Structural Divergence in SH2 Domains: Decoding STAT-type vs. Src-type for Targeted Therapeutics
This article provides a comprehensive analysis of the structural distinctions between STAT-type and Src-type Src Homology 2 (SH2) domains, modular protein domains critical for phosphotyrosine signaling.
Structural Divergence in SH2 Domains: Decoding STAT-type vs. Src-type for Targeted Therapeutics
Abstract
This article provides a comprehensive analysis of the structural distinctions between STAT-type and Src-type Src Homology 2 (SH2) domains, modular protein domains critical for phosphotyrosine signaling. Aimed at researchers and drug development professionals, it explores the foundational architecture of these domains, detailing how STAT-type-specific adaptations, such as the lack of βE and βF strands, facilitate unique functions like dimerization for transcription. The content covers advanced methodologies for studying these structures, addresses challenges in drug discovery, including the impact of disease-associated mutations, and validates insights through comparative analysis with other SH2 domain families. The review concludes by synthesizing how this structural knowledge informs the development of targeted therapies, such as small-molecule inhibitors, for cancers and immune disorders driven by aberrant SH2 domain signaling.
Architectural Blueprints: Unveiling the Core Structural Frameworks of STAT and Src SH2 Domains
The Src Homology 2 (SH2) domain is a foundational modular protein domain that plays a critical role in cellular signal transduction by specifically recognizing phosphotyrosine (pTyr) motifs [1] [2]. Since its discovery in the v-Src oncoprotein of Rous sarcoma virus in 1986, the SH2 domain has become a cornerstone concept for understanding how reversible post-translational modifications regulate protein-protein interactions and intracellular signaling networks [1] [2]. This ~100 amino acid domain serves as a key mediator in tyrosine kinase signaling pathways, enabling the assembly of specific signaling complexes in response to extracellular stimuli [1] [3]. Despite significant sequence variation across the human SH2 domain proteome (which includes approximately 110-120 SH2 domains in 111 human proteins), the three-dimensional structure of this domain remains remarkably conserved [3] [4]. This structural conservation maintains the fundamental phosphotyrosine-binding function while allowing for precise specificity in ligand recognition, a feature essential for the proper routing of intracellular signals [1]. Within this conserved structural framework, however, lies important variation that has enabled the evolution of distinct SH2 domain subtypes, most notably the structural differences between STAT-type and Src-type SH2 domains that form the core focus of current research in this field [5] [6].
The Conserved Architecture of the SH2 Domain
Core Secondary Structure Elements
The canonical SH2 domain fold consists of a highly conserved "αβββα" structural core composed of a central anti-parallel β-sheet flanked by two α-helices [3] [6]. This core structure is remarkably consistent across the SH2 domain family, with some family members sharing as little as 15% pairwise sequence identity while maintaining nearly identical three-dimensional folds [7]. The central β-sheet typically comprises three major strands (βB, βC, βD), while the two α-helices (αA and αB) position themselves on either side of this sheet [3] [2]. The N-terminal region of the SH2 domain, containing the βB strand and its highly conserved FLVR motif, shows particularly strong structural conservation, while the C-terminal region demonstrates greater variability that contributes to functional diversity [3] [7].
Table 1: Core Secondary Structure Elements of the Canonical SH2 Domain
| Structural Element | Position | Key Features | Functional Role |
|---|---|---|---|
| αA Helix | N-terminal region | Flanks one side of central β-sheet | Forms part of pTyr binding pocket |
| βB Strand | Early in sequence | Contains conserved FLVR motif | Critical for pTyr coordination |
| βC Strand | Central β-sheet | Part of anti-parallel sheet | Contributes to structural stability |
| βD Strand | Central β-sheet | Longest strand; divides domain | Separates pTyr and specificity pockets |
| αB Helix | C-terminal region | Flanks opposite side of β-sheet | Forms part of specificity pocket |
The Phosphotyrosine and Specificity Binding Pockets
The SH2 domain employs a "two-pronged plug" binding mechanism that engages phosphotyrosine-containing peptides through two adjacent binding pockets on either side of the central β-sheet [2] [4]. The phosphotyrosine (pTyr) binding pocket is located on one side of the central βD strand and specializes in recognizing the phosphorylated tyrosine residue itself [1] [6]. This pocket contains a highly conserved arginine residue at position βB5 (within the FLVRES sequence motif) that forms a critical salt bridge with the phosphate moiety of the phosphotyrosine [3] [2]. This single interaction contributes substantially to the binding energy, with mutation of this arginine resulting in up to a 1000-fold reduction in binding affinity [2].
The second binding pocket, located on the opposite side of the βD strand, is termed the specificity pocket or pY+3 pocket, as it typically recognizes the amino acid at the +3 position relative to the phosphotyrosine [1] [4]. The structural characteristics and residue composition of this pocket determine the sequence specificity of each SH2 domain, allowing different SH2 domains to recognize distinct pTyr-containing motifs [1]. For example, Src family kinases preferentially bind pYEEI motifs, while the SH2 domain of Grb2 recognizes pYXNX sequences [1] [8]. This specificity pocket is formed by residues from the αB helix, βG strand, and the BG and EF loops, which show greater sequence variation across SH2 domains [3] [7].
Structural Classification: STAT-Type versus Src-Type SH2 Domains
C-Terminal Structural Variations
While all SH2 domains share the conserved αβββα core, they can be classified into two major subgroups based on distinctive structural features at their C-terminal: STAT-type and Src-type SH2 domains [5] [6]. This structural divergence represents an important evolutionary adaptation that correlates with functional specialization.
Src-type SH2 domains, representative of the majority of SH2 domains, contain additional β-strands (βE, βF, and βG) following the core αβββα structure [5] [7]. These extra strands contribute to the overall stability of the domain and participate in forming the specificity pocket. The presence of these β-strands is characteristic of SH2 domains found in cytoplasmic signaling proteins such as kinases, phosphatases, and adaptor proteins [3].
In contrast, STAT-type SH2 domains exhibit a distinct C-terminal architecture characterized by a split αB helix (forming αB and αB' helices) and the absence of the βE and βF strands typically found in Src-type domains [5] [6]. This structural adaptation is particularly suited to the STAT protein function, as the αB' helix participates in critical protein-protein interactions required for STAT dimerization and nuclear translocation following activation [6]. The absence of the βE and βF strands in STAT-type domains creates a more compact structure that may facilitate the specific dimerization interface required for STAT transcriptional function.
Table 2: Comparative Features of STAT-type versus Src-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| C-terminal Structure | Split αB helix (αB and αB') | Additional β-strands (βE, βF, βG) |
| βE and βF Strands | Absent | Present |
| Representative Proteins | STAT family transcription factors | Src, Abl, PLCγ, p120RasGAP |
| Dimerization Mechanism | SH2-pTyr interaction between STAT monomers | Various, including domain-domain interactions |
| Evolutionary Origin | Ancient, predating animal multicellularity | More recent diversification |
Functional Implications of Structural Differences
The structural distinctions between STAT-type and Src-type SH2 domains have direct functional consequences. STAT-type SH2 domains are specialized for homo- and heterodimerization between STAT proteins following receptor recruitment and phosphorylation [6]. This dimerization occurs through reciprocal SH2-phosphotyrosine interactions between two STAT monomers, creating functional transcription factors that can translocate to the nucleus [6]. The unique architecture of the STAT-type SH2 domain, particularly the αB' helix and the surrounding regions, facilitates this specific dimerization interface while maintaining the ability to recognize phosphorylated receptor chains during initial activation.
Src-type SH2 domains display greater functional diversity, participating in various signaling contexts including membrane recruitment, substrate targeting, and allosteric regulation [1] [3]. The presence of additional β-strands in Src-type domains may contribute to this functional versatility by providing additional interaction surfaces and stability. For example, the SH2 domains in enzymes like phospholipase Cγ (PLCγ) and GTPase activating proteins (GAPs) often employ their SH2 domains for both recruitment to specific phosphorylated sites and for intramolecular interactions that regulate catalytic activity [1].
Experimental Approaches for SH2 Domain Structural Analysis
Crystallization of SH2 Domain-Phosphopeptide Complexes
X-ray crystallography has been instrumental in elucidating the structural principles of SH2 domain function. The following protocol for co-crystallizing SH2 domains with phosphopeptides is adapted from established methodologies [4]:
Protein Purification: Express and purify recombinant SH2 domain protein (typically comprising 100-150 amino acids) using standard bacterial expression systems and affinity chromatography. The protein should be in a storage buffer such as 20 mM Tris-HCl (pH 8.0) with 150 mM NaCl.
Phosphopeptide Preparation: Obtain synthetic phosphopeptides corresponding to known binding motifs, typically 7-15 residues in length with the phosphotyrosine positioned near the center. Peptides should be HPLC-purified to >98% purity and modified with acetyl and amide groups at N- and C-termini, respectively, to neutralize charge and improve stability [4].
Complex Formation: Mix purified SH2 domain protein with phosphopeptide at a 1:1.2 molar ratio in a low-salt buffer. Incubate on ice for 30-60 minutes to allow complex formation.
Crystallization: Use the hanging drop vapor diffusion method by mixing 1-2 μL of protein-peptide complex solution with an equal volume of reservoir solution. Suitable reservoir conditions vary by SH2 domain but often include PEG-based solutions (e.g., 15-20% PEG 10,000) with appropriate salts and pH buffers.
Data Collection and Analysis: Harvest crystals, cryoprotect as needed, and collect X-ray diffraction data. Molecular replacement using known SH2 domain structures typically enables phasing.
This approach has revealed both canonical binding modes, as observed in the p120RasGAP N-SH2 domain complex, and atypical binding interactions, such as those discovered in the p120RasGAP C-SH2 domain where the FLVR arginine does not directly coordinate the phosphotyrosine [4].
Research Reagent Solutions for SH2 Domain Studies
Table 3: Essential Research Reagents for SH2 Domain Structural Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Expression Systems | E. coli BL21(DE3) | Recombinant SH2 domain protein production |
| Purification Tools | Ni-NTA resin (for His-tagged proteins), GST resin | Affinity purification of recombinant SH2 domains |
| Crystallization Kits | Hampton Research Crystal Screens, PEG-Ion Screen | Initial crystallization condition screening |
| Phosphopeptides | pTyr-1105: EEENI(pY)SVPHDST, pTyr-1087: DpYAEPMD | SH2 domain binding partners for complex formation |
| Chromatography | Size exclusion chromatography (Superdex 75) | Final purification step to obtain monodisperse protein |
| Crystallization Plates | VDXm Crystallization Plate with sealant | Vapor diffusion crystallization setup |
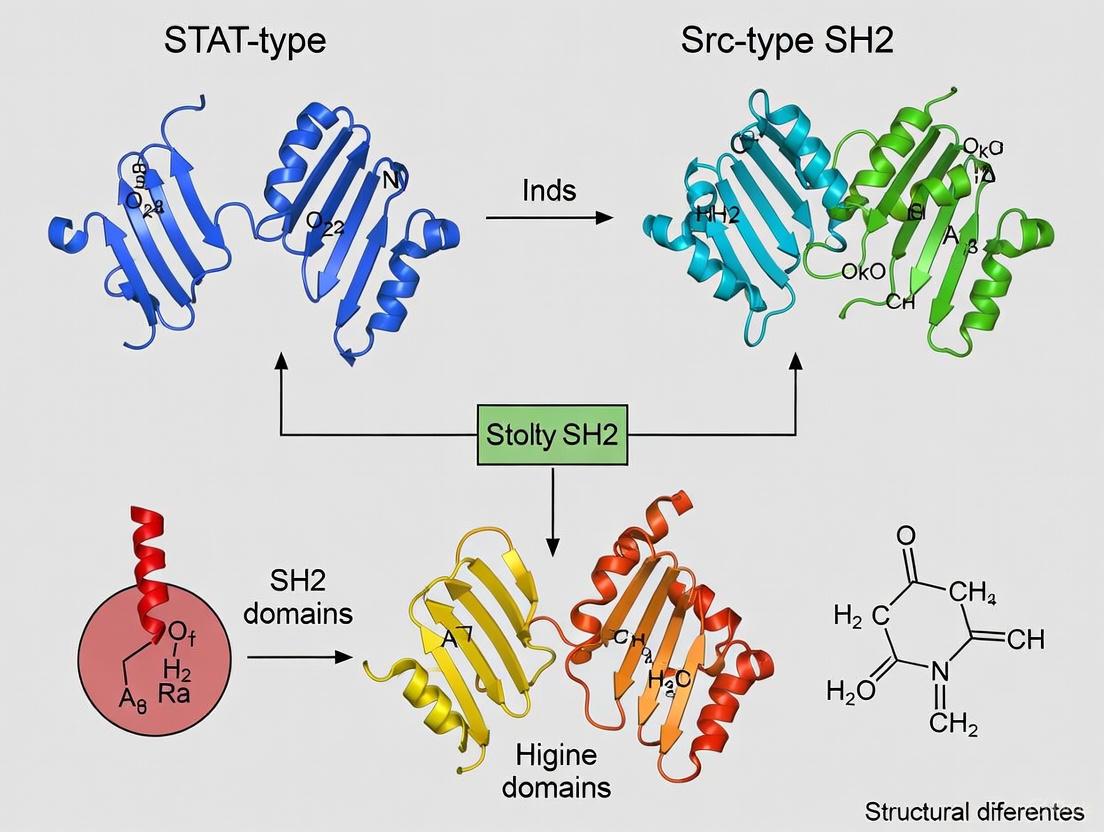
Visualization of SH2 Domain Architecture and Classification
The following diagram illustrates the core secondary structure organization of the canonical SH2 domain fold and highlights the key differences between STAT-type and Src-type SH2 domains.
SH2 Domain Structure and Classification
This structural visualization highlights how the conserved core architecture diverges into distinct C-terminal structures between STAT-type and Src-type SH2 domains, with functional implications for their respective roles in cellular signaling.
The canonical SH2 domain fold represents a remarkable evolutionary solution to the challenge of specific phosphotyrosine recognition in eukaryotic signal transduction. The conserved αβββα core structure provides a stable scaffold that maintains the essential phosphotyrosine-binding function across diverse signaling contexts, while variations in key regions—particularly the C-terminal structural elements that distinguish STAT-type from Src-type SH2 domains—enable functional specialization [5] [6]. The structural differences between these two SH2 domain classes directly correlate with their distinct biological roles: STAT-type domains are optimized for the specific dimerization requirements of transcription factors, while Src-type domains exhibit greater versatility in their signaling applications [7] [6].
Ongoing research continues to reveal unexpected complexities in SH2 domain function, including non-canonical binding modes, regulation by secondary interfaces, and roles in liquid-liquid phase separation [3] [7]. The deep structural understanding of the conserved SH2 fold and its variations provides a foundation for targeted therapeutic intervention in diseases driven by aberrant tyrosine kinase signaling, particularly through the development of small molecules that disrupt specific SH2 domain interactions in both STAT and Src family proteins [3] [6]. As structural biology techniques advance, our understanding of this fundamental signaling domain continues to evolve, revealing new layers of complexity in one of cell signaling's most conserved interaction modules.
Signal transducer and activator of transcription (STAT) proteins represent crucial signaling molecules in metazoan cells, functioning as both signal transducers and transcription factors. Central to their function is the Src Homology 2 (SH2) domain, a module of approximately 100 amino acids that specifically recognizes phosphorylated tyrosine motifs. STAT-type SH2 domains belong to a specialized subclass that diverges structurally and functionally from the more widely studied Src-type SH2 domains. These domains emerged approximately 600 million years ago within metazoan signaling pathways and are integral to phosphotyrosine-mediated signal transduction [6] [9]. In STAT proteins, the SH2 domain performs the critical dual function of mediating receptor recruitment through phosphotyrosine binding and facilitating STAT dimerization—an essential step for nuclear translocation and transcriptional activation [6]. The unique structural adaptations of STAT-type SH2 domains reflect their specialized role in directly linking extracellular signals to transcriptional responses, distinguishing them from SH2 domains in other protein families that primarily serve scaffolding or regulatory roles within cytoplasmic signaling networks.
Table 1: Core Characteristics of SH2 Domains
| Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| C-terminal Structure | α-helix (αB') | β-sheet (βE and βF) |
| Representative Proteins | STAT1, STAT3, STAT5, STAT6 | Src, Grb2, SHP2, PLCγ |
| Primary Functional Role | Dimerization & transcriptional activation | Scaffolding & signal relay |
| Presence in Unicellular Organisms | Limited or absent | Limited or absent |
| Domain Architecture | Often conjugated with linker domain | Variable domain combinations |
Structural Biology of STAT-type SH2 Domains
Core Folding Motifs and Architecture
The STAT-type SH2 domain maintains the fundamental SH2 fold—a central anti-parallel β-sheet flanked by two α-helices, creating an αβββα motif [6] [7]. The central β-sheet (comprising strands βB, βC, and βD) partitions the domain into two functionally distinct subpockets: the phosphotyrosine (pY) binding pocket and the pY+3 specificity pocket [6]. The pY pocket, formed by the αA helix, BC loop, and one face of the central β-sheet, contains conserved residues that directly coordinate the phosphotyrosine moiety of target peptides. The pY+3 pocket, created by the opposite face of the β-sheet along with residues from the αB helix and CD and BC* loops, determines binding specificity by accommodating residues C-terminal to the phosphotyrosine [6].
Despite this conserved core architecture, STAT-type SH2 domains exhibit distinctive structural adaptations. Most notably, they possess a split αB helix and lack the βE and βF strands characteristic of Src-type SH2 domains [7]. The C-terminal region of the pY+3 pocket, termed the evolutionary active region (EAR), contains an additional α-helix (αB') in STAT-type SH2 domains, contrasting with the β-sheet elements (βE and βF) found in Src-type domains [6]. This structural divergence likely represents an evolutionary adaptation facilitating STAT dimerization, a critical step in STAT-mediated transcriptional regulation [7]. Additionally, STAT-type SH2 domains typically feature shorter connecting loops compared to enzymatic SH2 domain-containing proteins, with the CD-loop length varying depending on protein family classification [7].
Molecular Determinants of Phosphopeptide Recognition
STAT SH2 domains recognize phosphorylated tyrosine motifs through a combination of conserved binding interactions and domain-specific features. The phosphotyrosine residue inserts into the pY pocket where it forms critical contacts with conserved residues, including an invariant arginine at position βB5 that directly coordinates the phosphate moiety through a salt bridge [7]. Residues C-terminal to the phosphotyrosine extend across the SH2 domain surface, with side chains at positions pY+1 through pY+5 contributing to binding affinity and specificity through interactions with the pY+3 pocket [10].
Structural studies reveal that STAT SH2 domains exhibit significant flexibility, particularly in the pY pocket, where accessible volume varies dramatically even on sub-microsecond timescales [6]. This inherent dynamics complicates drug discovery efforts, as crystal structures may not capture all accessible conformational states. Beyond primary sequence determinants, STAT SH2 domains recognize contextual sequence information in their peptide ligands, integrating both permissive residues that enhance binding and non-permissive residues that oppose binding through steric clash or charge repulsion [10]. This complex recognition mechanism allows STAT SH2 domains to distinguish subtle differences in peptide ligands, significantly expanding the information content embedded in relatively short linear motifs.
Figure 1: STAT Protein Activation Pathway Mediated by SH2 Domain Interactions. The SH2 domain facilitates receptor recruitment, dimerization, and nuclear translocation essential for transcriptional activation.
Functional Consequences of the STAT-type SH2 Structure
Dimerization and Nuclear Translocation
The specialized architecture of STAT-type SH2 domains directly enables their cardinal function: mediating STAT dimerization through reciprocal SH2-phosphotyrosine interactions. Upon phosphorylation of a specific C-terminal tyrosine residue by receptor-associated kinases, two STAT monomers form parallel dimers through interaction between one monomer's SH2 domain and the phosphotyrosine of its partner [6]. This dimeric configuration is essential for nuclear accumulation and represents the transcriptionally active form of STAT proteins. The unique features of STAT-type SH2 domains, particularly the αB' helix in the EAR region, facilitate critical cross-domain interactions that stabilize the dimeric configuration [6]. This dimerization mechanism stands in contrast to Src-type SH2 domains, which typically mediate transient protein-protein interactions rather than stable homodimerization.
Beyond facilitating dimerization, the STAT SH2 domain participates in multiple protein-protein interactions throughout the activation cycle. Initially, it mediates recruitment to activated cytokine and growth factor receptors by binding to specific phosphotyrosine motifs within receptor cytoplasmic domains [6]. Following dimerization, the SH2 domain may contribute to interactions with nuclear import machinery, though this function is less well characterized. The multi-functional nature of STAT SH2 domains underscores their strategic importance in STAT signaling pathways and explains why this domain represents a hotspot for pathogenic mutations across various diseases.
Structural Basis of Disease-Associated Mutations
Sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in STAT proteins, particularly STAT3 and STAT5B [6]. These mutations can produce either gain-of-function or loss-of-function phenotypes, sometimes at identical amino acid positions, highlighting the delicate structural balance required for proper STAT regulation. For instance, the S614R mutation in STAT3 is associated with T-cell large granular lymphocytic leukemia (T-LGLL) and represents an activating mutation, while mutations at nearby positions (S614G, K591E/M, R609G) cause loss-of-function and are linked to immunological deficiencies like autosomal-dominant Hyper IgE Syndrome (AD-HIES) [6].
Table 2: Disease-Associated Mutations in STAT3 and STAT5B SH2 Domains
| Mutation | Structural Location | Domain Region | Associated Pathology | Functional Effect |
|---|---|---|---|---|
| STAT3 S614R | BC loop | pY pocket | T-LGLL, NK-LGLL | Activating |
| STAT3 K591E/M | αA helix | pY pocket | AD-HIES | Loss-of-function |
| STAT3 S611N | βB strand | pY pocket | AD-HIES | Loss-of-function |
| STAT3 E616K | BC loop | pY pocket | NKTL | Activating |
| STAT5B N642H | βB strand | pY pocket | Lymphoma, Leukemia | Activating |
Loss-of-function mutations in STAT3 typically disrupt phosphopeptide binding or dimerization capacity, impairing nuclear translocation and transcriptional activation. This manifests clinically as AD-HIES, characterized by diminished Th17 T-cell responses, recurrent infections, eczema, and eosinophilia [6]. Conversely, gain-of-function mutations enhance dimer stability, prolong nuclear retention, or increase DNA-binding affinity, leading to constitutive transcriptional activity that drives proliferative diseases like leukemias and lymphomas. The location of these mutations within structurally critical regions underscores the functional importance of specific SH2 domain elements—the pY pocket, the phosphopeptide binding groove, and the dimerization interface—in maintaining physiological STAT signaling.
Comparative Analysis: STAT-type versus Src-type SH2 Domains
Structural Divergence and Functional Specialization
The evolutionary divergence between STAT-type and Src-type SH2 domains represents a fascinating case of structural adaptation to distinct physiological roles. While both share the conserved αβββα core fold, they differ substantially in their C-terminal structural elements. STAT-type domains feature a split αB helix and lack the βE and βF strands present in Src-type domains [7]. Instead, they contain an αB' helix in the evolutionary active region (EAR) of the pY+3 pocket, an adaptation that likely facilitates STAT dimerization [6]. This structural difference reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity, as observed in Dictyostelium, which employs SH2 domain/phosphotyrosine signaling for transcriptional regulation [7].
Functional differences between these SH2 domain classes mirror their structural distinctions. Src-type SH2 domains typically mediate transient protein-protein interactions that assemble signaling complexes or regulate enzymatic activity through intramolecular interactions. For example, in SHP2 phosphatase, the N-SH2 domain allosterically regulates catalytic activity by switching between inhibitory and activating conformations [11]. In contrast, STAT-type SH2 domains specialize in mediating stable homodimerization or heterodimerization between STAT family members, creating the DNA-binding competent transcription factors. This functional specialization explains why STAT proteins represent one of the most ancient SH2 domain-containing families, with homologs identified in plants that predate the plant-animal divergence [5].
Evolutionary Trajectory and Genomic Distribution
SH2 domains first emerged in unicellular eukaryotes, with their expansion closely coupled to the development of tyrosine kinases and tyrosine phosphatases in metazoans [9]. Analysis across 21 eukaryotic species reveals that SH2 domains co-evolved with protein tyrosine kinases (PTKs), with their numbers expanding rapidly in the choanoflagellate and metazoan lineages [9]. The correlation between the percentage of PTKs and SH2 domains in genomes is remarkably high (0.95), indicating their coordinated evolution [9]. STAT-type SH2 domains represent an ancient lineage within this expansion, with the linker-SH2 domain of STAT serving as a template for continuing SH2 domain evolution [5].
The human genome encodes approximately 110 SH2 domain-containing proteins housing 121 SH2 domains, which can be classified into 38 subfamilies based on phylogenetic analysis [6] [9]. STAT proteins constitute one of these subfamilies, with seven members in humans (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) [12]. This expansion of SH2 domain proteins, primarily through gene duplication and domain shuffling events, allowed for increased complexity in phosphotyrosine signaling networks that likely contributed to metazoan diversification and specialization.
Research Methodologies and Experimental Approaches
Techniques for Characterizing SH2 Domain Structure and Function
Research into STAT-type SH2 domains employs multidisciplinary approaches to elucidate structure-function relationships. X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy provide high-resolution structural information, revealing atomic-level details of phosphopeptide binding and dimerization interfaces [6]. Structural studies often involve co-crystallizing SH2 domains with phosphopeptide ligands corresponding to physiological binding motifs, such as those from cytokine receptor cytoplasmic domains [13]. These approaches have revealed that STAT SH2 domains exhibit significant conformational flexibility, particularly in the pY pocket, underscoring the importance of accounting for protein dynamics in drug discovery efforts [6].
Biophysical techniques including fluorescence polarization, isothermal titration calorimetry, and surface plasmon resonance quantify binding affinities and kinetic parameters for SH2 domain-phosphopeptide interactions [10]. These methods typically employ purified recombinant SH2 domains and synthetic phosphopeptides, allowing precise determination of dissociation constants (Kd), which generally range from 0.1–10 μM for physiological SH2 domain interactions [7]. For STAT proteins, dimerization assays using co-immunoprecipitation, size-exclusion chromatography, and analytical ultracentrifugation provide functional validation of mutations affecting SH2 domain function.
Figure 2: Experimental Workflow for Characterizing STAT-type SH2 Domain Function. Integrated approaches from structural biology to cellular validation provide comprehensive functional assessment.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for STAT-type SH2 Domain Studies
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Recombinant SH2 Domains | GST-STAT3-SH2, His-STAT5B-SH2 | Binding assays, structural studies | Tagged for purification, wild-type vs mutant |
| Phosphopeptide Libraries | SPOT membranes, oriented peptide libraries | Specificity profiling, motif identification | Addressable arrays, physiological sequences |
| Binding Assay Reagents | Fluorescent probes, biosensor chips | Affinity measurements, kinetic analysis | High sensitivity, real-time monitoring |
| Cellular Expression Systems | STAT-deficient cell lines, reconstitution models | Functional validation, signaling studies | Controlled genetic background |
| Disease-Associated Mutants | STAT3 S614R, STAT3 K591E, STAT5B N642H | Pathophysiological mechanism studies | Gain-of-function and loss-of-function variants |
Experimental investigation of STAT-type SH2 domains relies on specialized reagents and methodologies. Recombinant SH2 domains, typically expressed as glutathione S-transferase (GST) or polyhistidine (His) fusions in Escherichia coli, provide purified protein for biophysical and structural studies [10]. Phosphopeptide libraries, including those synthesized using SPOT methodology, enable high-throughput specificity profiling by testing interactions with large sets of physiological tyrosine phosphopeptides [10]. For cellular studies, STAT-deficient cell lines allow functional characterization of wild-type and mutant STAT proteins in controlled genetic backgrounds, while reconstitution models assess signaling output and transcriptional activity.
Advanced computational approaches, including molecular dynamics simulations and enhanced sampling techniques, complement experimental methods by providing insights into conformational dynamics and allosteric regulation [11]. These approaches are particularly valuable for studying the flexible nature of STAT SH2 domains and understanding how disease-associated mutations affect structural stability and signaling output. For drug discovery efforts, virtual screening of compound libraries against SH2 domain structures identifies potential inhibitors that disrupt pathological protein-protein interactions.
Therapeutic Targeting and Future Perspectives
The strategic position of STAT-type SH2 domains in signaling pathways controlling cell proliferation, survival, and immune function makes them attractive therapeutic targets for cancer, autoimmune disorders, and immunodeficiencies. Despite this potential, no clinical candidates directly targeting STAT SH2 domains have yet reached approval, reflecting the challenges inherent in targeting protein-protein interactions [6]. Most drug discovery efforts have focused on the pY and pY+3 pockets due to their well-defined features and conserved residues [6]. However, the shallow, dynamic nature of these binding surfaces and the high affinity for natural phosphopeptide ligands present significant obstacles for small molecule inhibitor development.
Emerging strategies include targeting allosteric sites, developing stabilized peptides or macrocyclic compounds, and exploiting unique features of pathogenic mutant SH2 domains. Recent research has also revealed that nearly 75% of SH2 domains interact with lipid molecules, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3), suggesting potential alternative targeting strategies [7]. Additionally, the role of SH2 domains in liquid-liquid phase separation (LLPS) and intracellular condensate formation presents novel regulatory mechanisms that might be therapeutically exploited [7]. As structural and mechanistic understanding of STAT-type SH2 domains continues to advance, so too will opportunities for developing targeted interventions that modulate their function in disease contexts.
Src homology 2 (SH2) domains represent a critical class of protein interaction modules that specifically recognize phosphotyrosine (pY) motifs, thereby establishing specificity in intracellular signaling networks. Among these, Src-type SH2 domains serve as the architectural standard-bearers, characterized by their conserved structural framework and versatile specificity pockets that enable selective ligand recognition. This review comprehensively examines the structural determinants of Src-type SH2 domains, contrasting them with STAT-type variants, and elucidates their mechanistic roles in cellular signaling processes. We detail experimental methodologies for investigating SH2 domain interactions and present current targeting strategies for therapeutic intervention. Through integrated structural, functional, and pharmacological perspectives, this analysis establishes Src-type SH2 domains as fundamental components of phosphotyrosine signaling circuitry with emerging significance in drug discovery.
Src homology 2 (SH2) domains are approximately 100 amino acid modular protein domains that specifically recognize and bind to phosphorylated tyrosine residues, facilitating protein-protein interactions in intracellular signaling pathways [3] [14]. The human genome encodes approximately 110-120 SH2 domain-containing proteins, which are functionally classified into diverse groups including enzymes, adaptor proteins, docking proteins, transcription factors, and cytoskeletal proteins [3] [15]. These domains form a crucial part of the protein-protein interaction network involved in cellular processes spanning development, homeostasis, immune responses, and cytoskeletal rearrangement [3].
SH2 domains primarily function to recruit host polypeptides to specific tyrosine-phosphorylated sites on target proteins, thereby inducing proximity between tyrosine kinases, tyrosine phosphatases, and their substrates [3] [10]. This selective recognition establishes signaling specificity downstream of tyrosine phosphorylation events. Beyond their canonical role in phosphotyrosine recognition, emerging research indicates that nearly 75% of SH2 domains also interact with membrane lipids, particularly phosphoinositides such as phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [3] [7]. Additionally, SH2 domain-containing proteins are increasingly implicated in liquid-liquid phase separation (LLPS), where multivalent interactions drive formation of intracellular signaling condensates [3].
The SH2 domain family bifurcates into two major structural subgroups: Src-type and STAT-type SH2 domains. Src-type domains represent the standard architectural framework with versatile specificity pockets, while STAT-type domains exhibit distinct structural adaptations suited for their role in transcription factor dimerization [7]. This review focuses specifically on Src-type SH2 domains as the paradigmatic fold, examining their structural features, ligand recognition mechanisms, functional diversity, and emerging therapeutic targeting strategies.
Structural Architecture of Src-type SH2 Domains
Consensus Fold and Conservation Patterns
Src-type SH2 domains adopt a conserved protein fold consisting of a central anti-parallel β-sheet flanked by two α-helices, forming a characteristic "sandwich" structure [3] [7] [14]. The core structural elements follow the arrangement αA-βB-βC-βD-αB, where a three-stranded antiparallel beta-sheet is flanked on each side by an alpha helix [3]. Most Src-type SH2 domains contain additional secondary structural elements, including beta strands E, F, and G, creating a total of seven motifs [3]. The N-terminal region of the SH2 domain exhibits high conservation, while the C-terminal region demonstrates considerable variability across family members [3].
Despite significant sequence divergence among family members (with pairwise identity as low as ~15%), all SH2 domains maintain nearly identical three-dimensional folds, suggesting evolutionary optimization for phosphotyrosine recognition [3] [7]. Structural conservation is particularly evident in the phosphotyrosine-binding pocket, where key residues remain invariant across most SH2 domains [14]. This structural preservation amidst sequence diversity enables both conserved binding function and specialized recognition specificities across different Src-type SH2 domains.
Table 1: Core Structural Elements of Src-type SH2 Domains
| Structural Element | Description | Functional Role |
|---|---|---|
| αA helix | N-terminal alpha helix | Structural stability, phosphate coordination |
| βB strand | Central beta strand | Houses invariant arginine for pY binding |
| βC strand | Central beta strand | Structural integrity |
| βD strand | Central beta strand | Contains conserved histidine for phosphate coordination |
| αB helix | C-terminal alpha helix | Structural stability, contributes to specificity pocket |
| EF loop | Connects βE and βF strands | Controls access to ligand specificity pockets |
| BG loop | Connects αB helix and βG strand | Determines binding selectivity |
The Phosphotyrosine Recognition Pocket
The N-terminal region of Src-type SH2 domains contains a deep pocket within the βB strand that specifically recognizes the phosphotyrosine moiety [3] [14]. This pocket harbors an invariant arginine residue at position βB5 (according to strand B, position 5), which forms part of the FLVR motif conserved across almost all SH2 domains [3]. This arginine directly coordinates the phosphate group of phosphotyrosine through a salt bridge interaction [3] [14]. Additional coordination is provided by conserved residues at positions αA2 and βD4, typically a histidine, which further stabilize phosphate binding through hydrogen bonding and electrostatic interactions [14].
The phosphotyrosine-binding pocket provides approximately half of the total binding energy for SH2 domain-ligand interactions, explaining its high conservation across the protein family [14]. Mutational studies confirm the critical importance of these conserved residues, as substitutions in either Arg βB5 or His βD4 abolish phosphotyrosine-specific binding [14]. The primarily electrostatic nature of these interactions enables rapid association and dissociation kinetics, facilitating dynamic signaling responses in cellular environments.
Specificity Pockets and Ligand Recognition
Beyond the phosphotyrosine pocket, Src-type SH2 domains contain additional binding clefts that determine ligand selectivity by recognizing residues C-terminal to the phosphotyrosine [14]. These specificity pockets display considerable structural diversity across different SH2 domains, enabling recognition of distinct peptide motifs [3] [10]. The binding surface is divided into two primary recognition clefts separated by the core β-sheet: the first cleft binds the phosphotyrosine moiety, while the second, more variable cleft engages residues at the +1 to +5 positions C-terminal to the phosphotyrosine [14].
