (e.g., B220, CD19), then fix, permeabilize, and stain intracellularly for IgA and the master plasma cell regulator BLIMP1 (encoded by Prdm1).
- Identify IgA+BLIMP1+ antibody-secreting plasma cells by flow cytometry [40].
Identifying and Phenotyping Antigen-Specific BRM Cells
Experimental Objective: To precisely identify, enumerate, and characterize the phenotype of antigen-specific BRM cells within complex tissue environments.
Detailed Protocol:
- Tetramer Staining for Antigen-Specific B Cells:
- Generate fluorescently labeled tetramers by conjugating recombinant antigen (e.g., SARS-CoV-2 RBD) to streptavidin with a 4:1 molar ratio.
- Pre-incubate single-cell suspensions from tissues with Fc block to reduce non-specific binding.
- Stain cells with antigen-specific tetramers and a panel of surface antibodies for 30-60 minutes on ice. Key markers include:
- Lineage: B220, CD19
- Memory/Switching: CD38, GL7, IgD, IgM
- Tissue-Residency: CD69, CD49a, CXCR3 [40]
- Include viability dye to exclude dead cells.
- Flow Cytometry and Cell Sorting:
- Analyze stained cells using a high-parameter flow cytometer. Antigen-specific BRM are typically identified as B220+CD19+tetramer+CD38+GL7−CD69+.
- For downstream functional assays or transcriptomic analysis, sort this population to high purity into RNase-free buffer or culture media.
- Parabiosis for Confirming Tissue-Residency:
- Surgically join a CD45.2 congenic mouse (immunized) with a CD45.1 congenic naive mouse, allowing them to share a circulatory system for 2+ weeks to achieve blood cell equilibration [40].
- Administer a mucosal booster to one parabiont.
- Analyze tissues from both partners. Genuine tissue-resident cells (BRM, TRM) will be overwhelmingly (>90%) derived from the host (CD45.2+ in the immunized mouse), while circulating cells will be a mix of both CD45.1 and CD45.2 [40]. This definitively distinguishes resident from recirculating populations.
The following diagram illustrates the core experimental workflow for processing samples to identify and validate bona fide BRM cells, integrating the key protocols described above.
The Scientist's Toolkit: Essential Research Reagents
Successful interrogation of BRM correlates of protection relies on a specific toolkit of high-quality reagents and assays.
Table 2: Essential Research Reagents for BRM Studies
| Reagent / Assay | Specific Example | Function in BRM Research |
|---|---|---|
| Recombinant Antigens | SARS-CoV-2 Spike/RBD protein [40] | Used to generate tetramers for identifying antigen-specific B cells and as coating antigens in ELISA/ELISpot. |
| MHC-II Tetramers | I-A(b)/SARS-CoV-2 epitope tetramers [40] | Directly label and identify antigen-specific CD4+ T cells that provide help to BRM. |
| Fluorophore-Conjugated Antibodies | Anti-B220, CD19, CD38, CD69, CD49a, IgA, BLIMP1 [40] | Enable phenotyping of BRM and plasma cells via flow cytometry and intracellular staining. |
| Chemokine Receptor Assays | Anti-CXCR3 antibody [40] | Detect receptor crucial for B cell recruitment to lung via CXCL9/CXCL10 signaling. |
| In Vivo Model Systems | Parabiosis surgery [40] | The gold-standard experiment for conclusively demonstrating tissue residency versus circulation. |
| Single-Cell RNA Sequencing | 10x Genomics Platform | Uncover the complete transcriptional profile and heterogeneity of BRM populations. |
Mucosal vs. Systemic Vaccination: A Paradigm Shift in Eliciting BRM
The fundamental difference in immune outcomes between mucosal and systemic vaccination underscores the importance of BRM-focused correlates. Intramuscular (IM) vaccination, while effective at raising systemic IgG and circulating memory, is inherently limited in its ability to establish protective mucosal immunity [27]. In contrast, intranasal (IN) boosting, even with an unadjuvanted protein following IM priming, potently "retools" the immune response, converting pre-existing systemic immunity into robust local mucosal protection [40].
This paradigm, often called "prime and spike," leverages the systemic priming phase to generate a pool of memory lymphocytes. The subsequent mucosal booster then acts as a homing beacon, recruiting these cells to the respiratory tract via chemokine signals like CXCL9 and CXCL10, which engage CXCR3 on memory B cells [40]. Once in the tissue, these cells differentiate into BRM and local IgA-secreting plasma cells, establishing a durable defensive barrier. This mechanistic understanding reveals that CoPs like serum IgG, while valuable for predicting protection from severe systemic disease, are insufficient correlates for the sterilizing immunity sought in next-generation vaccines. The field must therefore adopt and standardize the measurement of local IgA, antigen-specific BRM frequency, and tissue-residency markers to fully evaluate a vaccine's potential to block transmission and prevent initial infection at the mucosal frontier.
Overcoming Hurdles: Key Challenges and Optimization Strategies in Mucosal Vaccine Development
The durability of immune responses presents a central challenge in modern vaccinology. While conventional intramuscular (IM) vaccines can generate robust systemic immunity, their capacity to elicit long-lasting protection at mucosal surfaces—the primary entry points for most pathogens—is limited [6]. This is particularly true for the secretory IgA (SIgA) antibodies and specialized tissue-resident memory B cells (BRM) that constitute the first line of defense at these barriers [35] [1]. A growing body of evidence suggests that vaccination strategies which directly target the mucosal tissues themselves are superior for inducing these local, durable responses [42] [40] [43]. This review objectively compares the persistence of immune responses elicited by mucosal versus systemic vaccination platforms, focusing on quantitative measures of durability and the underlying mechanistic insights that inform next-generation vaccine design.
Comparative Durability of Immune Responses: Mucosal vs. Systemic Vaccination
Direct comparison of immune response kinetics reveals stark differences between vaccination routes. The key metrics of durability—antigen-specific IgA levels and the persistence of memory B cells in mucosal tissues—consistently favor mucosal approaches, particularly those employing a prime-pull strategy (systemic prime followed by mucosal boost).
Table 1: Comparative Durability of Mucosal Immune Responses Following Different Vaccination Strategies
| Vaccination Strategy | Key Immune Components | Persistence/Durability | Experimental Model |
|---|---|---|---|
| Intranasal (Live-attenuated) | Upper airway HA-specific IgG+ and IgA+ memory B cells | Remained elevated at 6 months post-vaccination [42] | Human adenoid tissue |
| Heterologous Prime-Pull (IM prime + IN boost) | Lung spike-specific CD4+ and CD8+ T cells; RBD-specific IgA+ Antibody Secreting Cells (ASCs) | Peak responses at 21 days post-priming; sustained local IgA [40] | Mouse model (C57BL/6J) |
| Homologous Intramuscular (mRNA) | Systemic antibody and memory B cells | Minimal local mucosal B-cell responses detected [42] | Human and Mouse models |
| Intranasal ChAdOx1 boost post-IM | Spike-specific IgA and tissue-resident T cells | Protection in upper respiratory tract maintained at 12 weeks, unlike IM-only [43] | Mouse model |
Table 2: Quantitative Comparison of Immune Cell Phenotypes Induced by Mucosal Vaccination
| Immune Cell Type | Phenotype/Function | Induction by Mucosal Vaccination | Role in Durability |
|---|---|---|---|
| Tissue-Resident Memory B Cells (BRM) | CD38+GL7-IgM-IgD- class-switched; rapidly differentiate into ASCs upon rechallenge [40] [1] | Robustly induced by intranasal vaccination [42] [1] | Provides a local, rapid-response reservoir for long-term protection [1] |
| Tissue-Resident Memory T Cells (TRM) | CD69+CD103+; remain in non-lymphoid tissues [35] [6] | Effectively induced by mucosal vaccination [6] [43] | Critical for limiting viral replication upon re-exposure; can persist for extended periods [35] |
| IgA+ Antibody Secreting Cells (ASCs) | icIgA+BLIMP1+; source of mucosal IgA [40] | Substantially increased in lungs after intranasal booster [40] | Maintains levels of secretory IgA (SIgA), the predominant antibody at mucosal sites [35] [6] |
Detailed Experimental Protocols for Assessing Mucosal Durability
To ensure reproducible results in mucosal immunology, standardized and detailed protocols are essential. The following section outlines key methodologies used to generate the comparative data presented above.
Protocol 1: Longitudinal Tracking of Mucosal B Cell Memory in Humans
This protocol, adapted from a 2025 preprint, details the method for directly sampling and analyzing human upper airway mucosal immunity [42].
- Objective: To longitudinally compare the durability and phenotype of influenza-specific B cell responses in the human upper respiratory tract following intranasal (FluMist) versus intramuscular vaccination.
- Key Materials:
- Nasopharyngeal Swabs: For longitudinal sampling of adenoid tissue and resident immune cells.
- Major Histocompatibility Complex (MHC) Class II Tetramers: For identifying hemagglutinin (HA)-specific B cells.
- Flow Cytometry Panel: Antibodies against human CD19, CD27, CD21, IgG, and IgA for phenotyping B cells.
- Methodology:
- Participant Groups: Enroll adults receiving either seasonal intranasal live-attenuated (FluMist) or intramuscular inactivated influenza vaccine.
- Sample Collection: Collect nasopharyngeal swabs at baseline (pre-vaccination) and at multiple time points post-vaccination (e.g., 1, 3, and 6 months).
- Cell Processing: Isolate mononuclear cells from swab samples.
- Flow Cytometric Analysis:
- Stain cells with HA-specific tetramers and fluorescently-labeled antibodies.
- Identify and quantify HA-specific IgG+ and IgA+ memory B cells.
- Analyze activation markers (CD27+CD21-) to determine differentiation status.
- Outcome Measures: Frequency and absolute numbers of HA-specific memory B cells in the upper airway over time, with durability defined as significant elevation above baseline at 6 months [42].
Protocol 2: Prime-Pull Immunization and Mucosal Recall Response in Mice
This protocol, from a seminal Nature Immunology 2025 study, describes a murine model to mechanistically dissect how a mucosal booster converts systemic immunity into durable mucosal protection [40].
- Objective: To investigate the cellular mechanisms by which an unadjuvanted intranasal protein booster, following intramuscular mRNA priming, elicits durable mucosal IgA recall responses.
- Key Materials:
- mRNA-LNP Vaccine: Pfizer/BioNTech BNT162b2 (1 µg dose) for intramuscular priming.
- Recombinant SARS-CoV-2 Spike Protein: Unadjuvanted, for intranasal boosting (1 µg dose).
- Aicda-ERT2-Cre-Rosa26-tdTomato Prdm1-EYFP Mice: Transgenic model for fate-mapping B cells and plasma cells.
- Parabiosis Surgery Setup: For connecting circulatory systems of CD45.1 and CD45.2 mice to track cell migration.
- Methodology:
- Priming: Immunize C57BL/6J or fate-mapping mice intramuscularly with 1 µg of mRNA-LNP vaccine at day 0.
- Boosting: At day 14 or 35, administer 1 µg of unadjuvanted spike protein intranasally to anesthetized mice.
- Sample Collection: At designated endpoints, collect bronchoalveolar lavage fluid (BALF), serum, lung tissue, and lymphoid organs.
- Immune Analysis:
- ELISA: Quantify S1-specific IgA and IgG in BALF and serum.
- Flow Cytometry: Use RBD-specific tetramers and intracellular staining for IgA and BLIMP1 to identify antigen-specific B cells and plasma cells in lungs.
- Parabiosis: Surgically join CD45.2 (vaccinated) and CD45.1 (naïve) mice. After circulatory equilibration, boost one partner intranasally and track donor-derived immune cells in the lung.
- Outcome Measures: Quantification of spike-specific T cells, RBD-specific IgA+ ASCs, and S1-specific IgA in mucosal sites; determination of the origin (circulating vs. locally induced) of these responses [40].
Mechanisms and Pathways Underlying Durable Mucosal Immunity
The superior durability observed with mucosal vaccination strategies is governed by specific cellular behaviors and molecular pathways that promote long-term residency and rapid recall in mucosal tissues.
The Prime-Pull Mechanism: Recruiting Systemic Immunity to Mucosal Sites
The heterologous "prime-pull" strategy leverages pre-existing systemic immunity established by a parenteral prime and actively recruits it to the mucosal tissue via a localized booster. The following diagram illustrates this coordinated mechanism.
