Spectrophotometry vs. UFLC-DAD: A Comprehensive Guide for Method Selection in Pharmaceutical Analysis
This article provides a systematic comparison between spectrophotometric and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) methods for pharmaceutical analysis.
Spectrophotometry vs. UFLC-DAD: A Comprehensive Guide for Method Selection in Pharmaceutical Analysis
Abstract
This article provides a systematic comparison between spectrophotometric and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) methods for pharmaceutical analysis. Aimed at researchers and drug development professionals, it explores the fundamental principles, guides method selection and application, offers troubleshooting and optimization strategies, and details validation protocols. By synthesizing current research, including a direct comparative study on metoprolol tartrate, this review delivers a practical framework for choosing the appropriate analytical technique based on cost, sensitivity, specificity, and environmental impact, ultimately supporting robust quality control and research outcomes.
Core Principles: Understanding Spectrophotometry and UFLC-DAD Fundamentals
The Role of Analytical Method Validation in Pharmaceutical Development
Analytical method validation is a critical, systematic process to demonstrate that a laboratory measurement technique is appropriate for its intended purpose, providing reliable data to ensure the identity, purity, potency, and safety of pharmaceutical products throughout their lifecycle [1] [2]. This process confirms that analytical methods are suitably optimized and controlled to obtain consistent, accurate, and precise results, forming the foundation for quality control, regulatory compliance, and decision-making in drug development [1] [2]. The International Council for Harmonisation (ICH) guidelines establish standardized validation parameters—including accuracy, precision, specificity, linearity, and sensitivity—that methods must fulfill to be deemed valid for regulatory submission and commercial quality control [1] [2].
Within pharmaceutical development, two prominent analytical techniques are Ultra-Fast Liquid Chromatography with Diode-Array Detection (UFLC-DAD) and UV-Vis spectrophotometry. The selection between these methods represents a strategic balance between analytical needs and practical constraints. UFLC-DAD offers superior separation capabilities and specificity for complex analyses, while UV-Vis spectrophotometry provides simplicity, cost-effectiveness, and rapid analysis for appropriate applications [1] [2]. This guide provides a comparative framework to inform method selection based on scientific requirements, regulatory considerations, and operational efficiency.
Comparative Analysis: Spectrophotometry vs. UFLC-DAD
Fundamental Principles and Technical Capabilities
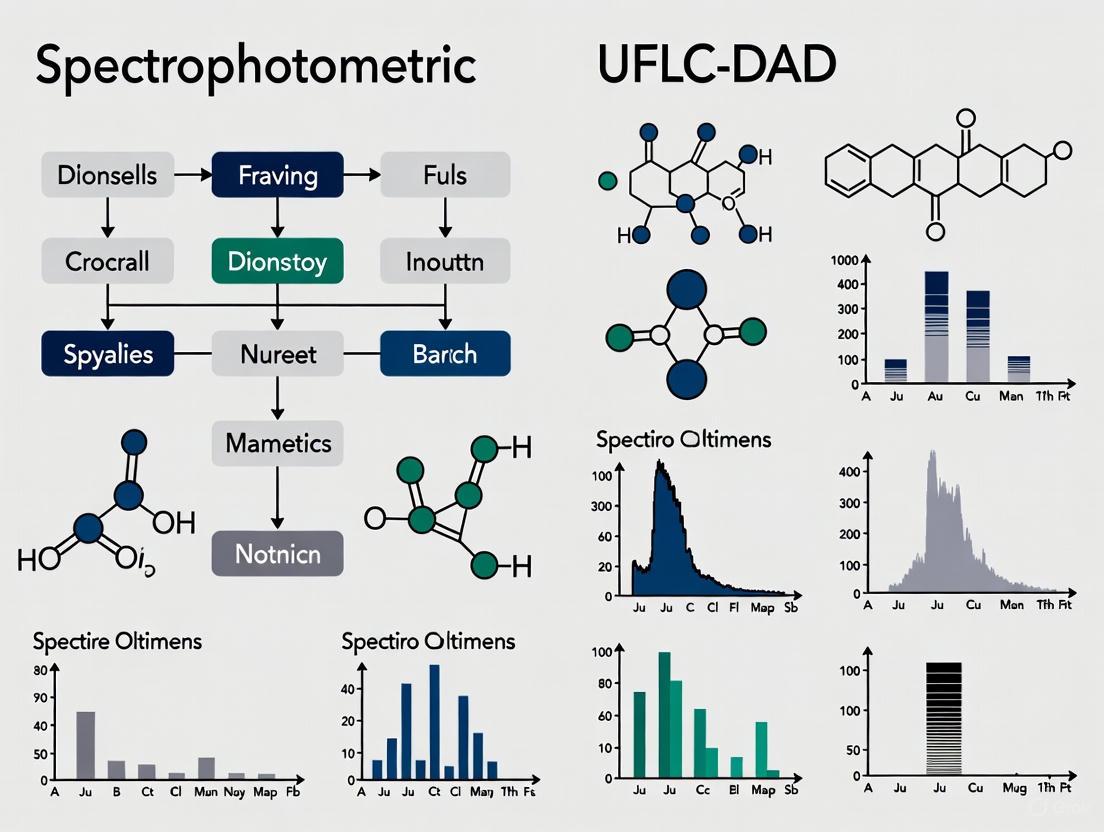
UV-Vis Spectrophotometry operates on the Beer-Lambert Law principle, which states that the absorbance of light by a substance is directly proportional to its concentration in solution [3]. The technique measures light absorption at specific wavelengths (typically the maximum absorption wavelength, λmax) to quantify analytes. For pharmaceuticals lacking inherent chromophores, various reagents—including complexing agents (e.g., ferric chloride for phenolic compounds), oxidizing/reducing agents (e.g., ceric ammonium sulfate), pH indicators, and diazotization reagents—are employed to induce measurable color changes [3]. Despite its simplicity, classical spectrophotometry faces limitations with overlapping spectral bands from multiple analytes or excipients, which can complicate quantitative analysis without prior separation [1].
UFLC-DAD (Ultra-Fast Liquid Chromatography with Diode-Array Detection) combines high-efficiency chromatographic separation with full-spectrum UV-Vis detection. UFLC systems utilize columns with smaller particle sizes (e.g., 1.7 μm) and higher operating pressures compared to conventional HPLC, resulting in improved resolution, shorter analysis times, and reduced solvent consumption [1] [4]. The DAD detector simultaneously captures absorbance data across multiple wavelengths, enabling peak purity assessment and method specificity verification [1] [5]. This hyphenated technique effectively separates analytes from complex matrices, including degradation products and excipients, before quantification [4] [5].
Direct Performance Comparison
Table 1: Comparative Analysis of Spectrophotometric and UFLC-DAD Methods
| Parameter | UV-Vis Spectrophotometry | UFLC-DAD |
|---|---|---|
| Cost & Equipment | Low cost; simple instrumentation [2] | High cost; complex instrumentation requiring skilled operation [1] [2] |
| Selectivity/Specificity | Limited; prone to spectral overlaps and matrix interference [1] [3] | High; excellent separation of complex mixtures and degradation products [1] [5] |
| Sensitivity | Good for simple assays; LOD/LOQ in μg/mL range [6] [7] | Superior; detects lower concentrations with LOD/LOQ in ng/mL range [8] [4] |
| Sample Throughput | Fast analysis time (minutes) [2] | Moderate; method lengths vary but typically faster than conventional HPLC [4] |
| Solvent Consumption | Minimal [2] | Reduced compared to HPLC but higher than spectrophotometry [4] |
| Multi-Component Analysis | Limited without sophisticated mathematical processing [1] [5] | Excellent for simultaneous quantification of multiple analytes [4] [5] |
| Greenness Assessment | Generally more environmentally friendly [1] | Lower greenness scores due to higher solvent consumption [1] |
| Primary Applications | Routine QC of simple formulations, dissolution profiling, single-component assay [3] [2] | Complex formulations, stability-indicating methods, impurity profiling, bioanalysis [8] [5] |
Table 2: Validation Performance Examples from Literature
| Analytical Method | Analyte | Linearity Range | Precision (%RSD) | Accuracy (% Recovery) | LOD/LOQ | Reference |
|---|---|---|---|---|---|---|
| UPLC/DAD | Empagliflozin & related substances | 50-700 ng/mL | N/S | ≥96.97% | 15/50 ng/mL | [8] |
| UV-Spectrophotometry | Vildagliptin | 5-60 μg/mL | Intraday: 1.263% Interday: 1.162% | 98-101% | 0.951/2.513 μg/mL | [6] |
| UFLC-DAD | Metoprolol tartrate | Validated for 50 mg & 100 mg tablets | N/S | N/S | N/S | [1] |
| HPLC-DAD vs. UV | Lychnopholide | HPLC: 2-25 μg/mL UV: 5-40 μg/mL | Low RSD values | HPLC: 98-101% UV: 96-100% | HPLC more sensitive | [7] |
| UPLC | Proton Pump Inhibitors | 0.75-200 μg/mL | Intraday: ≤0.21% Interday: ≤5% | Confirmed | 0.23-0.59/0.71-1.78 μg/mL | [4] |
Experimental Protocols and Methodologies
Representative UFLC-DAD Method for Anti-Diabetic Drugs
A validated UPLC/DAD method for empagliflozin determination in spiked human plasma demonstrates typical UFLC-DAD protocol [8]. The methodology employed an Acquity UPLC BEH C18 column (50 mm × 2.1 mm i.d, 1.7 μm particle size) with a mobile phase consisting of aqueous trifluoroacetic acid (0.1%, pH 2.5) and acetonitrile (60:40, v/v) at a flow rate of 0.5 mL/min. The method utilized tetrahydrofuran as a novel protein precipitating agent and dapagliflozin as an internal standard. Key validation parameters included linearity (50-700 ng/mL with r² = 0.9994-0.9999), accuracy (mean recoveries ≥96.97%), and sensitivity (LOD 15 ng/mL, LOQ 50 ng/mL for empagliflozin). The optimized method achieved a run time of less than 1.2 minutes with significantly reduced solvent consumption (0.36 mL acetonitrile per run), highlighting the efficiency advantages of UPLC approaches [8].
Representative UV-Spectrophotometric Method for Gastric Medium Analysis
For vildagliptin estimation in gastric medium, researchers developed a simple UV-spectrophotometric method using 0.1N HCl as the solvent medium with detection at 210 nm [6]. The method demonstrated linear response over 5-60 μg/mL (r² = 0.999) with accuracy between 98-101%. Precision studies showed intraday and interday %RSD values of 1.263 and 1.162, respectively, within acceptable limits. The method achieved LOD and LOQ values of 0.951 μg/mL and 2.513 μg/mL, respectively. The straightforward sample preparation involved direct dissolution of the drug in acidic medium without derivatization or complex extraction procedures, making it suitable for routine quality control applications with minimal resource requirements [6].
Comparative Validation Study Protocol
A direct comparison study evaluated spectrophotometric and UFLC-DAD methods for metoprolol tartrate (MET) analysis in commercial tablets [1]. For spectrophotometry, absorbance was measured at λmax = 223 nm after appropriate sample preparation. For UFLC-DAD, method optimization preceded validation, with both techniques assessed for specificity/selectivity, sensitivity, linearity, dynamic range, LOD, LOQ, accuracy, precision, and robustness. Statistical analysis using ANOVA at 95% confidence level determined no significant difference between concentrations obtained by both methods. Additionally, the Analytical GREEnness (AGREE) metric approach compared environmental impact, revealing the spectrophotometric method's superior greenness profile [1].
Method Selection Workflow
The following diagram illustrates the decision-making process for selecting between spectrophotometric and UFLC-DAD methods based on analytical requirements and practical constraints:
Validation Parameter Relationships
The interrelationships between key validation parameters and their role in establishing method validity are depicted below:
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Spectrophotometric and UFLC-DAD Analysis
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Complexing Agents (e.g., Ferric chloride, Potassium permanganate) | Form stable, colored complexes with pharmaceutical analytes to enhance absorbance | Detection of phenolic drugs like paracetamol; metal-containing drug analysis [3] |
| Oxidizing/Reducing Agents (e.g., Ceric ammonium sulfate, Sodium thiosulfate) | Modify oxidation state of analytes to create measurable color changes | Analysis of drugs lacking chromophores; ascorbic acid determination [3] |
| Diazotization Reagents (e.g., Sodium nitrite + HCl, N-(1-naphthyl)ethylenediamine) | Convert primary amines to diazonium salts forming colored azo compounds | Sulfonamide antibiotics analysis; drugs with primary aromatic amines [3] |
| UPLC C18 Columns (1.7-1.8 μm particle size, 50-100 mm length) | High-efficiency chromatographic separation under high pressure | Empagliflozin related substances; proton pump inhibitors separation [8] [4] |
| Protein Precipitating Agents (e.g., Tetrahydrofuran, Acetonitrile) | Remove proteins from biological samples prior to analysis | Sample preparation for spiked human plasma analysis [8] |
| Derivatization Reagents (e.g., AQC, OPA, FMOC) | Enhance detection sensitivity for compounds with poor UV absorption | Amino acid analysis in feed and pharmaceutical products [9] |
| pH Indicators (e.g., Bromocresol green, Phenolphthalein) | Color change based on solution pH for acid-base equilibria studies | Analysis of acid/base pharmaceuticals; formulation pH optimization [3] |
Regulatory Considerations and Future Directions
The validation of analytical methods must comply with rigorous regulatory standards outlined in ICH guidelines (Q2(R1)), USP, EP, and other regulatory frameworks [1] [2]. These guidelines establish minimum requirements for validation parameters regardless of the analytical technique employed. For UFLC-DAD methods, regulatory submissions typically require extensive forced degradation studies to demonstrate method specificity and stability-indicating capabilities [5]. Spectrophotometric methods may face greater scrutiny regarding specificity when applied to complex formulations, potentially requiring additional validation data to prove the absence of interference [1] [3].
Future directions in pharmaceutical analysis emphasize green analytical chemistry principles, with recent studies incorporating the Analytical GREEnness (AGREE) metric approach to evaluate environmental impact [1] [5]. Method validation practices are evolving to incorporate quality by design (QbD) principles, real-time analytical technologies, and computational modeling to enhance method robustness and transferability [2]. The integration of artificial intelligence for method development and validation represents an emerging frontier, with recent applications including insilico assessment of potential drug-drug interactions in combination therapy analysis [5].
The comparative analysis demonstrates that both spectrophotometry and UFLC-DAD provide distinct advantages for pharmaceutical analysis when properly validated. The selection between these techniques should be guided by analytical requirements, regulatory expectations, and practical constraints, with both methods playing complementary roles in ensuring drug quality throughout the development lifecycle.
Spectrophotometry is a foundational analytical technique that measures how much light a substance absorbs, transmits, or reflects at specific wavelengths. This method is built upon the fundamental principle that molecules interact with light in predictable ways, allowing researchers to determine concentration, assess purity, and identify chemical properties. The versatility of spectrophotometry has cemented its role across diverse scientific fields, including pharmaceutical research, clinical diagnostics, environmental monitoring, and material science [10] [11].
This technique operates across various regions of the electromagnetic spectrum, with UV-Visible (UV-Vis) spectrophotometry being one of the most prevalent forms. UV-Vis instruments measure absorption in the ultraviolet (190–400 nm) and visible (400–700 nm) ranges, making them suitable for analyzing a vast array of organic compounds, biological macromolecules, and inorganic ions [11] [12]. The technique's widespread adoption stems from its direct applicability to quantitative analysis, where it provides rapid, sensitive, and non-destructive measurements for both research and quality control purposes [13].
In the context of modern analytical laboratories, spectrophotometry often serves as a benchmark against which newer, more complex techniques are compared. Its inherent simplicity and cost-effectiveness make it an attractive first-line analytical tool, though its limitations in handling complex mixtures have led to the development and integration of more specialized methods like Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) [7] [14]. Understanding the core principles, advantages, and limitations of spectrophotometry is therefore essential for researchers and drug development professionals seeking to select the most appropriate analytical strategy for their specific applications.
Core Principles of Spectrophotometry
The Beer-Lambert Law
The quantitative foundation of spectrophotometry is the Beer-Lambert Law, which establishes the mathematical relationship between a substance's concentration and its light absorption. This law states that the absorbance (A) of light by a solution is directly proportional to the concentration (c) of the absorbing species and the path length (l) of the light through the sample [10] [11]. The law is expressed by the equation:
A = ε × l × c
Where:
- A is the measured absorbance (no units)
- ε is the molar absorptivity or extinction coefficient (L·mol⁻¹·cm⁻¹)
- l is the path length of the light through the sample (cm)
- c is the concentration of the absorbing species (mol·L⁻¹)
The molar absorptivity (ε) is a compound-specific constant that represents how strongly a chemical species absorbs light at a particular wavelength. The linear relationship described by the Beer-Lambert Law enables the determination of unknown concentrations by measuring absorbance, provided the values of ε and l are known [10]. This principle forms the bedrock of quantitative analysis in spectrophotometry, allowing researchers to construct calibration curves from standards of known concentration and use these to interpolate values for unknown samples.
Instrumentation and Mechanism
A spectrophotometer operates through a coordinated sequence of events involving several key components. The basic design consists of: a light source that emits polychromatic light (typically a deuterium lamp for UV and a tungsten lamp for visible regions); a monochromator (prism or diffraction grating) that separates the light into specific wavelengths; a sample holder (cuvette) where the sample is placed; a detector that measures the intensity of transmitted light; and a readout device that displays the results as absorbance or transmittance [11] [12].