The structural diversity of specificity pockets arises from variations in loop regions between secondary structural elements, particularly the EF loop (joining βE and βF strands) and the BG loop (joining αB helix and βG strand) [7]. These loops control accessibility to the ligand specificity pockets and directly contact peptide side chains, thereby dictating binding preferences for specific amino acid residues at positions C-terminal to the phosphotyrosine [7]. This architectural arrangement enables Src-type SH2 domains to recognize specific sequence motifs, such as the pYEEI motif preferentially bound by c-Src SH2 domains [16] [14].
Figure 1: Src-type SH2 domain recognition mechanism. The domain features two key binding pockets: a conserved phosphotyrosine pocket and a variable specificity pocket that recognizes C-terminal residues.
Comparative Analysis: Src-type versus STAT-type SH2 Domains
Structural and functional distinctions between Src-type and STAT-type SH2 domains reflect their divergent biological roles in cellular signaling. While Src-type domains serve as versatile recognition modules in multidomain signaling proteins, STAT-type domains specialize in facilitating transcription factor dimerization and nuclear transport [7].
Table 2: Structural and Functional Comparison of SH2 Domain Types
| Characteristic | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Overall Structure | Complete αA-βB-βC-βD-αB fold with additional βE, βF, βG strands | Lack βE and βF strands; αB helix split into two helices |
| Specificity Pockets | Versatile pockets with diverse selectivity profiles | Adapted for reciprocal phosphotyrosine exchange in dimerization |
| Biological Function | Modular recognition in signaling proteins | Transcription factor dimerization for nuclear transport |
| Evolutionary Origin | Metazoan signaling adaptation | Predates animal multicellularity (observed in Dictyostelium) |
| Loop Characteristics | Longer loops in enzymatic proteins; variable lengths | Shorter loops optimized for reciprocal binding |
| Representative Proteins | SRC, ABL, PLCγ1, PIK3R2 | STAT1, STAT3, STAT5 |
STAT-type SH2 domains lack the βE and βF strands present in Src-type domains and feature a split αB helix, structural adaptations that facilitate their specialized role in mediating transcription factor dimerization [7]. This structural simplification likely represents an evolutionary adaptation for reciprocal phosphotyrosine exchange between STAT monomers, a critical step in JAK-STAT signaling pathway activation. The observation that STAT-type SH2 domains predate animal multicellularity, evidenced by their presence in Dictyostelium for transcriptional regulation, suggests an ancestral SH2 domain function that was subsequently elaborated in metazoan Src-type domains [7].
In contrast, Src-type SH2 domains exhibit more complex loop structures and versatile specificity pockets, reflecting their adaptation for diverse signaling contexts. Enzymatic SH2 domain-containing proteins tend to feature longer loops compared to non-enzymatic proteins, potentially accommodating more complex regulatory interactions [7]. This structural versatility enables Src-type domains to participate in the formation of heterogeneous signaling complexes with precise specificity determinants.
Ligand Recognition Mechanisms and Specificity Determinants
Affinity and Kinetic Parameters
SH2 domain interactions with phosphotyrosine ligands are characterized by moderate binding affinities typically ranging from 0.1-10 μM, balancing specificity with reversibility to permit dynamic signaling responses [7] [17]. This affinity range enables sensitive response to phosphorylation status while allowing timely complex disassembly upon signal termination. Quantitative studies using purified recombinant SH2 domains have demonstrated nanomolar affinities for specific physiological ligands, such as the interaction between SH2 domains from rasGAP and p85 with the tyrosine-phosphorylated epidermal growth factor receptor [17].
The moderate affinity of SH2 domain interactions facilitates competition between different signaling proteins for limited phosphorylated sites, creating regulatory networks capable of integrating multiple inputs [17]. This competitive binding paradigm allows contextual signal processing based on expression levels, subcellular localization, and post-translational modifications of SH2 domain-containing proteins.
Contextual Sequence Recognition
Src-type SH2 domains achieve ligand specificity through integration of both permissive residues that enhance binding and non-permissive residues that oppose binding in positions surrounding the phosphotyrosine [10]. This contextual recognition capability allows SH2 domains to distinguish subtle sequence variations that are not captured by simple binding motifs [10]. The recognition mechanism involves complex integration of various permissive and non-permissive factors in a context-dependent manner, substantially increasing the information content accessible from peptide ligands [10].
Structural analyses reveal that neighboring positions within peptide ligands influence one another, making local sequence context a critical determinant of binding specificity [10]. This contextual dependence explains why prediction algorithms based solely on optimal binding motifs perform poorly when predicting interactions with physiological peptide sequences, which frequently deviate from ideal consensus motifs [16] [10]. The sophisticated recognition capacity of SH2 domains enables discrimination between highly similar peptide sequences, ensuring fidelity in signaling network activation.
Non-Canonical Binding Functions
Beyond phosphopeptide recognition, Src-type SH2 domains engage in non-canonical interactions that expand their functional repertoire. Approximately 75% of SH2 domains interact with membrane lipids, particularly phosphoinositides such as PIP2 and PIP3 [3] [7]. These interactions often involve cationic regions near the phosphotyrosine-binding pocket, typically flanked by aromatic or hydrophobic amino acid side chains [3]. Lipid binding modulates SH2 domain signaling by facilitating membrane recruitment or altering conformational dynamics.
Table 3: Lipid Interactions of Selected Src-type SH2 Domains
| Protein Name | Lipid Moieties | Functional Role of Lipid Association |
|---|---|---|
| SYK | PIP3 | PIP3-dependent membrane binding required for SYK scaffolding function |
| ZAP70 | PIP3 | Facilitates and sustains ZAP70 interactions with TCR-ζ chain |
| LCK | PIP2, PIP3 | Modulates LCK interaction with binding partners in TCR signaling |
| ABL | PIP2 | Membrane recruitment and modulation of Abl activity |
| VAV2 | PIP2, PIP3 | Modulates VAV2 interaction with membrane receptors (e.g., EphA2) |
Additionally, Src-type SH2 domains participate in liquid-liquid phase separation (LLPS) processes, where multivalent interactions drive formation of membrane-free intracellular condensates [3]. For example, interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation that enhances T-cell receptor signaling [3]. In podocyte kidney cells, phase separation increases the ability of adapter NCK to promote N-WASP–Arp2/3–mediated actin polymerization by extending membrane dwell times of actin regulatory complexes [3].
Experimental Methods for Analyzing SH2 Domain Interactions
Fluorescence Polarization Assays
Fluorescence polarization (FP) provides a robust solution-phase method for quantitatively measuring SH2 domain-phosphopeptide interactions with sensitivity for detecting low-affinity binding events [16]. This approach involves titrating purified SH2 domains against a fixed concentration of fluorescently labeled phosphopeptide and measuring changes in polarization values as the complex forms [16]. FP assays enable determination of dissociation constants (Kd) through nonlinear regression analysis of binding isotherms, providing quantitative interaction data under physiological solution conditions [16].
The technical protocol involves expressing SH2 domains as GST fusion proteins in E. coli, purifying them using glutathione-Sepharose chromatography, and dialyzing to remove glutathione [16]. Synthetic phosphopeptides corresponding to physiological tyrosine phosphorylation sites are labeled with fluorescent dyes such as fluorescein. Measurements are performed in buffer systems containing appropriate salts and detergents (e.g., 50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100) to maintain protein stability and prevent nonspecific interactions [16]. This method successfully identified over 1,000 novel peptide-protein interactions when applied to 93 human SH2 domains against phosphopeptides from receptor tyrosine kinases and signaling adapters [16].
SPOT Peptide Array Analysis
SPOT peptide array synthesis provides a complementary approach for semiquantitative analysis of SH2 domain binding specificities across large numbers of peptide sequences [10]. This technique involves synthesizing peptides directly on cellulose membranes using automated SPOT synthesis, with each peptide occupying a discrete spatial location [10]. Membranes are blocked with non-fat milk, incubated with purified SH2 domains, washed, and detected using anti-GST or domain-specific antibodies with chemiluminescent or colorimetric substrates [10].
SPOT arrays enable medium-throughput specificity profiling by testing binding against 192 or more physiological peptides in parallel, generating comprehensive interaction maps for SH2 domain families [10]. The method successfully identified contextual sequence preferences and non-permissive residues that oppose binding, revealing sophisticated recognition capabilities beyond simple motif recognition [10]. While less quantitative than FP assays, SPOT arrays provide valuable insights into binding selectivity and have been used to develop improved prediction algorithms for SH2 domain interactions [16] [10].
Figure 2: Experimental methodologies for SH2 domain interaction analysis. Fluorescence polarization provides quantitative binding constants, while SPOT arrays enable medium-throughput specificity profiling.
Structural Biology Approaches
X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy have provided foundational insights into Src-type SH2 domain architecture and ligand recognition mechanisms. To date, structures of 70 SH2 domains have been experimentally determined with varying resolution [3]. Crystallographic analyses of SH2 domains complexed with phosphopeptide ligands reveal the molecular details of phosphotyrosine coordination and specificity pocket interactions [14].
Emerging computational approaches complement experimental structural biology. Molecular dynamics simulations elucidate conformational flexibility and allosteric regulation mechanisms, as demonstrated in studies of SHP2 phosphatase activation [11]. Specialized databases like SH2db provide curated structural information, phylogenetic relationships, and ready-to-use structural files for all human SH2 domains, facilitating comparative analyses and structural modeling [18]. These resources employ generic numbering systems that enable residue-to-residue comparisons across different SH2 domains, identifying key functional positions despite sequence variation [18].
Research Reagent Solutions
Table 4: Essential Research Reagents for SH2 Domain Studies
| Reagent / Resource | Specifications | Research Application |
|---|---|---|
| SH2 Domain Constructs | GST-tagged human SH2 domains (93 of 120 human SH2 domains available) | Protein expression and purification for binding assays |
| Phosphopeptide Libraries | 11-mer peptides with central phosphotyrosine; 192+ physiological sequences | Specificity profiling using FP or SPOT assays |
| Expression System | E. coli BL21 with pGEX-2TK vector | High-yield protein production for structural and biophysical studies |
| Detection Antibodies | Anti-GST, anti-phosphotyrosine (4G10, pY20) | Western blotting and array detection |
| Structural Database | SH2db database with PDB and AlphaFold models | Structural comparisons and modeling |
| Fluorescent Probes | Fluorescein-labeled phosphopeptides | Fluorescence polarization binding assays |
Therapeutic Targeting of Src-type SH2 Domains
The critical role of Src-type SH2 domains in signaling pathways dysregulated in disease states, particularly cancer and immune disorders, makes them attractive therapeutic targets. Multiple strategies have emerged for inhibiting SH2 domain function, including small molecules that target phosphotyrosine pockets, allosteric inhibitors, and compounds that disrupt protein phase separation [3].
Conventional approaches have focused on developing phosphotyrosine mimetics that compete with natural ligands for binding to the conserved pY pocket. However, recent strategies leverage more sophisticated mechanisms, including targeting lipid-binding sites adjacent to the pY pocket [3]. For example, nonlipidic inhibitors of Syk kinase have been developed that specifically target lipid-protein interactions, potentially yielding potent, selective, and resistance-resistant inhibitors for various SH2 domain-containing kinases [3].
Notably, allosteric regulation represents a promising frontier in SH2 domain pharmacology. Studies of SHP2 phosphatase reveal complex autoinhibitory mechanisms where the N-SH2 domain blocks the catalytic site in the basal state, with activation involving conformational rearrangement upon engagement of bisphosphorylated ligands [11]. Pathogenic mutations such as the E76K variant in SHP2 disrupt autoinhibition, leading to constitutive activation and disease pathogenesis [11]. Understanding these regulatory mechanisms enables development of allosteric inhibitors that stabilize inactive conformations, providing enhanced specificity compared to active-site directed compounds.
Src-type SH2 domains represent paradigmatic modular interaction domains that establish specificity in phosphotyrosine signaling networks through their versatile specificity pockets and conserved structural framework. Their ability to integrate both permissive and non-permissive sequence determinants enables precise recognition of physiological ligands within complex cellular environments. Ongoing structural and biophysical studies continue to reveal unexpected complexities in SH2 domain function, including roles in membrane lipid binding, liquid-liquid phase separation, and allosteric regulation. These emerging insights, coupled with advanced targeting strategies, position Src-type SH2 domains as promising therapeutic targets for diverse disease pathologies, particularly in oncology and immunology. Future research will undoubtedly continue to elucidate the sophisticated mechanisms through which these domains orchestrate cellular signaling and enable therapeutic intervention.
The Src Homology 2 (SH2) domain, comprising approximately 100 amino acids, serves as a crucial modular domain in intracellular signal transduction by specifically recognizing phosphotyrosine (pTyr) motifs [3] [19]. These domains are found in approximately 110-120 human proteins, including enzymes, adaptors, and transcription factors, where they facilitate the assembly of signaling complexes in response to tyrosine phosphorylation [3] [19]. All SH2 domains share a conserved structural fold featuring a central antiparallel β-sheet flanked by two α-helices, forming a characteristic "αβββα" motif [3] [6]. Despite this conserved architecture, SH2 domains exhibit remarkable diversity in ligand specificity, primarily determined by variations in two critical regions: the phosphotyrosine (pTyr)-binding pocket and the specificity loops [20] [21].
This guide focuses on the key structural determinants that differentiate two major SH2 domain classes: the Src-type (represented by Src, Fyn, and other cytoplasmic signaling proteins) and the STAT-type (found in Signal Transducers and Activators of Transcription proteins). Understanding these differences is paramount for drug development professionals targeting specific SH2 domain families, particularly given that the STAT SH2 domain represents a hotspot for disease-associated mutations in conditions such as cancers and immunodeficiencies [6]. The structural variations between these classes influence their binding preferences, regulatory mechanisms, and potential as therapeutic targets.
The Canonical SH2 Fold
The SH2 domain core structure consists of a central three-stranded β-sheet (strands βB, βC, βD) sandwiched between two α-helices (αA and αB) [3] [2] [6]. This scaffold creates two primary binding surfaces: a highly conserved pTyr-binding pocket and a more variable specificity pocket that recognizes residues C-terminal to the phosphotyrosine. The binding interaction with phosphopeptides occurs in an extended conformation across the β-sheet, often described as a "two-pronged plug" model where the pTyr residue anchors into its dedicated pocket while C-terminal residues engage the specificity-determining regions [2].
Classification of SH2 Domains
SH2 domains are broadly classified based on structural and phylogenetic characteristics. Structurally, they are divided into:
- Src-type SH2 domains: Characterized by an additional β-sheet (βE or βE-βF motif) at the C-terminus [5] [6].
- STAT-type SH2 domains: Feature an α-helix (αB') instead of the C-terminal β-sheets found in Src-type domains [5] [6].
Phylogenetic analysis further categorizes SH2 domain-containing proteins into 38 sub-families, while functional studies have classified them based on the critical fifth residue in the βD strand, which significantly influences phosphopeptide selectivity [6] [22]. These classifications reflect evolutionary adaptations that have tuned different SH2 domains for specific signaling contexts while maintaining the core pTyr-binding function.
Table 1: Fundamental Classification of SH2 Domains
| Classification Basis | Major Categories | Defining Characteristics | Representative Proteins |
|---|---|---|---|
| Structural Features | Src-type | C-terminal β-sheet (βE/βF strands) | Src, Fyn, LCK, GRB2 |
| STAT-type | C-terminal α-helix (αB') | STAT1, STAT3, STAT5 | |
| Functional Groups | Group IA/IB/IIA/IIB | Prefer hydrophobic residue at pY+3 | Src, FYN, ABL1, VAV, PI3K-p85α |
| Group IC | Prefer Asn at pY+2 | GRB2, GADS, GRB7, GRB14 | |
| Group IIC | Prefer hydrophobic residue at pY+4 | BRDG1, BKS, CBL |
The Phosphotyrosine (pTyr)-Binding Pocket
Conserved Features Across SH2 Domains
The pTyr-binding pocket is a deeply buried cavity that provides the fundamental binding energy for SH2-phosphopeptide interactions. This pocket is formed by residues from the αA helix, βB strand, and the BC loop (connecting βB and βC strands) [2] [6]. Several conserved molecular features define this pocket across most SH2 domains:
- FLVR Arginine (Arg βB5): This strictly conserved residue at the fifth position of the βB strand serves as the structural floor of the pTyr pocket, forming a critical salt bridge with the phosphate moiety of pTyr [2] [23]. Mutational studies demonstrate that Arg βB5 contributes approximately 50% of the total binding free energy, with its mutation causing a 1,000-fold reduction in binding affinity [23].
- Complementary Basic Residues: Additional basic residues at positions αA2 and/or βD6 form a "clamp" around the phenolic ring of pTyr and provide additional phosphate coordination [23] [6]. The presence of basic residues at these positions defines two major pTyr-binding modes: Src-like (basic residue at αA2) and SAP-like (basic residue at βD6) [2].
- Hydrogen Bond Donors: Residues such as Ser βB7 and Thr BC2 provide additional hydrogen bonding to the phosphate group, stabilizing the pTyr interaction [23].
Comparative Analysis: Src-type vs. STAT-type pTyr Pockets
While the fundamental architecture of the pTyr pocket is conserved, important distinctions exist between Src-type and STAT-type SH2 domains:
Table 2: Comparison of pTyr-Binding Pockets in Src-type vs. STAT-type SH2 Domains
| Structural Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Conservation | High conservation of FLVR motif | High conservation of FLVR motif |
| Arg βB5 Role | Contributes ~50% of binding free energy; essential for pTyr specificity over pSer/pThr | Similarly critical for pTyr binding and STAT dimerization |
| Additional Basic Residues | Typically features Arg αA2 as part of pTyr clamp | Conserved basic residues but may exhibit different spatial arrangements |
| Unique Characteristics | Sometimes contains unique residues (e.g., Cys βC3 in Src) that modulate affinity | Greater structural flexibility with pTyr pocket accessibility varying dramatically even on sub-microsecond timescales |
| Disease Associations | Mutations can disrupt kinase regulation and signaling | Hotspot for mutations in cancer and immunodeficiencies (e.g., STAT3 R609G corresponding to βB5 position) |
The pTyr pocket in STAT-type SH2 domains exhibits remarkable conformational flexibility, with the accessible volume varying significantly even on sub-microsecond timescales [6]. This dynamic behavior presents both challenges and opportunities for drug discovery efforts targeting STAT SH2 domains.
Specificity Loops and Their Structural Determinants
The Role of Loops in Defining SH2 Specificity
While the pTyr pocket provides the fundamental binding energy, the specificity of different SH2 domains for distinct peptide motifs is primarily determined by surface loops that control access to subsidiary binding pockets. These loops shape the binding surface and determine which residues C-terminal to the pTyr can be accommodated [20] [21]. The key loops involved in specificity determination include:
- EF Loop: Connects βE and βF strands; plays a critical role in controlling access to the pY+3 binding pocket [20].
- BG Loop: Connects βG strand and αB helix; works in concert with the EF loop to form the hydrophobic specificity pocket [20].
- BC Loop: Part of the pTyr-binding pocket but can influence specificity through its positioning.
- CD and BC* Loops: Contribute to the pY+3 pocket formation in STAT-type SH2 domains [6].
Mechanism of Loop-Mediated Specificity
The loops dictate specificity through several mechanisms. First, they can physically block certain binding subsites - for instance, in Grb2 SH2 domain (group IC), a bulky tryptophan in the EF loop occupies the P+3 binding pocket, forcing the peptide to adopt a β-turn conformation and shifting specificity toward Asn at P+2 [20]. Second, loops define the shape and chemical environment of binding pockets - hydrophobic residues in these loops create cavities that preferentially accommodate hydrophobic amino acids at specific positions [20]. Third, in STAT-type SH2 domains, the BC* loop (connecting αB and αC helices) participates in both phosphopeptide binding and STAT dimerization, creating a dual functional role not typically observed in Src-type domains [6].
Engineering SH2 Domain Specificity Through Loops
The critical role of loops in determining specificity has been demonstrated through protein engineering approaches. Studies show that combinatorial mutations in just the EF and BG loops of the Fyn SH2 domain can encode a wide spectrum of specificities, including all three major specificity classes (pY+2, pY+3, and pY+4) [21]. This loop flexibility suggests a plausible evolutionary mechanism whereby SH2 domains acquired diverse specificities through loop variation with minimal disturbance to the conserved core fold [21].
Table 3: Specificity Determinants in Major SH2 Domain Classes
| SH2 Class | Key Specificity Loops | Preferred Motif | Structural Basis of Specificity |
|---|---|---|---|
| Src-type (Group IA/IB) | EF, BG loops | pYEEI (hydrophobic at pY+3) | Deep hydrophobic pocket formed by EF and BG loops accommodates Ile/Val at pY+3 |
| GRB2-type (Group IC) | EF loop (Trp residue) | pYxN (Asn at pY+2) | Bulky Trp in EF loop blocks pY+3 pocket, forces β-turn conformation enabling Asn recognition at pY+2 |
| BRDG1-type (Group IIC) | EF, BG loops | pYxxxψ (hydrophobic at pY+4) | Open binding pocket with unobstructed access to pY+4 position; "pentagon basket" of hydrophobic residues |
| STAT-type | CD, BC* loops | pYxxQ (Gln at pY+3 in STAT1) | Unique pY+3 pocket architecture; BC* loop participates in both peptide binding and STAT dimerization |
Experimental Approaches for Characterizing pTyr Pockets and Specificity Loops
Methodologies for Binding Affinity and Specificity Assessment
Several well-established experimental techniques enable quantitative analysis of SH2 domain binding properties:
Isothermal Titration Calorimetry (ITC) ITC provides direct measurement of binding thermodynamics by quantifying heat changes upon ligand binding [23]. This method was instrumental in demonstrating that the pTyr residue contributes approximately 50% of the total binding free energy for Src SH2 domain, with Arg βB5 accounting for the majority of this interaction energy [23].
Protocol Overview:
- Purified SH2 domain is placed in the sample cell.
- Phosphopeptide solution is titrated into the cell in precise increments.
- Measured heat changes are used to calculate binding constants (Kd), stoichiometry (n), enthalpy (ΔH), and entropy (ΔS).
- Control experiments with dephosphorylated peptides and phosphoserine-containing peptides establish phosphorylation-dependent binding.
Phage Display and Peptide Library Screening This approach identifies specificity determinants by screening SH2 domains against vast libraries of potential phosphopeptide ligands [21].
Protocol Overview:
- Create phage-displayed peptide libraries with fixed pTyr and randomized C-terminal residues.
- Incubate library with immobilized SH2 domains.
- Wash away non-binding phage; elute and amplify specifically bound phage.
- Repeat panning cycles to enrich high-affinity ligands.
- Sequence enriched phage to determine consensus binding motif.
Surface Plasmon Resonance (SPR) SPR enables real-time monitoring of binding interactions without labeling requirements, providing kinetic parameters (kon, koff) in addition to affinity measurements.
Structural Determination Methods
X-ray Crystallography This method provides high-resolution structures of SH2 domains in complex with phosphopeptides, revealing atomic-level details of pTyr pocket and loop conformations [20] [23].
Protocol Overview:
- Purify SH2 domain protein to homogeneity.
- Co-crystallize with phosphopeptide ligands.
- Collect X-ray diffraction data and solve structure by molecular replacement.
- Analyze binding interfaces and conformational changes.
NMR Spectroscopy NMR offers solution-state structural information and can capture dynamics and flexibility, particularly valuable for studying conformational changes in specificity loops [22] [6].
Protocol Overview:
- Prepare isotopically labeled (^15N, ^13C) SH2 domain protein.
- Collect multidimensional NMR spectra.
- Assign chemical shifts and calculate solution structure.
- Monitor chemical shift perturbations upon peptide binding.
- Measure dynamics through relaxation experiments.
Visualization of SH2 Domain Architecture and Binding Relationships
SH2 Domain Structural Architecture and Binding
SH2 Domain Binding Specificity Determinants
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 4: Essential Research Tools for SH2 Domain Characterization
| Tool/Reagent | Specifications | Research Application | Key References |
|---|---|---|---|
| Recombinant SH2 Domains | ~100 aa constructs; GST/His-tagged; wild-type and mutant variants | Binding assays, structural studies, specificity profiling | [20] [23] |
| Oriented Peptide Array Libraries (OPAL) | Positional scanning libraries with fixed pTyr and randomized flanking residues | High-throughput specificity profiling; consensus motif identification | [20] [22] |
| Phosphopeptide Libraries | Synthetic pTyr peptides with systematic variation at C-terminal positions | Affinity measurements; specificity determinants; competition assays | [21] [23] |
| ITC Instrumentation | Microcalorimeters with high sensitivity (nanoWatts) | Thermodynamic characterization; binding constants; stoichiometry | [23] |
| NMR Isotope Labeling | ^15N, ^13C-labeled SH2 domains in bacterial expression systems | Solution structure determination; dynamics studies; binding interface mapping | [22] [6] |
| Crystallization Screens | Sparse matrix screens optimized for SH2 domain-peptide complexes | X-ray crystallography for high-resolution structural data | [20] [23] |
| Phage Display Libraries | M13-based libraries with random peptide inserts displayed on pIII protein | Selection of high-affinity ligands; specificity profiling | [21] |
| Phosphospecific Antibodies | Antibodies recognizing specific pTyr motifs | Validation of physiological interactions; cellular signaling studies | [24] |
Implications for Drug Discovery and Therapeutic Targeting
The structural differences between STAT-type and Src-type SH2 domains have significant implications for drug development. STAT SH2 domains, particularly those of STAT3 and STAT5, represent attractive therapeutic targets due to their central role in oncogenic signaling and their identification as mutation hotspots in various cancers [6]. However, the high degree of conservation in the pTyr-binding pocket across all SH2 domains presents challenges for developing selective inhibitors.
Several strategies have emerged to overcome these challenges. First, targeting the unique features of STAT-type specificity pockets, particularly the region containing the αB' helix and the more flexible BC* loop, may enable development of STAT-selective compounds [6]. Second, the observed conformational dynamics of STAT SH2 domains, with the pTyr pocket exhibiting significant volume fluctuations, suggests opportunities for allosteric inhibitors that stabilize inactive conformations [6]. Third, targeting disease-associated mutant forms of STAT SH2 domains that exhibit altered binding properties may provide a pathway to personalized therapeutics [6].
Recent research has also explored non-traditional approaches, including targeting the lipid-binding capabilities of some SH2 domains or developing engineered high-affinity SH2 variants that can act as competitive antagonists of endogenous signaling [3] [19]. As structural characterization of both canonical and atypical SH2 domains continues to advance, new opportunities for therapeutic intervention in SH2-mediated signaling pathways will undoubtedly emerge.
The Src Homology 2 (SH2) domain represents a fundamental protein interaction module that specifically recognizes phosphorylated tyrosine (pTyr) residues, serving as a critical component in eukaryotic signal transduction networks. Comprising approximately 100 amino acids, SH2 domains emerged within metazoan signaling pathways and are involved in protein regulation across multiple pleiotropic cascades [25] [3]. These domains facilitate the assembly of specific protein complexes in response to tyrosine phosphorylation, thereby enabling precise spatiotemporal control of cellular processes including development, homeostasis, immune responses, and transcription [3]. The human proteome encodes roughly 110 SH2 domain-containing proteins classified into diverse functional categories including enzymes, adaptor proteins, docking proteins, and transcription factors [3] [7]. Among these, the STAT-type SH2 domain represents a distinctive structural and functional subclass with unique evolutionary origins and mechanistic properties that have proven essential for metazoan signaling complexity.
This review examines the evolutionary emergence of STAT-type SH2 domains, focusing on their structural specialization, functional divergence from Src-type counterparts, and implications for therapeutic targeting. We trace their phylogenetic origins from primordial precursors through metazoan diversification, highlighting how structural variations underpin specialized functions in transcriptional regulation. Through comprehensive analysis of structural data, evolutionary patterns, and clinical mutations, we elucidate the molecular mechanisms by which STAT-type SH2 domains have shaped complex signaling networks in metazoans.
Evolutionary Origins and Phylogenetic Distribution
Deep Evolutionary Roots of SH2 Domains
The evolutionary history of SH2 domains extends deep into eukaryotic lineage, predating the emergence of metazoans. Genomic analyses reveal that SH2 domains co-evolved with protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) to coordinate cellular and organismal complexity throughout the evolution of the unikont branch of eukaryotes [26]. The most ancient SH2 domain identified to date resides in SPT6, an essential transcription elongation protein conserved from yeast to humans [2]. This ancestral SH2 domain exhibits a near-canonical phospho-binding pocket but recognizes phosphorylated serine and threonine residues in RNA polymerase II rather than phosphotyrosine, representing an evolutionary stepping stone toward pTyr recognition [2]. The tandem SH2 domains in SPT6 pack against one another and recognize extended phosphorylated peptides, illustrating the early structural versatility of this fold.
Comparative genomics of the choanoflagellate Monosiga brevicollis, the closest known unicellular relative of metazoans, has provided unprecedented insights into the premetazoan repertoire of signaling domains. The M. brevicollis genome encodes 78 protein domains previously thought to be exclusive to metazoans, including numerous components involved in cell adhesion and signaling [27]. This finding demonstrates that many critical molecular components required for metazoan multicellularity evolved before the origin of metazoans themselves. The genome of this protist contains a surprisingly elaborate and diverse tyrosine kinase signaling network, more complex than found in any known metazoan, suggesting substantial signaling complexity predated multicellularity [26].
Emergence of the STAT-type SH2 Domain
The STAT-type SH2 domain represents one of the most ancient and fully developed functional domains, serving as an evolutionary template for the continuing evolution of SH2 domain functionality [5]. Phylogenetic analysis using secondary structural alignment rather than primary sequence comparison has enabled the classification of SH2 domains into two major groups: Src-type and STAT-type [5]. This structural approach revealed that the linker domain-conjugated SH2 domain in STAT contains distinctive structural elements, notably the αB' motif, while Src-type SH2 domains contain extra β-strands (βE or βE-βF motif) [5].
Remarkably, genes carrying the STAT-type linker-SH2 domain have been identified in a wide array of vascular and nonvascular plants, indicating that this domain evolved prior to the divergence of plants and animals [5]. The discovery of these genes, designated STAT-type linker-SH2 domain factors (STATL), in Arabidopsis and other plants demonstrates the deep evolutionary provenance of the STAT-type SH2 domain architecture. This evolutionary perspective reveals that STAT-type SH2 domains represent one of the most ancient functional templates for phosphotyrosine signal transduction [5].
Table 1: Evolutionary Distribution of SH2 Domains Across Eukaryotes
| Organismal Group | SH2 Domain Presence | STAT-type SH2 Examples | Key Evolutionary Significance |
|---|---|---|---|
| Yeast (S. cerevisiae) | 2 SH2 domains (in SPT6) | None | Most ancestral SH2 domains; recognize pSer/pThr |
| Plants (A. thaliana) | Present | STATL genes | STAT-type SH2 predates plant-animal divergence |
| Choanoflagellates (M. brevicollis) | Abundant (>100) | Not specified | Elaborate pTyr signaling predates metazoans |
| Dictyostelium | Present | Not specified | Employ SH2 domain/pTyr signaling for transcriptional regulation |
| Metazoans | ~110 in humans | STAT1-6 | Full diversification of STAT-type SH2 functions |
Structural Divergence Between STAT-type and Src-type SH2 Domains
Canonical SH2 Domain Architecture
The fundamental architecture of SH2 domains consists of a central sandwich structure formed by a three-stranded antiparallel beta-sheet flanked on each side by an alpha helix, forming a characteristic αA-βB-βC-βD-αB topology [3] [7]. This conserved fold creates two critical binding sites: a deep basic pocket that binds the phosphotyrosine moiety, and a specificity pocket that recognizes residues C-terminal to the pTyr, particularly the amino acid at the +3 position [2]. This "two-pronged plug" interaction mechanism is largely conserved across most SH2 domains and provides both high specificity toward cognate pY ligands with moderate binding affinity (Kd typically 0.1-10 μM) [7].
The most critical conserved feature of SH2 domains is the FLVR motif (also called "FLVRES"), which contains an invariant arginine at position βB5 that directly coordinates the phosphate moiety of phosphotyrosine [2]. This arginine residue is conserved in all but three of the 120+ human SH2 domains and provides the structural basis for pTyr specificity over pSer/pThr [2]. Other conserved residues that frequently contribute to pTyr coordination include basic residues at positions αA2 and βD6, with the presence of these residues helping to define the Src-like (basic at αA2) and SAP-like (basic at βD6) subclasses of SH2 domains [2].
Distinctive Structural Features of STAT-type SH2 Domains
STAT-type SH2 domains exhibit several distinctive structural characteristics that differentiate them from Src-type SH2 domains and enable their specialized functions in transcription factor activation:
Absence of βE and βF Strands: Unlike Src-type SH2 domains that contain extra β-strands (βE or βE-βF motifs), STAT-type SH2 domains lack these structural elements [7]. This structural simplification represents an ancestral feature that facilitates STAT dimerization.
Split αB Helix: The αB helix in STAT-type SH2 domains is characteristically split into two helices, a structural adaptation that facilitates the reciprocal dimerization critical for STAT-mediated transcriptional regulation [7].
Linker Domain Integration: STAT-type SH2 domains are uniquely conjugated with a linker domain that contains the αB' motif, a feature not found in Src-type SH2 domains [5]. This linker-SH2 domain integration represents the evolutionary origin of the SH2 domain functionality.
Shorter CD-Loops: STAT-type SH2 domains typically have shorter CD-loops compared to enzymatic SH2 domain-containing proteins, reflecting their specialization for dimerization rather than catalytic function [7].
These structural differences represent evolutionary adaptations that optimize STAT-type SH2 domains for their primary function: mediating reciprocal dimerization between STAT monomers following phosphorylation, thereby enabling nuclear translocation and DNA binding [25] [7].
Table 2: Structural Comparison Between Src-type and STAT-type SH2 Domains
| Structural Feature | Src-type SH2 Domains | STAT-type SH2 Domains | Functional Implications |
|---|---|---|---|
| Core Structure | αA-βB-βC-βD-αB with additional elements | αA-βB-βC-βD-αB (split) | Conserved pTyr binding function |
| Additional β-strands | βE or βE-βF motifs present | Absent | Adaptation for dimerization in STATs |
| αB Helix | Single continuous helix | Split into two helices | Facilitates reciprocal STAT dimerization |
| Linker Domain | Not conjugated | Conjugated with αB' motif | Evolutionary ancient feature |
| CD-loop Length | Longer in enzymatic proteins | Shorter | Specialization for non-enzymatic function |
| Primary Function | Substrate recruitment, enzyme regulation | STAT dimerization, nuclear accumulation | Distinct signaling roles |
Structural Basis for Dimerization and Nuclear Accumulation
In STAT proteins, SH2 domain interactions are critical for molecular activation and nuclear accumulation of phosphorylated STAT dimers to drive transcription [25]. The structural adaptations of STAT-type SH2 domains enable reciprocal interactions where the SH2 domain of one STAT monomer binds the phosphorylated tyrosine residue of another, forming stable dimers that translocate to the nucleus [25] [7]. This dimerization mechanism represents a specialized function that diverges from the typical role of Src-type SH2 domains in recruiting signaling proteins to phosphorylated receptors or adaptors.
The structural biology of STAT-type SH2 domains reveals how their unique features facilitate this dimerization function. The absence of the βE and βF strands and the split αB helix create a structural environment optimized for stable dimer interface formation rather than the transient interactions characteristic of many Src-type SH2 domain complexes [7]. This structural specialization reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity, as observed in Dictyostelium, which employs SH2 domain/phosphotyrosine signaling for transcriptional regulation [7].