Diagram 1: Prime-pull mechanism for durable mucosal immunity.
As depicted, the process begins with an intramuscular prime, which generates a pool of antigen-specific memory B and T cells in the systemic circulation and lymph nodes [40]. The critical event is the subsequent intranasal boost, which triggers the local production of chemokines CXCL9 and CXCL10 in the lung. These chemokines bind to the receptor CXCR3 on circulating memory B cells, pulling them into the mucosal tissue [40]. Once in the lung, these recruited B cells rapidly differentiate into IgA-secreting plasma cells, establishing a durable source of mucosal antibody. This mechanism explains how a mucosal booster can "retool" pre-existing systemic immunity without the need for a strong adjuvant, enhancing both safety and efficacy [40].
Signaling Pathways in Mucosal Memory B Cell Development and Function
The development and maintenance of long-lived BRM and IgA responses are dependent on specific signaling pathways and cellular interactions within the mucosal microenvironment.
Diagram 2: Key pathways for mucosal B cell differentiation.
The germinal center (GC) reaction in mucosal-associated lymphoid tissue (MALT) is the cornerstone of BRM development. As shown, this process is CD40-dependent, requiring cognate help from T follicular helper (Tfh) cells [1]. The cytokine TGF-β provides a critical signal for B cells to undergo class-switch recombination to IgA, the dominant antibody isotype in mucosa [40]. These coordinated signals give rise to two key populations: BRM that reside in the tissue and can rapidly respond to reinfection, and long-lived plasma cells that continuously produce dimeric IgA (dIgA). The dIgA is then transported across the epithelium by the polymeric immunoglobulin receptor (pIgR) to be released as SIgA, which neutralizes pathogens at the mucosal surface [6]. This intricate coordination ensures sustained local protection.
The Scientist's Toolkit: Essential Reagents for Mucosal Immunology Research
Advancing research into durable mucosal immunity requires a specific set of reagents and model systems. The following table details key tools used in the cited studies.
Table 3: Essential Research Reagents for Investigating Mucosal Immunity Durability
| Reagent / Model | Specific Example | Function/Application | Reference |
|---|---|---|---|
| MHC Tetramers | HA-specific (Influenza) and RBD-specific (SARS-CoV-2) tetramers | Identification and phenotyping of antigen-specific B and T cells via flow cytometry. | [42] [40] |
| Fate-Mapping Mouse Models | Aicda-ERT2-Cre-Rosa26-tdTomato Prdm1-EYFP | Lineage tracing of activated B cells and their differentiation into plasma cells in vivo. | [40] |
| Parabiosis Surgery Model | Surgical pairing of CD45.1 and CD45.2 mice | Distinguishing between locally induced and recruited circulating immune cells. | [40] |
| Mucosal Adjuvants | T-vant (OMV-based adjuvant) | Enhancing immunogenicity of mucosally delivered subunit vaccines safely. | [44] |
| Specimen Collection Tools | Nasopharyngeal swabs; Bronchoalveolar Lavage (BAL) | Longitudinal sampling of immune cells and antibodies from human or murine respiratory tract. | [42] [40] |
The collective evidence demonstrates that mucosal vaccination strategies, particularly heterologous prime-pull regimens, are fundamentally superior to homologous intramuscular vaccination for achieving durable mucosal immunity. The critical differentiator is the ability to generate and maintain a local arsenal of tissue-resident memory B cells (BRM) and a sustained source of protective IgA directly at the site of pathogen entry [42] [40] [1]. While the current data is compelling, future research must focus on standardizing correlates of protection for mucosal immunity and optimizing safe, effective adjuvants and delivery platforms for human use [6] [44]. Overcoming the durability dilemma requires a paradigm shift in vaccine design—from solely preventing severe disease to effectively blocking infection and transmission at the source. The continued development of mucosal vaccines represents the most promising path toward achieving this goal.
Mucosal tissues serve as the primary entry point for most infectious pathogens, yet they also represent a sophisticated immunological barrier capable of mounting potent first-line defense mechanisms. The paradigm in vaccine development is increasingly shifting toward mucosal immunization strategies that can elicit robust tissue-resident memory responses, particularly tissue-resident memory B cells (Brm), which provide localized protection at pathogen entry sites [17]. Unlike systemic vaccination, which primarily generates circulating antibodies and memory cells, mucosal vaccination establishes resident immune populations that can respond immediately upon pathogen encounter [1]. However, this approach presents unique challenges in adjuvant and antigen design, where the imperative for potency must be carefully balanced against safety considerations in delicate mucosal environments. This comparison guide examines current approaches, their immunological mechanisms, and experimental evidence supporting their efficacy in generating protective mucosal immunity.
Foundational Concepts: Mucosal versus Systemic Immunity
The immune system employs distinct strategies at mucosal surfaces compared to systemic compartments. The mucosal immune system operates as a body-wide interconnected lymphatic network defending against pathogen entry at all internal-external secretory interfaces, while the systemic immune system activates when pathogens breach these barriers and enter internal tissues [45]. This fundamental difference necessitates tailored vaccine approaches.
Tissue-resident memory B cells (Brm) have emerged as crucial mediators of mucosal protection. These non-circulating cells persist in mucosal tissues and provide rapid, localized antibody responses during secondary pathogen exposure [1]. Unlike conventional memory B cells that recirculate, Brm reside permanently in tissues and express characteristic markers including CD69, often with CXCR3 and CD44 [17]. Their induction typically requires local antigen encounter, making mucosal vaccination particularly effective for generating these populations [17] [1].
Table 1: Key Differences Between Mucosal and Systemic Vaccination
| Parameter | Mucosal Vaccination | Systemic Vaccination |
|---|---|---|
| Primary Immune Response | Local secretory IgA, tissue-resident memory cells | Serum IgG, circulating memory cells |
| Tissue-Resident Memory Generation | Strong induction of Brm and Trm | Limited tissue-resident memory |
| Administration Routes | Intranasal, aerosol, oral | Intramuscular, subcutaneous |
| Protection at Entry Sites | Strong first-line barrier protection | Limited mucosal protection |
| Safety Considerations | Risk of local inflammation, neurotropism concerns | Primarily systemic reactogenicity |
Adjuvant Design Strategies for Mucosal Tissues
Particulate Adjuvants and Size Considerations
Particle size represents a critical design parameter for mucosal adjuvants, directly influencing antigen delivery, cellular interactions, and immune polarization. Nanoemulsion-based adjuvants demonstrate how precisely controlled particle dimensions can enhance mucosal immunity while maintaining safety profiles.
Table 2: Impact of Adjuvant Particle Size on Immune Responses
| Particle Size | Cellular Uptake Mechanism | Dominant Immune Response | Mucosal Transport |
|---|---|---|---|
| 20-200 nm | Endocytosis by immune cells | Cellular immunity (CD8+ T cells) | Efficient across mucosal barriers |
| 200-500 nm | Endocytosis/phagocytosis | Balanced cellular/humoral | Moderate transport |
| >500 nm | Phagocytosis | Humoral immunity | Limited transport |
Research on squalene-based nanoemulsions highlights how size optimization enhances adjuvant function. A monodisperse nanoemulsion with ~200 nm particles (PELC@MN) induced the emergence of membranous epithelial cells and natural killer cells in nasal mucosal tissues, facilitated protein antigen delivery across the nasal mucosal membrane, and drove broad-spectrum antigen-specific T-cell immunity in both nasal tissues and spleen [46]. Crucially, these nanoemulsions generated potent antigen-specific CD8+ cytotoxic T-lymphocyte responses that provided 50% survival in tumor-bearing mice 40 days after tumor inoculation and significantly attenuated lung metastasis [46].
Immunostimulatory Adjuvants and PRR Targeting
Pattern recognition receptor (PRR) agonists constitute another major adjuvant class being developed for mucosal applications. These molecules directly activate innate immune pathways to orchestrate subsequent adaptive responses. The largest group explored to date targets Toll-like receptors (TLRs), which are strategically located on mucosal surfaces [47].
TLR agonists show particular promise when combined with delivery systems. For example, monophosphoryl lipid A (MPL), a TLR4 agonist, has been incorporated into adjuvant systems such as AS04 by adsorption onto aluminum salts, creating synergistic effects that enhance immune responses while maintaining acceptable safety profiles [47]. The intracellular TLRs (TLR3, TLR7, TLR8, TLR9) that recognize microbial nucleic acids have also been investigated, with their ligands serving as potential mucosal adjuvants that can bias toward desirable T-helper responses [47].
Figure 1: Mucosal Adjuvant Mechanism of Action: This diagram illustrates the sequential immunological events initiated by adjuvanted mucosal vaccines, from pattern recognition receptor (PRR) activation to the generation of tissue-resident memory B cells (Brm) and secretory IgA production.
Antigen Design Considerations for Mucosal Vaccination
Epitope Selection and Cross-Reactivity
Antigen design significantly influences the quality and durability of mucosal immune responses. Optimal epitope selection can enhance tissue-resident memory formation and cross-protection against variant pathogens. Research on SARS-CoV-2 has demonstrated that conserved epitopes, particularly in the receptor-binding domain (RBD) of the spike protein, elicit more durable and cross-reactive antibodies [48]. Similar principles apply to mucosal vaccine design, where epitope conservation across pathogen strains becomes crucial for broad protection.
Computational approaches for epitope selection have advanced significantly. One COVID-19 vaccine candidate employed a multi-neoepitope peptide vaccine approach using computationally selected SARS-CoV-2 neoepitopes, which induced peripheral viral-specific CD8+ T cells expressing tissue-residency markers (CD103, CD49a) in spleen and draining lymph nodes [17]. This strategy highlights how rational epitope selection can program tissue-homing characteristics in vaccine-induced lymphocytes.
Stabilization and Conformational Preservation
Antigen structure preservation represents another critical design parameter, particularly for viral glycoproteins that exist in meta-stable conformations. The SARS-CoV-2 spike protein exemplifies this principle, where stabilization in prefusion conformation through proline substitutions (S-2P) maintains important neutralizing epitopes that would otherwise be lost in postfusion configurations [48]. Such structural preservation proves especially important for mucosal vaccines, where rapid neutralization at entry sites depends on antibodies targeting functional epitopes.
Comparative Experimental Data: Mucosal Vaccine Approaches
Nanoemulsion Adjuvant Systems
The PELC (squalene-based) nanoemulsion system provides compelling experimental evidence for size-optimized mucosal adjuvants. In a direct comparison between polydisperse submicron emulsion (PELC@PE) and monodisperse nanoemulsion (PELC@MN), the nanoformulation demonstrated superior mucosal immunogenicity [46].
Table 3: Experimental Outcomes of Nanoemulsion Adjuvantation
| Parameter | PELC@PE (Polydisperse) | PELC@MN (200 nm Monodisperse) |
|---|---|---|
| Mucosal Transport | Limited antigen delivery | Enhanced antigen transport across nasal mucosa |
| T-cell Responses | Moderate antigen-specific T cells | Broad-spectrum antigen-specific T-cell immunity |
| Tumor Protection | Limited survival benefit | 50% survival at 40 days post-tumor challenge |
| Metastasis Control | Minimal reduction | Significant attenuation of lung metastasis |
| Immune Cell Recruitment | Standard mucosal recruitment | Emergence of membranous epithelial and NK cells |
Route of Administration Comparisons
The vaccination route profoundly impacts tissue-resident memory generation. Comparative studies in simian immunodeficiency virus (SIV) models demonstrated that aerosolized adenovirus serotype 5 (rAd5) vaccination induced strong cellular responses in the lung and systemic humoral responses equivalent to intramuscular delivery [15]. However, the mucosal vaccination route provided superior protection against mucosal challenge.
Similarly, intranasal administration of live-attenuated influenza virus induced development of CD4 and CD8 tissue-resident memory T cells (Trm) in lungs, whereas systemic immunization failed to generate comparable Trm responses [17]. This route-dependent effect extends to Brm generation, though the mechanisms are less fully characterized [17].
Experimental Protocols for Mucosal Immunity Evaluation
Nanoemulsion Preparation and Characterization
Protocol: PELC Nanoemulsion Formulation
- Initial Emulsification: Combine 120 mg diblock copolymer PEG-b-PLACL, 0.8 mL PBS, 0.935 mL squalene, and 0.165 mL Span85.
- Homogenization: Emulsify using a homogenizer (Polytron PT 2500E) at high speed for 3-5 minutes.