The mechanism of analysis involves the interaction of light with matter at the molecular level. When light passes through a sample, specific molecules absorb photons at particular wavelengths, promoting electrons to higher energy states. The transmitted light, which represents the portion that passes through without being absorbed, is measured by the detector. The instrument compares this intensity to the initial light intensity, calculating the absorbance according to the Beer-Lambert Law [10]. This process can be visualized in the following workflow:
Different types of spectrophotometers are designed for specific applications. Single-beam instruments use a single light path for both reference and sample measurements, requiring manual calibration between readings. Double-beam spectrophotometers employ two separate beams (sample and reference) simultaneously, enhancing accuracy and stability by compensating for source fluctuations. UV-Vis spectrophotometers cover both ultraviolet and visible regions, while IR spectrophotometers analyze molecular vibrations in the infrared region, providing structural information about materials [10] [12].
Advantages of Spectrophotometry
Operational Benefits
Spectrophotometry offers numerous operational advantages that contribute to its enduring popularity in research and industrial settings:
High Sensitivity and Accuracy: Modern spectrophotometers are highly sensitive instruments capable of detecting minute changes in light absorption, enabling accurate quantification of analytes even at low concentrations. This sensitivity allows for precise determination of substance concentration in a sample, which is crucial for applications requiring high precision such as drug formulation and environmental pollutant detection [10] [13].
Non-Destructive Nature: A significant advantage of spectrophotometric analysis is its non-destructive character. Samples remain unchanged after measurement, allowing for repeated analyses on the same material. This is particularly valuable when working with precious or limited samples, such as in pharmaceutical research or forensic analysis, where sample preservation is essential for additional testing [10] [13].
Rapid Analysis and Efficiency: Spectrophotometers provide almost immediate results after sample introduction, making them ideal for high-throughput environments. This speed facilitates quick decision-making in quality control laboratories and enables real-time monitoring of chemical reactions or biological processes, significantly enhancing research efficiency [13].
Cost-Effectiveness: Compared to other analytical techniques like HPLC, GC, or FTIR, basic UV-Vis spectrophotometers represent a more affordable investment. The operational costs are also relatively low, as they typically require minimal consumables beyond inexpensive cuvettes and common solvents, making the technique accessible to laboratories with varying budget constraints [13].
Application Versatility
The widespread adoption of spectrophotometry across diverse scientific disciplines underscores its remarkable versatility:
Pharmaceutical Applications: In drug development and quality control, spectrophotometry is used to assess drug purity, stability, and concentration. It ensures the effectiveness and safety of pharmaceutical products by quantifying active ingredients and detecting potential impurities [10] [14].
Clinical Diagnostics: Spectrophotometric assays are routinely employed in clinical laboratories for diagnostic purposes. These include measuring specific biomarkers or metabolites in blood and urine, analyzing respiratory gases in hospital settings, and supporting biomarker discovery and quantification [10] [11].
Environmental Monitoring: The technique plays a vital role in environmental testing, particularly in water quality analysis. It enables detection and quantification of pollutants such as heavy metals, organic compounds, and nutrients, contributing to environmental impact assessment and regulatory compliance [10] [11].
Biological Research: In life sciences, spectrophotometry is indispensable for DNA, RNA, and protein quantification, enzyme kinetics studies, cell density measurements (OD600), and monitoring various biochemical reactions [10] [11] [12].
Material Science and Semiconductor Industry: Applications in these fields include colorimetry, thin film characterization, monitoring photodegradation of dyes, determining magnetic material concentrations, and measuring trace anionic contaminants in high-purity water used in semiconductor manufacturing [10].
Inherent Limitations and Challenges
Despite its numerous advantages, spectrophotometry faces several inherent limitations that affect its application in certain analytical scenarios. Understanding these constraints is crucial for researchers to determine when the technique is appropriate and when alternative methods might be preferable.
Susceptibility to Interference: Spectrophotometric measurements can be compromised by various forms of interference. Stray light—any light that reaches the detector without passing through the sample—can distort spectra and lead to inaccurate measurements, particularly at low absorbance levels [13]. Additionally, samples with turbidity or particulate matter can scatter light, affecting absorption accuracy. The presence of multiple absorbing components in a mixture can cause overlapping absorption bands, making it difficult to discern individual substances without prior separation [13].
Sample Preparation Requirements: For accurate results, spectrophotometry often demands careful sample preparation. Samples may need to be dissolved in specific solvents, filtered to remove particulates, or diluted to fall within the optimal absorbance range (typically 0.1-2.0 absorbance units) [10] [13]. Measurements outside this range can lead to non-linear behavior, with values below 0.1 suffering from low sensitivity and those exceeding 2.0 potentially causing saturation effects that compromise quantitative accuracy [10].
Limited Structural Information: Unlike techniques such as NMR or mass spectrometry, spectrophotometry provides limited information about molecular structure. While absorption spectra can serve as fingerprints for compound identification, they offer little insight into detailed molecular architecture or functional group arrangement [13]. This restriction makes spectrophotometry less suitable for de novo compound characterization.
Restricted Dynamic Range: The useful analytical range of spectrophotometry is constrained by the linear response described by the Beer-Lambert Law, which typically holds only for relatively dilute solutions (often below 0.01 M). At higher concentrations, molecular interactions can lead to deviations from linearity, necessitating sample dilution and introducing additional processing steps [11].
The following table summarizes these key limitations and their practical implications for analytical work:
| Limitation | Description | Impact on Analysis |
|---|---|---|
| Stray Light Interference | Light reaching detector without passing through sample [13] | Decreased linearity, inaccurate low-concentration measurements [13] |
| Sample Complexity | Multiple absorbing components with overlapping bands [13] | Difficulty discerning individual substances in mixtures [13] |
| Limited Dynamic Range | Optimal absorbance range typically 0.1-2.0 AU [10] | Requires sample dilution at high concentrations, potentially introducing error |
| Limited Structural Information | Primarily measures absorption/transmission [13] | Cannot provide detailed molecular structure or functional group data [13] |
| Turbidity Interference | Particulate matter scattering light [13] | Inaccurate absorbance readings, requires filtration or clarification |
Comparative Analysis: Spectrophotometry vs. UFLC-DAD
Performance Comparison in Pharmaceutical Analysis
When evaluating analytical techniques for drug development, direct comparison between spectrophotometry and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) reveals distinct performance characteristics suited to different applications. A study developing analytical methods for Filgrastim (a biosimilar drug) demonstrated that while both techniques can be validated according to ICH guidelines, their capabilities differ significantly [14].
The UV-spectrophotometry method for Filgrastim analysis showed a linear range of 1-3 μg/mL with a correlation coefficient of 0.9994, while the RP-UFLC method exhibited a wider linear range of 5-15 μg/mL with a correlation coefficient of 0.999 [14]. This illustrates UFLC-DAD's advantage in handling a broader concentration range while maintaining excellent linearity. Additionally, the UFLC method provided superior specificity by separating Filgrastim from potential degradants and excipients through chromatographic separation—a critical advantage in stability-indicating methods [14].
Another study comparing methods for quantifying lychnopholide in nanocapsule dosage forms found that while both HPLC-DAD and UV-spectrophotometry were validated successfully, the HPLC-DAD method offered greater sensitivity with a lower limit of detection (2-25 μg/mL for HPLC-DAD vs. 5-40 μg/mL for spectrophotometry) [7]. This enhanced sensitivity enables more precise drug release kinetic studies, particularly important for nanocarrier systems where controlled release profiles are essential for therapeutic efficacy.
Methodological Considerations
The fundamental distinction between these techniques lies in their operational principles. Spectrophotometry provides a composite measurement of all light-absorbing species in a sample, whereas UFLC-DAD first separates mixture components chromatographically before performing spectrophotometric detection on individual analytes [7] [14]. This separation step in UFLC-DAD dramatically reduces interference issues that commonly plague direct spectrophotometric analysis of complex mixtures.
For vitamin C quantification in fruit juices, a comparative study revealed that spectrophotometric methods demonstrated superior performance over titrimetric approaches, with lower LOD (0.002 mg/mL) and LOQ (0.010 mg/mL) values, and greater accuracy (error <5%) particularly in colored solutions where visual endpoint determination in titration was compromised [15]. However, neither spectrophotometry nor titration could match the specificity of chromatographic methods for analyzing complex matrices without prior sample clean-up.
The following table provides a quantitative comparison of both techniques based on experimental data from the cited studies:
| Parameter | UV-Spectrophotometry | UFLC-DAD | Experimental Context |
|---|---|---|---|
| Linear Range | 1-3 μg/mL [14] | 5-15 μg/mL [14] | Filgrastim analysis [14] |
| Correlation Coefficient (R²) | 0.9994 [14] | 0.999 [14] | Filgrastim analysis [14] |
| Limit of Detection | 0.002 mg/mL [15] | 0.3-1.0 μg/L [16] | Vitamin C analysis [15], PDE-5 inhibitors [16] |
| Accuracy | 98-101% [7] | 81.6-118.9% [16] | Lychnopholide in nanocapsules [7], PDE-5 inhibitors [16] |
| Precision (RSD) | <5% error [15] | ≤4.2-5.2% RSD [16] | Vitamin C analysis [15], PDE-5 inhibitors [16] |
| Analysis Time | Fast (minutes) [13] | Moderate (10-30 min) [16] | General operation [13], multi-analyte screening [16] |
Essential Research Reagent Solutions
Successful implementation of spectrophotometric methods requires specific reagents and materials tailored to the analytical application. The following table outlines key research reagent solutions commonly employed in spectrophotometric analyses, particularly in pharmaceutical and biological contexts:
| Reagent/Material | Function | Application Example |
|---|---|---|
| Quartz Cuvettes | Sample holder with high UV transmission | UV analysis below 350 nm [12] [14] |
| Methanol & Acetonitrile (HPLC Grade) | Mobile phase components, solvent for samples | RP-UFLC analysis, sample preparation [14] [16] |
| Formic Acid/Acetic Acid | Mobile phase modifier, improves chromatography | Enhancing peak shape in UFLC [16] |
| Buffer Salts (Phosphate, Acetate) | Maintain pH for stable analyte measurement | Biological samples, protein analyses [14] |
| Reference Standards | Calibration and method validation | Quantification of target analytes [14] |
| Filter Membranes (0.22/0.45 μm) | Sample clarification, mobile phase degassing | Removing particulates that cause light scattering [14] |
Methodological Protocols
Standard UV-Spectrophotometry Protocol
Based on validated methods for pharmaceutical analysis [14], the following protocol outlines the general steps for UV-spectrophotometric quantification:
Instrument Calibration: Turn on the spectrophotometer and allow it to stabilize for 15-20 minutes. Select the appropriate wavelength specific to the analyte of interest (e.g., 215 nm for proteins like Filgrastim) [14].
Blank Measurement: Prepare a blank solution containing all components except the analyte. For aqueous samples, this is typically double-distilled water. Place the blank in a clean quartz or glass cuvette (depending on wavelength range) and calibrate the instrument to zero absorbance or 100% transmittance [12] [14].
Standard Preparation: Prepare a series of standard solutions of known concentration covering the expected range of the sample. For Filgrastim, concentrations of 1, 1.4, 1.8, 2.2, 2.6, and 3 μg/mL were used to construct a calibration curve [14].
Sample Measurement: Fill a clean cuvette with the test sample, ensuring no air bubbles are present and the optical surfaces are free of fingerprints or smudges. Insert the cuvette into the sample holder and record the absorbance value [12].
Data Analysis: Construct a calibration curve by plotting absorbance versus concentration for the standard solutions. Use the resulting equation to calculate the concentration of unknown samples from their measured absorbance values [14].
This workflow can be visualized as follows:
Integrated UFLC-DAD Protocol
For complex mixtures requiring separation prior to detection, the UFLC-DAD method offers enhanced specificity. A typical protocol based on pharmaceutical applications includes [14]:
Mobile Phase Preparation: Prepare appropriate mobile phase mixtures based on analyte characteristics. For protein analysis like Filgrastim, a mixture of acetonitrile and water (80:20 v/v) is commonly used. Filter and degas the mobile phase before use [14].
Chromatographic Conditions: Set the UFLC system parameters including flow rate (typically 0.5-1.0 mL/min), column temperature, injection volume, and detection wavelength. For Filgrastim, a Phenomenex C4 column (25 cm × 0.46 cm, 15 μ) with detection at 215 nm provided optimal results [14].
System Equilibration: Allow the chromatographic system to equilibrate until a stable baseline is achieved, typically requiring 10-15 column volumes of mobile phase.
Standard and Sample Analysis: Inject standards of known concentration to establish retention times and calibration curves. Follow with unknown samples, using peak areas or heights for quantification relative to the calibration curve [14].
Method Validation: Perform validation according to ICH guidelines, assessing parameters including linearity, precision, accuracy, limit of detection (LOD), limit of quantification (LOQ), and robustness [14].
Spectrophotometry remains an indispensable analytical technique with broad applicability across numerous scientific disciplines, particularly in pharmaceutical research and drug development. Its strengths lie in its simplicity, cost-effectiveness, rapid analysis time, non-destructive nature, and excellent quantitative capabilities for well-characterized systems obeying the Beer-Lambert Law [10] [13]. These advantages ensure its continued relevance as a first-line analytical tool in both research and quality control environments.
However, the inherent limitations of spectrophotometry—including susceptibility to interference from complex matrices, limited structural information, and restricted dynamic range—necessitate complementary techniques like UFLC-DAD for more challenging analytical scenarios [7] [14]. The comparative data presented in this guide demonstrates that while spectrophotometry offers superior accessibility and speed for routine analyses, UFLC-DAD provides enhanced specificity, sensitivity, and the ability to resolve complex mixtures through chromatographic separation prior to detection.
For researchers and drug development professionals, the choice between these techniques should be guided by specific analytical requirements. Spectrophotometry serves as an excellent tool for quantitative analysis of pure substances, routine quality checks, and educational applications, while UFLC-DAD is better suited for method development, complex mixture analysis, and regulatory submissions requiring comprehensive validation. Understanding the principles, advantages, and limitations of each approach enables informed methodological decisions that optimize analytical outcomes in scientific research and pharmaceutical development.
Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) represents a significant advancement in analytical separation science, offering improved resolution, speed, and detection capabilities compared to conventional HPLC systems. This technology combines the high-speed separation power of ultra-fast liquid chromatography with the versatile detection capabilities of diode array detectors, making it particularly valuable in pharmaceutical analysis, bioanalytical applications, and quality control laboratories where efficiency and reliability are paramount.
The Prominence UFLC XR system exemplifies this technology, featuring ultra high-pressure endurance up to 66 MPa which provides a wider variety of applications for UFLC, enabling both higher resolution and higher sensitivity across diverse analytical scenarios [17]. The balance between an appropriately small particle-sized column media and the column length is an important factor in improving overall column performance, and UFLC systems are specifically engineered to optimize this relationship. Through the use of longer columns, mobile phases containing methanol, and analysis under slightly lower room temperature conditions, Prominence UFLC XR provides enhanced resolution while maintaining the ability to easily transfer existing analytical methods to high-speed, high-resolution conditions [17].
Technology Mechanism and Components
Core System Architecture
The UFLC-DAD system comprises several integrated components that work in concert to achieve rapid separations with comprehensive detection capabilities. The system typically includes a binary or quaternary solvent delivery system capable of maintaining precise gradients at high pressures, an advanced autosampler with temperature control capabilities, a thermostatted column compartment for maintaining optimal separation efficiency, and the diode array detector itself with a high-pressure flow cell [17]. The Shimadzu Prominence UFLC XR system, for instance, incorporates the SIL-20A XR/20AC XR autosampler with an injection volume setting range of 0.1 to 50 μL (standard) or 1 to 100 μL (optional), capable of withstanding pressures up to 66 MPa, which enables precise sample introduction even at extreme operating pressures [17].
The diode array detector operates on the principle of parallel detection across multiple wavelengths simultaneously. Unlike conventional UV-Vis detectors that monitor at fixed wavelengths, DAD captures the complete spectrum from 190 to 640 nm (depending on the specific instrument) for each data point during the chromatographic run [18]. This is achieved through an array of hundreds of individual photodiodes, each measuring a narrow bandwidth of the spectrum. Critical to this detection method is the use of optofluidic waveguides that improve light transmission through the flow cell, enabling full spectral detection with high sensitivity [18]. The detector employs a high data-sampling rate of up to 240 Hz, making it capable of accurately tracking even very narrow chromatographic peaks with widths as small as 450 ms, which is particularly advantageous in UFLC where peak widths are substantially reduced compared to conventional HPLC [18].
Separation Mechanism
The separation power of UFLC systems derives from the use of stationary phases with smaller particle sizes (typically sub-2μm) and optimized fluidic pathways that can withstand the elevated pressures generated by these efficient packing materials. The relationship between particle size, backpressure, and efficiency follows the van Deemter equation, where smaller particles provide higher efficiency with minimal loss at increased flow rates. The UFLC XR system leverages this principle through columns packed with smaller particle size media, which provide higher chromatographic efficiencies at substantially lower system backpressures compared to conventional HPLC systems [19].
The mobile phase delivery system in UFLC is engineered for high-pressure operation, with the Prominence UFLC XR capable of maintaining stable flow rates against backpressures up to 66 MPa [17]. This enables the use of longer columns for increased resolution when needed, as demonstrated in the analysis of polycyclic aromatic compounds where a 150 mmL. × 3.0 mmI.D. column provided excellent separation of 9 different compounds in a single run [17]. The system's ability to handle high-viscosity mobile phases and operate at lower temperatures further expands the methodological possibilities for challenging separations [17].
