The Charge Effect: How Protein Net Charge Governs Migration and Resolution in Native PAGE
This article provides a comprehensive analysis of how a protein's intrinsic net charge dictates its electrophoretic mobility in Native Polyacrylamide Gel Electrophoresis (PAGE).
The Charge Effect: How Protein Net Charge Governs Migration and Resolution in Native PAGE
Abstract
This article provides a comprehensive analysis of how a protein's intrinsic net charge dictates its electrophoretic mobility in Native Polyacrylamide Gel Electrophoresis (PAGE). Tailored for researchers and drug development professionals, we explore the fundamental principles of charge-based separation, detail methodological setups for different protein types, address common troubleshooting scenarios related to charge, and validate findings through comparative analysis with denaturing techniques. The guide synthesizes foundational knowledge with advanced applications to empower the study of native protein complexes, enzyme activity, and protein-protein interactions.
The Core Principles: Understanding the Interplay of Charge, Size, and Shape in Native PAGE
Native polyacrylamide gel electrophoresis (Native PAGE) is a powerful analytical technique that separates proteins based on their net charge, size, and three-dimensional shape while maintaining their native conformation [1] [2]. Unlike denaturing SDS-PAGE, which disrupts protein structure and renders proteins inactive, Native PAGE preserves protein complexes in their functional state, allowing researchers to study quaternary structure, enzymatic activity, and protein-protein interactions under non-denaturing conditions [1] [3]. This technique is particularly valuable for investigating membrane proteins, protein complexes, and enzymes whose functional properties must be retained for subsequent analysis.
The fundamental principle of Native PAGE relies on the fact that most proteins carry a net negative charge in alkaline running buffers, causing them to migrate toward the anode when an electric field is applied [1]. The rate of migration is determined by the protein's charge density (number of charges per molecular mass) and the frictional force exerted by the gel matrix, which creates a molecular sieving effect [2]. Proteins with higher negative charge density migrate faster, while larger proteins experience greater frictional forces that slow their progression through the gel [1]. This dual dependence on both charge and physical dimensions makes Native PAGE uniquely suited for analyzing proteins in their biologically active states.
Fundamental Principles of Protein Separation in Native PAGE
The Role of Protein Charge in Electrophoretic Migration
In Native PAGE, protein migration is governed primarily by the protein's intrinsic charge at the pH of the running buffer [2] [4]. Unlike SDS-PAGE, where SDS confers a uniform negative charge to all proteins, Native PAGE preserves the protein's native charge distribution, making separation dependent on the protein's isoelectric point (pI) relative to the buffer pH [5]. Proteins with pI values below the buffer pH become negatively charged and migrate toward the anode, while proteins with pI values above the buffer pH carry a positive charge and would theoretically migrate toward the cathode [6]. However, in standard Native PAGE systems using alkaline buffers, most proteins exhibit a net negative charge and migrate toward the anode [1].
The relationship between protein charge and migration can be described by the equation for electrophoretic mobility (μ): μ = ZeffQp/6πRsη where Zeff represents the effective valence (unitless ratio of Coulombic charge to elementary proton charge), Qp is the proton fundamental charge, Rs is the Stokes radius of the protein, and η is the solvent viscosity [5]. This equation highlights how both charge (Zeff) and size (Rs) jointly determine a protein's migration rate through the gel matrix.
Research has demonstrated significant disparities between theoretically calculated protein charges and experimentally measured values. For cytochrome c, the measured valence at pH 7.0 was approximately 2-fold lower than predicted from primary structure analysis, highlighting the complex nature of charge-charge interactions on polyelectrolyte surfaces [5]. These interactions are influenced by buffer composition, ionic strength, and preferential binding of specific ions to the protein surface [5].
Molecular Sieving and the Influence of Protein Size and Shape
The polyacrylamide gel matrix creates a porous network that acts as a molecular sieve, imparting size-dependent frictional resistance to migrating proteins [2] [4]. Smaller proteins navigate the pores more easily and migrate farther, while larger proteins are impeded by the gel matrix [7]. The pore size of the gel is controlled by the acrylamide concentration, with lower percentages (e.g., 6-8%) creating larger pores suitable for high molecular weight complexes, and higher percentages (12-16%) creating smaller pores for better resolution of lower molecular weight proteins [1].
A protein's three-dimensional shape further influences its migration by affecting its hydrodynamic size [7]. A compact, globular protein will migrate faster than an elongated protein of the same molecular weight due to differences in frictional drag [7]. This shape sensitivity enables Native PAGE to detect conformational changes in proteins that alter their hydrodynamic properties without affecting their molecular weight.
Buffer Systems and pH Considerations
The choice of buffer system and pH is critical in Native PAGE as it determines the net charge on proteins and their direction of migration [1] [4]. Different buffer systems operate at specific pH ranges:
Table 1: Native PAGE Buffer Systems and Their Characteristics
| Gel System | Operating pH Range | Key Features | Recommended Applications |
|---|---|---|---|
| Tris-Glycine | 8.3-9.5 | Traditional Laemmle system; preserves native net charge | Studying smaller molecular weight proteins (20-500 kDa) [1] |
| Tris-Acetate | 7.2-8.5 | Better resolution of larger molecular weight proteins | Analyzing proteins >150 kDa [1] |
| NativePAGE Bis-Tris | ~7.5 | Uses Coomassie G-250 to impart negative charge; enables separation regardless of pI | Membrane proteins, hydrophobic proteins, or when separation by molecular weight is desired [1] |
For basic proteins with high pI values, the standard alkaline buffer systems may cause these proteins to carry a net positive charge and migrate toward the cathode, potentially being lost from the gel [6] [4]. Specialized protocols using low pH gel systems or charge-shifting molecules like Coomassie G-250 are employed to ensure anodic migration of basic proteins [1] [4].
Native PAGE Methodologies and Technical Approaches
Blue Native PAGE (BN-PAGE)
Blue Native PAGE (BN-PAGE) is a specialized variant that uses the anionic dye Coomassie Blue G-250 to bind to protein surfaces and confer a net negative charge while maintaining proteins in their native state [1] [6]. This technique, developed by Schägger and von Jagow, overcomes the limitation of traditional native gel electrophoresis by providing a near-neutral operating pH and detergent compatibility [1]. In BN-PAGE, Coomassie G-250 is present in the cathode buffer, providing a continuous flow of dye into the gel during electrophoresis [1].
The binding of Coomassie G-250 offers two significant advantages: (1) it converts basic proteins (pI >7.5) to a net negative charge, allowing them to migrate toward the anode, and (2) it reduces aggregation of membrane proteins and proteins with significant surface-exposed hydrophobic areas by binding nonspecifically to hydrophobic sites and converting them to negatively charged sites [1]. BN-PAGE is particularly valuable for studying membrane protein complexes and mitochondrial complexes, with a separation range from 10 kDa to 10 MDa [6].
Clear Native PAGE (CN-PAGE) and High-Resolution Clear Native PAGE (hrCNE)
Clear Native PAGE (CN-PAGE) and its high-resolution variant (hrCNE) are non-colored alternatives to BN-PAGE that differ primarily in cathode buffer composition [6]. In CN-PAGE, proteins migrate according to their intrinsic isoelectric point without charged compounds that could bind and cause a charge shift [6]. However, CN-PAGE is limited to acidic soluble proteins (pI <7.5), as basic proteins migrate toward the cathode and are lost [6].
High-Resolution Clear Native PAGE (hrCNE) represents an improvement over traditional CN-PAGE. The cathode buffer for hrCNE contains mixed anionic micelles formed from a neutral and an anionic detergent that can bind to hydrophobic proteins and some soluble proteins, similar to Coomassie dye in BN-PAGE [6]. This charge-shift technique makes hrCNE particularly suitable for analyzing fluorescently labeled proteins or conducting in-gel catalytic activity assays where dye interference would be problematic [6].
Comparative Analysis of Native PAGE Variants
Table 2: Comparison of Native PAGE Methodologies
| Method | Charge Modification | Resolution | Advantages | Limitations |
|---|---|---|---|---|
| Traditional Native PAGE | None; relies on intrinsic protein charge | Moderate | Preserves native charge; simple protocol | Limited to acidic proteins in standard alkaline buffers [6] |
| BN-PAGE | Coomassie Blue G-250 binding | High | Compatible with membrane proteins; separates basic and acidic proteins | Dye may interfere with some downstream applications [1] [6] |
| hrCNE | Mixed anionic micelles | High | No dye interference; ideal for fluorescent tags and activity assays | Less robust than BN-PAGE [6] |
Native PAGE separation principles determine the optimal choice of methodology for different protein types and research applications.
Experimental Protocol for Native PAGE
Gel Preparation
The following protocol describes the preparation of a basic non-denatured discontinuous gel for separating acidic proteins [4]:
Separating Gel (17%, 10 mL):
- 4.25 mL 40% Acr-Bis solution (Acr:Bis = 19:1)
- 2.5 mL 4× Separating Gel Buffer (1.5 mol/L Tris-HCl, pH 8.8)
- 3.2 mL Deionized Water
- 35 μL 10% Ammonium Persulfate (APS)
- 15 μL TEMED
Stacking Gel (4%, 5 mL):
- 0.5 mL 40% Acr-Bis solution (Acr:Bis = 19:1)
- 1.25 mL 4× Stacking Gel Buffer (0.5 mol/L Tris-HCl, pH 6.8)
- 3.2 mL Deionized Water
- 35 μL 10% APS
- 15 μL TEMED
The separating gel components (except APS and TEMED) are mixed and degassed before adding polymerization catalysts. After adding APS and TEMED, the solution is poured to about 3/4 of the gel cassette height and overlaid with isopropanol or water to create a flat interface. Once polymerized (approximately 30 minutes), the stacking gel is prepared similarly and poured over the polymerized separating gel. A sample comb is inserted, and polymerization proceeds for another 30 minutes [4].
Sample Preparation
Proper sample preparation is critical for successful Native PAGE separation. For standard Native PAGE, samples are typically mixed with Native Sample Buffer (e.g., Tris-Glycine Native Sample Buffer) containing glycerol to increase density and a tracking dye [1]. It is essential to avoid denaturing agents such as SDS, urea, or reducing agents like β-mercaptoethanol [7].
For BN-PAGE, samples are mixed with NativePAGE Sample Buffer and NativePAGE 5% G-250 Sample Additive [1]. The Coomassie G-250 dye in the sample additive binds to proteins prior to electrophoresis, initiating the charge-shift process. For membrane proteins, solubilization with appropriate detergents (dodecylmaltoside, Triton X-100, or digitonin) is necessary to extract protein complexes while preserving native structure [6].
Electrophoresis Conditions
The polymerized gel is placed in an electrophoresis chamber filled with appropriate running buffer. For Tris-Glycine gels, the running buffer typically consists of 25 mM Tris and 192 mM glycine at pH 8.3-9.5 [1]. For BN-PAGE, specialized cathode and anode buffers are used, with Coomassie G-250 included in the cathode buffer [1] [3].
Samples are loaded into wells, and electrophoresis is performed at constant voltage or current. Typical conditions for a mini-gel system are 100 V constant voltage for approximately 20 minutes until the tracking dye enters the separation gel, then increased to 160 V constant voltage for the remainder of the separation (approximately 60-80 minutes total) [4]. It is crucial to maintain cool temperatures during electrophoresis, often by using a cooling system or performing the run in a cold room, as heating can denature proteins and alter migration patterns [4].
Research Reagent Solutions for Native PAGE
Table 3: Essential Reagents for Native PAGE Experiments
| Reagent | Function | Application Notes |
|---|---|---|
| Acrylamide-Bis Solution | Forms the porous gel matrix | Total concentration (T%) determines pore size; crosslinker ratio (C%) affects gel elasticity [4] |
| Tris-HCl Buffers | Maintains pH during electrophoresis | Different pH for stacking (pH 6.8) and separating (pH 8.8) gels in discontinuous systems [4] |
| Ammonium Persulfate (APS) | Polymerization initiator | Provides free radicals for acrylamide polymerization [4] |
| TEMED | Polymerization catalyst | Accelerates the polymerization reaction initiated by APS [4] |
| Coomassie G-250 | Charge-shift molecule in BN-PAGE | Binds to proteins imparting negative charge without denaturation [1] |
| Glycine | Leading ion in discontinuous systems | Facilitizes stacking of proteins at the stack-separating gel interface [1] |
| Non-ionic Detergents | Solubilizes membrane proteins | Dodecylmaltoside, Triton X-100, or digitonin used for extracting membrane complexes [6] |
| Glycerol | Increases sample density | Added to sample buffer to prevent diffusion from wells during loading [4] |
| Tracking Dyes | Visualize migration progress | Bromophenol blue or Ponceau S used to monitor electrophoresis progression [6] [4] |
Troubleshooting and Optimization Strategies
Common Issues and Solutions
Poor Resolution: Optimize acrylamide concentration gradient for the protein size range of interest. For broad molecular weight ranges, use gradient gels (e.g., 4-16% or 3-12%) [1] [7].
Protein Aggregation: For membrane or hydrophobic proteins, increase detergent concentration or use charge-shift methods like BN-PAGE to reduce aggregation [1] [6].
Altered Migration Patterns: Ensure consistent buffer pH and ionic strength between experiments. Check for proteolysis by including protease inhibitors in sample preparation [7].
In-Gel Artifacts: Run electrophoresis at 4°C to minimize heating effects that can denature proteins. Use fresh ammonium persulfate solutions to ensure complete gel polymerization [4].
Method Selection Guidelines
Choosing the appropriate Native PAGE variant depends on the protein properties and research objectives:
For acidic soluble proteins with pI <7.0: Traditional Native PAGE with Tris-Glycine or Tris-Acetate systems [1]
For basic proteins with pI >7.5: BN-PAGE or specialized low-pH systems with reversed electrode configuration [6] [4]
For membrane protein complexes: BN-PAGE with appropriate detergents (dodecylmaltoside for individual complexes, digitonin for supercomplexes) [6]
For enzymatic activity assays: hrCNE to avoid dye interference with activity measurements [6]
For maximum resolution of complex mixtures: 2D-PAGE combining Native PAGE in the first dimension with SDS-PAGE in the second dimension [8]
Native PAGE remains an indispensable technique in the protein researcher's toolkit, offering unique capabilities for analyzing proteins in their native, functional states. The separation mechanism, dependent on both protein charge and size/shape, provides information complementary to denaturing electrophoretic methods. Through various methodological adaptations—including BN-PAGE, CN-PAGE, and hrCNE—researchers can tailor the technique to diverse protein types and experimental goals.
Understanding how protein charge affects migration in Native PAGE is fundamental to proper experimental design and data interpretation. The effective valence of proteins in electrophoretic conditions often differs significantly from theoretical predictions due to ion-binding and charge-shift phenomena [5]. By selecting appropriate buffer systems, pH conditions, and potential charge-modifying agents, researchers can optimize separation for their specific protein systems, advancing our understanding of protein structure-function relationships in both basic research and drug development applications.
In the realm of protein analysis, gel electrophoresis stands as a fundamental technique for separating complex protein mixtures. At its core, this technique relies on a simple yet powerful principle: charged molecules migrate when subjected to an electric field. Proteins, as amphoteric molecules containing both positively and negatively charged amino acid residues, possess a net charge that varies with their environment. This net charge constitutes the primary driving force behind their electrophoretic mobility in native polyacrylamide gel electrophoresis (PAGE). Unlike denaturing techniques such as SDS-PAGE that obscure intrinsic charge characteristics, native PAGE preserves proteins in their natural conformation, enabling separation based on the combined effects of charge, size, and shape [2] [9]. For researchers in drug development and proteomics, understanding the precise role of net charge is essential for interpreting experimental results, optimizing separation conditions, and studying protein function in near-physiological states.
The direction and speed of a protein's migration in native PAGE are direct reflections of its electrostatic properties at a given pH. When an electric field is applied, proteins with a net negative charge migrate toward the positively charged anode, while those with a net positive charge move toward the negatively charged cathode [10] [2]. The magnitude of this net charge determines the protein's mobility through the gel matrix—proteins with higher charge density experience greater electrostatic pull and migrate faster, assuming similar sizes and shapes. This fundamental relationship between charge and mobility provides the theoretical foundation for native PAGE and forms the basis for its application in characterizing native protein complexes, studying protein-protein interactions, and analyzing functional enzymatic activity [2] [9].
The Biochemical Basis of Protein Charge
Amino Acid Composition and Ionizable Groups
A protein's net charge originates from the ionizable side chains of its constituent amino acids. The specific combination of acidic residues (aspartic acid and glutamic acid), which carry negatively charged carboxylate groups (-COO⁻) at neutral pH, and basic residues (lysine, arginine, and histidine), which carry positively charged groups (-NH₃⁺ for lysine, guanidinium for arginine, and imidazolium for histidine) at neutral pH, determines its overall charge signature [10]. At any given pH, the protein's net charge represents the sum of all these positive and negative charges. Each ionizable group has a characteristic acid dissociation constant (pKₐ) that influences its protonation state across the pH spectrum. Understanding this biochemical foundation is crucial for predicting electrophoretic behavior and designing appropriate buffer systems for separation.
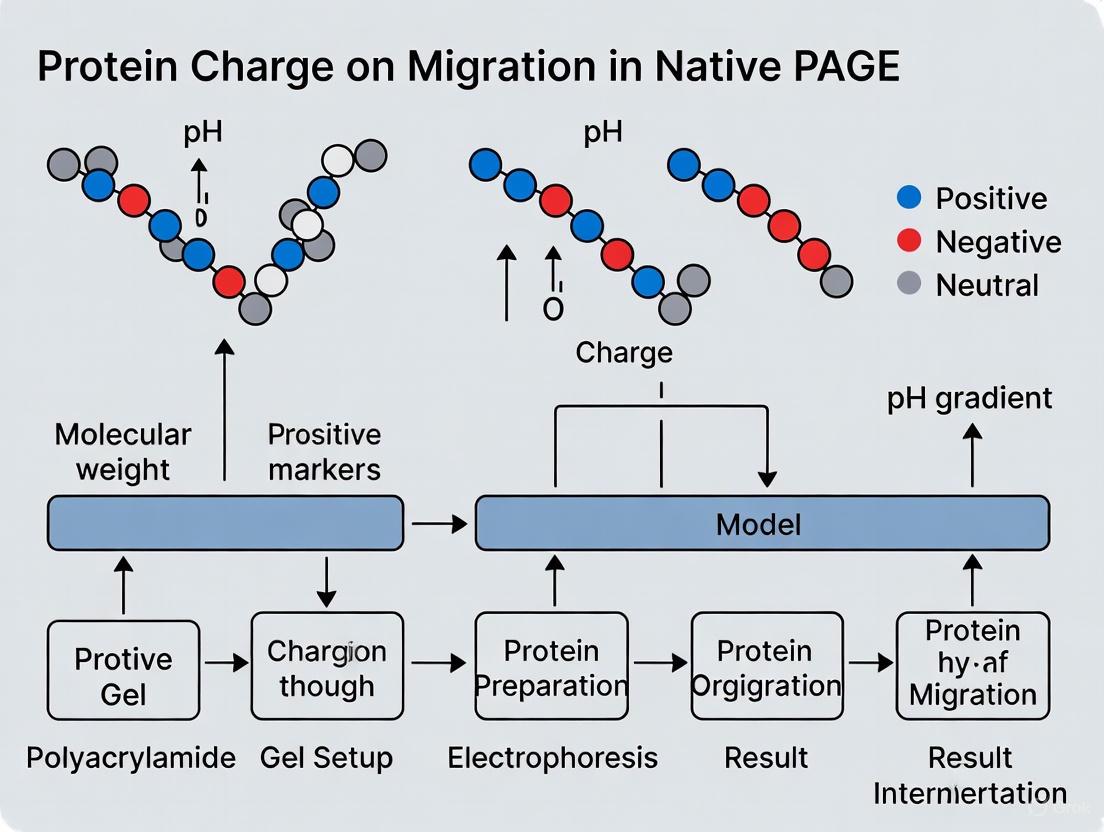
The relationship between a protein's net charge and the surrounding pH is quantified by its isoelectric point (pI), defined as the specific pH at which the protein carries no net electrical charge [10]. This fundamental property serves as a critical predictor of electrophoretic behavior:
- Below its pI: A protein exists in an environment where the pH is lower than its pI, resulting in protonation of basic groups and the protein carrying a net positive charge [10]
- Above its pI: A protein exists in an environment where the pH is higher than its pI, resulting in deprotonation of acidic groups and the protein carrying a net negative charge [10]
Table 1: Relationship Between Buffer pH, Protein Charge, and Migration Direction
| Buffer pH Relative to Protein pI | Net Protein Charge | Migration Direction |
|---|---|---|
| pH < pI | Positive | Toward cathode (-) |
| pH = pI | Zero | No migration |
| pH > pI | Negative | Toward anode (+) |
The Role of Buffer pH in Determining Net Charge
In native PAGE, the buffer pH is the critical experimental parameter that determines a protein's net charge by establishing the protonation state of its ionizable groups [10]. This relationship makes electrophoresis a powerful tool for characterizing protein properties, as researchers can manipulate separation outcomes by simply adjusting the buffer conditions. For example, a protein with a pI of 5.5 will carry a net negative charge in a Tris-glycine buffer at pH 8.3 and migrate toward the anode, whereas the same protein would carry a net positive charge and migrate toward the cathode in a citrate buffer at pH 4.5.
The buffer pH not only determines the direction of migration but also affects its rate. According to Ohm's law, the electrophoretic mobility (μ) of a protein is proportional to its net charge (Q) and inversely proportional to the frictional coefficient (f), which relates to the protein's size and shape: μ = Q/f [2]. Thus, at a constant pH, proteins with higher net charge will migrate faster through the gel matrix than those with lower net charge, assuming similar sizes and shapes. This charge-based separation is the hallmark of native PAGE and distinguishes it from SDS-PAGE, where the uniform charge imparted by SDS binding makes separation primarily dependent on molecular size [2] [9].
Native PAGE: Separation Based on Native Charge Properties
Fundamental Principles and Comparison with SDS-PAGE
Native PAGE represents a fundamental approach in protein separation that preserves the protein's higher-order structure and biological activity. Unlike denaturing electrophoresis methods, native PAGE employs non-denaturing buffers without SDS or reducing agents, maintaining the protein's native conformation, quaternary structure, and post-translational modifications [9]. This preservation enables researchers to study proteins in a state that closely resembles their physiological condition, providing unique insights into functional properties that would be lost under denaturing conditions.
The separation mechanism in native PAGE is multifaceted, depending on the protein's intrinsic charge, molecular size, and three-dimensional shape [2] [9]. The intrinsic charge dictates the electrostatic driving force, while the size and shape collectively determine the frictional resistance experienced as the protein migrates through the porous gel matrix. This complex interplay of factors means that native PAGE can resolve protein species that would co-migrate in SDS-PAGE, such as different oligomeric states of the same polypeptide or proteins with similar mass but different charge characteristics.
Table 2: Key Differences Between Native PAGE and SDS-PAGE
| Feature | Native PAGE | SDS-PAGE |
|---|---|---|
| Separation Basis | Size, shape, and intrinsic charge | Molecular weight (polypeptide chain length) |
| SDS | Absent | Present (denatures and imparts uniform charge) |
| Protein State | Native conformation | Denatured, unfolded |
| Protein Complexes | Preserved | Dissociated |
| Biological Activity | Often retained | Lost |
| Resolution | Lower, more complex interpretation | Higher, simpler interpretation |
| Typical Uses | Studying protein complexes, enzyme activity, interactions | Determining molecular weight, analyzing purity |
Experimental Considerations for Native PAGE
Successful implementation of native PAGE requires careful attention to several experimental parameters. The buffer system must maintain a pH that preserves protein structure while promoting optimal separation. Common buffer systems include Tris-glycine (pH 8.3-8.8) for basic separations and other specialized formulations for specific applications [11]. The polyacrylamide concentration determines the gel pore size and thus the separation range—lower percentages (e.g., 7-10%) are suitable for higher molecular weight proteins, while higher percentages (12-20%) provide better resolution for smaller proteins [2].
Temperature control is another critical factor, as the absence of denaturants makes proteins more vulnerable to heat-induced aggregation or conformational changes. Running native PAGE at 4°C is a common practice to minimize these effects [9]. Additionally, the ionic strength of the buffer must be optimized—too low and proteins may not solubilize properly; too high and excessive current and heating may occur during electrophoresis. Sample preparation is also crucial, requiring gentle lysis conditions with appropriate protease and phosphatase inhibitors to prevent degradation while maintaining native structure [12].
Methodologies and Experimental Protocols
Standard Native PAGE Protocol
The following protocol provides a standardized approach for native PAGE separation, optimized for preserving protein structure and function while achieving high-resolution separation based on native charge properties:
Gel Preparation: Prepare a non-denaturing polyacrylamide gel (typically 6-10% acrylamide depending on protein size) using Tris-HCl or Tris-glycine buffer at the desired pH (commonly pH 8.8 for the resolving gel). Omit SDS from all solutions. Add ammonium persulfate (APS) and TEMED to initiate polymerization. Pour the gel between glass plates and allow it to polymerize completely (approximately 30 minutes) [2] [13].
Sample Preparation: Lyse cells or tissues using a non-denaturing lysis buffer (e.g., Tris-HCl, NP-40, or CHAPS-based buffers) containing protease inhibitors. Maintain samples at 4°C throughout preparation. Centrifuge at 10,000 × g for 10 minutes to remove insoluble debris. Determine protein concentration using a compatible assay (Bradford or BCA). Mix protein sample with native sample buffer (typically containing glycerol, Tris-HCl, and tracking dye) without heating [12].
Electrophoresis: Assemble the gel apparatus and fill both chambers with native running buffer (e.g., Tris-glycine, pH 8.3). Load samples into wells alongside native molecular weight markers. Run electrophoresis at constant voltage (typically 100-150 V for mini-gels) at 4°C until the tracking dye reaches the bottom of the gel [2].
Post-Electrophoresis Analysis: Following separation, proteins can be visualized using compatible staining methods (Coomassie Blue, Silver Stain) or transferred to membranes for western blotting. For functional assays, proteins can be eluted from excised gel bands using appropriate buffers [2] [12].
Specialized Native Electrophoresis Techniques
Beyond standard native PAGE, several specialized techniques offer unique advantages for specific applications:
Blue Native PAGE (BN-PAGE): This technique utilizes Coomassie G-250 dye, which binds to proteins and confers additional negative charges, allowing the separation of membrane protein complexes and high molecular weight assemblies. BN-PAGE is particularly valuable for studying mitochondrial complexes, nuclear pore complexes, and other large macromolecular assemblies [11].
Semi-Native PAGE: This hybrid approach involves separating partially denatured protein samples in gels containing low concentrations of SDS. It enables separation based on differences in structural stability and can be used to screen protein-ligand interactions, particularly with metallocomplexes, as it preserves some native structure while allowing for size-based separation [14].
Isoelectric Focusing (IEF): While not strictly a native technique, IEF separates proteins based solely on their isoelectric points in a pH gradient, providing high-resolution charge-based separation. IEF is often used as the first dimension in two-dimensional gel electrophoresis (2D-PAGE), where proteins are subsequently separated by SDS-PAGE in the second dimension [2] [11].
The Researcher's Toolkit: Essential Reagents and Materials
Successful native PAGE experimentation requires specific reagents and materials optimized for preserving protein structure and function while enabling charge-based separation. The following table details essential components for native PAGE workflows:
Table 3: Essential Research Reagents for Native PAGE
| Reagent/Material | Function/Application |
|---|---|
| Acrylamide/Bis-acrylamide | Forms the porous gel matrix for molecular sieving; concentration determines pore size |
| Tris-based Buffers | Maintains stable pH during electrophoresis; different formulations (Tris-glycine, Bis-Tris) available |
| TEMED and Ammonium Persulfate | Catalyzes acrylamide polymerization to form the polyacrylamide gel matrix |
| Protease Inhibitor Cocktails | Prevents protein degradation during sample preparation, crucial for maintaining native structure |
| Non-ionic Detergents | (e.g., NP-40, Triton X-100) Solubilizes membrane proteins while maintaining native conformation |
| Glycerol | Increases density of protein samples for easier loading into gel wells |
| Native Marker Standards | Proteins of known molecular weight and charge for comparison and calibration |
| Coomassie Blue Stain | Visualizes separated protein bands after electrophoresis; compatible with native structures |
Commercial precast native gels offer convenience and reproducibility for researchers. Companies such as Thermo Fisher Scientific provide specialized native gel systems including NativePAGE Bis-Tris gels, Novex Tris-Glycine gels, and NuPAGE Tris-Acetate gels, each optimized for specific separation needs and protein characteristics [11]. These pre-cast systems eliminate gel-to-gel variability and are particularly valuable for comparative studies and high-throughput applications.
Current Research Applications and Case Study
Investigation of PSME3 in Myoblast Differentiation
A 2025 study investigating the role of the proteasome regulator PSME3 in myoblast differentiation provides an excellent example of native PAGE application in contemporary research [15]. Researchers employed native electrophoresis techniques to characterize PSME3-containing protein complexes and their dynamics during muscle cell differentiation. This approach was crucial for maintaining the native interactions between PSME3 and its binding partners, which would have been disrupted under denaturing conditions.
The study revealed that PSME3 forms specific complexes with proteins including RPRD1A and NUDC, interactions that regulate the levels of adhesion-related proteins and influence cell migration and differentiation [15]. By using co-immunoprecipitation followed by native PAGE analysis, researchers could demonstrate that these complexes remain stable under native conditions, providing evidence for their physiological relevance. This research highlights how charge-based separation techniques continue to enable discoveries in cell biology, particularly in understanding the macromolecular complexes that govern cellular processes.
Technical Workflow for Protein Complex Analysis
The following diagram illustrates a generalized experimental workflow for analyzing protein complexes using native PAGE, as applied in current research:
The migration of proteins in an electric field represents a fundamental phenomenon with profound implications for protein research and biotechnology applications. The net charge of a protein, determined by its amino acid composition and the buffer pH, serves as the primary driver of electrophoretic mobility in native PAGE systems. This charge-based separation principle enables researchers to study proteins in their native, functional states—preserving complex quaternary structures, enzymatic activities, and protein-protein interactions that would be lost under denaturing conditions.