- Dilution: Redisperse 200 μL stock emulsion into 1800 μL PBS with gentle mixing (5 rpm) for 1 hour.
- Size Optimization (PELC@MN): Pass suspension through polycarbonate membranes (1.0, 0.4, 0.2, 0.1 μm sequentially) using a mini-extruder with 11 passages.
- Characterization: Measure particle size distribution via dynamic light scattering (Nano ZS analyzer). Verify monodispersity and morphology by transmission electron microscopy [46].
Mucosal Immune Response Assessment
Protocol: Tissue-Resident B Cell Analysis
- Tissue Processing: Collect mucosal tissues (nasal, pulmonary) and process into single-cell suspensions using enzymatic digestion.
- Intravascular Staining: Administer fluorescently-labeled anti-CD45 antibody intravenously 3-5 minutes before sacrifice to discriminate circulating (CD45+) from tissue-resident (CD45-) lymphocytes.
- Surface Staining: Incubate cells with antibodies against CD19, CD69, CD38, CD27, CD73, CXCR3, and IgA.
- Flow Cytometry Gating: Identify Brm as CD19+CD69+ cells, further subtyped by CD73 expression and immunoglobulin isotype.
- Functional Assessment: Sort Brm cells and stimulate with TLR agonists or antigens for 5-7 days. Measure antibody secretion by ELISA and assess differentiation into antibody-secreting cells by ELISpot [17] [1].
Figure 2: Brm Characterization Workflow: This experimental flowchart outlines the key steps for identifying and characterizing tissue-resident memory B cells from mucosal tissues, including critical intravascular staining to distinguish resident from circulating populations.
Table 4: Key Research Reagents for Mucosal Immunology Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Adjuvant Systems | Squalene nanoemulsions (PELC), MPL, AS04 | Enhance mucosal immunogenicity |
| Cell Isolation | Collagenase/DNase digestion cocktails, density gradient media | Tissue processing for lymphocyte isolation |
| Flow Cytometry | Anti-CD19, CD69, CD73, CD38, CD27, IgA, CXCR3 | Brm phenotyping and characterization |
| ELISA/ELISpot | IgA/IgG detection antibodies, antigen-coated plates | Antibody secretion quantification |
| Animal Models | BALB/c, C57BL/6 mice | In vivo vaccine efficacy testing |
| Challenge Models | OVA-expressing tumor cells (EG7, B16-F10-OVA) | Protective efficacy evaluation |
The evolving understanding of tissue-resident immunity continues to reshape adjuvant and antigen design principles for mucosal vaccination. Current evidence strongly supports the superiority of mucosal routes for generating protective tissue-resident memory B cell responses, with nanoemulsion systems demonstrating how physical parameters like particle size can be optimized to enhance efficacy and safety. Future work should focus on elucidating the specific signals that promote Brm differentiation and retention, developing novel delivery systems that target mucosal inductive sites, and identifying conserved epitopes that elicit broad protection against antigenically variable pathogens. As these advances mature, mucosal vaccination may ultimately provide more effective first-line defense against respiratory, gastrointestinal, and sexually transmitted infections that initiate at mucosal surfaces.
The fundamental goal of vaccination—to elicit protective immunological memory—is not achieved through a one-size-fits-all approach. The immunological history of an individual creates a distinct landscape upon which subsequent vaccines act, making the choice between priming a naive immune system and boosting an experienced one a critical consideration in vaccine design. This is particularly true in the burgeoning field of tissue-resident memory B cells (BRM cells), which reside in mucosal tissues like the airways and provide a first line of defense against invading pathogens [1]. These cells rapidly differentiate into antibody-secreting cells upon re-encounter with a pathogen, providing a robust, localized response that is often more effective than systemic immunity alone [1] [49].
The immune environment in barrier tissues is complex, and the strategies to populate it with protective memory cells differ dramatically depending on whether the host is encountering an antigen for the first time or has pre-existing immunity. Mucosal vaccines have emerged as a powerful tool for inducing this localised immunity, as they can stimulate the production of secretory IgA (SIgA) and engage the common mucosal immune system to promote the homing of activated immune cells to distant mucosal tissues [6]. However, their efficacy is heavily influenced by prior antigen exposure. This review will objectively compare the strategies for priming naive individuals versus boosting experienced ones, with a specific focus on the generation of BRM cells, and will provide the supporting experimental data and methodologies essential for research and drug development.
Fundamental Immunological Differences Between Naive and Experienced Hosts
The immune system's response to a vaccine is dictated by the pre-existing repertoire of memory B cells, T cells, and long-lived plasma cells. The differences between naive and experienced hosts are not merely quantitative but are qualitative, affecting the speed, magnitude, and character of the immune response.
In naive hosts, the immune system must undergo de novo priming. This process requires the activation of naive B and T cells, a clonal expansion, and the initial differentiation into effector and memory cells. This process is comparatively slower and depends on the full priming of the immune system, often resulting in weaker initial responses [6]. The establishment of germinal centers in mucosal-associated lymphoid tissue (MALT) is critical for this process, where B cells undergo somatic hypermutation and class-switch recombination before differentiating into plasma cells that secrete dimeric IgA (dIgA) [6].
In experienced hosts, the presence of pre-existing memory cells allows for a rapid recall response. Memory B cells are transcriptionally and epigenetically distinct from naive B cells, enabling them to respond more rapidly and vigorously upon antigen re-encounter [49]. Studies have shown that intranasal vaccination, for instance, induces significantly higher nasal IgA and IgG responses in individuals previously exposed to SARS-CoV-2 compared to naive subjects [6]. This recall response is a hallmark of adaptive immunity and is the fundamental principle behind booster vaccinations.
Impact on Tissue-Resident Memory: The pathway to generating BRM cells is also influenced by immune history. In a primary response, memory B cells are first generated in secondary lymphoid organs before being recruited to barrier tissues like the lungs, where they establish residence [49]. In contrast, upon re-challenge in an experienced host, local BRM cells can be directly reactivated and may also undergo further affinity maturation in inducible tertiary lymphoid structures at the site of infection [49].
Table 1: Core Immunological Differences Between Naive and Experienced Hosts
| Feature | Naive Host | Experienced Host |
|---|---|---|
| Key Responding Cells | Naive B and T cells | Memory B cells, Memory T cells, Long-lived plasma cells |
| Response Kinetics | Slower (days to weeks) | Rapid (hours to days) |
| Antibody Characteristics | Lower affinity, primarily IgM | Higher affinity, class-switched (e.g., IgG, IgA) |
| Germinal Center Reaction | Required for primary response | Can be rapidly re-established for further affinity maturation |
| Tissue-Resident Memory | Must be established from scratch through recruitment | Pre-existing pool can be directly reactivated and expanded |
Comparative Analysis of Vaccination Strategies
The strategic choice between priming and boosting, and the modalities used for each, have profound and measurable effects on the resulting immune protection. The data below, derived from recent studies, highlight these differences.
Mucosal vs. Systemic Antibody Responses
A critical distinction between vaccine platforms is their ability to induce mucosal IgA, a key correlate of protection for many respiratory pathogens.
Table 2: Mucosal vs. Systemic Vaccination in Naive and Experienced Hosts
| Vaccination Strategy | Host Status | Key Systemic Response | Key Mucosal Response | Protective Outcome |
|---|---|---|---|---|
| Intramuscular (IM) Prime | Naive | Strong serum IgG & neutralizing antibodies [50] | Negligible mucosal IgA [51] | Protects against severe disease; limited protection against infection/transmission [6] |
| Intranasal (IN) Boost | Experienced (IM-primed) | Enhanced & broadened serum neutralization [50] | Induces variant-specific IgA in nasal wash and BALF [50] | Potentially reduces viral replication and transmission [50] |
| Heterologous IM-IN "Prime and Spike" | Experienced | Broad, cross-variant serum neutralizing antibodies [50] | Robust mucosal IgA and tissue-resident memory [50] | Superior protection against reinfection and transmission in animal models [50] |
| Multiple Infections (Mucosal Exposure) | Experienced | Strong systemic immunity | High-magnitude, long-lasting mucosal IgA (≥22 months) [51] | Strong association with protection against infection [51] |
Impact on Tissue-Resident Memory B Cells
The generation of BRM cells is a key objective for next-generation vaccines, as these cells are positioned at the site of pathogen entry.
Table 3: Strategies for Inducing Tissue-Resident Memory B Cells
| Strategy | Host Context | Effect on BRM Cells | Supporting Evidence |
|---|---|---|---|
| Live Infection | Naive or Experienced | Establishes BRM cell population in tissues like the lungs [49] | Influenza infection induces virus-specific BRM cells in lungs; clonally related to GC B cells in LNs [49] |
| Systemic (IM) Vaccination | Naive | Limited induction of BRM cells | Conventional vaccines often fail to induce tissue-resident lymphocytes, relying on circulating antibodies [49] |
| Mucosal (e.g., IN) Vaccination | Naive | Can induce BRM cell formation | Intranasal vaccination induces memory B cells and TRM cells in the respiratory tract that persist for months [6] |
| Heterologous IM-IN Regimen | Experienced | Leverages systemic immunity to induce robust mucosal responses | "Prime and Spike" strategy uses IM mRNA prime followed by IN protein boost to induce mucosal immunity without adjuvant [50] |
A large clinical cohort study (n=879 healthcare workers) provided critical insights into how the sequence of antigen exposure shapes mucosal immunity. It found that repeated mucosal exposures (infections) elicited enhanced and long-lasting mucosal IgA responses. However, repeated systemic vaccinations were associated with a lower magnitude of subsequent mucosal IgA following infection. Furthermore, the timing mattered: participants who were infected before receiving systemic vaccination developed higher mucosal IgA levels compared to those whose first viral encounter was a breakthrough infection after vaccination [51]. This suggests that systemic vaccination may influence the subsequent generation of mucosal immunity, potentially by reducing viral load and inflammation at the mucosa during infection.
Experimental Protocols for Key Studies
To facilitate replication and further research, below are detailed methodologies from pivotal studies cited in this review.
Protocol 1: Heterologous Prime-Boost in Murine Model
This protocol is adapted from the study investigating "Prime and Spike" strategies [50].
- Objective: To investigate the impact of heterologous variant prime-boost regimens, focusing on intramuscular (IM) and intranasal (IN) routes, on systemic and mucosal humoral immunity.
- Model: Female, 8-week-old BALB/c mice.
- Immunization Regimen:
- Prime (Day 1): Intramuscular injection in the left quadriceps with 1 μg of Pfizer-Comirnaty monovalent (Wu-1) or bivalent (Wu-1 + BA.4/5) mRNA-LNP vaccine.
- Boost (Day 21): Intranasal administration of 25 μL per nare, delivering a total dose of 1 μg or 10 μg of unadjuvanted spike protein (Wu-1, BA.4/5, or Wu-1 + BA.4/5).
- Sample Collection: Plasma samples collected periodically via the submandibular vein, with terminal samples via cardiac puncture. Mucosal samples (nasal wash, bronchoalveolar lavage fluid - BALF) collected at endpoint.
- Immune Assays:
- Serology: ELISA for total anti-spike IgG and IgA.
- Neutralization: Pseudovirus or live virus neutralization assays against a panel of Variants of Concern (VOCs).
- Mucosal Antibodies: Measurement of antigen-specific IgA in nasal wash and BALF.
Protocol 2: Longitudinal Clinical Cohort Study of Mucosal IgA
This protocol is based on the COMMUNITY study of healthcare workers [51].
- Objective: To explore the impact of vaccinations and prior infections on the magnitude of SARS-CoV-2 spike-specific IgA in nasal secretions.
- Cohort: 879 healthcare workers with well-defined SARS-CoV-2 immune histories via linkage to national vaccine and infection registries and regular serological monitoring.
- Study Design: Longitudinal, with follow-ups every four months since April 2020. The analysis focused on samples from a specific follow-up in October 2022.
- Sample Collection:
- Nasal Secretions: Collected using nasal swabs.
- Serum: Collected concurrently.
- Laboratory Analysis:
- Mucosal IgA: Measurement of SARS-CoV-2 spike-specific IgA in nasal secretions by ELISA.
- Serum IgG: Measurement of spike-specific and nucleocapsid-specific IgG to confirm infection and vaccination status.
- Total IgA: Measurement to ensure sample quality.