Table 1: Key Technical Specifications of Shimadzu Prominence UFLC XR System
| Component | Specification | Performance Capability |
|---|---|---|
| Solvent Delivery System | Maximum pressure endurance: 66 MPa | Enables use of columns packed with smaller particles for higher efficiency |
| Autosampler (SIL-20A XR/20AC XR) | Injection volume: 0.1-50 μL (standard), 1-100 μL (option) | Precise sample introduction at high operating pressures |
| Column Oven | Temperature control capability | Maintains optimal separation efficiency |
| Diode Array Detector | High data-sampling rate up to 240 Hz | Capable of tracking peaks with widths as narrow as 450 ms |
| Sample Capacity | 175 (1mL vials), 70 (1.5 mL vials), 50 (4 mL vials) | High throughput capability for extended runs |
Separation Power and Performance
Enhanced Resolution and Speed
The separation power of UFLC-DAD systems manifests primarily in two dimensions: chromatographic resolution and analysis speed. The technology enables faster separations without compromising resolution, or alternatively, higher resolution separations in comparable timeframes to conventional HPLC. This is achieved through a combination of reduced particle size in stationary phases, optimized system volumes to minimize extracolumn band broadening, and detectors capable of high-speed data acquisition.
In practical applications, UFLC-DAD has demonstrated exceptional performance across diverse analytical scenarios. For instance, in the analysis of catechins in Japanese green tea, the UFLC XR system successfully achieved complete separation using both water/acetonitrile and water/methanol mobile phases, showcasing its versatility in handling different solvent systems while maintaining separation efficiency [17]. Similarly, in the analysis of a mixture of 9 polycyclic aromatic compounds, the system provided high-resolution separation with excellent peak symmetry, enabling more accurate quantification through precise separation of closely eluting compounds [17].
The kinetic performance of UFLC-DAD systems represents a significant advancement over traditional HPLC. The combination of Core-Shell technology columns with the low-dispersion fluidic pathways of UFLC instruments allows for substantially faster analyses while maintaining resolution. As demonstrated in the development of a stability-indicating method for ritlecitinib, UHPLC-DAD-MS/MS enabled fast separation of the drug substance and its degradation products, with the method showing high specificity, sensitivity (LOD: 0.04 µg/mL; LOQ: 0.14 µg/mL), precision (RSD ≤ 0.15%), and accuracy (99.9-100.3%) [20] [19]. The use of reversed-phase C18 columns with sub-3 μm particles provided higher hydrophobic retention suitable for ritlecitinib and its non-polar degradation products, highlighting the importance of stationary phase selection in maximizing UFLC performance [19].
Method Transfer and Flexibility
A key advantage of UFLC-DAD systems is their method transfer flexibility, allowing seamless transition of existing HPLC methods to faster UFLC methods with minimal modification. The instrumental design accommodates a wide range of column dimensions and particle sizes, facilitating method optimization for either speed or resolution as required by specific applications. The Prominence UFLC XR specifically addresses this through compatibility with various column configurations and mobile phase compositions, enabling straightforward method transfer from conventional HPLC to high-speed, high-resolution conditions [17].
This flexibility extends to the detection capabilities as well, with the DAD component providing comprehensive spectral information that can be re-interrogated post-analysis without reinjection. The ability to monitor multiple wavelengths simultaneously while capturing full UV-Vis spectra for each analyte makes UFLC-DAD particularly valuable in method development and peak purity assessment. As demonstrated in the analysis of menaquinone-4 in spiked rabbit plasma, UFLC-DAD enabled detection in the range of 190-600 nm with 269 nm set as a reference wavelength, providing both specificity for quantification and spectral confirmation of compound identity [21].
Detector Capabilities
Diode Array Detection Technology
The diode array detector in UFLC-DAD systems represents a significant advancement in detection technology, offering capabilities beyond those of conventional single-wavelength UV detectors. The fundamental operating principle involves passing polychromatic light through the sample flow cell, after which the transmitted light is dispersed onto an array of individual photodiode elements, each measuring a specific narrow wavelength band [18]. This parallel detection approach enables full spectrum acquisition for every data point during the chromatographic run, creating a three-dimensional data matrix (time, absorbance, wavelength) that contains substantially more information than single-wavelength detection.
The technical implementation of DAD in modern UFLC systems addresses several critical challenges in high-speed chromatography. The detector incorporates advanced thermal management systems to maintain stability under varying operating conditions, often utilizing positive temperature coefficient (PTC) thermistors as heaters for energy efficiency and protection against overheating [18]. Additionally, the flow path is designed for high inertness to prevent analyte adsorption or absorption, which is particularly important for sensitive pharmaceutical applications [18]. The cartridge cell design with optofluidic waveguides improves light transmission efficiency, contributing to the high sensitivity of these detection systems [18].
Spectral Information and Applications
The rich spectral data provided by DAD detection enables multiple advanced applications that are not possible with conventional detectors. Peak purity assessment is significantly enhanced through the comparison of spectra across different regions of a chromatographic peak, allowing detection of co-eluting impurities with similar retention times but distinct spectral characteristics. Spectral matching against library references provides additional confirmation of compound identity, complementing retention time information for enhanced confidence in identification.
In the quantification of menaquinone-4 in spiked rabbit plasma, UFLC-DAD demonstrated excellent performance with a linear range of 0.374 to 6 µg/mL (R² = 0.9934), accuracy with % RSD <15%, and inter and intraday precisions <10% [21]. The method employed protein precipitation for sample preparation and chromatographic separation using Isopropyl Alcohol and Acetonitrile (50:50 v/v) as mobile phase on a C-18 column with 1ml/min flow rate and 10-minute run time, showcasing the efficiency of UFLC-DAD for bioanalytical applications [21].
Diagram 1: DAD Detection Mechanism - from Light Source to Spectral Output
Comparative Analysis with Alternative Technologies
UFLC-DAD vs. Conventional HPLC
When compared to conventional HPLC systems, UFLC-DAD offers substantial improvements in analysis speed, resolution, and detection capabilities. The reduced particle size of stationary phases in UFLC (typically sub-2μm versus 3-5μm in conventional HPLC) directly translates to higher efficiency according to the van Deemter equation, allowing either faster separations or improved resolution in comparable timeframes. The market analysis data confirms the growing preference for advanced liquid chromatography systems, with the global HPLC market projected to grow from USD 5.01 billion in 2024 to USD 7.74 billion by 2032, registering a steady CAGR of 5.64%, driven largely by adoption of UHPLC and UFLC technologies [22].
The detection advantage of DAD compared to single-wavelength UV detectors in conventional HPLC systems is particularly significant for method development and complex sample analysis. While conventional HPLC with single-wavelength detection may be sufficient for simple quality control applications, the comprehensive spectral information provided by DAD is invaluable for peak identification, purity assessment, and method development. As demonstrated in the analysis of traditional Chinese medicines and complex pharmaceutical formulations, the ability to monitor multiple wavelengths simultaneously and obtain full spectra for each analyte provides a level of characterization not possible with conventional detection [23].
Table 2: Performance Comparison: UFLC-DAD vs. Conventional HPLC-UV
| Parameter | UFLC-DAD | Conventional HPLC-UV |
|---|---|---|
| Analysis Speed | Significantly faster (up to 5-10x reduction in run time) | Standard analysis times |
| Chromatographic Resolution | Higher efficiency due to smaller particle sizes | Moderate efficiency with larger particles |
| Detection Capability | Full spectral information for all peaks | Single or dual wavelength monitoring |
| Peak Purity Assessment | Possible through spectral comparison | Limited or not available |
| Method Development Flexibility | High - post-acquisition reprocessing | Limited - requires reinjection for different wavelengths |
| Pressure Capability | High (up to 66 MPa for Prominence UFLC XR) | Moderate (typically <40 MPa) |
| Data Richness | 3D data (time, absorbance, wavelength) | 2D data (time, absorbance at fixed wavelength) |
UFLC-DAD vs. Spectrophotometric Methods
The comparison between UFLC-DAD and direct spectrophotometric methods reveals even more dramatic differences in capability and application range. While spectrophotometric methods offer simplicity and low cost, they suffer from significant limitations in specificity and accuracy, particularly in complex matrices. A comparative study between titrimetric and spectrophotometric methods for quantification of vitamin C found that the spectrophotometric method was more sensitive, with lower values for LOD (0.002mg/mL) and LOQ (0.010mg/mL), and more accurate with error less than 5% while results from the titrimetric method were affected by the juice color, which generated errors in excess of 15% [15]. However, even with this advantage, spectrophotometric methods cannot match the separation power and specificity of chromatographic techniques.
In the determination of lychnopholide in nanocapsule dosage forms, both HPLC-DAD and UV-spectrophotometry methods were validated, but the HPLC-DAD method demonstrated superior sensitivity with a linear range of 2-25 µg/mL compared to 5-40 µg/mL for spectrophotometry [7]. The sensitivity of the HPLC-DAD method also enabled studies of drug release/dissolution in sink conditions, revealing that while pure LYC presented 100% dissolution after 24 h, only 60% of LYC was released from the nanocapsule dosage form, with no burst effect - findings that would not be accessible through spectrophotometric methods alone [7].
UFLC-DAD vs. Other Detection Techniques
While DAD provides valuable spectral information, other detection techniques offer complementary capabilities that may be advantageous for specific applications. Mass spectrometric detection coupled with UFLC (UFLC-MS) provides superior sensitivity and definitive structural identification, particularly valuable in metabolite identification and trace analysis. As demonstrated in the forced degradation study of ritlecitinib, the combination of UHPLC-DAD with tandem mass spectrometry enabled not only separation and quantification but also identification of four novel degradation products [20] [19].
Fluorescence detection offers higher sensitivity for compounds with native fluorescence or those that can be derivatized, while electrochemical detection provides selectivity for electroactive compounds. The DAD's advantage lies in its universal applicability to chromophoric compounds, non-destructive nature (allowing coupling with other detectors in series), and the rich spectral information it provides for method development and peak identification [18].
Experimental Protocols and Methodologies
Standard UFLC-DAD Method Protocol
A representative UFLC-DAD method for pharmaceutical analysis typically includes the following components, based on the methodology developed for menaquinone-4 quantification in rabbit plasma [21]:
Sample Preparation Protocol:
- Protein precipitation using appropriate solvents (e.g., acetonitrile, methanol)
- Centrifugation at high speed (typically 10,000-15,000 × g) for 10-15 minutes
- Collection of supernatant for analysis
- Possible dilution or concentration steps depending on analyte levels
Chromatographic Conditions:
- Column: Reversed-phase C18 (e.g., 150 × 4.6 mm, sub-2μm particles)
- Mobile Phase: Binary or ternary mixtures optimized for specific separation (e.g., Isopropyl Alcohol and Acetonitrile 50:50 v/v for menaquinone-4)
- Flow Rate: 0.8-1.0 mL/min (depending on column dimensions and pressure limitations)
- Column Temperature: 25-40°C (controlled by thermostat)
- Injection Volume: 1-10 μL (optimized for sensitivity and resolution)
- Run Time: Typically 5-15 minutes for fast separations
DAD Detection Parameters:
- Wavelength Range: 190-600 nm (full spectrum acquisition)
- Reference Wavelength: Set based on analyte chromophores (e.g., 269 nm for menaquinone-4)
- Spectral Acquisition Rate: 10-20 Hz (balancing data richness and file size)
- Slit Width: 1-4 nm (optimizing sensitivity and resolution)
Method Validation Parameters
Comprehensive method validation for UFLC-DAD methods follows ICH guidelines and typically includes assessment of the following parameters, as demonstrated in the ritlecitinib stability-indicating method [20] [19] and the menaquinone-4 bioanalytical method [21]:
- Linearity: Evaluation across relevant concentration range with correlation coefficient (R²) > 0.995
- Accuracy: Typically 98-101% for pharmaceutical applications
- Precision: Intra-day and inter-day precision with RSD ≤ 0.15% for active pharmaceutical ingredients
- Specificity: Resolution from potential interferents and degradation products
- Sensitivity: Limit of Detection (LOD) and Limit of Quantification (LOQ) determined based on signal-to-noise ratio
- Robustness: Assessment of method performance under deliberate variations in chromatographic conditions
Diagram 2: UFLC-DAD Method Development Workflow - from Sample to Validation
Essential Research Reagent Solutions
The effective implementation of UFLC-DAD methods requires specific reagents and materials optimized for high-performance liquid chromatography. The following table details key research reagent solutions essential for successful UFLC-DAD analysis:
Table 3: Essential Research Reagent Solutions for UFLC-DAD Analysis
| Reagent/Material | Function/Purpose | Application Example |
|---|---|---|
| High-Purity Acetonitrile (HPLC Grade) | Mobile phase component providing efficient elution | Reverse-phase separation of pharmaceutical compounds [19] |
| Mass Spectrometry Grade Water | Aqueous component of mobile phase | Essential for preparing mobile phases with minimal UV-absorbing impurities |
| Formic Acid (LC-MS Grade) | Mobile phase additive to improve peak shape and ionization | Acidic modifier for separation of basic compounds like ritlecitinib [19] |
| Ammonium Acetate (HPLC Grade) | Buffer salt for controlling mobile phase pH | Used in mass spectrometry-compatible methods at concentrations typically 1-10 mM |
| Reverse-Phase C18 Columns (sub-2μm) | Stationary phase for high-efficiency separations | Core-Shell technology columns for fast separations with high resolution [19] |
| Protein Precipitation Reagents | Sample clean-up for biological matrices | Acetonitrile or methanol used for plasma sample preparation [21] |
| Reference Standards | Method development, calibration, and quality control | Certified reference materials for quantification and identification |
UFLC-DAD technology represents a significant advancement in analytical chromatography, offering substantially improved separation power, detection capabilities, and analytical efficiency compared to conventional HPLC and spectrophotometric methods. The combination of ultra-fast liquid chromatography with diode array detection provides researchers with a powerful tool for method development, quality control, and complex sample analysis across pharmaceutical, biomedical, and environmental applications.
The separation mechanism of UFLC, leveraging smaller particle sizes and higher pressure capabilities, enables faster analyses with improved resolution, while the detection capabilities of DAD provide comprehensive spectral information for peak identification, purity assessment, and method flexibility. As demonstrated in numerous applications from pharmaceutical stability testing to bioanalytical quantification, UFLC-DAD delivers performance parameters that meet the demanding requirements of modern analytical laboratories, with sensitivity (LOD typically 0.04-0.002 µg/mL), precision (RSD ≤ 0.15-10%), and accuracy (98-101%) appropriate for even regulated environments.
While alternative technologies such as direct spectrophotometry offer simplicity and LC-MS provides superior structural elucidation capabilities, UFLC-DAD occupies a unique position in the analytical toolbox, balancing separation power, detection versatility, and operational practicality. As the field continues to evolve, UFLC-DAD is poised to remain a cornerstone technique for analytical scientists seeking to maximize information content while maintaining efficiency in their chromatographic analyses.
In the realm of analytical chemistry, the selection of an appropriate method is pivotal for obtaining reliable and meaningful data. This guide provides a comprehensive theoretical overview comparing spectrophotometric methods with Ultra-Fast Liquid Chromatography-Diode Array Detection (UFLC-DAD), two prominent techniques utilized across pharmaceutical, environmental, and food sciences. The distinction between these methods is foundational; spectrophotometry generally offers a macroscopic view of total analyte content, whereas UFLC-DAD provides a microscopic, separated perspective of individual components within a mixture. Framing this comparison within a broader thesis on analytical techniques reveals how the complementary strengths and weaknesses of each method dictate their application in research and quality control, guiding professionals toward informed methodological choices.
Fundamental Principles and Instrumentation
Spectrophotometry
Spectrophotometry is a conventional technique based on the measurement of light absorption by a chemical substance in solution. The fundamental principle, governed by the Beer-Lambert law, states that the absorbance of a solution at a specific wavelength is directly proportional to its concentration and the path length of the light through it. This method typically employs a single wavelength or a narrow range to quantify the total concentration of an analyte. Its instrumentation is relatively straightforward, consisting of a light source, a monochromator to select the wavelength, a sample holder (cuvette), and a photodetector to measure the intensity of the transmitted light. Its primary application is the rapid determination of total analyte content, such as total capsaicinoids in peppers or total polyphenols in plant extracts, where the collective measurement is sufficient [24] [25].
UFLC-DAD (Ultra-Fast Liquid Chromatography-Diode Array Detection)
UFLC-DAD is a hyphenated technique that combines a high-efficiency separation mechanism with sophisticated detection. The chromatographic component (UFLC) separates a complex mixture into its individual constituents as they pass through a column under high pressure with a optimized mobile phase. The subsequent detection component (DAD) then identifies and quantifies each separated compound by acquiring its full ultraviolet-visible absorption spectrum in real-time. Unlike a simple UV detector set at a fixed wavelength, the DAD can monitor multiple wavelengths simultaneously and provide spectral confirmation of each peak's identity, enhancing the method's specificity and reliability [25] [16]. This makes it indispensable for analyzing complex matrices like herbal formulations [25] or counterfeit drugs [16], where resolving individual components is critical.
Comparative Strengths and Weaknesses
The choice between spectrophotometry and UFLC-DAD is a trade-off between speed, cost, and informational depth. The table below summarizes their core operational and performance characteristics.