For drug development professionals and research scientists, understanding the role of net charge in protein migration is not merely academic; it provides a practical foundation for experimental design, result interpretation, and method selection. The continuing development of specialized native electrophoresis techniques, including Blue Native PAGE and semi-native approaches, expands the toolbox available for characterizing challenging protein samples. As proteomics research increasingly focuses on macromolecular complexes and functional interactions, charge-based separation methods will remain indispensable for elucidating the complex machinery of biological systems.
Native polyacrylamide gel electrophoresis (PAGE) is a fundamental technique in protein research that separates proteins under non-denaturing conditions, preserving their higher-order structure and biological activity. Unlike denaturing SDS-PAGE, which separates proteins primarily by mass, native PAGE resolves protein mixtures based on the complex interplay of three intrinsic properties: net charge, size, and three-dimensional shape [1]. This multi-parameter separation is crucial for researchers and drug development professionals studying native protein complexes, oligomeric states, and functional proteoforms, as it provides information that is lost in denaturing techniques.
In native PAGE, electrophoretic migration occurs because most proteins carry a net negative charge in alkaline running buffers. The rate of migration is governed by both the protein's charge density and the frictional force imposed by the gel matrix [1]. Proteins with higher negative charge density migrate more rapidly toward the anode, while the gel matrix creates a sieving effect that retards movement according to the protein's size and three-dimensional structure [1]. This dual mechanism enables the separation of complex protein mixtures while maintaining their native conformations, subunit interactions, and enzymatic activities—critical considerations for drug development where functional protein characterization is essential.
Core Separation Mechanisms in Native PAGE
The Interplay of Charge, Size, and Shape
The fundamental separation mechanism in native PAGE relies on three interdependent factors that collectively determine a protein's electrophoretic mobility:
Net Charge and Charge Density: A protein's net charge, determined by the ionization state of its surface amino acid residues at the running buffer pH, creates the electrophoretic driving force. However, charge density—the number of charges per unit mass—is often more significant than absolute charge, as it determines how effectively the electric field propels the molecule through the gel matrix [1]. Proteins with higher negative charge density migrate faster toward the anode.
Molecular Size and Mass: The polyacrylamide gel acts as a molecular sieve, creating frictional resistance that opposes protein migration. Larger proteins experience greater frictional forces and therefore migrate more slowly than smaller proteins with similar charge characteristics [1].
Three-Dimensional Shape and Conformation: A protein's native conformation significantly impacts its mobility through the gel matrix. Compact, globular proteins encounter less resistance than extended or irregularly shaped proteins of equivalent molecular weight, leading to different migration patterns despite similar mass and charge [1].
Because no denaturants are used in native PAGE, subunit interactions within multimeric proteins are generally preserved, allowing researchers to gain information about quaternary structure and complex formation [1]. This preservation of native structure also enables the recovery of enzymatically active proteins following separation, making the technique invaluable for functional studies and preparative applications.
Comparative Analysis of Native PAGE Gel Systems
Three primary gel chemistry systems are available for native PAGE separation, each with distinct operational characteristics and applications. The selection of an appropriate system depends on protein properties and research objectives, as no universal chemistry is ideal for all proteins in their native state [1].
Table 1: Native PAGE Gel Chemistry Systems and Their Applications
| Gel System | Operating pH Range | Key Features | Optimal Use Cases |
|---|---|---|---|
| Novex Tris-Glycine | 8.3 - 9.5 | Traditional Laemmle system; preserves native net charge | Studying smaller MW proteins (20-500 kDa); when maintaining native charge is critical [1] |
| NuPAGE Tris-Acetate | 7.2 - 8.5 | Enhanced resolution for larger molecular weight proteins | Analyzing proteins >150 kDa; maintaining native charge structure [1] |
| NativePAGE Bis-Tris | ~7.5 | Uses Coomassie G-250 dye for charge shifting; resolves proteins by MW regardless of pI | Membrane proteins, hydrophobic proteins, separation primarily by molecular weight [1] |
The Tris-Glycine and Tris-Acetate systems maintain proteins' native net charges, while the Bis-Tris system employs a different mechanism based on the blue native PAGE (BN-PAGE) technique developed by Schägger and von Jagow [1]. This system uses Coomassie G-250 dye to bind proteins and confer a net negative charge while maintaining native conformation, overcoming limitations of traditional native gel electrophoresis through near-neutral pH operation and detergent compatibility [1].
Table 2: Technical Specifications of Native PAGE Systems
| Parameter | Tris-Glycine Gels | Tris-Acetate Gels | NativePAGE Bis-Tris Gels |
|---|---|---|---|
| Available Gel Concentrations | 6%, 8%, 10%, 12%, 14%, 16%, 4–12%, 4–20%, 8–16%, 10–20% | 7%*, 3–8% | 3–12%, 4–16% |
| Recommended Sample Buffer | Tris-Glycine Native Sample Buffer | Tris-Glycine Native Sample Buffer | NativePAGE Sample Buffer, NativePAGE 5% G-250 Sample Additive |
| Recommended Running Buffer | Tris-Glycine Native Running Buffer | Tris-Glycine Native Running Buffer | NativePAGE Running Buffer, NativePAGE Cathode Buffer Additive |
| Shelf Life | Up to 12 months | Up to 8 months | Up to 6 months |
| Storage Conditions | 2-8°C | Room temperature | Room temperature |
*7% polyacrylamide is only available in the mini size [1]
Methodologies and Experimental Protocols
Standard Native PAGE Procedure Using Tris-Glycine System
Sample Preparation:
- Dilute protein samples in Tris-Glycine Native Sample Buffer to desired concentration [1].
- Avoid denaturing agents (SDS, urea, β-mercaptoethanol) and reducing agents that disrupt disulfide bonds.
- Centrifuge at 10,000-15,000 × g for 5-10 minutes to remove insoluble material.
- Maintain samples at 4°C until loading to preserve native state.
Gel Electrophoresis:
- Select appropriate polyacrylamide concentration based on target protein size (refer to Table 2).
- Pre-run gel for 15-30 minutes at constant voltage (recommended: 125V) to establish pH gradient.
- Load samples (typically 10-60 μL per well) alongside native protein standards.
- Run at constant voltage (125V for mini-gels) for approximately 90 minutes or until dye front reaches bottom [1].
- Maintain temperature at 4°C during run using cooling apparatus if necessary.
Post-Electrophoresis Analysis:
- Proteins may be visualized using Coomassie Blue, silver stain, or specialized activity stains.
- For western blotting, use PVDF membranes with Tris-Glycine Transfer Buffer [1].
- Nitrocellulose membranes are not recommended due to poor compatibility with native conditions [1].
Blue Native PAGE (BN-PAGE) Protocol
Based on the technique developed by Schägger and von Jagow, BN-PAGE has become instrumental for analyzing membrane protein complexes and oxidative phosphorylation (OXPHOS) systems [16]:
Membrane Protein Solubilization:
- Solubilize membrane proteins using mild, nonionic detergents such as n-dodecyl-β-d-maltoside [16].
- Include zwitterionic salt 6-aminocaproic acid in extraction buffer to support solubilization without affecting electrophoresis [16].
- Add Coomassie blue G-250 to extracted samples prior to electrophoresis and to cathode buffer [16].
Electrophoresis Conditions:
- Use linear gradient polyacrylamide gels (typically 3-12% or 4-16%) [16].
- The anionic blue dye binds to hydrophobic protein surfaces, imposing a negative charge shift that forces even basic proteins to migrate toward the anode at pH 7.0 [16].
- The induced negative surface charge prevents aggregation of hydrophobic proteins and maintains solubility during electrophoresis [16].
Downstream Applications:
- For supercomplex analysis (e.g., respiratory chain complexes), use digitonin instead of n-dodecyl-β-d-maltoside for milder solubilization [16].
- Combine with second dimension SDS-PAGE for comprehensive complex analysis (BN/SDS-PAGE) [16].
- Perform in-gel enzyme activity staining for functional assessment [16].
- Use clear-native PAGE (CN-PAGE) with mixed detergents instead of Coomassie dye when residual dye interferes with activity staining [16].
Critical Parameter Optimization
pH Considerations:
- Select running pH based on protein stability and isoelectric point (pI).
- Proteins with pI below running buffer pH will carry negative charge and migrate toward anode.
- Proteins with pI above running buffer pH may require charge-shifting systems like BN-PAGE.
Detergent Selection:
- For membrane proteins, choose detergents that maintain native structure without disrupting protein complexes.
- n-dodecyl-β-d-maltoside provides balanced solubilization for many membrane proteins [16].
- Digitonin offers milder solubilization for preserving supercomplexes [16].
Gel Concentration:
- Lower percentage gels (4-8%) better resolve high molecular weight complexes (>500 kDa).
- Higher percentage gels (10-20%) optimal for smaller proteins and subunits (<100 kDa).
- Gradient gels (e.g., 4-16%) provide broad separation range for complex mixtures.
Visualization of Native PAGE Separation Mechanisms
The following workflow diagram illustrates the key separation principles and experimental considerations in native PAGE:
Native PAGE Separation Workflow and Key Parameters
Essential Research Reagents and Materials
Successful native PAGE experimentation requires careful selection of reagents and materials that preserve protein native structure while enabling effective separation.
Table 3: Essential Research Reagents for Native PAGE
| Reagent/Material | Function | Application Notes |
|---|---|---|
| NativePAGE Bis-Tris Gels (3-12%, 4-16%) | Provides polyacrylamide matrix for separation | Commercial pre-cast gels; compatible with BN-PAGE; room temperature storage [1] |
| Coomassie G-250 Dye | Charge-shifting agent for BN-PAGE | Binds hydrophobic protein surfaces; confers negative charge; prevents aggregation [1] [16] |
| n-dodecyl-β-d-maltoside | Nonionic detergent for membrane protein solubilization | Maintains native protein complexes; compatible with activity assays [16] |
| Digitonin | Mild detergent for supercomplex analysis | Preserves labile protein interactions; used for respiratory chain complexes [16] |
| 6-aminocaproic acid | Zwitterionic solubilization aid | Supports membrane protein extraction; zero net charge at pH 7.0 [16] |
| NativeMark Unstained Protein Standard | Native molecular weight standards | Essential for accurate size estimation under native conditions |
| PVDF Membrane | Western blot transfer membrane | Required for NativePAGE gels; nitrocellulose incompatible due to G-250 binding [1] |
| NativePAGE Running Buffer | Electrolyte system for BN-PAGE | Maintains near-neutral pH (7.5); contains cathode buffer additive [1] |
Advanced Applications and Research Implications
The unique ability of native PAGE to separate proteins based on charge, size, and shape simultaneously makes it invaluable for advanced research applications, particularly in drug development and structural proteomics. The technique provides critical insights into protein complex stoichiometry, assembly pathways, and higher-order structures that are often disrupted under denaturing conditions [16].
For researchers studying mitochondrial oxidative phosphorylation (OXPHOS) systems, BN-PAGE has become an indispensable tool for analyzing respiratory supercomplexes and assembly intermediates [16]. The combination of one-dimensional BN-PAGE with second-dimension denaturing electrophoresis (BN/SDS-PAGE) enables comprehensive mapping of complex subunits and identification of assembly defects in metabolic disorders [16]. Furthermore, the preservation of enzymatic activity following native PAGE separation allows direct functional assessment through in-gel activity staining for Complexes I, II, IV, and V [16].
In biopharmaceutical development, native PAGE techniques support characterization of therapeutic proteins, including monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs), by monitoring charge heterogeneity and aggregation states—Critical Quality Attributes (CQAs) mandated by regulatory authorities [17]. The technique's compatibility with downstream mass spectrometry analysis further enhances its utility for comprehensive biotherapeutic characterization [18].
Native PAGE represents a powerful separation methodology that extends beyond simple charge-based separation to incorporate the sieving effects of protein size and three-dimensional shape. The technique's ability to preserve native protein structure and activity provides researchers with unique insights into protein complexes, oligomeric states, and functional characteristics that are essential for both basic research and drug development applications. Through appropriate selection of gel chemistry, buffer systems, and experimental parameters, scientists can leverage the full potential of native PAGE to address complex questions in structural biology and biopharmaceutical analysis.
In native polyacrylamide gel electrophoresis (PAGE), a protein's migration is not solely dictated by its mass but by the complex interplay between its intrinsic isoelectric point (pI) and the pH of the electrophoresis buffer. This relationship directly determines the protein's net charge at run time, thereby influencing its electrophoretic mobility. This technical guide delves into the core principles of how protein charge governs migration in native PAGE. We provide a detailed framework for predicting migration behavior, summarize key native PAGE systems, and present established and novel experimental protocols for the charge-based analysis of proteins, including the determination of pI and the characterization of membrane protein complexes.
Core Principles: pI, Buffer pH, and Net Charge
The fundamental parameter governing protein migration in native PAGE is the net charge the molecule carries during electrophoresis. This net charge is not a fixed property but is dynamically determined by the buffer environment.
- The Isoelectric Point (pI): The pI is the specific pH at which a protein carries no net electrical charge. At this pH, the number of positively charged groups (e.g., from lysine and arginine) equals the number of negatively charged groups (e.g., from aspartic acid and glutamic acid). [19]
- Net Charge in a Running Buffer: When the same protein is placed in an electrophoresis buffer with a pH different from its pI, it will possess a net charge. In a buffer with a pH below the protein's pI, the protein's basic groups become protonated, resulting in a net positive charge. Conversely, in a buffer with a pH above the protein's pI, the protein's acidic groups are deprotonated, resulting in a net negative charge. [20]
The following diagram illustrates this deterministic relationship:
Diagram 1: The relationship between pI, buffer pH, and net charge.
In native PAGE, the applied electric field causes charged proteins to migrate through the porous gel matrix. The direction of migration is determined by the sign of the net charge: positively charged proteins (cationic) migrate toward the cathode (negative electrode), while negatively charged proteins (anionic) migrate toward the anode (positive electrode). The speed of migration is influenced by the charge density (net charge relative to size and shape) and the frictional force imposed by the gel. A protein with a high charge density and compact structure will migrate faster than a large, low-charge protein. [1]
Native PAGE Systems and Their Operational pH
There is no universal native PAGE system ideal for all proteins. The choice of gel chemistry and buffer system is critical and depends on the protein's stability, isoelectric point, and molecular weight. The table below summarizes the characteristics of three common commercial systems.
Table 1: Comparison of Common Native PAGE Gel Chemistries and Their Properties [1]
| Gel System | Operating pH Range | Key Features | Optimal Use Cases |
|---|---|---|---|
| Novex Tris-Glycine | 8.3 - 9.5 | Traditional Laemmle system; proteins retain their native charge. | Studying smaller proteins (20-500 kDa); general native analysis. |
| NuPAGE Tris-Acetate | 7.2 - 8.5 | Provides better resolution for larger molecular weight proteins. | Analyzing large proteins (>150 kDa). |
| NativePAGE Bis-Tris | ~7.5 | Uses Coomassie G-250 dye to impart negative charge; resolves proteins by molecular weight regardless of intrinsic pI. | Membrane proteins, hydrophobic proteins, or when separation by molecular weight is desired in a native state. |
The NativePAGE Bis-Tris system deserves special attention as it uses a unique mechanism to overcome the limitations of intrinsic protein charge. In this system, the anionic Coomassie G-250 dye binds non-specifically to hydrophobic patches on proteins, conferring a relatively uniform negative charge density. This allows even basic proteins (pI > buffer pH) to migrate consistently toward the anode, enabling separation primarily by size and shape. [1]
Experimental Protocols
Protocol 1: Determining the pI of a Native Protein
This protocol outlines the use of vertical isoelectrofocusing (IEF) under non-denaturing conditions to determine the isoelectric point of a native protein from a biological sample, followed by immunoblotting for identification. [19] [21]
Workflow Overview:
Diagram 2: Workflow for native protein pI determination.
Detailed Methodology:
- Sample Preparation: Prepare the biological sample (e.g., cell lysate, tissue homogenate) in a non-denaturing lysis buffer without SDS, reducing agents, or urea to preserve the native state of the proteins. [19]
- Gel Equilibration: Rehydrate a commercial immobilized pH gradient (IPG) strip with the appropriate pH range (e.g., 3-10, 5-8) according to the manufacturer's instructions.
- Sample Loading: Load the prepared native protein sample onto the IPG strip.
- Isoelectrofocusing (IEF): Perform IEF using a vertical electrophoresis unit under pre-optimized conditions (e.g., stepwise increasing voltage). During IEF, a protein with a net charge will migrate through the pH gradient. It will stop migrating at the point in the gradient where the pH equals its pI and it becomes neutrally charged. [19]
- Immunoblotting: Following IEF, proteins are transferred from the IPG gel to a membrane (e.g., PVDF or nitrocellulose) via western blotting under non-denaturing conditions.
- pI Determination: Identify the protein of interest using a specific antibody. The pI is determined by correlating the band's position on the membrane with the known pH gradient of the IPG strip. [19] [21]
Protocol 2: Native PAGE for GPCR-Mini-G Protein Coupling
This membrane protein native PAGE assay is a powerful method to visualize and biochemically characterize agonist-dependent coupling of detergent-solubilized G protein-coupled receptors (GPCRs) to purified mini-G proteins. [22]
Workflow Overview:
Diagram 3: Workflow for GPCR-mini-G protein coupling assay.
Detailed Methodology:
- Receptor Expression and Preparation: Transiently express an EGFP-tagged GPCR in a mammalian cell line (e.g., HEK293S GnT1-). Prepare crude membranes from the harvested cells and solubilize the membrane proteins using a suitable detergent, such as lauryl maltose neopentyl glycol (LMNG), to extract the GPCR while maintaining its native conformation. [22]
- Complex Formation: Incubate the solubilized receptor with the agonist of interest and a purified mini-G protein. Mini-G proteins are engineered, minimal Gα subunits that stabilize the active state of the GPCR. [22]
- High-Resolution Clear Native Electrophoresis (hrCNE): Load the samples onto a native polyacrylamide gel. The hrCNE method is compatible with fluorescently-labeled proteins and detergents. Electrophoresis is then carried out.
- Detection and Analysis: Visualize the EGFP-tagged receptor using in-gel fluorescence imaging. The formation of a stable GPCR-mini-G protein complex will result in a distinct upward mobility shift (slower migration) compared to the receptor alone, due to the increased size and potential alteration in charge of the complex. This assay can be adapted to a quantitative format to determine apparent binding affinities. [22]
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of native electrophoresis experiments requires specific reagents. The following table details key solutions and their functions.
Table 2: Essential Research Reagent Solutions for Native Electrophoresis
| Reagent / Material | Function / Purpose | Key Considerations |
|---|---|---|
| Coomassie G-250 | Charge-shift molecule in NativePAGE Bis-Tris gels; binds proteins to confer net negative charge while maintaining native state. [1] [3] | Added to cathode buffer and sample; enables analysis of basic proteins. Preferable to SDS for native conditions. |
| Mild Detergents (e.g., LMNG) | Solubilizes membrane proteins while preserving native protein-protein interactions and functional state. [22] | Critical for studying membrane protein complexes like GPCRs; choice of detergent is experiment-dependent. |
| Native Sample Buffer | Prepares protein sample for loading without denaturation; typically contains glycerol, tracking dye, and a compatible buffer. | Lacks SDS and reducing agents (DTT, β-mercaptoethanol). May include Coomassie G-250 for certain systems. [1] |
| PVDF Membrane | Recommended blotting membrane for western blotting following NativePAGE. [1] | Binds Coomassie G-250 less tightly than nitrocellulose, making it compatible with the destaining and fixing steps required. |
| Mini-G Proteins | Surrogate, engineered G protein alpha subunits used to trap and stabilize active-state GPCRs for biochemical studies. [22] | Enhance stability of GPCR complexes in detergent solution; available for different G protein families (Gs, Gi/o, Gq). |
| Tris-Based Buffers | Form the basis of most running and sample buffers; provide a constant pH in the continuous buffer system. [20] | Effective pH range can be shifted to ~10.0; high buffering capacity is essential for stable pH during runs. |
In native polyacrylamide gel electrophoresis (Native PAGE), proteins are separated in their folded, functional state, allowing for the analysis of complex quaternary structures, enzymatic activity, and protein-protein interactions. Unlike denaturing SDS-PAGE, which separates proteins primarily by mass, Native PAGE separates proteins according to their charge-to-size ratio within a gel matrix. This technique is indispensable for researchers studying multimeric protein complexes, such as those involved in mitochondrial oxidative phosphorylation (OXPHOS) and fatty acid metabolism, as it preserves native structural and functional characteristics [16].
The fundamental parameter governing migration in Native PAGE is a protein's intrinsic charge, which is determined by its amino acid composition and the pH of the running buffer. In the absence of denaturing detergents like SDS, a protein's surface charge dictates its electrophoretic mobility. When an electric field is applied, negatively charged proteins migrate toward the anode, while positively charged proteins move toward the cathode. The porous gel matrix then acts as a molecular sieve, allowing smaller proteins to migrate faster than larger ones. Consequently, the final position of a protein band reflects a combination of its inherent charge and its hydrodynamic size—the compactness of its three-dimensional structure [3].
Key Methodological Variations in Native Electrophoresis
Several variants of Native PAGE have been developed to optimize separation for different applications. The table below summarizes the core characteristics of the three main techniques.
Table 1: Core Native Electrophoresis Techniques Compared
| Technique | Separation Principle | Key Feature | Primary Application |
|---|---|---|---|
| Blue-Native PAGE (BN-PAGE) | Charge & Size | Uses Coomassie G-250 dye to impart negative charge [16] | Analysis of membrane protein complexes and supercomplexes [16] |
| Clear-Native PAGE (CN-PAGE) | Charge & Size | Uses mixed micelles of detergents instead of dye for charge shift [23] [16] | In-gel enzyme activity assays without dye interference [23] |
| Native SDS-PAGE (NSDS-PAGE) | Size (Minimal Denaturation) | Uses greatly reduced SDS concentration and no heating [3] | Retaining enzymatic activity and metal cofactors while achieving high resolution [3] |
BN-PAGE is particularly powerful for membrane proteins. The anionic Coomassie blue dye binds to hydrophobic protein surfaces, conferring a uniform negative charge that drives electrophoretic migration and prevents protein aggregation [16]. In contrast, CN-PAGE, which replaces the dye with mild detergents, is advantageous for subsequent in-gel activity assays because the absence of blue dye eliminates potential interference with colorimetric detection [23] [16]. The NSDS-PAGE method offers a hybrid approach, providing the high resolution接近SDS-PAGE while maintaining protein function for many enzymes [3].
Experimental Protocol: A Step-by-Step Guide
This section provides a detailed methodology for conducting a high-resolution clear-native PAGE (hrCN-PAGE), adapted for analyzing the medium-chain acyl-CoA dehydrogenase (MCAD) enzyme, a homotetrameric flavoprotein [23].
Sample Preparation
- Protein Extraction: Solubilize mitochondrial or cellular membrane proteins using a mild, non-ionic detergent such as n-dodecyl-β-d-maltoside or digitonin. The choice and concentration of detergent are critical for maintaining the integrity of protein complexes while ensuring sufficient solubilization [16]. The addition of 6-aminocaproic acid, a zwitterionic salt, helps stabilize proteins and prevents aggregation during extraction [16].
- Sample Buffer Preparation: Mix the solubilized protein sample with a native sample buffer. For the hrCN-PAGE method used for MCAD, this buffer typically contains 100 mM Tris HCl, 150 mM Tris base, 10% glycerol, and tracking dyes, with a final pH of 8.5 [23] [3]. Crucially, the sample is not heated to preserve native structure.
Gel Electrophoresis
- Gel Casting: Manually cast or use commercial linear gradient gels, typically between 4-16% acrylamide, to achieve optimal separation across a broad molecular weight range [23] [16]. A gradient gel allows smaller proteins to migrate freely in the lower-percentage regions while effectively resolving larger complexes in the higher-percentage regions.
- Electrophoresis Conditions:
- Running Buffer: For CN-PAGE, the cathode buffer contains a mixture of anionic and neutral detergents to induce the necessary charge shift on proteins, replacing Coomassie blue [23] [16].
- Running Conditions: Load the prepared samples onto the gel. Run electrophoresis at a constant voltage (e.g., 150-200V) at 4°C to prevent heat-induced denaturation, until the dye front migrates to the bottom of the gel [23] [3].
Downstream Applications: In-Gel Activity Assay
A significant advantage of native electrophoresis is the ability to detect enzymatic activity directly within the gel.
- Activity Staining: After electrophoresis, incubate the gel in a reaction mixture specific to the enzyme of interest.
- For MCAD, the gel is stained in a solution containing its physiological substrate, octanoyl-CoA, and an electron acceptor, nitro blue tetrazolium chloride (NBT). MCAD activity oxidizes the substrate, leading to the reduction of NBT and the formation of an insoluble, purple-colored diformazan precipitate at the location of the active enzyme band [23].
- Quantification: The intensity of the resulting activity bands can be quantified using densitometry. This assay has been shown to be sensitive enough to quantify the activity of less than 1 µg of protein and can linearly correlate with the amount of protein and its FAD cofactor content [23].
The Scientist's Toolkit: Essential Research Reagents
Successful native PAGE requires specific reagents designed to maintain protein structure and function. The following table details key materials and their roles.
Table 2: Essential Reagents for Native PAGE Experiments
| Reagent / Solution | Function / Purpose | Example from Literature |
|---|---|---|
| n-Dodecyl-β-d-maltoside | Mild, non-ionic detergent for solubilizing membrane proteins without dissociating complexes [16]. | Used to extract intact OXPHOS complexes from mitochondrial membranes [16]. |
| Digitonin | Very mild, non-ionic detergent used to preserve weak protein-protein interactions in supercomplexes [16]. | Used for the analysis of respiratory chain supercomplexes (respirasomes) [16]. |
| Coomassie Blue G-250 | Anionic dye that binds protein surfaces, imparting negative charge and enhancing solubility during BN-PAGE [16]. | A key component in the cathode buffer and sample mix for BN-PAGE [16]. |
| 6-Aminocaproic Acid | Zwitterionic salt used as a stabilizing agent in extraction buffers; prevents protein aggregation [16]. | Included in the sample extraction buffer to support protein solubilization [16]. |
| Nitro Blue Tetrazolium (NBT) | Colorimetric electron acceptor; reduces to an insoluble purple formazan precipitate upon enzyme activity [23]. | Used in the in-gel activity stain to visualize active MCAD tetramers [23]. |
| Octanoyl-CoA | Physiological substrate for the MCAD enzyme; acts as an electron donor in the activity assay [23]. | The reductant in the in-gel activity stain, enabling specific detection of MCAD function [23]. |
Data Interpretation and the Impact of Protein Charge
Interpreting native PAGE results requires understanding how protein properties influence migration. A protein with a higher negative surface charge will migrate faster toward the anode. However, a larger hydrodynamic size (or molecular mass) will retard migration. The quaternary structure profoundly impacts this balance. For example, a compact tetramer might migrate faster than a more loosely structured dimer of similar mass due to its smaller effective size.
Pathogenic mutations offer a clear view of charge and structure effects. Research on MCAD deficiency (MCADD) reveals that some missense variants (e.g., p.R206C) do not significantly alter the monomer's molecular weight on denaturing gels but cause a measurable shift in migration on native gels [23]. This shift indicates a change in the protein's global conformation or surface charge, which can destabilize the homotetramer, leading to fragmentation into inactive, lower-mass species or formation of aggregates, all of which are separable and identifiable via native PAGE [23].
Workflow for Native PAGE and In-Gel Activity Assay
Factors Influencing Protein Migration in Native PAGE
Native PAGE is a powerful and versatile technique that provides unique insights into the functional state of proteins and their complexes. By separating proteins based on their intrinsic charge, size, and quaternary structure, it allows researchers to move beyond simple molecular weight determination to analyze enzymatic activity, protein-protein interactions, and the structural consequences of genetic variations. The continued refinement of methods like BN-PAGE, CN-PAGE, and NSDS-PAGE ensures that this foundational technology will remain a cornerstone of biochemical and biomedical research, directly contributing to our understanding of cellular mechanisms and the molecular basis of disease.
Practical Applications: Method Design and Buffer Selection for Charge-Based Separation
In native polyacrylamide gel electrophoresis (PAGE), a protein's migration behavior is fundamentally governed by its intrinsic charge, size, and three-dimensional structure, unlike denaturing SDS-PAGE where charge differences are masked by SDS. The separation occurs because most proteins carry a net negative charge in alkaline running buffers, causing them to migrate toward the anode. A protein's charge density (the number of charges per unit mass) directly determines its electrophoretic mobility—the higher the negative charge density, the faster a protein migrates. Simultaneously, the gel matrix creates a sieving effect that retards movement according to proteins' size and shape [24].
The choice of gel chemistry—Tris-Glycine, Bis-Tris, or Tris-Acetate—directly manipulates the electrostatic environment, profoundly impacting which protein properties dominate the separation and ultimately determining the success of the experiment [24]. This technical guide provides researchers and drug development professionals with a comprehensive framework for selecting the optimal native PAGE system based on their specific experimental requirements.
Comparative Analysis of Native PAGE Gel Systems
Technical Specifications and Operating Parameters
Table 1: Core Characteristics of Native PAGE Gel Systems
| Parameter | Tris-Glycine Gels | Tris-Acetate Gels | Bis-Tris Gels (NativePAGE) |
|---|---|---|---|
| Operating pH Range | 8.3 - 9.5 [24] | 7.2 - 8.5 [24] | ~7.5 [24] |
| Key Separation Basis | Net charge, size, & shape [24] | Net charge, size, & shape [24] | Molecular weight (using Coomassie G-250) [24] |
| Optimal Protein Size Range | 20 - 500 kDa [24] | >150 kDa [24] | 15 - 10,000 kDa [25] |
| Key Features | Traditional Laemmli system [24] | Better resolution of high molecular weight proteins [24] | Near-neutral pH; detergent compatible; overcomes pI limitations [24] |
| Recommended Sample Buffer | Tris-Glycine Native Sample Buffer [24] | Tris-Glycine Native Sample Buffer [24] | NativePAGE Sample Buffer + 5% G-250 Additive [24] |
| Recommended Running Buffer | Tris-Glycine Native Running Buffer [24] | Tris-Glycine Native Running Buffer [24] | NativePAGE Running Buffer + Cathode Buffer Additive [24] |
| Shelf Life & Storage | Up to 12 months at 2-8°C [24] | Up to 8 months at room temperature [24] | Up to 6 months at room temperature [24] |
Mechanism of Protein Separation and Charge Manipulation
Figure 1: Mechanism of Protein Separation in Native PAGE Systems
The fundamental separation mechanisms differ significantly between the systems, particularly in how they handle protein charge:
Tris-Glycine & Tris-Acetate Systems: These systems rely on the protein's intrinsic net charge at the operating pH. In the alkaline environment of Tris-Glycine gels (pH 8.3-9.5), most proteins carry a net negative charge and migrate toward the anode. However, proteins with basic isoelectric points (pI >9.5) may carry a net positive charge and migrate in the opposite direction or not enter the gel, making them unsuitable for these systems [24].