- Statistical Analysis: Regression models were developed to assess the influence of the number and timing of vaccinations and infections on mucosal IgA levels.
Signaling Pathways and Immune Mechanisms
The following diagram illustrates the key signaling pathways involved in the generation of memory B cells, a process central to both priming and boosting, and how these pathways are engaged by different vaccination strategies.
The diagram outlines the critical decision points in B cell activation and differentiation. The CD40-CD40L interaction between B cells and T follicular helper (Tfh) cells is a master regulator. The duration and strength of this signal, in conjunction with cytokines like IL-21, determine whether a B cell becomes an early, pre-germinal center (GC) memory B cell or enters the GC to undergo affinity maturation and become a high-affinity, class-switched memory B cell or plasma cell [26]. These memory B cells can then be recruited to tissues via specific chemokines (e.g., CXCR3, CCR6 ligands) and establish residence as BRM cells, expressing markers like CD69 [49] [2]. Upon re-exposure to antigen, these BRM cells can rapidly differentiate into local antibody-secreting cells, providing immediate protection at the portal of pathogen entry [1] [49].
The Scientist's Toolkit: Essential Reagents and Models
This section details key reagents, models, and technologies used in the cited research to study naive and experienced immune responses.
Table 4: Key Research Reagent Solutions for Immune Profiling
| Reagent / Model / Technology | Function and Application | Context from Search Results |
|---|---|---|
| BALB/c Mouse Model | A standard inbred mouse strain used for immunization and challenge studies to model immune responses. | Used to test heterologous prime-boost regimens with mRNA and intranasal protein vaccines [50]. |
| Parabiosis Surgery | A surgical technique joining the circulation of two mice to definitively identify tissue-resident cells (which do not recirculate). | A direct method to identify tissue-resident immune cells (TRICs) like BRM and TRM cells [2]. |
| Intravascular Staining | An antibody-based technique administered intravenously to label circulatory cells, allowing distinction from tissue-resident cells in analysis. | Used to identify BRM and TRM cells in tissues, which are protected from labeling [2]. |
| scRNA-seq & TCR/BCR Sequencing | High-throughput single-cell technologies to analyze transcriptional profiles and clonal relationships of immune cells. | Used to explore phenotypic profiles of TRICs and show clonal overlap between lung memory B cells and germinal center B cells in LNs [49] [2]. |
| Recombinant Spike Proteins (Wu-1, BA.4/5) | Purified viral antigens used in subunit vaccines or to probe immune responses in immunoassays. | Used as unadjuvanted intranasal boost in heterologous prime-boost studies [50]. |
| mRNA-LNP Vaccines (e.g., BNT162b2) | Lipid nanoparticle-formulated mRNA vaccines that encode viral antigens and induce strong systemic immunity. | Used as the intramuscular priming agent in heterologous regimens [50]. |
| ELISA for Mucosal IgA | Enzyme-linked immunosorbent assay adapted to measure antigen-specific IgA in mucosal secretions like nasal wash or BALF. | Critical for quantifying the mucosal antibody response, a key endpoint in mucosal vaccine studies [50] [51]. |
| Virus Neutralization Assay | A functional assay measuring the ability of serum or mucosal antibodies to neutralize live virus or pseudovirus. | Used to assess the breadth and potency of serum responses against Variants of Concern (VOCs) [50]. |
The evidence clearly demonstrates that tailoring vaccination strategies to immune background is not a mere refinement but a necessity for optimizing protection, particularly at the mucosal barriers where most infections begin. Priming the naive immune system requires robust strategies, often involving potent adjuvants or specific platforms like live-attenuated vaccines, to establish a foundational pool of systemic and tissue-resident memory. In contrast, boosting the experienced host offers a powerful opportunity to leverage pre-existing immunity, where heterologous regimens—especially those combining systemic priming with mucosal boosting—can elicit exceptionally broad and potent immunity at the site of infection.
The critical role of tissue-resident memory B cells (BRM cells) is a through-line in this comparison. Effective priming strategies must create avenues for their initial generation, while boosting strategies should aim to reactivate and expand these local sentinels. Future vaccine development, particularly against respiratory pathogens like influenza, SARS-CoV-2, and RSV, must prioritize the direct induction of mucosal immunity. As the field advances, a deeper understanding of the specific cues that govern the homing, maintenance, and reactivation of BRM cells will be indispensable for designing the next generation of vaccines that can achieve sterilizing immunity and disrupt transmission chains.
The generation of durable and high-quality antibody responses is a cornerstone of effective vaccination and immunity against pathogens. This process relies critically on the intricate dynamics within germinal centers (GCs) and the specialized help provided by T follicular helper (Tfh) cells. In the context of vaccine development, a key paradigm shift involves comparing the effectiveness of mucosal vaccination, which aims to generate local tissue-resident memory, against conventional systemic vaccination. This guide provides a comparative analysis of the cellular mechanisms, experimental data, and methodological approaches that define robust GC and Tfh cell responses, offering a framework for researchers and drug development professionals to overcome inherent immune regulation and enhance vaccine efficacy.
Comparative Mechanisms: Mucosal vs. Systemic Vaccination
The route of vaccine administration fundamentally shapes the ensuing immune response by engaging different anatomical sites and cellular players. The table below compares the key immunological features of mucosal and systemic vaccination strategies.
| Feature | Mucosal Vaccination | Systemic Vaccination |
|---|---|---|
| Primary Induction Site | Mucosal-associated lymphoid tissues (MALT), Inducible Bronchus-Associated Lymphoid Tissue (iBALT) [6] | Secondary Lymphoid Organs (SLOs) like spleen and draining lymph nodes [52] |
| Key Antibody Type | Secretory IgA (SIgA) in the lumen; dimeric IgA (dIgA) [35] [6] | Serum IgG [6] [45] |
| Key T Helper Cells | T follicular helper (Tfh) cells in MALT; T peripheral helper (Tph) cells in tissues [52] [6] | Canonical Tfh cells in SLOs [53] [52] |
| Key B Cell Population | Tissue-resident memory B cells (BRM) in the airway [1] [2] | Memory B cells (BMem) and long-lived plasma cells in bone marrow [52] |
| Key T Memory Population | Tissue-resident memory T cells (TRM, CD69⁺CD103⁺) [6] | Circulating memory T cells [6] |
| Primary Function | Barrier immunity; first line of defense at the portal of pathogen entry; can block infection and transmission [35] [6] | Systemic protection; prevents severe disease and viremia [6] [45] |
Experimental Data and Protocols for Assessing GC/Tfh Responses
A critical challenge in vaccine research is the direct monitoring of GC reactions, which typically requires longitudinal studies and specialized techniques. The following section summarizes key experimental findings and the methodologies used to obtain them.
Key Experimental Findings
Table: Quantitative Findings from Key GC/Tfh Cell Studies
| Experimental Model | Key Intervention | Major Finding | Impact on Antibody Response |
|---|---|---|---|
| Rhesus macaques (NHP) [54] | Slow-delivery escalating-dose HIV Env immunization vs. bolus prime | Robust, prolonged GC-Tfh responses with distinct Tfh cell states (Tfh-1, Tfh-2, Tfh-17) were observed over 63 weeks. | Associated with enhanced development of autologous tier-2 neutralizing antibodies. |
| Mouse model (H2b-mCherry) [55] | Immunization with NP-OVA or SARS-CoV-2 vaccine | High-affinity GC B cells underwent more divisions but had a lower somatic hypermutation (SHM) rate per division (pmut). | Optimized antibody affinity maturation by protecting high-affinity B cell lineages from deleterious mutations. |
| Human / Translational Research [6] | Intranasal vaccination (e.g., against influenza, SARS-CoV-2) | Induction of airway TRM and BRM cells, and production of SIgA. | Correlated with reduced viral replication and potentially greater protection against initial infection. |
Detailed Experimental Protocol
The following is a detailed methodology based on the non-human primate study that successfully tracked antigen-specific Tfh cells over an extended period [54].
1. Animal Model and Immunization:
- Model: Rhesus macaques (RMs), n=8 per group.
- Immunogen: HIV Env trimer protein (MD39) with SMNP adjuvant.
- Groups: Bolus prime vs. slow-delivery escalating-dose (ED) immunization, administered bilaterally in the thighs.
- Schedule: Prime at week 0, boosts at weeks 10 and 24 (or week 24 only). The study duration was 63 weeks.
2. Sample Collection and Processing:
- Longitudinal Lymph Node (LN) Aspirates: Repeatedly collected from multiple sites (e.g., inguinal, axillary, iliac) throughout the study.
- Cell Isolation: LN mononuclear cells (LNMCs) were isolated for downstream analysis.
3. Antigen-Specific Tfh Cell Analysis:
- Stimulation: LNMCs were stimulated with pools of MD39 peptides.
- Surface & Intracellular Staining: Cells were stained for surface markers (CD3, CD4, CXCR5, PD-1, CCR7) and intracellular cytokines (IL-21, IFN-γ, IL-17) after stimulation.
- Flow Cytometry & Sorting: Antigen-specific Tfh cells were identified as CD3⁺CD4⁺CXCR5⁺PD-1⁺ and cytokine-positive. These cells were sorted for single-cell RNA sequencing (scRNA-seq).
4. B Cell and Antibody Response Assessment:
- Neutralizing Antibody Assays: Serum was used to measure autologous tier-2 neutralizing antibody titers against HIV.
- Memory B Cell Analysis: Antigen-specific memory B cells were evaluated via flow cytometry.
5. Data Integration:
- The phenotypic and functional data from antigen-specific Tfh cells were correlated with the magnitude and quality of the neutralizing antibody response.
Visualizing Key Concepts and Workflows
Germinal Center Dynamics and Affinity Maturation
This diagram illustrates the cyclic process of affinity maturation within the germinal center, highlighting the critical role of Tfh cells and the recently discovered mechanism that protects high-affinity B cell lineages.
Tissue-Resident Immunity After Mucosal Vaccination
This diagram outlines the induction of tissue-resident memory following mucosal vaccination, showcasing the key cells involved in localized protection at the respiratory barrier.
The Scientist's Toolkit: Essential Research Reagents
This table catalogs key reagents and their applications for studying GC and Tfh cell biology, as derived from the cited experimental protocols.
Table: Key Research Reagent Solutions
| Reagent / Assay | Function / Specificity | Experimental Application |
|---|---|---|
| Anti-CXCR5 & Anti-PD-1 Antibodies [54] | Surface markers for identifying Tfh cells (CXCR5⁺PD-1⁺). | Flow cytometry staining and fluorescence-activated cell sorting (FACS) of Tfh cells from lymphoid tissues. |
| Peptide MHC Class II Tetramers [54] | Directly identify antigen-specific CD4⁺ T cells, including Tfh. | Tracking and characterizing vaccine-specific Tfh cell responses without the need for in vitro stimulation. |
| H2b-mCherry Reporter Mouse Model [55] | A DOX-sensitive system to track and quantify cell divisions in vivo. | Studying the relationship between GC B cell division cycles and somatic hypermutation rates. |
| SMNP Adjuvant [54] | A nanoparticle-based adjuvant used in NHP studies. | Enhancing and prolonging the GC reactions to protein immunogens, such as HIV Env trimers. |
| scRNA-seq with BCR/TCR sequencing [54] [55] | High-resolution analysis of transcriptional states and clonal relationships. | Defining Tfh cell heterogeneity, B cell clonal dynamics, and affinity maturation pathways. |
| Intravascular Staining [2] | Differentiates circulating immune cells from tissue-resident cells. | Identifying bona fide tissue-resident memory lymphocytes (TRM, BRM) in tissues like the lung. |
Ensuring robust and durable germinal center and T follicular helper cell responses is a multifaceted challenge at the forefront of modern vaccinology. The comparative data and methodologies presented here underscore that the route of immunization—mucosal versus systemic—fundamentally dictates the quality, location, and longevity of adaptive immunity. While systemic vaccines excel at generating strong serum IgG, mucosal strategies offer the superior advantage of eliciting a first line of defense at the portal of entry via tissue-resident memory B and T cells and secretory IgA. Cutting-edge research reveals that the quality of the antibody response is not merely a function of the magnitude of the GC reaction, but is finely regulated by mechanisms such as affinity-dependent somatic hypermutation. The experimental tools and protocols detailed in this guide provide a roadmap for researchers to dissect these complex processes and design next-generation vaccines capable of overcoming immune regulation to provide sterilizing immunity and superior protection.