Functional Implications and Clinical Relevance
Role in Disease and Mutational Hotspots
Sequencing analyses of patient samples have revealed the SH2 domain as a hotspot in the mutational landscape of STAT proteins, particularly STAT3 and STAT5 [25]. The functional impact for the vast majority of these mutations remains poorly characterized, despite their clinical significance. Mutations within the STAT SH2 domain can result in either gain-of-function or loss-of-function phenotypes, sometimes occurring at identical residues, underscoring the delicate evolutionary balance of wild-type STAT structural motifs in maintaining precise levels of cellular activity [25].
Disease-associated mutations in STAT SH2 domains have been implicated in numerous pathological conditions, including T-cell large granular lymphocytic leukemia, T-cell prolymphocytic leukemia, autosomal-dominant hyper IgE syndrome, growth hormone insensitivity syndrome, and inflammatory hepatocellular adenomas [25]. Understanding the molecular and biophysical impact of these mutations has revealed convergent mechanisms of action for mutations localized within the STAT SH2 domain, providing insights for targeted therapeutic interventions [25].
Emerging Therapeutic Targeting Strategies
The structural and functional specialization of STAT-type SH2 domains presents unique opportunities for therapeutic targeting. Several innovative approaches are currently being explored:
Small Molecule Inhibitors: Development of compounds that directly target the SH2 domain to disrupt pathological STAT dimerization or protein interactions. These inhibitors aim to achieve high specificity by exploiting unique structural features of STAT-type SH2 domains [3] [28].
PROTAC Applications: SH2 domains are being exploited in the development of Proteolysis-Targeting Chimeras (PROTACs) for targeted protein degradation. This approach uses the SH2 domain's binding specificity to recruit E3 ubiquitin ligases to target proteins, facilitating their degradation [28].
Lipid-Binding Disruption: Emerging evidence indicates that nearly 75% of SH2 domains interact with membrane lipids, particularly PIP2 and PIP3, and that disease-causing mutations often localize within lipid-binding pockets [3] [7]. Targeting these lipid-protein interactions represents a promising avenue for developing selective inhibitors.
Phase Separation Modulation: Recent research has linked SH2 domain-containing proteins to the formation of intracellular condensates via liquid-liquid phase separation (LLPS) [3] [7]. Small molecules that modulate these phase separation processes offer potential therapeutic strategies for disrupting pathological signaling hubs.
Table 3: Therapeutic Targeting Approaches for SH2 Domain-Containing Proteins
| Therapeutic Approach | Molecular Target | Development Stage | Key Challenges |
|---|---|---|---|
| Small Molecule Inhibitors | pTyr-binding pocket | Preclinical to clinical | Achieving specificity among similar SH2 domains |
| PROTAC Technology | SH2 domain surface | Early development | Identifying suitable E3 ligase recruits |
| Lipid-Binding Disruption | Lipid-binding pocket | Experimental | Membrane localization requirements |
| Phase Separation Modulation | Multivalent interactions | Theoretical | Specificity for pathological condensates |
Experimental Approaches and Research Methodologies
Structural Characterization Techniques
The elucidation of STAT-type SH2 domain structure and function has relied on multiple complementary experimental approaches:
X-ray Crystallography: This technique has provided high-resolution structures of SH2 domains in complex with phosphopeptide ligands, revealing the molecular details of the "two-pronged plug" binding mechanism [2]. To date, the structures of approximately 70 SH2 domains have been experimentally solved with varying degrees of resolution [3] [7].
Secondary Structural Alignment: Conventional primary sequence alignment often fails to identify distantly related SH2 domains due to sequence divergence. Two-dimensional structural alignment based on predicted secondary structure has proven essential for identifying STAT-type SH2 domains across diverse species [5].
Mutational Analysis: Systematic point mutations, particularly of the conserved FLVR arginine residue, have been used to interrogate SH2 domain function and validate structural predictions [2]. Mutagenesis studies have confirmed that the βB5 arginine contributes approximately half of the free energy of binding, with mutation resulting in up to 1000-fold reduction in binding affinity [2].
Functional Assays and Binding Studies
Competitive Binding Assays: Eurofins Discovery has developed SH2 domain binding assays that utilize competitive binding principles similar to KINOMEscan technology to support drug discovery initiatives targeting SH2 domain-mediated protein-protein interactions [28].
Phosphopeptide Array Screening: Proteome-derived peptide arrays have been employed to map SH2 domain binding specificity and identify novel interaction partners [26].
Biophysical Characterization: Isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) have been used to quantify binding affinities and thermodynamic parameters of SH2 domain-phosphopeptide interactions, typically revealing Kd values in the 0.1-10 μM range [7].
Visualizing STAT-type SH2 Domain Evolution and Structure
The following diagrams illustrate key concepts in STAT-type SH2 domain evolution, structure, and function.
Evolutionary Origins of STAT-type SH2 Domains
Structural Comparison: STAT-type vs Src-type SH2 Domains
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 4: Key Research Reagents and Experimental Resources for SH2 Domain Studies
| Reagent/Resource | Type | Primary Application | Key Features |
|---|---|---|---|
| SH2 Domain Binding Assays (Eurofins) | Biochemical assay | Drug discovery screening | Competitive binding format; adaptable for high-throughput screening |
| Phosphopeptide Arrays | Peptide library | Binding specificity profiling | Proteome-derived; enables mapping of interaction networks |
| Recombinant SH2 Domains | Protein reagents | Structural & biophysical studies | Often expressed as GST-fusions for purification |
| FLVR Mutants (R→K) | Mutagenesis tools | Functional validation | Disrupts pTyr binding; confirms SH2-dependent effects |
| STAT Reporter Cell Lines | Cellular assays | Functional signaling analysis | Luciferase-based readout of STAT pathway activation |
| SH2 Domain Crystallization Kits | Structural biology | Protein crystallization | Optimized for obtaining SH2 domain crystals |
The emergence of STAT-type SH2 domains represents a pivotal event in the evolution of metazoan signaling complexity. Their deep phylogenetic roots, extending beyond the metazoan lineage into plants and protists, demonstrate the ancient provenance of this specialized protein interaction module. The unique structural features of STAT-type SH2 domains—including the absence of βE and βF strands, the split αB helix, and integration with linker domains—represent evolutionary adaptations that optimize them for their specialized role in transcription factor dimerization and nuclear signaling.
The clinical significance of STAT-type SH2 domains is underscored by their identification as mutational hotspots in diverse pathologies, from immunodeficiencies to hematological malignancies. The delicate structural balance within these domains means that mutations at identical residues can produce either activating or deactivating phenotypes, highlighting the precision of evolutionary optimization. Future research directions include elucidating the full spectrum of disease-associated mutations, developing increasingly selective therapeutic agents that exploit structural vulnerabilities, and understanding the role of SH2 domains in emerging paradigms such as liquid-liquid phase separation.
As structural biology techniques continue to advance, particularly in cryo-electron microscopy and computational prediction, our understanding of STAT-type SH2 domain dynamics and their interactions within complex signaling networks will deepen. These insights will not only illuminate fundamental mechanisms of metazoan signaling evolution but also provide foundation for novel therapeutic strategies targeting the pathological consequences of dysregulated SH2 domain function.
From Structure to Function: Techniques for Probing SH2 Domain Mechanisms and Interactions
The Src Homology 2 (SH2) domain, identified in 1986, serves as a fundamental modular unit that specifically recognizes phosphotyrosine (pTyr) motifs, enabling the assembly of complex signaling networks in eukaryotic cells [2] [29]. These approximately 100-amino-acid domains are crucial for propagating signals from protein tyrosine kinases (PTKs) by mediating specific, phosphorylation-dependent protein-protein interactions [3] [12]. The human proteome encodes approximately 110 proteins containing SH2 domains, which are broadly classified into enzymes, adaptors, regulators, docking proteins, and transcription factors [3] [7]. A key structural and functional dichotomy exists within the SH2 family, primarily between Src-type and STAT-type domains, which have evolved distinct structural features to support their specific biological roles [5] [2]. This technical guide examines how modern structural biology tools, particularly X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy, have been deployed to elucidate the architecture, binding mechanics, and dynamic properties of these critical signaling domains, providing a foundation for targeted therapeutic intervention.
Structural Classification of SH2 Domains: Src-type vs. STAT-type
Canonical Architecture and the FLVR Motif
All SH2 domains share a conserved core fold despite significant sequence divergence in some family members (~15% pairwise identity) [3] [7]. The fundamental structure consists of a central three-stranded antiparallel beta-sheet (βB-βC-βD) flanked by two alpha helices (αA and αB), forming a characteristic "βαββββαβ" sandwich [3] [29]. The N-terminal region is highly conserved and contains a deep pocket that binds the phosphate moiety of phosphotyrosine. This pocket invariably contains a critical arginine residue at position βB5 that forms part of the FLVR motif (or FLVRES motif), which directly coordinates the phosphate group through a salt bridge interaction and provides specificity for pTyr over phosphoserine or phosphothreonine [3] [2]. The C-terminal region is more variable and contains the specificity pocket that recognizes residues C-terminal to the phosphotyrosine, typically the +3 position, creating a "two-pronged plug" binding mechanism [2] [29].
Distinguishing Src-type and STAT-type SH2 Domains
Recent structural analyses have revealed that SH2 domains can be divided into two major subgroups based on their secondary structure elements and functional adaptations [5].
Table 1: Structural and Functional Comparison of Src-type vs. STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | Conserved βαββββαβ fold with extra β-strands (βE, βF) | Lacks βE and βF strands; αB helix split into two helices (αB and αB') |
| Specificity Pocket | Hydrophobic pocket for residue at pY+3 position | Adapted for dimerization and nuclear transport |
| Conserved Motifs | FLVR motif with Arg βB5; basic residue at αA2 (Src-like) or βD6 (SAP-like) | FLVR motif with Arg βB5; specialized for DNA binding and transcriptional regulation |
| Primary Function | Signal transduction through reversible protein-protein interactions | Transcription factor activation through dimerization and nuclear localization |
| Representative Proteins | Src, Fyn, Lck, Grb2, PLCγ1 [3] | STAT1, STAT2, STAT3, STAT4, STAT5, STAT6 [3] [5] |
The STAT-type SH2 domains lack the βE and βF strands present in most Src-type domains and feature a split αB helix [7] [5]. This structural adaptation facilitates SH2 domain-mediated dimerization, a critical step in STAT protein activation and nuclear translocation for transcriptional regulation [7]. Evolutionary studies suggest this architecture represents an ancestral form, predating animal multicellularity and observed in organisms like Dictyostelium that employ SH2 domain/phosphotyrosine signaling for transcriptional control [7] [5].
X-ray Crystallography of SH2 Domain Complexes
Technical Approaches and Historical Developments
X-ray crystallography has provided the majority of high-resolution structural data for SH2 domains, with over 70 unique SH2 domain structures experimentally determined to date [3] [7]. The first cohort of SH2 domain structures determined in 1992-1993 revealed the conserved fold and established the fundamental "two-pronged plug" binding mechanism for phosphopeptide recognition [2]. Crystallography excels at visualizing precise atomic interactions within the binding pocket and has been instrumental in mapping the molecular determinants of binding specificity.
Recent methodological advances have addressed specific challenges in SH2 domain crystallography:
- Ligand Soaking: Complex formation through soaking pre-formed SH2 domain crystals with phosphopeptide solutions
- Co-crystallization: Growing crystals of pre-formed SH2 domain-ligand complexes to capture native binding conformations
- Engineering Strategies: Utilizing surface entropy reduction mutations to improve crystallization propensity without disrupting functional regions
Key Structural Insights from Crystallographic Studies
Crystallographic analyses have revealed several fundamental principles of SH2 domain function:
Specificity Determinants: Structures of Src SH2 domain complexed with pYEEI ligand revealed how the domain recognizes both the phosphotyrosine and the +3 isoleucine residue through complementary binding pockets [8] [29]. The ThrEF1 residue in Src SH2 forms part of the hydrophobic pocket that accommodine the Ile(pY+3) side chain [8].
Specificity Switching: A landmark crystallographic study demonstrated that mutating ThrEF1 to tryptophan in the Src SH2 domain physically occludes the pY+3 binding pocket while providing additional surface area to accommodate Asn(pY+2), effectively switching the specificity to resemble that of Grb2 SH2 domain [8]. This structural plasticity demonstrates how novel SH2 domain specificities can evolve through single amino acid substitutions.
Domain Swapping: The crystal structure of the interleukin-2 tyrosine kinase (Itk) SH2 domain revealed a domain-swapped dimer similar to those observed in Grb2 and Nck SH2 domains, where the β-meander region exchanges between monomers [30]. This quaternary arrangement suggests potential regulatory mechanisms beyond canonical phosphopeptide binding.
Table 2: Representative SH2 Domain Crystal Structures and Key Findings
| SH2 Domain | Ligand/Context | Resolution (Å) | PDB Code | Key Structural Insight |
|---|---|---|---|---|
| LCK SH2 | pYEEI peptide | 1.5-2.0 | 1LCJ | Established "two-pronged plug" binding mode [2] |
| Src SH2 (T→W mutant) | pYVNV peptide | 2.1-2.5 | - | Demonstrated specificity switching mechanism [8] |
| Grb2 SH2 | pYVNV peptide | 2.0-2.3 | - | Revealed β-turn conformation for Asn(pY+2) recognition [29] |
| Itk SH2 | Domain-swapped dimer | 2.35 | - | Illustrated alternative oligomerization states [30] |
| SPT6 N-SH2 | pThr-X-Tyr motif | 2.4-2.8 | - | Revealed ancestral stepping-stone to pTyr recognition [2] |
NMR Spectroscopy of SH2 Domain Complexes
Technical Approaches and Methodological Advances
NMR spectroscopy provides complementary insights into SH2 domain structure, dynamics, and binding mechanisms, particularly for capturing conformational heterogeneity and transient states that may be obscured in crystal structures. Recent methodological improvements have significantly enhanced the quality of NMR-derived SH2 domain structures:
Hydrogen Bond Restraints: A recent study of the SH2B1 SH2 domain demonstrated that systematic inclusion of hydrogen bond restraints significantly improves structure quality and accuracy [31]. The protocol involves:
- Initial structure calculation without hydrogen bond restraints
- Analysis of initial ensembles to identify persistent hydrogen bonds
- Iterative refinement with gradually introduced hydrogen bond restraints
- Validation using ANSURR (Analysis of NMR Structures Using RCI and Random Coil Index) to determine optimal stopping point
Backbone Assignment: Sequential assignment of ^15N-^1H and ^13C-^1H resonances using triple-resonance experiments (HNCA, HNCOCA, CBCACONH, HNCACB) Distance Constraints: Collection of through-space correlations using ^15N- and ^13C-edited NOESY experiments Dynamic Information: Analysis of ^15N relaxation parameters (T1, T2, heteronuclear NOE) to characterize ps-ns timescale motions
Key Dynamic Insights from NMR Studies
NMR has revealed several critical aspects of SH2 domain behavior that complement crystallographic data:
Prolyl Isomerization: Solution NMR studies of the Itk SH2 domain identified cis-trans isomerization of the Asn286-Pro287 imide bond, with approximately 65% of molecules adopting the trans conformation and 35% the cis conformation in solution [30]. This dynamic equilibrium was not observable in the crystal structure of the same domain, highlighting NMR's unique capability to detect conformational switching with potential functional significance.
Binding Interface Dynamics: Combined NMR and molecular dynamics simulations of Src SH2 complexed with pYEEI and constrained analogs revealed that despite nearly identical average structures, subtle chemical shift perturbations across the binding interface correlate with enthalpic penalties in constrained ligands [32]. This demonstrates how NMR can detect energetically significant perturbations that escape crystallographic detection.
Entropy-Enthalpy Compensation: Studies of preorganized phosphotyrosine mimics bound to Src SH2 revealed expected entropic gains but unexpected enthalpic penalties, with NMR chemical shifts identifying subtle geometric alterations in hot spot interactions that explain the compensation mechanism [32].
Integrated Structural Workflow for SH2 Domain Analysis
The following experimental workflow represents a comprehensive approach for determining SH2 domain structures and characterizing their interactions, integrating both crystallographic and NMR methodologies:
Diagram 1: Integrated structural biology workflow for SH2 domain analysis, combining crystallographic and NMR approaches.
Research Reagent Solutions for SH2 Domain Studies
Table 3: Essential Research Reagents for SH2 Domain Structural Studies
| Reagent Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Expression Constructs | Mouse Itk SH2 (residues 231-338) [30]; pp60 v-Src SH2 (residues 144-249) [32] | Provides defined SH2 domain boundaries for structural studies | N-terminal GST tags with thrombin cleavage sites facilitate purification |
| Isotope Labeling | ^15N-enriched ammonium chloride; ^13C-glucose; selenomethionine | Enables NMR studies and MAD phasing for crystallography | Modified M9 minimal media for bacterial expression systems |
| Phosphopeptide Ligands | pYEEI (Src SH2 canonical); pYVNV (Grb2 SH2 canonical) [29] [32] | For complex formation and binding studies | Acetylated N-terminus, unblocked C-terminus from commercial suppliers (e.g., SynPep) |
| Pseudopeptide Analogs | cpYEEI (constrained); fpYEEI (flexible control) [32] | Investigating preorganization effects on binding energetics | Trisubstituted cyclopropyl moiety as rigid pY replacement |
| Crystallization Reagents | Commercial sparse matrix screens; HEPES buffer (50 mM, pH 7.4) | Crystal formation and optimization | Requires 150 mM NaCl, 2 mM DTT, 0.02% NaN₃ for SH2 domain stability |
| NMR Buffers | Sodium phosphate (50 mM, pH 7.2); DTT; NaN₃ | Maintain protein stability during data collection | 75 mM NaCl for physiological ionic strength |
Implications for Drug Discovery and Therapeutic Targeting
The structural insights gained from crystallographic and NMR studies of SH2 domains have direct implications for pharmaceutical development, particularly in oncology and immunology. Several targeting strategies have emerged:
Small Molecule Inhibitors: Structure-based drug design has yielded inhibitors targeting pathogenic SH2 interactions, particularly in oncology [3] [29]. The high conservation of the pTyr binding pocket presents challenges for achieving specificity, necessitating detailed structural information to exploit subtle differences in neighboring regions.
Lipid-Binding Pocket Targeting: Recent research indicates that approximately 75% of SH2 domains interact with membrane lipids such as PIP2 and PIP3, with cationic regions near the pTyr-binding pocket serving as lipid interaction sites [3] [7]. Non-lipidic small molecules targeting these interfaces have shown promise, as demonstrated with Syk kinase inhibitors [3].
Phase Separation Modulation: SH2 domain-containing proteins increasingly are recognized as participating in liquid-liquid phase separation (LLPS) through multivalent interactions [3]. In T-cell receptor signaling, interactions among GRB2, Gads, and LAT receptors drive condensate formation that enhances signaling output [3] [7]. Structural insights into these multivalent networks may enable new therapeutic strategies.
The integration of X-ray crystallography and NMR spectroscopy has provided a comprehensive understanding of SH2 domain structure and function, revealing both the conserved framework that defines this protein family and the specialized adaptations that enable diverse signaling roles. The structural dichotomy between Src-type and STAT-type SH2 domains exemplifies how evolutionary pressures have shaped a conserved fold to support distinct biological functions—from membrane-proximal signaling events to nuclear transcriptional regulation.
Future structural studies will likely focus on several emerging areas:
- Multidomain Architectures: Investigating how SH2 domains function within full-length proteins containing multiple modular domains
- Transient Complexes: Applying solution methods to characterize weak, transient interactions that govern signaling network dynamics
- Therapeutic Targeting: Leveraging structural insights to develop allosteric inhibitors and protein-protein interaction disruptors
- Phase Separation Mechanisms: Elucidating the structural basis for SH2 domain involvement in biomolecular condensate formation
As structural biology methodologies continue to advance, particularly with developments in cryo-electron microscopy and integrative modeling approaches, our understanding of SH2 domain biology will further deepen, enabling more sophisticated therapeutic interventions targeting these critical signaling modules.
Src Homology 2 (SH2) domains are approximately 100-amino-acid protein modules that specifically recognize and bind phosphorylated tyrosine (pTyr) motifs, forming crucial components of intracellular signaling networks that regulate cell proliferation, survival, differentiation, and immune responses [3]. These domains facilitate the assembly of specific signaling complexes by recruiting proteins to activated receptor tyrosine kinases, thereby initiating downstream signaling cascades. The human proteome contains approximately 110 SH2 domain-containing proteins, which can be broadly classified into several functional categories including enzymes, adaptor proteins, docking proteins, transcription factors, and cytoskeletal proteins [3] [7]. Among these, the STAT-type and Src-type SH2 domains represent two major structural subgroups with distinct functional characteristics and therapeutic implications, particularly in cancer therapy [7].
The ability of SH2 domains to recognize specific pTyr-containing sequences makes them attractive targets for therapeutic intervention in diseases characterized by aberrant signaling, such as cancer. STAT3, for instance, is a transcription factor whose SH2 domain facilitates dimerization—a critical step in its activation—making it a promising target for cancer therapy [33]. Similarly, the SH2 domain-containing phosphatase SHP2 plays a regulatory role in multiple intracellular signaling cascades and is known to be oncogenic in certain contexts [34]. This technical guide explores the computational approaches, particularly free energy calculations and molecular dynamics (MD) simulations, that are advancing our understanding of SH2 domain structure, function, and inhibition within the context of drug discovery.
Structural and Functional Divergence Between STAT-type and Src-type SH2 Domains
Fundamental Structural Differences
Despite sharing a common fold, STAT-type and Src-type SH2 domains exhibit significant structural variations that underlie their distinct functional roles. All SH2 domains assume a conserved "sandwich" fold consisting of a three-stranded antiparallel beta-sheet flanked on each side by an alpha helix, arranged in an αA-βB-βC-βD-αB configuration [3] [7]. However, STAT-type SH2 domains are characterized by several distinctive structural features that set them apart from Src-type domains.
Table 1: Structural Comparison Between STAT-type and Src-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| Beta Strands | Lacks βE and βF strands | Contains additional βE, βF, and βG strands |
| Alpha Helix B | Split into two helices | Single continuous αB helix |
| CD-loop Length | Typically shorter | Varies, often longer in enzymatic proteins |
| Ancestral Function | Adapted for dimerization | Diverse recognition functions |
| Representative Proteins | STAT1, STAT2, STAT3, STAT4, STAT5, STAT6 | SRC, FYN, LCK, GRB2, ABL1 |
STAT-type SH2 domains lack the βE and βF strands present in most other SH2 domains, including Src-type domains [7]. Additionally, the αB helix in STAT domains is split into two separate helices. This structural disparity likely represents an adaptation that facilitates dimerization, which is a critical step in STAT-mediated transcriptional regulation [7]. This specialization reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity, as observed in organisms like Dictyostelium, which employ SH2 domain/phosphotyrosine signaling for transcriptional regulation [7].
Ligand Recognition and Specificity Determinants
SH2 domain binding is characterized by a combination of high specificity toward cognate pTyr ligands with moderate binding affinity (Kd typically ranging from 0.1–10 µM) [7]. This balance allows for specific yet reversible interactions, a defining characteristic of most cell signaling mediators. The pY binding pocket of SH2 domains is divided into three sub-pockets referred to as the pY+X (hydrophobic side), pY+0 (binds to pY705), and pY+1 (binds to L706) pockets [33].
The N-terminal region of the SH2 domain contains a deep pocket located within the βB strand that binds the phosphate moiety. This pocket harbors an invariable arginine (R) at position βB5, which is part of the FLVR motif found in most SH2 domains [3] [7]. This arginine directly binds to the pY residue within peptide ligands through a salt bridge, providing a fundamental recognition mechanism conserved across most SH2 domains. The C-terminal region contains additional structural elements that contribute to ligand specificity, with the EF and BG loops playing crucial roles in determining binding selectivity by controlling access to the ligand specificity pockets [7].
Computational Methodologies for SH2 Domain Analysis
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations have emerged as a powerful technique for studying the structural dynamics, conformational changes, and binding mechanisms of SH2 domains at atomic resolution. MD simulations solve Newton's equations of motion for all atoms in a molecular system, generating a trajectory that describes how the positions and velocities of atoms change over time. This approach provides insights into processes that are difficult to capture experimentally, such as transient conformational states, allosteric mechanisms, and the dynamics of ligand binding.
In a recent study investigating the selective mechanism of a monobody inhibitor (Mb13) to the phosphatase domain of SHP2, researchers conducted extensive MD simulations of the Mb13–SHP2-PTP and Mb13–SHP1-PTP systems [35]. The simulations employed multiple analysis techniques, including cluster analysis, principal component analysis, free energy landscape evaluation, cross-correlation matrix analysis, and binding free energy calculations. Results demonstrated that Mb13 bound more stably to SHP2-PTP compared to SHP1-PTP, with the SHP2 complex exhibiting conformational stability and reduced flexibility, indicating a more substantial interaction [35]. Specific residues within SHP2-PTP formed more robust interactions with Mb13, enhancing the complex's overall stability and revealing the molecular basis for selective inhibition.
Another MD investigation focused on the N-SH2 domain of SHP2 phosphatase revealed that the crystallographic environment can significantly influence the structure of the isolated domain, leading to potentially misleading interpretations [34]. Using a combination of NMR spectroscopy and MD simulations, researchers determined that the apo N-SH2 domain in solution primarily adopts a conformation with a fully zipped central β-sheet, contrary to earlier reports based on crystallographic data. The simulations further demonstrated that partial unzipping of this β-sheet is promoted by binding of either phosphopeptides or even phosphate/sulfate ions, revealing an allosteric mechanism for regulation of SHP2 activity [34].
Table 2: Key Parameters for MD Simulations of SH2 Domains
| Parameter | Typical Settings | Application Context |
|---|---|---|
| Force Field | OPLS3e, AMBER | Protein-ligand interactions, solvent models |
| Simulation Time | 100 ns - 1 µs | Conformational sampling, binding events |
| Water Model | TIP3P, SPC | Explicit solvent environment |
| Temperature Control | 300 K, Nose-Hoover | Physiological conditions |
| Pressure Control | 1 bar, Parrinello-Rahman | Isotropic pressure coupling |
| Analysis Methods | RMSD, RMSF, PCA, H-bond analysis | Stability, flexibility, conformational changes |
Binding Free Energy Calculations
Accurate prediction of binding affinities is crucial for computational drug discovery targeting SH2 domains. The Molecular Mechanics Generalized Born Surface Area (MM-GBSA) method has become a widely used approach for calculating binding free energies from MD trajectories. This method combines molecular mechanics calculations with implicit solvation models to estimate the free energy of binding.
In a comprehensive study screening natural compounds targeting the SH2 domain of STAT3, researchers employed the Prime MM-GBSA module to determine binding free energy (ΔG Binding) for protein-ligand complexes [33]. The calculations utilized the OPLS3e force field and VSGB solvent model, with the binding free energy calculated using the equation:
ΔGBinding = ΔGComplex - (ΔGReceptor + ΔGLigand)
where ΔGBinding, ΔGReceptor, and ΔGLigand denote the total binding energy of the complex, free receptor, and unbound ligand, respectively [33]. More negative values indicate stronger binding potential. In this study, MM-GBSA calculations helped identify ZINC255200449, ZINC299817570, ZINC31167114, and ZINC67910988 as potential STAT3 inhibitors, with ZINC67910988 demonstrating superior stability in subsequent analyses [33].
Molecular Docking and Virtual Screening
Molecular docking serves as a fundamental computational approach for predicting how small molecules bind to SH2 domains and for virtually screening large compound libraries to identify potential inhibitors. Docking algorithms sample possible binding orientations (poses) and score them based on energy functions that estimate binding affinity.
In the STAT3 SH2 domain inhibitor study, researchers screened 182,455 natural compounds from the ZINC15 database using a multi-step docking protocol implemented in the GLIDE tool [33]. The screening employed successively more precise docking modes: High-Throughput Virtual Screening (HTVS) for initial screening of all compounds, Standard Precision (SP) docking for the top 55,872 molecules from HTVS, and finally Extra Precision (XP) docking for the most promising candidates (cut-off at -6.5 kcal/mol) [33]. This tiered approach balanced computational efficiency with prediction accuracy, successfully identifying several natural compounds with high binding affinity for the STAT3 SH2 domain.
Experimental Protocols for Key Computational Analyses
Protocol: Molecular Dynamics Simulation of SH2 Domain-Ligand Complexes
Objective: To characterize the dynamic behavior, stability, and interaction mechanisms of SH2 domain-ligand complexes.
Software Requirements: Desmond, AMBER, GROMACS, or similar MD simulation package; molecular visualization software.
Procedure:
- System Preparation: Obtain the initial coordinates of the SH2 domain-ligand complex from crystallography, NMR, or docking studies. Use the Protein Preparation Wizard (Schrödinger) or similar tools to add hydrogen atoms, assign bond orders, and optimize side-chain conformations.
- Force Field Selection: Apply an appropriate force field (e.g., OPLS3e, CHARMM36, AMBER ff19SB) for the protein and a compatible force field for the ligand parameters.
- Solvation and Ion Addition: Solvate the system in an orthorhombic water box (e.g., TIP3P water model) with a minimum 10 Å buffer distance from the protein surface. Add ions (e.g., NaCl) to neutralize the system and achieve physiological concentration (0.15 M).
- Energy Minimization: Perform steepest descent and conjugate gradient minimization to relieve steric clashes and optimize the system geometry.
- System Equilibration: Conduct a multi-step equilibration process:
- NVT ensemble (constant Number, Volume, Temperature): 100 ps at 300 K using a Berendsen thermostat.
- NPT ensemble (constant Number, Pressure, Temperature): 100 ps at 1 bar using a Berendsen barostat.
- Production MD Run: Perform the production MD simulation for 100 ns to 1 µs, saving coordinates every 100 ps for analysis. Use the NPT ensemble with a Nosé-Hoover thermostat and Parrinello-Rahman barostat.
- Trajectory Analysis: Analyze the trajectory using:
- Root Mean Square Deviation (RMSD) to assess structural stability.
- Root Mean Square Fluctuation (RMSF) to identify flexible regions.
- Hydrogen bond analysis to determine persistent interactions.
- Principal Component Analysis (PCA) to identify essential dynamics.
Protocol: MM-GBSA Binding Free Energy Calculations
Objective: To calculate the binding free energy for SH2 domain-ligand complexes from MD trajectories.
Software Requirements: Schrödinger Prime, AMBER MMPBSA.py, or similar MM-GBSA implementation.
Procedure:
- Trajectory Preparation: Extract snapshots from the equilibrated portion of the MD trajectory (typically every 100-200 ps).
- Complex, Receptor, and Ligand Selection: Define the components for calculation: the complex, the SH2 domain alone (receptor), and the ligand alone.
- Energy Calculation: For each snapshot, calculate:
- The gas-phase molecular mechanics energy (EMM), including bonded and non-bonded terms.
- The solvation free energy (Gsolv) using the Generalized Born (GB) model for the polar component and surface area (SA) for the nonpolar component.
- Binding Free Energy Computation: Compute the binding free energy for each snapshot using: ΔGbind = Gcomplex - (Greceptor + Gligand) where G = EMM + Gsolv - TS (with entropy term often omitted for relative comparisons).
- Statistical Analysis: Calculate the mean and standard error of ΔGbind across all snapshots. Compare results across different ligands or mutations.
Protocol: Virtual Screening for SH2 Domain Inhibitors
Objective: To identify potential small-molecule inhibitors of SH2 domains through computational screening.
Software Requirements: Schrödinger GLIDE, AutoDock Vina, or similar docking software; compound library (e.g., ZINC15).
Procedure:
- Protein Preparation: Obtain the SH2 domain structure (e.g., PDB ID 6NJS for STAT3). Preprocess using Protein Preparation Wizard: add hydrogens, assign bond orders, fill missing side chains, and optimize hydrogen bonding networks. Perform constrained energy minimization.
- Grid Generation: Define the binding site using the Receptor Grid Generation tool. Center the grid on the known ligand binding site or functional epitope (e.g., pY binding pocket). Set the grid box size to encompass the entire binding site (e.g., 20 Å × 20 Å × 20 Å).
- Ligand Library Preparation: Download and prepare compounds from a database (e.g., 182,455 natural compounds from ZINC15). Use LigPrep to generate 3D structures, possible tautomers, stereoisomers, and protonation states at physiological pH (7.4 ± 0.5).
- Hierarchical Docking: Perform multi-stage docking:
- HTVS Mode: Rapid initial screening of the entire library.
- SP Mode: Standard precision docking of top compounds from HTVS (e.g., top 30%).
- XP Mode: Extra precision docking of the most promising candidates from SP for accurate pose prediction and scoring.
- Post-docking Analysis: Visually inspect top-scoring poses for conserved interactions (e.g., with Arg βB5 in the pY pocket). Filter based on docking scores, interaction patterns, and chemical properties.
Visualization of Computational Workflows and Signaling Pathways
Figure 1: Computational Workflow for SH2 Domain-Drug Discovery
Figure 2: SH2 Domain-Mediated Signaling Pathways
Research Reagent Solutions for SH2 Domain Studies
Table 3: Essential Research Reagents for Computational and Experimental SH2 Domain Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| SH2 Domain Structures | STAT3 SH2 (PDB: 6NJS), GRB2-SH2 (PDB: 1BMB), SHP2 N-SH2 | Structural templates for docking/MD; resolution: 2.70 Å for 6NJS [33] [36] |
| Compound Libraries | ZINC15 natural compounds (182,455 compounds screened) | Virtual screening for inhibitors [33] |
| Computational Software | Schrödinger Suite (2024-2), Desmond, AMBER, GROMACS | MD simulations, docking, MM-GBSA calculations [33] [36] |
| Force Fields | OPLS3e, AMBER ff19SB, CHARMM36 | Molecular mechanics parameters for simulations [33] [34] |
| Analysis Tools | WaterMap, Prime MM-GBSA, PCA, FEL | Energetic analysis, solvation effects, conformational sampling [33] [35] |
| Experimental Validation | NMR spectroscopy, Isothermal Titration Calorimetry | Validation of computational predictions [36] [34] |
Computational approaches, particularly free energy calculations and molecular dynamics simulations, have become indispensable tools for studying SH2 domain structure, function, and inhibition. These methods provide atomic-level insights into the dynamic behavior of SH2 domains, their mechanisms of ligand recognition, and the structural differences between STAT-type and Src-type domains that underlie their distinct biological functions. The integration of hierarchical docking protocols with MD simulations and MM-GBSA calculations has proven effective for identifying and optimizing SH2 domain inhibitors, as demonstrated by the discovery of natural compounds targeting the STAT3 SH2 domain [33].
Future developments in this field will likely focus on enhanced sampling techniques to access longer timescales, more accurate force fields for phosphorylated residues and drug-like molecules, and machine learning approaches to predict binding affinities and specificities. Additionally, the emerging role of SH2 domains in liquid-liquid phase separation [3] presents new challenges and opportunities for computational methods to model these mesoscale assemblies. As these computational approaches continue to evolve, they will undoubtedly accelerate the discovery and optimization of therapeutic agents targeting SH2 domains in cancer and other diseases.