Bis-Tris (NativePAGE) System: This system uses Coomassie G-250 dye as a charge-shift molecule, fundamentally altering the separation paradigm. The dye binds non-specifically to proteins, conferring a net negative charge on all proteins regardless of their intrinsic pI. This allows even basic proteins to migrate toward the anode and enables separation primarily by molecular weight and shape, similar to SDS-PAGE but without denaturation [24]. This mechanism is particularly valuable for membrane proteins and complexes with significant hydrophobic surfaces, as the dye binding reduces aggregation by converting hydrophobic sites to negatively charged sites [24].
Detailed Experimental Protocols
NativePAGE Bis-Tris Gel Electrophoresis Protocol
The NativePAGE Bis-Tris system, based on blue native PAGE (BN-PAGE) technology, provides a robust method for analyzing membrane protein complexes and soluble native proteins [25].
Sample Preparation:
- Dilute protein samples with NativePAGE Sample Buffer.
- Add NativePAGE 5% G-250 Sample Additive to the samples prior to loading [24]. This additive provides the Coomassie dye necessary for imparting charge.
Gel Electrophoresis:
- Prepare anode (clear) and cathode (dark blue) buffers according to manufacturer instructions [24].
- Load samples onto a NativePAGE 3-12% Bis-Tris gel [25].
- Assemble the gel in the Mini Gel Tank, filling the front cathode chamber with dark blue cathode buffer and the back anode chamber with clear anode buffer [25].
- Run at constant voltage until the dye front has migrated appropriately (typically ~1/3 of the way through the gel for transfer applications).
- For western transfer applications: Pause the run, replace the cathode buffer with light blue cathode buffer, and resume electrophoresis [25].
Critical Considerations:
- Membrane Selection: PVDF membranes are required for western blotting with NativePAGE gels. Nitrocellulose is incompatible as it tightly binds the Coomassie G-250 dye [24].
- Power Supply Settings: During NativePAGE runs, current may drop below 1 mA, triggering "No Load" errors on some power supplies. Disable the load check feature to prevent automatic shutdown [25].
- Protein Standards: Use NativeMark Unstained Protein Standard (Cat. No. LC0725) for accurate molecular weight estimation under native conditions [25].
Tris-Glycine and Tris-Acetate Native Electrophoresis
Sample Preparation:
- Prepare protein samples in Tris-Glycine Native Sample Buffer [24].
- Avoid reducing agents unless specifically required, as they may disrupt native structure.
Gel Electrophoresis:
- Use the appropriate gel percentage based on target protein size (refer to Table 1).
- Prepare Tris-Glycine Native Running Buffer [24].
- Load samples and run at constant voltage recommended by the manufacturer.
- For Tris-Acetate gels, follow similar procedures but use the specified buffers and conditions for that system [24].
Transfer Considerations for Basic Proteins: Proteins with pI higher than the transfer buffer pH may require modified transfer conditions:
- Increase Tris-Glycine transfer buffer pH to 9.2 to transfer proteins with pI up to 9.2 [26].
- Place a membrane on both sides of the gel to capture proteins migrating in either direction [26].
- Pre-incubate the gel in transfer buffer containing 0.1% SDS to provide additional charge for transfer, though this may cause partial denaturation [26].
Advanced Applications and Integrative Techniques
SMA-PAGE: A Novel Approach for Membrane Protein Complexes
Recent advancements have integrated native PAGE with novel encapsulation technologies for studying membrane protein complexes. The SMA-PAGE method combines styrene maleic acid lipid particles (SMALPs) with native gel electrophoresis to separate membrane proteins in their native state while maintaining their lipid environment [27].
Key Advantages:
- Preserved Quaternary Structure: SMA-PAGE provides an excellent measure of protein quaternary structure within the membrane [27].
- Lipid Environment Analysis: The surrounding lipid environment can be probed using mass spectrometry [27].
- Compatibility with Downstream Techniques: Intact membrane protein-SMALPs extracted from gels can be visualized using electron microscopy [27].
- Immunoblotting Compatibility: The method complements traditional immunoblotting techniques [27].
This innovative approach demonstrates how native PAGE systems continue to evolve, particularly for challenging targets like membrane protein complexes that are crucial in drug development research.
Troubleshooting Common Native PAGE Issues
Poor Resolution or Smearing:
- Cause: Protein aggregation, especially common with hydrophobic or membrane proteins.
- Solution: Use NativePAGE Bis-Tris system with Coomassie G-250, which reduces aggregation by binding to hydrophobic sites [24].
Gel Run Stops Prematurely:
- Cause: Current drops below 1 mA in NativePAGE, triggering power supply safety features.
- Solution: Disable the "Load Check" feature on the power supply [25].
Bands Appear Narrowed or "Funneled":
- Cause: Excessive reducing agent (BME, DTT) in samples, causing over-reduction and charge repulsion.
- Solution: Optimize reducing agent concentration or omit unless essential for native structure [26] [25].
High Background in Western Blotting:
- Cause: Membrane contamination or insufficient blocking.
- Solution: Wear gloves, handle membranes carefully, optimize blocking conditions, and ensure proper antibody concentrations [25].
Essential Research Reagent Solutions
Table 2: Key Reagents for Native PAGE Experiments
| Reagent/Catalog Item | Function/Application | Compatible System(s) |
|---|---|---|
| NativePAGE Bis-Tris Gels (3-12%, 4-16%) | High-resolution separation of native proteins & membrane complexes | NativePAGE Bis-Tris [24] |
| NativePAGE Sample Buffer & 5% G-250 Additive | Provides charge shift for proteins via Coomassie binding | NativePAGE Bis-Tris [24] |
| Tris-Glycine Native Sample Buffer | Maintains proteins in native state without charge modification | Tris-Glycine, Tris-Acetate [24] |
| NativeMark Unstained Protein Standard (LC0725) | Accurate molecular weight estimation under native conditions | All native systems [25] |
| HiMark Prestained/Unstained Standards (LC5699/LC5688) | Molecular weight standards for high molecular weight proteins | Tris-Acetate [26] |
| PVDF Transfer Membrane | Required for blotting with NativePAGE systems; compatible with Coomassie dye | NativePAGE Bis-Tris [24] |
Choosing between Tris-Glycine, Tris-Acetate, and Bis-Tris native PAGE systems requires careful consideration of experimental goals and protein properties:
- Select Tris-Glycine when studying smaller proteins (20-500 kDa) and maintaining native charge is essential for interpretation [24].
- Choose Tris-Acetate for superior resolution of large protein complexes (>150 kDa) while preserving native charge states [24].
- Opt for NativePAGE Bis-Tris when working with membrane proteins, hydrophobic proteins, basic proteins (high pI), or when molecular weight estimation is desired under native conditions [24].
The integration of native PAGE with emerging technologies like SMA-PAGE demonstrates the continued evolution of these separation systems, offering researchers powerful tools for probing protein complex structure and function in drug development and basic research.
Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) represents a powerful technique for separating native protein complexes according to their molecular mass while maintaining their structural integrity and enzymatic activity. Unlike denaturing SDS-PAGE, which disrupts protein-protein interactions, BN-PAGE preserves the quaternary structure of protein assemblies, enabling researchers to study intact complexes and supercomplexes. The core innovation enabling this technique lies in the strategic use of the dye Coomassie Brilliant Blue G-250, which imposes a uniform negative charge shift upon proteins, facilitating their migration under native conditions. This charge-shift mechanism is particularly crucial for investigating how intrinsic protein charge influences electrophoretic mobility in native systems, revealing that migration depends not merely on innate charge but on a dye-imposed uniform charge density that enables separation primarily by size and shape.
The fundamental challenge in native electrophoresis of membrane proteins stems from their hydrophobic nature and varying intrinsic charges. Without a standardized charge carrier, these proteins would migrate unpredictably based on their inherent charge properties rather than their molecular dimensions. Coomassie G-250 resolves this limitation by binding nonspecifically to protein surfaces, converting diverse protein charge profiles into a uniformly negative state [1]. This transformation allows even basic proteins to migrate toward the anode at near-neutral pH (approximately 7.5) [28], enabling separation based primarily on molecular size and structural conformation rather than intrinsic charge characteristics.
The Biochemical Mechanism of Coomassie G-250 Binding
Chemical Properties and Molecular Interactions
Coomassie Brilliant Blue G-250 is an anionic synthetic dye belonging to the triphenylmethane family, characterized by its three phenyl rings [29]. The dye exists in different ionic forms depending on pH conditions, which directly influences its coloring and binding behavior. Critically for BN-PAGE applications, at the operating pH of approximately 7.5 used in NativePAGE Bis-Tris systems, Coomassie G-250 forms an anion and appears blue (590 nm absorption) [29]. This anionic form is essential for its function in BN-PAGE, as the negatively charged sulfonic acid groups of the dye molecule facilitate binding to protein surfaces.
The binding mechanism between Coomassie G-250 and proteins involves multiple molecular interactions. The dye binds primarily through heteropolar bonding to basic amino acid residues (lysine, arginine, and histidine) and through hydrophobic interactions with surface-exposed hydrophobic protein regions [29]. This dual binding mechanism is particularly advantageous for membrane proteins, which typically contain substantial hydrophobic surfaces. The binding of numerous Coomassie G-250 molecules converts hydrophobic regions into negatively charged sites, significantly reducing the tendency of membrane proteins to aggregate during electrophoresis [1]. This comprehensive dye coating effectively eliminates the native charge disparities between different proteins, creating a uniform negative charge landscape that enables separation based primarily on molecular dimensions rather than intrinsic charge properties.
Comparative Charge-Shift Mechanisms in Electrophoresis
Table 1: Charge-Shift Mechanisms in Different Electrophoresis Systems
| Electrophoresis System | Charge-Shift Molecule | Effect on Protein Structure | Primary Separation Basis | Operating pH Range |
|---|---|---|---|---|
| SDS-PAGE | Sodium Dodecyl Sulfate (SDS) | Denatures proteins; disrupts quaternary structure | Molecular mass (linear relationship) | Alkaline (varies) |
| BN-PAGE | Coomassie Brilliant Blue G-250 | Maintains native structure; preserves complexes | Size and shape of native complexes | ~7.5 [1] |
| Traditional Native PAGE | None (relies on intrinsic protein charge) | Maintains native structure | Net charge, size, and shape | 8.3-9.5 (Tris-Glycine) [1] |
| Tris-Acetate Native PAGE | None (relies on intrinsic protein charge) | Maintains native structure | Net charge, size, and shape | 7.2-8.5 [1] |
The unique advantage of Coomassie G-250 becomes evident when comparing different electrophoretic techniques. In standard SDS-PAGE, SDS denatures proteins and confers a strong uniform negative charge, but at the cost of disrupting native structure and protein-protein interactions [1]. Traditional native PAGE methods maintain protein structure but suffer from inconsistent migration patterns due to proteins' varying intrinsic charges at alkaline pH. BN-PAGE with Coomassie G-250 bridges this divide by providing uniform charge modification while preserving native conformations and complex interactions, effectively creating an ideal system for analyzing intact protein assemblies.
Experimental Methodology for BN-PAGE with Coomassie G-250
Sample Preparation and Solubilization Protocols
Proper sample preparation is critical for successful BN-PAGE separation, particularly when investigating membrane protein complexes. The initial step involves isolating the protein source, such as mitochondria from mouse liver tissue as described in a detailed protocol [28]. Approximately 30 mg of liver tissue is homogenized in ice-cold isolation buffer using a Wheaton Glass Potter-Elvehjem Tissue Grinder at 1500 rpm for 20 strokes [28]. The homogenate is centrifuged at 600 × g for 10 minutes at 4°C to separate cell debris from the mitochondrial extract [28]. This mitochondrial fraction then serves as the starting material for protein complex solubilization.
The choice of detergent for solubilizing membrane protein complexes significantly impacts the results obtained from BN-PAGE. Different detergents vary in their ability to preserve native protein interactions while effectively solubilizing membranes:
Table 2: Detergents for Solubilizing Protein Complexes in BN-PAGE
| Detergent | Strength | Applications | Impact on Protein Complexes |
|---|---|---|---|
| n-Dodecylmaltoside | Moderate | General membrane protein solubilization | May dissociate supercomplexes; yields individual complexes |
| Triton X-100 | Moderate | General membrane protein solubilization | Similar to dodecylmaltoside; may not preserve supercomplexes |
| Digitonin | Mild | Preservation of supercomplexes | Maintains associations between complexes (e.g., respirasomes) |
| Brij 96 | Mild | Alternative for sensitive complexes | Can preserve specific interactions |
| Saponin | Mild | Similar to digitonin | Natural detergent mixture; requires optimization |
The detergent selection directly influences the experimental outcomes and biological interpretations. For instance, using dodecylmaltoside or Triton X-100 typically reveals only individual respiratory complexes (I, II, III, IV, and V), supporting a "fluid state model" of randomly diffusing complexes. In contrast, digitonin solubilization preserves stoichiometric associations between complexes, revealing supercomplexes or "respirasomes" that support a "solid state model" of organized respiratory chains [30]. This distinction highlights how methodological choices in BN-PAGE can fundamentally shape our understanding of protein organization in biological systems.
Electrophoresis Conditions and Staining Procedure
Following solubilization, the protein extract is mixed with Coomassie G-250 sample additive before loading onto the BN-PAGE gel [28]. The BN-PAGE system typically uses Bis-Tris based gels with an acrylamide gradient ranging from 3-5% at the top to 13-16% at the bottom to maximize resolution of protein complexes across a broad size range [30]. The cathode buffer contains Coomassie G-250 to provide a continuous supply of dye during electrophoresis, maintaining the charge shift throughout the separation process [1].
For specific staining of protein complexes after electrophoresis, a highly sensitive colloidal Coomassie G-250 staining protocol can be employed. This method utilizes a specially formulated staining solution containing:
- 0.02% (w/v) Coomassie G-250
- 5% (w/v) aluminum sulfate hydrate
- 10% (v/v) ethanol (96%)
- 2% (v/v) orthophosphoric acid (85%) [31]
The sequential addition of components is crucial: first dissolving aluminum sulfate in water, then adding ethanol, followed by Coomassie G-250, and finally adding phosphoric acid [31]. This specific order promotes the formation of colloidal dye particles essential for high-sensitivity detection. After electrophoresis, gels are washed three times with Milli-Q water to remove SDS that might interfere with dye binding, then incubated in the Coomassie staining solution for 2-12 hours with agitation [31]. The stained gels are destained briefly with a solution containing 10% ethanol and 2% phosphoric acid, followed by rinsing with water [31]. This protocol enables detection of as little as 4-8 ng of protein per band [31], making it suitable for analytical applications.
Diagram 1: BN-PAGE Experimental Workflow
Research Applications and Practical Implementation
Analysis of Mitochondrial Supercomplexes
BN-PAGE with Coomassie G-250 has proven indispensable for investigating the structural organization of mitochondrial respiratory complexes. Research has revealed that rather than existing as isolated entities in the inner mitochondrial membrane, respiratory complexes form defined supramolecular assemblies called supercomplexes or "respirasomes" [28]. These supercomplexes include various stoichiometric combinations of Complexes I, III, and IV, such as I+III₂+IV₂ (SC 3) and I+III₂+IV₃ (SC 4) [28]. The formation of these higher-order structures demonstrates the critical importance of preserving native protein interactions during electrophoresis—a capability uniquely provided by the BN-PAGE technique.
The functional significance of supercomplex organization extends beyond structural considerations. Evidence suggests that association of mitochondrial complexes into supercomplexes may offer structural and functional advantages, including prevention of destabilization and degradation, enhancement of electron transport efficiency through substrate channeling, and reduction of electron or proton leakages [28]. This organization creates structural interdependence among individual OXPHOS complexes, explaining why mutations affecting one complex often lead to pleiotropic deficiencies in other complexes in human mitochondrial diseases [28]. For example, pathogenic mutations in Complex III subunits can lead to combined deficiencies of Complex I and IV, likely due to disrupted supercomplex assembly that normally stabilizes the individual complexes [28].
The Scientist's Toolkit: Essential Reagents for BN-PAGE
Table 3: Essential Research Reagents for BN-PAGE Experiments
| Reagent/Category | Specific Examples | Function in BN-PAGE |
|---|---|---|
| Detergents | n-Dodecylmaltoside, Digitonin, Triton X-100 | Solubilize membrane proteins while preserving native interactions [30] |
| Dye Solutions | Coomassie Brilliant Blue G-250 | Impose uniform negative charge; prevent protein aggregation [1] |
| Buffer Systems | NativePAGE Bis-Tris Buffer, HEPES, Aminocaproic acid | Maintain appropriate pH and ionic strength for native conditions [28] [30] |
| Gel Matrices | 3-12% or 4-16% Bis-Tris Acrylamide Gradient Gels | Size-based separation of protein complexes [1] [30] |
| Protease Inhibitors | Commercial protease inhibitor cocktails | Prevent protein degradation during isolation [28] |
| Centrifugation Media | Sucrose-based isolation buffers | Organelle and membrane isolation [28] |
Successful BN-PAGE experiments require careful selection of reagents that maintain protein complexes in their native state while enabling effective separation. The choice between detergents represents one of the most critical decisions, as different detergents can reveal distinct aspects of protein organization. For instance, while dodecylmaltoside may solubilize individual complexes effectively, digitoin has been instrumental in revealing the existence of respiratory supercomplexes that fundamentally changed our understanding of mitochondrial organization [30]. This toolkit provides the foundation for investigating protein-protein interactions in their native context, enabling researchers to explore the functional implications of complex formation beyond simple protein identification.
Diagram 2: Protein Charge Shift Mechanism by Coomassie G-250
Technical Considerations and Methodological Optimization
Strain-Specific and Tissue-Specific Variations
The application of BN-PAGE to different biological systems has revealed important methodological considerations. Studies in mouse models have demonstrated significant strain-specific differences in supercomplex formation due to natural variations in assembly factors. Specifically, C57BL/6 mice—the most commonly used laboratory strain—possess a mutated version of the Cox7a2l protein (also known as SCAFI), which impairs the formation of specific supercomplexes containing multiple copies of Complex IV [28]. In contrast, strains like DBA, CBA, CD1, 129, and NZB have functional Cox7a2l and can form the full repertoire of supercomplexes [28]. This genetic variation profoundly impacts the interpretation of mitochondrial function studies and underscores the importance of considering genetic background when designing experiments.
Tissue-specific differences further complicate BN-PAGE analysis. The assembly of respiratory supercomplexes appears strongly tissue-dependent, with certain supercomplexes forming in tissues like heart even in the absence of functional Cox7a2l, though to a much lesser extent than in Cox7a2l-proficient strains [28]. These observations highlight that optimal BN-PAGE conditions may require optimization for different tissue types, potentially involving adjustments to detergent concentrations, solubilization conditions, or electrophoresis parameters. Researchers must therefore carefully consider both genetic background and tissue source when designing experiments and interpreting BN-PAGE results.
Troubleshooting Common Experimental Challenges
Several technical challenges commonly arise in BN-PAGE experiments that can affect result quality and interpretation:
Protein Aggregation: Incomplete solubilization or insufficient Coomassie G-250 binding can lead to protein aggregation and poor resolution. This can be addressed by optimizing detergent-to-protein ratios, ensuring fresh Coomassie dye solutions, and including appropriate salts like aminocaproic acid to support solubilization [30].
Weak Band Intensity: Faint protein bands after staining may result from insufficient protein loading, incomplete dye binding, or over-staining that masks detection [29]. Increasing protein load, optimizing staining duration, and ensuring thorough washing before staining can improve results.
High Background Staining: Excessive background noise often stems from incomplete washing of gels before staining or interference from SDS and salts [29]. Increasing the number and duration of washing steps with appropriate solutions can reduce background interference.
Uneven Staining: Inadequate stain coverage or inconsistent agitation during staining can create uneven staining patterns [29]. Ensuring gels are fully immersed in staining solution with consistent agitation promotes uniform dye distribution.
For specialized applications like western blotting following BN-PAGE, PVDF membranes are recommended rather than nitrocellulose, as nitrocellulose binds Coomassie G-250 dye very tightly and is incompatible with alcohol-containing destaining solutions [1]. This technical detail highlights the importance of adapting downstream applications to accommodate the unique properties of BN-PAGE separations.
The strategic implementation of Coomassie G-250 in BN-PAGE represents a sophisticated solution to the fundamental challenge of separating native protein complexes based on size rather than intrinsic charge. By imposing a uniform negative charge shift through specific hydrophobic and ionic interactions, this dye enables the resolution of intact macromolecular assemblies that would otherwise be inaccessible to electrophoretic analysis. This capability has proven particularly transformative for membrane protein complexes, whose hydrophobic nature and charge heterogeneity traditionally complicated native separation techniques.
The implications of this methodology extend far beyond technical convenience, fundamentally advancing our understanding of cellular organization. The discovery of respiratory supercomplexes through BN-PAGE has reshaped models of mitochondrial electron transport, moving from a "fluid state" paradigm of freely diffusing individual complexes to a "dynamic aggregate" model incorporating both isolated complexes and organized supercomplex assemblies [28] [30]. This structural reorganization has functional consequences for metabolic efficiency, complex stability, and disease pathogenesis, illustrating how methodological innovations can drive conceptual advances in biological understanding. As BN-PAGE continues to be adapted for new biological systems and complex types, the controlled charge manipulation afforded by Coomassie G-250 remains central to elucidating the intricate protein networks underlying cellular function.
In Native Polyacrylamide Gel Electrophoresis (Native PAGE), the migration of proteins is not based on molecular weight alone, as in denaturing SDS-PAGE, but on a complex interplay between the protein's intrinsic charge, its size and shape, and the pore size of the gel matrix. The protein's charge, determined by the ionization state of its amino acid side chains, is directly manipulated by the pH of the electrophoresis buffers. Selecting the optimal buffer pH is therefore not a mere technical step; it is a critical strategic decision that governs the stability, mobility, and resolution of proteins in their native state. This guide details the principles and methodologies for buffer pH selection to optimize protein separation, with a specific focus on how protein charge density dictates electrophoretic migration.
Theoretical Foundation: Charge, pI, and Electrophoretic Mobility
Protein Charge and Isoelectric Point (pI)
A protein's net charge is the sum of all its positive (e.g., from Lys, Arg, His) and negative (e.g., from Asp, Glu) groups at a given pH. The isoelectric point (pI) is the specific pH at which a protein carries no net charge. The buffer pH selected for a Native PAGE experiment relative to the protein's pI determines its net charge and, consequently, its direction and rate of migration [32].
- Buffer pH < Protein pI: The protein carries a net positive charge and will migrate towards the cathode (negative electrode).
- Buffer pH > Protein pI: The protein carries a net negative charge and will migrate towards the anode (positive electrode).
- Buffer pH = Protein pI: The protein has zero net charge and will not migrate into the gel.
Most proteins have a pI in the acidic to slightly basic range (approximately pH 3–8). For these, the standard Tris-glycine running buffer (pH ~8.3) will impart a net negative charge, causing migration towards the anode [32]. A critical exception must be noted: if a protein's pI is highly basic (e.g., >8.5), the polarity of the electrodes must be reversed to drive the positively charged protein into the gel [32].
The Imperative of Native Conditions
The fundamental goal of Native PAGE is to preserve the protein's higher-order structure and, by extension, its biological activity. This is achieved by using non-reducing, non-denaturing sample buffers that omit agents like SDS, urea, or beta-mercaptoethanol. The success of this approach is demonstrated by techniques like Native SDS-PAGE (NSDS-PAGE), where the removal of SDS and EDTA from buffers and the omission of a heating step allowed for high-resolution separation while retaining enzymatic activity and bound metal ions in metalloproteins [3]. This underscores that meticulous buffer design is the key to balancing separation resolution with functional integrity.
Optimizing Buffer pH: A Practical Guide
The core challenge is selecting a buffer pH that provides all proteins in a mixture with a strong, uniform charge polarity to ensure they migrate into the gel, while also maintaining their stability.
Standard Buffer Systems for Native PAGE
The most common system uses a discontinuous chloride and glycine ion front, similar to SDS-PAGE, to stack and then separate polypeptides [32].
Table 1: Common Native PAGE Buffer Compositions
| Buffer Component | Composition | Function |
|---|---|---|
| Sample Buffer (2X) [32] | 62.5 mM Tris-HCl, pH 6.825% Glycerol1% Bromophenol Blue | Provides initial loading environment; glycerol adds density for well loading. |
| Running Buffer [32] | 25 mM Tris192 mM Glycine~pH 8.3 | Creates the pH environment for separation; the pH should not be adjusted. |
| Stacking Gel [32] | 0.375 M Tris-HCl, pH 8.8 | Initiates the stacking of proteins into a sharp band before entering the separating gel. |
| Separating Gel [32] | 0.375 M Tris-HCl, pH 8.8 | Provides the resolving environment where proteins separate based on charge-to-mass ratio. |
A Comparative View of Electrophoresis Methods
Different PAGE techniques manipulate buffer conditions to achieve distinct outcomes, highlighting the critical role of pH and detergents.
Table 2: Buffer Composition Comparison Across PAGE Methods
| Component | SDS-PAGE [3] | BN-PAGE [3] | NSDS-PAGE [3] |
|---|---|---|---|
| Sample Buffer | Contains SDS & EDTA; sample is heated | Contains NaCl, Ponceau S; no SDS | No SDS or EDTA; contains Coomassie G-250; sample not heated |
| Running Buffer | MOPS/Tris with 0.1% SDS & EDTA | BisTris/Tricine with Coomassie in cathode buffer | MOPS/Tris with reduced SDS (0.0375%); no EDTA |
| Key Outcome | Denaturation; separation by mass | Native state; separation of oligomeric complexes | Native state; high-resolution separation; retains metal ions & activity |
Experimental Protocol: pH Selection and Gel Casting
This protocol provides a detailed methodology for preparing and running a Native PAGE gel based on a standard Tris-glycine system [32].
Part A: Gel Preparation
- Prepare Separating Gel: Choose an acrylamide percentage (typically 6-12%) based on your target protein size. Mix the appropriate volumes of acrylamide/bis-acrylamide solution and 0.375 M Tris-HCl (pH 8.8) in a beaker.
- Catalyze Polymerization: Add 10% Ammonium Persulfate (AP) and TEMED to the separating gel solution. Swirl gently to mix. Note: These catalysts initiate rapid polymerization and must be added immediately before use.
- Cast the Gel: Pipette the solution into the gap between the glass plates. Carefully overlay with water or isopropanol to create a flat interface. Allow 20-30 minutes for complete polymerization.
- Prepare and Cast Stacking Gel: While the separating gel sets, prepare the stacking gel solution. Add AP and TEMED, pour out the overlay, pipette the stacking gel solution, and insert the comb. Allow 20-30 minutes to polymerize.
Part B: Sample Preparation and Electrophoresis
- Prepare Sample: Mix your protein sample with an equal volume of 2X non-reducing, non-denaturing sample buffer. CRITICAL: Do not heat the sample [32].
- Load and Run: Load the sample mixture into the wells. Fill the tank with running buffer. Run the electrophoresis at a constant voltage. Recommendation: Place the apparatus on ice or in a cold room and avoid excessively high voltage to prevent protein denaturation from Joule heating.
- Post-Electrophoresis Analysis: Upon completion, the gel can be stained (e.g., Coomassie blue) or used for immuno-blotting (Western blotting) or activity assays.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Native PAGE Experiments
| Reagent | Function | Technical Notes |
|---|---|---|
| Tris-HCl Buffer | Standard buffering agent for gel and running buffers; maintains pH in the 8-9 range. | The pH of the stacking and separating gels (8.8) is critical for establishing the discontinuous ion front. |
| Glycine | Component of the running buffer; acts as a trailing ion in the discontinuous system. | The running buffer pH (~8.3) should not be adjusted to ensure proper stacking and separation dynamics. |
| Acrylamide/Bis-acrylamide | Forms the porous polyacrylamide gel matrix for size-based separation. | The percentage (e.g., 6%, 8%, 10%, 12%) determines the gel pore size and resolution range. |
| Ammonium Persulfate (AP) & TEMED | Catalysts for the free-radical polymerization of acrylamide. | Must be added last, just before casting the gel. TEMED is hazardous and should be handled with care. |
| Glycerol | Added to the sample buffer to increase density for easy loading into wells. | Does not interact with or denature proteins. |
| Coomassie G-250 | Used in some native methods (e.g., NSDS-PAGE, BN-PAGE) to impart a slight negative charge. | Helps maintain protein solubility and can improve resolution without full denaturation [3]. |
Workflow Visualization: From pH Selection to Analysis
The following diagram illustrates the logical workflow and decision-making process for optimizing buffer pH in a Native PAGE experiment.
Buffer pH is a powerful tool that directly controls a protein's charge density, the fundamental driver of its migration in Native PAGE. A methodical approach to pH selection—informed by the protein's isoelectric point and balanced with the need for structural stability—enables researchers to achieve high-resolution separation of complex protein mixtures while preserving their native conformation and biological function. This optimization is essential for applications ranging from the study of protein-protein interactions and oligomeric state analysis to the functional characterization of enzymes and metalloproteins.
The decision to apply heat during sample preparation for native polyacrylamide gel electrophoresis (Native PAGE) is a critical methodological crossroads that directly impacts experimental outcomes. Unlike denaturing SDS-PAGE, where heating with SDS and reducing agents is standard practice, Native PAGE aims to preserve proteins in their biologically active states, making heat application generally unsuitable. This technical guide examines the fundamental principles governing this decision, focusing on how protein charge—a key determinant of electrophoretic migration in native systems—is compromised by heat-induced denaturation. By exploring established protocols and emerging methodologies, this review provides researchers with a structured framework for optimizing sample preparation to maintain native protein structure, charge characteristics, and biological function throughout electrophoretic separation.