Head-to-Head Protection: Comparative Efficacy of Mucosal vs. Systemic Vaccination
The strategic induction of adaptive immunity at portal of entry represents a paradigm shift in vaccinology. For pathogens that invade via mucosal surfaces, the presence of local immune sentries—specifically, tissue-resident memory B cells (BRM) and secretory IgA (SIgA)—can determine the success or failure of the host's frontline defense [56] [57]. This guide provides a direct objective comparison between mucosal and intramuscular (i.m.) vaccination routes for their capacity to generate these crucial mucosal immune components. The data, derived from recent immunological research, underscores a fundamental principle: the route of antigen administration is a critical determinant in positioning functional immune memory at the site of potential infection.
Quantitative Comparison of Key Immune Outcomes
The table below summarizes experimental data from animal and human studies, directly comparing immune outcomes following mucosal (intranasal or aerosol) versus intramuscular vaccination.
Table 1: Comparative Immune Outcomes of Mucosal vs. Intramuscular Vaccination
| Immune Parameter | Mucosal Vaccination | Intramuscular Vaccination | Supporting Evidence |
|---|---|---|---|
| SIgA in Respiratory Mucosa | Induced (superior for luminal secretion) [58] | Not induced/Low (relies on serum exudation) [58] [59] | Mouse influenza model: IgA in BAL fluid only after intranasal, not intraperitoneal, immunization [58]. |
| Tissue-Resident Memory B Cells (BRM) | Robust induction in lung tissue [58] [1] | Minimal to none in lung tissue [17] [58] | Parabiosis and IV labeling in mice confirmed residency of IgA+ B cells after intranasal immunization [58]. |
| Pathogen-Specific IgA in BAL Fluid | High titers [60] [58] | Low/Undetectable titers [58] | COVID-19 mucosal vaccine study: Orally aerosolized vaccine showed 65% IgA positive conversion rate in sputum [60]. |
| Systemic IgG Response | Induced [58] [57] | Robustly induced (often the primary response) [57] [59] | Comparable serum virus-specific antibody levels after intranasal vs. intraperitoneal immunization in mice [58]. |
| Protection Against Heterologous Challenge | Superior (correlated with local IgA and BRM) [58] | Limited (primarily protects against systemic disease) [59] | Local IgA secretion in lung correlated with better protection against secondary homologous and heterologous virus infection [58]. |
Underlying Mechanisms and Signaling Pathways
The disparate outcomes between vaccination routes are governed by distinct cellular trafficking, priming, and differentiation events.
Cellular Mechanisms of Mucosal Immunity Induction
Mucosal Vaccination: Antigen delivery to mucosal surfaces (e.g., intranasal, aerosol) allows for direct uptake by antigen-presenting cells in the Nasal-Associated Lymphoid Tissue (NALT) or Bronchus-Associated Lymphoid Tissue (BALT) [57]. This local encounter is a prerequisite for the establishment of long-lived BRM and IgA-secreting plasma cells within the lung parenchyma [17] [58]. A key mechanism is the "prime and pull" effect, where systemically primed B cells are recruited to the lung mucosa upon intranasal boosting via CXCR3–CXCL9/CXCL10 signaling [40]. These recruited B cells then differentiate into antigen-specific IgA-secreting plasma cells, a process dependent on CD40 and TGF-β signaling and facilitated by resident memory CD4+ T cells acting as a natural adjuvant [56] [40].
Intramuscular Vaccination: This route primarily primes immune responses in the draining lymph nodes and spleen, generating strong systemic IgG and circulating memory B cells [57] [59]. However, without a local inflammatory or antigenic "pull" signal, these circulating cells do not efficiently extravasate and take up residence in mucosal tissues. Consequently, i.m. vaccination fails to establish a significant pool of BRM or drive the local differentiation of IgA-secreting plasma cells in the respiratory mucosa [17] [58]. The absence of local SIgA means protection relies on circulating antibodies diffusing into the mucosa, which is less effective at preventing initial infection [49] [57].
The following diagram illustrates the key signaling pathway and cellular interactions that drive the formation of mucosal IgA following an intranasal booster, converting systemically primed immunity into local protection.
Detailed Experimental Protocols from Key Studies
To facilitate replication and critical evaluation, this section details the methodologies from pivotal studies cited in this comparison.
Table 2: Key Experimental Protocols for Evaluating Mucosal Immunity
| Study Objective | Model & Immunization Protocol | Key Analytical Methods | Critical Reagents & Tools |
|---|---|---|---|
| Assess route-dependent IgA & BRM induction [58] | Mouse model. Immunization: Intranasal (i.n.) vs. Intraperitoneal (i.p.) with A/PR/8/34 (H1N1) influenza virus. | - Antibody measurement: ELISA on BronchoAlveolar Lavage (BAL) fluid and serum.- Cell residency: Intravenous (i.v.) anti-CD45 antibody labeling to distinguish circulating vs. tissue-resident cells.- Parabiosis: Surgical joining of immunized and naïve mice to confirm tissue residency. | - A/PR/8/34 influenza virus.- Anti-mouse CD45 antibody (for i.v. labeling).- Flow cytometry antibodies: Anti-B220, CD138, IgA. |
| Define mechanism of unadjuvanted nasal booster [40] | Mouse model. Prime: I.m. with BNT162b2 mRNA-LNP. Boost: Day 14, i.n. with unadjuvanted SARS-CoV-2 spike protein. | - Tetramer staining: MHC-I/II tetramers for spike-specific T cells; RBD tetramers for B cells.- Intracellular cytokine staining: For IgA and BLIMP1 in Antibody Secreting Cells (ASCs).- Genetic fate mapping: Aicda-ERT2-Cre-Rosa26-tdTomato Prdm1-EYFP mice to track B cell lineage. | - BNT162b2 mRNA-LNP vaccine.- Recombinant SARS-CoV-2 spike protein.- MHC-I/II and RBD tetramers.- Antibodies: Anti-CD38, GL7, IgA, BLIMP1. |
| Compare COVID-19 mucosal vaccines in humans [60] | Human clinical study. 40 participants immunized with either orally aerosolized Ad5-nCoV or intranasal dNS1-RBD COVID-19 vaccine. | Longitudinal sampling: Nasal secretions and sputum at multiple time points post-immunization.IgA subtyping: Measurement of IgA1 and IgA2 titers against SARS-CoV-2 RBD/Spike.Cytokine profiling. | - Authorized mucosal vaccines: Ad5-nCoV, dNS1-RBD.- ELISA/Specific assays for IgA1/IgA2 subclasses. |
The Scientist's Toolkit: Essential Research Reagents
This table catalogs key reagents and their applications for studying BRM and SIgA responses, as utilized in the cited literature.
Table 3: Essential Research Reagents for Mucosal Immunity Studies
| Reagent / Tool | Primary Function in Research | Example Application |
|---|---|---|
| Fluorescently-labeled anti-CD45 Antibody | Intravenous (i.v.) staining to distinguish circulating (labeled) from tissue-resident (unlabeled) immune cells. | Gold-standard validation of BRM and TRM residency in tissues like lung [58]. |
| Antigen-Specific Tetramers (MHC, RBD) | Identification and phenotypic characterization of antigen-specific T and B cell populations by flow cytometry. | Tracking spike-specific CD4+/CD8+ T cells and RBD-specific B cells after SARS-CoV-2 vaccination [40]. |
| Parabiosis Surgery Model | Creation of a shared circulatory system between two mice to definitively prove long-term tissue residency of lymphocytes. | Confirming that IgA+ plasma cells in the lung are non-circulating and tissue-resident [58]. |
| CXCR3-Knockout Mice | Genetic model to investigate the essential role of the CXCR3 chemokine receptor in lymphocyte homing. | Demonstrating that BRM establishment and IgA production in the lung require CXCR3 [58] [61]. |
| Aicda-ERT2-Cre Fate-Mapping Mice | Genetic lineage tracing of germinal center-derived B cells and their differentiation into plasma cells. | Determining that IgA+ plasma cells after i.n. boost originate from B cells primed during i.m. immunization [40]. |
The collective experimental data provides a clear and consistent narrative: mucosal vaccination strategies are unequivocally superior to intramuscular immunization for the establishment of protective tissue-resident memory B cells (BRM) and secretory IgA at respiratory mucosal surfaces. While intramuscular vaccination remains the gold standard for inducing robust systemic IgG and circulating memory, it largely fails to populate mucosal tissues with the resident lymphocytes and antibodies that constitute the critical first line of defense. The emerging mechanistic understanding of how mucosal boosters, even without adjuvant, can "retool" systemic immunity into local protection via pathways like CXCR3 signaling and CD40/TGF-β collaboration [40] opens new avenues for vaccine design. For researchers and drug developers aiming to block infection and transmission at the portal of entry, the evidence strongly advocates for the continued development and deployment of mucosal vaccination platforms.
The primary goal of any vaccine is to prevent disease, but the strategic imperative for controlling pandemics lies not only in reducing disease severity but also in effectively interrupting viral transmission within populations [35]. While traditional intramuscular vaccines have demonstrated excellent efficacy in inducing systemic immunity and reducing severe illness, their protection of the respiratory mucosa remains limited, especially in blocking the initial stages of infection and subsequent viral shedding [6] [62]. This fundamental limitation became particularly evident during the COVID-19 pandemic, where breakthrough infections occurred frequently among vaccinated individuals, permitting continued viral circulation and the emergence of new variants [62].
Mucosal vaccines represent a paradigm shift in vaccinology by focusing on inducing immunity at the initial site of pathogen entry. These vaccines are administered via non-invasive routes such as intranasal or oral delivery, and their superiority in preventing transmission stems from their unique capacity to elicit a localized immune response at mucosal barriers during the earliest stages of infection [6] [63]. By stimulating the production of secretory IgA (SIgA) antibodies and establishing tissue-resident memory lymphocytes directly at the portal of viral entry, mucosal vaccines provide a critical first line of defense that can prevent pathogen adherence, initial replication, and subsequent shedding to other individuals [63] [62]. This review examines the immunological basis for this superiority, presents comparative efficacy data, and explores the experimental approaches driving this innovative vaccine strategy, with particular focus on the role of tissue-resident memory B cells.
Immunological Mechanisms: Tissue-Resident Memory and Mucosal Defense
The respiratory tract possesses a sophisticated mucosal immune system that functions independently from systemic immunity. When pathogens enter through the nasal passage or airways, this localized immune apparatus mounts a rapid response that can neutralize threats before they establish infection or disseminate to other hosts.
Key Players in Mucosal Immunity
Secretory IgA (SIgA) represents the predominant antibody isotype in mucosal secretions and serves as the first line of adaptive defense. Unlike serum antibodies, SIgA is in a polymeric form (particularly dimeric or tetrameric) and contains a secretory component that enables non-inflammatory neutralization of pathogens [62]. SIgA antibodies effectively inhibit viral binding to mucosal surfaces, thereby preventing pathogen entry into cells and subsequent infection establishment [62]. The concentration of SIgA in mucosal secretions exceeds that of IgG by approximately 2.5-fold, highlighting its dominance at these entry sites [62].
Tissue-resident memory B cells (B~RM~) represent a recently characterized subset of memory B cells that reside permanently in mucosal tissues without recirculating [1]. These cells play a crucial role in generating robust and localized immune responses to respiratory infections, particularly during secondary challenges, by rapidly differentiating into antibody-secreting cells [1]. Their strategic positioning at the site of potential infection allows for a response that is both faster and more targeted than that of their circulating counterparts.
Tissue-resident memory T cells (T~RM~) similarly persist long-term in mucosal tissues and respond more rapidly than systemic memory T cells upon reinfection [6]. These cells are regulated by cytokine signaling within the mucosal microenvironment – with IL-13 derived from innate lymphoid cells (ILC2) promoting T~RM~ differentiation, while IL-17 from ILC3 supports their persistence in mucosal tissues [6].
The following diagram illustrates the coordinated immune response in the respiratory mucosa following vaccination:
The Advantage of Localized Immune Memory
The strategic positioning of tissue-resident memory cells provides mucosal vaccines with a decisive advantage in preventing transmission. Whereas intramuscular vaccination primarily generates circulating antibodies and systemic memory cells that must be recruited to sites of infection, mucosal vaccination establishes permanent sentinels at the precise locations where pathogens initiate infection [62]. Upon encountering the pathogen for a second time, these tissue-resident memory cells quickly exert effector functions, thereby limiting disease progression and halting viral replication before significant shedding occurs [35]. Studies have demonstrated that intranasal vaccination can induce the formation of tissue-resident memory T cells (such as CD69⁺CD103⁺ variants), germinal center reactions, and memory B cells in the respiratory tract that persist locally for at least six months and play a pivotal role in limiting viral replication and transmission [6].