Src homology 2 (SH2) domains are protein modules of approximately 100 amino acids that serve as crucial readers of phosphotyrosine-based cellular information [3] [19]. These domains specifically recognize and bind to phosphorylated tyrosine residues on target proteins, thereby facilitating the formation of complex signaling networks that govern critical cellular processes including development, homeostasis, immune responses, and cytoskeletal rearrangement [3] [37]. The human proteome encodes approximately 110 SH2 domain-containing proteins, which can be broadly classified into several functional categories including enzymes, adaptor proteins, signaling regulators, docking proteins, transcription factors, and cytoskeletal proteins [3] [7].
SH2 domains function as key components in phosphotyrosine signaling networks by inducing proximity between protein tyrosine kinases (PTKs), protein tyrosine phosphatases (PTPs), and their specific substrates and signaling effectors [3] [7]. This selective recognition of proteins containing phosphotyrosine (pY) peptide binding motifs enables precise spatiotemporal control of cellular signaling events. Recent research has revealed that SH2 domains exhibit remarkable functional diversity beyond canonical phosphopeptide binding, including interactions with lipid molecules and participation in the formation of intracellular condensates through liquid-liquid phase separation (LLPS) [3] [7].
The structural and functional differences between the two major SH2 domain subgroups—Src-type and STAT-type—provide a critical framework for understanding their specialized roles in cellular signaling and present unique challenges and opportunities for computational prediction and classification [3] [7]. This technical guide explores the integration of deep learning and bioinformatics approaches to address these challenges, with particular emphasis on their application within drug discovery pipelines.
Structural Basis of SH2 Domain Specificity
Conserved Architecture and Recognition Principles
All SH2 domains share a conserved structural fold consisting of a central antiparallel β-sheet flanked by two α-helices, forming a characteristic "sandwich" structure [3] [7] [37]. Despite significant sequence variation (as low as ~15% pairwise identity among some family members), the three-dimensional architecture remains remarkably conserved, reflecting evolutionary optimization for phosphotyrosine recognition [3] [7].
The phosphopeptide binding mechanism employs a conserved "two-pronged plug" interaction [2]. A deep basic pocket binds the phosphotyrosine residue, while an adjacent specificity pocket engages residues C-terminal to the pY, particularly the +3 position [2] [37]. The phosphotyrosine binding pocket contains a critically conserved arginine residue at position βB5 within the FLVR motif, which forms bidentate hydrogen bonds with the phosphate moiety and provides substantial binding energy [3] [2]. Additional conserved basic residues at positions αA2 and βD6 further contribute to phosphate coordination [2].
Structural and Functional Dichotomy: Src-type versus STAT-type SH2 Domains
SH2 domains are structurally and evolutionarily divided into two major subgroups with distinct characteristics and functional implications, as summarized in Table 1.
Table 1: Structural and Functional Comparison of Src-type and STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB with additional βE, βF, βG strands | αA-βB-βC-βD-αB' split helix; lacks βE and βF strands |
| Terminal Regions | Conserved N-terminal region, variable C-terminal region | Adapted for dimerization in transcriptional regulation |
| Loop Characteristics | Longer CD-loops in enzymatic proteins | Shorter loops in non-enzymatic proteins like STATs |
| Evolutionary Origin | Derived form | Considered one of the most ancient and fully developed functional templates |
| Primary Function | Diverse signaling roles including kinase regulation | Specialized for STAT-mediated transcriptional regulation |
The STAT-type SH2 domains represent an ancestral form that predates animal multicellularity and appears to have served as an evolutionary template for SH2 domain development [5] [7]. This structural divergence directly impacts their functional specialization, with STAT-type domains optimized for the dimerization required for transcriptional activation, while Src-type domains have evolved for diverse signaling contexts including lipid membrane interactions and complex formation with multiple partners [3] [7].
Figure 1: Structural classification and functional specialization of SH2 domains
Quantitative Models for SH2 Domain Specificity and Binding Affinity
From Specificity Profiling to Energy-Based Prediction
Traditional approaches to understanding SH2 domain specificity relied on degenerate peptide libraries and position-specific scoring matrices (PSSMs) [10] [38]. While these methods identified broad binding motifs, they often missed contextual sequence information and inhibitory residues that oppose binding [10]. The limitations of these approaches became increasingly apparent as research revealed that SH2 domains achieve remarkable selectivity through complex integration of both permissive residues (that enhance binding) and non-permissive residues (that oppose binding) in a context-dependent manner [10].
Recent advances have transformed SH2 domain specificity profiling from classification to quantitative affinity prediction [38]. Integration of bacterial peptide display, enzymatic phosphorylation of displayed peptides, affinity-based selection, and next-generation sequencing (NGS) has enabled the development of accurate sequence-to-affinity models [38]. The ProBound computational framework, initially developed for protein-DNA interactions, has been successfully adapted to model SH2 domain binding, generating additive models that predict binding free energy across the full theoretical ligand sequence space [38].
These quantitative models represent a significant advancement because they:
- Predict binding affinity in biophysically meaningful units (ΔΔG)
- Cover the complete theoretical sequence space beyond library constraints
- Jointly analyze data from multi-round selection experiments
- Accommodate extremely sparse coverage of highly complex libraries [38]
Experimental Methodologies for Specificity Profiling
Table 2: Key Experimental Methods for SH2 Domain Specificity Profiling
| Method | Throughput | Key Measurements | Applications | Limitations |
|---|---|---|---|---|
| Bacterial Peptide Display + NGS | 10^6-10^7 sequences | Binding free energy (ΔΔG) | Full sequence-space affinity models | Requires specialized computational analysis |
| SPOT Peptide Arrays | 10^2-10^3 peptides | Semi-quantitative interaction strength | Specificity profiling against defined physiological peptides | Membrane-based, limited quantitative accuracy |
| Fluorescence Polarization | 10^1-10^2 peptides | Equilibrium dissociation constant (K_D) | Validation of binding affinity and specificity | Lower throughput, requires peptide synthesis |
| Oriented Peptide Libraries | 10^4-10^5 sequences | Binding motifs and selectivity | Initial domain characterization and comparison | Misses contextual and non-permissive residues |
The integration of these complementary approaches provides a comprehensive framework for establishing quantitative binding models. Bacterial peptide display with NGS enables exploration of vast sequence spaces, while fluorescence polarization and SPOT arrays offer validation against physiologically relevant peptides [10] [38].
Deep Learning Approaches for SH2 Motif Prediction and Classification
Architecture Design for SH2 Domain Bioinformatics
Modern deep learning approaches for SH2 domain prediction leverage several neural network architectures optimized for different aspects of the classification and prediction problem:
Convolutional Neural Networks (CNNs) applied to sequence data can identify conserved binding motifs and structural patterns characteristic of SH2 domain subtypes. These networks excel at detecting local sequence patterns that correspond to critical structural features such as the FLVR motif, specificity pocket residues, and loop regions that differentiate Src-type and STAT-type domains [38].
Recurrent Neural Networks (RNNs), particularly Long Short-Term Memory (LSTM) and Gated Recurrent Unit (GRU) architectures, capture contextual dependencies within SH2 domain sequences that influence structural stability and binding specificity. These models are particularly effective for modeling the relationships between spatially separated but functionally coupled residues [38].
Attention Mechanisms and Transformer Architectures enable the identification of critical residues contributing to subclass specificity and binding energy. These approaches excel at weighing the importance of different sequence positions for accurate classification and affinity prediction [38].
Multi-task learning frameworks simultaneously predict SH2 domain classification, binding specificity, and structural features, leveraging shared representations to improve performance across related tasks despite limited training data for any single objective [38].
Implementation Workflow for SH2 Domain Analysis
Figure 2: Integrated computational and experimental workflow for SH2 domain analysis
Experimental Protocols for SH2 Domain Binding Characterization
Bacterial Peptide Display with Next-Generation Sequencing
Objective: Quantitative profiling of SH2 domain binding specificity across highly diverse peptide libraries.
Materials:
- SH2 domain of interest (cloned into pGEX-2TK or similar expression vector)
- Escherichia coli strain BL21 for protein expression
- Random peptide library (complexity 10^6-10^7 sequences)
- Glutathione-Sepharose for affinity purification
- NGS platform (Illumina or similar)
Procedure:
- Library Construction: Generate random peptide library with central tyrosine residue flanked by degenerate sequences. Ensure library diversity exceeds 10^6 unique members.
- Bacterial Display: Express peptide library on bacterial surface using appropriate display system.
- Enzymatic Phosphorylation: Treat displayed peptides with purified tyrosine kinases to phosphorylate tyrosine residues.
- Affinity Selection: Incubate phosphorylated peptide library with purified SH2 domain. Perform multiple rounds of selection with increasing stringency.
- Next-Generation Sequencing: Sequence input and selected populations after each selection round.
- Data Analysis: Apply ProBound or similar computational framework to model binding energy as a function of peptide sequence.
Critical Considerations:
- Maintain library diversity throughout selection process
- Include appropriate controls for non-specific binding
- Normalize for variations in phosphorylation efficiency
- Account for potential biases in amplification and sequencing [38]
SPOT Membrane Peptide Array Analysis
Objective: Semi-quantitative assessment of SH2 domain binding to defined physiological peptides.
Materials:
- Nitrocellulose membrane for peptide synthesis
- Automated peptide synthesizer (e.g., Intavis MultiPep)
- Recombinant SH2 domain (GST-tagged for detection)
- Anti-GST antibody conjugated to horseradish peroxidase
- Enhanced chemiluminescence detection reagents
Procedure:
- Peptide Synthesis: Synthesize 11-amino acid peptides directly on membrane with phosphotyrosine at position 5.
- Membrane Processing: Block membrane with 5% non-fat dry milk in TBST.
- SH2 Domain Incubation: Apply purified SH2 domain (0.1-1 μM) and incubate for 2 hours at room temperature.
- Detection: Incubate with anti-GST-HRP antibody, develop with ECL reagent.
- Quantification: Analyze spot intensity using densitometry software.
Applications:
- Validation of computationally predicted interactions
- Assessment of binding specificity across physiological ligands
- Identification of non-permissive residues through systematic mutagenesis [10]
Table 3: Key Research Reagents for SH2 Domain Investigations
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Expression Systems | pGEX-2TK vector, E. coli BL21 | Recombinant SH2 domain production with GST tag for purification |
| Purification Resins | Glutathione-Sepharose | Affinity purification of GST-tagged SH2 domains |
| Peptide Library Platforms | Cellulose membrane arrays, bacterial display systems | High-throughput binding specificity assessment |
| Detection Reagents | Anti-phosphotyrosine antibodies (4G10, pY20), anti-GST-HRP | Detection of phosphorylated peptides and domain binding |
| Computational Tools | ProBound framework, PSSM generators | Binding affinity prediction and specificity modeling |
| Reference Databases | Protein Data Bank, SH2 domain resource (sh2.uchicago.edu) | Structural information and specificity data |
Applications in Drug Discovery and Therapeutic Development
The integration of deep learning approaches with structural insights into SH2 domain differences has significant implications for drug discovery. Several targeting strategies have emerged:
Direct SH2 Domain Inhibition: Small molecules that block phosphopeptide binding pockets represent a promising approach for modulating signaling pathways. Structural differences between Src-type and STAT-type domains enable the development of selective inhibitors [3] [7]. STAT3 SH2 domain inhibitors, for example, have shown promise in preclinical cancer models by disrupting STAT3 dimerization and nuclear translocation [3].
Lipid-Binding Interface Targeting: Recent research indicates that nearly 75% of SH2 domains interact with membrane lipids, particularly PIP2 and PIP3 [3] [7]. These interactions play crucial roles in membrane recruitment and activation of SH2-containing proteins. The development of nonlipidic inhibitors targeting these interfaces, as demonstrated for Syk kinase, offers an alternative to traditional active-site inhibitors [3].
Allosteric Modulation: The discovery of interdomain communications and dynamic unfolding events in SH2 domains suggests opportunities for allosteric regulation [39]. Hydrogen exchange mass spectrometry studies have revealed that SH2 and SH3 domains influence each other's dynamics when expressed in tandem constructs, providing potential targets for allosteric control [39].
Phase Separation Manipulation: Emerging evidence links SH2 domain-containing proteins to liquid-liquid phase separation (LLPS) in signaling condensates [3]. In T-cell receptor signaling, interactions among GRB2, Gads, and LAT receptors contribute to LLPS formation, enhancing signaling efficiency [3]. Small molecules that modulate these phase separation behaviors represent a novel therapeutic approach.
The integration of deep learning with structural bioinformatics has dramatically advanced our ability to predict SH2 domain binding motifs and classify domain subtypes. The structural and functional differences between Src-type and STAT-type SH2 domains provide a critical framework for understanding their specialized roles in cellular signaling and developing targeted therapeutic interventions.
Future advancements in this field will likely focus on several key areas:
- Integration of structural dynamics data into predictive models
- Expansion of multi-domain interaction predictions
- Application of geometric deep learning to incorporate 3D structural information
- Development of generative models for designing novel SH2 domain binders
As these computational approaches continue to evolve, coupled with experimental validation through the methodologies described herein, they will increasingly enable researchers to decipher the complex language of SH2-mediated signaling and exploit this knowledge for therapeutic benefit.
In cellular signaling networks, Src homology 2 (SH2) domains serve as critical "readers" of phosphotyrosine (pTyr) signals, directing the formation of protein complexes that control fundamental processes including proliferation, differentiation, and apoptosis. The human genome encodes approximately 120 SH2 domains distributed across 111 proteins, yet all share a conserved structural fold despite recognizing distinct pTyr-containing sequences [37]. This specificity paradox—how highly conserved structures achieve diverse ligand recognition—is resolved through structural variations in two critical surface loops: the EF loop (connecting β-strands E and F) and the BG loop (connecting the α-helix B and β-strand G) [40] [20]. These loops function as molecular gatekeepers that control access to binding pockets, thereby defining the sequence specificity of different SH2 domains.
Within the broader classification of SH2 domains, STAT-type and Src-type domains represent two major structural and functional subgroups. STAT-type SH2 domains, which facilitate dimerization and nuclear translocation in signal transducers and activators of transcription, lack the βE and βF strands characteristic of Src-type domains and feature a split αB helix [7]. This structural adaptation enables STAT SH2 domains to participate in reciprocal phosphopeptide-mediated dimerization with another STAT molecule, a critical step in JAK-STAT signaling pathways. In contrast, Src-type domains, which include those in kinases, phosphatases, and adaptor proteins, maintain the complete set of secondary structural elements and typically engage in transient signaling interactions with various binding partners. Understanding how EF and BG loops encode specificity in both domain types provides fundamental insights for developing therapeutic strategies targeting SH2-mediated interactions in disease.
Structural Architecture of SH2 Domains
Conserved SH2 Domain Fold
All SH2 domains adopt a conserved structural framework consisting of a central anti-parallel β-sheet flanked by two α-helices, forming a compact sandwich-like structure. The core elements follow the pattern: αA-βB-βC-βD-αB, with most SH2 domains containing additional β-strands (A, E, F, and G) to complete the characteristic seven-stranded sheet [7] [37]. This conserved architecture provides the structural scaffold upon which specificity determinants are built. The N-terminal region of the domain contains a deeply conserved pTyr-binding pocket formed primarily by residues in the βB strand, which harbors an invariant arginine residue (Arg175 in v-Src) that forms critical bidentate hydrogen bonds with the phosphate moiety of pTyr [20] [37]. This interaction provides approximately half of the total binding free energy and is essential for phosphopeptide recognition.
STAT-type versus Src-type SH2 Domains
STAT-type SH2 domains exhibit distinct structural adaptations that differentiate them from Src-type domains. Unlike Src-type domains, STAT SH2 domains lack the βE and βF strands and the connecting loops, and feature a split αB helix [7]. This structural simplification likely represents an adaptation for reciprocal dimerization between STAT molecules, where one STAT molecule provides a phosphopeptide ligand while its SH2 domain engages a phosphopeptide from its partner. This arrangement facilitates the formation of stable dimers that translocate to the nucleus and regulate transcription. The absence of the EF loop in STAT SH2 domains eliminates one of the key structural elements that control specificity in Src-type domains, resulting in different mechanisms of ligand selection.
In contrast, Src-type SH2 domains maintain the complete complement of secondary structural elements, including the EF and BG loops that serve as critical determinants of ligand specificity. These domains typically recognize phosphorylated proteins in extended conformations, with the peptide binding perpendicular to the central β-sheet [41] [37]. The presence of both EF and BG loops in Src-type domains allows for more complex regulation of binding pocket accessibility and enables greater diversity in sequence recognition, which is essential for their roles in transient signaling complexes and dynamic cellular processes.
Table 1: Key Structural Features Differentiating STAT-type and Src-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| βE and βF strands | Absent | Present |
| EF loop | Not present | Present and variable |
| BG loop | Present but modified | Present and variable |
| αB helix | Split into two helices | Single continuous helix |
| Primary function | Reciprocal dimerization | Transient signaling interactions |
| Specificity mechanism | Reduced loop involvement | EF and BG loop controlled access |
Molecular Mechanisms of Specificity Determination
The Loop-Controlled Access Model
The EF and BG loops govern SH2 domain specificity through a mechanism termed "loop-controlled access," where these surface loops physically regulate ligand entry to binding pockets. Structural analyses of diverse SH2 domains reveal that all SH2 domains potentially contain three binding pockets that can accommodate residues at the P+2, P+3, and P+4 positions C-terminal to the phosphotyrosine [20]. However, in any individual SH2 domain, only one of these pockets is typically accessible for ligand binding, while the others are blocked by specific residues from the EF and BG loops. This selective pocket accessibility creates distinct specificity classes among SH2 domains: P+2, P+3, and P+4 binders [40].
For example, in the Src SH2 domain (a P+3 class binder), the P+4 pocket is plugged by a residue from the BG loop, while in the Grb2 SH2 domain (a P+2 class binder), both P+3 and P+4 pockets are blocked [20]. Conversely, in BRDG1 SH2 domain (a P+4 class binder), the P+3 pocket is occupied by an EF loop residue that prevents ligand binding at this position. This loop-mediated control of pocket accessibility explains how a conserved structural scaffold can generate diverse specificities without compromising structural integrity. The molecular basis for this mechanism lies in the sequence variability of the EF and BG loops, which, despite their differences, maintain conserved structural features that preserve the overall SH2 fold while enabling functional specialization.
Key Interactions in the N-SH2 Domain of SHP2
The N-SH2 domain of SHP2 exemplifies how EF and BG loops contribute to specificity determination. Molecular dynamics simulations reveal that in addition to the essential pTyr interaction, the complex is stabilized by hydrophobic insertion of residues at P+1, P+3, and P+5 into an apolar groove of the domain, along with interaction of residue P-2 with both the pY and a protein surface residue [41]. Additional stabilization comes from hydrogen bonds formed by the backbone of residues at P-1, P+1, P+2, and P+4. Particularly important for SHP2 N-SH2 specificity are electrostatic interactions between negatively charged residues at positions P+2 and P+4 and two lysine residues (Lys89 and Lys91) that are specific to this domain [41]. These interactions work in concert with the loop-mediated control of pocket accessibility to define the binding preferences for this critical regulatory domain.
Table 2: Specificity Determinants in SH2 Domain-Peptide Interactions
| Position Relative to pY | Chemical Property | Structural Role | Domain Region Involved |
|---|---|---|---|
| P-2 | Hydrophobic (A, L, I, V, M, F, P) | Interacts with pY and protein surface | pY binding pocket |
| pY | Phosphotyrosine | Primary anchoring interaction | Conserved pY pocket (βB strand) |
| P+1 | Hydrophobic | Hydrophobic insertion | Apolar groove |
| P+2 | Acidic (D, E) in SHP2 N-SH2 | Electrostatic interactions | Lys89/Lys91 (SHP2-specific) |
| P+3 | Hydrophobic | Hydrophobic insertion | EF/BG loop region |
| P+4 | Acidic (D, E) in SHP2 N-SH2 | Electrostatic interactions | Lys89/Lys91 (SHP2-specific) |
| P+5 | Hydrophobic | Hydrophobic insertion | Apolar groove |
Experimental Approaches for Profiling SH2 Specificity
Phage Display with Engineered SH2 Libraries
Phage display technology enables comprehensive profiling of SH2 domain specificity by screening combinatorial libraries of SH2 variants against phosphopeptide targets. This approach involves randomizing the EF and BG loops of a template SH2 domain (such as Fyn SH2) to create library diversity, followed by panning against biotinylated pY peptides immobilized on streptavidin-coated plates [40]. After multiple rounds of selection, enriched phage pools are isolated and subjected to DNA sequencing to identify SH2 variants with specific binding characteristics. This method has demonstrated that the EF and BG loops can encode a wide spectrum of specificities, including all three major specificity classes (P+2, P+3, and P+4) found in natural SH2 domains [40].
The experimental workflow begins with library construction using Kunkel mutagenesis to introduce diversity into the EF and BG loop regions. The resulting library is then incubated with target phosphopeptides immobilized on solid supports. After washing to remove non-specific binders, specifically bound phages are eluted and amplified for subsequent rounds of selection. This iterative process enriches for SH2 variants with high affinity and specificity for the target sequence. Finally, individual clones are characterized using phage ELISA and DNA sequencing to determine their sequence and binding properties. This approach has revealed that SH2 variants can employ diverse structural solutions to achieve the same specificity, highlighting the flexibility and adaptability of the EF and BG loops in conferring binding preferences.
Peptide Array-Based Profiling
Peptide arrays provide a high-throughput platform for assessing SH2 domain binding specificity across large sets of potential ligands. In this method, biotin-labeled peptides are incubated with neutravidin in solution and then printed onto activated glass slides to create spatially addressable arrays [40]. The arrays are then probed with purified GST-tagged SH2 domains, followed by detection with anti-GST antibodies and fluorescently labeled secondary antibodies. Laser scanning and quantification of fluorescence signals enable quantitative assessment of binding interactions across hundreds or thousands of peptide sequences simultaneously.
This technique has been particularly valuable for defining the specificity of SH2 domains using Oriented Peptide Array Library (OPAL) approaches, where degenerate peptide libraries systematically vary residues at each position relative to the phosphotyrosine [20]. Analysis of binding patterns reveals position-specific amino acid preferences that define the recognition motif for each SH2 domain. Peptide arrays have confirmed that most SH2 domains exhibit primary specificity for residues at the P+2, P+3, or P+4 positions, with the particular preference determined by the structural features of the EF and BG loops [20]. This method provides comprehensive specificity data that facilitates the construction of position-specific scoring matrices for predicting novel SH2 binding sites in proteomic datasets.
Quantitative Affinity Profiling with Next-Generation Sequencing
Recent advances combine bacterial display of genetically encoded peptide libraries with next-generation sequencing (NGS) to quantitatively profile SH2 domain binding affinity across extremely diverse sequence spaces. This approach involves displaying random peptide libraries on the surface of bacteria, followed by affinity-based selection using purified SH2 domains [38]. The key innovation lies in using NGS to count sequence abundance before and after selection, enabling quantitative measurement of enrichment ratios for thousands to millions of distinct peptide sequences.
Computational analysis of these data using methods like ProBound allows construction of sequence-to-affinity models that predict binding free energy for any peptide sequence within the theoretical space covered by the library [38]. This free-energy regression approach assumes additivity of binding contributions across peptide positions and can accurately predict binding affinity across multiple orders of magnitude. This method represents a significant advance over classification-based approaches, as it provides biophysically interpretable parameters (ΔΔG values) that quantify the contribution of each amino acid at each peptide position to the overall binding energy. For SH2 domains profiled in this manner, the resulting models can predict novel phosphosite targets and assess the impact of disease-associated mutations on binding affinity.
Diagram 1: Experimental workflows for determining EF/BG loop-mediated SH2 specificity. The diagram illustrates how different experimental approaches leverage SH2 domain structural features to determine binding specificity and affinity.
Research Reagent Solutions for SH2 Domain Studies
Table 3: Essential Research Reagents for SH2 Domain Specificity Studies
| Reagent Category | Specific Examples | Applications | Key Features |
|---|---|---|---|
| Engineered SH2 Libraries | Fyn SH2 EF/BG loop randomization [40] | Phage display selection | Combinatorial diversity in specificity-determining regions |
| Peptide Synthesis Systems | Fmoc-based solid-phase peptide synthesis [40] | Peptide array production | Incorporation of biotin/fluorescent labels with spacer arms |
| Display Platforms | M13 phage display [40]; Bacterial display [38] | Library selection | High diversity (10^6-10^7 variants); compatibility with NGS |
| Detection Reagents | Anti-GST antibodies [40]; DyLight 649-labeled secondary antibodies [40] | Fluorescence-based detection | Compatible with microarray scanning |
| Computational Tools | ProBound [38]; SMALI [20] | Specificity prediction | Free-energy regression; binding pocket analysis |
| Immobilization Matrices | Streptavid-coated plates [40]; Activated glass slides [40] | Peptide presentation | High binding capacity; low background |
Implications for Drug Discovery and Therapeutic Targeting
The critical role of EF and BG loops in determining SH2 domain specificity presents attractive opportunities for therapeutic intervention. As key mediators of phosphotyrosine signaling, SH2 domains contribute to numerous disease processes, including cancer, immune disorders, and developmental syndromes. In particular, SHP2, which contains two SH2 domains (N-SH2 and C-SH2), represents a promising target for cancer therapy, as it functions as a central node in oncogenic signaling and drug resistance mechanisms [41]. Mutations in PTPN11, the gene encoding SHP2, are associated with juvenile myelomonocytic leukemia and Noonan syndrome, further highlighting its clinical relevance [41].
Targeting the EF and BG loops offers a potential strategy for developing specific inhibitors that disrupt pathological SH2-mediated interactions while sparing physiological signaling. The structural diversity of these loops across different SH2 domains provides a basis for achieving selectivity in pharmacological targeting. Emerging approaches include the development of non-lipidic small molecules that target lipid-binding pockets adjacent to the pY-binding site in SH2 domains, as demonstrated for Syk kinase inhibitors [7]. Additionally, the discovery that many disease-causing mutations localize to lipid-binding pockets of SH2 domains [7] further validates these regions as therapeutic targets.
Beyond traditional inhibition, understanding EF and BG loop function enables engineering of modified SH2 domains with altered specificities for therapeutic applications. Such engineered domains could potentially act as molecular decoys that sequester pathological phosphoproteins or redirect signaling pathways toward beneficial outcomes. The flexibility and adaptability of the EF and BG loops, as revealed by phage display studies [40], suggest that substantial retargeting of SH2 domain specificity is achievable through rational design or directed evolution approaches.
The EF and BG loops serve as critical structural determinants that encode binding specificity across the SH2 domain family. Through a combination of steric hindrance, pocket occlusion, and direct interaction with peptide ligands, these loops control access to binding subsites and define whether an SH2 domain preferentially recognizes residues at P+2, P+3, or P+4 positions C-terminal to phosphotyrosine. The loop-controlled access mechanism explains how a conserved structural scaffold can generate the remarkable diversity of specificities observed among SH2 domains, enabling precise decoding of phosphotyrosine signals in cellular signaling networks.
Advanced experimental approaches, including phage display, peptide arrays, and bacterial display coupled with next-generation sequencing, have provided detailed insights into how sequence variations in EF and BG loops translate to distinct binding preferences. These methods enable quantitative profiling of SH2 specificity and the construction of predictive models that can identify novel binding sites and assess the functional impact of sequence variations. For researchers investigating STAT-type versus Src-type SH2 domains, these tools offer powerful means to explore how structural differences translate to functional specialization in signaling pathways.
The strategic importance of EF and BG loops in determining SH2 domain specificity positions them as attractive targets for therapeutic intervention in diseases driven by aberrant phosphotyrosine signaling. As structural insights into these loops continue to accumulate, so too will opportunities for developing targeted therapies that modulate specific SH2-mediated interactions with precision and selectivity.
The Src Homology 2 (SH2) domain has long been recognized as a quintessential modular domain specializing in phosphotyrosine (pTyr) recognition, facilitating critical protein-protein interactions in cellular signaling networks [2]. However, emerging research reveals that SH2 domains participate in biological functions extending far beyond this canonical role. This technical guide examines two significant non-canonical mechanisms—direct lipid interaction and participation in biomolecular condensates via liquid-liquid phase separation (LLPS)—and frames these findings within the structural and functional divergence between the two major SH2 subfamilies: Src-type and STAT-type. For researchers and drug development professionals, understanding these mechanisms provides novel insights into signal transduction complexity and reveals potential therapeutic targets for cancer and other diseases. The structural differences between SH2 domain types, summarized in Table 1, form the foundation for their divergent non-canonical functions.
Table 1: Structural and Functional Comparison of Src-type and STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB sandwich with additional βE, βF, βG strands | αA-βB-βC-βD-αB sandwich; lacks βE and βF strands |
| αB Helix | Single continuous helix | Split into two helices (αB and αB') |
| C-terminal Region | Contains β strands E, F, G and adjoining loops | Simplified C-terminal structure |
| CD-loop Length | Generally longer | Generally shorter |
| Primary Functional Adaptation | Diverse phosphopeptide recognition | Dimerization for transcriptional regulation |
| Evolutionary Origin | Later evolution from STAT-type template | Considered more ancient; ancestral form |
Structural Basis of SH2 Domains: Src-type versus STAT-type
The classical SH2 domain fold consists of a central antiparallel β-sheet flanked by two α-helices, forming a conserved structure that specifically recognizes phosphorylated tyrosine residues through a "two-pronged plug" mechanism [2]. This binding involves a deep pocket that engages the phosphotyrosine and a specificity pocket that typically recognizes residues at the +3 position C-terminal to the pTyr [2].
Despite this conserved core architecture, significant structural variations distinguish Src-type and STAT-type SH2 domains. Src-type domains, found in signaling adapters and kinases, contain extra secondary structural elements including β-strands E, F, and G, creating a more complex C-terminal region [7] [5]. These domains often feature longer CD-loops, which may facilitate diverse peptide recognition capabilities essential for their roles in complex signaling networks [7].
In contrast, STAT-type SH2 domains exhibit a simplified architecture adapted for dimerization, a critical step in STAT-mediated transcriptional activation. These domains lack the βE and βF strands and possess a split αB helix [7] [5]. Evolutionary studies suggest that STAT-type SH2 domains represent a more ancient form, with the linker-SH2 domain of STAT proteins potentially serving as the evolutionary template for the entire SH2 family [5]. This structural divergence underpins the differential engagement of these domain types in non-canonical functions such as lipid binding and phase separation.
Lipid Interactions with SH2 Domains
Mechanisms and Functional Consequences
Recent biochemical and biophysical studies have revealed that approximately 75% of SH2 domains interact with membrane lipids, particularly phosphoinositides such as phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [7] [3]. These interactions occur through cationic regions adjacent to the phosphotyrosine-binding pocket, typically flanked by aromatic or hydrophobic amino acid side chains that facilitate membrane association [7].
These lipid interactions significantly modulate cellular signaling by affecting membrane recruitment, enzymatic activity, and scaffolding functions of SH2-containing proteins. For example, the PIP3 binding activity of the TNS2 SH2 domain is essential for regulating insulin receptor substrate-1 (IRS-1) phosphorylation in insulin signaling pathways [7] [3]. Similarly, lipid binding by SYK, ZAP70, and LCK SH2 domains is crucial for their functions in immune receptor signaling [7].
Table 2: Functional Roles of Lipid Binding by SH2 Domain-Containing Proteins
| Protein | Lipid Moisty | Functional Role of Lipid Association |
|---|---|---|
| SYK | PIP3 | PIP3-dependent membrane binding required for activation of SYK scaffolding function, leading to noncatalytic activation of STAT3/5 |
| ZAP70 | PIP3 | Facilitates and sustains ZAP70 interactions with TCR-ζ chain in T-cell receptor signaling |
| LCK | PIP2, PIP3 | Modulates interaction of LCK with binding partners in the TCR signaling complex |
| ABL | PIP2 | Mediates membrane recruitment and modulation of Abl kinase activity |
| VAV2 | PIP2, PIP3 | Modulates interaction of VAV2 with membrane receptors such as EphA2 |
| C1-Ten/Tensin2 | PIP3 | Regulates Abl activity and IRS-1 phosphorylation in insulin signaling |
Structural Determinants of Lipid Binding
The structural basis for lipid recognition involves conserved motifs within the SH2 domain. The FLVR (Phe-Leu-Val-Arg) motif, particularly the invariant arginine at position βB5, is crucial for both phosphotyrosine and lipid phosphate group coordination [7] [2]. In Src-type SH2 domains, additional basic residues at position αA2 create an extended cationic surface that facilitates membrane association, while in SAP-like SH2 domains, a basic residue at position βD6 may serve a similar function [2].
Disease-associated mutations frequently localize within lipid-binding pockets of SH2 domains, underscoring the physiological importance of these interactions [7]. Targeting these lipid-protein interactions represents a promising therapeutic strategy, as demonstrated by the development of non-lipidic inhibitors against SYK kinase that disrupt its membrane association and scaffolding functions [7] [3].
SH2 Domains in Biomolecular Condensates
Phase Separation Mechanisms
Biomolecular condensates are membrane-less organelles formed through liquid-liquid phase separation (LLPS), enabling spatial and temporal organization of cellular components [42] [43]. SH2 domains contribute to condensate formation through multivalent interactions with other modular domains (e.g., SH3 domains) and phosphorylated signaling proteins [7] [3].
These multivalent interactions drive the assembly of higher-order structures that reduce the critical concentration required for phase separation. In T-cell receptor signaling, interactions among GRB2, Gads, and the LAT adapter protein undergo LLPS, enhancing signaling efficiency by concentrating components [7]. Similarly, in kidney podocytes, phase separation of adapter protein NCK increases membrane dwell time of N-WASP and Arp2/3 complexes, promoting actin polymerization [7].
Post-translational modifications, particularly phosphorylation, dynamically regulate condensate assembly and disassembly [7] [42]. Phosphotyrosine-driven protein condensation can couple with membrane lipid phase transitions, creating organized signaling platforms that enhance signaling specificity and efficiency [42].
Table 3: SH2 Domain-Containing Proteins in Biomolecular Condensates
| Condensate Complex | SH2-Containing Proteins | Biological Role |
|---|---|---|
| FGFR2:SHP2:PLCγ1 | SHP2, PLCγ1 | Enhances RTK signaling activity |
| LAT-GRB2-SOS1 | ZAP70, LCK, GRB2, PLCγ1 | Promotes T-cell activation and phosphorylation |
| N-WASP–NCK | NCK | Facilitates actin polymerization in T-cell signaling and kidney podocytes |
| SLP65, CIN85 | SLP65 | Mediates B-cell receptor signaling |
Regulatory Mechanisms
Cellular membranes serve as nucleation sites for biomolecular condensates, reducing the critical concentration for phase separation by orders of magnitude—from micromolar levels in bulk solution to nanomolar concentrations at membrane surfaces [42]. This enhancement occurs through several mechanisms: membrane confinement effects that elevate local protein concentrations, specific binding interactions between condensate-forming proteins and membrane lipids, and cooperative stabilization of protein assemblies via membrane anchoring [42].
The thermodynamic coupling between protein phase separation and lipid domain formation represents a critical regulatory mechanism. Protein condensates can induce or enhance lipid phase separation by locally concentrating membrane-binding proteins, while lipid domains provide organized platforms that reduce energetic barriers for protein condensation [42]. Environmental factors such as pH, ionic strength, and calcium concentration further modulate these interactions by influencing both protein-protein interactions and membrane physical properties [42].