Native PAGE represents a cornerstone technique for analyzing proteins in their non-denatured, functionally active states. Unlike SDS-PAGE, which separates proteins primarily by molecular weight under denaturing conditions, Native PAGE separates proteins based on a combination of their intrinsic charge, size, and three-dimensional shape [1] [33]. This preservation of native structure enables researchers to study physiological protein properties, including enzymatic activity, subunit interactions within multimeric complexes, and ligand binding capabilities [1] [34]. The central dilemma in sample preparation—whether to apply heat—stems from this fundamental objective of preserving native conformation.
The effect of heat on protein structure is a critical consideration. While heating in SDS-PAGE intentionally denatures proteins to linearize them and impart a uniform charge-to-mass ratio, applying similar heating for Native PAGE typically disrupts the delicate balance of non-covalent interactions maintaining tertiary and quaternary structures [35]. This disruption alters the very parameters—native size, shape, and intrinsic charge—upon which separation depends, potentially compromising experimental results. For researchers investigating protein-protein interactions, oligomeric states, or native enzymatic function, inappropriate heating can destroy the biological phenomena under investigation, leading to erroneous conclusions about protein behavior in physiological conditions.
The Role of Protein Charge in Native PAGE Migration
Fundamental Principles of Electrophoretic Separation
In Native PAGE, electrophoretic migration occurs because proteins carry a net charge in the running buffer environment. The migration velocity depends on the protein's charge density (number of charges per unit mass), with higher negative charge density resulting in faster migration toward the anode [1]. Simultaneously, the gel matrix exerts a sieving effect, creating frictional forces that regulate movement according to protein size and three-dimensional shape [1]. Small, compact proteins encounter less resistance and migrate faster, while larger proteins or complexes with more extensive structures face greater frictional forces [1] [36]. This dual dependence on both charge and physical dimensions creates the unique separation profile of Native PAGE, where proteins with different charge-to-size ratios can be resolved even with identical molecular weights.
The operating pH of the electrophoresis system profoundly influences protein charge, as it determines the ionization state of amino acid side chains. Different Native PAGE systems utilize different pH ranges to optimize separation:
Table 1: Native PAGE Systems and Their Operating Parameters
| Gel System | Operating pH Range | Charge Modification | Optimal Protein Size Range |
|---|---|---|---|
| Tris-Glycine | 8.3 - 9.5 | Relies on intrinsic protein charge | 20 - 500 kDa |
| Tris-Acetate | 7.2 - 8.5 | Relies on intrinsic protein charge | >150 kDa |
| Bis-Tris (NativePAGE) | ~7.5 | Coomassie G-250 imparts negative charge | All sizes, including membrane proteins |
Advanced Separation Mechanisms
Beyond basic charge and size considerations, the migration of proteins in Native PAGE is influenced by higher-order structural characteristics. The theory of reptation describes how macromolecules move through the gel matrix in a snake-like fashion through virtual tubes in the polymer network [36]. According to this model, electrophoretic mobility depends on the mean-square end-to-end distance of the macromolecule, meaning that more compact, folded proteins migrate faster than unfolded or extended conformations of equivalent molecular weight [36]. This phenomenon explains why Native PAGE can resolve different conformational states of the same protein, as the gel mobility reflects the protein's compactness rather than just its mass.
In the Ogston sieving model, particularly relevant for proteins approaching the pore size of the gel (typically 1-8 nm for 4-20% polyacrylamide gels), mobility decreases with increasing gel concentration according to the relationship log μ = log μ₀ - Kᵣc, where μ is mobility, μ₀ is free solution mobility, Kᵣ is a retardation coefficient, and c is gel concentration [36]. This relationship allows researchers to use Ferguson plots (log mobility versus gel concentration) to extract information about protein size and conformation. The presence of distinct conformational states can be detected when proteins in slow exchange appear as multiple bands with different mobilities, while those in fast exchange manifest as a single band with averaged mobility [36].
The Case Against Heating: Preserving Native Structure
General Principles and Rationale
The primary argument against heating samples for Native PAGE centers on preserving the native structural integrity of proteins. Heating, particularly at temperatures exceeding 60-70°C, disrupts the weak non-covalent interactions—including hydrogen bonds, hydrophobic interactions, and van der Waals forces—that maintain the tertiary and quaternary structure of proteins [35]. This structural perturbation has several consequential effects on electrophoretic separation:
- Altered Migration Patterns: Denaturation typically unfolds proteins, increasing their hydrodynamic radius and changing their interaction with the gel matrix, which slows migration and can produce smeared or anomalous bands [36].
- Loss of Biological Activity: Enzymatic function and binding capabilities depend on precise three-dimensional structures that are heat-labile [1] [33].
- Disruption of Protein Complexes: Multimeric proteins and macromolecular assemblies may dissociate into subunits, eliminating information about quaternary structure [1] [37].
The preservation of these structural features enables applications such as analyzing oligomeric states, detecting conformational changes, and isolating functionally active proteins for downstream assays [1] [38].
Specific Risks for Membrane and Temperature-Sensitive Proteins
Membrane proteins represent a particularly vulnerable category where heating introduces substantial risks. Due to their hydrophobic nature, membrane proteins tend to aggregate and form dimers or multimers when exposed to high temperatures [35]. These large aggregates may precipitate directly, potentially failing to enter the gel, or they may become trapped in the wells, resulting in complete assay failure [35]. Many antibody manufacturers specifically caution against heating membrane protein samples beyond 70°C, with some recommendations suggesting even lower temperature thresholds [35].
For particularly thermolabile proteins, including certain plant plasma membrane proteins, heating at 70°C may already be excessive. Research indicates that some target proteins are best detected when samples are denatured at ambient temperature (approximately 30°C) or even without any heating [35]. This temperature sensitivity necessitates empirical determination of optimal conditions for each protein system under investigation.
Specialized Native PAGE Systems and Their Sample Requirements
Blue Native PAGE (BN-PAGE)
Blue Native PAGE employs Coomassie G-250 dye as a charge-shifting molecule that binds non-covalently to proteins, conferring a net negative charge while maintaining proteins in their native state without denaturation [1]. In this system, the G-250 dye is present in the cathode buffer, providing a continuous flow into the gel during electrophoresis, and is also added to samples containing non-ionic detergent prior to loading [1]. This approach offers distinct advantages:
- Charge Manipulation: Proteins with basic isoelectric points (pI) that would normally carry a net positive charge at neutral pH are converted to net negative charge by G-250 binding, enabling consistent migration toward the anode [1].
- Reduced Aggregation: Membrane proteins and those with significant surface-exposed hydrophobic regions are less prone to aggregation because G-250 binds nonspecifically to hydrophobic sites, converting them to negatively charged surfaces [1].
- Detergent Compatibility: The system maintains functionality in the presence of non-ionic detergents necessary for solubilizing membrane protein complexes [1].
Heating samples for BN-PAGE is contraindicated as it would disrupt the native protein-dye interactions and potentially denature protein complexes, defeating the purpose of this specialized technique.
Clear Native PAGE (CN-PAGE)
Clear Native PAGE represents a dye-free variant of native electrophoresis where protein migration depends predominantly on the intrinsic charge of the protein at the system's pH, without the charge-shifting effect of Coomassie dye [39] [38]. This technique offers advantages for specific applications but has inherent limitations:
- Advantages: CN-PAGE eliminates dye-related quenching of fluorescence (90-95% quenching by Coomassie), making it compatible with fluorescent assays and detection methods [39]. It also provides milder conditions that can retain labile supramolecular assemblies that might dissociate under BN-PAGE conditions [38].
- Limitations: Without charge-shifting dyes, CN-PAGE is effectively limited to acidic proteins (pI < 7), as basic proteins would migrate toward the cathode and be lost from the gel [39] [38]. The resolution is typically lower than BN-PAGE unless modified as high-resolution CN-PAGE (hrCNE) with mild anionic detergents [39].
Table 2: Comparison of Specialized Native PAGE Techniques
| Parameter | BN-PAGE | CN-PAGE | High-Resolution CN-PAGE |
|---|---|---|---|
| Charge Source | Coomassie G-250 dye | Intrinsic protein charge | Intrinsic charge + mild detergents |
| Optimal pH | ~7.5 | Varies with protein pI | Varies with system |
| Protein Compatibility | All pI values | pI < 7 (acidic proteins) | Improved for membrane proteins |
| Fluorescence Compatibility | Low (dye quenches) | High | High |
| Resolution | High | Moderate | Improved |
| Heating Recommended? | No | No | No |
Experimental Protocols and Methodological Considerations
Standard Native PAGE Protocol Without Heating
The following protocol outlines a standardized approach for Native PAGE sample preparation without heating, applicable to most experimental scenarios:
Sample Preparation:
- Harvesting and Lysis: Use gentle, non-denaturing lysis buffers containing appropriate salts (e.g., 150 mM NaCl), buffering agents (e.g., 50 mM Tris-HCl, pH 7-8), and protease inhibitors [39]. Avoid ionic detergents like SDS; instead, use non-ionic (e.g., Triton X-100, digitonin) or zwitterionic detergents for membrane proteins.
- Clarification: Centrifuge lysates at 10,000-20,000 × g for 10-20 minutes at 4°C to remove insoluble debris.
- Sample Buffer Preparation: Prepare native sample buffer by combining:
- 50-100 mM buffer (Tris, Bis-Tris, or HEPES) matching the gel system pH
- 10-20% glycerol (for loading density)
- 0.001-0.01% non-ionic detergent if needed
- Tracking dye (e.g., Bromophenol Blue) optional
- Sample Mixing: Combine clarified sample with native sample buffer typically in a 3:1 to 5:1 ratio. For BN-PAGE, add 2-5% Coomassie G-250 additive to the sample [1].
- Incubation: Incubate samples on ice for 10-30 minutes. DO NOT HEAT.
- Loading and Electrophoresis: Load samples onto pre-cast or hand-cast native gels. Run at constant voltage (typically 100-150 V for mini-gels) at 4°C to minimize heat-induced artifacts [33].
Electrophoresis Conditions:
- Temperature Control: Maintain temperature at 4°C throughout electrophoresis using a cooled chamber or cold room [36] [33].
- Running Buffer: Use appropriate native running buffer without SDS or reducing agents.
- Voltage/Current Settings: Begin electrophoresis at 100 V for 15-20 minutes to allow sample entry into the gel, then increase to 150-200 V until separation is complete [39].
Specialized Protocol for Temperature-Sensitive Proteins
For particularly labile proteins, including some membrane proteins and multi-enzyme complexes, additional modifications are necessary:
- Mild Solubilization: Use digitonin or glyco-diosgenin (GDN) for membrane protein extraction, as these detergents better preserve native interactions than harsher detergents [38].
- Lower Temperature Incubation: For proteins where standard preparation causes aggregation, experiment with incubation temperatures ranging from ambient (20-25°C) down to 4°C [35].
- Polymer-Based Extraction: Utilize charged polymers like Glyco-DIBMA for membrane protein extraction, which forms nanodiscs that maintain a native-like lipid environment and prevent aggregation without requiring heat [39].
- Electrophoresis Modifications: Consider very low voltage (1-8 V/cm) separations for extremely labile complexes, extending run times to compensate for reduced migration rates [34].
Alternative Electrophoretic Approaches and Emerging Methods
Mild Detergent-Based Systems
For specific applications where completely native conditions are insufficient but full denaturation is undesirable, alternative electrophoretic methods offer intermediate approaches:
05SAR-PAGE utilizes a very low concentration (0.05% w/v) of the mild anionic detergent sarkosyl instead of SDS or Coomassie dyes [40]. This system enables separation of protein dimerization states and modification patterns while maintaining some native structural features [40]. The method has been successfully applied to analyze the dimerization of bacterial response regulator proteins like PhoBN directly in cell lysates without heating [40].
Partially Denaturing Systems incorporating reduced SDS concentrations (as low as 0.1%) or specialized additives can provide a balance between maintaining protein interactions and achieving sufficient separation resolution. These systems may involve controlled heating at specific temperatures (e.g., 37-45°C) rather than boiling, representing a middle ground for challenging separations.
Nanodisc-Based Electrophoresis
Recent advances in native electrophoresis incorporate charged polymer-encapsulated nanodiscs for analyzing membrane proteins [39]. In this approach:
- Membrane proteins are extracted using negatively charged amphiphilic copolymers like Glyco-DIBMA, which spontaneously form nanodiscs harboring the target protein within a native-like lipid-bilayer environment [39].
- These nanodiscs are then subjected to detergent-free clear native electrophoresis, where migration distance increases with the number of protomers due to higher charge densities [39].
- The nanodiscs remain intact throughout electrophoresis, and proteins show negligible aggregation, making them immediately suitable for downstream structural and functional studies [39].
This method represents a significant advancement for membrane protein analysis, as it avoids both detergent-mediated disruption and heat-induced aggregation while maintaining the proteins in a near-physiological environment.
Research Reagent Solutions for Native PAGE
Table 3: Essential Reagents for Native PAGE Experiments
| Reagent/Category | Specific Examples | Function & Importance |
|---|---|---|
| Gel Systems | Novex Tris-Glycine, NuPAGE Tris-Acetate, NativePAGE Bis-Tris | Provide different pH environments (7.2-9.5) optimized for various protein classes [1] |
| Charge-Shifting Agents | Coomassie Brilliant Blue G-250 | Imparts negative charge to proteins while maintaining native state in BN-PAGE [1] |
| Mild Detergents | Dodecyl maltoside (DDM), Digitonin, Triton X-100 | Solubilize membrane proteins while preserving native complexes [39] [38] |
| Protein Extraction Polymers | Glyco-DIBMA, SMA copolymers | Form native-like lipid nanodiscs around membrane proteins, preventing aggregation [39] |
| Protease Inhibitors | PMSF, AEBSF, EDTA-free cocktails | Prevent protein degradation during sample preparation without introducing denaturants [37] |
| Membrane Materials | PVDF membranes | Recommended for western blotting after NativePAGE; nitrocellulose binds Coomassie dye too tightly [1] |
| Molecular Weight Markers | Native protein standards | Specialized markers required as SDS-PAGE markers are inappropriate for native conditions [39] |
The question of whether to heat samples for Native PAGE finds its definitive answer in the fundamental purpose of the technique: to preserve proteins in their native, functional states. With few exceptions, heating is incompatible with this objective, as it disrupts the precise interplay of protein charge, size, and shape that governs separation in native systems. The migration of proteins in Native PAGE directly reflects their native charge characteristics, which are intimately tied to their three-dimensional structure and biological function. By adhering to non-heating protocols and selecting appropriate Native PAGE variants—whether BN-PAGE for comprehensive charge shifting or CN-PAGE for maintaining intrinsic charge properties—researchers can ensure the integrity of their experimental results while advancing our understanding of protein behavior in physiological contexts.
In the analysis of protein complexes, such as dimers, Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) provides separation primarily based on molecular mass by denaturing proteins and masking their intrinsic charge. In contrast, Native PAGE (Native Polyacrylamide Gel Electrophoresis) separates proteins according to their native charge, size, and shape, making it the essential technique for studying oligomeric states and functional protein complexes [2]. This case study is framed within broader research on how protein charge governs electrophoretic migration, focusing on resolving a protein dimer to demonstrate the practical application and interpretation of Native PAGE.
The migration of a protein in Native PAGE is determined by its electrophoretic mobility, which depends on the protein's net negative charge density (charge-to-mass ratio), the frictional force exerted by the gel matrix (related to protein size and shape), and the properties of the electrophoresis system [2]. A fundamental understanding of these principles is crucial for accurately interpreting Native PAGE results, particularly when distinguishing between monomers, dimers, and higher-order complexes.
Theoretical Foundation: Principles of Native PAGE Separation
Key Determinants of Protein Migration
In Native PAGE, the protein's intrinsic charge and the gel's sieving effect work in concert to achieve separation. The core principles governing migration are:
- Net Charge and Charge Density: At the alkaline pH (typically ~8.8) of Native PAGE running buffers, most proteins carry a net negative charge and migrate toward the positively charged anode. The charge density (net charge per unit mass) is a primary determinant of migration speed; a higher negative charge density results in faster migration [2] [41].
- Size and Shape (Frictional Force): The gel matrix acts as a molecular sieve, creating a frictional force that regulates protein movement. Larger proteins and complexes experience greater frictional resistance and migrate more slowly than smaller, more compact proteins of similar charge [2].
- Matrix Properties: The pore size of the polyacrylamide gel, controlled by the percentage of acrylamide and the cross-linker bisacrylamide, determines the effective separation range. Lower percentage gels (e.g., 7-8%) are suitable for separating large complexes, while higher percentages (e.g., 12%) provide better resolution for smaller proteins [2].
Table 1: Factors Influencing Protein Migration in Native PAGE
| Factor | Effect on Migration | Experimental Control |
|---|---|---|
| Net Charge (Charge Density) | Higher negative charge density increases migration rate | Buffer composition and pH |
| Protein Size & Mass | Larger size/mass decreases migration rate | Gel percentage (pore size) |
| Protein Shape/Structure | Compact shapes migrate faster than extended forms | Gel percentage; buffer additives |
| Gel Pore Size | Lower % acrylamide speeds up large complex migration | Acrylamide/bisacrylamide concentration |
Comparison of Electrophoresis Techniques
Understanding what Native PAGE does not do is as important as understanding its function. The table below contrasts it with other common techniques.
Table 2: Comparison of Key Protein Electrophoresis Techniques
| Technique | Separation Basis | Protein State | Key Applications |
|---|---|---|---|
| Native PAGE | Mass/Charge/Shape ratio [2] | Native, functional | Analysis of oligomeric states, complexes, enzymatic activity |
| SDS-PAGE | Molecular mass [2] | Denatured, linearized | Molecular weight estimation, purity check, subunit analysis |
| Blue Native (BN)-PAGE | Mass & native charge [3] [6] | Native, complexes preserved | Separation of membrane protein complexes, molecular mass estimation |
| 2D-PAGE | 1st: pI (IEF); 2nd: Mass (SDS-PAGE) [2] | Denatured | High-resolution proteomic analysis, separation of protein isoforms |
Case Study: Resolving a Hypothetical Protein Dimer
Experimental Scenario and Objectives
Consider a scenario where a purified protein, "Protein X," when analyzed by SDS-PAGE under reducing conditions, runs as a single band at 25 kDa. However, when analyzed by Size Exclusion Chromatography (SEC), a major peak appears consistent with a size larger than 25 kDa, suggesting a potential oligomeric state. The objective is to use Native PAGE to confirm or refute the presence of a dimer and to investigate how the protein's intrinsic charge influences its migration behavior.
Methodology and Workflow
A detailed Native PAGE protocol was followed, adapted from standard methodologies [2] [3]. The workflow is as follows:
Diagram 1: Native PAGE experimental workflow.
Sample Preparation
- Protein Samples: Protein X was prepared in a non-denaturing buffer (50 mM Tris-Cl, pH 7.4). No SDS or reducing agents were used [3].
- Native Protein Standards: A set of known native proteins (e.g., Thyroglobulin (669 kDa), Apoferritin (443 kDa), Amylase (200 kDa), Albumin (66 kDa)) was used for molecular weight calibration [6].
Gel Electrophoresis Conditions
- Gel Composition: A 8% polyacrylamide resolving gel (pH 8.8) was cast without SDS. A 4% polyacrylamide stacking gel (pH 6.8) was used to concentrate the sample before entry into the resolving gel [2] [41].
- Running Buffer: Tris-Glycine buffer, pH 8.3, without SDS or EDTA [41] [3].
- Electrophoresis: Conducted at a constant voltage of 150V for approximately 90 minutes, with the apparatus kept in a cold room (4°C) to minimize denaturation and proteolysis [2].
Detection
- Coomassie Staining: The gel was stained with Coomassie Brilliant Blue to visualize total protein.
- In-gel Activity Assay: A separate gel was subjected to a specific activity stain to confirm the functionality of the separated protein bands [2] [3].
The Scientist's Toolkit: Essential Reagents
Table 3: Key Research Reagents for Native PAGE
| Reagent/Category | Specific Examples | Function & Importance |
|---|---|---|
| Matrix Components | Acrylamide, Bisacrylamide, TEMED, Ammonium Persulfate (APS) | Forms the cross-linked polyacrylamide gel matrix that acts as a molecular sieve [2]. |
| Running Buffers | Tris-Glycine (pH 8.3) | Conducts current and maintains pH; glycine's zwitterionic nature is key to the discontinuous buffer system [41]. |
| Sample Buffer | Tris-HCl, Glycerol, tracking dye (e.g., Bromophenol Blue) | Provides ions for conductivity, density for loading, and a visible migration front [3]. |
| Native MW Standards | Thyroglobulin, Apoferritin, Albumin (non-denatured) | Calibrates the gel for molecular weight estimation of native complexes [6]. |
Results and Interpretation
Data Analysis and Observed Migration
After electrophoresis and staining, the following results were observed:
- Lane 1 (Native Standards): Showed a ladder of bands with migration distances inversely proportional to their molecular weights.
- Lane 2 (Protein X): Showed a single, dominant band.
- The migration distance of Protein X was plotted on the standard curve generated from the native markers.
Interpretation in the Context of Charge and Mass
The interpretation of the results is critical:
- Apparent Molecular Weight: The standard curve indicated an apparent native molecular weight of approximately 50 kDa for Protein X.
- Comparison with SDS-PAGE: Since SDS-PAGE showed a single band at 25 kDa, the 50 kDa complex in Native PAGE strongly suggests that Protein X exists as a homodimer under native conditions.
- Role of Charge: The migration of the dimer is not solely a function of its mass (50 kDa). Its net negative charge at the running pH is the other critical factor driving its migration ahead of other 50 kDa proteins with different charge densities. If the dimer were highly acidic, it might migrate faster than expected for its mass; if it were less charged or basic, it might migrate more slowly [2] [42]. The observed position confirms a charge-to-mass ratio consistent with a 50 kDa particle.
Advanced Techniques and Considerations
Troubleshooting and Methodological Refinements
Several factors can impact the accuracy and reliability of Native PAGE:
- Protein Charge Anomalies: Proteins with unusually high contents of acidic amino acids (Asp and Glu) can exhibit retarded migration in SDS-PAGE, appearing larger than predicted [42]. In Native PAGE, such proteins would display anomalously fast migration due to their increased negative charge density.
- Retaining Native Function: The successful application of an in-gel activity assay on the Protein X dimer band confirms that the oligomer is functional and that the Native PAGE conditions successfully preserved its tertiary and quaternary structure [2] [3].
- Buffer Optimization: For delicate proteins or metalloenzymes, minor modifications to standard Native PAGE can be employed. The Native SDS-PAGE (NSDS-PAGE) technique uses drastically reduced SDS and omits heating and EDTA, allowing for high-resolution separation while retaining enzymatic activity and bound metal ions in many cases [3].
This case study demonstrates that Native PAGE is a powerful, indispensable tool for confirming the oligomeric state of a protein dimer. The experiment successfully distinguished the 50 kDa dimeric form of Protein X from its 25 kDa monomeric subunit, which was only possible by analyzing the protein in its native state. The results underscore the fundamental principle that electrophoretic migration in Native PAGE is a function of both molecular size and intrinsic charge.
For researchers in drug development, where targeting functional protein complexes is paramount, Native PAGE provides a critical assay for studying complex formation, stability, and function under various biochemical conditions or in the presence of candidate therapeutic molecules.
Troubleshooting Migration: Resolving Common Issues in Native PAGE Analysis
Protein electrophoresis, a cornerstone technique in molecular biology, provides critical insights into protein purity, size, and oligomeric state. However, researchers frequently encounter unexpected band patterns that deviate from theoretical predictions, presenting significant interpretive challenges. In native polyacrylamide gel electrophoresis (PAGE), protein migration depends on a complex interplay of intrinsic charge, size, shape, and conformational stability, making anomaly interpretation particularly demanding. These aberrant patterns can stem from various physicochemical properties, including surface charge distribution, hydrophobic characteristics, and aggregation propensity, which collectively influence protein behavior under electrophoretic conditions.
Understanding these anomalies is not merely an academic exercise—it has profound implications for accurate protein characterization in both basic research and drug development. Misinterpretation of band patterns can lead to incorrect conclusions about protein identity, purity, oligomeric state, and functional integrity. This technical guide examines the fundamental principles governing protein migration in native PAGE, with particular emphasis on how protein charge and aggregation phenomena contribute to unexpected electrophoretic patterns, and provides structured experimental approaches for their systematic investigation and resolution.
Fundamental Principles of Native PAGE Migration
Unlike denaturing SDS-PAGE, which separates proteins primarily by molecular weight, native PAGE separates proteins based on both charge and size, preserving protein structure and function throughout the electrophoretic process. The migration velocity of a protein in native PAGE depends on its charge-to-size ratio, with more highly charged proteins migrating faster toward the oppositely charged electrode, while larger proteins experience greater frictional resistance within the gel matrix. This fundamental relationship, however, represents a simplification of a more complex reality where protein shape, conformational stability, and buffer conditions significantly influence migration behavior.
The intrinsic charge of a protein derives from the ionization state of its amino acid side chains, which varies with pH according to their respective pKa values. At a pH above its isoelectric point (pI), a protein carries a net negative charge and migrates toward the anode, while below its pI, it carries a net positive charge and migrates toward the cathode. Critically, the distribution of charges across the protein surface, not merely the net charge, affects electrophoretic mobility due to local interactions with the gel matrix. Furthermore, proteins with anomalous migration often exhibit deviations from idealized spherical structures, with elongated or asymmetric shapes creating greater frictional drag than compact globular proteins of equivalent molecular weight. These fundamental principles establish the theoretical framework for understanding normal electrophoretic behavior, yet numerous factors can disrupt this expected migration, yielding the unexpected band patterns that complicate protein analysis.
Key Mechanisms Behind Anomalous Band Patterns
Charge Distribution and Surface Properties
The arrangement of charged residues on a protein's surface significantly influences its electrophoretic mobility beyond what would be predicted from net charge alone. Proteins with clustered charge regions may exhibit different mobilities compared to proteins with evenly distributed charges, even when their net charges are identical. This phenomenon occurs because localized charge concentrations can create distinct electrostatic interactions with the gel matrix, effectively altering migration rates. Additionally, surface hydrophobicity can impact mobility through transient interactions with the gel matrix, although this effect is typically more pronounced in SDS-PAGE systems where hydrophobic binding to SDS is a fundamental aspect of the technique.
Post-translational modifications further complicate migration patterns by altering protein charge. Phosphorylation adds negative charges, glycosylation can mask positive charges and increase hydrodynamic volume, and acetylation eliminates positive charges. These modifications create charge microenvironments that deviate from predictions based solely on amino acid sequence. The buffer system itself also plays a crucial role; different buffer ions can bind to proteins to varying degrees, effectively modulating their apparent charge and thus their electrophoretic mobility. This complex interplay between intrinsic protein properties and experimental conditions creates a fertile ground for anomalous band patterns that require careful interpretation.
Aggregation and Oligomeric States
Protein aggregation represents a major source of electrophoretic anomalies, producing high-molecular-weight complexes that manifest as smears or unexpected bands near the gel top. As research on neurodegenerative diseases has elucidated, protein aggregates exist on a spectrum from soluble oligomers to insoluble fibrils, with oligomeric species being particularly relevant to native PAGE analysis due to their persistence under non-denaturing conditions. These β-sheet-rich oligomeric species represent a critical intermediate in the aggregation pathway and can exhibit unexpected migration due to their altered hydrodynamic radii and surface properties compared to native monomers or structured oligomers [43].
The aggregation process itself follows a nucleation-polymerization mechanism wherein a slow nucleation phase is followed by rapid growth through monomer addition. In native PAGE, different points along this aggregation pathway can be visualized as discrete bands or smears, reflecting various oligomeric states. Importantly, certain pathological variants can destabilize native quaternary structures, leading to fragmentation into lower molecular weight forms or aberrant aggregation. For instance, studies on medium-chain acyl-CoA dehydrogenase (MCAD) have shown that specific variants (R206C and K329E) cause tetramer fragmentation, resulting in additional bands at lower molecular mass ranges despite maintaining identical monomeric molecular weights [44] [23]. This dissociation of multimeric proteins under non-denaturing conditions can produce complex band patterns that reflect equilibrium between different oligomeric states.
Table 1: Types of Protein Aggregates and Their Electrophoretic Signatures
| Aggregate Type | Structure | Native PAGE Manifestation | Biological Relevance |
|---|---|---|---|
| Soluble Oligomers | Amorphous, β-sheet-rich | Diffuse bands/smears at high molecular weights | High neurotoxicity, implicated in neurodegenerative diseases [43] |
| Amyloid Fibrils | Cross-β structure, insoluble | Often too large to enter gel, material at well bottom | Pathological hallmark of >50 human diseases [45] |
| Off-pathway Oligomers | Non-fibrillar, metastable | Discrete bands at unexpected positions | May inhibit fibril formation [43] |
| Fragmented Oligomers | Dissociated multimers | Bands at lower molecular weights than native form | Result from destabilizing variants (e.g., MCAD R206C) [44] |
Hydrophobicity and Membrane Proteins
Hydrophobicity represents a particularly potent source of electrophoretic anomalies, especially for membrane proteins with extensive transmembrane domains. These proteins frequently exhibit marked deviations between their theoretical and apparent molecular weights due to abnormal SDS binding and altered conformational states during electrophoresis. The SLC7A11 protein provides a compelling case study, migrating at approximately 37 kDa despite having a theoretical molecular mass of 55.4 kDa [46]. This significant discrepancy was systematically investigated through knockdown and overexpression experiments, which confirmed the protein's identity while excluding proteolytic cleavage as an explanatory mechanism.
Research has demonstrated that the high hydrophobicity of SLC7A11, evidenced by its elevated GRAVY (Grand Average of Hydropathicity) score and abundant non-polar amino acid residues, fundamentally underlies its anomalous migration. This abnormal migration pattern proved responsive to experimental manipulations—substituting hydrophobic isoleucine residues with polar asparagine residues reduced overall hydrophobicity and restored normal migration at the predicted 55 kDa molecular weight [46]. Additionally, the gel concentration itself modulated this effect, with higher acrylamide concentrations (12-15%) eliminating the aberrant migration observed at lower concentrations. This concentration-dependent behavior suggests that hydrophobicity may influence the protein's interaction with the gel matrix itself, independent of its SDS-binding characteristics, highlighting the complex interplay between protein physicochemical properties and electrophoretic conditions.