Table 1: Core Characteristics of Spectrophotometry and UFLC-DAD
| Feature | Spectrophotometry | UFLC-DAD |
|---|---|---|
| Analytical Principle | Measures total light absorption of a sample without separation. | Separates components first, then identifies and quantifies each individually. |
| Information Obtained | Total content of a target compound or class. | Individual concentration and spectral identity of multiple analytes in a mixture. |
| Selectivity/Specificity | Low to Moderate. Susceptible to interference from other absorbing substances. | High. Separation minimizes interference; spectral confirmation enhances specificity [15]. |
| Sensitivity | Generally lower. Limited by background interference. | Generally higher. Lower limits of detection and quantification (e.g., at ng/mL level) are common [25]. |
| Analytical Speed | Very Fast (minutes per sample). | Slower (tens of minutes per run), but offers more comprehensive data. |
| Cost & Operational Complexity | Low cost, simple operation, minimal training required. | High initial and maintenance cost, requires significant expertise to operate and maintain. |
| Sample Throughput | High, suitable for routine analysis of large sample batches. | Lower, but automation can facilitate sequential runs. |
| Primary Application | Quality control, total content analysis, quick screening. | Research, method development, analysis of complex mixtures, regulatory testing. |
Detailed Discussion of Key Differentiators
Selectivity and Interference: The most significant weakness of direct spectrophotometry is its vulnerability to interference. For instance, in the quantification of vitamin C in colored juices, the sample's inherent pigmentation can lead to significant inaccuracies, with errors exceeding 15% in titrimetric methods affected by color [15]. UFLC-DAD circumvents this issue through physical separation; even compounds with similar chromophores elute at different times, allowing for clean quantification of each.
Sensitivity and Limits of Detection: The sensitivity of UFLC-DAD is typically superior. A validated method for polyphenols achieved limits of detection and quantification at the nanogram per milliliter (ng/mL) level [25]. In contrast, the sensitivity of a spectrophotometer is constrained by the molar absorptivity of the analyte and the baseline noise of the unseparated matrix. While techniques like derivative spectrophotometry can enhance performance by resolving overlapping peaks [26], they generally do not match the fundamental sensitivity boost provided by chromatographic separation and focused detection.
Analysis Time and Cost-Effectiveness: This dimension highlights the primary strength of spectrophotometry. For applications where a single parameter, like total capsaicinoid content, is the quality marker, a quick spectrophotometric assay is profoundly more efficient and economical [24]. The high throughput and low operational cost make it ideal for routine industrial quality control. UFLC-DAD, while slower and more expensive, delivers a wealth of information that is necessary for method development, stability studies, and confirming product authenticity in complex scenarios [16].
Experimental Data and Methodological Protocols
Case Study: Quantification of Capsaicinoids in Chiltepin Pepper
A direct comparison was performed to determine the total capsaicinoids in Chiltepin pepper using both UV-spectrophotometry and HPLC-DAD [24].
Experimental Protocol for Spectrophotometry:
- Extraction: Capsaicinoids were extracted from pepper fruit using acetonitrile.
- Measurement: The absorbance of the diluted extract was measured on a spectrophotometer at 280 nm.
- Quantification: The concentration was calculated using the predetermined molar absorptivity coefficients for capsaicin and dihydrocapsaicin.
Experimental Protocol for HPLC-DAD:
- Extraction: The same acetonitrile extract was used.
- Chromatographic Separation: Analysis was performed using an HPLC system equipped with a column. The mobile phase and gradient were optimized to separate capsaicin, dihydrocapsaicin, and nordihydrocapsaicin.
- Detection & Quantification: Detection was via a DAD at 280 nm. Quantification was achieved by comparing peak areas against those of external standards.
The results demonstrated a strong correlation (R = 0.91) between the two methods, confirming that the simpler spectrophotometric approach can serve as a reliable alternative for total capsaicinoid analysis in certain quality control contexts [24].
Table 2: Quantitative Results from Capsaicinoid Analysis [24]
| Method | Measured Pungency (Scoville Heat Units) | Key Findings |
|---|---|---|
| Spectrophotometry | Correlation of 0.91 with HPLC | Suitable for routine total capsaicinoids analysis and quality control. |
| HPLC-DAD | 29.55 to 129 mg/g | Identified that capsaicin, dihydrocapsaicin, and nordihydrocapsaicin comprised up to 98% of total capsaicinoids. |
Case Study: Analysis of Polyphenols in Cecropia Species
An optimized and validated HPLC-DAD method was developed for the simultaneous quantification of chlorogenic acid, total flavonoids, and flavonolignans in leaf extracts of Cecropia species [25].
- Extraction Optimization: A fractional factorial design (FFD) and central composite design (CCD) were used to optimize ultrasound-assisted extraction. Key variables like methanol fraction, extraction temperature, and number of extractions were fine-tuned to maximize yield.
- HPLC-DAD Analysis: The separation was achieved on a reversed-phase C18 column with a gradient mobile phase of water and acetonitrile (both acidified). The DAD collected spectra for peak identification and purity assessment.
- Validation Results: The method was validated per ICH guidelines, showing excellent specificity, linearity (R² > 0.999), precision (RSD < 5%), and accuracy (98-102%). The limits of quantification were at the ng/mL level, demonstrating high sensitivity [25].
This case underscores the necessity of UFLC-DAD for complex analytical tasks requiring the specific quantification of multiple chemical markers in a natural product, a feat unattainable by direct spectrophotometry.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents and materials essential for executing the experiments described in the cited studies, along with their critical functions.
Table 3: Key Research Reagent Solutions and Materials
| Item | Function/Application |
|---|---|
| Acetonitrile / Methanol (HPLC Grade) | Primary solvent for extracting analytes from solid samples and as a component of the mobile phase in HPLC [24] [25]. |
| C18 Reversed-Phase Chromatography Column | The stationary phase for HPLC separation, separating compounds based on their hydrophobicity [25] [16]. |
| Chlorogenic Acid, Capsaicin, etc. (Reference Standards) | High-purity compounds used to create calibration curves for accurate identification and quantification of analytes in unknown samples [24] [25]. |
| Diode Array Detector (DAD) | A detector that captures the full UV-Vis spectrum of an eluting compound, allowing for peak purity assessment and spectral confirmation [25]. |
| Formic Acid / Ammonium Format Buffer | Mobile phase additives used to control pH and improve chromatographic peak shape and separation efficiency [25] [16]. |
| Thiobarbituric Acid (TBA) | A derivatizing reagent used in spectrophotometric methods (e.g., TBA test) to detect malondialdehyde (MDA), a marker for lipid oxidation [26]. |
| Ultrasound-Assisted Extraction (UAE) Apparatus | Uses ultrasonic energy to enhance the extraction efficiency of target compounds from plant or food matrices, reducing time and solvent consumption [25]. |
Visualized Workflows and Logical Pathways
The following diagrams illustrate the fundamental operational workflows for both spectrophotometry and UFLC-DAD, highlighting their core differences.
Diagram 1: Spectrophotometry workflow. This is a direct, single-step measurement process.
Diagram 2: UFLC-DAD workflow. This is a multi-stage process involving separation followed by detection, yielding rich, component-specific data.
The selection of an appropriate analytical method is a critical first step in pharmaceutical development and research, forming the foundation for reliable data, regulatory compliance, and efficient resource utilization. Within the context of comparative analysis between spectrophotometric and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) methods, researchers must navigate a complex landscape of technical capabilities and practical constraints. Method selection represents a strategic decision point that influences all subsequent phases of analytical development. The fundamental principles of spectroscopy and chromatography provide distinct advantages and limitations that must be carefully balanced against project requirements.
This guide provides an objective comparison between these established techniques, focusing on key performance metrics drawn from experimental data across diverse pharmaceutical applications. The analytical landscape for pharmaceutical compounds requires methods that can accurately quantify active ingredients amidst complex matrices, and the choice between spectrophotometric and chromatographic approaches significantly impacts method development timelines, operational costs, and data quality. As analytical technologies continue to evolve, understanding the core performance differentiators between these platforms becomes increasingly essential for researchers tasked with developing robust, fit-for-purpose analytical methods.
Performance Comparison: Spectrophotometry vs. UFLC-DAD
The selection between spectrophotometric and UFLC-DAD methods requires careful consideration of multiple performance parameters. The following comparison synthesizes experimental data from validation studies across various pharmaceutical applications, highlighting the distinct operational characteristics of each technique.
Table 1: Direct Performance Comparison Between Spectrophotometric and UFLC-DAD Methods
| Performance Parameter | UV-Vis Spectrophotometry | UFLC-DAD |
|---|---|---|
| Typical Linear Range | 2-70 μg/mL [27] [7] | 0.5-400 μg/mL [27] |
| Limit of Detection | 0.060 μg/mL (Methotrexate) [28] | 0.014 μg/mL (Methotrexate) [28] |
| Limit of Quantification | 0.010 mg/mL (Vitamin C) [15] | Not explicitly quantified in sources |
| Accuracy (Recovery) | 89.5-105.5% [28] | 89.5-105.5% [28] |
| Precision (RSD) | <2.90% [28] | <3.39% [27] [28] |
| Analysis Time | Rapid (minutes) | Moderate to Fast (varies with method) |
| Multi-Component Analysis | Limited, requires derivative methods [27] [29] | Excellent for complex mixtures [27] |
| Selectivity/Specificity | Moderate, susceptible to interference [15] | High, with spectral confirmation [27] [7] |
| Equipment & Operational Costs | Lower | Higher |
| Solvent Consumption | Lower (green solvent potential) [27] | Higher, but reduced with UFLC |
Table 2: Application-Based Method Performance Examples
| Analyte/Application | Spectrophotometric Performance | UFLC-DAD Performance |
|---|---|---|
| Capsaicinoids in Peppers | Correlation of 0.91 with HPLC; sufficient for total content [24] | Gold standard for individual capsaicinoid quantification [24] |
| Vitamin C in Juices | Superior to titration with LOD 0.002 mg/mL; less affected by color [15] | Not specifically tested in retrieved studies |
| Malondialdehyde in Milk | Comparable results to HPLC with proper clean-up [26] | High reproducibility and precision with clean-up [26] |
| Ternary Drug Mixture (Analgin, Caffeine, Ergotamine) | Possible with derivative methods (DDRD, RDW) [27] | Excellent separation with gradient elution and dual detection [27] |
Experimental Protocols and Methodologies
Spectrophotometric Method Protocols
UV-Vis Spectrophotometry for Single-Component Analysis The fundamental protocol for quantifying paracetamol exemplifies single-component spectrophotometric analysis. The method involves dissolving the analyte in an appropriate solvent such as methanol or water, followed by scanning in the UV-Vis range (typically 200-400 nm) to identify the maximum absorption wavelength (λmax). Quantification employs the Beer-Lambert law, where absorbance is proportional to concentration. Method validation establishes linearity across the working range, precision through replicate measurements, accuracy via recovery studies, and determination of LOD/LOQ values. This approach is characterized by its simplicity, rapid implementation, and minimal solvent consumption [29].
Derivative Spectrophotometry for Multi-Component Analysis For complex mixtures where analyte spectra overlap, derivative techniques enhance selectivity without physical separation. The double divisor ratio spectra derivative (DDRD) and ratio dual wavelength (RDW) methods have been successfully applied to ternary mixtures such as analgin, caffeine, and ergotamine. The DDRD method enabled determination of ergotamine at 355 nm and caffeine at 268 nm using third and first derivatives, respectively. The RDW method utilized amplitude differences for simultaneous determination of caffeine and analgin. These mathematical manipulations of spectral data resolve overlapping absorbances, though they require careful method development and validation [27].
UFLC-DAD Method Protocols
Chromatographic Separation for Ternary Mixture Analysis A validated HPLC-DAD method for simultaneous determination of analgin, caffeine, and ergotamine exemplifies the chromatographic approach. Separation was achieved using an Inertsil-C8 column with gradient elution of acetonitrile and ammonium format buffer (pH 4.2). The DAD provided detection at multiple wavelengths: 280 nm for analgin, 254 nm for caffeine, while ergotamine was detected fluorometrically (λexc = 310 nm, λemm = 360 nm). The method demonstrated linearity across 50-400 μg/mL for analgin, 25-200 μg/mL for caffeine, and 0.5-10 μg/mL for ergotamine, with precision RSD values below 3.39%. This approach highlights the superior separation capability of chromatographic methods for complex mixtures [27].
UFLC-DAD for Lychnopholide in Nanocapsules An UFLC-DAD method for lychnopholide quantification in polymeric nanocapsules utilized an RP C18 column with isocratic elution (methanol:water, 60:40 v/v) at 0.8 mL/min flow rate and detection at 265 nm. Validation confirmed linearity (r² > 0.999) within 2-25 μg/mL, with accuracy of 98-101% and precision RSD below 1.41%. This method's sensitivity enabled dissolution studies under sink conditions, revealing that 100% of pure lychnopholide dissolved within 24 hours, while only 60% was released from nanocapsules without burst effect. The method proved suitable for determining drug loading and encapsulation efficiency in complex delivery systems [7].
Analytical Workflow Visualization
The following diagram illustrates the key decision points and methodological pathways in selecting and implementing spectrophotometric versus UFLC-DAD methods:
Analytical Method Selection Workflow
Essential Research Reagent Solutions
The following table details key reagents and materials essential for implementing the discussed analytical methods, along with their specific functions in experimental protocols.
Table 3: Essential Research Reagents and Materials for Analytical Method Development
| Reagent/Material | Function in Spectrophotometry | Function in UFLC-DAD |
|---|---|---|
| HPLC-Grade Solvents (Acetonitrile, Methanol) | Sample dissolution and dilution [29] | Mobile phase component for compound separation [27] [7] |
| Buffer Salts (Ammonium format, acetate) | Generally not required | Mobile phase modifier for pH control and peak shaping [27] [16] |
| Derivatization Agents (DNPH, TBA) | Specific compound detection (e.g., MDA with TBA) [26] | Enhanced detection of non-UV absorbing compounds [26] |
| Stationary Phases (C8, C18 columns) | Not applicable | Core separation media; selection critical for resolution [27] [7] |
| Reference Standards | Calibration curve construction [29] | System calibration and compound identification [27] [28] |
Strategic Application Scenarios
Method Selection Guidelines
When to Prioritize Spectrophotometry Spectrophotometric methods are ideally suited for routine analysis of single components or simple mixtures where high throughput and cost efficiency are primary concerns. The technique excels in applications such as raw material identification, dissolution testing of immediate-release formulations, and quality control in stability studies. The green chemistry advantages of spectrophotometry, including reduced solvent consumption and minimal waste generation, align with increasingly important sustainability goals in analytical laboratories [27]. Furthermore, in resource-limited settings or educational environments, spectrophotometry provides accessibility without significant compromise in data quality for appropriate applications.
When to Require UFLC-DAD UFLC-DAD becomes essential when analyzing complex mixtures, requiring high sensitivity, or needing to confirm analyte identity through spectral matching. Applications such as stability-indicating methods, impurity profiling, and bioanalytical studies typically demand the separation power of chromatography. The diode array detector provides an additional dimension of data through full spectral acquisition, enabling peak purity assessment and method specificity verification. In regulated environments, the robustness and defensibility of chromatographic data often make UFLC-DAD the preferred choice despite higher operational costs and method development complexity [27] [7].
Emerging Trends and Convergent Approaches
The historical dichotomy between spectrophotometric and chromatographic methods is increasingly blurred by technological advancements. Hyphenated techniques that combine separation power with spectroscopic detection continue to evolve, while multivariate calibration methods expand the capabilities of spectrophotometry for complex mixtures. Modern method scoping must also consider emerging approaches such as handheld spectrophotometers for field testing and UHPLC systems that further reduce analysis time and solvent consumption [16]. The optimal foundation for method development increasingly involves a hybrid strategy, leveraging the complementary strengths of both platforms throughout the product lifecycle from early development to commercial quality control.
Practical Implementation: Method Development and Real-World Applications
In the pharmaceutical sciences, the development of robust analytical methods is fundamental for drug quantification, quality control, and regulatory approval. This guide presents a comparative analysis of two cornerstone techniques: Ultraviolet-Visible (UV-Vis) Spectrophotometry and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD). The focus is on the critical aspects of wavelength selection and calibration, which are paramount for ensuring method accuracy, sensitivity, and specificity. Spectrophotometry is celebrated for its simplicity, cost-effectiveness, and rapid analysis, making it a staple in many quality control laboratories [29]. In contrast, UFLC-DAD offers superior separation power and specificity, capable of handling complex mixtures with ease [30] [27]. The ensuing comparison, grounded in experimental data and structured protocols, is designed to equip researchers and drug development professionals with the information necessary to select the most appropriate analytical technique for their specific application.
Fundamental Principles and Key Concepts
The Beer-Lambert Law and Quantification
The quantitative foundation of UV-Vis spectrophotometry is the Beer-Lambert Law. This law states that the absorbance (A) of a solution is directly proportional to the concentration (c) of the absorbing species and the path length (b) of the radiation through the sample [29]. It is mathematically expressed as A = a b c, where 'a' is the absorptivity coefficient. This relationship is the cornerstone of calibration, where the absorbance of unknown samples is measured and their concentration is determined by comparison with standards of known concentration [29]. Common quantification methods include using a standard calibration curve, single-point standardization, or standard absorptivity values.
Critical Method Parameters
- Wavelength of Maximum Absorption (λmax): This is the wavelength at which an analyte has its strongest absorption. Selecting this wavelength for quantification ensures maximum sensitivity and minimizes errors due to slight instrumental wavelength drifts [29].