Experimental Approaches for Investigating Non-Canonical SH2 Functions
Methodologies for Lipid Interaction Studies
Surface Plasmon Resonance (SPR) SPR provides detailed kinetics of SH2 domain binding to membrane lipids. The experimental workflow involves:
- Sensor Chip Preparation: Create supported lipid bilayers (SLBs) on sensor chips using lipids such as DOPC (neutral) or DOPC:DOPG mixtures (anionic) [44].
- Protein Purification: Express and purify SH2 domains or regulatory elements (e.g., Src N-terminal regulatory element - SNRE) in systems co-expressing N-myristoyltransferase for proper lipidation [44].
- Binding Kinetics: Inject purified proteins at varying concentrations (3-20 μM) and monitor association/dissociation in real-time [44].
- Mutational Analysis: Perform alanine scanning mutagenesis of basic residues to identify lipid-binding motifs [44].
Atomic Force Microscopy (AFM) AFM visualizes protein-lipid interactions and condensate formation:
- Sample Preparation: Form SLBs on mica substrates [44].
- Imaging: Scan protein-bound membranes in liquid using tapping mode [44].
- Topographical Analysis: Identify protein clusters and condensates based on height differences [44].
Diagram Title: Lipid Binding Assay Workflow
Phase Separation Characterization Techniques
Fluorescence Recovery After Photobleaching (FRAP) FRAP assesses dynamics within biomolecular condensates:
- Sample Preparation: Express fluorescently tagged SH2 proteins (e.g., GFP fusions) in cells or purify for in vitro studies [43].
- Photobleaching: Apply high-intensity laser to bleach fluorescence in a small region of the condensate [43].
- Recovery Monitoring: Track fluorescence recovery over time using low-intensity laser [43].
- Quantitative Analysis: Calculate recovery half-time and mobile fraction to determine liquid-like properties [43].
OptoDroplet Assay This optogenetics approach probes phase separation propensity:
- Construct Design: Fuse protein of interest to CRY2PHR domain [43].
- Light Activation: Expose cells to blue light (380-500 nm) to induce CRY2 oligomerization [43].
- Condensate Quantification: Monitor condensate formation microscopically and quantify number, size, and kinetics [43].
- Variant Comparison: Test wild-type versus mutant proteins to identify domains critical for phase separation [43].
Diagram Title: Phase Separation Analysis Methods
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for Investigating Non-Canonical SH2 Functions
| Reagent / Technique | Specific Application | Key Function |
|---|---|---|
| Supported Lipid Bilayers (SLBs) | SPR and AFM studies | Mimics native membrane environment for protein-lipid interaction studies |
| Myristoyl-CoA & N-Myristoyltransferase | Protein lipidation | Enables proper N-terminal myristoylation of SH2-containing proteins for membrane association studies |
| Alanine Scanning Mutagenesis | Lipid-binding motif mapping | Identifies critical basic residues required for lipid interactions |
| CRY2 OptoDroplet System | Phase separation induction | Light-controllable system to probe phase separation propensity of SH2 domains |
| FRAP (Fluorescence Recovery After Photobleaching) | Condensate dynamics | Quantifies liquid-like properties and molecular mobility within SH2-containing condensates |
| Phase Diagrams | Condensate characterization | Maps conditions (pH, temperature, concentration) promoting SH2-mediated phase separation |
Therapeutic Implications and Future Perspectives
The emerging roles of SH2 domains in lipid interactions and phase separation open new avenues for therapeutic intervention. Targeting non-canonical SH2 functions offers potential for developing more selective inhibitors with reduced off-target effects compared to traditional kinase inhibitors [7] [43].
Membrane-anchored condensates formed by SH2-containing proteins represent particularly promising targets. For Src kinase, self-association via a conserved lysine cluster in its SH4 domain facilitates condensate formation that modulates its transforming capacity [44]. Disrupting these condensates through small molecules that target lipid-binding interfaces could provide new strategies for inhibiting oncogenic signaling.
Similarly, the development of non-lipidic inhibitors against SYK kinase demonstrates the feasibility of targeting SH2-lipid interactions [7] [3]. These compounds specifically disrupt PIP3-dependent membrane binding required for SYK's scaffolding function, effectively blocking its non-catalytic activation of STAT3/5 signaling pathways [7].
Future research directions should focus on:
- Structural Characterization: High-resolution studies of SH2 domains in complex with membrane lipids.
- Dynamic Monitoring: Real-time visualization of SH2 domain recruitment to biomolecular condensates in live cells.
- Selective Modulation: Development of chemical probes that specifically disrupt pathological phase separation without affecting canonical SH2 functions.
- Computational Modeling: Predictive models for how SH2 domain mutations affect lipid binding and phase separation propensity.
Understanding the structural differences between STAT-type and Src-type SH2 domains will be crucial for developing targeted therapeutic approaches that exploit their distinct lipid binding and phase separation characteristics. As our knowledge of these non-canonical functions expands, so too will opportunities for innovative therapeutic strategies in cancer and other diseases driven by aberrant signaling.
Navigating Complexities: Challenges in SH2 Domain Research and Therapeutic Targeting
Addressing Moderate Binding Affinities and Transient Interaction Dynamics
Src Homology 2 (SH2) domains are modular protein domains of approximately 100 amino acids that serve as crucial "readers" of phosphotyrosine-based cellular signals [3] [45]. These domains specifically recognize and bind to phosphorylated tyrosine (pTyr) residues on target proteins, thereby facilitating the assembly of multiprotein signaling complexes that drive essential cellular processes including development, homeostasis, immune responses, and cytoskeletal rearrangement [3] [7]. The human proteome encodes approximately 110 proteins containing SH2 domains, which can be broadly classified into several functional categories including enzymes, adaptor proteins, docking proteins, transcription factors, and cytoskeletal proteins [3].
SH2 domain-mediated interactions characteristically exhibit moderate binding affinities (typically in the 0.1-10 μM range) and transient dynamics [7]. These properties are not limitations but rather sophisticated adaptations that enable rapid, reversible assembly and disassembly of signaling complexes in response to cellular stimuli [7]. This technical guide examines the structural mechanisms underlying these binding characteristics, with particular emphasis on the distinctions between STAT-type and Src-type SH2 domains, and explores contemporary methodological approaches for investigating these dynamic interactions.
Structural Basis of SH2 Domain Classification and Function
Conserved Architecture and Divergent Lineages
All SH2 domains share a highly conserved tertiary structure consisting of a central anti-parallel β-sheet flanked by two α-helices, forming a compact globular domain [3] [7]. Despite this structural conservation, SH2 domains can be divided into two major evolutionary and structural subgroups: Src-type and STAT-type SH2 domains [7].
Table 1: Comparative Structural Features of Src-type and STAT-type SH2 Domains
| Structural Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB with additional β-strands | αA-βB-βC-βD-αB' without βE/βF strands |
| Additional Elements | Contains βE, βF, and βG strands | Lacks βE and βF strands |
| αB Helix Configuration | Single continuous αB helix | Split into two helices (αB') |
| Representative Proteins | SRC, ABL, ZAP70, PLCγ1 | STAT1, STAT3, STAT5 |
| Functional Specialization | Diverse signaling adaptors and enzymes | Transcription factors requiring dimerization |
The Src-type SH2 domains represent the canonical SH2 structure, containing extra secondary structural elements including beta strands E, F, and G in addition to the core fold [7]. These domains typically feature longer connecting loops, particularly in enzymatic proteins, which contribute to their ligand specificity [7]. In contrast, STAT-type SH2 domains lack the βE and βF strands and exhibit a split αB helix (designated αB') [7]. This structural adaptation is believed to facilitate the dimerization process essential for STAT-mediated transcriptional activation and reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity [5] [7].
Molecular Determinants of Phosphotyrosine Recognition
The primary function of SH2 domains is to specifically recognize and bind phosphorylated tyrosine residues within protein sequences. This recognition occurs through a conserved "two-pronged plug" mechanism involving two adjacent binding sites on the SH2 domain surface [2]:
Phosphotyrosine Binding Pocket: A deep basic pocket located within the βB strand that coordinates the phosphorylated tyrosine residue. This pocket contains a highly conserved arginine residue at position βB5 (part of the "FLVR" motif) that forms a critical salt bridge with the phosphate moiety of pTyr [7] [2]. Mutation of this arginine typically results in a 1,000-fold reduction in binding affinity, highlighting its essential role in pTyr recognition [2].
Specificity Pocket: An adjacent pocket that recognizes amino acids C-terminal to the phosphorylated tyrosine, typically with strong preference for residues at the +3 position relative to pTyr [7] [2]. The structural composition of this pocket varies among SH2 domains and constitutes the primary determinant of binding specificity.
Table 2: Key Residues in SH2 Domain Phosphotyrosine Recognition
| Structural Position | Conserved Feature | Role in pTyr Binding |
|---|---|---|
| βB5 (FLVR motif) | Arg in nearly all human SH2 domains | Forms salt bridge with phosphate group; provides ~50% of binding energy |
| αA2 | Basic residue (Arg/Lys) in Src-like SH2 domains | Coordinates phosphate moiety |
| βD6 | Basic residue (Arg/Lys) in SAP-like SH2 domains | Alternative phosphate coordination site |
| BG and EF Loops | Variable length and sequence | Determine specificity pocket architecture and ligand selectivity |
The combination of high conservation in the pTyr binding pocket with variability in the specificity pocket enables SH2 domains to achieve both universal recognition of phosphotyrosine and specific selection of particular peptide sequences, resulting in the moderate affinities and transient interactions ideal for dynamic cellular signaling [7].
Methodological Approaches for Investigating SH2 Domain Interactions
High-Throughput Affinity Profiling Using Display Technologies
Recent advances in peptide display technologies coupled with next-generation sequencing have revolutionized the quantitative profiling of SH2 domain binding specificities [38] [46]. These approaches enable systematic measurement of binding affinities across vast sequence spaces, providing comprehensive datasets for modeling SH2-ligand interactions.
Bacterial Peptide Display Protocol:
Library Construction: Generate plasmid libraries encoding random peptide sequences with either fixed phosphorylated tyrosine (X(5)YX(5) design) or fully randomized sequences (X(_{11}) design) [38] [46]. The theoretical diversity of these libraries can exceed 10(^13) sequences, with practical diversity typically around 10(^6)-10(^7) variants.
Bacterial Surface Display: Express the peptide library on the surface of bacteria, typically using anchoring domains such as the Aga2p adhesion subunit or other surface proteins [38].
Enzymatic Phosphorylation: Treat the displayed peptide library with tyrosine kinases to phosphorylate tyrosine residues present in the randomized sequences [38] [46].
Affinity Selection: Incubate the phosphorylated peptide library with the SH2 domain of interest (often conjugated to a capture tag such as streptavidin) and isolate bound peptides through multiple rounds of selection under controlled conditions [38].
Deep Sequencing: Use next-generation sequencing to quantitatively compare peptide abundances before and after selection, generating enrichment values for each sequence [38] [46].
Workflow for SH2 Binding Profiling
Quantitative Modeling of Binding Energetics
The enrichment data generated through peptide display experiments can be analyzed using computational frameworks such as ProBound to develop quantitative sequence-to-affinity models [38] [46]. This approach employs free-energy regression to estimate the contribution of each amino acid at each position to the overall binding free energy (ΔΔG).
ProBound Analysis Workflow:
Data Integration: Combine sequencing data from multiple selection rounds and different library designs (e.g., X(5)YX(5) and X(_{11})) to maximize sequence space coverage [38] [46].
Model Training: Use maximum likelihood estimation to learn a free-energy matrix that predicts binding affinity for any peptide sequence within the theoretical sequence space [38]. The model accounts for all possible binding offsets and includes a non-specific binding term to correct for background selection.
Model Validation: Compare predicted affinities with experimental measurements for known SH2 ligands to validate model accuracy [38]. The ProBound framework has demonstrated superior robustness to library design biases compared to simple enrichment-based scoring methods [46].
Biological Application: Apply trained models to predict novel physiological binding partners, identify the impact of disease-associated mutations on SH2-mediated interactions, and guide the design of optimized binding peptides [38].
Extended Functional Capabilities of SH2 Domains
Lipid Binding and Membrane Recruitment
Beyond phosphotyrosine recognition, approximately 75% of SH2 domains have been shown to interact with membrane lipids, particularly phosphoinositides such as phosphatidylinositol-4,5-bisphosphate (PIP(2)) and phosphatidylinositol-3,4,5-trisphosphate (PIP(3)) [3] [47]. These interactions occur through cationic surface patches distinct from the pTyr-binding pocket and are typically flanked by aromatic or hydrophobic side chains [3] [7].
Table 3: Functional Lipid Interactions of Selected SH2 Domains
| SH2 Domain Protein | Lipid Specificity | Functional Role |
|---|---|---|
| ZAP70 | PIP(_3) | Facilitates and sustains interactions with TCR-ζ in T cell signaling |
| SYK | PIP(_3) | Required for scaffolding function and non-catalytic STAT3/5 activation |
| LCK | PIP(2), PIP(3) | Modulates interaction with binding partners in TCR signaling complex |
| ABL | PIP(_2) | Mediates membrane recruitment and modulates Abl activity |
| VAV2 | PIP(2), PIP(3) | Modulates interaction with membrane receptors (e.g., EphA2) |
| TENSIN2 | PIP(_3) | Regulates Abl activity and IRS-1 phosphorylation in insulin signaling |
Lipid binding enables spatiotemporal control of SH2 domain localization and function, particularly in immune cell signaling where recruitment to membrane microdomains is essential for signal propagation [47]. Disease-causing mutations frequently localize within these lipid-binding pockets, highlighting their physiological importance [3].
Role in Biomolecular Condensate Formation
SH2 domain-containing proteins have recently been implicated in the formation of intracellular condensates through liquid-liquid phase separation (LLPS) [3] [7]. Multivalent interactions between SH2 domains and their binding partners drive the assembly of these membrane-less organelles, which enhance signaling efficiency by increasing local concentration of signaling components [3].
Notable examples include:
- T-cell Receptor Signaling: Interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation, enhancing T-cell receptor signaling [3] [7].
- Actin Polymerization: In kidney podocyte cells, phase separation of adapter NCK increases membrane dwell time of N-WASP and Arp2/3 complexes, promoting actin polymerization [3].
Post-translational modifications, including phosphorylation, dynamically regulate the assembly and disassembly of these condensates, adding another layer of control to SH2-mediated signaling networks [3].
Research Reagent Solutions for SH2 Domain Studies
Table 4: Essential Research Tools for SH2 Domain Investigation
| Reagent/Tool | Specifications | Research Application |
|---|---|---|
| Random Peptide Libraries | X(5)YX(5) or X(_{11}) designs; diversity 10(^6)-10(^7) variants | High-throughput specificity profiling |
| Bacterial Display System | Aga2p or other surface anchors; inducible expression | Peptide library presentation and selection |
| Recombinant SH2 Domains | GST- or His-tagged constructs; point mutants (e.g., FLVR Arg) | Binding assays, structural studies, inhibitor screening |
| Tyrosine Kinase Sources | Recombinant kinases (e.g., c-Src); optimized reaction conditions | Peptide library phosphorylation prior to selection |
| Phosphopeptide Arrays | Cellulose-bound peptide spots; comprehensive proteome coverage | Specificity validation and focused screening |
| ProBound Software | Free-energy regression algorithms; multi-round data integration | Quantitative modeling of binding energetics |
The moderate binding affinities and transient interaction dynamics characteristic of SH2 domains represent sophisticated functional adaptations rather than limitations. These properties enable rapid, reversible assembly of signaling complexes essential for cellular responsiveness to environmental cues. The structural divergence between STAT-type and Src-type SH2 domains illustrates evolutionary specialization for distinct physiological roles, from transcriptional regulation to adaptive immune signaling.
Contemporary methodological approaches, particularly high-throughput peptide display coupled with quantitative modeling, have dramatically enhanced our ability to probe the sequence determinants of SH2 binding specificity and affinity. These techniques, combined with growing appreciation of non-canonical SH2 functions in lipid binding and phase separation, provide powerful tools for deciphering the complex role of SH2 domains in health and disease. Furthermore, the increasing recognition of SH2 domains as therapeutic targets underscores the translational importance of understanding their binding energetics and interaction dynamics [3] [48] [7].
Signal Transducer and Activator of Transcription (STAT) proteins are critical transcription factors that mediate cellular responses to cytokines, growth factors, and hormones. The Src Homology 2 (SH2) domain is arguably the most crucial functional module within STAT proteins, serving dual essential roles: it facilitates phosphotyrosine-dependent recruitment to activated receptors and mediates STAT dimerization through reciprocal phosphotyrosine-SH2 interactions. This dimerization is prerequisite for nuclear translocation and DNA binding. The SH2 domain achieves this through a highly conserved structure that specifically recognizes phosphorylated tyrosine motifs, making it a nexus for STAT regulation and a hotspot for pathogenic mutations in human disease [7] [49].
STAT proteins encompass both STAT-type and Src-type SH2 domains, which have evolved distinct structural characteristics reflecting their specialized functions. STAT-type SH2 domains, which lack the βE and βF strands found in Src-type domains and feature a split αB helix, are structurally adapted for the specific dimerization requirements of STAT transcription factors [7]. This structural divergence from Src-type SH2 domains represents an important evolutionary adaptation for STAT-specific functions in transcriptional regulation.
This technical review examines the disease implications of mutations within the SH2 domains of STAT3 and STAT5, with particular focus on their structural consequences, experimental methodologies for functional characterization, and emerging therapeutic strategies targeting these critical domains.
Structural Architecture of STAT SH2 Domains
Conserved Structure and Ligand Recognition
All SH2 domains share a conserved fold comprised of a central three-stranded antiparallel beta-sheet flanked by two alpha helices, forming a compact structure of approximately 100 amino acids. The fundamental architecture follows an αA-βB-βC-βD-αB pattern, though many SH2 domains contain additional secondary structural elements [7].
The phosphotyrosine (pY) binding pocket is located within the βB strand and features a nearly invariant arginine residue at position βB5 (part of the FLVR motif) that forms a critical salt bridge with the phosphate moiety of phosphorylated tyrosine residues. This specific interaction provides the fundamental binding energy and specificity for SH2 domain interactions [7].
Table 1: Key Structural Features of STAT-type vs. Src-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB1-αB2 | αA-βB-βC-βD-αB |
| βE and βF Strands | Absent | Present |
| αB Helix | Split into two helices | Single continuous helix |
| C-terminal Loops | Simplified | More extensive |
| Primary Function | STAT dimerization | Diverse signaling interactions |
| Representative Proteins | STAT1, STAT3, STAT5A, STAT5B | SRC, ABL, ZAP70, SYK |
Determinants of Binding Specificity
SH2 domain binding is characterized by high specificity for cognate pY-containing ligands with moderate binding affinity (Kd typically 0.1-10 μM). This affinity range allows for specific but transient interactions suitable for dynamic signaling processes. Specificity is determined by interactions between the SH2 domain and the three to five amino acid residues C-terminal to the phosphotyrosine, with particular importance placed on the +3 residue for STAT SH2 domains [7].
The structural elements governing specificity include the EF loop (joining β-strands E and F) and the BG loop (joining α-helix B and β-strand G), which control access to ligand specificity pockets. These regions show considerable variation among different STAT proteins, contributing to their distinct signaling specificities [7].
STAT5B SH2 Domain Mutations: Y665 as a Critical Hotspot
Functional Impact of Y665 Mutations
Tyrosine 665 within the STAT5B SH2 domain represents a critical mutation hotspot identified in human T-cell leukemias. Research has characterized two specific missense mutations at this residue: substitution with phenylalanine (Y665F) or histidine (Y665H). Despite their proximity, these mutations produce strikingly different functional consequences [50] [51].
The Y665F mutation functions as a gain-of-function (GOF) variant, leading to enhanced STAT5 phosphorylation, increased DNA binding capacity, and elevated transcriptional activity following cytokine activation. In contrast, the Y665H mutation acts as a loss-of-function (LOF) variant, impairing normal STAT5 activation and resembling a null phenotype in functional assays [51].
Table 2: Functional Characterization of STAT5B SH2 Domain Mutations
| Parameter | STAT5BY665F (GOF) | STAT5BY665H (LOF) | Wild-type STAT5B |
|---|---|---|---|
| Tyrosine Phosphorylation | Enhanced | Diminished | Normal |
| DNA Binding | Increased | Impaired | Normal |
| Transcriptional Activity | Elevated | Reduced | Normal |
| Enhancer Establishment | Accelerated | Impaired | Normal |
| CD8+ T-cell Population | Expanded | Diminished | Normal |
| CD4+ Treg Population | Expanded | Diminished | Normal |
| Mammary Gland Development | Accelerated | Failed (initial pregnancy) | Normal |
| Lactation Capability | Normal | Impaired (initial pregnancy) | Normal |
In Vivo Pathophysiological Consequences
Mouse models harboring these human mutations demonstrate their profound physiological impact. STAT5BY665F knock-in mice exhibit accelerated mammary gland development during pregnancy and altered immune populations characterized by accumulation of CD8+ effector/memory T cells and CD4+ regulatory T cells. Conversely, STAT5BY665H knock-in mice fail to develop functional mammary tissue during initial pregnancy, resulting in lactation failure, and show diminished CD8+ effector/memory and CD4+ regulatory T cells [50] [51].
Notably, the STAT5BY665H phenotype demonstrates plasticity, as persistent hormonal stimulation through two successive pregnancies can establish functional enhancer structures, restore gene expression programs, and enable successful lactation. This recovery highlights the resilience of developmental programs despite initial SH2 domain impairment [50].
STAT3 SH2 Domain Mutations in Human Disease
Mutation Spectrum and Functional Consequences
The STAT3 SH2 domain represents another major mutation hotspot in human disease. Both germline and somatic mutations within this domain have been associated with diverse pathological conditions including autosomal-dominant hyper-IgE syndrome (AD-HIES), large granular lymphocytic leukemia (LGL), and other immunodeficiencies [52].
A specific five-amino-acid deletion in the SH2 domain (STAT3G656_M660del) has been investigated in mouse models designed to mimic human AD-HIES (Job's syndrome). Surprisingly, while this deletion resulted in frequency changes in several immune populations measured by complete blood count and flow cytometry, it did not recapitulate the characteristic hyper-IgE phenotype of human AD-HIES. This suggests significant species-specific differences in STAT3 function and highlights the limitations of animal models for certain human STAT pathologies [52].
Structural analysis of the STAT3G656_M660del mutation reveals significant alterations in protein architecture that potentially affect the neighboring Y705 phosphorylation site, which is critical for STAT3 activation and dimerization. This structural disruption likely explains the immune dysregulation observed in both human patients and mouse models [52].
Sexual Dimorphism in STAT3 Mutation Effects
The STAT3G656_M660del mouse model demonstrates sexually dimorphic immune dysregulation, with differential effects between male and female animals. This sexual dimorphism highlights the complex interplay between STAT signaling and endocrine factors, an important consideration for understanding the variable presentation of STAT-associated diseases in human populations [52].
Experimental Approaches for SH2 Domain Mutation Analysis
In Silico Modeling and Structural Analysis
Computational approaches provide powerful tools for initial characterization of SH2 domain mutations. For STAT5B mutations, in silico modeling can predict divergent energetic effects on homodimerization with a range of pathogenicity. Molecular dynamics simulations enable detailed analysis of mutation-induced structural perturbations and their functional consequences [53] [51].
For example, molecular dynamics simulations of SH2 domain-containing phosphatase 2 (SHP2) have revealed how specific mutations affect protein stability and dynamics. Similar approaches can be applied to STAT SH2 domains, examining root-mean-square deviations (RMSD), root-mean-square fluctuations (RMSF), and dynamic cross-correlation matrices (DCCM) to quantify mutation-induced structural changes [53].
In Vitro Functional Characterization
Primary T-cell assays represent a crucial experimental system for evaluating STAT5B SH2 domain mutations. Introducing mutant STAT5B into primary T cells enables comprehensive assessment of phosphorylation kinetics, DNA binding capacity, and transcriptional activity in response to cytokine stimulation. These assays directly demonstrate the GOF nature of Y665F and LOF characteristics of Y665H [51].
Advanced transcriptomic and epigenomic analyses, including RNA-seq and ChIP-seq for H3K27ac and STAT5 binding, identify how SH2 domain mutations alter enhancer establishment and function. These approaches have revealed that STAT5BY665H impairs enhancer formation and alveolar differentiation, while STAT5BY665F elevates enhancer formation and accelerates mammary development [50].
Genetically Engineered Mouse Models
Knock-in mouse models with precise human mutations represent the gold standard for investigating the pathophysiological impact of STAT SH2 domain mutations in vivo. These models enable comprehensive analysis of developmental, immunological, and oncological consequences in multiple tissue contexts [50] [52] [51].
For STAT5B, knock-in models have revealed tissue-specific phenotypes in mammary gland development and immune system function. The surprising divergence between human STAT3 mutation phenotypes and mouse models highlights the importance of species-specific considerations when extrapolating results from murine systems to human pathology [52].
Research Reagent Solutions for STAT SH2 Domain Studies
Table 3: Essential Research Reagents for STAT SH2 Domain Investigation
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Engineered Cell Lines | STAT-deficient cells, Reporter gene assays (Luciferase), Primary T cells | Functional characterization of STAT mutations, High-throughput inhibitor screening | Ensure proper cytokine responsiveness, Validate STAT dependency |
| Animal Models | STAT knock-in mice (e.g., STAT5BY665F/Y665H), Tissue-specific conditional knockouts | In vivo pathophysiological analysis, Developmental studies, Therapeutic testing | Monitor strain-specific backgrounds, Consider sexual dimorphism |
| Antibodies | Phospho-STAT specific antibodies, Total STAT antibodies, ChIP-validated antibodies | Western blotting, Immunofluorescence, Flow cytometry, Chromatin immunoprecipitation | Verify specificity for target STAT, Validate for specific applications |
| Recombinant Proteins | Wild-type and mutant SH2 domains, Full-length STAT proteins | Structural studies, Biophysical binding assays, Crystallography | Maintain phosphorylation status, Ensure proper folding |
| Computational Tools | Molecular dynamics simulations, Docking algorithms, Free energy calculations | Predicting mutation effects, Inhibitor design, Structural analysis | Validate force fields, Use experimental structures as templates |
Targeting Strategies for SH2 Domain Pathologies
Emerging Therapeutic Approaches
Targeting SH2 domains therapeutically represents a significant challenge due to the relatively flat and extensive protein-protein interaction surfaces involved. However, several innovative strategies are emerging:
Monobody technology has shown promise for specific targeting of SH2 domain-containing proteins. The monobody Mb11 demonstrates exceptional binding affinity (Kd = 2.7 nM) for the SHP2 phosphatase domain, with significantly reduced affinity for the C459S mutant (Kd = 120 nM). Similar approaches could be developed for STAT SH2 domains [53].
Small molecule inhibitors that target lipid-binding capabilities of SH2 domains represent another innovative approach. Nonlipidic small molecules have been developed that specifically and potently inhibit lipid protein interactions, potentially producing selective inhibitors for various kinases possessing SH2 domains [7].
Stabilization-based inhibitors that exploit the autoinhibitory conformations of proteins like SHP2 may also be applicable to STAT proteins. Such approaches could potentially stabilize inactive states and prevent pathological activation [53].
Experimental Pathway Analysis
The SH2 domains of STAT3 and STAT5 represent critical functional modules whose mutational disruption leads to diverse human diseases including immunodeficiencies, developmental disorders, and hematologic malignancies. The precise structural location and nature of SH2 domain mutations determine their functional consequences, with specific residues like STAT5B Y665 capable of producing either gain-of-function or loss-of-function phenotypes depending on the substituting amino acid.
Comprehensive characterization of these mutations requires integrated approaches combining in silico predictions, in vitro functional assays, and in vivo animal models, while acknowledging potential species-specific differences in STAT function. Emerging targeting strategies including monobodies, small molecule inhibitors, and stabilization approaches offer promising avenues for therapeutic intervention against pathological SH2 domain signaling.
Future research should focus on elucidating the structural determinants of mutation-specific phenotypes, developing more refined animal models that better recapitulate human disease, and advancing innovative therapeutic strategies that can specifically target pathological STAT signaling while preserving essential physiological functions.
Protein-protein interactions (PPIs) represent a formidable frontier in drug discovery, often characterized by large, relatively flat binding surfaces that lack defined pockets for conventional small-molecule binding [54]. Among these challenging targets, Src Homology 2 (SH2) domains stand as archetypical "readers" of phosphotyrosine (pY) signaling, playing pivotal roles in cellular processes including development, homeostasis, cytoskeletal rearrangement, and immune responses [3] [12]. These compact protein modules of approximately 100 amino acids specifically recognize sequences containing phosphorylated tyrosine, thereby facilitating phosphorylation-dependent PPIs that propagate critical cellular signals [3] [55].
The human proteome encodes roughly 110 proteins containing SH2 domains, which can be broadly classified into several functional groups including enzymes, adaptor proteins, docking proteins, and transcription factors [3] [12]. What makes SH2 domains particularly fascinating from a structural and therapeutic perspective is their division into two major subgroups: Src-type and STAT-type SH2 domains. These subgroups exhibit significant structural differences that dictate their functional mechanisms and present distinct challenges and opportunities for therapeutic targeting [7]. This review examines the structural basis of these differences and explores emerging strategies to overcome the inherent challenges of targeting these critical PPIs.
Structural Biology of SH2 Domains: STAT-type vs. Src-type
Conserved Architecture and Key Differences
All SH2 domains share a conserved structural fold despite having as little as ~15% pairwise sequence identity among family members [3] [7]. The fundamental architecture consists of a three-stranded antiparallel beta-sheet flanked on each side by an alpha helix, forming an αA-βB-βC-βD-αB sandwich structure [7]. The N-terminal region contains a deep pocket within the βB strand that binds the phosphate moiety of phosphotyrosine, featuring an invariant arginine residue (at position βB5) that directly engages pY through a salt bridge [3] [7].
The critical structural divergence between STAT-type and Src-type SH2 domains lies in their C-terminal regions. STAT-type SH2 domains lack the βE and βF strands and their adjoining loops that are present in Src-type domains [7]. Additionally, the αB helix in STAT domains is split into two separate helices. This structural simplification is likely an adaptation that facilitates the dimerization essential for STAT-mediated transcriptional regulation [7]. In contrast, Src-type domains typically contain more extensive secondary structures including beta strands E, F, and G, with variable loop regions that contribute to ligand binding specificity [7].
Table 1: Structural and Functional Comparison of Src-type vs. STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB sandwich with additional β strands | αA-βB-βC-βD-αB sandwich without βE/βF strands |
| βE and βF Strands | Present | Absent |
| αB Helix | Single continuous helix | Split into two helices |
| CD-loop Length | Variable; longer in enzymatic proteins | Typically shorter |
| Primary Function | Diverse signaling roles | Dimerization for transcriptional regulation |
| Binding Affinity (Kd) | 0.1-10 μM range [7] | 0.1-10 μM range [7] |
| Evolutionary Origin | Animal multicellularity | Predate animal multicellularity (e.g., Dictyostelium) [7] |
Specificity Determinants and Binding Characteristics
Both SH2 domain types recognize their ligands through a combination of conserved phosphotyrosine engagement and sequence-specific interactions C-terminal to the phosphorylated tyrosine. The binding is characterized by moderate affinity (Kd typically 0.1-10 μM) with high specificity toward cognate pY ligands [7]. This balance allows for specific yet reversible interactions suitable for dynamic cellular signaling.
The EF loop (joining β-strands E and F) and BG loop (joining α-helix B and β-strand G) play crucial roles in determining binding selectivity in Src-type SH2 domains by controlling access to ligand specificity pockets [7]. In STAT-type domains, which lack these structural elements, alternative mechanisms for specificity determination have evolved.
Diagram 1: Structural classification of SH2 domains
Experimental Approaches for SH2 Domain Profiling and Targeting
Specificity Profiling Technologies
Deciphering the phosphotyrosyl peptide motif recognized by an SH2 domain is essential for understanding its cellular function and developing targeted inhibitors [56]. Several high-throughput experimental approaches have been developed to define SH2 domain specificity:
Oriented Peptide Array Library (OPAL) Screening: This classical approach involves screening SH2 domains against arrays of immobilized phosphopeptides. Researchers have successfully cloned all 120 SH2 domains identified in the human genome and determined the phosphotyrosyl peptide binding properties of 76 SH2 domains using this method [56]. The technique enabled definition of selectivity for 43 SH2 domains and refinement of binding motifs for another 33 SH2 domains, revealing novel binding motifs such as the BRDG1 SH2 domain that specifically selects for a bulky, hydrophobic residue at P+4 relative to the phosphotyrosine [56].
Bacterial Peptide Display with Next-Generation Sequencing: This innovative method combines bacterial display of genetically-encoded peptide libraries, enzymatic phosphorylation of displayed peptides, affinity-based selection, and next-generation sequencing (NGS) [38]. The approach employs multi-round affinity selection on random phosphopeptide libraries to generate NGS data suitable for training quantitative models that predict binding free energy across the full theoretical ligand sequence space [38].
Integrated Experimental-Computational Framework using ProBound: A coordinated strategy employs the ProBound statistical learning method to build sequence-to-affinity models from peptide display data [38]. This method can learn a model that predicts binding free energy relative to the optimal sequence for any peptide sequence, assuming additivity of binding free energy over all residue positions in the peptide [38].
Table 2: Key Experimental Techniques for SH2 Domain Binding Characterization
| Technique | Throughput | Key Measured Parameter | Applications | Limitations |
|---|---|---|---|---|
| Oriented Peptide Array Library (OPAL) | 76 SH2 domains profiled [56] | Binding specificity motifs | SMALI prediction algorithm [56] | Semi-quantitative, surface-bound peptides |
| Bacterial Peptide Display + NGS | 10^6-10^7 sequences/library [38] | Relative binding free energy (ΔΔG) | Predicting impact of phosphosite variants [38] | Requires specialized library construction |
| ProBound Modeling | Full theoretical sequence space [38] | Quantitative affinity prediction | Novel phosphosite target identification [38] | Model dependent (assumes additivity) |
| Surface Plasmon Resonance | Low to medium | Binding kinetics (Kd, Kon, Koff) | Validation of putative interactions | Lower throughput, requires purified components |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for SH2 Domain Studies
| Reagent / Tool | Function / Application | Key Features / Examples |
|---|---|---|
| SH2 Domain Constructs | Recombinant protein production | 120 human SH2 domains cloned [56]; various expression systems |
| Phosphopeptide Libraries | Specificity profiling | Oriented peptide arrays [56]; random peptide libraries for display [38] |
| Non-hydrolyzable pY Mimetics | Inhibitor design | pTyr bioisosteres; phosphonodifluoromethyl phenylalanine [55] |
| ProBound Software | Data analysis and modeling | Free-energy regression; predicts binding for any sequence [38] |
| SMALI Algorithm | Binding partner prediction | Web-based program; correlates score with binding energy [56] |
| Lipid Vesicles | Membrane interaction studies | PIP2/PIP3-containing membranes; 75% of SH2 domains bind lipids [3] |
Diagram 2: High-throughput SH2 specificity profiling workflow
Therapeutic Targeting Strategies for SH2 Domains
Conventional Approaches and Challenges
Traditional efforts to develop SH2 domain inhibitors have faced several formidable challenges. The highly peptidic nature of early lead compounds and the requirement for phosphotyrosine (pTyr) for high-affinity binding presented significant obstacles to developing cellularly active inhibitors [55]. Additionally, the presence of multiple SH2-containing proteins in cells creates selectivity problems, as off-target effects could disrupt multiple signaling pathways [55].