Figure 1: Mechanism of Hydrophobicity-Induced Migration Anomalies. This pathway illustrates how high protein hydrophobicity leads to altered conformation and reduced SDS binding, resulting in faster electrophoretic mobility and lower apparent molecular weight. Experimental interventions include increasing gel concentration or substituting hydrophobic residues with polar ones to restore normal migration [46].
Experimental Approaches for Anomaly Investigation
Methodological Framework for Systematic Analysis
When confronted with unexpected band patterns in native PAGE, researchers should adopt a systematic investigative approach that progresses from simple verification experiments to more complex mechanistic studies. The initial phase should focus on confirming the protein's identity through specific detection methods such as Western blotting with validated antibodies, coupled with mass spectrometric analysis of excised bands when necessary. These confirmatory steps are particularly crucial given that various antibodies against the same protein may suggest different molecular masses due to non-specific binding, as observed in studies of SLC7A11 where some antibodies indicated 55 kDa while others detected the protein at 37 kDa [46].
Following identity confirmation, researchers should explore methodological modifications to resolve anomalies, including varying gel concentrations, adjusting buffer pH to alter protein charge states, and incorporating different detergent concentrations. The SLC7A11 case study clearly demonstrates the utility of this approach, where increasing acrylamide concentration from standard levels to 12-15% eliminated the aberrant migration [46]. For suspected aggregation phenomena, complementary techniques such as size-exclusion chromatography, analytical ultracentrifugation, and dynamic light scattering provide orthogonal size characterization independent of electrophoretic mobility. Furthermore, structural investigations using circular dichroism to assess secondary structure, thermal shift assays to evaluate stability, and cross-linking experiments to stabilize transient oligomers can provide mechanistic insights into the molecular basis of observed anomalies.
Table 2: Troubleshooting Electrophoretic Anomalies in Native PAGE
| Anomaly Type | Diagnostic Experiments | Expected Outcomes | Interpretation |
|---|---|---|---|
| Charge-based anomalies | Vary pH conditions; Isoelectric focusing | Altered migration with pH; Identification of pI | Net charge or charge distribution differs from prediction |
| Aggregation | Size-exclusion chromatography; Cross-linking | High-molecular-weight species; Stabilized oligomers | Presence of oligomeric states or aggregates |
| Hydrophobicity effects | Vary gel concentration; Modify detergent | Altered migration with gel density; Improved resolution | Hydrophobic interactions with gel matrix |
| Conformational changes | Limited proteolysis; Chemical denaturation | Altered fragmentation pattern; Shift to expected MW | Non-native protein conformation affecting mobility |
| Oligomer dissociation | Vary protein concentration; Cross-linking | Concentration-dependent band intensity; Stabilized complexes | Equilibrium between oligomeric states |
Advanced Techniques: In-Gel Activity Assays
For enzymatic proteins, in-gel activity assays provide a powerful method to distinguish functional oligomeric states from non-functional aggregates or degradation products. This approach was elegantly applied in studies of medium-chain acyl-CoA dehydrogenase (MCAD), where a colorimetric assay coupling octanoyl-CoA oxidation to nitro blue tetrazolium reduction allowed specific visualization of active tetramers separate from other protein forms [44] [23]. The assay demonstrated linear correlation between protein amount, FAD content, and enzymatic activity, enabling quantification of even subtle functional differences between variants.
The application of this methodology to disease-associated MCAD variants revealed critical insights into structure-function relationships. While wild-type MCAD and the Y67H variant presented predominantly as active tetramers, the R206C and K329E variants showed additional bands at lower molecular mass ranges, suggesting tetramer fragmentation [44]. Importantly, these lower molecular weight species displayed no enzymatic activity despite maintaining FAD binding capacity, highlighting their structural and functional deficiency. Interestingly, the R206C variant tetramers migrated at an apparent lower molecular mass than wild-type despite identical monomeric molecular weights, suggesting conformational differences affecting electrophoretic mobility rather than changes in oligomeric state [23]. This case study illustrates how combining separation with functional assessment can decode complex band patterns that would be otherwise difficult to interpret.
Figure 2: In-Gel Activity Assay Workflow for Analyzing Functional Oligomeric States. This experimental approach separates protein complexes by high-resolution clear native electrophoresis followed by activity staining, enabling distinction between active tetramers and inactive fragmented forms in MCAD variant studies [44] [23].
Research Reagent Solutions for Electrophoretic Analysis
Table 3: Essential Reagents for Investigating Electrophoretic Anomalies
| Reagent/Category | Specific Examples | Application Purpose | Technical Considerations |
|---|---|---|---|
| Electrophoresis Matrices | Polyacrylamide (varying concentrations); Agarose | Separation matrix; Molecular sieving | Higher acrylamide concentrations (12-15%) can resolve hydrophobicity anomalies [46] |
| Detection Reagents | Coomassie Blue; Silver stain; Amytracker (pFTAA) | Total protein staining; Aggregate-specific detection | Amytracker specifically detects amyloid-like aggregates [47] |
| Activity Assay Components | Nitro blue tetrazolium (NBT); Octanoyl-CoA; FAD | In-gel enzymatic activity detection | Linear correlation with protein amount and FAD content [44] |
| Molecular Chaperones | Hsp70; GroEL/ES | Suppressing aggregation in sample preparation | Maintain native state during electrophoresis |
| Cross-linking Agents | Glutaraldehyde; DSS/BS3; Formaldehyde | Stabilizing oligomeric complexes | Identifies weak interactions and transient complexes |
| Charge Modifiers | CTAB; DEAE-dextran; Different buffer systems | Altering electrophoretic mobility | Reveals charge-based anomalies |
Implications for Drug Development and Research
Decoding electrophoretic anomalies has profound implications for pharmaceutical development, particularly in the characterization of biologic therapeutics and understanding disease mechanisms. The propensity for protein aggregation represents a critical quality attribute for biopharmaceuticals, as aggregates can reduce efficacy and increase immunogenicity risk. The ability to distinguish between active oligomers and inactive aggregates, as demonstrated in the MCAD studies, directly informs developability assessments and formulation strategies for protein-based therapeutics [44] [23]. Furthermore, understanding charge-based anomalies ensures accurate characterization of charge variants, which is essential for maintaining product consistency and stability.
In neurodegenerative disease research, where protein aggregation is a hallmark pathological feature, electrophoretic techniques capable of resolving different aggregate species provide invaluable tools for investigating disease mechanisms and evaluating therapeutic interventions. The link between β-sheet-rich oligomers and neurotoxicity highlights the importance of techniques that can distinguish these species from less pathogenic forms [43]. Additionally, the discovery that antimicrobial peptides can induce co-translational aggregation of bacterial membrane proteins suggests novel antibiotic mechanisms that could be exploited therapeutically [47]. As electrophoretic methodologies continue to evolve, their application in decoding anomalous band patterns will remain essential for advancing both basic research and therapeutic development across multiple disease areas.
Unexpected band patterns in native PAGE present both challenges and opportunities for researchers seeking to understand protein function and dysfunction. Through systematic investigation of charge-based phenomena, aggregation states, and hydrophobic interactions, these anomalies can be decoded to reveal critical information about protein structure, stability, and function. The integration of complementary techniques—including in-gel activity assays, structural probes, and methodological modifications—provides a powerful framework for resolving electrophoretic discrepancies. As research continues to elucidate the complex relationships between protein sequence, structure, and electrophoretic behavior, the interpretation of these patterns will become increasingly sophisticated, further enhancing the utility of native PAGE in both basic research and applied pharmaceutical development.
In native polyacrylamide gel electrophoresis (Native PAGE), the migration of proteins is governed by a delicate interplay of their intrinsic charge, size, and three-dimensional structure. Unlike denaturing techniques such as SDS-PAGE, which separates proteins primarily by molecular weight, Native PAGE preserves proteins in their native state, maintaining their biological activity, quaternary structure, and conformational integrity. This technique is therefore indispensable for studying functional protein complexes, enzymatic activity, and native protein-protein interactions. The buffer system—specifically its pH, ionic strength, and additives—serves as the fundamental experimental parameter that controls protein charge and electrophoretic behavior.
The net charge of a protein in solution is not a fixed property but is dynamically regulated by the buffer pH relative to the protein's isoelectric point (pI). A protein will carry a net positive charge below its pI and a net negative charge above its pI [48]. During Native PAGE, this net charge dictates the direction and speed of migration toward the oppositely charged electrode. Consequently, precise control of buffer conditions is not merely a technical detail but a prerequisite for achieving high-resolution separation, accurate interpretation of results, and meaningful biological insights. This guide provides a comprehensive framework for optimizing these critical parameters to control protein charge and migration within the context of Native PAGE research.
Theoretical Foundation: Protein Charge and Electrophoretic Migration
Fundamental Principles of Protein Charge
Proteins are amphoteric molecules; their net surface charge arises from the ionization of amino acid side chains and terminal groups. Positively charged groups include the side chains of lysine, arginine, and histidine (at lower pH), as well as the N-terminus. Negatively charged groups comprise the side chains of aspartate and glutamate, along with the C-terminus [48]. The pH of the surrounding environment determines the protonation state of these ionizable groups. The isoelectric point (pI) is the specific pH at which a protein carries no net charge [48]. This relationship between pH and charge is the primary lever for controlling protein migration in Native PAGE.
The Combined Influence of Charge, Size, and Shape
In Native PAGE, a protein's electrophoretic mobility is determined by its charge density (net charge relative to its mass), its size, and its three-dimensional shape [1] [49]. A protein with a high negative charge density will migrate rapidly toward the anode. Simultaneously, the gel matrix exerts a sieving effect, retarding larger proteins more than smaller ones [1]. Crucially, the native conformation of a protein can mask or expose charged regions, and a compact protein may migrate faster than a larger, more extended protein of identical mass [49]. This multi-factor dependence is what makes Native PAGE a powerful tool for analyzing native protein states, but also necessitates careful optimization of running conditions to achieve desired separations.
Core Buffer Parameters and Their Optimization
pH Selection and Control
The buffer pH is the most critical parameter for controlling protein charge and migration direction. Selecting an appropriate pH ensures that all target proteins carry a sufficient net charge to migrate in the desired direction.
Key Considerations for pH Selection:
- For Standard Native PAGE (Tris-Glycine): The operating pH range is 8.3–9.5 [1]. At this alkaline pH, most proteins (those with a pI < 8.3) carry a net negative charge and will migrate toward the anode. Proteins with a pI above the buffer pH will carry a net positive charge and migrate toward the cathode [49].
- For Charge-Shift Techniques (NativePAGE Bis-Tris): This system operates at a near-neutral pH of ~7.5 and uses Coomassie G-250 dye to bind proteins and confer a net negative charge, allowing even basic proteins to migrate toward the anode [1].
- Predicting Migration Direction: A simple decision tree can be used: If buffer pH > protein pI, the protein is negatively charged and migrates toward the anode. If buffer pH < protein pI, the protein is positively charged and migrates toward the cathode. If buffer pH = protein pI, the protein has no net charge and will not migrate [49] [48].
Table 1: Comparison of Common Native PAGE Gel Chemistries and Their pH Ranges
| Gel System | Operating pH Range | Key Features | Ideal Use Cases |
|---|---|---|---|
| Novex Tris-Glycine | 8.3 – 9.5 [1] | Traditional Laemmli system [1] | Keeping native net charge; studying smaller proteins (20–500 kDa) [1] |
| NuPAGE Tris-Acetate | 7.2 – 8.5 [1] | Better resolution of larger proteins [1] | Keeping native net charge; studying larger proteins (>150 kDa) [1] |
| NativePAGE Bis-Tris | ~7.5 [1] | Uses G-250 dye for charge-shift; resolves proteins by mass regardless of pI [1] | Membrane/hydrophobic proteins; separating by molecular weight [1] |
Ionic Strength and Buffer Composition
The ionic strength of the buffer, determined by the concentration of salts and ions, plays a dual role. It is essential for maintaining buffering capacity and protein stability, but it must be carefully controlled to prevent adverse effects.
Effects of Ionic Strength:
- Optimal Conductivity: Adequate ionic strength provides the necessary conductivity for efficient electrophoretic runs.
- Shielding and Disruption: High ionic strength can shield protein charges, weakening the electric field within the gel and leading to slower migration, band spreading, and Joule heating [7]. Excessive salts (e.g., NaCl) can also disrupt native protein structures.
- Practical Guideline: Salt concentrations in samples should generally be kept below 500 mM to prevent smearing and distorted bands [7]. For running buffers, follow manufacturer recommendations, which are typically in the range of 25-200 mM.
Volatile Buffers for MS Compatibility: For downstream analysis like native mass spectrometry, volatile buffers such as ammonium acetate (AmAc), ammonium bicarbonate (AmBc), and ammonium formate (AmFo) are preferred due to their compatibility with MS systems [48]. Non-volatile salts and buffers like Tris, HEPES, or PBS can interfere with ionization and should be exchanged prior to MS analysis [18].
Functional Additives
Additives are used to enhance protein solubility, stabilize native structures, and prevent aggregation, which is particularly important for membrane proteins and large complexes.
Table 2: Common Additives for Native PAGE Optimization
| Additive Category | Examples | Function | Application Notes |
|---|---|---|---|
| Charge-Shift Agents | Coomassie G-250 [1] | Binds proteins hydrophobically, conferring negative charge; prevents aggregation [1] | Used in NativePAGE Bis-Tris system; added to cathode buffer and sample [1] |
| Mild Detergents | Dodecyl maltoside (DDM), Decyl maltoside (DM) [39] | Solubilizes membrane proteins while preserving native complexes | Critical for extracting and analyzing membrane proteins; used below critical micelle concentration |
| Non-Ionic Detergents | Triton X-100, Nonidet P-40 | Solubilizes proteins without denaturing | Used in sample preparation to maintain solubility |
| Reducing Agents | DTT, β-Mercaptoethanol | Breaks disulfide bonds to control oligomerization | Use fresh to maintain efficacy; can be omitted to preserve quaternary structure [7] |
| Protease Inhibitors | EDTA-free protease inhibitor cocktails [39] | Prevents protein degradation during sample prep | Essential for working with sensitive or precious samples |
| Polymeric Nanodisc Agents | Glyco-DIBMA, SMA [39] | Encapsulates membrane proteins in a native-like lipid environment | Prevents aggregation; enables analysis of intact membrane protein complexes [39] |
Experimental Protocols for Buffer Optimization
Systematic Screening of Wash and Elution Buffers
An automated, microscale purification workflow can efficiently screen multiple buffer parameters for optimal protein purification before Native PAGE analysis. This approach is highly relevant for optimizing conditions for specific protein targets.
Methodology:
- Setup: Implement a dispersive solid-phase extraction pipette tip on an automated liquid handler to screen resin, wash, and elution parameters [50].
- Wash Buffer Optimization: Test wash buffers with varying additives. For instance, sodium chloride can be added to minimize host cell protein (HCP) contamination while preserving monomeric yields [50].
- Elution pH Screening: Systematically vary the elution pH across a physiologically relevant range (e.g., pH 3.0–5.0). Monitor for performance differences between resins, as some may maintain higher yields at higher elution pH (>pH 4.2) [50].
- Analysis: Evaluate outcomes based on target protein yield, HCP clearance, and the presence of aggregates or monomers [50].
High-Resolution Clear Native Electrophoresis (hrCNE) for Membrane Proteins
This protocol is designed for the analysis of membrane protein complexes, which are prone to aggregation.
Workflow:
- Membrane Protein Extraction and Solubilization:
- Resuspend the membrane fraction in a standard buffer (e.g., 150 mM NaCl, 50 mM Tris-HCl, pH 8.0).
- Solubilize overnight at 18–20°C using a charged polymer like Glyco-DIBMA (in a ratio of 2:1 to 1:2 polymer to membrane protein), supplemented with 4 mM MgCl₂ [39]. This forms nanodiscs that preserve a native-like lipid bilayer environment.
- Purification: Purify the target protein using affinity chromatography (e.g., Ni²⁺-NTA for His-tagged proteins) followed by size-exclusion chromatography [39].
- Gel Electrophoresis:
- Use a pre-cast or hand-casted gradient polyacrylamide gel.
- For hrCNE, the cathode buffer may contain mixed micelles of mild anionic and neutral detergents (e.g., sodium deoxycholate and dodecyl maltoside) to impose a negative charge shift and increase resolution without dyes [39].
- Load samples mixed with a glycerol-based solution (e.g., containing 0.1% Ponceau S for visualization).
- Run the gel at 4°C. Start at 100 V for 15–20 minutes for stacking, then increase to a maximum of 160 V until the dye front approaches the gel bottom [39].
- Post-Electrophoresis Analysis:
- Visualize protein bands using in-gel fluorescence (if pre-labeled) or other native-compatible staining.
- For functional studies, excise bands, resuspend in an appropriate buffer overnight to extract proteins, and analyze [39].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Native PAGE Buffer Optimization
| Reagent / Solution | Function / Purpose | Technical Notes |
|---|---|---|
| Tris-Glycine Native Running Buffer | Provides high-pH (8.3-9.5) environment for charge-based separation [1] | Standard for traditional Native PAGE; compatible with various gel types [1] |
| NativePAGE Running Buffer & Cathode Additive | Creates charge-shift environment with Coomassie G-250 at ~pH 7.5 [1] | Essential for NativePAGE Bis-Tris system; allows separation by mass [1] |
| Coomassie G-250 Dye | Charge-shift molecule; binds proteins to confer negative charge [1] | Prevents aggregation of membrane/hydrophobic proteins [1] |
| Glyco-DIBMA Polymer | Charged amphiphilic copolymer for membrane protein nanodisc formation [39] | Preserves native lipid bilayer; prevents aggregation during CNE [39] |
| Dodecyl Maltoside (DDM) | Mild non-ionic detergent for membrane protein solubilization [39] | Maintains native protein complexes; used in hrCNE cathode buffers [39] |
| Protease Inhibitor Cocktails (EDTA-free) | Inhibits proteolytic degradation during sample preparation [39] | Critical for preserving full-length proteoforms; EDTA-free is MS-compatible |
| Volatile Salts (AmAc, AmBc, AmFo) | MS-compatible buffers for IEC and other hyphenated techniques [48] | Enable direct coupling of separation to native mass spectrometry [48] |
Advanced and Integrated Techniques
Hyphenated Techniques for Charge Variant Analysis
Ion Exchange Chromatography-Mass Spectrometry (IEC-MS) is a powerful non-denaturing technique for analyzing protein charge heterogeneity. It is particularly useful for characterizing charge variants arising from post-translational modifications (PTMs) like deamidation, phosphorylation, and sialylation [48].
Key Developments:
- pH Gradient Elution: Modern IEC-MS methods often use volatile buffer systems to create controlled linear pH gradients, replacing traditional salt gradients that rely on non-volatile additives incompatible with MS [48].
- Application: This technique can resolve and identify diverse therapeutic protein variants, including monoclonal antibodies (mAbs), antibody-drug conjugates (ADCs), and complex biological samples, providing a direct link between charge heterogeneity and molecular identity [48].
Native Top-Down Mass Spectrometry for Modification Mapping
Software platforms like precisION leverage native top-down mass spectrometry (nTDMS) to discover "hidden" protein modifications within intact protein complexes [51]. This technique preserves the native state of proteins and their complexes, allowing researchers to directly link PTMs (e.g., phosphorylation, glycosylation) to their effects on protein structure and function [51]. The workflow involves soft ionization of the native protein complex, mass analysis, and subsequent fragmentation to localize modifications, all under conditions that maintain non-covalent interactions.
The optimization of buffer conditions—pH, ionic strength, and additives—is a foundational activity in Native PAGE research that directly controls protein charge and migration. A deep understanding of the relationship between buffer pH and protein pI is essential for predicting and controlling electrophoretic behavior. The strategic use of additives like detergents, charge-shift dyes, and stabilizing polymers is critical for handling challenging proteins, particularly membrane protein complexes. By applying the systematic optimization protocols and leveraging the advanced toolkit outlined in this guide, researchers can design robust and reproducible Native PAGE experiments. This enables the accurate characterization of proteins in their native, functional states, thereby advancing research in structural biology, biopharmaceutical development, and molecular pathology.
In native polyacrylamide gel electrophoresis (PAGE), a protein's migration is not solely dependent on its mass but is governed by a complex interplay of its intrinsic charge, hydrodynamic size, and three-dimensional shape. This stands in stark contrast to SDS-PAGE, where SDS treatment confers a uniform negative charge and denatures proteins, making separation dependent primarily on molecular weight [7] [2]. The preservation of native structure is a double-edged sword; while it allows for the analysis of functional protein complexes, quaternary structure, and enzymatic activity, it also introduces significant challenges in obtaining sharp, reproducible bands. artefacts such as smeared bands, aberrant migration, and loss of resolution frequently arise from two critical, and often overlooked, technical factors: inadequate temperature control and incomplete gel polymerization. This guide details the core principles and methodologies for managing these variables to ensure the integrity of native PAGE data, with a specific focus on how they influence the fundamental relationship between native protein charge and electrophoretic mobility.
The Core Principle: Protein Charge and Migration in Native PAGE
A protein's net charge in a native gel is determined by the pH of the running buffer relative to the protein's isoelectric point (pI). In the standard alkaline Tris-Glycine buffer (pH ~8.3-8.8), most proteins carry a net negative charge and will migrate towards the anode [2]. The resulting mobility is a function of both this charge density (net charge per unit mass) and the frictional force, or sieving effect, imposed by the gel matrix [2]. A protein with a high negative charge density will migrate rapidly, while a larger, more globular protein will experience greater resistance. This charge-mass-shape relationship is the basis of separation in native PAGE.
Consequently, any factor that alters the protein's native conformation, its effective charge, or the consistency of the gel matrix will disrupt this relationship and introduce artefacts. Incomplete polymerization creates a heterogeneous gel with uneven pore sizes, leading to distorted band shapes and inconsistent migration. Poor temperature control can induce protein aggregation, alter buffer pH, and cause convective mixing within the gel, all of which manifest as smeared or "smiling" bands [7].
Section 1: Managing Temperature to Preserve Native Charge and Conformation
Temperature is a critical parameter that, if unmanaged, can degrade separation quality through multiple mechanisms. Joule heating can generate localized hot spots, denaturing proteins and disrupting their charge profile. It can also alter buffer pH and ionic strength, directly impacting the protein's net charge and migration rate [7].
Standard Temperature Control Protocols
For conventional native PAGE, maintaining a cool and consistent temperature is paramount. Standard practice involves running gels in a cold room (4°C) or using a recirculating cooling apparatus [2]. This minimizes Joule heating effects, helps maintain protein stability, and prevents the gel buffer from shifting pH. The phenomenon of "smiling" bands, where bands curve upward at the edges, is a classic indicator of excessive heat, typically caused by running the gel at too high a voltage or with an improperly formulated buffer [7].
Advanced Thermal Gel Electrophoresis
Emerging technologies are redefining the role of temperature in electrophoresis. Thermal Gel Transient Isotachophoresis (TG-tITP) utilizes temperature-responsive polymers like Pluronic F-127, which can reversibly change viscosity by over 1000-fold in response to temperature [52]. This allows temperature to be used as an active separation parameter rather than a variable to be suppressed.
- Methodology: A microfluidic device is loaded with a thermal gel containing the protein sample. Electrolytes are optimized for tITP to preconcentrate proteins before separation. The device is then placed on a Peltier stage for precise thermal control [52].
- Application of Temperature Gradients: Research has demonstrated that applying a spatial or temporal temperature gradient during a run can dynamically alter the gel's viscosity. This maximizes separation resolution across a wide mass range (6–464 kDa) by fine-tuning the sieving properties of the matrix during the separation process [52].
- Performance: This advanced method has been shown to provide a two-fold higher resolution than traditional native PAGE while using 15,000-fold less sample and achieving a five-fold faster analysis time [52].
Table 1: Temperature Management Techniques and Their Applications
| Technique | Principle | Optimal Conditions | Impact on Separation |
|---|---|---|---|
| Standard Cooled PAGE | Minimizes Joule heating to preserve protein structure and buffer pH. | Run in cold room (4°C) or with external cooling; use correct voltage. | Prevents band smiling, smearing, and protein aggregation artefacts. |
| Thermal Gel tITP | Uses temperature to dynamically control gel matrix viscosity. | Employ a temperature gradient (e.g., 10°C to 25°C) during electrophoresis [52]. | Enhances resolution; allows separation of a wide mass range in a single run. |
The following workflow contrasts the setup for standard cooled native PAGE with the advanced thermal gel tITP method:
Section 2: Ensuring Complete Polymerization for a Uniform Matrix
A consistently polymerized polyacrylamide gel is a non-negotiable prerequisite for high-resolution native PAGE. Incomplete or uneven polymerization creates a heterogeneous matrix with varying pore sizes, which distorts protein migration and leads to smeared or skewed bands. This is especially critical in native PAGE because proteins are separated based on their ability to navigate these pores according to their native size and shape.
Gel Polymerization Components and Protocol
The gel matrix is formed by the copolymerization of acrylamide monomers and bisacrylamide cross-linkers, a reaction typically initiated by ammonium persulfate (APS) and catalyzed by TEMED (N,N,N',N'-Tetramethylethylenediamine) [2].
- Standard Protocol for Casting a Discontinuous Gel:
- Prepare the Resolving Gel: Mix acrylamide/bisacrylamide solution at the desired percentage (e.g., 7-12%), Tris-HCl buffer (pH 8.8), and water. Add 10% APS and TEMED, mix thoroughly but gently to avoid introducing oxygen, and pipette the solution into the gel cassette. Carefully overlay with water-saturated butanol or isopropanol to exclude oxygen and create a flat gel surface.
- Polymerization: Allow the gel to polymerize completely for 20-30 minutes at room temperature. A distinct schlieren line will appear between the gel and the overlay.
- Prepare the Stacking Gel: Once the resolving gel is set, pour off the overlay. Mix a lower percentage acrylamide solution (e.g., 4-5%) with Tris-HCl buffer (pH 6.8). Add APS and TEMED, pour over the resolving gel, and immediately insert a clean comb.
- Polymerization: Allow the stacking gel to polymerize for 15-20 minutes before carefully removing the comb [2].
Troubleshooting Incomplete Polymerization
- Old or Degraded Reagents: APS solution decomposes quickly and should be made fresh weekly or stored as frozen aliquots. Acrylamide and bisacrylamide solutions can also hydrolyze over time, inhibiting polymerization.
- Insufficient APS/TEMED: An inadequate amount of initiator or catalyst will slow the reaction or prevent it from going to completion.
- Oxygen Inhibition: Oxygen is a potent inhibitor of the free-radical polymerization reaction. Overlaying the gel solution is a crucial step to create an anaerobic interface [2].
- Incorrect pH: The polymerization reaction is most efficient within a specific pH range. Ensure all buffers are prepared correctly.
Table 2: Essential Reagents for Gel Polymerization and Artefact Prevention
| Research Reagent / Material | Critical Function | Considerations for Native PAGE |
|---|---|---|
| Acrylamide / Bis-acrylamide | Monomer and cross-linker that form the porous gel matrix. | Concentration (%T) dictates pore size; must be optimized for target protein size [2]. |
| Ammonium Persulfate (APS) | Initiates the free-radical polymerization reaction. | Prepare fresh solution weekly; decomposition is a primary cause of incomplete gel formation. |
| TEMED | Catalyzes the production of free radicals from APS, accelerating polymerization. | Quantity affects polymerization speed; too little leads to slow, uneven gel formation. |
| Water-saturated Butanol | Overlay to exclude oxygen during resolving gel polymerization. | Critical for preventing oxygen inhibition and ensuring a straight, fully polymerized gel interface. |
| Tris-based Buffers | Provides the required pH for both polymerization and electrophoresis. | Use high-purity reagents; pH of stacking (∼6.8) and resolving (∼8.8) gels is critical for proper stacking [2]. |
| Pluronic F-127 Thermal Gel | Temperature-responsive matrix for advanced TG-tITP [52]. | Allows dynamic control of separation; requires precise temperature control equipment. |
The consequences of improper technique and the path to optimal results are summarized in the following troubleshooting flowchart:
The integrity of data derived from native PAGE is fundamentally dependent on rigorous technical execution. The migration of a native protein—a direct reflection of its intrinsic charge, size, and shape—is exquisitely sensitive to its electrophoretic environment. As detailed in this guide, failure to maintain a cool, stable temperature and to ensure the formation of a completely uniform gel matrix will inevitably introduce artefacts that obscure true protein behavior. By adopting the protocols for temperature management and gel polymerization outlined herein, researchers can mitigate these pervasive sources of error. This disciplined approach is essential for generating reliable, reproducible data that accurately reveals the properties of native proteins and their complexes, thereby strengthening conclusions in fundamental research and drug development.
In native polyacrylamide gel electrophoresis (PAGE), the migration of proteins is governed by a combination of their intrinsic charge, size, and three-dimensional shape. This stands in stark contrast to denaturing techniques like SDS-PAGE, where SDS binding confers a uniform negative charge and denatures proteins, causing separation based primarily on molecular mass [53] [9]. While invaluable for studying proteins in their native state, the multi-parameter dependence in native PAGE often leads to a significant analytical challenge: the co-migration of distinct proteins that possess similar charge-to-mass ratios, thereby compromising resolution and leading to misinterpretation of results.
This technical guide explores the fundamental principles behind this phenomenon and provides detailed methodologies to overcome it, framed within the context of advanced electrophoretic research.
Core Principles: Why Proteins Co-migrate in Native PAGE
The fundamental difference between SDS-PAGE and native PAGE lies in the treatment of the protein sample.
- In SDS-PAGE, proteins are denatured and uniformly coated with the negatively charged SDS detergent. This masks the protein's intrinsic charge and disrupts its higher-order structure, creating a near-linear relationship between migration distance and the logarithm of the polypeptide's molecular mass [53] [9].
- In native PAGE, proteins are not denatured. Consequently, their migration through the gel matrix is influenced by:
Therefore, two different proteins—for instance, a large, highly charged protein and a small, moderately charged protein—can experience the same net electrophoretic driving force and frictional drag, resulting in identical migration distances and a single, coalesced band. This co-migration obscures the true complexity of a sample and can invalidate quantitative and qualitative analyses.
The table below summarizes the key factors governing protein migration in different electrophoretic techniques.