- Validation Parameters: According to International Conference on Harmonization (ICH) guidelines, methods must be validated for parameters including linearity, precision, accuracy, limit of detection (LOD), and limit of quantitation (LOQ) [30] [29]. These parameters confirm that the method is suitable for its intended use.
Wavelength Selection Strategies
The selection of an appropriate analytical wavelength is a critical step that directly influences the method's sensitivity and reliability. The strategies for UV-Vis and UFLC-DAD differ significantly due to the latter's chromatographic separation step.
Wavelength Selection in UV-Vis Spectrophotometry
For single-component analysis, the process involves scanning the standard solution across the UV-Vis spectrum (e.g., 200-400 nm) to identify the wavelength of maximum absorption (λmax) [30] [29]. For example, repaglinide and paracetamol exhibit λmax at 241 nm and others at 280 nm, respectively [30] [29].
The challenge becomes more complex with multi-component mixtures where spectra overlap. Advanced mathematical techniques are then employed:
- Derivative Spectrophotometry: This technique enhances the resolution of overlapping spectra by converting the normal absorbance spectrum into its first, second, or third derivative. The third derivative method has been successfully used to resolve malondialdehyde (MDA) in complex matrices like infant formula, eliminating background interference [26].
- Genetic Algorithm-Based Wavelength Selection: This sophisticated chemometric approach uses a computational search algorithm to select an optimal set of wavelengths for Partial Least Squares (PLS) calibration. This is particularly useful for mixtures with nearly identical spectra, such as copper and zinc, leading to significantly lower prediction errors compared to full-spectrum PLS [31].
Wavelength Selection in UFLC-DAD
In UFLC-DAD, wavelength selection occurs after chromatographic separation, which physically resolves the mixture components. The DAD detector simultaneously records the absorbance of the eluting peaks across a range of wavelengths [27] [26]. The analyst can then:
- Extract the chromatogram at the λmax of each target analyte for optimal quantification.
- Use the full spectral data to confirm the identity and purity of each peak by comparing its spectrum against a reference standard [27].
Table 1: Comparison of Wavelength Selection for Different Analytical Challenges
| Analytical Challenge | UV-Vis Spectrophotometry Approach | UFLC-DAD Approach |
|---|---|---|
| Single Component | Direct scan to find λmax (e.g., 241 nm for repaglinide) [30] | Post-selection of λmax from DAD data after chromatographic separation |
| Multi-Component Mixtures | Use of derivative spectroscopy [26] or genetic algorithms for wavelength selection [31] | Intrinsic separation followed by individual wavelength selection for each pure compound |
| Identity Confirmation | Limited to comparison of λmax | Robust peak purity assessment by overlaying full UV spectra from the sample and standard [27] |
The following workflow outlines the decision-making process for selecting and developing an analytical method based on the sample composition and analytical requirements.
Calibration Methodologies
Calibration establishes the quantitative relationship between the analyte's concentration and the instrument's response, which is absorbance for spectrophotometry and peak area (or height) for UFLC-DAD.
UV-Vis Spectrophotometry Calibration
Calibration in UV-Vis is typically performed using a series of standard solutions across a concentration range that encompasses the expected unknown values [29].
- Calibration Curve: A graph of absorbance versus concentration is plotted. The data should ideally produce a linear relationship, and the unknown concentration is determined from the regression equation [29].
- Range and Linearity: The linear range must be established during validation. For instance, a method for repaglinide was linear in the range of 5-30 μg/mL [30], while a method for analgin, caffeine, and ergotamine showed linearity from 10-70 μg/mL for different components [27].
UFLC-DAD Calibration
The calibration principle is similar but is applied to chromatographic peak areas. The process involves:
- Injecting a series of standard solutions with known concentrations.
- Measuring the integrated peak area for the analyte at its specific retention time.
- Constructing a calibration curve of peak area versus concentration.
UFLC-DAD methods often have a wider linear dynamic range compared to spectrophotometry. For example, an HPLC method for repaglinide demonstrated linearity from 5-50 μg/mL [30].
Table 2: Exemplary Calibration and Validation Data from Comparative Studies
| Analyte / Method | Linear Range (μg/mL) | Correlation Coefficient (r²) | Precision (%RSD) | Accuracy (% Recovery) |
|---|---|---|---|---|
| Repaglinide (UV) [30] | 5 - 30 | > 0.999 | < 1.50 | 99.63 - 100.45 |
| Repaglinide (HPLC) [30] | 5 - 50 | > 0.999 | < 1.50 (more precise) | 99.71 - 100.25 |
| Analgin (HPLC-DAD) [27] | 50 - 400 | Not specified | Not specified | No significant difference from reported methods |
| Copper (GA-PLS) [31] | 0.05 - 1.8 | Not specified | RMSD: 0.0407 (with GA) | Good reliability in water samples |
| Malondialdehyde (Derivative) [26] | Nanogram scale | High reproducibility & precision | High reproducibility for both methods | Good overlapping with HPLC-DAD method |
Experimental Protocols for Method Comparison
To illustrate a direct comparison, here are summarized protocols from studies that evaluated both UV-Vis and chromatographic methods for the same analyte.
UV-Vis Spectrophotometry:
- Instrument: Double-beam UV-Vis spectrophotometer.
- Wavelength: 241 nm.
- Solvent: Methanol.
- Procedure: Standard solutions (5-30 μg/mL) were prepared and their absorbance measured against a methanol blank. The calibration graph was constructed and validated.
RP-HPLC Method:
- Instrument: HPLC with UV detector.
- Column: Agilent TC-C18 (250 mm × 4.6 mm, 5 μm).
- Mobile Phase: Methanol:Water (80:20 v/v, pH 3.5).
- Flow Rate: 1.0 mL/min.
- Detection: 241 nm.
- Procedure: Standard solutions (5-50 μg/mL) were injected, and the peak areas were recorded for calibration.
Third Derivative Spectrophotometry:
- Reaction: MDA was derivatized with thiobarbituric acid (TBA) to form a pink chromophore.
- Measurement: The third derivative of the absorbance spectrum was used to resolve MDA signals from background interference in milk and infant formula samples.
HPLC-DAD Method:
- Derivatization: MDA was reacted with 2,4-dinitrophenylhydrazine (DNPH).
- Column: C18 column (250 x 4.6 mm, 5μm).
- Detection: DAD was used for detection.
- Procedure: The derivatized samples were injected, separated, and quantified. The study highlighted the importance of the clean-up procedure for both methods.
The experimental data allows for a clear, objective comparison of the performance characteristics of spectrophotometric and UFLC-DAD methods.
Table 3: Overall Performance Comparison of Spectrophotometric vs. UFLC-DAD Methods
| Performance Characteristic | UV-Vis Spectrophotometry | UFLC-DAD |
|---|---|---|
| Simplicity & Cost | Simple, rapid, and low-cost [29] | Complex, requires trained operators, higher cost |
| Analysis Speed | Very fast (minutes) | Slower (per run) but can be automated |
| Specificity | Low for complex mixtures; requires chemometrics for resolution [31] [26] | Very high due to combined separation and spectral identification [30] [27] |
| Sensitivity | Good (e.g., μg/mL range) [30] | Excellent, often better LOD/LOQ [30] |
| Linear Dynamic Range | Limited range [30] | Wider linear range [30] |
| Tolerance to Matrix Effects | Low; prone to interference from other absorbing compounds | High; interferents are separated from the analyte |
| Ideal Application | Quality control of raw materials, single-component assays, and simple mixtures where analytes have distinct λmax [29] | Analysis of complex mixtures, stability-indicating methods, bioanalytical studies, and when unambiguous identification is required [30] [27] |
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key reagents and materials commonly used in the development and application of these analytical methods.
Table 4: Essential Research Reagents and Materials for Spectrophotometric and UFLC-DAD Analysis
| Item | Function / Application | Example from Literature |
|---|---|---|
| Methanol / Acetonitrile (HPLC Grade) | Common solvent for preparing standard and sample solutions; component of mobile phase in HPLC [30] | Used as solvent for repaglinide in UV and as mobile phase component in HPLC [30] |
| Buffer Salts (e.g., Ammonium Format) | Used to adjust and control the pH of the mobile phase in HPLC, improving peak shape and separation [27] | Ammonium format buffer (pH 4.2) used in gradient elution for ternary mixture analysis [27] |
| Reference Standards | Highly pure characterized material used to prepare calibration standards for accurate quantification [30] | Repaglinide reference standard used for calibration curve construction [30] |
| Derivatization Reagents (TBA, DNPH) | React with the target analyte to form a chromophore with better absorption or detection properties [26] | Thiobarbituric acid (TBA) for MDA detection in spectrophotometry; DNPH for HPLC-DAD analysis [26] |
| C18 Chromatographic Column | The stationary phase for reversed-phase HPLC; workhorse column for separating a wide range of compounds [30] [27] | Agilent TC-C18 column used for repaglinide and Inertsil-C8 for analgin/caffeine/ergotamine [30] [27] |
| Genetic Algorithm (GA) Software | Chemometric software for advanced wavelength selection in multivariate calibration for complex mixtures [31] | Used to select optimal wavelengths for PLS calibration of copper and zinc mixtures [31] |
The experimental workflow for developing and comparing these methods, from sample preparation to data analysis, can be summarized as follows.
The choice between UV-Vis spectrophotometry and UFLC-DAD is not a matter of one being universally superior to the other, but rather of selecting the right tool for the specific analytical problem. UV-Vis spectrophotometry offers an efficient, economical, and straightforward solution for the quantitative analysis of pure substances or simple mixtures, especially when combined with advanced chemometric techniques for wavelength selection. In contrast, UFLC-DAD provides an indispensable platform for analyzing complex mixtures, offering high specificity, sensitivity, and the ability to confirm analyte identity through peak purity assessment. The decision should be guided by factors such as sample complexity, required specificity, available resources, and the purpose of the analysis. Both methods, when developed and validated with careful attention to wavelength selection and robust calibration, form the bedrock of reliable pharmaceutical analysis.
Advanced Spectrophotometric Techniques for Complex Mixtures (e.g., Derivative, Ratio Spectra)
The analysis of complex pharmaceutical mixtures often presents a significant challenge in drug development and quality control, particularly when the components exhibit severely overlapping spectra in the ultraviolet-visible (UV-Vis) region. While chromatographic techniques like UFLC-DAD provide powerful separation capabilities, advanced spectrophotometric methods offer compelling alternatives through mathematical filtration techniques that resolve spectral overlaps without physical separation. These approaches are experiencing a renaissance due to their cost-effectiveness, simplicity, and reduced environmental impact compared to solvent-intensive chromatographic methods, positioning them as valuable tools for routine analysis in resource-constrained settings [32] [33].
The fundamental challenge arises from the additive nature of absorbance in multicomponent systems, where the combined spectrum masks the individual contributions of each component. This limitation of conventional spectrophotometry has been overcome through the application of signal processing algorithms and chemometric approaches that extract quantitative information from what appears to be undifferentiated spectral data [32] [33]. The development of these techniques represents an important advancement for pharmaceutical analysis, particularly for simultaneous quantification of drugs and their degradation products or multiple active ingredients in fixed-dose combinations.
Comparative Analysis of Advanced Spectrophotometric Techniques
Fundamental Principles and Mathematical Foundations
Advanced spectrophotometric techniques for analyzing complex mixtures operate on the principle of absorbance additivity while employing various mathematical transformations to extract component-specific information from overlapped spectra. These methods can be broadly categorized into derivative-based, ratio-based, and hybrid techniques that combine elements of both approaches. The underlying theory leverages the differences in spectral curvature, inflection points, and amplitude relationships between components to achieve analytical resolution [32] [34].
The derivative spectroscopy approach converts normal zero-order absorption spectra into first or higher-order derivatives, which enhances resolution by transforming shoulders and inflection points into easily measurable peaks and troughs. The ratio-based methods involve dividing the mixture spectrum by a carefully selected divisor spectrum, typically of one component, to generate a new ratio spectrum that can be further manipulated. Hybrid techniques such as derivative ratio spectroscopy combine both principles to achieve even greater specificity in cases of extreme spectral overlap [33] [35].
Comprehensive Technique Comparison
Different spectrophotometric approaches offer varying advantages depending on the nature of the spectral overlap, component concentration ratios, and analytical requirements. The table below provides a systematic comparison of the most widely used advanced spectrophotometric techniques:
Table 1: Comparison of Advanced Spectrophotometric Techniques for Complex Mixtures
| Technique | Fundamental Principle | Typical Applications | Key Advantages | Limitations |
|---|---|---|---|---|
| Derivative Spectroscopy [33] [35] | Conversion of zero-order spectra to higher-order derivatives to enhance resolution of overlapping bands | Determination of components with overlapping spectra; elimination of baseline drifts | Enhanced selectivity for overlapping bands; elimination of matrix interference | Reduced signal-to-noise ratio at higher derivative orders; requires precise wavelength selection |
| Dual Wavelength (DW) [36] [34] | Measurement of absorbance difference at two wavelengths where interferent shows equal absorbance | Binary mixtures with partially overlapping spectra; formulation analysis | Simple calculation; minimal data processing; high precision | Requires specific wavelength pairs where interferent absorbance is identical |
| Ratio Difference (RD) [36] [33] | Difference in amplitudes of ratio spectra at two selected wavelengths | Simultaneous determination of drugs and degradation products; stability-indicating assays | Effective for severely overlapping spectra; relatively simple computations | Dependent on proper divisor concentration selection |
| First Derivative Ratio (1DD) [36] [35] | First derivative of the ratio spectrum obtained using a divisor | Multicomponent analysis in pharmaceutical formulations; compounds with similar structural features | Enhanced resolution capability; minimizes background interference | Sensitive to noise amplification; requires optimization of derivative parameters |
| Mean Centering of Ratio Spectra (MCR) [36] | Successive mean centering of ratio spectra to amplify specific component signals | Complex mixtures with extensive spectral overlap; quality control applications | High specificity; excellent for resolving complex mixtures | Computationally intensive; requires specialized software |
| Absorbance Ratio Method [35] | Utilization of absorbance ratios at isosbestic and component-specific wavelengths | Binary mixtures with isosbestic points; formulation uniformity testing | Simple methodology based on Q-ratio analysis | Requires distinct isosbestic point in mixture spectra |
Quantitative Performance Metrics
The analytical performance of advanced spectrophotometric methods has been extensively validated across numerous pharmaceutical applications. The following table summarizes typical performance characteristics reported in recent literature:
Table 2: Performance Metrics of Spectrophotometric Techniques for Pharmaceutical Analysis
| Application Context | Technique(s) Employed | Linear Range (μg/mL) | LOD/LOQ (μg/mL) | Recovery (%) | Precision (RSD%) |
|---|---|---|---|---|---|
| Vericiguat and Alkaline Degradant [36] | DW, RD, 1DD, MCR | 5-50 (VER), 5-100 (ADP) | Not specified | 98-102% | <2% |
| Amiodipine and Telmisartan [33] | RD, 1DD, Derivative, Amplitude Factor | Variable by technique | 0.12-0.43 (AMLB), 0.08-0.56 (TEL) | 96-101% | <2% |
| Analgin, Caffeine, Ergotamine [27] | DDRD, RDW | 10-35 (ANA), 2-30 (CAF), 10-70 (ERG) | Not specified | Comparable to HPLC | Comparable to HPLC |
| Hydrocortisone Acetate and Fusidic Acid [34] | IDW, DWRT, AAM, IAM | 5-50 (HCA), 2-40 (FSA) | Not specified | 98-102% | <1.5% |
| Amoxicillin and Cloxacillin [35] | Absorbance Ratio, Compensation, Derivative | 60-140 (both) | Not specified | Not specified | <2% |
Experimental Protocols and Methodologies
Standard Solution Preparation and Instrumentation
The foundation of reliable spectrophotometric analysis lies in careful solution preparation and instrument calibration. Most protocols begin with preparation of stock solutions (typically 100-1000 μg/mL) using appropriate solvents, followed by serial dilutions to working concentrations. Methanol and water are commonly employed solvents, though green solvent alternatives like propylene glycol are increasingly favored for their reduced environmental impact [36] [33]. For the analysis of drug degradation products, alkaline degradant solutions are often prepared through controlled stress testing, such as heating the drug substance in alkaline medium followed by neutralization and extraction [36].
Modern double-beam UV-Vis spectrophotometers with high wavelength accuracy (typically ±1 nm) and matched quartz cells represent the standard instrumentation. Critical specifications include a narrow spectral bandwidth (1-2 nm) and high photometric precision (±0.001A) to ensure sufficient resolution for derivative and ratio-based techniques. Software capabilities for spectral storage, mathematical manipulation, and derivative calculation are essential for implementing these advanced methods [36] [33] [34]. Most contemporary systems include proprietary software that enables direct calculation of ratio spectra and derivatives with user-defined parameters.
Detailed Methodological Protocols
Ratio Difference Spectrophotometric Method
The Ratio Difference (RD) method has demonstrated excellent performance for analyzing binary mixtures with overlapping spectra. The protocol involves a systematic process:
Record absorption spectra of standard solutions for both components across the relevant concentration range, typically 200-400 nm.
Select an appropriate divisor concentration from one component (typically 10 μg/mL of the degradant or second component).
Generate ratio spectra by dividing the mixture spectra by the spectrum of the divisor solution.
Measure the amplitude differences in the ratio spectrum at two carefully selected wavelengths (e.g., 318 nm and 342 nm for Vericiguat).