The pTyr-binding pocket presents particular difficulties for drug design. This pocket contains a complex hydrogen-bonding network within a highly positively charged environment, making the development of effective pTyr bioisosteres particularly challenging [55]. Early strategies focused on incorporating non-peptide replacements amino-terminal to the pTyr, resulting in inhibitors with increased affinity relative to their cognate peptide sequences [55].
Significant progress has been made in targeting specific SH2 domains, particularly those of Src and Grb2. For Src, several non-peptide templates have been developed with high affinity, including compounds incorporating bone-targeting phosphotyrosine bioisosteres that have yielded in vivo active antiresorptive agents [55]. Similarly, high-affinity Grb2 SH2 inhibitors with novel phosphotyrosine replacements have demonstrated cellular activities consistent with anticancer agents [55].
Emerging Paradigms in SH2 Domain Targeting
Recent research has revealed novel aspects of SH2 domain biology that open alternative avenues for therapeutic intervention:
Targeting Lipid Interactions: Nearly 75% of SH2 domains interact with lipid molecules in the membrane, with particular tendency toward phosphatidylinositol-4,5-bisphosphate (PIP2) or phosphatidylinositol-3,4,5-trisphosphate (PIP3) [3]. Studies have identified cationic regions close to the pY-binding pocket as lipid-binding sites, often flanked by aromatic or hydrophobic amino acid side chains [3]. Disease-causing mutations frequently localize within these lipid-binding pockets, highlighting their functional importance [3]. Researchers have successfully developed nonlipidic inhibitors of Syk kinase that target these lipid-protein interactions, suggesting this approach could yield potent, selective inhibitors for various other SH2 domain-containing kinases [3].
Liquid-Liquid Phase Separation (LLPS) Modulation: Proteins with SH2 domains have increasingly been linked to the formation of intracellular condensates via protein phase separation [3]. Multivalent interactions between SH2 domains and their binding partners drive condensate formation, with phosphorylation modulating assembly and disassembly. For example, interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation that enhances T-cell receptor signaling [3]. In kidney podocyte cells, LLPS increases the ability of adapter NCK to promote actin polymerization by increasing membrane dwell time of protein complexes [3]. Targeting these phase separation processes represents a novel approach to modulating SH2 domain function.
Covalent Inhibition Strategies: Covalent inhibitors that bind irreversibly to target proteins through covalent bonds offer advantages of sustained inhibition and longer residence time compared to non-covalent inhibitors [54]. This approach has proven successful for other "undruggable" targets like KRAS, with the approval of sotorasib demonstrating the potential of covalent targeting for challenging PPIs [54]. While application to SH2 domains is still emerging, covalent strategies represent a promising frontier.
Computational and AI-Driven Approaches
Advanced computational methods are revolutionizing SH2 domain drug discovery:
Multimodal Deep Learning: Methods like MESM (Multimodal ESM) integrate multiple data sources including protein sequence information, structural data, and point cloud features through Variational Autoencoders to predict PPIs with significantly improved accuracy [57]. These approaches can extract both global and local features from PPI networks, enhancing prediction of SH2 domain interactions [57].
Graph Neural Networks (GNNs): GNN-based architectures including Graph Convolutional Networks (GCNs), Graph Attention Networks (GATs), and GraphSAGE adeptly capture local patterns and global relationships in protein structures [58]. Frameworks like AG-GATCN integrate GAT and temporal convolutional networks to provide robust solutions against noise interference in PPI analysis [58].
Surface-Based Prediction: Methods that learn from molecular surfaces can predict PPIs not found in nature, including interactions induced by small molecules [59]. This capability is particularly valuable for predicting how drug-like compounds might modulate SH2 domain interactions.
Targeting SH2 domains represents both the challenges and promises of PPI-focused drug discovery. The structural differences between STAT-type and Src-type SH2 domains illustrate how evolutionary adaptations have shaped specialized functions within a conserved structural framework. While conventional approaches have made incremental progress against these difficult targets, emerging strategies focusing on lipid interactions, phase separation phenomena, and covalent inhibition offer new therapeutic avenues.
The integration of advanced computational methods, particularly multimodal deep learning and graph neural networks, with high-throughput experimental profiling is accelerating our ability to predict and target SH2 domain interactions with unprecedented precision. As these technologies mature and our understanding of SH2 domain biology deepens, the pharmaceutical landscape for targeting these critical signaling modules is likely to transform, potentially yielding novel therapeutics for cancer, immune disorders, and other diseases driven by aberrant tyrosine kinase signaling.
The journey to effectively drug SH2 domains has exemplified the broader challenge of targeting protein-protein interfaces, but recent advances suggest that these obstacles are not insurmountable. With continued innovation in both experimental and computational approaches, the therapeutic targeting of SH2 domains may soon transition from formidable challenge to clinical reality.
Src Homology 2 (SH2) domains are protein interaction modules that recognize phosphorylated tyrosine (pTyr) motifs, playing a fundamental role in intracellular signal transduction. The human proteome encodes approximately 110 proteins containing around 120 SH2 domains, which are often highly conserved, presenting a significant challenge for developing selective inhibitors. Recent advances in structural biology and screening technologies have illuminated the precise molecular determinants of SH2 domain specificity, particularly the critical role of surface loops in controlling access to binding pockets. This whitepaper details the structural mechanisms governing SH2 domain selectivity, with a specific focus on the distinctions between Src-type and STAT-type SH2 domains. It further provides a comprehensive guide to modern methodologies for inhibitor discovery, including experimental protocols and computational approaches, offering a strategic framework for the development of next-generation therapeutics targeting SH2 domain-mediated interactions in cancer and immune disorders.
SH2 domains are approximately 100-amino-acid protein modules that specifically bind to phosphorylated tyrosine residues, facilitating the assembly of multiprotein signaling complexes [7] [12]. They are crucial for transmitting signals from receptor tyrosine kinases (RTKs) and other signaling molecules, thereby regulating essential cellular processes such as proliferation, differentiation, and survival [60]. The human genome encodes 120 SH2 domains distributed across 110 proteins, making them one of the largest families of phosphopeptide-binding modules [20] [12]. Dysregulation of SH2-mediated interactions is implicated in a wide range of diseases, particularly cancers and immune disorders, rendering them attractive therapeutic targets [7] [61].
A major obstacle in drug development is achieving selectivity when targeting individual SH2 domains within this large, conserved family. Promiscuous inhibitors risk disrupting multiple signaling pathways, leading to off-target effects. Success, therefore, hinges on a deep understanding of the subtle structural differences that confer unique binding specificities to each SH2 domain.
Structural Basis of SH2 Domain Specificity
Conserved Architecture and Variable Loops
All SH2 domains share a conserved core fold: a central three-stranded antiparallel β-sheet flanked by two α-helices, often described as a "sandwich" structure (αA-βB-βC-βD-αB) [7] [20]. The phosphotyrosine (pY) binding pocket is highly conserved and features an invariant arginine residue (at position βB5 in the FLVR motif) that forms a salt bridge with the phosphate moiety of the pY residue [7] [61].
Despite this conserved scaffold, different SH2 domains recognize distinct peptide sequences C-terminal to the pY residue. This specificity is primarily determined by surface loops that connect the secondary structural elements [20]. These loops, which exhibit significant sequence and conformational variability, control access to key specificity pockets that accommodate residues at the P+1, P+2, P+3, and P+4 positions relative to the pY.
Table 1: Key Specificity-Determining Loops in SH2 Domains
| Loop Name | Connects | Role in Specificity |
|---|---|---|
| EF Loop | β-strands E and F | Defines the shape and accessibility of the P+3 binding pocket; can physically block the pocket in some domains [20]. |
| BG Loop | α-helix B and β-strand G | Works in concert with the EF loop to form the hydrophobic P+3 pocket; can also be involved in P+4 binding [20]. |
| D'E Loop | β-strands D' and E | Contributes to binding site electrostatic environment; in Grb7 family, forms a highly acidic region of unknown function [60]. |
Src-type vs. STAT-type SH2 Domains: A Structural Classification
SH2 domains can be divided into two major subgroups based on their structural features: Src-type and STAT-type. This distinction is critical for understanding their evolutionary history and functional specialization [7] [5].
Table 2: Structural and Functional Comparison of Src-type and STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Representative Members | Src, Fyn, Grb2, SHIP1/2 [7] [62] | STAT1, STAT3, STAT5A [20] |
| Core Structure | αA-βB-βC-βD-αB, plus additional β-strands (βE, βF, βG) [7] | αA-βB-βC-βD-αB; lacks βE and βF strands [7] [5] |
| αB Helix | Single continuous helix [7] | Split into two helices (αB and αB') [7] [5] |
| Key Loops | Contain EF and BG loops that define P+3/P+4 pockets [20] | Lacks the EF loop; has a more open BG loop conformation [20] |
| Primary Function | Induce proximity in signaling cascades [7] | Facilitate dimerization and nuclear translocation for transcriptional activation [7] |
| Ligand Preference | Generally recognize pY followed by hydrophobic residues at P+3 [20] | Prefer pY followed by a Gln at P+3 (e.g., pYxxQ motif in STAT3) [20] |
The following diagram illustrates the key structural differences between these two SH2 domain subtypes:
The concept of loop-controlled access to binding pockets is a fundamental tenet of SH2 domain specificity [20]. For instance, in Group IA/IB SH2 domains, the EF and BG loops form an accessible hydrophobic pocket that engages a P+3 residue. In contrast, in Group IC SH2 domains like Grb2, a bulky tryptophan residue in the EF loop physically blocks the P+3 pocket, forcing the peptide ligand to adopt a β-turn conformation and allowing for specific recognition of an asparagine at the P+2 position [20]. For SH2 domains that recognize a hydrophobic residue at P+4 (e.g., BRDG1), the canonical P+3 pocket is occupied by an intramolecular residue, revealing an alternative "pentagon basket" pocket that accommodates the P+4 side chain [20].
Experimental and Computational Methodologies
Determining SH2 Domain Specificity and Kinetics
A critical first step in designing selective inhibitors is to comprehensively characterize the binding preferences and kinetics of the target SH2 domain.
Oriented Peptide Array Library (OPAL) Screening This high-throughput method determines the sequence specificity of an SH2 domain [20].
- Procedure: A library of immobilized peptides with a fixed phosphotyrosine and degenerate residues at downstream positions (e.g., pY-X-X-X) is synthesized. The SH2 domain of interest is applied to the array, and its binding is detected. The sequences of bound peptides are analyzed to derive a consensus binding motif [20].
- Output: A specificity profile revealing preferred residues at positions C-terminal to pY (e.g., P+1 to P+4).
Surface Plasmon Resonance (SPR) SPR is used to quantify the affinity and kinetics of SH2 domain interactions with phosphopeptides [62].
- Procedure: A phosphopeptide is immobilized on a sensor chip. Purified SH2 domain is flowed over the chip at varying concentrations. The instrument measures the association and dissociation rates in real-time.
- Output: Binding affinity (Kd), association rate (kₐₙ), and dissociation rate (kₐₜₜ) [62]. This can reveal critical kinetic differences; for example, the SHIP1 and SHIP2 SH2 domains have similar affinities, but SHIP2 exhibits slow-binding kinetics due to cis-trans proline isomerization [62].
The following diagram outlines a combined workflow for characterizing SH2 domain binding and leveraging the data for inhibitor design:
Structure-Based Inhibitor Design
X-ray Crystallography and NMR Spectroscopy These techniques provide atomic-resolution structures of SH2 domains, either alone or in complex with peptides or inhibitors [60] [63]. This reveals the precise geometry of the binding pocket, hydrogen-bonding networks, and hydrophobic contacts, which are essential for structure-based drug design.
Virtual Screening and Molecular Dynamics (MD)
- Molecular Docking: Computational tools like AutoDock Vina or Smina are used to screen large virtual libraries of small molecules against the target SH2 domain structure. The binding pose and predicted affinity of each compound are calculated [61].
- Molecular Dynamics (MD) Simulations: This technique simulates the physical movements of atoms over time, providing insights into the flexibility and stability of the SH2 domain and its complexes. It can reveal transient pockets and conformational changes not visible in static crystal structures [61].
- MM/PBSA Calculations: This method uses snapshots from MD trajectories to estimate binding free energies, helping to rank the potency of potential inhibitors [61]. A recent study targeting the N-SH2 domain of SHP2 used this combined approach to identify the drug Irinotecan as a promising candidate [61].
Emerging Therapeutic Targeting Strategies
Targeting the SH2 Domain of Bruton's Tyrosine Kinase (BTK)
Recent breakthroughs demonstrate the therapeutic potential of selectively targeting SH2 domains. Recludix Pharma has developed the first-in-class BTK SH2 domain inhibitor (BTK SH2i), representing a novel approach to treat B-cell and mast-cell-mediated diseases like chronic spontaneous urticaria and multiple sclerosis [64].
This inhibitor was developed using a platform combining custom DNA-encoded libraries (DELs), crystallographic structure-guided design, and proprietary biochemical assays [64]. The resulting compound exhibits:
- Exceptional Potency: Kd of 0.055 nM for the BTK SH2 domain.
- Unprecedented Selectivity: >8,000-fold selectivity over off-target SH2 domains, avoiding off-target effects such as TEC kinase inhibition associated with traditional BTK kinase-domain inhibitors [64].
- Durable Pathway Inhibition: A prodrug formulation enables sustained intracellular concentrations and prolonged target engagement [64].
Advanced Modalities for Inhibition
Beyond small molecules, other modalities are being explored to target SH2 domains with high specificity:
- Monobodies: These synthetic binding proteins have been engineered to bind with nanomolar affinity and high selectivity to the SH2 domains of Src-family kinases (SFKs). They can discriminate between the SrcA and SrcB subgroups and have been shown to modulate kinase activity and downstream signaling in cells [63].
- Cyclic Peptides: A high-throughput method for synthesizing and screening support-bound cyclic pY peptide libraries has yielded potent and selective inhibitors for SH2 domains such as Grb2, which show efficacy in disrupting cancer cell growth and actin cytoskeleton organization [62].
Table 3: Key Research Reagent Solutions for SH2 Domain Studies
| Reagent / Tool | Function / Application | Key Feature |
|---|---|---|
| Oriented Peptide Array Library (OPAL) | High-throughput determination of SH2 domain binding motif [20] | Defines consensus sequence C-terminal to pY |
| Surface Plasmon Resonance (SPR) | Label-free analysis of binding affinity and kinetics (Kd, kₐₙ, kₐₜₜ) [62] | Reveals critical differences in binding mechanisms |
| DNA-Encoded Library (DEL) | Discovery of novel small-molecule binders from vast chemical space [64] | Enables ultra-high-throughput screening |
| Monobodies | Engineered protein inhibitors for intracellular targeting [63] | Achieves high selectivity within SH2 subfamilies |
| Molecular Dynamics (MD) Simulations | Computational modeling of protein-ligand dynamics and stability [61] | Provides atomistic insight into binding events |
The strategic inhibition of SH2 domains represents a promising frontier in targeted therapy, particularly for cancer and immunological diseases. The path to achieving sufficient selectivity lies in a deep understanding of structural biology, particularly the distinctions between Src-type and STAT-type domains and the critical role of variable surface loops in controlling binding pocket access. By leveraging a toolkit of advanced methodologies—including OPAL screening, SPR kinetics, structural biology, and computational modeling—researchers can now design inhibitors with unprecedented precision. The recent success in developing a highly selective BTK SH2 domain inhibitor validates this approach and paves the way for a new class of therapeutics that overcome the limitations of traditional kinase inhibitors. Future efforts will likely focus on exploiting unique allosteric mechanisms and further refining our understanding of SH2 domain dynamics in the context of full-length proteins and cellular signaling networks.
Src homology 2 (SH2) domains have long been recognized as critical mediators of phosphotyrosine (pTyr) signaling, with their canonical "two-pronged plug" mechanism providing specificity for pTyr-containing protein ligands [2]. However, emerging research reveals a more complex picture of SH2 domain functionality, including non-canonical lipid-binding capabilities and allosteric regulatory mechanisms [3] [65] [47]. These findings are particularly significant when examined through the lens of structural differences between major SH2 domain subtypes—Src-type and STAT-type domains—which exhibit distinct architectural features that influence their signaling roles and targetability [7] [5]. The growing understanding of these non-canonical functions has opened new avenues for therapeutic intervention in cancers and other diseases driven by aberrant tyrosine kinase signaling. This whitepaper examines emerging strategies that leverage these novel targeting opportunities, focusing specifically on the structural contexts that differentiate SH2 domain subtypes and their implications for drug discovery.
Structural Divergence Between Src-type and STAT-type SH2 Domains
The fundamental structural differences between Src-type and STAT-type SH2 domains represent a critical framework for understanding their distinct biological functions and therapeutic targeting potential.
Comparative Architecture of SH2 Domain Subtypes
Table 1: Structural Comparison of Src-type and STAT-type SH2 Domains
| Structural Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB sandwich with additional β strands | Conserved core but lacking βE and βF strands |
| Beta Strand Composition | Contains βE and βF strands | Lacks βE and βF strands |
| C-terminal Region | βE, βF, βG strands present | Split αB helix (αB1 and αB2) |
| Dimerization Capability | Limited | Enhanced, facilitates STAT dimerization |
| Evolutionary Origin | Later evolution | More ancient, template for SH2 domain evolution |
The canonical Src-type SH2 domain adopts a "sandwich" structure consisting of a three-stranded antiparallel beta-sheet flanked by two alpha helices (αA-βB-βC-βD-αB), with most family members containing extra secondary structural elements including beta strands E, F, and G [3] [7]. In contrast, STAT-type SH2 domains are distinct in that they lack the βE and βF strands as well as the C-terminal adjoining loop, and feature a split αB helix [7]. This structural disparity represents an adaptation that facilitates dimerization, a critical step in STAT-mediated transcriptional regulation, reflecting the ancestral function of SH2 domain-containing proteins that predate animal multicellularity [7] [5].
Diagram 1: Structural and Functional Comparison of SH2 Domain Types
The N-terminal region of both SH2 domain types contains a deep pocket within the βB strand that binds the phosphate moiety, harboring the invariable arginine at position βB5 (part of the FLVR motif) that directly binds to pY residues through a salt bridge [3] [7] [2]. However, the C-terminal regions differ significantly, with Src-type domains containing β strands E, F, and G, while STAT-type domains exhibit modifications that facilitate their unique dimerization functions [7].
Non-Canonical Lipid Binding by SH2 Domains
Prevalence and Mechanisms of Lipid Interaction
Recent research has revealed that approximately 75-90% of SH2 domains interact with lipid molecules in the membrane, with a marked tendency toward phosphatidylinositol-4,5-bisphosphate (PIP2) or phosphatidylinositol-3,4,5-trisphosphate (PIP3) [3] [47]. These interactions occur through cationic surface patches separate from pTyr-binding pockets, allowing SH2 domains to bind lipids and pTyr motifs independently [47]. The lipid-binding sites are typically characterized by cationic regions close to the pY-binding pocket, usually flanked by aromatic or hydrophobic amino acid side chains [3].
Table 2: Lipid-Binding Properties of Selected SH2 Domain-Containing Proteins
| Protein Name | Lipid Specificity | Biological Function of Lipid Association |
|---|---|---|
| SYK | PIP3 | PIP3-dependent membrane binding required for activation of SYK scaffolding function |
| ZAP70 | PIP3 | Facilitates and sustains ZAP70 interactions with TCR-ζ chain |
| LCK | PIP2, PIP3 | Modulates interaction of LCK with binding partners in TCR signaling complex |
| ABL | PIP2 | Membrane recruitment and modulation of Abl activity |
| VAV2 | PIP2, PIP3 | Modulates interaction of VAV2 with membrane receptors (e.g., EphA2) |
| C1-Ten/Tensin2 | PIP3 | Regulation of Abl activity and phosphorylation of IRS-1 in insulin signaling |
Experimental Approaches for Characterizing SH2-Lipid Interactions
Method 1: Lipid Binding Assays Using Surface-Based Platforms
- Purpose: Quantitative assessment of SH2 domain binding to various lipid species
- Procedure: Immobilize purified lipids on membrane strips or glass-supported lipid bilayers; incubate with recombinant SH2 domains; detect binding via fluorescence, surface plasmon resonance, or immunoblotting; determine affinity constants (Kd) through concentration-dependent binding curves
- Key Applications: Mapping lipid specificity profiles, identifying cationic lipid-binding patches, evaluating competition between lipid and pTyr peptide binding [65] [47]
Method 2: Cellular Membrane Translocation Imaging
- Purpose: Visualize and quantify SH2 domain recruitment to cellular membranes
- Procedure: Express fluorescently tagged SH2 domains in live cells; stimulate with growth factors or receptor agonists; monitor subcellular localization via confocal or TIRF microscopy; quantify membrane recruitment kinetics using FRAP or ratio-metric imaging
- Key Applications: Establishing physiological relevance of lipid interactions, assessing spatiotemporal regulation of SH2 domain function [47]
Allosteric Regulation of SH2 Domain-Containing Proteins
Mechanisms of Allosteric Control in Multidomain Proteins
Allosteric regulation represents a sophisticated control mechanism for SH2 domain-containing proteins, particularly those with multiple interaction domains. The growth factor receptor-bound protein 2 (Grb2) exemplifies this principle, with recent investigations revealing that it utilizes intramolecular allosteric communication to modulate binding specificity rather than functioning merely as a passive bridge [66].
Experimental Approach: Double-Mutant Cycle Analysis
- Purpose: Quantitatively assess energetic coupling between domains and residues
- Procedure: Introduce single mutations at putative allosteric sites (X and Y); create double mutant (XY); measure binding affinities (KD) for all variants; calculate coupling energies (ΔΔΔGXY) using the equation: ΔΔΔGXY = ΔΔGP-XY→P - ΔΔGP-XY→P-Y - ΔΔGP-XY→P-X
- Interpretation: Non-zero ΔΔΔG values indicate energetic coupling between residues, revealing allosteric communication pathways [66]
Application of this approach to Grb2 demonstrated that ligand binding to the SH2 domain influences the interaction of the SH3 domain with Gab2 in a manner dependent on ligand identity. Surprisingly, while binding of an Irs-1 mimic to SH2 did not significantly alter SH3 binding kinetics, engagement with a Shp-2 mimicking peptide increased the dissociation constant (KD) of the SH3-Gab2 interaction from 2.3 ± 0.5 μM to 4.3 ± 0.8 μM, revealing ligand-specific allosteric effects [66].
Targeting Allosteric Sites in SHP2 Phosphatase
The protein tyrosine phosphatase SHP2 represents a prominent example of successful allosteric targeting, with its regulation involving complex interdomain interactions between two SH2 domains (N-SH2 and C-SH2) and a phosphatase domain (PTP) [67] [11]. Under basal conditions, SHP2 exists in an autoinhibited state with the N-SH2 domain blocking the PTP active site. Activation involves a rearrangement of the domains that makes the catalytic site accessible, coupled to association between the SH2 domains and cognate proteins containing phosphotyrosines [11].
Diagram 2: Allosteric Regulation and Inhibition of SHP2 Phosphatase
Experimental Approach: Irreversible Allosteric Inhibitor Development
- Purpose: Develop selective allosteric inhibitors targeting non-conserved cysteine residues
- Procedure: Screen compound libraries against wild-type SHP2 PTP domain and C333P mutant; identify leads showing selectivity for wild-type enzyme; optimize electrophilic warheads (cyanoacrylamide → acrylamide) for irreversible binding; evaluate dose-dependent inhibition (IC50 determination)
- Key Findings: Compound 12 (acrylamide derivative) inhibited wild-type SHP2 (IC50 ≈ 35 μM) with high selectivity over C333P mutant, demonstrating negligible inhibition even at 150 μM [67]
This approach successfully targets C333, a nonconserved cysteine residue that lies outside of the active site and represents the key selectivity determinant for SHP2 inhibition [67]. The structural instability of SHP2's active state in solution, with multiple interdomain arrangements being populated, creates opportunities for allosteric intervention that may remain functional regardless of the strength of the SH2/PTP domain interaction in particular SHP2 variants [11].
Emerging Therapeutic Targeting Strategies
Targeting Lipid-Binding Sites
The discovery that SH2 domains serve as lipid-binding modules suggests new therapeutic strategies focused on these non-canonical functions. Research indicates that targeting lipid binding in SH2 domain-containing kinases may offer a promising avenue for developing new small-molecule drugs [3]. Cologna and colleagues have successfully developed nonlipidic inhibitors of Syk kinase, demonstrating that nonlipidic small molecules are capable of specific and potent inhibition of lipid protein interactions (LPI) [3]. This approach could produce potent, selective, and resistance-resistant inhibitors for various other kinases possessing the SH2 domain.
Strategy: Nonlipidic Small Molecule Inhibitors
- Mechanism: Disrupt SH2 domain interaction with membrane lipids without competing for pTyr-binding pocket
- Advantages: Potential for greater selectivity, avoidance of charged pharmacophores that hinder cell permeability, potential to overcome resistance mechanisms
- Applications: SYK inhibition in immune signaling, ZAP70 targeting in T-cell activation [3] [47]
Allosteric Inhibitor Development
The challenges inherent in developing active-site-directed protein tyrosine phosphatase inhibitors have led to increased focus on allosteric approaches. Allosteric sites are typically much less strongly conserved than PTP active sites and do not necessarily privilege charged pharmacophores, offering significant advantages for drug development [67].
Strategy: Covalent Targeting of Nonconserved Cysteines
- Mechanism: Irreversible binding to nonconserved cysteine residues (e.g., C333 in SHP2) in allosteric sites
- Advantages: High selectivity over other PTP family members, potential for sustained target engagement, improved cellular activity
- Applications: SHP2 inhibition in Noonan syndrome, LEOPARD syndrome, and juvenile myelomonocytic leukemia [67]
Peptide-Based Inhibitors with Modified pTyr Mimetics
Recent developments in peptide inhibitors incorporating nonhydrolysable pTyr mimetics offer another approach to targeting SH2 domains. Research on SHP2 inhibitors has demonstrated that incorporation of the pTyr mimetic l-O-malonyltyrosine (l-OMT) results in robust binding affinity to the C-SH2 domain, while the widely used pTyr mimetic phosphonodifluoromethyl phenylalanine (F2Pmp) abolishes binding [68]. This challenges existing notions about pTyr mimetics and suggests they are not general binders of all SH2 domains.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Key Research Reagents and Methodologies for Investigating Non-Canonical SH2 Functions
| Reagent/Methodology | Function/Application | Key Considerations |
|---|---|---|
| Recombinant SH2 Domains | In vitro binding assays, structural studies | Ensure proper folding and phosphorylation state |
| Phosphoinositide Lipid Strips | Lipid binding specificity screening | Include positive and negative controls |
| Biacore/SPR Platforms | Quantitative binding kinetics | Immobilization strategy critical for data quality |
| Cyanoacrylamide Compound Libraries | Allosteric inhibitor screening | Monitor selectivity using mutant controls |
| Double-Mutant Cycle Methodology | Mapping allosteric communication | Requires comprehensive mutagenesis dataset |
| l-OMT Modified Peptides | SH2 domain inhibition studies | Superior to F2Pmp for certain SH2 domains |
| C333-Specific Inhibitors | Selective SHP2 targeting | Irreversible inhibition provides sustained effect |
The emerging strategies focusing on non-canonical lipid-binding and allosteric sites represent a paradigm shift in targeting SH2 domain-containing proteins for therapeutic intervention. The structural differences between Src-type and STAT-type SH2 domains provide important contextual frameworks for understanding their distinct functions and developing targeted approaches. As research continues to elucidate the complex roles of SH2 domains in cellular signaling, including their recently discovered involvement in liquid-liquid phase separation [3] [7], new opportunities will undoubtedly emerge for innovative targeting strategies. The integration of structural biology, biophysical analysis, and chemical biology approaches will be essential for translating these emerging strategies into clinically viable therapeutics for cancer, developmental disorders, and immune diseases driven by aberrant tyrosine phosphorylation signaling.
Context and Validation: Benchmarking STAT and Src SH2 Against the Broader Domain Family
Src homology 2 (SH2) domains represent a critical family of protein interaction modules that specifically recognize phosphotyrosine (pTyr) motifs, forming the backbone of eukaryotic cellular signaling networks. While sharing a conserved structural fold, SH2 domains exhibit remarkable functional and structural diversification into distinct classes, including the canonical Src-type, STAT-type, and atypical variants such as those in the Grb7 family. This technical analysis provides a comprehensive comparison of these SH2 domain subtypes, highlighting their distinctive structural features, binding mechanisms, and functional implications for targeted therapeutic development. Understanding these differences is paramount for exploiting SH2 domains as drug targets in oncology and other disease areas characterized by aberrant tyrosine kinase signaling.
SH2 domains are modular protein components of approximately 100 amino acids that serve as essential "readers" in phosphotyrosine-based signal transduction [37]. The human genome encodes approximately 120 SH2 domains distributed across 110 proteins, representing one of the largest families of specialized recognition modules [69] [36]. These domains function as critical intermediaries by binding with specificity to pTyr-containing motifs on activated receptor tyrosine kinases (RTKs) and other signaling molecules, thereby facilitating the assembly of multiprotein complexes that dictate cellular responses to extracellular stimuli [3] [7].
Despite their conserved primary function of pTyr recognition, SH2 domains have evolved significant structural variations that define their classification and functional specialization. The major classifications include:
- Src-type SH2 domains: Characterized by a complete αβββα fold with additional secondary structural elements, representing the canonical SH2 architecture found in adaptor proteins and cytoplasmic kinases.
- STAT-type SH2 domains: Feature structural adaptations that facilitate their unique role in transcription factor dimerization and nuclear signaling.
- Atypical SH2 domains: Include variants with sequence deviations that alter binding specificity or mechanism, such as those in the Grb7 family and other specialized members.
Table 1: Major SH2 Domain Classes and Their Characteristics
| SH2 Domain Class | Representative Members | Structural Features | Primary Cellular Functions |
|---|---|---|---|
| Src-type | Src, GRB2, PLCγ1 | Complete β-sheet (7 strands), two α-helices, conserved FLVR motif | Signal adaptor functions, kinase regulation, scaffold assembly |
| STAT-type | STAT1, STAT3, STAT5 | Lacks βE/βF strands, split αB helix, specialized for dimerization | Transcription factor activation, nuclear signaling, gene regulation |
| Grb7 Family | Grb7, Grb10, Grb14 | Canonical fold with specificity determinants at βD6 position | Specific RTK recognition, signaling complex assembly |
Structural Organization of Canonical SH2 Domains
Conserved Core Architecture
All typical SH2 domains share a fundamental structural scaffold centered on a central antiparallel β-sheet flanked by two α-helices, forming an αβββα motif [3] [7]. This core structure contains several invariant elements critical for phosphotyrosine recognition:
- A deep positively charged pocket within the βB strand that binds the phosphate moiety of pTyr
- A universally conserved arginine residue at position βB5 (part of the FLVRES motif) that forms bidentate salt bridges with the phosphate group
- A hydrophobic specificity pocket that engages residues C-terminal to the pTyr, primarily at the +1 to +4 positions
The N-terminal region (from αA to βD) is highly conserved across SH2 domains and provides the pTyr-binding pocket, while the C-terminal region (from βD to the C-terminus) exhibits greater structural variability and determines ligand specificity [3] [37]. This structural division allows for both conserved pTyr recognition and diversified target specificity within the same protein fold.
Structural Determinants of Phosphopeptide Recognition
SH2 domains bind their cognate phosphopeptides in an extended conformation that lies perpendicular to the central β-sheet [70] [37]. The binding interface involves two primary interaction sites:
- pTyr-binding pocket: Formed by residues from βB, βC, βD, αA, and the BC loop, with the conserved arginine (ArgβB5) serving as the primary anchor through direct salt bridge formation with the phosphate moiety. Additional positively charged residues (ArgαA2 and LysβD6 in Src SH2) provide supplementary stabilization.
- Specificity pocket: A largely hydrophobic cavity formed by the CD, DE, EF, and BG loops, along with βD and αB, that accommodates peptide residues C-terminal to the pTyr, particularly positions +1 to +3 [70].
The affinity of SH2 domains for their cognate phosphopeptides typically ranges from 0.1-10 μM, representing a balance between binding specificity and the reversibility required for dynamic signaling responses [70] [37].
Comparative Analysis of STAT-type versus Src-type SH2 Domains
Structural Divergence and Classification Criteria
The distinction between STAT-type and Src-type SH2 domains represents a fundamental evolutionary division within the SH2 superfamily. STAT-type SH2 domains lack the βE and βF strands present in Src-type domains and feature a split αB helix (designated αB' and αB) [5] [7]. This structural disparity is believed to be an adaptation that facilitates the dimerization necessary for STAT-mediated transcriptional activation, reflecting the ancestral function of SH2 domain-containing proteins that predate animal multicellularity [7].
Table 2: Structural and Functional Comparison of STAT-type vs. Src-type SH2 Domains
| Characteristic | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Secondary Structure | βA-βG (7 strands), αA, αB | βA-βD (4 core strands), αA, αB' + αB |
| βE/βF Strands | Present | Absent |
| αB Helix | Single continuous helix | Split into two helices (αB' + αB) |
| Loop Structures | Longer CD loops in enzymatic proteins | Adapted for dimerization interface |
| Primary Function | Signal transduction, kinase regulation | Transcription factor dimerization, nuclear transport |
| Representative Proteins | Src, GRB2, ABL1, PLCG1 | STAT1, STAT3, STAT5, STAT6 |
Functional Implications of Structural Differences
The structural variations between STAT-type and Src-type SH2 domains directly correlate with their distinct cellular functions. Src-type domains, with their complete structural elements, serve primarily in cytoplasmic signaling cascades as adaptors, enzymes, and regulators. In contrast, the specialized architecture of STAT-type domains facilitates their unique role in JAK-STAT signaling, where SH2 domain-mediated dimerization is essential for transcriptional activation [5] [7].
Evolutionary analysis suggests that the linker-SH2 domain of STAT represents one of the most ancient and fully developed functional domains, serving as a template for the continuing evolution of the SH2 domain essential for phosphotyrosine signal transduction [5]. This deep evolutionary conservation underscores the fundamental importance of these structural variations in metazoan signaling complexity.
Atypical SH2 Domains: The Grb7 Family and Beyond
Specificity Determinants in the Grb7 Family
The Grb7 family of adapter proteins (including Grb7, Grb10, and Grb14) contains SH2 domains with distinctive binding specificities that illustrate how subtle structural variations can dramatically alter recognition properties. While the Grb7 SH2 domain binds strongly to erbB2 receptors, the closely related Grb14 SH2 domain does not, despite their high sequence similarity [71].
The key determinant of this specificity difference resides at the βD6 position within the SH2 domain. In Grb7, this position is occupied by a leucine residue, while in Grb14 it is a glutamine. Remarkably, a single amino acid substitution (Gln to Leu) at the βD6 position in Grb14 imparts high-affinity erbB2 interaction, while the reverse mutation (Leu to Gln) in Grb7 abrogates binding [71]. This residue therefore represents a critical specificity determinant within the Grb7 family SH2 domains, highlighting how targeted variations in the SH2 fold can generate functional diversity.