Table 1: Fundamental Factors Governing Protein Migration in PAGE Techniques
| Factor | SDS-PAGE | Native PAGE | BN-/CN-PAGE |
|---|---|---|---|
| Primary Separation Basis | Molecular Mass | Size, Shape, & Net Charge | Native Mass & Shape |
| Protein Conformation | Denatured (Unfolded) | Native (Folded) | Native (Folded) |
| Quaternary Structure | Disrupted into subunits | Preserved | Preserved |
| Influence of Intrinsic Charge | Negligible (masked by SDS) | High | Moderate (affected by dye/detergent) |
| Typical Resolution | High | Variable; lower for similar charge/mass | High for protein complexes |
Advanced Methodologies to Enhance Resolution
When standard native PAGE yields poor resolution, several advanced techniques can be employed to separate co-migrating species.
High-Resolution Clear Native PAGE (hrCN-PAGE)
hrCN-PAGE is a refined technique designed to provide superior resolution of native protein complexes, particularly for in-gel functional assays. It improves upon traditional Blue Native PAGE (BN-PAGE), where the Coomassie dye used can interfere with fluorescence detection and enzymatic activity assays [54]. hrCN-PAGE substitutes the dye with non-colored mixtures of anionic and neutral detergents. These mixed micelles impose a charge shift on membrane proteins to enhance migration and improve solubility, thereby reducing aggregation and band broadening [54]. This method is ideal for subsequent in-gel activity staining.
Protocol: In-Gel Activity Assay for Medium-Chain Acyl-CoA Dehydrogenase (MCAD) following hrCN-PAGE [23]
This protocol demonstrates how hrCN-PAGE, coupled with a specific activity stain, can resolve active tetramers from other protein forms.
Key Research Reagent Solutions:
- hrCN-PAGE Gel: 4–16% gradient polyacrylamide gel for high-resolution separation.
- Reaction Buffer: Contains the physiological substrate, octanoyl-CoA, and the electron acceptor nitro blue tetrazolium chloride (NBT).
- Staining Solution: NBT is reduced to an insoluble, purple-colored diformazan precipitate upon accepting electrons, revealing the location of active enzyme bands.
Experimental Workflow:
- Sample Preparation: Isolate mitochondrial fractions or use purified recombinant protein. Maintain samples in a non-denaturing buffer.
- Electrophoresis: Perform hrCN-PAGE at 4°C to maintain protein activity, using an anode buffer without Coomassie dye.
- In-Gel Activity Staining: Incubate the gel in the reaction buffer containing octanoyl-CoA and NBT.
- Detection & Analysis: Monitor the formation of purple formazan bands, which indicates enzymatic activity. Use densitometry for quantification.
Application: This method was used to characterize clinically relevant MCAD variants, revealing that while some mutants formed tetramers, they exhibited altered migration and reduced activity, insights that would be lost in a standard activity assay [23].
Native SDS-PAGE (NSDS-PAGE)
NSDS-PAGE is a hybrid technique that aims to balance the high resolution of SDS-PAGE with the retention of native protein features. It involves significantly reducing the SDS concentration in the running buffer and eliminating the sample heating and denaturation steps [3].
Table 2: Buffer Composition Comparison for SDS-PAGE vs. NSDS-PAGE [3]
| Component | Standard SDS-PAGE | NSDS-PAGE |
|---|---|---|
| Sample Buffer | Contains LDS (a derivative of SDS) and EDTA | No SDS/EDTA; contains Coomassie G-250 & Phenol Red |
| Sample Treatment | Heated (70–100°C) | Not heated |
| Running Buffer | 0.1% SDS, 1 mM EDTA | 0.0375% SDS, no EDTA |
| Primary Outcome | Denatured polypeptides | Native enzymes with retained activity |
Protocol: NSDS-PAGE for Metalloprotein Analysis [3]
Key Research Reagent Solutions:
- NSDS Sample Buffer: 100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5.
- NSDS Running Buffer: 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7.
- Model Enzymes: e.g., Zn²⁺-proteins like alcohol dehydrogenase (ADH).
Experimental Workflow:
- Sample Preparation: Mix protein sample with 4X NSDS sample buffer without heating.
- Electrophoresis: Load onto a standard precast Bis-Tris gel and run with NSDS running buffer.
- Post-Electrophoresis Analysis:
- Activity Staining: Incubate gel in specific substrate/cofactor solutions to detect active enzymes.
- Metal Detection: Use laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) or in-gel staining with metal-binding fluorophores like TSQ.
Application: This method demonstrated a retention of 98% of Zn²⁺ bound in proteomic samples, and seven out of nine model enzymes retained their activity after separation, a feat impossible with standard SDS-PAGE [3].
Experimental Workflow and Decision Pathway
The following diagram illustrates a logical workflow for diagnosing poor resolution in native PAGE and selecting the most appropriate advanced method based on research objectives.
Addressing the challenge of co-migration in native PAGE requires a shift from standard protocols to more sophisticated, goal-oriented techniques. The co-migration of proteins with similar charge-to-mass ratios is not an insurmountable dead end but an indication that the sample's complexity demands a higher-resolution native separation method. By understanding the principles behind protein migration and implementing advanced methods like hrCN-PAGE for supreme functional analysis or NSDS-PAGE for a balance of high resolution and native state preservation, researchers can uncover critical details about protein complexes, oligomeric states, and enzymatic function that are essential for rigorous scientific discovery and therapeutic development.
The fundamental principle of native polyacrylamide gel electrophoresis (Native PAGE) is the separation of proteins based on their intrinsic properties—net charge, size, and three-dimensional shape—under non-denaturing conditions [1] [2]. Unlike SDS-PAGE, which imparts a uniform negative charge to all proteins, native PAGE preserves the protein's native structure, allowing for the analysis of multimeric complexes and enzymatic activity [3]. A protein's electrophoretic migration in this system is governed by its charge density (the ratio of net charge to molecular mass) and the sieving effect of the gel matrix [1]. Consequently, a protein's isoelectric point (pI), the pH at which it carries no net charge, becomes a critical determinant of its migratory behavior.
The central challenge addressed by polarity reversal arises from the standard alkaline running buffers (e.g., pH 8.3-9.5 for Tris-Glycine systems) used in native PAGE [1]. Under these conditions, most proteins carry a net negative charge and migrate toward the positively charged anode. However, highly basic proteins (pI > 9) retain a net positive charge even at these alkaline pH levels. In a standard setup, these positively charged proteins would either fail to enter the gel or migrate in the reverse direction toward the cathode, preventing effective separation and analysis [32]. The technique of reverse polarity, or "cathodal electrophoresis," elegantly solves this problem by physically swapping the electrical connections of the electrodes, thereby creating an attractive force that draws basic proteins into and through the gel matrix.
Core Principle: Why Reverse Polarity is Necessary for Basic Proteins
The Charge-Migration Relationship in Standard vs. Reverse Polarity
The direction and rate of a protein's migration in an electric field are direct consequences of its net charge at the running buffer's pH. Figure 1 illustrates this fundamental relationship and how it is manipulated for basic proteins.
Figure 1. Migration of Basic Proteins Under Standard and Reverse Polarity Conditions. In standard setups, the protein's positive charge causes migration toward the cathode, away from the gel matrix. Reversing the polarity makes the cathode the "top" electrode, drawing the protein into the gel.
In practice, the measured valence of a protein can differ significantly from its theoretical calculation. For instance, a study on cytochrome c (theoretical pI ~10) found its experimentally determined valence at pH 7.0 was approximately +4.1, roughly two-fold lower than the primary structure prediction of +7.8 [5]. This disparity underscores the importance of empirical data and the potential for specific ion interactions to modulate a protein's effective charge.
Alternative Strategies for Charge Modulation
While reverse polarity is a direct solution, other native PAGE methodologies modulate protein charge to achieve separation. The NativePAGE Bis-Tris system, for example, uses Coomassie G-250 dye, which binds non-specifically to hydrophobic protein patches [1]. This binding confers a net negative charge even on basic proteins, allowing them to migrate toward the anode in a standard setup [1]. This system is particularly beneficial for membrane proteins or those with significant hydrophobic surfaces, as the dye helps prevent aggregation [1]. However, for researchers requiring the use of specific gel chemistries like Tris-Glycine or needing to avoid dye-protein interactions for functional assays, reverse polarity remains the most straightforward and effective technique for basic proteins.
Detailed Experimental Protocol for Reverse Polarity Electrophoresis
This protocol is adapted from established native PAGE methods and the specific application of reverse polarity for protein recovery and analysis [55] [32]. The following section provides a step-by-step guide for the separation of a basic protein (pI > 9) using a standard vertical gel electrophoresis system.
Required Reagents and Materials
Table 1: Key Research Reagent Solutions for Reverse Polarity Native PAGE
| Item | Function/Description | Key Considerations |
|---|---|---|
| Tris-Glycine Native Gel (e.g., 8-16% gradient) [1] | Porous matrix for size-based separation. | Lower % acrylamide for larger proteins; higher % for smaller proteins. |
| Tris-Glycine Native Running Buffer (25 mM Tris, 192 mM Glycine, pH ~8.3) [32] | Conducts current and maintains pH for separation. | Do not adjust pH; its inherent alkalinity is key for charge determination [32]. |
| Non-Denaturing Sample Buffer (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 0.01% Bromophenol Blue) [32] | Loads sample without denaturation; glycerol adds density. | Crucially, do not heat the samples [32]. |
| Pre-stained Native Markers | Visualize migration and approximate separation. | Ensure markers are compatible with native conditions. |
| Standard Vertical Gel Electrophoresis Unit | Apparatus to hold gel and buffer for electrophoresis. | No specialized equipment beyond a standard slab gel system is required [55]. |
Step-by-Step Method
- Gel Preparation: Cast a discontinuous native polyacrylamide gel comprising a separating gel (e.g., 10-12%) and a stacking gel (e.g., 4%) using non-denaturing Tris-HCl buffers at pH 8.8 and 6.8, respectively [32]. Polymerize the gel completely.
- Sample Preparation: Mix the protein sample (containing your basic protein of interest) with an equal volume of 2X non-denaturing sample buffer. It is critical to avoid heating the samples to preserve native structure and activity [32].
- Apparatus Setup: Assemble the gel cassette into the electrophoresis tank. Fill the upper and lower buffer chambers with pre-chilled Tris-Glycine Native Running Buffer.
- Load Samples: Carefully load the prepared protein samples and native markers into the wells.
- Reverse Electrical Connections: This is the crucial step. To reverse the polarity, connect the positive (red) lead from the power supply to the upper buffer chamber electrode (where the wells and cathode are traditionally located), and the negative (black) lead to the lower buffer chamber electrode. This swap creates a cathode at the top, which will attract and draw the positively charged basic proteins into the gel.
- Electrophoresis: Run the gel at a constant voltage, typically between 100-150V, keeping the apparatus cool by using a cold room or a circulating water bath. Do not set a very high voltage to prevent protein degradation and overheating [32]. Continue electrophoresis until the tracking dye has migrated an appropriate distance through the gel.
- Post-Electrophoresis Analysis: Following separation, proteins can be recovered or analyzed by:
- Detection: Stain the gel with Coomassie Blue or use activity-specific stains for enzymatic detection [3].
- Protein Recovery: For high-yield recovery of native, biologically active proteins, the gel slice containing the protein of interest can be placed into an elution apparatus or a standard dialysis bag filled with buffer. Reverse polarity electrophoresis can then be applied again in a tube gel system to electro-elute the protein from the gel slice into the surrounding buffer, achieving recovery yields as high as 90% [55].
- Western Blotting: Note that standard Western blotting protocols assume negatively charged proteins. For basic proteins, the transfer conditions may need to be optimized to account for their positive charge [56]. PVDF membranes are recommended over nitrocellulose for native gels, especially if Coomassie dye is used [1].
Data Interpretation and Analysis
Interpreting results from a reverse polarity native PAGE experiment requires an understanding of how protein properties influence migration.
Table 2: Expected Migration Patterns in Reverse Polarity Native PAGE
| Protein Characteristic | Impact on Migration | Expected Observation |
|---|---|---|
| High Positive Net Charge | Increased migration rate toward the cathode. | Band appears further from the well. |
| Large Molecular Size/Shape | Increased frictional drag from gel matrix. | Band appears closer to the well (slower migration). |
| Protein Complex Formation | Alters both net charge and size/shape. | Shift in band position (e.g., slower migration for a larger complex) compared to monomer. |
| Bound Ligands or Cofactors | Can alter the net charge of the protein. | Subtle shift in band position or changes in detected activity. |
The successful separation and subsequent recovery of active protein is a key metric. The reverse polarity elution method has been demonstrated to effectively isolate a diverse range of proteins, from small polypeptides (9,000 daltons) to large complexes (186,000 daltons), with recovered proteins retaining their biological activity [55]. For example, this technique has been used to isolate the copper-zinc and manganese superoxide dismutases from crude soybean extracts and vitamin D-dependent calcium-binding proteins from rat tissues in a homogeneous and active form [55].
Troubleshooting Common Issues
- Protein Does Not Enter Gel: Confirm that the polarity has been correctly reversed. Verify the protein's pI is indeed basic and that the running buffer pH is appropriate.
- Poor Resolution or Smearing: Ensure the gel was thoroughly polymerized and that the samples were not heated or denatured. Run the gel at a lower voltage to minimize heat generation. Check for proteolytic degradation by adding protease inhibitors to the sample buffer.
- Low Protein Recovery after Elution: For electro-elution, ensure the gel slice is finely crushed to increase surface area and allow efficient elution. Use a clean dialysis membrane to prevent protein adsorption. Confirm the elution buffer is compatible with protein stability.
- No Enzymatic Activity after Recovery: Avoid harsh fixatives or organic solvents if activity assays are planned. Ensure that all steps, including electrophoresis and elution, are performed in cold buffers to maintain protein stability.
Validation and Technique Comparison: Native PAGE vs. SDS-PAGE and BN-PAGE
This technical guide provides an in-depth comparison of Native Polyacrylamide Gel Electrophoresis (PAGE) and Sodium Dodecyl Sulfate-PAGE (SDS-PAGE), focusing on their fundamental mechanisms, methodological considerations, and applications in protein research. Within the context of how protein charge affects migration in native PAGE, we explore the intricate relationship between a protein's intrinsic properties and its electrophoretic behavior. For researchers, scientists, and drug development professionals, understanding these distinctions is crucial for selecting the appropriate technique to answer specific biological questions, particularly those involving protein structure-function relationships, complex formation, and therapeutic development.
Protein electrophoresis is a standard laboratory technique by which charged protein molecules are transported through a solvent by an electrical field [2]. This method serves as a simple, rapid, and sensitive analytical tool for separating protein mixtures based on their physicochemical properties. Polyacrylamide gel electrophoresis (PAGE) represents one of the most widely employed electrophoretic techniques, with Native PAGE and SDS-PAGE constituting two principal variants that serve distinct purposes in protein characterization [57].
The core principle of electrophoresis relies on the fact that most biological molecules carry a net charge at any pH other than their isoelectric point and will migrate through a porous matrix at a rate proportional to their charge density when subjected to an electric field [2]. The mobility of a protein molecule during electrophoresis depends on several factors: field strength, net charge on the molecule, size and shape of the molecule, ionic strength, and properties of the matrix through which the molecule migrates [2]. Both Native PAGE and SDS-PAGE utilize polyacrylamide gels as a support matrix, which serves as a porous medium that behaves like a molecular sieve [2].
Core Separation Mechanisms: A Tale of Two Techniques
Fundamental Principles and Separation Criteria
The primary distinction between Native PAGE and SDS-PAGE lies in their treatment of protein structure and the consequent basis for separation:
Native PAGE: Separates proteins according to the net charge, size, and shape of their native structure [1] [2]. Electrophoretic migration occurs because most proteins carry a net negative charge in alkaline running buffers [1]. The higher the negative charge density (more charges per molecule mass), the faster a protein will migrate [1]. Simultaneously, the frictional force of the gel matrix creates a sieving effect, regulating movement according to size and three-dimensional shape [1].
SDS-PAGE: Separates proteins primarily by molecular mass [2]. The ionic detergent SDS denatures and binds to proteins in a constant weight ratio (approximately 1.4g SDS:1g polypeptide) [2], making them uniformly negatively charged [58]. This SDS binding masks intrinsic charges and unfolds proteins into linear chains [57], ensuring migration occurs strictly according to polypeptide size [2].
Table 1: Core Differences Between Native PAGE and SDS-PAGE
| Parameter | Native PAGE | SDS-PAGE |
|---|---|---|
| Separation Basis | Size, charge, and shape of native structure [1] [33] | Molecular weight only [58] [33] |
| Protein State | Native, folded conformation [33] [57] | Denatured, linearized [33] [57] |
| Detergent Usage | No SDS or non-denaturing detergents [33] | SDS present to denature proteins [33] |
| Sample Preparation | No heating; non-denaturing buffers [58] [33] | Heating with reducing agents [33] |
| Protein Function Post-Separation | Retained [1] [33] [57] | Lost [33] [57] |
| Primary Applications | Studying structure, complexes, function [33] [57] | Molecular weight determination, purity assessment [33] [57] |
The Critical Role of Protein Charge in Native PAGE Migration
In Native PAGE, a protein's migration pattern is intrinsically linked to its charge characteristics under the specific electrophoresis conditions. Unlike SDS-PAGE, where charge differences are masked by SDS binding, Native PAGE preserves the protein's intrinsic charge, making this parameter a critical determinant of electrophoretic mobility.
The relationship between protein charge and migration in Native PAGE can be understood through several key principles:
Charge Density: The higher the negative charge density (more charges per molecule mass), the faster a protein will migrate toward the anode [1]. This charge density depends on both the protein's amino acid composition and the pH of the running buffer relative to the protein's isoelectric point (pI) [2].
Isoelectric Point Considerations: Proteins with basic isoelectric points (pI) normally have a net positive charge at neutral or slightly basic pH, which would cause them to migrate toward the cathode rather than the anode [1]. Advanced Native PAGE systems address this limitation through the use of charge-shift molecules like Coomassie G-250, which binds to proteins and confers a net negative charge while maintaining native conformation [1].
Buffer pH Optimization: Different Native PAGE systems operate at different pH ranges to optimize protein stability and separation. Tris-Glycine systems operate at pH 8.3-9.5, Tris-Acetate at pH 7.2-8.5, and NativePAGE Bis-Tris at approximately pH 7.5 [1]. The choice of system depends on the protein's stability, resolution requirements, and isoelectric point.
Diagram 1: Comparative workflow of Native PAGE versus SDS-PAGE
Methodological Deep Dive: Experimental Protocols and Conditions
Native PAGE Methodologies and Buffer Systems
Native PAGE encompasses several distinct methodological approaches, each optimized for specific protein types and research questions. The selection of appropriate buffer systems and conditions is critical for maintaining protein stability and achieving optimal separation.
Native PAGE Buffer Systems and Operating Conditions
Three primary gel chemistry systems are available for Native PAGE separation, each with distinct characteristics and applications [1]:
Tris-Glycine System: This traditional Laemmle system operates at pH 8.3-9.5 and is suitable for studying smaller molecular weight proteins (20-500 kDa) while maintaining the native net charge of proteins [1].
Tris-Acetate System: Operating at pH 7.2-8.5, this system provides better resolution of larger molecular weight proteins (>150 kDa) while preserving native charge characteristics [1].
NativePAGE Bis-Tris System: This system operates at approximately pH 7.5 and utilizes Coomassie G-250 dye to resolve proteins by molecular weight regardless of isoelectric point, making it particularly suitable for membrane proteins or hydrophobic proteins [1].
Table 2: Native PAGE Buffer Systems and Specifications
| Parameter | Tris-Glycine | Tris-Acetate | NativePAGE Bis-Tris |
|---|---|---|---|
| Operating pH | 8.3-9.5 [1] | 7.2-8.5 [1] | ~7.5 [1] |
| Molecular Weight Range | 20-500 kDa [1] | >150 kDa [1] | Broad range [1] |
| Special Features | Traditional system [1] | Enhanced resolution for large proteins [1] | Compatible with detergents; uses G-250 dye [1] |
| Recommended Applications | Maintaining native charge; smaller proteins [1] | Larger molecular weight proteins [1] | Membrane proteins; hydrophobic proteins; MW-based separation [1] |
Detailed Native PAGE Protocol Using Bis-Tris Gels
The NativePAGE Bis-Tris system represents an advanced methodology based on the blue native polyacrylamide gel electrophoresis (BN-PAGE) technique developed by Schägger and von Jagow [1]. This protocol overcomes limitations of traditional native gel electrophoresis by providing a near-neutral operating pH and detergent compatibility [1].
Sample Preparation:
- Prepare protein samples in NativePAGE Sample Buffer [1]
- Add NativePAGE 5% G-250 Sample Additive [1]
- Do not heat samples to preserve native structure [33]
- Optional: Include non-ionic detergents for membrane proteins [1]
Gel Preparation and Electrophoresis:
- Use precast NativePAGE Novex 4-16% Bis-Tris 1.0 mm minigels [1]
- Prepare 1X NativePAGE Running Buffer [1]
- Add NativePAGE Cathode Buffer Additive containing Coomassie G-250 [1]
- Load samples and run at constant voltage (150V) at room temperature for approximately 90-95 minutes [3]
- Monitor migration until dye front reaches end of gel [3]
Post-Electrophoresis Analysis:
- For western blotting, use PVDF membranes (nitrocellulose is incompatible as it binds G-250 dye tightly) [1]
- Detect proteins using appropriate staining methods
- For functional studies, proteins can be recovered from gel by passive diffusion or electro-elution [2]
SDS-PAGE and Modified Native SDS-PAGE Protocols
Standard SDS-PAGE Protocol
Traditional SDS-PAGE follows well-established denaturing conditions optimized for molecular weight determination:
Sample Preparation:
- Mix protein sample with 4X LDS sample loading buffer [3]
- Include reducing agents (DTT or β-mercaptoethanol) to break disulfide bonds [33]
- Heat samples at 70°C for 10 minutes to ensure complete denaturation [3]
Gel Electrophoresis:
- Use precast NuPAGE Novex 12% Bis-Tris 1.0 mm minigels [3]
- Prepare 1X MOPS SDS running buffer [3]
- Load samples and Pre-stained SDS-PAGE Standards [3]
- Run at constant voltage (200V) at room temperature for approximately 45 minutes [3]
Native SDS-PAGE (NSDS-PAGE) Hybrid Protocol
Recent methodological advances have led to the development of Native SDS-PAGE (NSDS-PAGE), which aims to balance the high resolution of traditional SDS-PAGE with the functional preservation of Native PAGE [3]. This modified approach reduces denaturing conditions while maintaining effective protein separation.
Key Modifications from Standard SDS-PAGE:
- Remove SDS and EDTA from sample buffer [3]
- Omit the heating step during sample preparation [3]
- Reduce SDS concentration in running buffer from 0.1% to 0.0375% [3]
- Delete EDTA from running buffer [3]
NSDS-PAGE Buffer Composition:
- Sample Buffer: 100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5 [3]
- Running Buffer: 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 [3]
Procedure:
- Pre-run precast NuPAGE Novex 12% Bis-Tris gels at 200V for 30 minutes in ddH₂O to remove storage buffer and unpolymerized acrylamide [3]
- Mix 7.5 μL protein sample with 2.5 μL 4X NSDS sample buffer [3]
- Load samples and run at 200V for appropriate duration [3]
This modified protocol has demonstrated significant retention of metal ions (zinc retention increased from 26% to 98%) and enzymatic activity compared to standard SDS-PAGE [3].
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of Native PAGE and SDS-PAGE methodologies requires specific reagents and materials optimized for each technique. The following table summarizes key components and their functions:
Table 3: Essential Research Reagents for PAGE Techniques
| Reagent/Material | Function | Native PAGE Specifics | SDS-PAGE Specifics |
|---|---|---|---|
| Polyacrylamide Gels | Matrix for protein separation | Various percentages (e.g., 4-16%, 3-12%) [1] | Various percentages (e.g., 12% Bis-Tris) [3] |
| Charge-modifying Agent | Imparts charge for migration | Coomassie G-250 dye [1] | SDS detergent [58] |
| Sample Buffer | Prepares proteins for loading | Non-denaturing buffers [58] | Denaturing buffers with reducing agents [33] |
| Running Buffer | Conducts current; maintains pH | Tris-Glycine, Tris-Acetate, or Bis-Tris based [1] | MOPS SDS or similar SDS-containing buffers [3] |
| Molecular Weight Standards | Reference for size determination | NativeMark unstained standards [3] | Pre-stained or unstained SDS-PAGE standards [3] |
| Transfer Membrane | Protein immobilization for blotting | PVDF (nitrocellulose incompatible) [1] | Nitrocellulose or PVDF [1] |
Advanced Applications in Drug Development and Research
The distinct capabilities of Native PAGE and SDS-PAGE make them suitable for complementary applications in pharmaceutical research and development. Understanding their respective strengths enables researchers to select the optimal approach for specific investigative goals.
Native PAGE Applications in Therapeutic Development
Native PAGE provides unique advantages for drug discovery applications where maintaining protein structure and function is essential:
Protein-Protein Interaction Studies: Native PAGE preserves oligomeric states and protein complexes, allowing researchers to investigate protein-protein interactions under conditions that mimic the cellular environment [57]. This is particularly valuable for studying receptor-ligand interactions and multiprotein complexes targeted by therapeutic agents.
Enzymatic Activity Screening: Since Native PAGE preserves biological activity, it can be used to screen for enzymatic activity directly in gel fractions [1] [57]. This application is crucial for identifying and characterizing enzymes relevant to disease pathways or industrial processes.
Therapeutic Protein Characterization: For biologic therapeutics such as monoclonal antibodies and recombinant proteins, Native PAGE helps assess proper folding, complex formation, and stability under native conditions [57]. This information is critical for ensuring product quality and efficacy.
Membrane Protein Studies: The compatibility of certain Native PAGE systems with non-ionic detergents enables the analysis of membrane proteins and hydrophobic proteins that are challenging to study using denaturing methods [1]. This capability is particularly relevant for drug targets such as GPCRs and ion channels.
SDS-PAGE Applications in Biopharmaceutical Analysis
SDS-PAGE remains indispensable for specific aspects of therapeutic development requiring precise molecular weight determination and purity assessment:
Purity Analysis and Quality Control: SDS-PAGE provides high-resolution separation of proteins based on size, making it ideal for assessing the purity of protein therapeutics and identifying potential contaminants or degradation products [57].
Subunit Composition Determination: By disrupting non-covalent interactions, SDS-PAGE reveals the subunit composition of complex proteins, providing information about primary structure and potential heterogeneity [57].
Western Blotting Compatibility: The denatured, linearized proteins generated by SDS-PAGE are ideal for western blotting applications, enabling specific detection of target proteins using antibodies [57] [2]. This application is fundamental to target validation and biomarker discovery.
Post-Translational Modification Analysis: While not preserving native structure, SDS-PAGE can reveal size shifts caused by post-translational modifications such as glycosylation or phosphorylation, particularly when used in conjunction with specific enzymatic treatments [57].
Diagram 2: Factors affecting protein migration in Native PAGE, emphasizing charge relationships
Native PAGE and SDS-PAGE represent complementary techniques that serve distinct purposes in protein analysis. Native PAGE excels in applications requiring preservation of native structure, function, and interactions, with protein charge playing a critical role in separation efficiency [1] [2]. SDS-PAGE provides high-resolution separation based primarily on molecular weight, ideal for determining protein size, purity, and subunit composition [58] [2].
The development of hybrid techniques such as NSDS-PAGE [3] and the availability of specialized Native PAGE systems [1] continue to expand the toolbox available to researchers. For drug development professionals, strategic selection between these techniques—or their sequential application—can provide comprehensive insights into protein characteristics relevant to therapeutic discovery and development.
Understanding the fundamental role of protein charge in Native PAGE migration enables researchers to optimize experimental conditions, interpret results accurately, and extract maximum biological insight from their electrophoretic separations. As protein therapeutics and targeted therapies continue to advance, these electrophoretic techniques will remain essential components of the analytical toolkit in biomedical research and pharmaceutical development.
In protein analysis, encountering divergent results between electrophoretic techniques is not an anomaly but a critical source of structural information. A common observation—a protein migrating at 60 kDa on SDS-PAGE but at 120 kDa on Native PAGE—provides direct evidence of a multi-subunit native complex [33] [57]. This discrepancy arises from the fundamental principles governing each method: SDS-PAGE separates denatured polypeptide chains based almost exclusively on molecular weight, while Native PAGE separates native proteins based on the combined factors of size, intrinsic charge, and three-dimensional shape [33] [58] [59]. For researchers and drug development professionals, accurately interpreting this data is paramount for understanding protein quaternary structure, functional complexes, and interaction networks that define biological activity.
The core of this interpretation lies in understanding how protein charge affects migration in Native PAGE. Unlike SDS-PAGE, which masks intrinsic charge with a uniform negative charge coat, Native PAGE preserves the protein's native charge, making its isoelectric point (pI) and the buffer pH critical determinants of its electrophoretic mobility [33] [1]. This technical guide will explore the principles behind these techniques, provide detailed protocols, and explain how to reconcile apparent contradictions in molecular weight data within the context of a broader thesis on protein charge.
Fundamental Principles of SDS-PAGE and Native PAGE
SDS-PAGE: Separation by Molecular Weight
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) is a denaturing technique designed to separate proteins based solely on the molecular weight of their polypeptide chains [58] [60].
- Mechanism of Denaturation and Charge Manipulation: The anionic detergent SDS binds to hydrophobic regions of proteins at a ratio of approximately 1.4 g SDS per 1.0 g protein [33] [60]. This binding denatures the protein, disrupting its secondary, tertiary, and quaternary structures and unfolding it into a linear polypeptide chain. Critically, the negatively charged sulfate groups of SDS overwhelm the protein's intrinsic charge, conferring a uniform negative charge-to-mass ratio to all proteins [58] [59].
- Separation Criteria: Once denatured and uniformly charged, the protein-SDS complexes are loaded into a polyacrylamide gel matrix. Under an electric field, all complexes migrate toward the anode. The sieving effect of the gel pores means that smaller polypeptides migrate faster than larger ones, resulting in separation by molecular weight [60]. The presence of reducing agents like β-mercaptoethanol or DTT further breaks disulfide bonds, ensuring complete dissociation into individual subunits [33] [61].
Native PAGE: Separation by Charge, Size, and Shape
Native PAGE (Polyacrylamide Gel Electrophoresis) is a non-denaturing technique that separates proteins based on their native properties: their intrinsic charge, hydrodynamic size, and three-dimensional shape [33] [1] [57].