Construct calibration curves by plotting amplitude differences against component concentrations [36].
The selection of optimal wavelength pairs is critical and requires preliminary investigation of the ratio spectra to identify regions showing significant amplitude changes with concentration while maintaining minimal contribution from the interfering component.
First Derivative Ratio Method
The First Derivative Ratio (1DD) method enhances spectral resolution through derivative transformation of ratio spectra:
Prepare ratio spectra as described in the RD method using an optimally selected divisor concentration.
Calculate first derivatives of the ratio spectra using instrument software with defined Δλ values (typically 4-8 nm) and scaling factors.
Measure peak amplitudes at wavelengths where the component of interest shows maximum response while the interferent shows minimal contribution (e.g., 318 nm for Vericiguat).
Establish calibration curves by correlating peak amplitudes with concentrations [36].
This approach benefits from the signal enhancement properties of derivative spectroscopy while minimizing matrix effects through the initial ratio transformation.
Induced Dual Wavelength Method
For mixtures with complete spectral overlap where conventional dual wavelength methods fail, the Induced Dual Wavelength (IDW) method offers a solution:
Identify two wavelengths (λ1 and λ2) where the component of interest exhibits significant absorbance differences.
Calculate an equality factor (E.F.) by dividing the absorbance of the interferent at λ1 by its absorbance at λ2.
Multiply the mixture absorbance at λ2 by the E.F. to create a modified absorbance value.
Determine the concentration of the component of interest from the difference between the measured absorbance at λ1 and the modified absorbance at λ2 [34].
This innovative approach effectively induces a scenario where the interfering component shows equal absorbance at the two selected wavelengths, enabling selective quantification of the target analyte.
Method Validation Protocols
Validation of advanced spectrophotometric methods follows ICH guidelines and encompasses several key parameters:
Linearity: Established across specified concentration ranges with correlation coefficients (r²) typically >0.999 [36] [33].
Accuracy: Assessed through recovery studies (98-102%) using standard addition methods [36] [27].
Precision: Evaluated as intra-day and inter-day variations, with RSD values generally <2% [36] [34].
Specificity: Demonstrated through successful application to laboratory-prepared mixtures and pharmaceutical formulations [36] [33].
Robustness: Tested by deliberate variations in method parameters such as wavelength selection and derivative settings [34].
Comparative Analysis with UFLC-DAD Methods
Analytical Performance Comparison
When comparing advanced spectrophotometric techniques with UFLC-DAD methods, each approach demonstrates distinct advantages depending on the analytical context. The table below provides a systematic comparison:
Table 3: Spectrophotometric vs. UFLC-DAD Methods for Pharmaceutical Analysis
| Parameter | Advanced Spectrophotometric Methods | UFLC-DAD Methods |
|---|---|---|
| Analysis Time | Rapid (minutes per sample set) | Longer run times (typically 10-30 minutes per sample) |
| Cost Considerations | Lower equipment and operational costs | Higher capital investment and maintenance costs |
| Solvent Consumption | Minimal (mL range) | Significant (hundreds of mL per day) |
| Sample Throughput | High for routine analysis of known mixtures | Moderate, but capable of analyzing more complex mixtures |
| Sensitivity | Moderate (μg range) | High (ng-pg range) |
| Specificity | Mathematical separation | Physical separation followed by detection |
| Multi-component Analysis | Limited to 2-3 components with distinct spectra | Capable of analyzing complex mixtures with multiple components |
| Greenness Profile | Generally favorable with proper solvent selection | Lower due to higher organic solvent consumption |
Applications in Pharmaceutical Analysis
Advanced spectrophotometric methods have been successfully applied to numerous pharmaceutical analysis scenarios:
Stability-indicating assays for drugs and their degradation products, as demonstrated for Vericiguat and its alkaline degradation product [36].
Fixed-dose combination products containing two or more active ingredients, such as Amlodipine-Telmisartan and Amoxicillin-Cloxacillin combinations [33] [35].
Dissolution testing and release kinetics for novel dosage forms, including nanocapsules and other modified-release systems [7].
Content uniformity testing of pharmaceutical formulations, where rapid analysis of multiple dosage units is required [32] [27].
The complementary nature of spectrophotometric and chromatographic techniques is increasingly recognized, with each approach serving different needs within the pharmaceutical quality control environment.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Materials for Advanced Spectrophotometric Analysis
| Item | Specification | Application Context |
|---|---|---|
| Double-beam UV-Vis Spectrophotometer | 1-2 nm bandwidth, diode array detection, spectral software | All advanced spectrophotometric techniques |
| Matched Quartz Cells | 1 cm pathlength, high transmission quality | Absorbance measurements with minimal pathlength variation |
| Methanol (HPLC Grade) | High purity, low UV absorbance | Standard solvent for stock solution preparation |
| Green Solvents (e.g., Propylene Glycol) | High purity, environmentally favorable | Alternative solvents for green analytical chemistry |
| pH Buffers | Analytical grade, specific pH ranges | Mobile phase preparation or solubility enhancement |
| Standard Reference Materials | Certified purity (>98%) | Method development and calibration |
| Membrane Filters | 0.45 μm pore size, compatible with solvents | Sample clarification before analysis |
Method Selection Workflow and Signaling Pathways
The selection of an appropriate analytical technique for pharmaceutical mixtures depends on multiple factors, including the nature of the spectral overlap, component concentrations, and analytical requirements. The following diagram illustrates the decision-making pathway:
Diagram 1: Method Selection Workflow for Complex Mixture Analysis
Advanced spectrophotometric techniques represent powerful tools for the analysis of complex mixtures, particularly in pharmaceutical applications where cost-effectiveness, speed, and environmental impact are significant considerations. While UFLC-DAD methods offer superior separation capabilities for highly complex mixtures, spectrophotometric approaches provide viable alternatives for routine analysis of binary and ternary mixtures with overlapping spectra.
The continuing development of mathematical filtration techniques has substantially expanded the capabilities of spectrophotometry, enabling resolution of increasingly complex spectral overlaps. When selected appropriately based on the specific analytical challenge, these methods can deliver performance characteristics comparable to chromatographic techniques while offering advantages in terms of throughput, solvent consumption, and operational simplicity.
The future trajectory of these techniques will likely involve increased integration with multivariate calibration methods and further refinement of green analytical chemistry principles, ensuring their continued relevance in pharmaceutical analysis and quality control environments.
Ultra-Fast Liquid Chromatography coupled with Diode Array Detection (UFLC-DAD) represents a significant advancement in analytical separation science, offering improved resolution, speed, and detection capabilities compared to conventional HPLC. This technological evolution addresses the growing demand for high-throughput analysis in complex matrices such as pharmaceuticals, food products, and biological samples. The core advantage of UFLC systems lies in their ability to operate at higher pressures while utilizing smaller particle sizes in stationary phases, resulting in enhanced separation efficiency and reduced analysis time.
Within the broader context of comparative analytical research, UFLC-DAD frequently demonstrates superior performance against simpler spectrophotometric methods, particularly when analyzing multi-component mixtures or structurally similar compounds. While UV-spectrophotometry offers simplicity and cost-effectiveness for straightforward analyses, UFLC-DAD provides the separation power necessary for complex sample matrices, enabling specific quantification of individual components even in the presence of interfering substances. This comparison is crucial for researchers and drug development professionals who must select the most appropriate methodology based on their specific analytical requirements, balancing factors such as sensitivity, specificity, throughput, and operational complexity.
Critical Method Development Parameters
Stationary Phase Selection: C18 Columns as the Workhorse
The choice of column stationary phase fundamentally impacts the selectivity, resolution, and efficiency of UFLC-DAD methods. Among various options, C18 reversed-phase columns serve as the predominant choice for a wide range of applications, demonstrating exceptional versatility in separating compounds with diverse chemical properties.
Comparative Column Performance is well-illustrated by research on tocopherol and tocotrienol separation, where two different C18 columns were evaluated. The Luna Omega C18 column provided superior separation for some components compared to the Kinetex C18 column, though neither could resolve β- and γ- forms of tocopherols and tocotrienols without pre-column derivatization [37]. This limitation was overcome through an optimized pre-column esterification procedure using trifluoroacetic anhydride, which allowed satisfactory separation of these challenging compounds using conventional C18 chromatography [37] [38].
For specialized applications, different column chemistries may be employed. Chiral separations, such as determining the enantiomeric excess of 1-phenylethanol, require dedicated chiral stationary phases like the Chiralpak IA column, which successfully separated (R)- and (S)-enantiomers under isocratic conditions [39].
Table 1: Column Selection Guidelines for UFLC-DAD Method Development
| Application Type | Recommended Column | Separation Capabilities | Limitations |
|---|---|---|---|
| General Reversed-Phase | Conventional C18 (e.g., Phenomenex Luna) | Broad applicability for compounds of varying polarity | May not resolve structurally similar isomers |
| High-Efficiency Separations | C18 with smaller particles (e.g., Kinetex) | Improved resolution and faster analysis times | Higher backpressure requirements |
| Isomeric Separation | C18 with pre-column derivatization | Resolves β- and γ- forms of tocopherols/tocotrienols | Requires additional sample preparation steps [37] |
| Chiral Separations | Dedicated chiral columns (e.g., Chiralpak IA) | Separates enantiomers without derivatization | Limited to specific chiral applications [39] |
Mobile Phase Optimization: Composition and Gradient Profile
Mobile phase optimization represents perhaps the most critical aspect of UFLC-DAD method development, directly influencing retention behavior, peak shape, and overall separation quality. Both isocratic and gradient elution approaches offer distinct advantages depending on the analytical context.
Isocratic elution provides simplicity and stability, making it suitable for applications where the target analytes exhibit similar retention characteristics. For lychnopholide quantification in nanocapsules, a simple isocratic system employing methanol-water (60:40 v/v) at a flow rate of 0.8 mL/min demonstrated excellent performance with a linear response (r² > 0.999) within 2-25 µg/mL [7]. Similarly, trigonelline standardization in Trigonella foenum-graecum extracts utilized isocratic elution with methanol:water containing 0.5% acetic acid (60:40) at 1 mL/min, achieving good separation with a retention time of approximately 4 minutes [39].
Gradient elution offers superior capability for analyzing complex mixtures with components of widely varying polarities. The separation of tocopherols, tocotrienols, α-tocopheryl acetate, and cholesterol in diverse foods employed optimized binary gradient elution programs, allowing simultaneous quantification of these chemically diverse compounds in a single run [37] [38]. For more complex purification tasks, such as isolating α-ribazole-5′-phosphate, multi-step gradient programs proved essential, beginning with 3% acetonitrile and progressing through linear gradients to 40% and ultimately 80% acetonitrile to elute compounds of differing polarities at appropriate retention times [39].
Table 2: Mobile Phase Systems for Different UFLC-DAD Applications
| Application | Mobile Phase Composition | Elution Mode | Flow Rate | Key Separation Factors |
|---|---|---|---|---|
| Lychnopholide in Nanocapsules | Methanol-water (60:40 v/v) | Isocratic | 0.8 mL/min | Adequate for single compound quantification [7] |
| Tocopherols/Tocotrienols in Foods | Optimized binary gradient (varying methanol/water) | Gradient | Not specified | Simultaneous multi-component analysis [37] |
| α-ribazole-5′-phosphate Purification | Acetonitrile-ammonium acetate (pH 4.5) | Multi-step gradient | 0.8 mL/min | Complex gradient for challenging separation [39] |
| Trigonelline Standardization | Methanol:water with 0.5% acetic acid (60:40) | Isocratic | 1.0 mL/min | Acid modifier improves peak shape [39] |
Detection Optimization: Leveraging DAD Capabilities
The diode array detector represents a significant advancement over single-wavelength detection, enabling simultaneous monitoring at multiple wavelengths and providing spectral information for peak purity assessment and identity confirmation. Optimal wavelength selection depends on the absorbance characteristics of the target analytes.
For tocopherol and tocotrienol analysis, multiple detection approaches were evaluated. Fluorescence detection (FLD) generally provided better responses for these compounds, though DAD monitoring at 278 nm and 205 nm enabled the quantification of β- and γ-forms after esterification, while 195 nm was optimal for cholesterol analysis [37] [38]. In contrast, lychnopholide quantification utilized detection at 265 nm, which corresponded to its maximum absorbance [7], while α-ribazole-5′-phosphate was detected at 287 nm based on its spectral characteristics [39].
The DAD's ability to acquire full spectra also facilitates peak identity confirmation through spectral matching. In the analysis of Jatropha isabellei, the DAD provided UV spectra that helped identify chemical forms of tocols and confirm compound identity through retention time matching and standard addition [40].
Comparative Performance: UFLC-DAD vs. UV-Spectrophotometry
Direct comparison studies demonstrate the relative strengths and limitations of UFLC-DAD and UV-spectrophotometry, highlighting their appropriate application domains.
For lychnopholide quantification, both methods were validated and showed good performance, but with distinct advantages. UFLC-DAD provided superior sensitivity with a lower limit of detection (2-25 µg/mL range) compared to UV-spectrophotometry (5-40 µg/mL range) [7]. Both methods demonstrated excellent accuracy (98-101% for HPLC-DAD and 96-100% for UV-spectrophotometry) and precision, indicating that for well-characterized single-component systems, UV-spectrophotometry can offer a simpler, cost-effective alternative [7].
However, for complex samples, UFLC-DAD provides unambiguous advantages. In drug release kinetic studies, the sensitivity of the HPLC-DAD method enabled studies of lychnopholide release/dissolution in sink conditions, revealing that while 100% dissolution occurred after 24 hours, only 60% of lychnopholide was released from nanocapsule dosage forms, with no burst effect [7]. Such detailed release profiling would be challenging with conventional spectrophotometry due to potential interference from formulation components.
Table 3: Direct Comparison of UFLC-DAD and UV-Spectrophotometry for Lychnopholide Analysis
| Parameter | UFLC-DAD Performance | UV-Spectrophotometry Performance | Implications |
|---|---|---|---|
| Linear Range | 2-25 µg/mL [7] | 5-40 µg/mL [7] | UFLC-DAD offers better sensitivity |
| Accuracy | 98-101% [7] | 96-100% [7] | Comparable accuracy for both techniques |
| Application to Nanocapsules | Suitable for drug loading and release studies [7] | Suitable for drug loading assessment [7] | UFLC-DAD better for complex release kinetics |
| Selectivity | High (separates analytes from excipients) [7] | Moderate (potential for interference) [7] | UFLC-DAD superior for complex matrices |
Advanced Optimization Strategies
Automated Method Development and Response Functions
The complexity of UFLC-DAD method development has driven interest in automated optimization approaches. Recent advances employ retention modeling and sophisticated optimization algorithms to streamline the process of identifying optimal separation conditions. These approaches utilize chromatographic response functions (CRF) that mathematically encode separation quality metrics, enabling systematic evaluation of numerous method parameters without extensive manual experimentation [41].
Algorithm-assisted method development demonstrates particular value for gradient profile optimization, where the multidimensional parameter space (initial and final organic percentage, gradient time, curve shape) presents challenges for traditional one-factor-at-a-time approaches. By employing these computational strategies, researchers can efficiently navigate this complex parameter space to identify conditions that balance resolution, analysis time, and robustness [41].
Sample Preparation Techniques for Enhanced Separation
Effective UFLC-DAD analysis often requires tailored sample preparation to address matrix complexity and improve separation quality. Several advanced sample treatment strategies have demonstrated significant value in enhancing UFLC-DAD applications:
- Pre-column saponification effectively simplifies complex biological matrices like milk by removing glycerides that could interfere with analysis, though it may reduce tocol concentrations in oil samples [37] [38].
- Pre-column derivatization using reagents like trifluoroacetic anhydride enables separation of otherwise challenging compounds, successfully resolving β- and γ-forms of tocopherols and tocotrienols that cannot be separated using standard C18 chromatography [37].
- Solvent optimization studies identified propan-2-ol as the optimal solvent for oil-based samples, providing appropriate solubility and compatibility with the chromatographic system [38].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful UFLC-DAD method development requires carefully selected reagents, columns, and instruments. The following table details essential components for establishing robust UFLC-DAD methods based on the applications reviewed.
Table 4: Essential Research Reagents and Materials for UFLC-DAD Method Development
| Category | Specific Items | Function/Purpose | Application Examples |
|---|---|---|---|
| Chromatography Columns | C18 reversed-phase (e.g., Phenomenex Luna, Kinetex) | Primary separation medium for most applications | General reversed-phase separation [39] |
| Chiral stationary phases (e.g., Chiralpak IA) | Enantiomeric separation | Chiral analysis [39] | |
| Mobile Phase Modifiers | Trifluoroacetic anhydride | Pre-column derivatization for challenging separations | Separation of β- and γ-tocopherols [37] |
| Acetic acid | Improves peak shape for acidic compounds | Trigonelline analysis [39] | |
| Ammonium acetate (pH 4.5) | Buffer for controlling ionization | Nucleotide separation [39] | |
| Sample Preparation | Saponification reagents | Removes interfering glycerides | Milk sample preparation [37] |
| Solid-phase extraction cartridges (C18) | Sample clean-up and concentration | Pre-concentration of analytes [39] | |
| Solvents | HPLC-grade acetonitrile and methanol | Primary mobile phase components | Virtually all applications |
| Propan-2-ol | Optimal solvent for oil samples | Oil sample preparation [38] |
Experimental Workflows and Signaling Pathways
The development and optimization of UFLC-DAD methods follows a systematic workflow that integrates multiple decision points and parameter optimization steps. The following diagram illustrates this comprehensive process:
Diagram 1: UFLC-DAD Method Development Workflow - This diagram illustrates the systematic approach to developing and optimizing UFLC-DAD methods, highlighting key decision points and iterative optimization cycles.