Clinical and Preclinical Evidence: Efficacy Comparison
Substantial evidence from both clinical trials and preclinical studies demonstrates the superior ability of mucosal vaccines to prevent initial infection and reduce viral shedding compared to systemic vaccination approaches.
Meta-Analysis of Vaccine Efficacy
A comprehensive systematic review and meta-analysis evaluated the immunogenicity and protective efficacy of mucosal vaccines across multiple respiratory pathogens, providing robust comparative data [64]. The analysis included 65 studies with 229,614 participants and revealed important insights into the performance of mucosal vaccines across different platforms and delivery methods.
Table 1: Comparative Efficacy of Mucosal Vaccines from Meta-Analysis [64]
| Vaccine Target | Vaccine Type / Route | Efficacy / Immunogenicity Measure | Result | Comparison to Systemic Vaccines |
|---|---|---|---|---|
| COVID-19 | Mucosal (Overall) | Neutralizing Antibodies (Wild-type) | SMD = 2.48, 95% CI: 2.17–2.78 | Higher |
| COVID-19 | Mucosal (Overall) | Neutralizing Antibodies (Omicron) | SMD = 1.95, 95% CI: 1.32–2.58 | Higher |
| COVID-19 | Inhaled | Vaccine Efficacy | VE = 47%, 95% CI: 22–74% | N/A |
| COVID-19 | Intranasal | Vaccine Efficacy | VE = 17%, 95% CI: 0–31% | N/A |
| Influenza | Mucosal (Children) | Vaccine Efficacy | VE = 62%, 95% CI: 30–46% | Comparable |
| RSV | Mucosal | Seroconversion Rate | 73% | N/A |
| Pertussis | Mucosal | Seroconversion Rate | 52% | N/A |
The notably higher neutralizing antibody responses induced by mucosal COVID-19 vaccines, particularly against the challenging Omicron variant, demonstrate their enhanced capacity to block infection at the point of entry [64]. The variation in efficacy between inhaled and intranasal COVID-19 vaccines highlights the importance of delivery method and vaccine formulation in achieving optimal protection.
Direct Evidence of Transmission Blocking
Beyond statistical measures of efficacy, direct experimental evidence demonstrates the capacity of mucosal vaccines to reduce viral transmission:
NDV-Vectored SARS-CoV-2 Vaccine: Intranasal administration of a live Newcastle disease virus (NDV)-vectored vaccine expressing SARS-CoV-2 spike protein (NDV-HXP-S) provided protection against SARS-CoV-2 challenge and, crucially, prevented direct-contact transmission in hamsters [65]. This animal model provides direct evidence of transmission interruption, a key public health goal that has been difficult to achieve with systemic vaccines.
Virus-Like Vesicle (VLV) Platform: A COVID-19 vaccine candidate using a virus-like vesicle platform demonstrated that intranasal boosting significantly enhanced mucosal immunity, including IgA production and recruitment of CD4+ T, CD8+ T, and B cells in bronchoalveolar lavage fluid [63]. This localized immune cell recruitment correlates with enhanced protection against initial infection at the respiratory mucosa.
Multivalent Mucosal Vaccination: A trivalent live NDV-HXP-S vaccine (containing Wuhan, Beta, and Delta variants) induced more cross-reactive antibody responses against the phylogenetically distant Omicron variant than the ancestral vaccine alone [65]. Furthermore, intranasal trivalent NDV-HXP-S effectively boosted both systemic and mucosal immunity in mice pre-immunized with mRNA vaccine, creating a comprehensive immune defense that bridges systemic and mucosal compartments [65].
Experimental Approaches: Methodologies for Assessing Mucosal Immunity
To evaluate the efficacy of mucosal vaccines and their ability to prevent transmission, researchers employ specialized experimental protocols that assess both immunological parameters and functional protection.
Assessment of Mucosal Immune Responses
Table 2: Key Methodologies for Evaluating Mucosal Vaccine Efficacy
| Method | Experimental Details | Key Measurements | Application in Cited Studies |
|---|---|---|---|
| Antibody Measurement | ELISA of mucosal secretions (nasal washes, BALF); Neutralization assays | SIgA titers; Neutralizing capacity against specific variants | VLV-S-FL study showed enhanced mucosal IgA after intranasal boost [63] |
| Cell Isolation & Characterization | Flow cytometry of lung tissue, BALF; intracellular cytokine staining | T~RM~ (CD69⁺CD103⁺); B~RM~ populations; cytokine production | Identification of CD4+/CD8+ T cell recruitment in BALF [63] |
| Viral Challenge & Transmission Models | Hamster direct-contact model; SARS-CoV-2 challenge post-vaccination | Viral load in respiratory tissues; transmission to co-housed animals | NDV-HXP-S study demonstrated reduced transmission [65] |
| Biodistribution Studies | Bioluminescent imaging (rNDV-luc); tissue viral titers | Vaccine vector distribution; persistence at mucosal sites | Confined NDV replication to respiratory tract [65] |
The following diagram illustrates a typical experimental workflow for evaluating mucosal vaccine efficacy in animal models:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Mucosal Vaccine Development
| Reagent / Material | Function / Application | Specific Examples from Literature |
|---|---|---|
| Viral Vectors | Antigen delivery; immune activation | NDV-HXP-S [65]; Adenovirus vectors [8] |
| Virus-Like Vesicles (VLV) | Self-amplifying RNA platform; antigen presentation | VLV-S-FL (full-length spike) [63] |
| Animal Models | Vaccine efficacy; transmission studies | Golden Syrian hamsters [65]; C57BL/6J mice [63] |
| Detection Antibodies | Immune cell phenotyping; cytokine measurement | Anti-CD69/CD103 for T~RM~ [6]; anti-IgA for mucosal antibodies [63] |
| Molecular Imaging Tools | Vaccine biodistribution; tracking | rNDV-luciferase for in vivo imaging [65] |
The evidence comprehensively demonstrates that mucosal vaccines provide superior protection against initial infection and viral shedding compared to conventional systemic vaccines. This advantage stems from their unique capacity to induce tissue-resident memory B and T cells and generate secretory IgA directly at the site of pathogen entry, creating an early barrier that prevents establishment of infection and subsequent transmission [6] [1] [62]. The documented efficacy of mucosal vaccines across multiple platforms – including viral vectors, virus-like vesicles, and live attenuated designs – underscores the robustness of this approach [63] [64] [65].
For researchers and drug development professionals, the implications are substantial. First, the strategic focus on tissue-resident memory B cells (B~RM~) provides both a mechanistic explanation for mucosal vaccine superiority and a crucial correlate of protection for future vaccine development [1]. Second, the successful application of mucosal approaches across multiple pathogen targets – including COVID-19, influenza, and RSV – suggests this strategy may represent a universal platform for combating respiratory viruses with pandemic potential [6] [64]. Finally, the demonstrated ability of mucosal vaccines to boost pre-existing immunity from prior vaccination or infection offers a promising strategy for creating comprehensive protection that bridges both mucosal and systemic compartments [65].
As the field advances, the development of safe, effective, and broadly protective mucosal vaccines carries significant public health implications and represents a key trend in the evolution of vaccine technology [6]. Their potential to rapidly deploy during outbreaks, improve patient acceptance through needle-free administration, and ultimately block transmission chains positions mucosal vaccination as a cornerstone strategy for pandemic preparedness and the control of respiratory infectious diseases.
A paradigm shift is underway in vaccinology, moving beyond traditional intramuscular injections toward strategies that elicit immunity where pathogens first enter the body. Heterologous vaccination regimens, which combine different vaccine platforms and administration routes, are emerging as powerful tools to generate comprehensive immune protection, particularly against respiratory pathogens. These approaches strategically leverage the strengths of both systemic and mucosal immunization to overcome the limitations of homologous vaccination.
Central to this advancement is the understanding that while conventional intramuscular vaccines effectively generate circulating antibodies and systemic memory T cells, they often fail to induce robust responses at mucosal portals of entry, such as the respiratory tract [66]. This gap in protection has driven research into "prime-pull" strategies, where parenteral priming is followed by mucosal boosting to recruit immune cells to key barrier tissues [66]. The efficacy of these regimens is closely tied to their ability to generate tissue-resident memory B cells (BRM) and T cells that reside in mucosal tissues and provide rapid frontline defense against invading pathogens [49] [1].
This review synthesizes recent preclinical evidence comparing heterologous and homologous vaccination strategies, with a specific focus on their capacity to induce robust tissue-resident memory responses and overcome immunological barriers such as antigenic imprinting.
Comparative Analysis of Vaccination Regimens: Quantitative Immune Responses
Table 1: Comparison of immune responses across different vaccination regimens
| Vaccination Strategy | Antibody Responses | Mucosal Immunity | T Cell Responses | Protection Against Challenge |
|---|---|---|---|---|
| Heterologous (Systemic Prime + Mucosal Boost) | Significantly higher serum IgG and neutralizing antibodies [66] [67] | Robust mucosal IgA in NALT and BALF; induction of lung BRM cells [66] [68] | Enhanced lung CD4+ and CD8+ T cells; Th1/Th17 skewing [69] | Superior viral control; reduced lung pathology [66] [68] |
| Homologous (Mucosal Only) | Moderate serum antibody responses [69] | Moderate mucosal IgA [69] | Limited long-lived lung T cell responses [69] | Variable protection [69] |
| Homologous (Systemic Only) | High serum IgG but limited neutralizing capacity to variants [66] | Minimal mucosal antibodies [66] | Strong systemic T cells but limited lung residency [66] | Partial protection; reduced prevention of infection [66] |
Table 2: Specific heterologous regimen outcomes against respiratory pathogens
| Pathogen Target | Prime Strategy | Boost Strategy | Key Findings | Reference |
|---|---|---|---|---|
| SARS-CoV-2 | Adenoviral vector (IM) | Omicron spike vaccine (IN) | Overcame immune imprinting; induced de novo lung B cell responses | [66] |
| Influenza & SARS-CoV-2 | mRNA vaccine (IM) | Chimeric protein vaccine (IN) | Enhanced mucosal IgA in nasal washes and BALF; optimal protection against lethal challenge | [68] |
| Mycobacterium tuberculosis | H56+CAF01 (SC) | H56+mmCT (IN) | Induced long-lived Th1/Th17-skewed CD4+ T cell responses in lungs | [69] |
| Burkholderia pseudomallei | Capsular polysaccharide conjugate (SC) | Live attenuated vaccine (SC) | Significant protection against lethal aerosol challenge | [70] |
Mechanisms of Action: How Heterologous Regimens Generate Superior Immunity
Overcoming Immunological Imprinting
A significant challenge in vaccinology against rapidly evolving viruses like SARS-CoV-2 and influenza is "immunological imprinting," where pre-existing immunity to prior strains or vaccines can suppress de novo responses to new variants [66]. This phenomenon, also termed "primary addiction" or "negative interference," occurs when memory B cells generated against the original antigen dominate the response upon re-exposure, preventing the activation of naive B cells capable of recognizing novel epitopes on variant strains [66].
Mucosal boosting circumvents this limitation through spatial separation of immune activation. Research demonstrates that intranasal administration of Omicron-based vaccines bypasses suppressive antibody imprinting established by intramuscular priming with ancestral spike vaccines [66]. Unlike intramuscular boosting, which primarily recalls pre-existing memory responses, mucosal delivery enables the induction of de novo lung B cell responses against the variant strain while still engaging cross-reactive memory T cells [66]. This strategic approach allows for broadening of humoral immunity against emerging variants while maintaining the benefits of pre-existing cellular immunity.
Induction of Tissue-Resident Memory
The differential trafficking of immune cells following mucosal versus systemic vaccination explains the superior local immunity generated by heterologous regimens. Intramuscular vaccination typically induces effector cells that circulate systemically but do not efficiently populate mucosal tissues. In contrast, mucosal vaccination expresses specific homing receptors that direct cells to reside in the respiratory tract [49].