- Preservation of Native State: No denaturing agents are used. The protein sample is not heated, and the buffer contains no SDS or reducing agents [33]. This preserves the protein's native conformation, including secondary, tertiary, and, most importantly, quaternary structure [1]. Subunit interactions within a multimeric complex are retained.
- Complex Separation Criteria: Separation in Native PAGE is a function of two main factors:
- Net Charge: The protein's intrinsic net charge at the running buffer's pH determines its electrophoretic mobility and direction. Positively charged proteins (with pI > buffer pH) migrate toward the cathode, while negatively charged proteins (with pI < buffer pH) migrate toward the anode [1] [59].
- Size and Shape: The gel matrix exerts a sieving effect. A protein's hydrodynamic volume and shape (globular vs. elongated) determine the frictional force it encounters, thereby regulating its migration speed [1].
Table 1: Core Principles and Separation Criteria of SDS-PAGE and Native PAGE
| Feature | SDS-PAGE | Native PAGE |
|---|---|---|
| Primary Separation Basis | Molecular weight of polypeptide chains [58] [59] | Size, intrinsic charge, and shape of native structure [33] [57] |
| Gel Condition | Denaturing [33] [61] | Non-denaturing [33] [61] |
| Key Reagent | Sodium Dodecyl Sulfate (SDS) [33] [60] | Often Coomassie G-250 (in BN-PAGE) or no charge modifier [1] [3] |
| Sample Preparation | Heated with SDS and reducing agents [33] | Not heated; no denaturants or reducing agents [33] |
| Protein Structure | Denatured; primary structure only [57] | Native conformation retained (folded, functional) [33] [1] |
| Protein Function Post-Separation | Lost [33] | Often retained [33] [1] |
| Protein Recovery | Not typically recoverable for functional studies [33] | Can be recovered in active form [33] [57] |
The Role of Protein Charge in Native PAGE Migration
The protein's net charge is the engine of its migration in Native PAGE. This charge is not an inherent, fixed property but is context-dependent, determined by the interplay between the protein's isoelectric point (pI) and the pH of the running buffer.
- Charge Determination: A protein's net charge in a given buffer is its effective valence (Z~eff~). This can be measured experimentally using techniques like real-time electrophoretic mobility membrane confined electrophoresis (REM-MCE) and is often found to be significantly lower than the charge predicted from the sum of its ionizable residues due to counter-ion binding and other polyelectrolyte effects [5].
- Impact of Buffer Composition: The choice of buffer system and salt composition directly influences a protein's observed charge. For example, a study on cytochrome c demonstrated that its measured valence was highly salt-dependent, dropping to nearly zero in K~2~SO~4~ buffers due to specific sulfate ion binding, whereas it maintained a positive charge in KCl buffers [5]. This highlights that the local ionic environment and specific ion-protein interactions are critical in determining electrophoretic mobility.
- Charge-Shifting Dyes in BN-PAGE: A significant advancement in Native PAGE is Blue Native PAGE (BN-PAGE), which uses Coomassie Brilliant Blue G-250 to address the challenge of proteins with basic pIs [1] [3]. This dye binds non-specifically to hydrophobic patches on the protein surface, imparting a strong negative charge. This does the following:
- Allows all proteins, regardless of their native pI, to migrate toward the anode.
- Prevents aggregation of membrane and hydrophobic proteins by converting hydrophobic surfaces into charged, hydrophilic ones.
- Enables separation that more closely correlates with molecular size, while still maintaining the protein in a native state [1].
Experimental Protocols and Methodologies
Standard SDS-PAGE Protocol
The following protocol is adapted from common procedures using pre-cast Bis-Tris gels [3].
Sample Preparation:
Gel Setup:
- Use a pre-cast polyacrylamide gel (e.g., 12% Bis-Tris). Load prepared samples and a molecular weight marker into the wells.
Electrophoresis:
SDS-PAGE Experimental Workflow
Standard Native PAGE Protocol
This protocol outlines a general Native PAGE procedure, which can be adapted for different gel systems like Tris-Glycine or BN-PAGE [1] [3].
Sample Preparation:
Gel Setup:
- Use a pre-cast native gel (e.g., Tris-Glycine, Bis-Tris, or a gradient gel). Load the prepared native samples and appropriate native markers.
Electrophoresis:
Native PAGE Experimental Workflow
A Hybrid Approach: Native SDS-PAGE (NSDS-PAGE)
To bridge the gap between high resolution and native state preservation, a modified method called Native SDS-PAGE (NSDS-PAGE) has been developed [3]. This protocol reduces denaturing conditions to maintain some native properties while leveraging the resolution of SDS-PAGE systems.
Sample Preparation:
Electrophoresis:
- Use a standard SDS-PAGE gel apparatus.
- Use a running buffer with a significantly reduced SDS concentration (e.g., 0.0375% instead of 0.1%) and no EDTA [3].
- Run at standard voltages.
This method has been shown to retain Zn²⁺ in metalloproteins and preserve the activity of many enzymes while still providing high-resolution separation, demonstrating that functional properties can be maintained outside of strict native conditions [3].
Table 2: Key Research Reagent Solutions for PAGE Experiments
| Reagent / Material | Function in SDS-PAGE | Function in Native PAGE |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Denatures proteins and confers uniform negative charge [33] [60] | Not used |
| β-Mercaptoethanol / DTT | Reducing agent; breaks disulfide bonds [33] [61] | Not used |
| Coomassie G-250 Dye | Not typically used in sample prep | Charge-shifting molecule in BN-PAGE; imparts negative charge, prevents aggregation [1] [3] |
| Polyacrylamide Gel | Sieving matrix for size-based separation [33] | Sieving matrix for separation by size/shape [33] |
| Tris-Based Buffers | Provides conductive medium at specific pH (e.g., ~7.7) [3] | Provides conductive medium; pH choice (7.5-9.5) critical for defining protein charge [1] |
| Glycerol | Increases sample density for well loading [61] | Increases sample density for well loading [3] |
Interpretation of Divergent Results: The 60 kDa vs. 120 kDa Case
The scenario where a protein migrates at 60 kDa on SDS-PAGE and 120 kDa on Native PAGE is a classic indicator of a homodimeric complex in its native state.
- SDS-PAGE Result (60 kDa): Under denaturing conditions, the strong detergent and heat dissociate the complex into its individual subunits. Each subunit is a polypeptide chain with a molecular weight of 60 kDa. The SDS coats these subunits, and they migrate through the gel based solely on the mass of the individual polypeptide [33] [57].
- Native PAGE Result (120 kDa): Under non-denaturing conditions, the protein maintains its quaternary structure—in this case, a complex of two 60 kDa subunits. The migration rate is determined by the overall size (hydrodynamic radius of the ~120 kDa complex), the net charge of the entire complex, and its overall shape. The observed migration consistent with a 120 kDa standard suggests the complex is globular and that its charge-to-size ratio is similar to that of a standard protein of that mass [33] [57].
This interpretation is foundational. A hetero-oligomeric complex containing different subunits would show multiple bands on SDS-PAGE (e.g., 60 kDa and 45 kDa) but a single, larger band on Native PAGE. The specific migration on Native PAGE can also be influenced by the protein's non-globular shape or an atypical charge density, which would cause it to migrate at an apparent mass that does not match its true molecular weight.
Advanced Application: Two-Dimensional BN-/SDS-PAGE
For a comprehensive analysis of complex protein mixtures, researchers employ two-dimensional (2D) electrophoresis, which combines the strengths of both Native and Denaturing PAGE [62].
- First Dimension: Blue Native PAGE (BN-PAGE) separates intact protein complexes based on their native mass and charge [62].
- Complex Excision: A single lane from the BN-PAGE gel is excised.
- Second Dimension: The excised lane is incubated with SDS and reducing buffer, then placed horizontally on a standard SDS-PAGE gel. This second step denatures and dissociates the complexes isolated in the first dimension [62].
- Separation and Analysis: The second-dimension SDS-PAGE separates the complex into its constituent subunits by molecular weight.
This powerful technique allows researchers to determine both the native mass of a complex and the subunit composition of its constituents within a single experiment, providing an unambiguous link between a native complex and its dissociated subunits [62].
The apparent discrepancy of a 60 kDa band on SDS-PAGE versus a 120 kDa band on Native PAGE is not a contradiction but a coherent story of a dimeric protein complex. SDS-PAGE reveals the molecular weight of the constituent polypeptides, while Native PAGE reveals the size, charge, and oligomeric state of the functional, native complex. The protein's intrinsic charge, modulated by the buffer system and specific ion binding, is a primary director of its migration in Native PAGE. Mastery of these techniques and their interpretive principles is essential for any researcher aiming to characterize protein structure-function relationships, validate therapeutic targets, or purify bioactive complexes in drug development.
In-gel enzyme activity assays represent a powerful biochemical technique that combines electrophoretic separation with functional enzymatic analysis, enabling researchers to directly visualize and quantify activity within the native gel matrix. Unlike standard solution-based assays that measure total enzymatic activity in a sample, in-gel methods preserve the spatial separation of different protein forms achieved through electrophoresis, allowing for the specific detection of active enzyme complexes, isoforms, or oligomeric states. This capability is particularly valuable for studying multimeric enzymes and protein complexes where structural integrity is essential for function.
The fundamental principle underlying this technique involves separating proteins under non-denaturing conditions using native polyacrylamide gel electrophoresis (PAGE), which maintains protein structure and function, followed by incubation with specific substrates and detection reagents that produce a visible, often colored, precipitate at the site of enzymatic activity [23]. This approach provides a direct link between protein migration patterns and catalytic function, offering insights that would be lost in conventional denaturing electrophoresis or bulk solution assays.
For the broader context of protein charge research, in-gel activity assays serve as a critical validation tool, demonstrating how a protein's net charge, hydrodynamic size, and quaternary structure—factors governing its migration in native PAGE—directly correlate with its biological function [1] [7]. The preservation of enzymatic activity post-electrophoresis confirms that the native structure remains intact throughout the separation process, providing functional significance to the observed migration patterns.
Fundamental Principles of Native Electrophoresis
Separation Mechanisms in Native PAGE
In native PAGE, proteins are separated according to the net charge, size, and shape of their native structure, without the denaturing agents used in SDS-PAGE [1] [2]. Electrophoretic migration occurs because most proteins carry a net negative charge in alkaline running buffers, causing them to migrate toward the positive anode. The rate of migration is determined by both the protein's charge density (number of charges per mass unit) and the frictional force imposed by the gel matrix, which acts as a molecular sieve [1] [7]. A protein with high negative charge density will migrate rapidly toward the anode, while the gel matrix creates a sieving effect that retards larger proteins more than smaller ones [1].
The preservation of native structure is the defining characteristic of native PAGE, with several important implications. Multimeric proteins maintain their quaternary structure, allowing separation of different oligomeric states [1]. The physiological function, including enzymatic activity, is often retained, enabling functional assays directly in the gel [1] [2]. Additionally, a protein's migration position reflects its combined charge, size, and shape rather than mass alone [7]. This preservation enables researchers to correlate enzymatic function with specific protein complexes or oligomeric states separated within the gel matrix.
Protein Charge and Migration Behavior
A protein's net charge at the running buffer pH is a primary determinant of its migration in native PAGE. This net charge depends on the ionizable groups of its amino acid side chains and their protonation states relative to their isoelectric points (pI) [1]. Proteins with pI values below the buffer pH carry a net negative charge and migrate toward the anode, while those with pI above the buffer pH carry a net positive charge and would theoretically migrate toward the cathode, though this is often counteracted in modified native systems [1] [39].
The following diagram illustrates the key factors affecting protein migration in native PAGE and their relationship to the final separation result:
Factors Governing Protein Migration in Native PAGE
Advanced native techniques like blue native PAGE (BN-PAGE) and clear native PAGE (CN-PAGE) modify these charge-migration relationships. BN-PAGE uses Coomassie G-250 dye, which binds to hydrophobic protein regions and confers additional negative charge, allowing even basic proteins to migrate toward the anode while maintaining native structure [16] [1]. CN-PAGE, in contrast, typically uses mild detergents instead of dye, with migration depending more on intrinsic protein charge [16] [39]. These technical variations provide researchers with tools to optimize separation based on their specific experimental needs and the properties of the proteins being studied.
Native Electrophoresis Methodologies
Comparison of Native PAGE Systems
Several native PAGE systems have been developed, each with distinct advantages and applications. The choice between them depends on factors including protein properties, desired resolution, and downstream applications such as in-gel activity staining.
Table 1: Comparison of Native Electrophoresis Techniques
| Technique | Separation Basis | Key Components | Optimal Applications | Advantages | Limitations |
|---|---|---|---|---|---|
| Tris-Glycine Native PAGE [1] | Net charge, size, and shape at pH 8.3-9.5 | Tris-glycine buffer system | Smaller proteins (20-500 kDa); when maintaining native charge is critical | Simple, established methodology; maintains native charge | Limited resolution for membrane proteins; basic proteins may not migrate properly |
| Blue Native PAGE (BN-PAGE) [16] [1] | Mass with charge shift | Coomassie G-250 dye, n-dodecyl-β-d-maltoside | Membrane protein complexes, hydrophobic proteins; molecular weight estimation | Resolves basic proteins; reduces aggregation; high resolution for complexes | Dye may interfere with fluorescence assays; can quench 90-95% fluorescence [39] |
| Clear Native PAGE (CN-PAGE) [16] [39] | Intrinsic charge and size | Mixed detergents (e.g., sodium deoxycholate, DDM) | Acidic proteins (pI < 7); fluorescence detection; in-gel activity assays | No dye interference; compatible with fluorescent tags | Limited to acidic proteins; potential for protein aggregation |
| High-Resolution CN-PAGE [23] [39] | Charge, size with mild charge shift | Anionic/neutral detergent mixtures | Maximum resolution for activity assays; sensitive enzymes | Excellent resolution without dye interference; maintains enzymatic activity | Optimization required for different protein types |
Technical Workflow for Native Electrophoresis
The general workflow for native electrophoresis begins with sample preparation, which typically involves extraction with mild non-ionic detergents to solubilize proteins while preserving native structures and complexes [16]. For BN-PAGE, Coomassie G-250 dye is added to the sample, which binds to hydrophobic protein regions and provides negative charge for uniform migration [1]. For CN-PAGE, this step is omitted or replaced with mild detergents [39].
The following diagram outlines the key decision points and procedures in a standard native PAGE workflow:
Native PAGE Experimental Workflow
Gel casting follows standard polyacrylamide procedures, often using gradient gels (e.g., 4-16%) to resolve proteins across a broad size range [23] [1]. The electrophoresis is typically performed at 4°C to maintain protein stability, with running conditions varying by system [23] [63]. Following separation, proteins can be processed for various downstream applications including in-gel activity staining, western blotting (using PVDF membranes, as nitrocellulose binds Coomassie dye too tightly [1]), or protein recovery for further analysis.
In-Gel Activity Assay Methodologies
General Principles and Detection Strategies
In-gel activity assays leverage the preservation of enzymatic function after native PAGE to directly visualize catalytic activity within the gel matrix. The fundamental approach involves incubating the gel with a reaction mixture containing the enzyme's specific substrate along with necessary cofactors and detection reagents. The detection system typically produces an insoluble, colored precipitate at sites of enzymatic activity, forming visible bands that can be quantified by densitometry [23] [63].
Most assays use a coupled enzyme system where the primary enzyme catalyzes its native reaction, producing a product that serves as substrate for a detection reaction. For oxidoreductases, this often involves electron transfer to tetrazolium salts like nitro blue tetrazolium (NBT) or thiazolyl blue tetrazolium bromide (MTT), which form purple or blue formazan precipitates upon reduction [23] [63]. For example, in the MCAD activity assay, oxidation of octanoyl-CoA is coupled to reduction of NBT, forming a purple diformazan precipitate at active enzyme sites [23].
The sensitivity of these assays is remarkable, with linear detection ranges demonstrated for even sub-microgram protein quantities [23]. This sensitivity enables researchers to detect activity from multiple enzyme forms within a single sample, providing insights into isoform-specific activities, oligomeric state functionality, and the impact of post-translational modifications on enzyme function.
Specific Protocol: MCAD Dehydrogenase Activity Assay
The medium-chain acyl-CoA dehydrogenase (MCAD) in-gel activity assay demonstrates a well-optimized protocol for visualizing oxidoreductase activity [23]. After separation by high-resolution clear native PAGE (4-16% gradient gel), the gel is incubated in reaction mixture containing: 100-500 µM octanoyl-CoA (physiological substrate), 0.2-0.5 mg/mL nitro blue tetrazolium (NBT) as electron acceptor, and 100-200 µM phenazine methosulfate (PMS) as electron carrier in appropriate buffer (typically Tris-HCl or phosphate buffer, pH 7.0-8.0) [23].
The assay principle relies on MCAD catalyzing the oxidation of octanoyl-CoA, transferring electrons through FAD to PMS, which then reduces NBT to purple formazan precipitate at sites of MCAD activity. Incubation proceeds at room temperature or 37°C with gentle shaking until purple bands develop (typically 10-30 minutes), after which the reaction is stopped by transferring the gel to a fixing solution (e.g., 30-40% ethanol with 7-10% acetic acid) [23]. Bands can be quantified by densitometric analysis, with linear correlation between protein amount, FAD content, and in-gel activity demonstrated [23].
Specific Protocol: Aconitase Activity Assay
The aconitase in-gel activity assay employs a different detection strategy tailored to this iron-sulfur cluster enzyme [63]. Following native PAGE separation (using Tris-glycine or Tris-borate systems at pH 8.3-8.7), the gel is incubated in reaction mixture containing: 2.5 mM cis-aconitate (substrate), 0.5-1.0 mM NADP+, 2-4 U/mL NADP-dependent isocitrate dehydrogenase (coupling enzyme), 0.2-0.5 mg/mL MTT, and 0.05-0.1 mM phenazine methosulfate in appropriate buffer with magnesium and manganese cofactors [63].
In this coupled system, aconitase converts cis-aconitate to isocitrate, which is then oxidized by isocitrate dehydrogenase, reducing NADP+ to NADPH. PMS mediates electron transfer from NADPH to MTT, reducing it to purple formazan at sites of aconitase activity. The gel is incubated at 37°C in the dark until bands develop (30-60 minutes), allowing simultaneous detection of mitochondrial (ACO2) and cytosolic (ACO1) isoforms based on their distinct migration positions [63].
Specific Protocol: Antioxidant Enzyme Activity Assays
Antioxidant enzymes including catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPX) can be visualized using specialized in-gel assays [64]. For catalase activity after native PAGE, gels are washed with 0.01-0.03% H₂O₂ for 10-20 minutes, then stained with 2% potassium ferricyanide and 2% ferric chloride solution [64]. Active catalase bands appear as clear areas against a blue-green background due to catalase-mediated decomposition of H₂O₂ preventing the oxidation reaction.
For superoxide dismutase, gels are incubated in 2-5 mM nitro blue tetrazolium in potassium phosphate buffer for 20-30 minutes in the dark, followed by incubation in 0.1-0.5 mM riboflavin in 0.1% TEMED [64]. After brief illumination, active SOD bands appear as clear areas against a purple background due to SOD scavenging superoxide radicals needed for NBT reduction. These antioxidant enzyme assays have been successfully applied to ecotoxicological studies in marine organisms, demonstrating their utility in environmental research [64].
Research Reagent Solutions Toolkit
Table 2: Essential Reagents for In-Gel Activity Assays
| Reagent Category | Specific Examples | Function/Purpose | Application Notes |
|---|---|---|---|
| Electrophoresis Materials [1] | NativePAGE Bis-Tris Gels (3-12%, 4-16%), Tris-glycine gels, Tris-acetate gels | Matrix for protein separation under native conditions | Gradient gels (e.g., 4-16%) provide superior resolution across size ranges |
| Charge-Shift Molecules [16] [1] | Coomassie G-250 (BN-PAGE), Sodium deoxycholate/DDM mixtures (hrCN-PAGE) | Impart negative charge for controlled migration | Coomassie enables resolution of basic proteins; detergents reduce dye interference |
| Detection Reagents [23] [63] | Nitro blue tetrazolium (NBT), Thiazolyl blue tetrazolium (MTT), Phenazine methosulfate (PMS) | Electron acceptors/mediators for colorimetric detection | Form insoluble formazan precipitates at sites of enzymatic activity |
| Enzyme Substrates [23] [63] | Octanoyl-CoA (MCAD), cis-Aconitate (aconitase), Hydrogen peroxide (catalase) | Specific substrates for target enzymes | Physiological substrates preferred; alternatives used for optimization |
| Biological Samples [23] [16] | Recombinant purified proteins, Mitochondrial-enriched fractions, Cell homogenates, Tissue extracts | Source of enzymes for analysis | Extraction with mild detergents (e.g., n-dodecyl-β-d-maltoside) preserves complexes |
Applications and Data Interpretation
Research Applications
In-gel activity assays have enabled significant advances across multiple research domains, particularly in the study of mitochondrial disorders and metabolic diseases. The application to MCAD deficiency (MCADD) demonstrates how this technique provides insights beyond standard enzymatic assays [23]. By distinguishing active tetramers from fragmented or aggregated forms, researchers can determine whether pathogenic variants primarily affect catalytic activity or protein stability, with profound implications for understanding disease mechanisms and developing targeted therapies.
In structural biology, these assays help correlate quaternary structure with function. As demonstrated in the MCAD study, variants like R206C caused tetramers to migrate at apparently lower molecular masses despite unchanged monomeric mass, suggesting conformational changes affecting migration without complete loss of activity [23]. Such insights are invaluable for understanding structure-function relationships in multimeric enzymes.
The technique also finds application in ecotoxicology and environmental science, where it helps assess organismal stress responses. Studies on marine bivalves exposed to silver nanoparticles used in-gel activity assays for antioxidant enzymes like catalase, superoxide dismutase, and glutathione peroxidase to evaluate oxidative stress at the protein functional level [64]. This approach provides direct evidence of metabolic impacts from environmental contaminants.
Data Analysis and Quantification
Analysis of in-gel activity assays typically involves densitometric quantification of activity bands, with normalization to protein amount or reference standards. The linear correlation between protein amount and in-gel activity, demonstrated for MCAD across a range of 0.5-5 μg, enables semi-quantitative comparisons between samples [23]. This quantification can be complemented with spectrophotometric activity measurements from the same samples to calculate specific activity.
When interpreting results, several factors require consideration. Multiple active bands may represent different oligomeric states, isoforms, or post-translationally modified forms with distinct migration but similar function [23]. Altered migration patterns may indicate conformational changes rather than complete loss of function, as seen with the R206C MCAD variant [23]. Additionally, the presence of activity in higher molecular weight regions may suggest functional supercomplexes or aggregates, while lower molecular weight activity may indicate active protomers or fragments.
Technical Considerations and Limitations
While powerful, in-gel activity assays present several technical challenges that require consideration during experimental design. The absence of standardized molecular weight markers for native conditions complicates precise mass determination, though commercial soluble protein markers provide estimates [39]. Enzyme stability during electrophoresis is another concern, particularly for oxygen-sensitive or labile enzymes, necessitating cooled systems and rapid processing [63].
Not all enzymes are amenable to in-gel activity detection. Some complexes may not renature properly after electrophoresis, while others require membrane environments or specific lipid compositions that gel systems cannot provide [16]. For instance, in-gel activity staining for Complex III of the mitochondrial respiratory chain remains challenging [16] [65]. The sensitivity of in-gel assays, while sufficient for many applications, may not match solution-based assays for low-abundance enzymes, though improvements in detection chemistries continue to enhance sensitivity.
Optimization is typically required for each enzyme system, including testing different substrate concentrations, incubation times, and detection methods. The linear range of detection must be established for quantitative comparisons, and controls including heat-inactivated samples and specific inhibitors are essential to confirm specificity. Despite these challenges, the unique insights provided by correlating migration position with enzymatic function make in-gel activity assays an invaluable tool in functional proteomics.
In the realm of protein analysis, polyacrylamide gel electrophoresis (PAGE) serves as a fundamental tool for separating and characterizing complex protein mixtures. While one-dimensional separations provide valuable information, the integration of multiple techniques can unveil a more comprehensive picture of proteomic composition. Native PAGE stands apart from denaturing methods by preserving proteins in their folded, functional state. This technique separates proteins based on the net charge, size, and shape of their native structure [2]. In alkaline running buffers, most proteins carry a net negative charge, causing them to migrate toward the anode. The rate of migration is proportional to the protein's charge density (charge-to-mass ratio), while the gel matrix provides a sieving effect that retards movement according to size and three-dimensional structure [2]. This charge-dependent separation makes Native PAGE particularly valuable for studying functional protein properties, including oligomerization states, protein-protein interactions, and enzymatic activities that would be destroyed by denaturation [57].
The limitation of Native PAGE emerges when analyzing complex samples containing numerous proteins with similar charge densities, which may comigrate as unresolved species. This challenge is addressed by coupling Native PAGE with a second orthogonal separation method—SDS-PAGE—which separates proteins primarily by molecular weight under denaturing conditions [66]. This two-dimensional approach leverages the independent parameters of native charge and polypeptide size to achieve superior resolution, enabling the detection of thousands of protein components from complex biological sources [66].
Fundamental Principles: How Protein Charge Governs Native PAGE Migration
The Electrophoretic Basis of Charge-Dependent Separation
In Native PAGE, a protein's migration through the gel matrix is governed by its intrinsic electrochemical properties within the specific buffer system. Unlike SDS-PAGE, which masks intrinsic charge with a uniform negative charge from bound detergent, Native PAGE maintains the protein's natural charge distribution [57] [2]. Several interconnected factors determine electrophoretic mobility in this native state:
Net Charge and Charge Density: A protein's net charge at the running buffer pH represents the balance between its positively and negatively charged amino acid side chains. Proteins with higher negative charge density (more charge per unit mass) migrate faster toward the anode, while those with lower charge density migrate more slowly [2]. The frictional force of the gel matrix creates a sieving effect that further modulates mobility based on protein size and three-dimensional shape [2].
Isoelectric Point (pI) and Buffer pH: The relationship between a protein's pI (the pH at which it carries no net charge) and the buffer pH determines both the magnitude and sign of its net charge. In the standard alkaline running buffers (pH ~8.0-9.0) used in Native PAGE, most proteins acquire a net negative charge and migrate toward the anode [2]. Proteins with low pI values develop a strong negative charge, while those with pI values closer to the buffer pH carry less charge and migrate more slowly.
Molecular Size and Shape: The gel matrix acts as a molecular sieve, retarding the movement of larger proteins more than smaller ones. A protein's three-dimensional structure further influences migration compact globular proteins generally migrate faster than extended fibrous proteins of equivalent molecular weight [2].
The following table summarizes the key factors affecting protein migration in Native PAGE:
Table 1: Factors Governing Protein Migration in Native PAGE
| Factor | Effect on Migration | Basis of Separation |
|---|---|---|
| Net Charge | Higher negative charge → Faster migration | Electrophoretic mobility proportional to charge density [2] |
| Molecular Size | Larger size → Slower migration | Sieving effect of gel matrix [2] |
| 3D Structure/Shape | Compact shapes → Faster migration | Frictional resistance differences [2] |
| Buffer pH | Further from protein pI → Faster migration | Determines net charge magnitude [2] |
Comparative Separation Mechanisms: Native PAGE vs. SDS-PAGE
Understanding the distinction between Native PAGE and SDS-PAGE is crucial for appreciating their complementary nature in two-dimensional separations. These techniques operate on fundamentally different principles:
Table 2: Comparative Separation Mechanisms of Native PAGE and Denaturing SDS-PAGE
| Parameter | Native PAGE | SDS-PAGE |
|---|---|---|
| Protein State | Native, folded [57] | Denatured, linearized [57] [2] |
| Primary Separation Basis | Net charge, size, and shape [2] | Molecular weight [57] [2] |
| Charge State | Intrinsic protein charge | Uniform negative charge from SDS [57] [2] |
| Biological Activity | Often retained [57] [2] | Destroyed [57] |
| Protein Complexes | Maintained [2] | Dissociated into subunits [57] |
| Molecular Weight Determination | Not accurate due to charge/shape factors | Accurate with proper calibration [2] |
The following diagram illustrates how these orthogonal separation principles combine in two-dimensional electrophoresis:
Experimental Design and Workflow for 2D Native PAGE-SDS-PAGE
First-Dimension: Native PAGE Separation
The initial separation phase utilizes Native PAGE to resolve protein complexes based on their intrinsic properties. The following protocol details this critical first dimension:
Sample Preparation for Native PAGE:
- Prepare protein samples in non-denaturing buffers without SDS, β-mercaptoethanol, or other reducing agents [2]
- Maintain samples at 4°C throughout preparation to prevent denaturation or proteolysis [2]
- Avoid extreme pH conditions that might irreversibly denature or aggregate proteins [2]
- For complex samples like cell lysates, include protease inhibitors and nuclease treatment to minimize degradation [66]
Gel Composition and Electrophoresis Conditions:
- Use standard polyacrylamide gels with appropriate percentages for the target protein size range (typically 4-12% gradients) [2]
- Prepare gels in non-denaturing buffers without SDS or other denaturants [2] [3]
- Include a stacking gel with lower acrylamide concentration and pH to concentrate proteins before entering the resolving gel [2]
- Run electrophoresis at constant voltage (typically 100-150V) with cooling to maintain native conditions [2] [3]
- Use alkaline running buffers (e.g., Tris-glycine, pH ~8.3-8.8) to ensure most proteins carry negative charge [2]
Post-Electrophoresis Processing:
- After separation, carefully excise individual lanes from the Native PAGE gel
- Equilibrate gel strips in SDS-PAGE sample buffer to denature proteins and impart uniform negative charge [66]
- Incubate with gentle agitation for 15-30 minutes to ensure complete penetration of SDS
Second-Dimension: SDS-PAGE Separation
The second dimension separates denatured proteins from the first dimension by molecular weight, providing the orthogonal separation needed for high-resolution analysis.