The comparative analytical strategy between UFLC-DAD and UV-spectrophotometry involves parallel validation pathways with distinct consideration factors, as shown in the following relationship diagram:
Diagram 2: Method Selection Strategy - This diagram outlines the decision-making process for selecting between UFLC-DAD and UV-spectrophotometry based on specific analytical requirements and sample characteristics.
UFLC-DAD method development represents a sophisticated process requiring careful optimization of multiple interrelated parameters, with column selection, mobile phase composition, and gradient profile standing as the most critical factors determining analytical success. The comparative analysis with UV-spectrophotometry clearly demonstrates that while simpler spectroscopic methods suffice for straightforward applications, UFLC-DAD provides indispensable capabilities for complex samples, multi-component analysis, and challenging separations.
The ongoing development of automated optimization algorithms [41] and advanced sample preparation techniques [37] continues to enhance the efficiency and effectiveness of UFLC-DAD method development. These advancements, coupled with the fundamental separation power of ultra-fast liquid chromatography and the detection versatility of diode array detection, ensure that UFLC-DAD will remain a cornerstone technique in analytical laboratories addressing challenging separation problems across pharmaceutical, food, and biological applications.
For researchers and drug development professionals, the methodological principles and optimization strategies detailed in this guide provide a robust foundation for developing validated, reliable UFLC-DAD methods capable of meeting diverse analytical challenges while delivering the speed, sensitivity, and specificity demanded by modern analytical science.
Sample preparation is a critical step in analytical chemistry, serving as the foundation for accurate and reproducible results. The process of extraction and clean-up significantly influences method sensitivity, specificity, and reliability, particularly when analyzing complex matrices. Within pharmaceutical research and drug development, the selection of appropriate sample preparation strategies directly impacts the validity of subsequent analytical measurements. This guide examines sample preparation methodologies within the context of comparative analytical research, particularly focusing on the distinct requirements of spectrophotometric and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) methods. The fundamental principle driving sample preparation is that the complexity of the sample matrix must be adequately addressed to minimize interference during analysis [42]. Matrix effects—where sample components alter the analytical signal—can manifest as suppression or enhancement, ultimately compromising data integrity [43]. Therefore, the implementation of robust extraction and clean-up protocols is not merely preparatory but integral to the entire analytical workflow.
Fundamental Principles of Sample Preparation
The core objective of sample preparation is to isolate target analytes from interfering matrix components while ensuring adequate recovery and maintaining analyte integrity. The strategies employed depend heavily on the nature of the sample matrix and the detection technique utilized. Complex samples, such as biological fluids, environmental samples, or formulated products, contain numerous compounds that can co-elute with analytes or cause matrix effects [43] [42].
Matrix Effects present a substantial challenge, particularly in mass spectrometry, where co-eluting compounds can suppress or enhance ionization, leading to inaccurate quantification [43]. These effects are equally relevant in spectrophotometry and UFLC-DAD, where interfering compounds can absorb at similar wavelengths, causing spectral overlap and inaccurate results [3]. The overarching goal of sample clean-up is to reduce the complexity of the sample introduced into the analytical system, thereby mitigating these detrimental effects and improving the ruggedness of the method [44].
Various techniques are available for sample preparation, each with distinct advantages, limitations, and optimal application scenarios. The choice among them is guided by factors such as the physicochemical properties of the analyte, the complexity of the matrix, the required sensitivity, and the compatibility with the subsequent analytical technique.
Table 1: Comparison of Common Sample Preparation Techniques
| Technique | Principle | Best Suited For | Advantages | Limitations |
|---|---|---|---|---|
| Protein Precipitation | Uses organic solvents or acids to denature and precipitate proteins from biological samples [44]. | Quick clean-up of biological fluids (plasma, serum) for UFLC-DAD and LC-MS. | Rapid, simple, requires minimal expertise. | Limited clean-up, can concentrate other matrix components, may not be sufficient for complex matrices [44]. |
| Liquid-Liquid Extraction (LLE) | Partitioning of analytes between two immiscible liquids based on solubility [44]. | Extraction of non-polar to semi-polar analytes from aqueous matrices. | High capacity, selective based on pH and solvent, effective for a wide range of analytes. | Emulsion formation, requires large solvent volumes, not easily automated. |
| Solid-Phase Extraction (SPE) | Selective retention and elution of analytes from a liquid sample using a solid sorbent [42]. | Selective clean-up and pre-concentration of analytes from complex matrices (biological, environmental). | High selectivity, effective matrix removal, can be automated, pre-concentrates analytes. | Method development can be complex, sorbent cost, potential for cartridge clogging. |
| Solid-Phase Microextraction (SPME) | Equilibrium extraction of analytes using a fiber coated with stationary phase [42]. | Extraction of volatiles and semi-volatiles for GC or LC analysis, ideal for limited sample volumes. | Minimal or no solvent, integrates sampling and extraction, can be automated. | Fiber cost and fragility, limited sample capacity, potential for carryover. |
The following workflow diagram illustrates the decision-making process for selecting an appropriate sample preparation strategy based on the analytical requirements:
Figure 1: A decision workflow for selecting sample preparation techniques based on analytical requirements and constraints.
Sample Preparation for Spectrophotometric versus UFLC-DAD Methods
The choice of detection method imposes specific demands on sample preparation. While the core principles of extraction remain similar, the clean-up requirements differ significantly due to the varying susceptibility of each technique to matrix interferences.
Sample Preparation for Spectrophotometric Methods
Spectrophotometric methods, including UV-Vis spectroscopy, are based on the measurement of light absorption by the analyte at a specific wavelength [3]. The simplicity and cost-effectiveness of these methods make them attractive for routine analysis. However, their inherent lack of chromatographic separation means that the entire sample is measured at once, making them highly susceptible to interference from any co-extracted compounds that absorb at the same wavelength.
Consequently, sample preparation for spectrophotometry often requires a high degree of selectivity to ensure that the measured signal is specific to the target analyte. This is frequently achieved through derivatization, a process that uses chemical reagents to convert the analyte into a derivative with more desirable properties, such as enhanced absorbance or a shifted wavelength of maximum absorption [3].
Table 2: Common Reagent Types Used in Spectrophotometric Analysis
| Reagent Type | Function | Example Applications |
|---|---|---|
| Complexing Agents | Form stable, colored complexes with analytes, enabling quantification of compounds that lack inherent chromophores [3]. | Ferric chloride forms complexes with phenolic drugs like paracetamol [3]. |
| Oxidizing/Reducing Agents | Modify the oxidation state of the analyte, leading to a product with different absorbance properties [3]. | Ceric ammonium sulfate for determining ascorbic acid [3]. |
| pH Indicators | Exploit color changes dependent on the pH of the solution for the analysis of acid-base equilibria of drugs [3]. | Bromocresol green for the assay of weak acids [3]. |
| Diazotization Reagents | Convert primary aromatic amines into diazonium salts, which are then coupled to form highly colored azo compounds [3]. | Analysis of sulfonamide antibiotics using sodium nitrite and hydrochloric acid [3]. |
Sample Preparation for UFLC-DAD Methods
UFLC-DAD combines the separation power of chromatography with the qualitative and quantitative capabilities of diode array detection. The chromatographic step physically separates the analyte from many potential interferents before detection, which inherently reduces the sample clean-up burden compared to direct spectrophotometry. However, the DAD detector remains vulnerable to compounds that co-elute with the analyte and have overlapping UV spectra.
Therefore, the primary goal of sample preparation for UFLC-DAD is to protect the analytical column and prevent the introduction of particulates or strongly retained matrix components that could degrade chromatographic performance. For complex matrices, Solid-Phase Extraction (SPE) is a mainstream choice as it effectively removes proteins, phospholipids, and other interferences while allowing for pre-concentration of the analyte [44] [42]. The use of internal standards (IS), particularly stable isotopically labeled ones (e.g., ¹³C or ¹⁵N labeled), is a critical strategy in quantitative UFLC-DAD and LC-MS workflows. These ISs compensate for variability during sample preparation and analysis, improving accuracy and precision [42].
A practical example is the analysis of amitriptyline and its metabolites in serum. A comprehensive study comparing nine sample preparation methods found that phospholipid-removal SPE and certain LLE methods provided the best combination of analyte recovery and matrix removal, as evaluated by charged aerosol detection and LC-MS. This highlights that an optimized sample preparation method should be developed based not only on analyte recovery but also on the efficiency of matrix clean-up [44].
Comparative Experimental Data and Protocols
To illustrate the practical implications of sample preparation choice, data from comparative studies provides valuable insights. The performance of different techniques can be evaluated based on metrics like matrix removal efficiency and analyte recovery.
Table 3: Comparison of Sample Preparation Methods for Serum Analysis of a Model Drug (Amitriptyline) [44]
| Sample Preparation Method | Analyte Recovery | Matrix Removal Efficiency | Suitability for UFLC-DAD/LC-MS |
|---|---|---|---|
| Protein Precipitation (Acetonitrile) | Moderate to High | Low | Moderate (can cause ion suppression) |
| Liquid-Liquid Extraction (MTBE) | High | High | Good |
| Mixed-Mode Cation Exchange SPE | High | High | Excellent |
| Phospholipid-Removal SPE (PRP) | High | Very High (specific to phospholipids) | Excellent |
Example Protocol: Solid-Phase Extraction for Serum Samples
This generic protocol for a drug substance like amitriptyline is adapted from methodologies described in the literature [44].
- Conditioning: Condition a mixed-mode cation exchange SPE cartridge (e.g., 60 mg) sequentially with 2 mL of methanol and 2 mL of water.
- Sample Loading: Acidify 500 µL of serum sample with an equal volume of 0.1% formic acid in water. Load the diluted sample onto the conditioned cartridge.
- Washing: Wash the cartridge sequentially with 2 mL of water, 2 mL of 0.1% formic acid in water, and 2 mL of methanol. This step removes proteins, salts, and other polar and medium-polarity interferences.
- Drying: Centrifuge the cartridge or apply positive pressure for 1-2 minutes to dry the sorbent completely.
- Elution: Elute the analyte using 2 x 1 mL of a mixture of 5% ammonium hydroxide in methanol. Collect the entire eluate.
- Reconstitution: Evaporate the eluate to dryness under a gentle stream of nitrogen at 40°C. Reconstitute the dry residue in 250 µL of the initial mobile phase used for UFLC-DAD analysis. Vortex-mix and centrifuge before injection.
Example Protocol: Derivatization for Spectrophotometric Analysis
This protocol outlines the general steps for analyzing a drug containing a primary aromatic amine group using diazotization [3].
- Sample Preparation: Dissolve the pharmaceutical compound in an appropriate solvent (e.g., water or alcohol).
- Diazotization: To the sample solution, add 1 mL of hydrochloric acid (1 M) and 1 mL of sodium nitrite solution (0.1% w/v). Allow the reaction to proceed for 5 minutes with occasional mixing.
- Coupling: Add 1 mL of a coupling agent, such as N-(1-naphthyl)ethylenediamine dihydrochloride (0.1% w/v). A colored complex will form.
- Dilution and Measurement: Dilute the solution to a known volume with water. Allow the color to develop for 15-20 minutes, then measure the absorbance of the solution at the λmax of the formed azo dye (typically between 450-550 nm).
- Calibration: Construct a calibration curve using standard solutions of the drug treated identically.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and materials essential for implementing the sample preparation strategies discussed in this guide.
Table 4: Essential Research Reagent Solutions for Sample Preparation
| Item | Function | Application Context |
|---|---|---|
| Mixed-Mode SPE Cartridges | Combine reversed-phase and ion-exchange mechanisms for highly selective clean-up of ionizable analytes from complex matrices [44]. | UFLC-DAD/LC-MS analysis of drugs in biological fluids. |
| Phospholipid-Removal SPE Plates | Selectively remove phospholipids, a major source of matrix effects in ESI-MS, from biological samples [44]. | High-throughput bioanalysis by UFLC-MS/MS. |
| Stable Isotope-Labeled Internal Standards | Correct for analyte loss during preparation and matrix effects during analysis; ¹³C or ¹⁵N labels are preferred over deuterium to avoid isotopic effects [42]. | Quantitative UFLC-DAD and LC-MS in all matrices. |
| Diazotization Reagents (NaNO₂, HCl) | Convert primary aromatic amine groups into diazonium salts for subsequent coupling to form a colored complex [3]. | Spectrophotometric determination of sulfonamides and other amine-containing drugs. |
| Complexing Agents (e.g., Ferric Chloride) | Form colored complexes with specific functional groups (e.g., phenols) to enable spectrophotometric detection [3]. | Quantification of drugs like paracetamol in formulations. |
Selecting an optimal sample preparation strategy is a multidimensional decision that balances the nature of the sample matrix, the analytical technique, and the required data quality. As this guide demonstrates, spectrophotometric methods often demand highly selective clean-up or derivatization to overcome their inherent vulnerability to spectral interference. In contrast, UFLC-DAD methods, benefited by a separation step, can utilize more generalized clean-up protocols focused on column protection and removal of major interferents, with techniques like SPE providing robust solutions. The comparative data underscores that there is no universal "best" technique; rather, the choice must be validated in the context of the specific analytical method. For researchers, a thorough understanding of these principles and the available toolkit is fundamental to developing reliable, accurate, and robust analytical methods in drug development and beyond.
The quantitative analysis of multiple active pharmaceutical ingredients (APIs) within a single formulation presents a significant analytical challenge, demanding methods that are selective, precise, and efficient. This case study objectively compares two prominent analytical techniques: Ultraviolet-Visible Spectrophotometry (UV-Vis) and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD). The focus is on their application for the simultaneous determination of analytes in pharmaceutical products, providing researchers and drug development professionals with a clear comparison of performance characteristics, experimental protocols, and practical considerations. The analysis is framed within the broader thesis that while UFLC-DAD generally offers superior specificity and sensitivity, modern spectrophotometric methods present a compelling, cost-effective alternative for specific applications, particularly in resource-limited settings or for routine quality control of less complex matrices.
Principles and Instrumentation
Spectrophotometric Methods
UV-Vis spectrophotometry operates on the Beer-Lambert Law, which states that the absorbance (A) of a solution is directly proportional to the concentration (c) of the analyte and the path length (b) of the light through the solution (A = εbc, where ε is the molar absorptivity) [29] [3]. The technique measures the absorption of light in the ultraviolet or visible range by chromophores in the analyte molecules. For simultaneous determination of multiple components with overlapping spectra, techniques such as derivative spectrophotometry (e.g., first, second, or third derivative) and ratio-based methods (e.g., double divisor ratio spectra derivative) are employed to resolve the overlapping signals and enhance selectivity [27] [26] [3]. Reagents like complexing agents, oxidizing/reducing agents, and pH indicators are often used to induce measurable color changes in analytes that lack inherent chromophores [3].
UFLC-DAD Methods
UFLC is an advanced form of high-performance liquid chromatography that utilizes columns packed with smaller particles (typically less than 2 µm) and higher operating pressures to achieve superior separation efficiency, increased peak capacity, and shorter analysis times compared to conventional HPLC [1] [16]. The DAD detector is a critical component that measures the UV-Vis spectrum of each compound as it elutes from the column. This allows for the continuous monitoring of multiple wavelengths, the identification of compounds based on their spectral signature, and the assessment of peak purity by comparing spectra across a chromatographic peak [45] [1]. This hyphenated technique combines high-resolution separation with rich spectral data.
Performance Comparison
The following tables summarize the key performance characteristics and operational aspects of the two techniques, based on experimental data from published comparative studies.
Table 1: Analytical Performance Metrics for Spectrophotometry and UFLC-DAD
| Performance Parameter | Spectrophotometric Methods | UFLC-DAD Methods |
|---|---|---|
| Typical Linear Range | Relatively narrow (e.g., 2-70 µg/mL for analgin, caffeine, ergotamine) [27] | Broad (e.g., 0.5-400 µg/mL for analgin, caffeine, ergotamine) [27] |
| Limit of Detection (LOD) / Quantification (LOQ) | Higher LOD/LOQ; Spectrophotometric LOD for Vitamin C: 0.002 mg/mL [15] | Lower LOD/LOQ; More sensitive, suitable for trace analysis [27] |
| Precision (Repeatability) | High precision (RSD < 5% for Metoprolol assay) [1] | Very high precision (RSD typically < 1-2% for pharmaceutical assays) [45] [1] |
| Accuracy (Recovery) | Accurate (96-101% for Lychnopholide) [7] | Highly accurate (98-101% for Lychnopholide) [7] |
| Specificity/Selectivity | Moderate; can be improved with derivative techniques and reagents [1] [27] | Very high; achieved via chromatographic separation and spectral confirmation [45] [1] |
| Key Advantage | Simplicity, low cost, rapid analysis, high precision, green solvent usage [1] [27] | High sensitivity, superior selectivity, peak purity assessment, robust quantification in complex mixtures [45] [1] |
| Primary Limitation | Lower specificity; susceptible to interference from excipients or other analytes [1] | Higher instrumental and operational costs; requires more complex sample preparation [1] |
Table 2: Operational and Economic Considerations
| Consideration | Spectrophotometric Methods | UFLC-DAD Methods |
|---|---|---|
| Instrument Cost & Maintenance | Low cost, simple maintenance [1] [3] | High capital and operational cost, requires specialized maintenance [1] |
| Analysis Time | Very fast (minutes per sample) [3] | Fast, but longer than spectrophotometry (includes separation time) [1] [16] |
| Sample Throughput | High for single-analyte or simple mixtures | High, especially with automation, for complex mixtures |
| Sample & Solvent Consumption | Larger sample volumes may be required [1] | Lower sample volumes; reduced solvent consumption with UHPLC [1] [16] |
| Operator Skill Level | Minimal training required [3] | Requires significant technical expertise [1] |
| Environmental Impact (AGREE Score) | Generally higher greenness score [1] | Lower greenness score due to solvent use [1] |
Experimental Protocols
Detailed Methodologies from Case Studies
Case Study 1: Simultaneous Determination of Analgin, Caffeine, and Ergotamine [27]
- Objective: To develop and validate methods for the simultaneous determination of a ternary mixture in a migraine medication.