Following influenza infection, memory B cells establish residence in the lungs within the first two weeks, with CCR6 and CXCR3 identified as key chemokine receptors for this localization [49]. Type I interferon induced after viral infection stimulates lung fibroblasts to produce CXCL13, which recruits CXCR5-expressing B cells to the lungs [49]. These tissue-resident memory B cells (BRM) are positioned for rapid response upon pathogen encounter, capable of quickly differentiating into antibody-secreting cells that produce virus-specific immunoglobulins directly at the site of infection [49] [1].
Stromal and Innate Immune Programming
Recent single-cell transcriptomic analyses reveal that heterologous vaccination reprograms the local immune environment at injection sites. Adenoviral priming establishes a "pre-conditioned" innate immune environment that is dramatically amplified upon mRNA boosting, particularly through fibroblast-driven chemokine responses that promote immune cell recruitment [67]. This trained immunity effect, characterized by epigenetic and metabolic reprogramming of innate immune cells such as monocytes and macrophages, creates a more responsive environment upon subsequent antigen encounter [67].
The heterologous combination of adenoviral vectors and mRNA vaccines elicits higher neutralizing antibody titers and stronger CD8+ T cell responses against variants compared to homologous regimens [67]. This enhanced immunogenicity stems from the unique capacity of adenoviral vaccines to induce prolonged monocyte activation, resulting in enhanced cytokine production and antigen presentation capabilities that can persist for up to three months post-vaccination [67].
Experimental Models and Methodologies
Key Preclinical Models
Murine SARS-CoV-2 immunization and challenge model: Bissett et al. utilized a prime-pull approach where mice were primed intramuscularly with ChAdOx1 nCoV-19 (Ad-WT) encoding ancestral spike protein, followed by intranasal boosting with ChAdOx1-o (Ad-o) encoding Omicron BA.1 spike 4 weeks later [66]. Control groups received homologous IM or IN regimens. Five weeks post-boost, researchers collected systemic (serum, spleen) and mucosal (lungs, nasal-associated lymphoid tissue fluid, bronchoalveolar lavage fluid) samples for immunogenicity assessment before challenging with SARS-CoV-2 Omicron BA.1 [66].
Chimeric antigen immunization against influenza and COVID-19: Xing et al. developed mRNA and protein vaccines incorporating the HA stem of H1N1 influenza virus with the RBD from SARS-CoV-2 Delta variant [68]. Mice received intramuscular mRNA priming followed by intranasal protein boosting with PICKCa adjuvant (a complex of poly IC-kanamycin-Cacl2 that activates TLR3, NOD, and RIG-I receptors) [68]. Three weeks post-boost, mice were challenged with high-dose lethal influenza virus, with viral load measured in lungs and survival monitored for 14 days [68].
Mycobacterium tuberculosis heterologous immunization: Ghaem-Maghami et al. tested a parenteral prime-mucosal pull strategy using the H56 antigen (a fusion of three Mtb antigens) with different adjuvants [69]. Mice were subcutaneously primed with H56+CAF01 (liposomal adjuvant) twice, then intranasally boosted with H56 combined with either mmCT (multiple-mutated cholera toxin) or CTB-CpG (cholera toxin B subunit conjugated to CpG) [69]. Immune responses were assessed by measuring Th1 and Th17 cytokine production from lung and spleen cells [69].
Assessment Techniques
Humoral immunity quantification: ELISA measurements of antigen-specific IgG and IgA in serum, nasal washes, and BALF provide critical data on systemic and mucosal antibody responses [66] [68]. Neutralizing capacity is assessed through ACE-2 competition assays and pseudovirus neutralization assays [66].
Cellular immunity evaluation: Flow cytometry of lung homogenates and splenocytes characterizes T cell phenotypes and cytokine profiles (IFN-γ, IL-17, etc.) [69]. Intracellular cytokine staining after antigen re-stimulation identifies antigen-specific T cells [69].
Protection assessment: Viral challenge studies measure clinical outcomes including survival, weight loss, and viral load in lungs (quantified by TCID50 or plaque assays) [68]. Histopathological analysis of lung tissue evaluates inflammation and damage [68].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents for studying heterologous vaccination regimens
| Reagent Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| Vaccine Platforms | Adenoviral vectors (ChAdOx1), mRNA-LNPs, protein subunits | Antigen delivery | Different platforms leverage distinct immune activation pathways |
| Mucosal Adjuvants | mmCT (multiple-mutated cholera toxin), CTB-CpG, PICKCa, CAF01 | Enhancing mucosal immunogenicity | Promote antigen uptake, dendritic cell activation, and tissue-homing receptor expression |
| Immunoassay Reagents | ELISA kits for IgG/IgA, fluorescently-labeled antibodies for flow cytometry, MHC tetramers | Immune response quantification | Detect and characterize antibody and T cell responses |
| Animal Models | BALB/c mice, C57BL/6 mice, CB6F1/OlaHsd mice, hamster challenge models | Preclinical efficacy testing | Model human immune responses and disease pathogenesis |
| Challenge Strains | SARS-CoV-2 variants, influenza virus strains, Burkholderia pseudomallei | Protection assessment | Evaluate vaccine efficacy against relevant pathogens |
The accumulated evidence strongly supports heterologous regimens combining systemic priming with mucosal boosting as a superior strategy for generating comprehensive immunity against respiratory pathogens. These approaches uniquely address the limitations of conventional vaccination by overcoming immunological imprinting, establishing tissue-resident memory populations, and leveraging innate immune programming. The consistent findings across multiple pathogen models—SARS-CoV-2, influenza, and Mycobacterium tuberculosis—highlight the broad applicability of this strategy.
Future research should focus on optimizing antigen design for mucosal delivery, identifying ideal interval periods between prime and boost, and developing next-generation adjuvants specifically tailored for mucosal administration. Additionally, translational studies are needed to confirm these preclinical findings in human populations and establish correlates of protection mediated by tissue-resident memory cells. As respiratory pathogens continue to pose significant global health threats, heterologous vaccination regimens represent a promising frontier in the development of more effective vaccines that not only reduce disease severity but prevent infection and transmission altogether.
The pursuit of next-generation vaccines has brought mucosal immunization to the forefront of infectious disease research. Unlike conventional intramuscular vaccines that primarily elicit systemic immunity, mucosal vaccines target the initial sites of pathogen entry, inducing a specialized immune response characterized by secretory IgA (SIgA) and tissue-resident memory lymphocytes [6]. This localized immunity represents a critical barrier against infection and transmission of respiratory pathogens.
A key advancement in understanding mucosal protection has been the characterization of tissue-resident memory B cells (Brm) in the airways [1]. Although discovered more recently than their T cell counterparts, Brm have emerged as crucial mediators of localized protection against respiratory pathogens. These non-circulating cells persist in mucosal tissues and rapidly differentiate into antibody-secreting plasma cells upon re-encounter with pathogens, providing a first line of defense at the site of infection [17] [1]. This review synthesizes clinical and preclinical evidence for approved mucosal vaccines and late-stage candidates, with particular emphasis on their capacity to induce these specialized tissue-resident memory responses.
Currently Licensed Mucosal COVID-19 Vaccines
The rapid development of COVID-19 vaccines catalyzed unprecedented innovation in mucosal vaccine technology. As of 2025, five mucosal COVID-19 vaccines have received approval in various countries, though none yet from WHO-listed stringent regulatory authorities [71] [72]. These pioneering products provide the first clinical datasets on mucosal immunization against SARS-CoV-2.
Table 1: Licensed Mucosal COVID-19 Vaccines and Clinical Evidence
| Vaccine Name | Platform | Administration | Approving Countries | Key Clinical Findings |
|---|---|---|---|---|
| iNCOVACC | Viral Vector | Intranasal | India | Preclinical models showed induction of SIgA and tissue-resident T cells; associated with reduced viral replication and transmission [71]. |
| Convidecia Air | Viral Vector | Inhaled | China | Phase 4 trial (n=450) showed lower systemic antibodies but higher mucosal immunity vs. mRNA; significantly fewer adverse reactions (29% vs. 73%) [72]. |
| Sputnik V | Viral Vector | Intranasal* | Russia | Primate study showed waning immunity at 2-year follow-up for intranasal route, while injected version maintained immune responses [72]. |
| Pneucolin | Inactivated | Intranasal | China | Specific clinical data not publicly available; part of the approved mucosal vaccine portfolio [71]. |
| RAZI-COV PARS | Protein Subunit | Intranasal | Iran | Limited public clinical data; research emphasizes upregulation of SIgA and tissue-resident memory cells [71]. |
Note: Intranasal Sputnik V is in addition to the injectable version
The clinical evidence, while growing, highlights a significant challenge in the field: robust human efficacy data demonstrating infection and transmission blocking remains limited for most licensed mucosal vaccines [71]. The Convidecia Air Phase 4 trial represents one of the most comprehensive clinical datasets, revealing an intriguing dissociation between systemic antibody responses and mucosal protection [72].
Late-Stage Mucosal Vaccine Candidates in Clinical Development
Beyond currently licensed products, a robust pipeline of mucosal vaccine candidates is advancing through clinical trials, utilizing diverse technological platforms including viral vectors, protein subunits, and live attenuated viruses.
Table 2: Select Late-Stage Mucosal Vaccine Candidates in Clinical Development
| Vaccine Candidate | Platform | Administration | Development Phase | Key Findings & Status |
|---|---|---|---|---|
| MPV/S-2P (NIAID) | Viral Vector | Intranasal | Phase 1 | Trial recruiting 60 participants; completion expected 2025 [72]. |
| Vaxart Oral Vaccine | Viral Vector | Oral | Phase 2b | Trial announced September 2024; proceeding under Project NextGen [72]. |
| CyanVac/Blue Lake Biotech | Viral Vector | Intranasal | Phase 2b | Trial started December 2024; appears to be proceeding [72]. |
| Castlevax | Protein Subunit | Intranasal | Phase 2b (paused) | Grant paused and may be terminated [72]. |
| Codagenix Live Attenuated | Live Attenuated | Intranasal | Not yet initiated | Trial has not apparently started [72]. |
Globally, at least 34 mucosal COVID-19 vaccines have reached clinical trials, though some have been discontinued [72]. The majority are viral vector-based, reflecting the relative maturity of this platform for mucosal delivery [73]. These candidates are being evaluated not only as primary vaccines but also as boosters that can convert pre-existing systemic immunity into mucosal protection [40].
Quantitative Evidence: Meta-Analysis of Mucosal Vaccine Performance
A recent comprehensive meta-analysis evaluated the immunogenicity, safety, and protective efficacy of mucosal vaccines across multiple respiratory pathogens [74]. The analysis incorporated 65 studies with 229,614 participants, providing robust quantitative comparisons between mucosal and traditional intramuscular vaccines.
Table 3: Meta-Analysis of Mucosal Vaccine Performance Across Pathogens
| Pathogen | Neutralizing Antibody Response (SMD vs. IM) | Vaccine Efficacy | Seroconversion Rate | Key Findings |
|---|---|---|---|---|
| COVID-19 | Wild-type: SMD=2.48 (95% CI: 2.17-2.78); Omicron: SMD=1.95 (95% CI: 1.32-2.58) | Inhaled: VE=47% (95% CI: 22-74%); Intranasal: VE=17% (95% CI: 0-31%) | Not specified | Mucosal COVID-19 vaccines elicited higher neutralizing antibodies compared to intramuscular vaccines [74]. |
| Influenza | Not specified | VE=62% (95% CI: 30-46%) in children | Lower than intramuscular vaccines | Protected children well; I²=17.1% indicating low heterogeneity [74]. |
| RSV | Not specified | Not specified | 73% | High seroconversion rate [74]. |
| Pertussis | Not specified | Not specified | 52% | Moderate seroconversion rate [74]. |
The meta-analysis demonstrated that mucosal vaccines are generally immunogenic, safe, and protective, particularly for respiratory diseases [74]. However, the varying efficacy by administration route (inhaled vs. intranasal) highlights the importance of delivery optimization. The safety profile was comparable to intramuscular vaccines or placebo, with no detected publication bias [74].
Experimental Models: Decoding Mucosal Immune Mechanisms
Key Methodologies for Assessing Mucosal Immunity
Research into tissue-resident memory responses employs specialized experimental protocols designed to distinguish locally resident immune cells from their circulating counterparts:
Parabiosis Models: Surgical pairing of mice with conjoined circulatory systems enables researchers to distinguish between tissue-resident and circulating immune cells. CD45 congenic markers (CD45.1 vs. CD45.2) allow precise tracking of cell origin after mucosal booster immunization [40].