Non-Canonical Recognition Mechanisms
Beyond the Grb7 family, other atypical SH2 domains exhibit additional structural and functional specializations. For instance, a small subset of human SH2 domains (including those in RIN2, TYK2, and SH2D5) feature substitutions at the conserved arginine position in the FLVR motif, typically replaced by an aromatic residue [69]. These atypical domains recognize acidic residues other than pTyr (Glu or Asp) through non-canonical binding modes, expanding the functional repertoire of the SH2 superfamily beyond strict phosphotyrosine recognition.
Experimental Approaches for SH2 Domain Characterization
Structural Biology Methodologies
The structural characterization of SH2 domains relies on multiple complementary approaches that provide atomic-level insights into domain architecture and binding mechanisms:
X-ray Crystallography: Has yielded high-resolution structures of approximately 70 unique SH2 domains, primarily in complex with phosphopeptide ligands [3] [7]. This technique reveals the precise atomic coordinates of the SH2 fold and ligand interactions but may be limited by crystallization requirements.
Solution NMR Spectroscopy: Provides dynamic structural information and reveals conformational flexibility under physiological conditions. Recent application to the Drk-SH2 domain (a GRB2 homolog) demonstrated its common SH2 architecture consisting of three β strands imposed between two α helices, while also characterizing site-specific interactions with pY-containing peptides through titration experiments [36]. NMR relaxation experiments further enable analysis of domain dynamics and molecular recognition processes.
Hybrid and Computational Approaches: Emerging methods include:
- AlphaFold Prediction Integration: Structural databases like SH2db now incorporate AlphaFold models alongside experimental structures, enabling comprehensive analysis of SH2 domains lacking experimental characterization [69].
- Contact Mapping Pipelines: Tools like CoDIAC (Comprehensive Domain Interface Analysis of Contacts) systematically extract domain interfaces from experimental and predicted structures, mapping interactions with macromolecules and intraprotein interfaces [72].
- Molecular Dynamics Simulations: Provide insights into conformational dynamics and binding processes, complementing static structural data with temporal information [36].
Diagram 1: Experimental methodologies for SH2 domain characterization. SH2 domain research employs integrated structural, biophysical, functional, and computational approaches to comprehensively understand domain architecture and function.
Binding Affinity and Specificity Assays
Quantifying SH2 domain interactions requires specialized biochemical approaches that measure both affinity and kinetics:
Fluorescence Polarization (FP): Enables high-throughput analysis of SH2-phosphopeptide interactions. Recent studies have employed FP to empirically determine affinities between 93 human SH2 domains and phosphopeptides from receptor tyrosine kinases, revealing over 1000 novel interactions and significantly improving prediction algorithms for SH2 domain binding potentials [73].
Isothermal Titration Calorimetry (ITC): Provides complete thermodynamic profiles of binding interactions, including enthalpy (ΔH), entropy (ΔS), and binding constants (Kd). For example, the affinity of GRB2-SH2 for a pY-containing peptide (VPEpYINQSVPK) was determined to be 0.713 ± 0.145 μM by ITC [36].
Surface Plasmon Resonance (SPR): Measures binding kinetics in real-time, providing association (kon) and dissociation (koff) rates that are critical for understanding dynamic signaling processes.
Table 3: Key Experimental Techniques for SH2 Domain Analysis
| Technique | Key Applications | Information Obtained | Typical Throughput |
|---|---|---|---|
| X-ray Crystallography | Structure determination of SH2-ligand complexes | Atomic-resolution structure, binding interactions | Low |
| Solution NMR | Structure, dynamics, and binding studies | 3D structure, conformational dynamics, binding epitopes | Medium |
| Fluorescence Polarization | Binding affinity screens | Dissociation constants (Kd), specificity profiles | High |
| Isothermal Titration Calorimetry | Thermodynamic characterization | Kd, ΔG, ΔH, ΔS, stoichiometry | Low |
| Surface Plasmon Resonance | Kinetic analysis | Association/dissociation rates, affinity, specificity | Medium |
| Phage Display | Specificity profiling | Binding motif preferences, specificity determinants | High |
Table 4: Key Research Reagents and Resources for SH2 Domain Studies
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Structural Databases | SH2db [69], Protein Data Bank | Access to experimental structures, sequence alignments, generic numbering schemes |
| Prediction Tools | AlphaFold Models [69] [72], CoDIAC [72] | Structure prediction, contact mapping, interface analysis |
| Binding Assay Reagents | Phosphopeptide Libraries, Fluorescent Probes | Specificity profiling, affinity measurements, competition studies |
| Expression Systems | E. coli, Baculovirus, Mammalian Cells | Recombinant SH2 domain production for structural and biophysical studies |
| Specialized Software | Pymol, Maestro, CYANA [36] | Structure visualization, analysis, and calculation |
| NMR Resources | Isotopically Labeled Proteins (15N, 13C) | Resonance assignment, structure determination, dynamics studies |
Emerging Research Directions and Therapeutic Applications
Non-Canonical SH2 Domain Functions
Recent research has revealed unexpected roles for SH2 domains beyond traditional phosphopeptide recognition:
Membrane Lipid Interactions: Nearly 75% of SH2 domains interact with lipid molecules, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [3] [7]. These interactions modulate cellular signaling by facilitating membrane recruitment and influencing enzymatic activity. For example, the PIP3 binding activity of the TNS2 SH2 domain regulates phosphorylation of insulin receptor substrate-1 (IRS-1) in insulin signaling [3].
Liquid-Liquid Phase Separation (LLPS): SH2 domain-containing proteins increasingly link to intracellular condensate formation via multivalent interactions. Studies show that interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation, enhancing T-cell receptor signaling [3] [7]. In podocyte kidney cells, LLPS increases the ability of adapter NCK to promote N-WASP–Arp2/3-mediated actin polymerization [3].
SH2 Domains as Therapeutic Targets
The central role of SH2 domains in signaling pathways dysregulated in disease, particularly cancer, makes them attractive therapeutic targets. Several targeting strategies have emerged:
Traditional Orthosteric Inhibition: Developing small molecules that compete with phosphopeptide binding, though challenging due to the shallow, charged nature of the pTyr pocket.
Allosteric and Lipid-Targeted Approaches: Emerging strategies focus on targeting lipid-binding sites or allosteric regulatory mechanisms. For instance, nonlipidic inhibitors of Syk kinase have been developed that target lipid-protein interactions, potentially yielding potent, selective inhibitors for various SH2 domain-containing kinases [3] [7].
Structural-Based Design: Comprehensive structural databases and contact mapping approaches enable targeted inhibitor development based on precise molecular recognition features [69] [72].
Diagram 2: Therapeutic targeting strategies for SH2 domains. Multiple approaches are being developed to target SH2 domains, including traditional orthosteric inhibition, allosteric modulation, lipid-binding interference, and emerging protein degradation strategies.
The comparative analysis of STAT-type, Src-type, and atypical SH2 domains reveals both remarkable structural conservation and strategic functional diversification within this essential protein family. While maintaining a conserved core fold specialized for phosphotyrosine recognition, variations in secondary structure elements, loop configurations, and critical specificity determinants enable these domains to fulfill distinct roles in cellular signaling networks. The structural differences between STAT-type and Src-type domains reflect their divergent biological functions in nuclear signaling versus cytoplasmic signal transduction, respectively. Meanwhile, atypical domains like those in the Grb7 family demonstrate how targeted sequence variations can generate specialized binding properties. Continuing advances in structural characterization methods, binding profiling technologies, and computational approaches are rapidly expanding our understanding of SH2 domain biology and creating new opportunities for therapeutic intervention in cancer and other diseases driven by aberrant tyrosine kinase signaling.
Src Homology 2 (SH2) domains are modular protein domains approximately 100 amino acids in length that specifically recognize and bind to phosphorylated tyrosine (pY) motifs, thereby orchestrating a critical layer of the phosphotyrosine-dependent signaling network in eukaryotic cells [3]. The human proteome encodes roughly 110 proteins containing SH2 domains, which are functionally diversified into enzymes, adaptor proteins, docking proteins, transcription factors, and cytoskeletal proteins [3]. These domains achieve signaling specificity through their ability to recognize distinct amino acid sequences flanking the phosphotyrosine residue, enabling precise spatiotemporal control of cellular processes such as proliferation, differentiation, immune responses, and apoptosis [3] [38]. When mutations disrupt the delicate structure-function relationships within SH2 domains, they can precipitate pathological signaling cascades leading to cancer and immunodeficiency disorders. This technical guide provides a comprehensive framework for functionally validating SH2 domain mutations, with particular emphasis on the structural and functional differences between STAT-type and Src-type SH2 domains, and their implications for understanding disease mechanisms and developing targeted therapies.
Structural Biology of SH2 Domains: STAT-type versus Src-type
Conserved Core Structure and Differential C-terminal Features
All SH2 domains share a conserved structural fold characterized by a central antiparallel β-sheet flanked by two α-helices, forming an αβββα motif [74]. This core structure creates two primary binding pockets: the phosphotyrosine (pY) pocket that engages the phosphorylated tyrosine residue, and the specificity (pY+3) pocket that recognizes residues C-terminal to the pY, conferring binding specificity [74]. Despite this common scaffold, significant structural and functional distinctions exist between STAT-type and Src-type SH2 domains, primarily at their C-terminal regions.
Table 1: Structural and Functional Comparison of STAT-type versus Src-type SH2 Domains
| Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| C-terminal Structure | Contains an additional α-helix (αB') | Contains β-sheets (βE and βF) |
| Primary Function | Mediate STAT dimerization and nuclear translocation | Recruit signaling proteins to phosphorylated receptors |
| Dimerization Mode | Reciprocal phosphotyrosine-SH2 interactions between STAT monomers | Typically bind to phosphoproteins without self-dimerization |
| Domain Architecture | Embedded in transcription factors | Found in multidomain signaling proteins |
| Evolutionary Context | Specific to metazoan signal transduction | Widely distributed across signaling pathways |
The STAT-type SH2 domains contain an additional α-helix (αB') at the C-terminal region, whereas Src-type SH2 domains harbor β-sheets (βE and βF) in the equivalent position [74]. This structural distinction has profound functional implications. STAT-type SH2 domains facilitate the reciprocal phosphotyrosine-SH2 interactions that enable STAT dimerization following phosphorylation, which is essential for their nuclear translocation and transcriptional activity [74] [75]. In contrast, Src-type SH2 domains primarily function to recruit signaling proteins to specific phosphotyrosine sites on activated receptors or scaffold proteins, thereby assembling signaling complexes [3].
Structural Determinants of Phosphopeptide Recognition
The molecular basis for phosphopeptide recognition involves conserved structural elements across both STAT-type and Src-type SH2 domains. The pY pocket contains an invariant arginine residue (at position βB5) that forms a critical salt bridge with the phosphate moiety of the phosphotyrosine [3]. This arginine is part of the FLVR motif conserved across most SH2 domains, with only three known exceptions that feature an aromatic residue at this position instead [3]. The specificity (pY+3) pocket is formed by the opposite face of the central β-sheet along with residues from the αB helix and CD and BC* loops [74]. Within the pY+3 pocket lies the evolutionary active region (EAR), which displays greater sequence variability and contributes to binding specificity [74]. Additionally, a hydrophobic system comprising non-polar residues at the base of the pY+3 pocket helps stabilize the β-sheet conformation and maintain overall SH2 domain integrity [74].
Disease-Associated Mutations in SH2 Domains
STAT SH2 Domain Mutations in Disease
Sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in STAT proteins, particularly STAT3 and STAT5B, with profound implications for human disease [74]. These mutations can either enhance or diminish STAT activity, leading to diverse pathological manifestations.
Table 2: Disease-Associated Mutations in STAT SH2 Domains
| STAT Protein | Mutation Type | Associated Diseases | Molecular Consequence |
|---|---|---|---|
| STAT3 | Loss-of-function (LOF) | Autosomal-dominant Hyper IgE Syndrome (AD-HIES) | Reduced Th17 T-cell response, recurrent infections |
| STAT3 | Gain-of-function (GOF) | Autoimmune disorders, lymphoproliferative diseases | Th17 clonal expansion, suppressed Treg formation |
| STAT5B | Loss-of-function (LOF) | Growth hormone insensitivity syndrome (GHIS) | Postnatal growth impairment, immunological deficiencies |
| STAT5B | Gain-of-function (GOF) | Hematopoietic malignancies | Enhanced proliferation and survival signaling |
The functional impact of STAT SH2 domain mutations reflects the delicate evolutionary balance of wild-type STAT structural motifs in maintaining precise levels of cellular activity [74]. For instance, germline heterozygous LOF mutations in STAT3 cause AD-HIES due to impaired Th17 T-cell development, resulting in recurrent staphylococcal infections and elevated IgE levels [74]. Conversely, GOF mutations in STAT3 promote autoimmune manifestations through Th17 expansion and suppression of regulatory T-cell formation [74]. Interestingly, STAT3 GOF mutations can paradoxically mimic STAT5 LOF through compensatory upregulation of SOCS3, which inhibits hyperactivated STAT3 but also dampens STAT5 activity, leading to growth immunodeficiencies [74].
SHP2 SH2 Domain Mutations in Noonan Syndrome and Cancer
The protein tyrosine phosphatase SHP2 contains two SH2 domains (N-SH2 and C-SH2) that normally autoinhibit its catalytic domain [76]. In the basal state, the N-SH2 domain engages the PTP domain, maintaining SHP2 in a closed, autoinhibited conformation. Upon binding to phosphotyrosine motifs on receptors or scaffold proteins, SHP2 transitions to an open, active state [76]. Mutations disrupting the N-SH2/PTP interface can lead to constitutive SHP2 activation, causing Noonan syndrome and childhood hematopoietic cancers [76]. Deep mutational scanning of full-length SHP2 has revealed that disease-associated mutations cluster at key interdomain interfaces, particularly the N-SH2/PTP interface, with distinct mutational profiles observed across different cancer types [76].
Experimental Approaches for Functional Validation
Deep Mutational Scanning for Comprehensive Functional Analysis
Deep mutational scanning represents a powerful high-throughput approach for characterizing the functional consequences of thousands of protein variants in parallel [76]. This method combines selection assays on pooled mutant libraries with deep sequencing to profile mutational effects across entire protein domains.
Table 3: Key Methodological Components for SH2 Domain Functional Validation
| Method Category | Specific Technique | Key Application | Considerations |
|---|---|---|---|
| Library Generation | Saturation mutagenesis | Comprehensive coverage of variant space | MITE method for tile-based coverage |
| Selection System | Yeast growth rescue assay | Functional selection based on phosphatase activity | Kinase activity dictates selection pressure |
| Binding Profiling | Bacterial peptide display | Quantifying binding specificity across peptide libraries | Requires enzymatic phosphorylation of displayed peptides |
| Computational Analysis | ProBound with free-energy regression | Building sequence-to-affinity models | Assumes additivity of binding free energy |
| Biophysical Validation | Surface plasmon resonance | Direct measurement of binding kinetics and affinity | Low throughput but high quality data |
| Structural Analysis | X-ray crystallography, Cryo-EM | Determining atomic-level structural impacts | Resource-intensive but provides mechanistic insights |
For SHP2, a yeast viability assay has been successfully employed where cell growth depends on SHP2 catalytic activity [76]. In this system, yeast proliferation is arrested by expression of an active tyrosine kinase (v-Src or c-Src), but co-expression of an active tyrosine phosphatase rescues growth [76]. Saturation mutagenesis libraries for full-length SHP2 (divided into 15 sub-libraries) and the isolated phosphatase domain (divided into 7 sub-libraries) are constructed using the mutagenesis by integrated tiles (MITE) method [76]. Each sub-library is introduced into yeast cells alongside plasmids encoding either v-SrcFL or c-SrcKD. Following induction of kinase and phosphatase expression and a 24-hour outgrowth phase, SHP2-coding DNA is isolated and deep sequenced to calculate enrichment scores for each variant relative to wild-type SHP2 [76]. This approach has validated known mutational effects while identifying new mechanistic classes, including activating mutations in the N-SH2 domain core and inactivating mutations at the C-SH2/PTP interface [76].
Diagram 1: Experimental workflow for deep mutational scanning of SH2 domains
Quantitative Affinity Profiling Using Peptide Display
For characterizing the binding specificity of SH2 domains, bacterial display of genetically-encoded peptide libraries combined with next-generation sequencing provides a powerful platform for quantitative affinity profiling [38]. This approach involves creating highly diverse random peptide libraries (10^6-10^7 sequences) that are displayed on the bacterial surface. Following enzymatic phosphorylation of tyrosine residues in the displayed peptides, affinity-based selection is performed using purified SH2 domains [38]. The key innovation in this methodology is the application of ProBound, a statistical learning method that transforms sequencing data from multi-round selection experiments into quantitative sequence-to-affinity models [38]. ProBound employs free-energy regression to learn an additive model that accurately predicts binding free energy across the full theoretical ligand sequence space, effectively covering all possible amino acid combinations at each position in the peptide ligand [38]. This approach represents a significant advance over traditional position-specific scoring matrices (PSSMs) by providing biophysically interpretable parameters in meaningful energy units rather than arbitrary scores.
Structural Characterization of SH2 Domain Mutations
For clinically significant mutations identified through sequencing studies or functional screens, detailed structural characterization provides mechanistic insights into pathogenicity. X-ray crystallography and cryo-electron microscopy can reveal how mutations alter SH2 domain architecture and phosphopeptide binding capabilities [74]. Molecular dynamics simulations further complement experimental structures by capturing the flexibility and conformational dynamics of mutant SH2 domains [76]. STAT SH2 domains exhibit particularly flexible behavior even on sub-microsecond timescales, with the accessible volume of the pY pocket varying dramatically [74]. This inherent flexibility underscores the importance of accounting for protein dynamics in structure-function studies and drug discovery efforts targeting SH2 domains.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 4: Essential Research Reagents and Methodologies for SH2 Domain Studies
| Category | Reagent/Method | Specific Application | Key Utility |
|---|---|---|---|
| Library Resources | Saturation mutagenesis libraries | Comprehensive variant coverage | Deep mutational scanning |
| Random peptide libraries (10^6-10^7 diversity) | Binding specificity profiling | Bacterial/phage display | |
| Expression Systems | S. cerevisiae (yeast) growth rescue | Functional selection for phosphatase activity | High-throughput screening |
| Bacterial display system | Peptide library presentation | Affinity selection | |
| Computational Tools | ProBound software | Free-energy regression modeling | Quantitative affinity prediction |
| Molecular dynamics simulations | Studying domain flexibility and dynamics | Mechanism elucidation | |
| Analytical Methods | Next-generation sequencing | Variant frequency quantification | High-throughput readout |
| Surface plasmon resonance | Binding kinetics measurement | Validation of key interactions | |
| Specialized Reagents | Phosphospecific antibodies | Detection of phosphorylated proteins | Validation of signaling status |
| Active tyrosine kinases (v-Src, c-Src) | Selection pressure application | Functional screens |
Therapeutic Targeting of SH2 Domains
The critical role of SH2 domains in pathogenic signaling, coupled with their well-defined binding pockets, makes them attractive therapeutic targets. Several strategies have emerged for targeting SH2 domain-mediated interactions, with varying degrees of clinical success. Traditional approaches have focused on developing small-molecule inhibitors that target either the pY pocket or the specificity pocket to disrupt pathogenic protein-protein interactions [3]. More recently, novel targeting strategies have emerged, including the development of nonlipidic small molecules that inhibit lipid-protein interactions mediated by the cationic lipid-binding regions found in approximately 75% of SH2 domains [3]. Additionally, the role of SH2 domains in facilitating liquid-liquid phase separation (LLPS) and intracellular condensate formation presents new opportunities for therapeutic intervention [3]. For instance, interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation that enhances T-cell receptor signaling, while in kidney podocytes, LLPS increases the membrane dwell time of NCK-mediated actin polymerization complexes [3]. Understanding and targeting these phase separation mechanisms may offer new therapeutic avenues for modulating SH2 domain function in disease.
Functional validation of SH2 domain mutations requires a multidisciplinary approach integrating deep mutational scanning, quantitative biophysical measurements, structural analysis, and computational modeling. The distinct structural features of STAT-type versus Src-type SH2 domains dictate different validation strategies, particularly regarding their roles in transcription factor dimerization versus signal complex assembly, respectively. As new mechanisms of SH2 domain function continue to emerge, including their roles in lipid binding, phase separation, and non-canonical signaling, the framework for functional validation must similarly evolve. The experimental and computational approaches outlined in this technical guide provide a comprehensive foundation for linking structural mutations to disease phenotypes, ultimately facilitating the development of targeted therapeutic interventions for cancer and immunodeficiency disorders driven by SH2 domain dysregulation.
In phosphotyrosine signaling, Src homology 2 (SH2) domains are paramount for mediating specific protein-protein interactions by recognizing phosphorylated tyrosine (pTyr) residues within partner proteins. The human proteome encodes approximately 110 proteins containing SH2 domains, which are broadly classified into two major structural subgroups: Src-type and STAT-type [3] [7]. This classification is not merely structural but has profound functional implications for specificity profiling. Src-type SH2 domains, found in enzymes, adaptors, and regulators like Src family kinases and GRB2, typically bind peptides in an extended conformation and recognize residues C-terminal to the phosphotyrosine [3] [37]. In contrast, STAT-type SH2 domains, which facilitate the dimerization and nuclear translocation of signal transducers and activators of transcription, represent a more ancient structural lineage characterized by the absence of βE and βF strands and a split αB helix, adaptations that facilitate specific dimerization for transcriptional regulation [5] [7]. Understanding the distinct structural frameworks of these SH2 domain types is foundational to designing accurate experiments for validating peptide recognition motifs, such as the classic Src-binding motif pYEEI versus the STAT-preferred motif pYDKP.
Structural Determinants of SH2 Domain Specificity
Canonical Binding Mechanism and Specificity Pockets
The canonical SH2 domain fold consists of a central β-sheet flanked by two α-helices, forming a conserved structure that recognizes pTyr-containing peptides [37] [2]. The binding mechanism is fundamentally a "two-pronged plug" interaction [2]. The first "prong" is a deeply conserved phosphotyrosine-binding pocket located in the N-terminal region of the domain. This pocket invariably features a critical arginine residue (βB5) from the FLVR motif, which forms a salt bridge with the phosphate moiety of the pTyr, contributing up to half of the total binding free energy [3] [37] [2]. The second "prong" is a specificity pocket located in the more variable C-terminal region. This pocket typically engages residues located C-terminal to the pTyr, with the +3 position (three residues C-terminal to pTyr) being a major determinant for many SH2 domains, particularly those of the Src-type [77] [10] [37]. For example, the Src SH2 domain exhibits a strong preference for isoleucine at the +3 position (as in the pYEEI motif), which fits into a hydrophobic specificity pocket [8] [2].
Contextual Sequence Recognition and Selectivity
Beyond the canonical +3 position, SH2 domains exhibit a remarkable degree of selectivity by recognizing contextual sequence information [10]. Binding affinity is determined not only by "permissive" residues that enhance binding but also by "non-permissive" residues that oppose it through steric clash or charge repulsion [10]. The local sequence context matters, as the effect of a residue at one position can be influenced by neighboring residues. This complex linguistics allows SH2 domains to distinguish subtle differences in peptide ligands, substantially increasing the accessible information content embedded in short peptide sequences [10]. The EF and BG loops of the SH2 domain play crucial roles in controlling access to the specificity pockets and are primary contributors to this contextual recognition, with their composition and conformation varying significantly between different SH2 domains [7] [37].
Table 1: Key Structural Features Determining SH2 Domain Specificity
| Structural Element | Role in Specificity | Example |
|---|---|---|
| FLVR Arginine (βB5) | Essential for pTyr binding; forms salt bridge with phosphate moiety [3] [2] | Mutation reduces affinity ~1000-fold [2] |
| Specificity Pocket (+3) | Binds residue at pY+3 position; major selectivity determinant [77] [37] | Src SH2 prefers hydrophobic Ile (pYEEI) [8] |
| EF and BG Loops | Control ligand access to specificity pockets; confer contextual recognition [7] [37] | Variations explain differing specificities between SH2 domains [37] |
| Non-Permissive Residues | Inhibit binding through steric clash or charge repulsion [10] | Basic residues near pTyr can prevent ZAP-70 engagement [78] |
Methodologies for Specificity Profiling
High-Throughput Quantitative Profiling Technologies
Modern specificity profiling has moved beyond simple motif identification to quantitative models that can predict binding affinities across the theoretical sequence space. A powerful integrated approach combines bacterial surface display of peptide libraries with next-generation sequencing (NGS) and advanced computational modeling [38] [78].
Bacterial Peptide Display involves genetically encoding peptide libraries as fusions to surface proteins (e.g., eCPX) on E. coli [78]. For SH2 domain profiling, libraries are pre-phosphorylated using generic tyrosine kinases or contain genetically encoded phosphotyrosine. The SH2 domain of interest, typically tagged as a biotinylated bait protein, is used to pull down binding cells from the library using avidin-functionalized magnetic beads [78]. The bound populations are subsequently sequenced and quantified to determine enrichment ratios.
ccta90ee7a344a6d842c3b9e424b8727
The resulting NGS data from multiple selection rounds is analyzed using computational frameworks like ProBound, which employs a statistical learning method to build quantitative sequence-to-affinity models [38]. This approach can predict the binding free energy (ΔΔG) for any peptide sequence within the theoretical space covered by the library, moving beyond simple classification to accurate affinity prediction [38]. The model assumes additivity of binding free energy across residue positions, with relative affinity defined as exp(-ΔΔG/RT).
Supplementary Techniques for Validation
While high-throughput methods provide comprehensive datasets, orthogonal techniques remain valuable for validation:
- SPOT Synthesis and Peptide Array Analysis: Semi-quantitative approach where cellulose-bound peptide arrays are probed with purified SH2 domains to study interactions with hundreds to thousands of defined physiological peptides simultaneously [10].
- Fluorescence Polarization (FP): Solution-based technique that measures direct binding affinities between purified SH2 domains and fluorescently labeled phosphopeptides, providing precise Kd values [10].
- Isothermal Titration Calorimetry (ITC): Gold standard for determining thermodynamic parameters of binding (Kd, ΔH, ΔS, stoichiometry) through direct measurement of heat changes upon interaction [77].
- Yeast Surface Display: Enables estimation of dissociation constants (Kd) for monobody-SH2 interactions and rapid screening of selectivity profiles across multiple SH2 domains [77].
Experimental Protocol: Validating Motif Specificity Across SH2 Types
Profiling SH2 Domain Specificity Using Bacterial Display
This protocol details the steps for determining the specificity of a SH2 domain using bacterial peptide display, based on the methodology from [78].
Materials Required:
- eCPX Bacterial Display Vector: For peptide expression on E. coli surface.
- Random Peptide Library: X5-pY-X5 degenerate library (complexity 10^6-10^7 clones).
- Biotinylated SH2 Domain: Purified bait protein with high-affinity tag.
- Streptavidin Magnetic Beads: For pull-down of binding cells.
- TYR Host Strain: Non-specific tyrosine kinase expressing E. coli for library phosphorylation.
- NGS Platform: For sequencing input and output libraries.
Procedure:
- Library Transformation: Transform the X5-pY-X5 peptide library into the TYR E. coli strain for surface display.
- Induction and Phosphorylation: Induce peptide display with arabinose. Co-express a broad-specificity tyrosine kinase (or use purified kinase) to phosphorylate displayed peptides.
- Affinity Selection: Incubate the bacterial library with biotinylated SH2 domain. Capture binding cells with streptavidin magnetic beads.
- Wash and Elution: Wash beads to remove non-specific binders. Elute bound cells for regrowth and subsequent selection rounds (typically 2-3 rounds).
- Sequencing and Analysis: Isolate plasmid DNA from input and output populations. Prepare NGS libraries and sequence. Analyze data with ProBound to generate a sequence-to-affinity model.
Targeted Validation of pYEEI vs. pYDKP Motifs
Once initial profiling identifies candidate motifs, targeted validation is essential:
Design:
- Synthesize fluorescently labeled phosphopeptides: pYEEI (Src-type candidate) and pYDKP (STAT-type candidate).
- Purify SH2 domains of interest: Src-type (e.g., Src, Fyn) and STAT-type (e.g., STAT1, STAT3).
Fluorescence Polarization Assay:
- Prepare serial dilutions of each SH2 domain in binding buffer.
- Mix with fixed concentration of fluorescent phosphopeptide.
- Incubate in the dark for equilibrium (typically 30 minutes, room temperature).
- Measure fluorescence polarization for each sample.
- Fit data to a one-site binding model to determine Kd values.
Expected Results:
- Src-type SH2 domains should show nanomolar to low micromolar affinity for pYEEI and significantly weaker binding to pYDKP.
- STAT-type SH2 domains should show the reverse specificity, binding pYDKP with higher affinity.
Table 2: Expected Binding Affinities for Characterized SH2 Domains
| SH2 Domain | Type | pYEEI Motif Kd (μM) | pYDKP Motif Kd (μM) | Notes |
|---|---|---|---|---|
| Src | Src-type | 0.1 - 1.0 [8] | >10 [8] | Binds extended conformation; requires pY+3 Ile |
| GRB2 | Src-type | >10 | 0.1 - 1.0 [8] | Prefers pY+2 Asn (pYVNV) |
| STAT1 | STAT-type | >10 | 0.1 - 1.0 [5] | Adapted for dimerization |
| STAT3 | STAT-type | >10 | 0.1 - 1.0 [3] | Adapted for dimerization |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for SH2 Domain Specificity Profiling
| Reagent / Tool | Function | Example Application |
|---|---|---|
| Monobodies | Synthetic binding proteins; high-affinity SH2 inhibitors [77] | Selective perturbation of SFK SH2 domains in signaling studies |
| Position-Specific Scoring Matrix (PSSM) | Bioinformatics tool for predicting SH2 binding sites [38] [10] | Rapid scanning of protein sequences for potential SH2 ligands |
| Nonlipidic Small Molecule Inhibitors | Target lipid-binding pocket of SH2 domains [3] [7] | Inhibition of SYK kinase activity; potential therapeutic approach |
| ProBound Software | Statistical learning for sequence-to-affinity modeling [38] | Building quantitative models from NGS selection data |
| SPOT Membrane Arrays | Cellulose-bound peptide libraries for interaction screening [10] | Medium-throughput analysis of SH2 binding to physiological peptides |
Advanced Applications and Therapeutic Targeting
Understanding SH2 domain specificity has profound implications for therapeutic intervention. The development of highly selective monobodies against Src family kinase (SFK) SH2 domains demonstrates how specificity profiling can guide the creation of precision tools that discriminate between even closely related SH2 domains, achieving selectivity between SrcA (Yes, Src, Fyn, Fgr) and SrcB (Lck, Lyn, Blk, Hck) subfamilies [77]. These monobodies can selectively activate or inhibit kinase activity and proximal signaling events in cells, serving as excellent tools for dissecting SFK functions in normal and oncogenic signaling [77].
Additionally, emerging research shows that nearly 75% of SH2 domains interact with membrane lipids such as PIP2 and PIP3, with cationic regions near the pY-binding pocket serving as lipid-binding sites [3] [7]. This suggests a dual recognition mechanism where membrane recruitment via lipid binding works in concert with phosphopeptide specificity. Targeting these lipid-protein interactions with nonlipidic small molecules has shown promise for developing selective inhibitors resistant to resistance, as demonstrated for Syk kinase [3] [7].
Furthermore, SH2 domain-containing proteins are increasingly linked to the formation of intracellular condensates via liquid-liquid phase separation (LLPS) [3]. Multivalent interactions between SH2 domains and their partners drive the formation of these membrane-less organelles, enhancing signaling output in systems like the LAT-GRB2-SOS1 complex in T-cell receptor signaling [3]. This emerging role adds another dimension to specificity profiling, as the context of phase separation may influence motif recognition.
Specificity profiling of SH2 domains has evolved from simple motif identification to sophisticated quantitative models that account for contextual sequence information and structural variations between Src-type and STAT-type domains. The integration of high-throughput experimental approaches like bacterial peptide display with advanced computational modeling using frameworks like ProBound provides researchers with powerful tools to accurately predict SH2 domain specificities and affinities. These advances are not only refining our understanding of phosphotyrosine signaling networks but are also paving the way for novel therapeutic strategies that target specific SH2 interactions in disease contexts, particularly in cancer and immune disorders. As structural insights deepen and profiling technologies become more accessible, the precision with which we can map and manipulate SH2 domain interactions will continue to accelerate both basic research and drug development.
Src Homology 2 (SH2) domains are protein modules approximately 100 amino acids in length that specifically recognize and bind to phosphorylated tyrosine (pTyr) residues on target proteins [3]. These domains are fundamental components of intracellular signaling networks, enabling specific protein-protein interactions that regulate critical cellular processes including proliferation, differentiation, survival, and immune responses [3]. The human proteome contains roughly 110 proteins encoding SH2 domains, classified into various functional groups including kinases, phosphatases, adaptor proteins, and transcription factors [3]. Since their discovery in 1986, SH2 domains have emerged as promising therapeutic targets due to their essential role in propagating signals from receptor tyrosine kinases and their frequent dysregulation in diseases such as cancer and autoimmune disorders [79] [2]. The highly conserved nature of the pTyr binding pocket across SH2 domains, coupled with sequence variations that confer specificity, presents both challenges and opportunities for drug development. This assessment provides a comprehensive analysis of the current clinical development landscape for SH2 domain inhibitors, with particular emphasis on the structural distinctions between major SH2 domain classes.
Structural Biology of SH2 Domains
Conserved Architecture and Binding Mechanism
All SH2 domains share a conserved structural fold characterized by a central antiparallel β-sheet flanked by two α-helices in an αβββα configuration [6] [3]. This scaffold creates two primary binding pockets: a phosphotyrosine (pY) pocket that recognizes the phosphate moiety of phosphorylated tyrosine, and a specificity (pY+3) pocket that engages residues C-terminal to the pTyr, typically determining sequence selectivity [6]. The pY pocket contains a highly conserved arginine residue (βB5) that forms critical salt bridges with the phosphate group of pTyr [3]. This arginine is part of the signature FLVR (Phe-Leu-Val-Arg) motif found in most SH2 domains, which provides approximately half of the binding free energy for phosphopeptide interactions [2].
Table 1: Key Structural Features of SH2 Domains
| Structural Element | Components | Functional Role |
|---|---|---|
| Central β-sheet | βB, βC, βD strands | Forms backbone for peptide binding |
| Flanking α-helices | αA, αB | Contribute to pY and pY+3 pocket formation |
| pY Pocket | FLVR motif (particularly Arg βB5), αA2, βD6 | Binds phosphate moiety of pTyr |
| pY+3 Pocket | αB helix, EF loop, BG loop | Determines sequence specificity |
| Variable Loops | BC, CD, BG, EF loops | Provide binding diversity and specificity |
Structural Differences Between STAT-Type and Src-Type SH2 Domains
STAT-type and Src-type SH2 domains represent two major classes with distinct structural characteristics that influence their functions and drug targeting potential. STAT-type SH2 domains contain an additional α-helix (αB') at the C-terminus within the evolutionary active region (EAR), whereas Src-type domains feature β-sheets (βE and βF) in this region [6]. This structural distinction has profound implications for dimerization and phosphopeptide binding. STAT SH2 domains primarily facilitate homo- or heterodimerization between STAT molecules through reciprocal phosphotyrosine-SH2 domain interactions, followed by nuclear translocation and gene transcription activation [6]. In contrast, Src-type SH2 domains typically mediate interactions between signaling proteins in cascades initiated by receptor tyrosine kinases.