Gel Casting and Lane Transfer:
- Cast denaturing SDS-polyacrylamide gels appropriate for the target molecular weight range [2] [66]
- For broad molecular weight separation, use gradient gels (e.g., 4-12% or 10-20%) [2]
- Prepare standard SDS-PAGE running buffer containing Tris, glycine, and 0.1% SDS [2] [66]
- Place the equilibrated Native PAGE gel strip horizontally on top of the SDS-PAGE gel
- Seal with agarose (1% in SDS sample buffer) to prevent bubble formation and ensure electrical continuity [66]
Electrophoresis and Detection:
- Run electrophoresis at constant voltage (typically 150-200V) until the dye front reaches the gel bottom [2] [66]
- Following separation, detect proteins using sensitive staining methods compatible with downstream analysis
- Coomassie Brilliant Blue provides detection in the microgram range [66]
- Silver staining offers nanogram-level sensitivity [67]
- Fluorescent stains (e.g., SYPRO Ruby) enable quantitative analysis and mass spectrometry compatibility [67]
- For specific detection, transfer proteins to membranes for western blotting with targeted antibodies
The complete experimental workflow, from sample preparation to final analysis, is summarized below:
Research Reagent Solutions and Technical Specifications
Successful implementation of 2D Native PAGE-SDS-PAGE requires specific reagents and materials optimized for preserving protein native states while enabling high-resolution separation. The following table details essential research reagents and their functions:
Table 3: Essential Research Reagents for 2D Native PAGE-SDS-PAGE
| Reagent/Category | Function/Purpose | Technical Specifications |
|---|---|---|
| Native PAGE Buffers | Maintain native protein structure during first-dimension separation [2] | Tris-glycine (pH 8.3-8.8) or Bis-Tris (pH 6.8-7.2) systems; No SDS or denaturants [2] [3] |
| SDS-PAGE Buffers | Denature proteins and impart uniform charge for second-dimension separation [2] [66] | Tris-glycine-SDS (pH 8.3-8.8) or MOPS-SDS (pH 7.7) systems; 0.1% SDS [2] [66] |
| Sample Denaturation Buffer | Denature proteins after Native PAGE for second-dimension separation [66] | Contains SDS (2-4%), glycerol (5-10%), reducing agent (DTT or β-mercaptoethanol), and tracking dye [66] |
| Gel Matrix Components | Form porous polyacrylamide network for size-based separation [2] | Acrylamide-bisacrylamide (29:1 to 37.5:1 ratios); Concentrations from 4-20% depending on target protein size [2] |
| Protein Stains | Visualize separated protein spots after 2D separation [67] | Coomassie Blue (>7 ng), Silver Stain (0.3 ng), SYPRO Ruby (0.25-1 ng); compatibility with downstream MS analysis [67] |
Advanced Applications and Methodological Considerations
Quantitative Analysis and Detection Sensitivity
The resolving power of 2D Native PAGE-SDS-PAGE makes it particularly valuable for detecting subtle changes in protein complexes and post-translational modifications. The sensitivity of this technique enables detection of low-abundance proteins when combined with appropriate detection methods:
Table 4: Detection Methods and Sensitivities for 2D Gels
| Detection Method | Sensitivity Range | Compatibility with Downstream Analysis | Key Applications |
|---|---|---|---|
| Coomassie Staining | >7 ng/protein spot [67] | Moderate (requires destaining) | General protein profiling, abundance estimation |
| Silver Staining | 0.3 ng/protein spot [67] | Limited (requires special protocols) | High-sensitivity detection of low-abundance proteins |
| SYPRO Ruby Staining | 0.25-1 ng/protein spot [67] | High (MS-compatible) | Quantitative proteomics, mass spectrometry analysis |
| Western Blotting | Variable (antibody-dependent) | Specific target analysis | Detection of specific proteins or modifications |
| Autoradiography | Single disintegration/min of ¹⁴C or ³⁵S [66] | Metabolic labeling studies | Metabolic labeling experiments, turnover studies |
Specialized Variants: Native SDS-PAGE (NSDS-PAGE)
Recent methodological advances have led to the development of Native SDS-PAGE (NSDS-PAGE), which represents a hybrid approach balancing resolution and native state preservation. This technique modifies standard SDS-PAGE conditions by reducing SDS concentration in running buffers from 0.1% to 0.0375%, eliminating EDTA from sample buffers, and omitting the heating step [3]. These modifications enable retention of enzymatic activity in seven of nine model enzymes tested, compared to complete denaturation in standard SDS-PAGE [3]. Additionally, metal retention significantly improves, with Zn²⁺ bound in proteomic samples increasing from 26% to 98% when shifting from standard to modified conditions [3]. This approach offers particular advantages for metalloprotein analysis and functional studies requiring partial preservation of protein structure.
Troubleshooting Common Experimental Challenges
Implementing 2D Native PAGE-SDS-PAGE presents unique technical challenges that require specific troubleshooting approaches:
Horizontal Streaking in Second Dimension: Often results from incomplete denaturation or reduction after first dimension separation. Ensure adequate equilibration time in SDS sample buffer (15-30 minutes with agitation) and include fresh reducing agents [66].
Poor Resolution in Native PAGE Dimension: May indicate inappropriate buffer pH or insufficient cooling during electrophoresis. Verify that running buffer pH is optimal for the target protein charge separation and implement active cooling during runs [2].
Protein Precipitation at Interface: Can occur when transferring proteins from native to denaturing conditions. Include non-ionic detergents in equilibration buffer and ensure gradual transition between buffer systems [66].
Low Sensitivity Detection: May reflect protein loss during processing or suboptimal staining. Implement sensitive fluorescent stains like SYPRO Ruby and consider protein fixation steps between dimensions for low-abundance proteins [67].
The integration of Native PAGE with second-dimension SDS-PAGE represents a powerful analytical approach that leverages the orthogonal separation parameters of native charge and molecular weight. This 2D system provides unprecedented resolution for analyzing complex protein mixtures while preserving information about native protein complexes and functional states that would be lost in fully denaturing systems. The charge-based separation mechanism of Native PAGE, combined with the molecular weight discrimination of SDS-PAGE, enables researchers to resolve thousands of protein components from biological samples [66]. As methodological advances like NSDS-PAGE [3] continue to enhance our ability to balance resolution with native state preservation, this integrated approach will remain indispensable for comprehensive protein characterization in basic research and drug development.
Protein electrophoresis stands as a cornerstone technique in biochemistry and molecular biology, enabling researchers to separate and analyze complex protein mixtures. The fundamental principle governing all electrophoretic techniques is that charged protein molecules migrate through a matrix under the influence of an electrical field [2]. However, the behavior of proteins within this field varies dramatically depending on whether they maintain their native conformation or are denatured, leading to two principal approaches: native PAGE and SDS-PAGE.
In native PAGE, proteins are separated according to the net charge, size, and shape of their native structure [1] [2]. This technique preserves protein function, including enzymatic activity and subunit interactions, but provides lower resolution because multiple factors influence migration [1]. Conversely, SDS-PAGE denatures proteins using sodium dodecyl sulfate, masking intrinsic charges and creating a uniform charge-to-mass ratio, thereby separating proteins almost exclusively by molecular weight with high resolution [68] [69] [70]. This denaturing approach, while excellent for molecular weight determination, destroys functional properties including non-covalently bound metal ions and enzymatic activity [3].
The emerging hybrid technique of Native SDS-PAGE (NSDS-PAGE) represents a significant methodological advancement that addresses the critical limitation of standard SDS-PAGE: the destruction of native protein structure and function. NSDS-PAGE achieves this breakthrough by systematically modifying the standard SDS-PAGE conditions to retain functional properties while largely preserving the high-resolution separation capability [3]. This technique occupies a unique position in the electrophoretic landscape, offering researchers a powerful tool for studying metalloproteins and other functionally sensitive proteins in their native state without sacrificing analytical resolution.
The Fundamental Challenge: Protein Charge in Native Electrophoresis
Principles of Native PAGE Separation
In native polyacrylamide gel electrophoresis (PAGE), proteins migrate according to their intrinsic properties without denaturation. The separation mechanism depends on three key characteristics: net charge, size, and three-dimensional structure [1] [2]. Electrophoretic migration occurs because most proteins carry a net negative charge in alkaline running buffers, causing them to migrate toward the positive anode [1]. The charge density (number of charges per molecule mass) significantly influences migration speed, with higher negative charge density resulting in faster migration [1]. Simultaneously, the gel matrix creates a sieving effect that retards movement according to protein size and shape [1]. Smaller proteins experience less frictional force and migrate faster, while larger proteins face greater resistance [1].
The critical importance of native PAGE lies in its preservation of protein function. Because no denaturants are used, subunit interactions within multimeric proteins are generally retained, providing information about quaternary structure [1]. Additionally, many proteins retain enzymatic activity following separation, enabling functional studies and preparation of purified, active proteins [1]. However, the technique faces inherent limitations in resolution and predictability due to its dependence on multiple migration factors that vary between proteins.
Limitations of Standard SDS-PAGE
Standard SDS-PAGE revolutionized protein analysis by simplifying separation to primarily molecular weight [68] [69]. The system employs sodium dodecyl sulfate (SDS), which denatures proteins and confers a uniform negative charge, and reducing agents that cleave disulfide bonds [68] [70]. SDS binds to the protein backbone at a relatively constant ratio of approximately 1.4g SDS per 1g of protein [70] [13], creating a consistent charge-to-mass ratio across different proteins [69]. This process unfolds proteins into linear chains, eliminating the influence of tertiary structure [68].
The significant limitation of this approach is the complete destruction of native protein structure and consequent loss of functional properties [3]. This denaturation prevents researchers from studying enzymatic activity, protein-protein interactions, and the presence of non-covalently bound cofactors, including metal ions essential for metalloprotein function [3]. For the growing field of metallomics and functional proteomics, this represents a critical analytical gap that limits comprehensive protein characterization.
Table 1: Comparative Separation Principles of Electrophoresis Techniques
| Parameter | Native PAGE | SDS-PAGE | NSDS-PAGE |
|---|---|---|---|
| Separation Basis | Net charge, size, and 3D structure [1] [2] | Primarily molecular weight [68] [70] | Molecular weight with native state preservation [3] |
| Protein State | Native/folded [1] | Denatured/unfolded [68] [70] | Native/folded [3] |
| Charge Source | Intrinsic protein charge [1] | SDS-derived uniform negative charge [69] | Reduced SDS with Coomassie G-250 assistance [3] |
| Functional Preservation | Yes (enzymatic activity, interactions) [1] | No [3] | Yes (partial, 7 of 9 enzymes in testing) [3] |
| Metal Retention | Preserved | Lost (26% retention in testing) [3] | High (98% retention in testing) [3] |
| Resolution | Lower due to multiple factors [3] | High [3] | High, comparable to SDS-PAGE [3] |
NSDS-PAGE: Principles and Mechanism
Conceptual Foundation and Development
NSDS-PAGE emerged from systematic investigations into modifying standard SDS-PAGE conditions to preserve native protein features without sacrificing resolution [3]. Researchers recognized that while blue-native (BN)-PAGE successfully retains native protein states, it falls short of the high resolution achieved with SDS-PAGE for complex proteomic mixtures [3]. The development of NSDS-PAGE was driven by the need for a technique that could finely resolve individual proteins while maintaining their native conformation and functional properties, particularly for metalloprotein analysis where metal cofactor retention is essential [3].
The methodological breakthrough came from carefully testing the impact of incremental changes to standard SDS-PAGE protocols. Researchers discovered that removing SDS and EDTA from the sample buffer while omitting the heating step had minimal effect on separation quality [3]. Further refinement involved reducing SDS concentration in the running buffer from the standard 0.1% to 0.0375% while also deleting EDTA [3]. These modifications created an environment where proteins could maintain essential structural elements while still undergoing electrophoretic separation with resolution comparable to traditional SDS-PAGE [3].
Key Modifications and Their Biochemical Rationale
NSDS-PAGE incorporates several critical modifications to the standard SDS-PAGE protocol that enable its unique capabilities:
Elimination of Denaturing Steps: The sample preparation in NSDS-PAGE excludes the standard heating step (typically 70-100°C) and uses a modified sample buffer without SDS, thereby preventing protein denaturation [3].
Reduced SDS Concentration: The running buffer SDS concentration is substantially reduced from 0.1% to 0.0375%, creating a mildly denaturing environment that preserves native structure while allowing electrophoretic migration [3].
Coomassie G-250 Supplementation: The sample buffer includes Coomassie G-250 dye (0.01875%), which binds nonspecifically to hydrophobic protein sites, converting them to negatively charged sites and facilitating migration toward the anode without full denaturation [3].
EDTA Removal: By eliminating EDTA from both sample and running buffers, NSDS-PAGE preserves metal ions non-covalently bound to metalloproteins, a crucial factor for studying metal-containing enzymes [3].
These modifications collectively create an electrophoretic environment that balances the need for charge-based migration with the preservation of native protein structure. The binding of Coomassie G-250 to proteins offers particular advantages, including enabling proteins with basic isoelectric points (which normally carry net positive charge) to acquire net negative charge and migrate toward the anode [3]. Additionally, membrane proteins and those with significant surface-exposed hydrophobic areas become less prone to aggregation as G-250 binds to hydrophobic sites, converting them to negatively charged sites [3].
Diagram 1: Sample preparation workflow comparison
Experimental Protocol and Methodological Framework
Buffer Composition and Gel Preparation
The successful implementation of NSDS-PAGE depends critically on precise buffer formulations. The modified buffers differ substantially from both standard SDS-PAGE and BN-PAGE systems, creating the unique environment that enables high-resolution native separation.
Table 2: Comparative Buffer Compositions for Electrophoresis Methods
| Buffer Component | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Sample Buffer | 106 mM Tris HCl, 141 mM Tris Base, 0.51 mM EDTA, 0.22 mM SERVA Blue G-250, 0.175 mM Phenol Red, 2% LDS, 10% Glycerol, pH 8.5 [3] | 50 mM BisTris, 50 mM NaCl, 16 mM HCl, 10% Glycerol, 0.001% Ponceau S, pH 7.2 [3] | 100 mM Tris HCl, 150 mM Tris Base, 0.01875% Coomassie G-250, 0.00625% Phenol Red, 10% Glycerol, pH 8.5 [3] |
| Running Buffer | 50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, 0.1% SDS, pH 7.7 [3] | Cathode: 50 mM BisTris, 50 mM Tricine, 0.02% Coomassie G-250, pH 6.8Anode: 50 mM BisTris, 50 mM Tricine, pH 6.8 [3] | 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 [3] |
| Critical Additives | High SDS (0.1%), EDTA [3] | Coomassie G-250 in cathode buffer [3] | Reduced SDS (0.0375%), no EDTA, Coomassie G-250 in sample [3] |
For gel preparation, NSDS-PAGE typically uses standard precast polyacrylamide gels, such as NuPAGE Novex 12% Bis-Tris 1.0 mm mini-gels [3]. Prior to sample loading, these gels should be pre-run at 200V for 30 minutes in double distilled water to remove storage buffer and any unpolymerized acrylamide that might interfere with native protein separation [3]. The gel percentage can be adjusted based on the target protein size range, following conventional SDS-PAGE principles where higher acrylamide concentrations (e.g., 12-15%) better resolve smaller proteins, while lower concentrations (e.g., 8-10%) are suitable for larger proteins [70] [71].
Sample Preparation and Electrophoresis Conditions
The sample preparation protocol for NSDS-PAGE significantly diverges from standard denaturing approaches:
Sample Buffer Mixing: Combine 7.5 μL of protein sample with 2.5 μL of 4X NSDS sample buffer (100 mM Tris HCl, 150 mM Tris base, 10% v/v glycerol, 0.0185% w/v Coomassie G-250, 0.00625% w/v Phenol Red, pH 8.5) [3].
No Heat Denaturation: Unlike standard SDS-PAGE, which requires heating at 70-100°C for 5-10 minutes [68] [72], NSDS-PAGE samples are not heated, preserving native structure [3].
Optional Centrifugation: Centrifuge samples at 15,000 rpm for 1 minute at 4°C to remove any particulate matter that might interfere with electrophoresis [68].
Sample Loading: Load prepared samples into wells of the pre-run gel alongside appropriate molecular weight markers [3].
For electrophoresis, standard equipment such as the Invitrogen Mini Gel Tank or XCell SureLock Mini-Cell can be used [1]. The electrophoresis conditions typically involve running at a constant voltage of 200V for approximately 45 minutes using the modified NSDS-PAGE running buffer until the dye front reaches the bottom of the gel [3]. The entire procedure is performed at room temperature, with no special cooling requirements under standard conditions.
Diagram 2: NSDS-PAGE experimental workflow
Research Reagent Solutions for NSDS-PAGE Implementation
Successful implementation of NSDS-PAGE requires specific reagents optimized for the technique's unique requirements. The following table details essential materials and their functions:
Table 3: Essential Research Reagents for NSDS-PAGE
| Reagent/Chemical | Function in NSDS-PAGE | Recommended Specifications |
|---|---|---|
| Coomassie G-250 | Binds hydrophobic protein sites, confers negative charge without denaturation, enables migration of basic proteins [3] | 0.01875% in sample buffer [3] |
| SDS | Provides mild denaturing environment at reduced concentration, facilitates electrophoretic migration [3] | 0.0375% in running buffer (vs. 0.1% in standard SDS-PAGE) [3] |
| Tris Buffers | Maintains pH stability throughout electrophoresis system [3] | 100 mM Tris HCl, 150 mM Tris Base in sample buffer, pH 8.5 [3] |
| Bis-Tris Gels | Provides neutral pH gel matrix compatible with native protein separation [3] | Precast NuPAGE Novex 12% Bis-Tris 1.0 mm mini-gels [3] |
| MOPS Running Buffer | Conducts current while maintaining appropriate pH and ionic strength [3] | 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 [3] |
| Glycerol | Increases sample density for improved well loading, stabilizes protein structure [3] | 10% in sample buffer [3] |
| Phenol Red | Migration tracking dye, visualizes electrophoresis progress [3] | 0.00625% in sample buffer [3] |
Analytical Applications and Validation
Functional Retention and Metalloprotein Analysis
The analytical validation of NSDS-PAGE demonstrates its significant advantages for studying functional proteins, particularly metalloproteins. Experimental results show remarkable metal retention of 98% in NSDS-PAGE compared to only 26% in standard SDS-PAGE when analyzing zinc-containing proteins [3]. This preservation of metal cofactors is crucial for subsequent functional analyses and represents a major advancement for metalloprotein research.
Enzymatic activity studies further validate the technique's preservation of native structure. In tests with nine model enzymes, including four zinc-containing proteins, seven retained activity following NSDS-PAGE separation, while all nine were denatured and lost activity in standard SDS-PAGE [3]. All nine enzymes also maintained activity in BN-PAGE, but with lower resolution than achieved with NSDS-PAGE [3]. This balance of high resolution and functional retention makes NSDS-PAGE particularly valuable for screening enzymatic activities in complex mixtures.
Advanced detection methods confirm the preservation of metal-protein associations. Laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) and in-gel zinc-protein staining using the fluorophore TSQ have verified metal retention after NSDS-PAGE separation [3]. These techniques provide sensitive, element-specific detection that unequivocally demonstrates the maintenance of metalloprotein integrity throughout the electrophoretic process.
Comparative Performance Metrics
When evaluated against both standard SDS-PAGE and BN-PAGE, NSDS-PAGE demonstrates distinctive performance characteristics:
Resolution: NSDS-PAGE provides separation quality comparable to standard SDS-PAGE, significantly superior to BN-PAGE for complex protein mixtures [3].
Separation Range: The technique effectively separates proteins across a molecular weight range similar to standard SDS-PAGE (approximately 5-250 kDa) [13], while maintaining native properties.
Reproducibility: NSDS-PAGE shows consistent banding patterns and migration distances comparable to standard SDS-PAGE when analyzing proteomic samples from cell cultures [3].
Detection Compatibility: Proteins separated by NSDS-PAGE remain compatible with standard detection methods including Coomassie staining, western blotting (using PVDF membranes, as nitrocellulose is incompatible due to Coomassie G-250 binding) [1], and specialized metalloprotein detection techniques.
The combination of these performance characteristics positions NSDS-PAGE as an ideal technique for applications requiring both high resolution separation and preservation of protein function, filling a critical methodological gap between fully denaturing and fully native electrophoretic approaches.
NSDS-PAGE represents a significant methodological advancement in protein electrophoresis, successfully bridging the gap between the high resolution of denaturing techniques and the functional preservation of native approaches. By systematically modifying standard SDS-PAGE conditions through reduced SDS concentration, elimination of denaturing steps, and strategic use of Coomassie G-250, this technique enables researchers to separate complex protein mixtures while maintaining enzymatic activity and metal cofactor integrity.
The implications for protein research are substantial, particularly in the growing field of metallomics, where understanding metal-protein interactions is essential. NSDS-PAGE offers researchers a powerful tool for studying metalloproteins, metal-containing enzymes, and other functionally sensitive proteins without sacrificing analytical resolution. As proteomic research increasingly focuses on functional characteristics rather than mere presence/absence of proteins, techniques like NSDS-PAGE that preserve biological activity while providing high-resolution separation will become increasingly valuable.
Future developments will likely refine buffer formulations, expand applications to additional protein classes, and integrate NSDS-PAGE with downstream analytical techniques such as mass spectrometry and functional assays. The principles established by NSDS-PAGE may also inspire further hybrid techniques that optimize the balance between resolution and native state preservation for specific research applications. As these methodologies evolve, they will undoubtedly enhance our understanding of protein function and facilitate drug development approaches that target functionally relevant protein states.
Conclusion
The migration of a protein in Native PAGE is a direct readout of its native physicochemical properties—primarily its net charge, but also its size and shape. Mastering the relationship between buffer pH and a protein's isoelectric point is paramount for predicting and controlling separation. This technique is indispensable for drug discovery and biomedical research as it uniquely preserves functional protein states, enabling the study of active enzymes, protein-protein interactions, and complex quaternary structures. Future directions will leverage these capabilities for profiling complex molecular machines like the mitochondrial respiratory chain and developing advanced hybrid methods such as NSDS-PAGE that push the boundaries of resolution while maintaining native functionality.
References
- 1. Native PAGE Gels | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-gel-electrophoresis/protein-gels/specialized-protein-gels/nativepage-bis-tris-gels.html]
- 2. Overview of Protein Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 3. Native SDS-PAGE: High Resolution Electrophoretic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4517606/]
- 4. Principle and Protocol of Native-PAGE [https://www.creativebiomart.net/resource/principle-protocol-principle-and-protocol-of-native-page-435.htm]
- 5. Protein charge determination and implications for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5275725/]
- 6. Mass Estimation of Native Proteins by Blue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2953912/]
- 7. Western blotting guide: Part 2, Protein separation by SDS- ... [https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blotting-guide-part-2/]
- 8. Implications for Analytical Applications in Biotechnology [https://internationalscholarsjournals.org/articles/387804042025]
- 9. Describe the differences between Native PAGE and SDS-PAGE. [https://www.truegeometry.com/api/exploreHTML?query=Describe%20the%20differences%20between%20Native%20PAGE%20and%20SDS-PAGE.]
- 10. Total charge of a protein in native gel is determined by the pH [https://www.letstalkacademy.com/total-charge-of-a-protein-in-native-gel-is-determined-by-the-ph/]
- 11. Precast Protein Gels | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-gel-electrophoresis/protein-gels.html]
- 12. Western Blot: The Complete Guide [https://www.antibodies.com/applications/western-blotting]
- 13. SDS-PAGE [https://en.wikipedia.org/wiki/SDS-PAGE]
- 14. A screening method for binding synthetic metallo ... [https://www.sciencedirect.com/science/article/pii/S0003269722002445]
- 15. PSME3 regulates migration and differentiation of myoblasts [https://pmc.ncbi.nlm.nih.gov/articles/PMC12179657/]
- 16. Validation of blue- and clear-native polyacrylamide gel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445495/]
- 17. Exploring imaged capillary isoelectric focusing parameters ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1536222/full]
- 18. Preanalytical Strategies for Native Mass Spectrometry ... [https://www.preprints.org/manuscript/202510.1566]
- 19. pI Determination of Native Proteins In Biological Samples [https://pubmed.ncbi.nlm.nih.gov/30702808/]
- 20. QPNC-PAGE [https://en.wikipedia.org/wiki/QPNC-PAGE]
- 21. pI Determination of Native Proteins In Biological Samples [https://pure.johnshopkins.edu/en/publications/pi-determination-of-native-proteins-in-biological-samples]
- 22. A Native PAGE Assay for the Biochemical Characterization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8720520/]
- 23. High-resolution native electrophoresis in-gel activity assay ... [https://www.nature.com/articles/s41598-025-24684-3]
- 24. Native PAGE Gels | Thermo Fisher Scientific - ZA [https://www.thermofisher.com/za/en/home/life-science/protein-biology/protein-gel-electrophoresis/protein-gels/specialized-protein-gels/nativepage-bis-tris-gels.html]
- 25. NativePAGE™ 3 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel ... [https://www.thermofisher.com/order/catalog/product/BN1001BOX/faqs]
- 26. NuPAGE™ 7%, Tris-Acetate, 1.0 mm, Mini Protein Gel, 15- ... [https://www.thermofisher.com/store/v3/products/faqs/EA03555BOX]
- 27. SMA-PAGE: A new method to examine complexes of ... [https://www.sciencedirect.com/science/article/pii/S0005273619301087]
- 28. Analysis of mitochondrial respiratory chain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4823378/]
- 29. Coomassie blue staining - Proteins and protein analysis [https://www.abcam.com/en-us/knowledge-center/proteins-and-protein-analysis/coomassie-blue-staining]
- 30. Blue-native PAGE in plants: a tool in analysis of protein ... [https://plantmethods.biomedcentral.com/articles/10.1186/1746-4811-1-11]
- 31. Fast and Sensitive Colloidal Coomassie G-250 Staining for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3149902/]
- 32. Native-PAGE [http://www.assay-protocol.com/molecular-biology/electrophoresis/native-page.html]
- 33. Differences between SDS PAGE and Native PAGE [https://www.geeksforgeeks.org/biology/differences-between-sds-page-and-native-page/]
- 34. Native Polyacrylamide Gel Electrophoresis - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/native-polyacrylamide-gel-electrophoresis]
- 35. Is Heating Sample Skippable for Western Blotting? [https://www.biossusa.com/blogs/news/is-heating-sample-skippable-for-western-blotting?srsltid=AfmBOooXIAkA7g_gRGbTr1wHrh1xfJ_aj9LdriXhiJnVlim4UUM7tldc]
- 36. Analysis of RNA Folding by Native Polyacrylamide Gel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6343852/]
- 37. Structural and functional comparison of native pentameric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2265887/]
- 38. Advantages and limitations of clear-native PAGE [https://pubmed.ncbi.nlm.nih.gov/16220535/]
- 39. Clear Native Gel Electrophoresis for the Purification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355471/]
- 40. 05SAR-PAGE: Separation of protein dimerization and ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267019314497]
- 41. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoqNPRDvVJGNITeDRCf2FdmPYyMLlle-wi1ubUmydNQ2ZQKZMI9M]
- 42. An equation to estimate the difference between ... [https://www.nature.com/articles/srep13370]
- 43. Current Understanding of Protein Aggregation in ... [https://www.mdpi.com/1422-0067/26/21/10568]
- 44. High-resolution native electrophoresis in-gel activity assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550018/]
- 45. Massive experimental quantification allows interpretable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12042874/]
- 46. Hydrophobicity causes anomalous migration of cystine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12127883/]
- 47. Co-translational protein aggregation and ribosome stalling ... [https://www.nature.com/articles/s41467-025-56873-z]
- 48. Developments in Ion Exchange Chromatography–Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12451099/]
- 49. Native Gel Electrophoresis: Videos & Practice Problems [https://www.pearson.com/channels/biochemistry/learn/jason/protein-techniques/native-page]
- 50. Automated optimization of resin selection, wash ... [https://www.sciencedirect.com/science/article/pii/S1871678425001128]
- 51. Uncovering hidden protein modifications with native top ... [https://www.nature.com/articles/s41592-025-02846-5]
- 52. Controlling the separation of native proteins with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10033466/]
- 53. Overview of Protein Electrophoresis [https://www.thermofisher.com/tr/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 54. High Resolution Clear Native Electrophoresis for In-gel ... [https://www.sciencedirect.com/science/article/pii/S153594762031690X]
- 55. Recovery of native proteins from preparative electrophoresis ... [https://pubmed.ncbi.nlm.nih.gov/6091091/]
- 56. Western blotting analysis of proteins separated by agarose ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813020349187]
- 57. How Is Native PAGE Different from SDS-PAGE? [https://synapse.patsnap.com/article/how-is-native-page-different-from-sds-page]
- 58. What is the difference between PAGE and SDS-PAGE? [https://www.aatbio.com/resources/faq-frequently-asked-questions/What-is-the-difference-between-PAGE-and-SDS-PAGE]
- 59. Gel Electrophoresis, PAGE, SDS - Biochemistry [https://www.medschoolcoach.com/gel-electrophoresis-page-sds-page-mcat-biochemistry/]
- 60. How SDS-PAGE Separates Proteins [https://azurebiosystems.com/blog/how-sds-page-separates-proteins/]
- 61. SDS PAGE vs Native PAGE [https://byjus.com/biology/difference-between-sds-page-and-native-page/]
- 62. Mechanism of 2D Blue Native/SDS-PAGE Protein Complex ... [https://www.mtoz-biolabs.com/mechanism-of-2d-blue-native-sds-page-protein-complex-analysis.html]
- 63. In-Gel Activity Assay of Mammalian Mitochondrial and ... [https://en.bio-protocol.org/en/bpdetail?id=5126&type=0]
- 64. In-Gel Assay to Evaluate Antioxidant Enzyme Response to ... [https://www.mdpi.com/2076-3417/12/6/2760]
- 65. Validation of blue- and clear-native polyacrylamide gel ... [https://pubmed.ncbi.nlm.nih.gov/40966204/]
- 66. High Resolution Two-Dimensional Electrophoresis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2874754/]
- 67. 2D Gel Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-gel-electrophoresis/protein-gels/specialized-protein-gels/2d-gel-electrophoresis.html]
- 68. The principle and method of polyacrylamide gel ... [https://www.mblbio.com/bio/g/support/method/sds-page.html]
- 69. How SDS-PAGE Works [https://bitesizebio.com/580/how-sds-page-works/]
- 70. SDS-PAGE: A Cornerstone of Protein Analysis in Modern ... [https://www.metwarebio.com/sds-page-protein-analysis/]
- 71. SDS-PAGE guide: Sample preparation to analysis [https://www.abcam.com/en-us/knowledge-center/western-blot/sds-page-guide]
- 72. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOopeQoO5nvjINVOHAZC2SHKQ-mZZUEfzfsLlwk1FvczxjIU0jRNQ]