Intravenous CD45 Labeling: Antibody-based labeling of circulating leukocytes before tissue collection distinguishes cells inside blood vessels (labeled) from those in tissues (unlabeled). This method is crucial for identifying genuine tissue-resident populations [17] [40].
Tetramer Staining for Antigen-Specific Cells: MHC-I and MHC-II tetramers loaded with pathogen-specific peptides (e.g., SARS-CoV-2 spike protein epitopes) enable identification and quantification of antigen-specific T cells in mucosal tissues [40].
B Cell Fate Mapping: Genetic models like Aicda-ERT2-Cre-Rosa26-tdTomato Prdm1-EYFP mice allow permanent labeling of activated B cells and their plasma cell descendants, tracing the lineage of mucosal IgA-producing cells back to their systemic priming [40].
Critical Signaling Pathways in Mucosal Immunity
Mucosal Booster Immune Conversion Pathway
This diagram illustrates the key mechanism by which an intranasal booster converts pre-existing systemically-primed immunity into mucosal protection, as demonstrated in mouse models [40]. The process involves coordinated chemokine signaling and lymphocyte recruitment, with CD4+ T cells functioning as natural adjuvants through their production of CXCL9 and CXCL10.
The Scientist's Toolkit: Essential Reagents for Mucosal Immunity Research
Table 4: Key Research Reagent Solutions for Mucosal Immunity Studies
| Reagent/Cell Type | Key Markers | Research Function | Experimental Application |
|---|---|---|---|
| Tissue-Resident Memory B Cells (Brm) | CD69+, CXCR3+, CD44+ [17] | Local antibody production | Identification in lung tissue by flow cytometry; functional assessment in challenge models [1]. |
| Tissue-Resident Memory T Cells (Trm) | CD69+, CD103+, CXCR3+ [17] | Rapid local pathogen control | Tetramer staining for antigen specificity; cytokine production assays [6] [40]. |
| MHC Tetramers | Pathogen-specific peptide-loaded | Antigen-specific cell detection | Identification of pathogen-specific T cells in mucosal tissues by flow cytometry [40]. |
| CD45 Congenic Markers | CD45.1 vs. CD45.2 | Cell origin tracking | Parabiosis models to distinguish tissue-resident vs. circulating cells [40]. |
| Aicda-ERT2-Cre-Rosa26-tdTomato | tdTomato+, EYFP+ | B cell fate mapping | Tracing the lineage of activated B cells to mucosal plasma cells [40]. |
These specialized reagents enable researchers to dissect the complex cellular dynamics of mucosal immunity, particularly the differentiation and maintenance of tissue-resident memory populations that constitute the hallmark of effective mucosal vaccination.
The accumulated clinical and preclinical evidence firmly establishes that mucosal vaccination represents a viable strategy for inducing protective tissue-resident memory responses against respiratory pathogens. The licensed COVID-19 mucosal vaccines and advanced candidates in clinical development provide proof-of-concept for this approach. However, significant challenges remain, including the variability in efficacy between different administration routes, the need for more durable mucosal immunity, and the requirement for more robust human efficacy data demonstrating infection and transmission blocking [74] [71] [72].
Future research priorities should include standardized assessment of mucosal immune correlates of protection, optimization of delivery platforms and adjuvants, and development of strategies to enhance the longevity of tissue-resident memory B and T cell responses. As the field advances, mucosal vaccines targeting not only SARS-CoV-2 but also other respiratory pathogens like influenza and RSV hold promise for transforming respiratory disease control by potentially blocking infection at its initial site of establishment.
Conclusion
The strategic induction of tissue-resident memory B cells (BRM) represents a paradigm shift in vaccinology, moving beyond systemic protection to achieve sterilizing immunity at the primary sites of pathogen entry. Mucosal vaccination stands as the most effective strategy to establish this localized defensive wall, capable of not only reducing disease severity but also curtailing community transmission. Future research must prioritize the standardization of BRM-specific correlates of protection, the development of novel and safe mucosal adjuvants, and the design of clinical trials that directly measure tissue-resident memory responses. Overcoming the current challenges will unlock the full potential of BRM-focused vaccines, paving the way for a new generation of immunizations that offer more robust and comprehensive protection against a wide array of mucosal pathogens.
References
- 1. Tissue resident memory B cells mediate protective ... [https://www.sciencedirect.com/science/article/pii/S0165247825001245]
- 2. Tissue-resident immune cells: from defining characteristics ... [https://www.nature.com/articles/s41392-024-02050-5]
- 3. BRM Promoter Polymorphisms and Survival of Advanced Non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5422138/]
- 4. Resident Memory B Cells in Barrier Tissues [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.953088/full]
- 5. Regulation of pulmonary plasma cell responses during ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11044945/]
- 6. Advances and prospects of respiratory mucosal vaccines [https://www.nature.com/articles/s41541-025-01280-0]
- 7. B cells modulate lung antiviral inflammatory responses via ... [https://www.nature.com/articles/s41590-025-02124-8]
- 8. Editorial: Mucosal Immunity after Vaccination [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1690399/abstract]
- 9. Germinal center [https://en.wikipedia.org/wiki/Germinal_center]
- 10. Molecular mechanism and function of CD40/CD40L ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3826168/]
- 11. Immune metabolism regulation of the germinal center ... [https://www.nature.com/articles/s12276-020-0392-2]
- 12. Evidence for a novel function of the CD40 ligand as ... [https://www.sciencedirect.com/science/article/pii/S0014579397013069]
- 13. Comparison of Systemic and Mucosal Immunization with ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0067574]
- 14. Tissue resident memory B cells mediate protective ... [https://pubmed.ncbi.nlm.nih.gov/40967276/]
- 15. Comparison of systemic and mucosal vaccination - PubMed [https://pubmed.ncbi.nlm.nih.gov/22031182/]
- 16. Systemic and Mucosal B and T Cell Responses Upon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7921309/]
- 17. Pivotal role of tissue-resident memory lymphocytes in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10518612/]
- 18. Prior vaccination enhances immune responses during SARS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9934532/]
- 19. Memory persistence and differentiation into antibody ... [https://elifesciences.org/articles/79254]
- 20. Antibody-secreting cell destiny emerges during the initial ... [https://www.nature.com/articles/s41467-020-17798-x]
- 21. Single-Cell Technologies for the Study of Antibody-Secreting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8841722/]
- 22. Comprehensive analyses of B-cell compartments across ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7731793/]
- 23. Multimodal profiling reveals tissue-directed signatures of ... [https://www.nature.com/articles/s41590-025-02241-4]
- 24. Simultaneous profiling of the blood and gut T and B cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447526/]
- 25. ViCloD, an interactive web tool for visualizing B cell ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10304752/]
- 26. B cell memory: from generation to reactivation [https://www.nature.com/articles/s41420-024-01889-5]
- 27. Advances and prospects of respiratory mucosal vaccines [https://pmc.ncbi.nlm.nih.gov/articles/PMC12612081/]
- 28. Nasal Vaccines and the Future of Immunization [https://asm.org/articles/2025/september/nasal-vaccines-future-immunization]
- 29. Oral Vaccines: A Better Future of Immunization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10383709/]
- 30. Safety Assessment of a Sublingual Vaccine Formulated with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11945353/]
- 31. Intranasal vaccine combining adenovirus and trimeric ... [https://www.nature.com/articles/s41551-025-01517-2]
- 32. Modulation of oral vaccine efficacy by the gut microbiota [https://www.nature.com/articles/s41541-025-01240-8]
- 33. Intranasal Vaccine Trial Results and More on Preventing ... [https://absolutelymaybe.plos.org/2025/05/31/intranasal-vaccine-trial-results-and-more-on-preventing-covid-infection-nextgen-vax-update-29/]
- 34. A Bumper Update on Next Generation Covid Vaccines (No 34) [https://absolutelymaybe.plos.org/2025/11/06/a-bumper-update-on-next-generation-covid-vaccines-no-34/]
- 35. Mucosal immunity and vaccination strategies: current insights ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12364801/]
- 36. Secondary influenza challenge triggers resident memory B ... [https://www.sciencedirect.com/science/article/pii/S1074761322001261]
- 37. Current landscape and challenges in adjuvant and antigen ... [https://www.sciencedirect.com/science/article/pii/S2590136225001299]
- 38. Pivotal role of tissue-resident memory lymphocytes in the ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1216402/full]
- 39. T cell epitope mapping reveals immunodominance of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11527131/]
- 40. Mucosal unadjuvanted booster vaccines elicit local IgA ... [https://www.nature.com/articles/s41590-025-02156-0]
- 41. Tissue-resident immune cells in cervical cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC12053169/]
- 42. Local B-cell immunity and durable memory following live- ... [https://pubmed.ncbi.nlm.nih.gov/40791425/]
- 43. Intranasal booster induces durable mucosal immunity against ... [https://pubmed.ncbi.nlm.nih.gov/40624061/]
- 44. Heterologous prime-pull mucosal vaccination with an ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1673460/full]
- 45. Vaccination and SIDS: Mucosal vs. Systemic Immunity ... [https://esmed.org/vaccination-and-sids-mucosal-vs-systemic-immunity-insights/]
- 46. Nanoemulsion adjuvantation strategy of tumor-associated antigen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7549439/]
- 47. Advances in vaccine adjuvant development and future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12180328/]
- 48. B-cell and antibody responses to SARS-CoV-2 [https://www.nature.com/articles/s41423-023-01095-w]
- 49. Development and function of tissue-resident memory B cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701802/]
- 50. Systemic and Mucosal Antibody Responses to SARS-CoV ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12031244/]
- 51. Impact of systemic SARS-CoV-2 vaccination on mucosal IgA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12466159/]
- 52. Tissue‐Resident T Cells That Promote Humoral Immunity [https://pmc.ncbi.nlm.nih.gov/articles/PMC12402542/]
- 53. T follicular helper cells in primary immune regulatory disorders [https://pubmed.ncbi.nlm.nih.gov/40553897/]
- 54. Highly functional and prolonged germinal center T follicular ... [https://www.sciencedirect.com/science/article/abs/pii/S1074761325004625]
- 55. Regulated somatic hypermutation enhances antibody ... [https://www.nature.com/articles/s41586-025-08728-2]
- 56. Inducing mucosal IgA: A challenge for vaccine adjuvants and delivery systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5719502/]
- 57. Induction of protective immune responses at respiratory mucosal sites - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11221474/]
- 58. Intranasal priming induces local lung-resident B cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8762609/]
- 59. Induction of mucosal immunity through systemic immunization: Phantom or reality? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4962944/]
- 60. Kinetics of IgA Subtypes and Cytokines in Respiratory ... [https://pubmed.ncbi.nlm.nih.gov/41081494/]
- 61. IgA-producing B cells in lung homeostasis and disease [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1117749/full]
- 62. SARS-CoV-2 Vaccines: The Advantage of Mucosal ... [https://www.mdpi.com/2076-393X/12/7/795]
- 63. Immunization with virus-like vesicle-based COVID-19 ... [https://www.nature.com/articles/s41541-025-01260-4]
- 64. Immunogenicity, Safety, and Protective Efficacy of Mucosal ... [https://www.mdpi.com/2076-393X/13/8/825]
- 65. Mucosal multivalent NDV-based vaccine provides cross- ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1524477/full]
- 66. Heterologous mucosal vaccine boosting enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574661/]
- 67. Heterologous prime-boost vaccination drives stromal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12151836/]
- 68. Heterologous sequential immunization using chimeric ... [https://www.nature.com/articles/s41598-025-22414-3]
- 69. Heterologous prime-boost immunization combining ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1599713/full]
- 70. Comparison of homologous and heterologous vaccination ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1596265/full]
- 71. A review of currently licensed mucosal COVID-19 vaccines [https://www.sciencedirect.com/science/article/pii/S0264410X2500653X]
- 72. More New Covid Vaccine Candidates, Trials, and Preclinical ... [https://absolutelymaybe.plos.org/2025/08/31/more-new-covid-vaccine-candidates-trials-and-preclinical-studies-nextgen-update-32/]
- 73. Mucosal COVID-19 vaccines in clinical development [https://www.sciencedirect.com/science/article/pii/S0264410X25008990]
- 74. Immunogenicity, Safety, and Protective Efficacy of Mucosal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389829/]