The configuration of residues coordinating pTyr also differs between these classes. Src-like SH2 domains typically feature a basic residue at position αA2, while SAP-like SH2 domains (including STATs) often contain a basic residue at position βD6 [2]. These structural variations create differences in pocket geometries and chemical environments that can be exploited for selective inhibitor design. Additionally, STAT SH2 domains exhibit significant flexibility even on sub-microsecond timescales, with the accessible volume of the pY pocket varying dramatically, underscoring the importance of accounting for protein dynamics in drug discovery efforts [6].
Clinical Development Landscape of SH2 Domain Inhibitors
Approved and Clinical-Stage SH2-Targeting Therapies
The development of SH2 domain inhibitors has advanced significantly, with several candidates reaching clinical trials and a growing pipeline of preclinical assets. The most mature approaches target SH2 domains in specific signaling proteins implicated in disease pathways. Currently, no drugs specifically designed as SH2 domain inhibitors have received FDA approval, but multiple candidates have entered clinical development.
Table 2: SH2 Domain Inhibitors in Clinical Development
| Target | Compound | Developer | Stage | Indication | Key Characteristics |
|---|---|---|---|---|---|
| BTK | BTK SH2i (undisclosed) | Recludix Pharma | Preclinical | Chronic spontaneous urticaria, multiple sclerosis | First-in-class, exceptional selectivity (>8000-fold), prodrug delivery [80] [64] |
| SYK | Cevidoplenib dimesylate | Genosco | Phase II | Inflammatory diseases | Orally available, inhibits Fc receptor and B-cell receptor signaling [81] |
| SYK | HMPL-523 | Hutchison MediPharma | Phase II | Hematologic malignancies | Orally available, inhibits B-cell receptor signaling [81] |
| SYK | Entospletinib | Multiple | Phase II | Hematologic malignancies | Selective SYK inhibitor [81] |
| STAT3 | Undisclosed | Recludix Pharma | Preclinical | Inflammatory diseases | Targets STAT3 SH2 domain [80] |
| STAT6 | Undisclosed | Recludix Pharma/Sanofi | Preclinical | Inflammatory diseases | Partnership with Sanofi [80] |
| SHP2 | Irinotecan (repurposing) | Academic research | Preclinical | Cancer | Identified via virtual screening (CID 60838), binding free energy -64.45 kcal/mol [61] |
Novel SH2 Inhibitor Platforms and Technologies
Recent advances in SH2 inhibitor development have been facilitated by innovative platform technologies. Recludix Pharma has pioneered a comprehensive discovery platform integrating custom DNA-encoded libraries (DELs), SH2-targeted crystallographic structure-guided design, proprietary biochemical screening assays, and prodrug delivery modalities to enhance intracellular exposure [64]. This approach has yielded highly selective SH2 inhibitors with exceptional biochemical potency (BTK Kd = 0.055 nM) and minimal cytotoxicity (>10,000 nM EC50 in Jurkat cells) [64].
Another emerging strategy involves targeting lipid-binding sites adjacent to SH2 domains. Nearly 75% of SH2 domains interact with membrane lipids, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [3] [7]. Cologna and colleagues have successfully developed nonlipidic inhibitors of Syk kinase that target these lipid-protein interactions, demonstrating that this approach can produce potent, selective inhibitors resistant to common resistance mechanisms [3].
Experimental Approaches for SH2 Inhibitor Development
Structure-Based Drug Design Methodologies
Structure-based drug design has emerged as a powerful approach for developing SH2 domain inhibitors. A representative protocol for identifying SH2 inhibitors involves multiple computational and experimental stages [61]:
Target Preparation and Binding Site Identification
- Retrieve crystal structure from Protein Data Bank (e.g., SHP-2, PDB ID: 2SHP)
- Prepare protein structure using PDBFixer to add missing residues and hydrogens
- Identify druggable pockets using Fpocket tool
- Visually inspect binding sites with molecular visualization software (e.g., PyMol)
Virtual Screening and Molecular Docking
- Curate compound libraries (e.g., Broad's Drug Repurposing Hub: 13,553 compounds; ZINC15 in-trials subset: 5,900 compounds)
- Prepare ligand structures using RDKit for 3D structure generation and energy minimization
- Perform molecular docking studies using Smina (Autodock Vina variant)
- Define grid box coordinates to encompass binding site with adequate sampling
- Set exhaustiveness parameter to 16 for thorough conformational sampling
Molecular Dynamics Simulations and Binding Affinity Calculations
- Conduct MD simulations using Gromacs with OPLS-AA/M force field and SPC216 water model
- Generate ligand topologies using LigParGen server
- Perform MM/PBSA binding free energy calculations using g_mmpbsa
- Use 200 configurations from MD trajectories (1 ns spaced snapshots)
- Set parameters: grid space 0.5 Å, salt concentration 0.150 M, solute dielectric constant 2
The Scientist's Toolkit: Essential Research Reagents and Methods
Table 3: Essential Research Reagents and Methods for SH2 Inhibitor Development
| Reagent/Method | Function/Application | Examples/Specifications |
|---|---|---|
| Protein Data Bank Structures | Structural templates for drug design | SHP-2 (2SHP), STAT SH2 domains, Src-family structures |
| Compound Libraries | Source of potential inhibitors | Broad Repurposing Hub (13,553 compounds), ZINC15 in-trials subset (5,900 compounds) |
| Molecular Docking Software | Prediction of ligand binding poses | Smina, Autodock Vina (exhaustiveness: 16) |
| Molecular Dynamics Packages | Simulation of protein-ligand dynamics | Gromacs (ver. 2021.03), OPLS-AA/M force field |
| Binding Affinity Calculation Tools | Quantification of protein-ligand interactions | g_mmpbsa, MM/PBSA methods |
| DNA-Encoded Libraries (DELs) | High-throughput screening platform | Custom DELs for SH2 domain targeting [64] |
| Structure Visualization Software | Analysis of binding interactions | PyMol, Chimera |
| Lipid Binding Assays | Characterization of membrane interactions | PIP2/PIP3 binding assays [3] |
Challenges and Future Perspectives
Selectivity and Specificity Hurdles
Achieving selectivity among highly conserved SH2 domains remains a significant challenge in inhibitor development. The human genome encodes approximately 110 SH2 domain-containing proteins, many with structurally similar pY binding pockets [3]. However, recent advances demonstrate that exceptional selectivity is achievable. Recludix's BTK SH2 inhibitor shows >8000-fold selectivity over off-target SH2 domains, far exceeding the selectivity profile of traditional kinase domain inhibitors [80] [64]. This enhanced selectivity translates to improved safety profiles, as demonstrated by the avoidance of TEC kinase inhibition-associated platelet dysfunction that plagues many BTK kinase inhibitors [64].
Emerging Opportunities and Therapeutic Applications
Beyond the established targets in oncology and immunology, emerging research has revealed novel functions of SH2 domains that expand their therapeutic potential. Recent evidence indicates that SH2 domains participate in liquid-liquid phase separation (LLPS), facilitating the formation of membrane-free intracellular condensates that enhance signaling efficiency [3] [7]. For example, interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation, enhancing T-cell receptor signaling [3]. This discovery opens new avenues for modulating signal transduction by targeting the multivalent interactions that drive condensate formation.
The development of allosteric inhibitors targeting unique structural features of specific SH2 domains represents another promising direction. STAT-type SH2 domains possess distinctive structural motifs, including extended surface areas beyond the canonical pY pocket, that can be exploited for selective targeting [6]. As structural biology techniques advance, including cryo-EM and time-resolved crystallography, new opportunities will emerge for designing next-generation SH2 inhibitors with improved pharmacological properties.
The clinical development of SH2 domain inhibitors is transitioning from concept to reality, with multiple candidates demonstrating compelling preclinical efficacy and safety profiles. As these innovative therapeutics progress through clinical trials, they hold significant promise for addressing unmet medical needs across oncology, autoimmune diseases, and inflammatory disorders.
Src Homology 2 (SH2) domains represent a critical family of protein interaction modules that specifically recognize phosphotyrosine (pTyr) motifs, thereby orchestrating a vast network of intracellular signaling pathways. The human proteome encodes over 110 proteins containing SH2 domains, which are functionally diversified into enzymes, adaptors, regulators, docking proteins, and transcription factors [3]. These approximately 100-amino-acid domains emerged in early eukaryotes and expanded alongside protein tyrosine kinases (PTKs) and tyrosine phosphatases during metazoan evolution, with their numbers correlating strongly with biological complexity [9]. Despite a conserved structural fold, SH2 domains exhibit significant functional specialization. Among these, the STAT-type and Src-type SH2 domains represent two major evolutionary lineages with distinct structural features and biological roles [5]. This review places the differences between these two groups within the broader context of human SH2 protein diversity, providing structural, functional, and methodological insights for research and therapeutic targeting.
Classification and Evolution of SH2 Domains
Genomic Distribution and Phylogenetic Analysis
SH2 domains first appeared in unicellular eukaryotes and underwent substantial expansion in the choanoflagellate and metazoan lineages. Comparative genomic analysis across 21 eukaryotic species reveals that SH2 domain-containing proteins proliferated alongside tyrosine kinases, with a correlation coefficient of 0.95 between PTK and SH2 domain numbers across genomes [9]. This co-expansion facilitated the increasing sophistication of cell-cell communication networks in multicellular organisms. The human genome encodes SH2 domains within 38 distinct sub-families based on phylogenetic relationships [9].
Table: SH2 Domain-Containing Proteins by Functional Category in Humans
| Function | Representative Proteins |
|---|---|
| Enzymes | ABL1, JAK2, PIK3R2, PLCG1, PTPN11, SRC, SYK |
| Regulator (GTPase activity activator) | CHN1, RASA1, VAV1, RIN1 |
| Adaptor proteins | CRK, GRB2, NCK1, SHC1, SHB, SLAP |
| Docking proteins | BRDG1, SHC1, SH3BP2 |
| Transcription factor | STAT1, STAT2, STAT3, STAT4, STAT5, STAT5B, STAT6 |
| Cytoskeletal protein | TNS1, TENS2, TNS3, TNS4 |
Structural Classification: STAT-type vs. Src-type SH2 Domains
SH2 domains are primarily classified into two major groups based on structural characteristics: STAT-type and Src-type [5]. This division is founded on key differences in their C-terminal structural elements. The Src-type SH2 domain contains the basic "αβββα" structure with an extra β-strand (βE or βE-βF motif), while the STAT-type SH2 domain is characterized by a linker domain-conjugated SH2 domain containing the αB' motif [5]. Beyond this primary classification, phylogenetic analysis categorizes SH2 domain-containing proteins into 38 different sub-families, while functional activity-based screens stratify them into four categories based on the identity of the fifth residue in the βD strand, a critical determinant in phospho-peptide selectivity [82].
Structural Comparison of STAT-type and Src-type SH2 Domains
Core Architecture and Binding Pockets
All SH2 domains share a conserved central fold consisting of a three-stranded antiparallel beta-sheet (βB-βD) flanked by two alpha helices (αA and αB), forming an αβββα motif [3] [82]. This structure creates two primary binding subpockets: a deep pY (phosphate-binding) pocket that recognizes the phosphotyrosine residue, and a pY+3 (specificity) pocket that binds the residue three positions C-terminal to the pTyr, conferring sequence specificity [82].
The pY pocket is formed by the αA helix, BC loop, and one face of the central β-sheet, and contains highly conserved residues critical for pTyr coordination. Most notably, an invariable arginine at position βB5 (part of the FLVR motif) serves as a key recognition element, forming a salt bridge with the phosphate moiety of pTyr [3]. The pY+3 pocket is created by the opposite face of the β-sheet along with residues from the αB helix and CD and BC* loops [82].
Distinguishing Structural Elements
Despite this conserved core architecture, STAT-type and Src-type SH2 domains exhibit significant structural differences, particularly in their C-terminal regions:
STAT-type SH2 Domains: Feature an αB' helix in the evolutionary active region (EAR) of the pY+3 pocket [82]. This α-helical structure replaces the β-sheets found in Src-type domains and participates in SH2-mediated STAT dimerization, forming important cross-domain interactions [82].
Src-type SH2 Domains: Contain βE and βF strands in the C-terminal region rather than the αB' helix found in STAT domains [5] [82]. These additional β-strands contribute to the structural scaffold but do not participate in the dimerization interfaces characteristic of STAT proteins.
Table: Comparative Structural Features of STAT-type vs. Src-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| Core Structure | αβββα motif | αβββα motif |
| C-terminal Element | αB' helix | βE-βF β-strands |
| Representative Proteins | STAT1, STAT3, STAT5 | SRC, LCK, FYN |
| Dimerization Role | Critical for STAT dimerization | Not typically involved in dimerization |
| pY+3 Pocket | Contains EAR with αB' helix | Contains EAR with β-sheets |
Figure 1: Structural classification of STAT-type and Src-type SH2 domains, highlighting key differences in C-terminal elements and functional roles.
Structural Plasticity and Dynamics
Recent research has revealed that SH2 domains exhibit significant flexibility, particularly in their binding pockets. Molecular dynamics simulations show that the accessible volume of the pY pocket varies dramatically even on sub-microsecond timescales [82]. This structural plasticity presents challenges for drug discovery, as crystal structures may not preserve targetable pockets in accessible states. STAT SH2 domains demonstrate particularly flexible behavior, underscoring the importance of accounting for protein dynamics in therapeutic development [82].
Functional Implications of Structural Differences
STAT SH2 Domains in Dimerization and Transcriptional Activation
The unique structural features of STAT-type SH2 domains directly facilitate their central role in phosphorylated STAT dimerization and nuclear translocation. In canonical STAT activation, cytokine or growth factor stimulation triggers SH2 domain-mediated recruitment of STAT proteins to receptor cytoplasmic domains, followed by tyrosine phosphorylation [82]. The phosphorylated STATs then form stable homo- or heterodimers through reciprocal SH2-pTyr interactions, with the αB' helix and surrounding regions contributing critical contacts for dimer stability [82].
This dimerization is essential for nuclear accumulation and DNA binding, enabling STATs to drive transcription of genes involved in proliferation, survival, and immune responses. The structural organization of the STAT SH2 domain thus directly connects extracellular signaling to transcriptional regulation, with mutations frequently disrupting this precise coordination and causing disease.
Src-type SH2 Domains in Scaffolding and Proximity Regulation
Src-type SH2 domains primarily function in signal complex assembly and subcellular targeting. Rather than mediating stable dimerization like STAT SH2 domains, they typically recruit binding partners to specific subcellular locations or facilitate transient interactions within larger signaling complexes. For example, the Src SH2 domain can recognize specific phosphorylated motifs in activated receptors such as the PDGFβ receptor, localizing Src kinase to its substrates [83].
Recent research has revealed that many SH2 domain-containing proteins participate in liquid-liquid phase separation (LLPS), forming membrane-free condensates that enhance signaling specificity and efficiency. In T-cells, interactions among GRB2, Gads, and the LAT receptor contribute to LLPS formation, enhancing T-cell receptor signaling [3]. The multivalent interactions afforded by SH2 domains and other modular domains drive condensate formation, creating highly localized signaling hubs that optimize kinase-substrate interactions while preventing inappropriate cross-talk between pathways.
Lipid Binding and Membrane Interactions
Beyond phosphotyrosine recognition, approximately 75% of SH2 domains interact with lipid molecules, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [3]. These interactions involve cationic regions near the pY-binding pocket, typically flanked by aromatic or hydrophobic residues. Lipid binding modulates membrane localization and enzymatic activity, as demonstrated by the PIP3 binding activity of the TNS2 SH2 domain, which regulates phosphorylation of insulin receptor substrate-1 (IRS-1) in insulin signaling [3].
This lipid-binding capacity represents an additional layer of functional diversification among SH2 domains, with implications for subcellular targeting and regulation. Many disease-causing mutations in SH2 domains localize within these lipid-binding pockets, highlighting their physiological importance [3].
Methodologies for SH2 Domain Research
Structural Biology Techniques
X-ray Crystallography
Protocol: SH2 Domain Crystallization in Complex with Phosphopeptides
Protein Expression and Purification: Clone the SH2 domain (residues 1-111 for Crkl-SH2) into a pET28b+ vector with an N-terminal His-tag for purification. Transform into E. coli BL21 (DE3) cells and grow in LB medium with kanamycin (30 μg/mL) at 37°C until OD600 = 0.7-0.8. Induce expression with 0.5 mM IPTG and grow overnight at 25°C [84].
Protein Purification: Resuspend cell pellet in buffer (50 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole, pH 7.5) with protease inhibitors. Sonicate and centrifuge to collect soluble fraction. Purify using nickel-affinity chromatography with imidazole gradient elution (0-1 M). Desalt into appropriate buffer (50 mM Tris-HCl, 300 mM NaCl, pH 7.5) [84].
Crystallization: Complex the purified SH2 domain with a 2-3 molar excess of phosphopeptide. Set up crystallization screens using commercial kits. For Src SH2 domain bound to phosphorylated peptide (PDB 1NZV), crystals grew in conditions containing tetraethylene glycol and chloride ions [83].
Data Collection and Structure Determination: Collect X-ray diffraction data at synchrotron sources. Solve structure using molecular replacement with existing SH2 domain structures as search models. Refine iteratively using programs like CNS [83].
Nuclear Magnetic Resonance (NMR) Spectroscopy
Protocol: Solution Structure Determination of SH2-Phosphopeptide Complexes
Sample Preparation: Prepare isotopically labeled (15N, 13C) SH2 domain expressed in minimal media. Complex with phosphopeptide at 1:1.2 molar ratio in appropriate NMR buffer (e.g., 20 mM phosphate, 50 mM NaCl, pH 6.5). Concentrate to ~0.5-1 mM in 500 μL [85].
Data Collection: Acquire multidimensional NMR experiments including 1H-15N HSQC, HNCA, HNCO, HNCACB, CBCACONH for backbone assignment, and 13C-edited NOESY for structural constraints. For the human Src SH2 domain (1HCS), 2072 experimental restraints were derived from multifrequency/multidimensional NMR data [85].
Structure Calculation: Generate initial structures using distance geometry and simulated annealing protocols. Incorporate NOE-derived distance restraints, dihedral constraints from chemical shifts, and hydrogen bond restraints from slow-exchange amides.
Structure Refinement: Refine against experimental restraints using dynamical simulated annealing. Analyze a family of structures (e.g., 23 structures for 1HCS) to assess precision and identify well-defined regions [85].
Biophysical and Functional Characterization
Folding and Binding Kinetics
Protocol: Stopped-Flow Fluorescence Binding Assays
Experimental Setup: Perform experiments on an Applied Photophysics sequential-mixing stopped-flow apparatus in single mixing mode. Excite samples at 280 nm and monitor fluorescence emission above 475 nm using a cutoff filter [84].
Binding Measurements: Conduct pseudo-first-order experiments by mixing constant concentration of dansyl-labeled peptide (2 μM) with varying concentrations of SH2 domain (2-10 μM). Perform under different pH and ionic strength conditions to characterize electrostatic contributions.
Data Analysis: Fit observed rate constants to appropriate binding models. For Crkl-SH2 binding to Paxillin peptide, analysis revealed the critical role of a conserved histidine residue (His60) in binding stabilization through protonation-dependent interactions [84].
Equilibrium Unfolding Studies
Protocol: Guanidine-HCl Denaturation Experiments
Sample Preparation: Prepare SH2 domain at constant concentration (2 μM) in series of guanidine-HCl solutions (0-6 M range) in appropriate buffer. Include 0.15 M sodium sulfate for pH-dependent studies [84].
Fluorescence Measurements: Record emission spectra (300-400 nm) with excitation at 280 nm using a spectrofluorometer. Monitor changes in intrinsic fluorescence as function of denaturant concentration.
Data Fitting: Fit unfolding transitions to a two-state or three-state model depending on system. For Crkl-SH2, data revealed the presence of a folding intermediate, with stability maximum around pH 5.0-5.5 [84].
Figure 2: Experimental workflow for comprehensive SH2 domain investigation, integrating structural, biophysical, and functional approaches.
Research Reagent Solutions
Table: Essential Reagents for SH2 Domain Research
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Expression Systems | pET28b+ vector, E. coli BL21(DE3) | Recombinant protein production with His-tag purification |
| Purification Tools | Nickel-charged HisTrap columns, desalting columns | Affinity purification and buffer exchange |
| Structural Biology | Crystallization screens (commercial kits) | Crystal formation for X-ray diffraction |
| Biophysical Probes | Dansyl-labeled phosphopeptides | Fluorescence-based binding measurements |
| Kinetic Instruments | Stopped-flow apparatus (Applied Photophysics) | Rapid kinetic measurements of folding/binding |
| SH2 Domain Constructs | Crkl-SH2 (residues 1-111), Src SH2, STAT SH2 | Comparative studies of different SH2 types |
| Site-Directed Mutagenesis Kits | QuikChange mutagenesis kit | Introduction of point mutations for mechanistic studies |
Therapeutic Targeting and Clinical Implications
Disease-Associated Mutations in SH2 Domains
Sequencing studies have identified the SH2 domain as a mutational hotspot in STAT proteins, with profound clinical consequences. In STAT3, heterozygous germline mutations cause autosomal-dominant Hyper IgE Syndrome (AD-HIES), characterized by recurrent infections, eczema, and skeletal abnormalities due to impaired Th17 T-cell differentiation [82]. These loss-of-function mutations cluster in critical regions of the SH2 domain, including:
- pY binding pocket: Mutations at K591, R609, S611, and S614 disrupt phosphopeptide binding
- BC loop: E616 and G617 mutations impair structural integrity and signaling capacity
- Dimerization interface: Mutations affecting STAT3-STAT3 interactions
Conversely, somatic gain-of-function mutations in STAT3 and STAT5 SH2 domains drive oncogenesis in various hematologic malignancies, including T-cell large granular lymphocytic leukemia (T-LGLL) and natural killer T-cell lymphoma (NKTL) [82]. The same residue can harbor either activating or inactivating mutations depending on the specific amino acid substitution, underscoring the delicate evolutionary balance in SH2 domain structure.
SH2 Domain-Targeted Therapeutic Development
The central role of SH2 domains in disease has made them attractive therapeutic targets. Several strategies have emerged for targeting SH2 domain-mediated interactions:
Traditional Small Molecules: Focused on disrupting the pY pocket or specificity pocket with competitive inhibitors. Challenges include achieving sufficient affinity to compete with natural ligands and ensuring selectivity among similar SH2 domains.
Allosteric Inhibitors: Target regions outside the primary binding pocket, such as the dimerization interfaces unique to STAT SH2 domains. These offer potential for greater specificity but require detailed structural knowledge.
Non-lipidic Inhibitors of Lipid-Protein Interactions: Novel approach exemplified by inhibitors of Syk kinase that target its SH2 domain lipid-binding capacity, showing promise for developing selective, resistance-resistant inhibitors [3].
Stabilizers of Inactive Conformations: Exploit the dynamic nature of SH2 domains to stabilize inactive states, particularly effective for pathogenic gain-of-function mutants.
Table: Clinical Mutations in STAT3 and STAT5 SH2 Domains
| Mutation | Location | Pathology | Molecular Consequence |
|---|---|---|---|
| STAT3 S614R | BC loop (pY pocket) | T-LGLL, NKTL | Gain-of-function, enhanced signaling |
| STAT3 K591E/M | αA2 (pY pocket) | AD-HIES | Loss-of-function, impaired peptide binding |
| STAT3 R609G | βB5 (FLVR motif) | AD-HIES | Disrupted pTyr coordination |
| STAT5B N642H | SH2 domain | T-cell leukemia | Gain-of-function, constitutive activation |
The diversification of SH2 domains into STAT-type and Src-type lineages represents a fundamental evolutionary adaptation that enabled sophisticated signaling capabilities in metazoans. The structural differences in C-terminal elements directly correlate with their distinct biological functions: STAT-type SH2 domains with their αB' helices facilitate stable dimerization required for transcriptional activation, while Src-type domains with additional β-strands provide versatile scaffolding for transient signaling complexes.
Future research directions should focus on several key areas. First, elucidating the full structural diversity of less-characterized SH2 families will provide a more complete understanding of this protein interaction domain family. Second, exploiting advances in targeted protein degradation could provide new therapeutic avenues for targeting SH2 domain-containing proteins that have proven difficult to drug with conventional approaches. Finally, integrating knowledge of SH2 domain dynamics and allosteric regulation will enable development of next-generation therapeutics that modulate rather than completely inhibit SH2 function, potentially achieving better therapeutic indices.
The continued investigation of SH2 domain diversity, from atomic-level structural details to systems-level network analysis, will undoubtedly yield new insights into cellular signaling mechanisms and innovative approaches for therapeutic intervention in cancer, immune disorders, and other human diseases.
Conclusion
The structural dichotomy between STAT-type and Src-type SH2 domains underpins their specialized roles in cellular signaling, from transcriptional regulation to kinase activity control. The unique architecture of STAT-type domains, lacking standard βE and βF strands, is exquisitely tailored for dimerization and nuclear function, whereas the canonical Src-type fold supports diverse scaffolding and enzymatic roles. Understanding these differences at an atomic level, facilitated by advanced computational and structural methods, is no longer just an academic pursuit. It directly enables the rational design of targeted therapies. Future research must focus on elucidating the full spectrum of SH2 domain functions, including their roles in liquid-liquid phase separation and non-canonical lipid binding, to develop next-generation, high-specificity inhibitors for cancers, immune disorders, and other diseases driven by faulty phosphotyrosine signaling.
References
- 1. Molecular Mechanisms of SH2- and PTB-Domain-Containing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3839611/]
- 2. SH2 Domain Binding: Diverse FLVRs of Partnership [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2020.575220/full]
- 3. Update on Structure and Function of SH2 Domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC12470777/]
- 4. Structure determination of SH2–phosphopeptide complexes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11059313/]
- 5. Identification of the Linker-SH2 Domain of STAT as ... [https://www.sciencedirect.com/science/article/pii/S1535947620338639]
- 6. Structural Implications of STAT3 and STAT5 SH2 Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6895964/]
- 7. Update on Structure and Function of SH2 Domains [https://www.mdpi.com/1422-0067/26/18/9060]
- 8. Structural Basis for Specificity Switching of the Src SH2 ... [https://www.sciencedirect.com/science/article/pii/S1097276500802695]
- 9. The SH2 Domain–Containing Proteins in 21 Species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4255630/]
- 10. SH2 Domains Recognize Contextual Peptide Sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2984226/]
- 11. Discriminating between competing models for the allosteric ... [https://www.sciencedirect.com/science/article/pii/S2001037021004633]
- 12. SH2 Domains: Folding, Binding and Therapeutical Approaches [https://pmc.ncbi.nlm.nih.gov/articles/PMC9783222/]
- 13. Structural basis for the interaction of the free SH2 domain EAT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC125701/]
- 14. SH2 Domain - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/sh2-domain]
- 15. Evolution of SH2 domains and phosphotyrosine signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3415846/]
- 16. Enhanced Prediction of Src Homology 2 (SH2) Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4083110/]
- 17. Quantitative analysis of SH2 domain binding. Evidence for ... [https://www.sciencedirect.com/science/article/pii/S0021925819496890]
- 18. SH2db - Documentation [http://sh2db.ttk.hu/documentation]
- 19. SH2 domain [https://en.wikipedia.org/wiki/SH2_domain]
- 20. Loops Govern SH2 Domain Specificity by Controlling Access ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6590088/]
- 21. Surface Loops in a Single SH2 Domain Are Capable of ... [https://www.sciencedirect.com/science/article/pii/S1535947620318776]
- 22. Structural Insight into the Binding Diversity between the Tyr ... [https://www.sciencedirect.com/science/article/pii/S0021925820675706]
- 23. Investigation of phosphotyrosine recognition by the SH2 ... [https://www.sciencedirect.com/science/article/abs/pii/S002228369993190X]
- 24. Tyrosine Phosphorylation of the Lyn Src Homology 2 (SH2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4349988/]
- 25. Structural Implications of STAT 3 and STAT 5 SH Mutations 2 Domain [https://pubmed.ncbi.nlm.nih.gov/31717342/]
- 26. of Evolution and phosphotyrosine signalling networks SH 2 domains [https://www.academia.edu/1865504/Evolution_of_SH2_domains_and_phosphotyrosine_signalling_networks]
- 27. The genome of the choanoflagellate Monosiga brevicollis and... | Nature [https://www.nature.com/articles/nature06617?error=cookies_not_supported&code=25f4c475-8f27-46fc-a4e1-fd89720a053f]
- 28. SH2 domains as a target class in cancer and inflammation [https://www.drugtargetreview.com/webinar/110968/sh2-domains-as-a-target-class-in-cancer-and-inflammation/]
- 29. Review The SH2 domain: versatile signaling module and ... [https://www.sciencedirect.com/science/article/abs/pii/S1570963904002882]
- 30. Structure of the interleukin-2 tyrosine kinase Src homology 2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3274390/]
- 31. Improved methodology for protein NMR structure ... [https://www.sciencedirect.com/science/article/pii/S0969212623001685]
- 32. Constraining binding hot spots: NMR and MD simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2943377/]
- 33. Computational screening and molecular dynamics of natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12638358/]
- 34. Phosphopeptide binding to the N-SH2 domain of tyrosine ... [https://www.sciencedirect.com/science/article/pii/S2001037024000473]
- 35. Decoding the selective mechanism behind a monobody ... [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00211g]
- 36. Structure and Dynamics of Drk-SH2 Domain and Its Site- ... [https://www.mdpi.com/1422-0067/25/12/6386]
- 37. Phosphotyrosine recognition domains: the typical, the atypical ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-10-32]
- 38. Accurate affinity models for SH2 domains from peptide binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12521615/]
- 39. Comparison of SH3 and SH2 domain dynamics when ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283699926190]
- 40. Surface Loops in a Single SH2 Domain Are Capable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6356082/]
- 41. Structural Determinants of Phosphopeptide Binding to the N ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8007070/]
- 42. Lipid-Mediated Assembly of Biomolecular Condensates [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467959/]
- 43. Emerging regulatory mechanisms and functions of ... [https://www.nature.com/articles/s41392-024-02070-1]
- 44. Lipid-driven Src self-association modulates its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11909415/]
- 45. SH2 Domains: Folding, Binding and Therapeutical ... [https://www.mdpi.com/1422-0067/23/24/15944]
- 46. Accurate sequence-to-affinity models for SH2 domains from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11703206/]
- 47. SH2 Domains Serve as Lipid-Binding Modules for pTyr- ... [https://www.sciencedirect.com/science/article/pii/S109727651600054X]
- 48. Impact of structural biology and the protein data bank on us ... [https://www.nature.com/articles/s41388-024-03077-2]
- 49. Update on Structure and Function of SH2 Domains [https://pubmed.ncbi.nlm.nih.gov/41009621/]
- 50. Disease-Associated Mutations of the STAT5B SH2 Domain ... [https://pubmed.ncbi.nlm.nih.gov/40163145/]
- 51. STAT5B leukemic mutations, altering SH2 tyrosine 665 ... [https://www.life-science-alliance.org/content/8/7/e202503222]
- 52. Mouse Model of STAT3 Mutation Resulting in Job's ... [https://pubmed.ncbi.nlm.nih.gov/40868996/]
- 53. Computational Elucidation of a Monobody Targeting the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11853358/]
- 54. Recent advances in targeting the “undruggable” proteins [https://www.nature.com/articles/s41392-023-01589-z]
- 55. Review SH2 domain inhibition: a problem solved? [https://www.sciencedirect.com/science/article/abs/pii/S1367593100002222]
- 56. Defining the Specificity Space of the Human Src Homology ... [https://www.sciencedirect.com/science/article/pii/S1535947620312007]
- 57. integrating multi-source data for high-accuracy protein ... [https://pubmed.ncbi.nlm.nih.gov/40784875/]
- 58. Recent advances in deep learning for protein-protein interaction [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-025-00457-6]
- 59. Review Machine learning to predict de novo protein– ... [https://www.sciencedirect.com/science/article/pii/S0167779925001581]
- 60. Solution structure of the human Grb14–SH2 domain and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2280013/]
- 61. Determination of promising inhibitors for N-SH2 domain of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11612003/]
- 62. Investigation of SH2 Domains: Ligand Binding, Structure ... [https://rave.ohiolink.edu/etdc/view?acc_num=osu1259766230]
- 63. Selective Targeting of SH2 Domain–Phosphotyrosine ... [https://www.sciencedirect.com/science/article/pii/S0022283617301390]
- 64. Recludix Unveils First BTK SH2 Domain Inhibitor [https://www.dermatologytimes.com/view/recludix-unveils-first-btk-sh2-domain-inhibitor]
- 65. Novel Roles of SH2 and SH3 Domains in Lipid Binding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8152464/]
- 66. Allosteric modulation of Grb2 drives ligand-dependent signal ... [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-025-00656-5]
- 67. The Allosteric Site on SHP2's Protein Tyrosine Phosphatase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6275946/]
- 68. Development of a Peptide Inhibitor Targeting the C-SH2 ... [https://pubmed.ncbi.nlm.nih.gov/40318117/]
- 69. SH2db, an information system for the SH2 domain - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10320075/]
- 70. Specificity and regulation of phosphotyrosine signaling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7337045/]
- 71. Structural Determinants of the Interaction between ... [https://www.sciencedirect.com/science/article/pii/S0021925818355029]
- 72. CoDIAC: A comprehensive approach for interaction analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11291013/]
- 73. Enhanced Prediction of Src Homology 2 (SH2) Domain ... [https://www.sciencedirect.com/science/article/pii/S1535947620343395]
- 74. Structural Implications of STAT3 and STAT5 SH2 Domain ... [https://www.mdpi.com/2072-6694/11/11/1757]
- 75. STAT proteins: a kaleidoscope of canonical and non ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-021-01214-y]
- 76. Deep mutational scanning of the multi-domain ... [https://www.nature.com/articles/s41467-025-60641-4]
- 77. Selective Targeting of SH2 Domain–Phosphotyrosine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5417323/]
- 78. High-throughput profiling of sequence recognition by ... [https://elifesciences.org/articles/82345]
- 79. Progress towards the development of SH2 domain inhibitors [https://pubs.rsc.org/en/content/articlelanding/2013/cs/c3cs35449k]
- 80. Bruton's tyrosine kinase (BTK) SH2 inhibitor reduced skin ... [https://recludixpharma.com/recludix-pharma-presents-preclinical-data-on-first-in-class-btk-sh2-domain-inhibitor-demonstrating-powerful-btk-inhibition-exceptional-selectivity-and-encouraging-efficacy-in-a-model-of-chronic-spon/]
- 81. SYK Inhibitors Pipeline Outlook Report 2025: DelveInsight ... [https://www.barchart.com/story/news/36314021/syk-inhibitors-pipeline-outlook-report-2025-delveinsight-analyzes-key-rd-progressions-powering-next-generation-therapies]
- 82. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6895964/]
- 83. 1NZV: Crystal Structure of Src SH2 domain bound to ... [https://www.rcsb.org/structure/1NZV]
- 84. Folding and Binding Mechanisms of the SH2 Domain from Crkl [https://pmc.ncbi.nlm.nih.gov/articles/PMC9332313/]
- 85. 1HCS: NMR STRUCTURE OF THE HUMAN SRC SH2 ... [https://www.rcsb.org/structure/1HCS]