- UFLC-DAD Protocol:
- Chromatography: An Inertsil-C8 column was used with a gradient elution of acetonitrile and ammonium format buffer (pH 4.2).
- Detection: Ultraviolet detection for analgin and caffeine was performed at 280 nm and 254 nm, respectively. Ergotamine was detected using fluorometric detection (λexc = 310 nm, λemm = 360 nm) due to its higher sensitivity.
- Validation: The method was validated over the concentration ranges of 50-400 µg/mL for analgin, 25-200 µg/mL for caffeine, and 0.5-10 µg/mL for ergotamine.
- Spectrophotometric Protocol:
- Methods: Two novel methods were employed: the Double Divisor Ratio Spectra Derivative (DDRD) method for ergotamine (at 355 nm) and caffeine (at 268 nm), and the Ratio Dual Wavelength (RDW) method for caffeine and analgin.
- Validation: The spectrophotometric methods were validated over the ranges of 10-35 µg/mL for analgin, 2-30 µg/mL for caffeine, and 10-70 µg/mL for ergotamine.
Case Study 2: Determination of Metoprolol Tartrate (MET) [1]
- Objective: To optimize and validate methods for quantifying MET in commercial tablets and compare the results from both techniques.
- Sample Preparation: MET was extracted from tablets into ultrapure water. Solutions were protected from light.
- UFLC-DAD Protocol:
- The UFLC conditions were optimized before validation. The DAD was used for detection.
- Validation Parameters: Specificity, sensitivity, linearity, LOD, LOQ, accuracy, precision, and robustness were determined.
- Spectrophotometric Protocol:
- Absorbance was recorded at the maximum absorption wavelength of MET (λ = 223 nm).
- The method was validated for the same parameters as the UFLC-DAD method.
- Statistical Comparison: Results from both methods were compared using Analysis of Variance (ANOVA) at a 95% confidence level, showing no significant difference.
Case Study 3: Determination of Lychnopholide in Nanocapsules [7]
- Objective: To develop methods for quantifying lychnopholide (LYC) in a nanocapsule dosage form and study its release kinetics.
- HPLC-DAD Protocol:
- Chromatography: An RP C18 column with an isocratic mobile phase of methanol-water (60:40 v/v) at a flow rate of 0.8 mL/min.
- Detection: Detection was at 265 nm.
- Linearity: The method was linear (r² > 0.999) from 2-25 µg/mL.
- UV-Spectrophotometry Protocol:
- The method was linear from 5-40 µg/mL.
- Outcome: Both methods were successfully applied to determine drug loading and encapsulation efficiency, with the HPLC-DAD method being sensitive enough for release studies in sink conditions.
Workflow Diagrams
The following diagrams illustrate the generalized experimental workflows for sample analysis using both techniques.
Diagram 1: Analytical Workflows Comparison
Diagram 2: Method Selection Logic
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key reagents and materials used in the development and application of these analytical methods, as derived from the cited case studies.
Table 3: Key Research Reagent Solutions and Materials
| Reagent/Material | Function & Principle | Example Application |
|---|---|---|
| Complexing Agents (e.g., Ferric Chloride, Potassium Permanganate) | Form stable, colored complexes with analytes, enhancing absorbance for compounds with weak inherent chromophores. [3] | Analysis of phenolic drugs like paracetamol. [3] |
| Diazotization Reagents (e.g., Sodium Nitrite, Hydrochloric Acid) | Convert primary aromatic amines in drugs to diazonium salts, which couple to form highly colored azo compounds for detection. [3] | Determination of sulfonamide antibiotics and other amine-containing drugs. [3] |
| Ultra-Pure Water (UPW) | Serves as a dissolution and dilution solvent to minimize background interference in spectrophotometric and chromatographic analysis. [1] | Preparation of standard solutions and sample extracts for Metoprolol analysis. [1] |
| UFLC Mobile Phase Buffers (e.g., Ammonium Formate) | Control the pH and ionic strength of the mobile phase to optimize chromatographic separation, peak shape, and reproducibility. [27] | Gradient elution for separation of analgin, caffeine, and ergotamine. [27] |
| Derivatization Agents (e.g., 2,4-Dinitrophenylhydrazine - DNPH) | React with target analytes to form derivatives with more favorable detection properties (e.g., stronger UV absorption). [26] | Detection of malondialdehyde (MDA) in milk and infant formula prior to HPLC-DAD analysis. [26] |
| C18 Reverse-Phase Chromatography Column | The stationary phase for UFLC separation, separating compounds based on their hydrophobicity. | Used in nearly all cited UFLC-DAD protocols for analyte separation. [27] [7] [26] |
This case study demonstrates that both spectrophotometry and UFLC-DAD are capable of accurately determining multiple analytes in formulations, yet they serve different purposes within the drug development and quality control workflow. UFLC-DAD is unequivocally more powerful for the analysis of complex mixtures, offering superior separation, definitive identification via spectral matching, and robust quantification of trace components, making it the preferred choice for method development, stability-indicating assays, and impurity profiling. Spectrophotometry, particularly with advanced mathematical processing, remains a highly valuable, cost-effective, and efficient technique for routine quality control of formulations with a limited number of analytes, especially in environments with budgetary constraints. The choice between them should be guided by a balanced consideration of the required specificity, sample complexity, available resources, and the intended purpose of the analysis.
In modern pharmaceutical development, the choice of analytical technique is critical for ensuring drug safety, efficacy, and quality. Two predominant methodologies have emerged as fundamental tools: Ultraviolet-Visible (UV-Vis) spectrophotometry and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD). These techniques represent different points on the spectrum of analytical capability, from straightforward API quantification to sophisticated stability-indicating methods capable of resolving complex mixtures and degradation products. UV-Vis spectrophotometry offers simplicity, rapid implementation, and cost-effectiveness for basic quantitative analysis, making it suitable for routine quality control applications where specificity is not a primary concern. In contrast, UFLC-DAD provides superior separation power, specificity, and sensitivity, enabling comprehensive characterization of complex pharmaceutical formulations under forced degradation conditions.
The fundamental distinction between these methodologies lies in their operational principles. UV-Vis spectrophotometry measures the absorption of ultraviolet or visible radiation by a substance in solution, following the Beer-Lambert law which establishes that absorbance is proportional to concentration [29]. This technique provides a composite spectral signature of all light-absorbing compounds in a sample without separation. UFLC-DAD, however, incorporates a separation dimension through liquid chromatography coupled with full-spectrum ultraviolet detection, allowing for the resolution of multiple components within a single analysis [46]. This hyphenated approach enables both quantitative determination and qualitative assessment through spectral comparison, making it particularly valuable for stability-indicating methods where drug substances must be resolved from their degradation products.
Theoretical Foundations and Operational Principles
UV-Visible Spectrophotometry Fundamentals
UV-Visible spectrophotometry operates on the principle that molecules absorb specific wavelengths of light in the ultraviolet (200-400 nm) and visible (400-750 nm) regions of the electromagnetic spectrum [29]. When a molecule absorbs this electromagnetic radiation, electrons transition from ground state to excited state, resulting in characteristic absorption spectra. The Beer-Lambert law forms the quantitative foundation for this technique, mathematically expressed as A = εbc, where A represents absorbance, ε is the molar absorptivity coefficient, b is the path length of the radiation through the sample, and c is the concentration of the solute [29]. This linear relationship between absorbance and concentration enables quantitative analysis of pharmaceutical compounds.
The instrumentation for UV-Vis spectrophotometry typically consists of a light source, wavelength selector (monochromator or filters), sample holder (cuvette), and photodetector. Modern instruments are predominantly double-beam designs that simultaneously measure sample and reference light paths to compensate for source fluctuations and solvent absorption. For analytical measurements, the wavelength of maximum absorption (λmax) is typically selected for quantification, as this provides maximum sensitivity and minimizes errors due to slight wavelength miscalibration [29]. The technique can be enhanced through derivative spectroscopy, which helps resolve overlapping spectral bands and improves specificity in analyzing complex mixtures without physical separation [26].
UFLC-DAD System Fundamentals
UFLC-DAD represents an advanced liquid chromatographic technique that couples high-efficiency separation with full-spectrum ultraviolet detection. The "ultra-fast" aspect refers to the use of stationary phases with smaller particle sizes (typically below 2μm) and higher operating pressures, enabling faster separations with improved resolution compared to conventional HPLC [47]. The diode array detection component utilizes an array of photodiodes to simultaneously monitor multiple wavelengths, capturing complete UV-Vis spectra throughout the chromatographic run.
The operational principle involves the separation of components based on their differential partitioning between a stationary phase and a mobile phase under high pressure. As compounds elute from the chromatographic column, they pass through a flow cell where they are exposed to deuterium or tungsten lamp radiation. The transmitted light is dispersed onto the diode array, capturing absorbance spectra typically from 190 to 800 nm [46]. This dual-dimensional data (retention time and spectral information) provides both quantitative data through peak area integration and qualitative information through spectral matching. The gradient elution capability, where mobile phase composition is systematically changed during the analysis, further enhances separation efficiency for complex mixtures with diverse polarities [48].
Comparative Performance Data
Table 1: Direct comparison of UV-Vis spectrophotometry and UFLC-DAD performance characteristics
| Performance Parameter | UV-Vis Spectrophotometry | UFLC-DAD |
|---|---|---|
| Linear Range | 5-40 μg/mL for lychnopholide [7]; 5-60 μg/mL for vildagliptin [6] | 2-25 μg/mL for lychnopholide [7]; 2-100 μg/mL for antiretroviral drugs [48] |
| Limit of Detection (LOD) | 0.951 μg/mL for vildagliptin [6]; 0.060 μg/mL for methotrexate [28] | 0.014 μg/mL for methotrexate [28]; 0.28 μg/mL for dolutegravir [48] |
| Limit of Quantification (LOQ) | 2.513 μg/mL for vildagliptin [6] | 0.94 μg/mL for dolutegravir [48]; 6.73 μg/mL for abacavir [48] |
| Precision (RSD) | Intra-day: 1.263% for vildagliptin [6]; <2.90% for methotrexate [28] | Intra-day: <1% for lychnopholide [7]; <3.39% for methotrexate [28] |
| Accuracy (% Recovery) | 96-101% for lychnopholide [7]; 98-101% for vildagliptin [6] | 98-101% for lychnopholide [7]; 89.5-105.5% for methotrexate [28] |
| Analysis Time | Minutes (rapid) | 10-20 minutes (chromatographic run times) [48] |
| Multi-component Analysis | Limited, requires mathematical processing [27] | Excellent, physical separation of components [48] |
Table 2: Application-specific comparison for different pharmaceutical analyses
| Application Type | UV-Vis Spectrophotometry Approach | UFLC-DAD Approach |
|---|---|---|
| Single API Quantification | Direct measurement at λmax [6] [29] | Retention time identification with spectral confirmation [7] |
| Fixed-Dose Combinations | Derivative spectroscopy or absorbance ratio methods [27] | Gradient elution with selective wavelength monitoring [27] [48] |
| Stability-Indicating Methods | Limited capability without separation | Comprehensive forced degradation studies with peak purity assessment [48] [47] |
| Nanoparticulate Systems | Determination of encapsulation efficiency [7] | Drug release kinetics under sink conditions [7] |
| Biocompatible Media Analysis | Direct analysis in gastric medium [6] | Requires sample preparation and clean-up [26] |
Detailed Experimental Protocols
Protocol for UV-Vis Spectrophotometric API Quantification
The quantification of vildagliptin in gastric medium demonstrates a standardized UV-Vis spectrophotometric approach [6]. For method development, a stock solution of vildagliptin reference standard is prepared in 0.1N HCl to simulate gastric conditions. Working standards across the concentration range of 5-60 μg/mL are prepared by serial dilution. The absorption maximum (λmax) is determined by scanning from 200-400 nm, identified at 210 nm for vildagliptin. For validation, linearity is established by analyzing standards in triplicate and plotting absorbance versus concentration. The method is evaluated for precision using intra-day (repeatability within same day) and inter-day (variability between different days) measurements at three concentration levels. Accuracy is determined through standard addition recovery studies, spiking pre-analyzed samples with known amounts of reference standard [6].
Sample preparation for tablet formulations involves powdering twenty tablets, weighing an amount equivalent to one tablet, and dissolving in 0.1N HCl with sonication for 30 minutes. The solution is filtered, diluted to appropriate concentration, and analyzed against a solvent blank. For method validation specificity, placebo formulations containing all excipients except the active ingredient are analyzed to confirm absence of interference at the analytical wavelength [6]. The robustness is evaluated by deliberate variations in analytical parameters such as slight changes in wavelength (±2 nm) and different instrument sources.
Protocol for UFLC-DAD Stability-Indicating Method
The development of a stability-indicating UFLC-DAD method for abacavir, dolutegravir, and lamivudine illustrates a comprehensive approach [48]. Chromatographic separation is achieved using a Kinetex C18 column (250 mm × 4.6 mm, 5μm) with gradient elution combining acetonitrile and water. The mobile phase composition is programmed from 20% acetonitrile to 60% over 5 minutes, maintained for 3 minutes, then returned to initial conditions over 2 minutes, with a total run time of 15 minutes including re-equilibration. The flow rate is maintained at 1.0 mL/min with detection at 258 nm. Standard solutions are prepared in diluent (ACN-water 60:40 v/v) at concentrations of 60, 5, and 30 μg/mL for abacavir, dolutegravir, and lamivudine, respectively [48].
Forced degradation studies are conducted to demonstrate method specificity under stress conditions. Acid degradation employs 3N hydrochloric acid at 60°C for 1 hour, while alkaline degradation uses 2N sodium hydroxide under the same conditions. Oxidative degradation involves 30% hydrogen peroxide at 60°C for 1 hour, and thermal degradation is performed by exposing solid drug substances to 105°C for 72 hours. Photostability is assessed according to ICH guidelines by exposing drug solutions to UV light (200 Watt-hours/m²). All degradation samples are neutralized where appropriate and analyzed alongside untreated controls to demonstrate separation of degradation products from parent compounds [48]. Method validation includes determination of system suitability parameters (theoretical plates, tailing factor, resolution), specificity (peak purity index using DAD), and robustness to deliberate variations in flow rate (±0.1 mL/min), mobile phase composition (±2%), and column temperature.
Figure 1: Experimental workflow comparison between UV-Vis spectrophotometry and UFLC-DAD methodologies
Critical Reagent and Material Solutions
Table 3: Essential research reagents and materials for analytical method development
| Reagent/Material | Function/Purpose | Application Examples |
|---|---|---|
| Acetonitrile (HPLC grade) | Mobile phase component for chromatographic separation | Reversed-phase LC separation of APIs [48] |
| Methanol (HPLC grade) | Solvent for sample preparation and mobile phase | Extraction of drugs from formulations [48] |
| Buffer Salts (e.g., ammonium acetate, phosphate buffers) | Mobile phase modification for pH control | Separation of ionizable compounds [27] [48] |
| Reference Standards | Quantitative calibration and method validation | API quantification against certified references [6] |
| Stationary Phases (C8, C18, F5 columns) | Chromatographic separation media | Resolution of complex mixtures [48] [47] |
| Acid/Base Reagents (HCl, NaOH) | Forced degradation studies | Stress testing for stability-indicating methods [48] |
| Oxidizing Agents (H₂O₂) | Oxidative stress studies | Forced degradation under oxidative conditions [48] |
| Derivatization Agents (DNPH) | Analyte chemical modification for enhanced detection | HPLC analysis of malondialdehyde [26] |
Application Spectrum and Decision Framework
Figure 2: Application spectrum and methodology selection framework
The selection between UV-Vis spectrophotometry and UFLC-DAD depends on multiple factors including analytical requirements, sample complexity, and development stage. UV-Vis spectrophotometry is optimally employed for simple API quantification in bulk drug substances and single-component formulations where specificity is not a concern [6] [29]. Its implementation is particularly advantageous in quality control laboratories requiring rapid, cost-effective analysis with minimal method development time. The technique also finds application in dissolution testing where continuous monitoring of API release is necessary, and in the analysis of biocompatible media such as gastric fluid for drug stability assessment [6].
UFLC-DAD demonstrates superior capability for fixed-dose combination products containing multiple active ingredients with overlapping spectral profiles [27] [48]. The chromatographic separation prior to detection enables accurate quantification of individual components despite similar structural characteristics. This technique is indispensable for stability-indicating methods where drug substances must be resolved from their degradation impurities [48] [47] [46]. The diode array detector provides additional peak purity assessment by comparing spectra across the peak profile. UFLC-DAD is equally essential for characterizing complex drug delivery systems including polymeric nanoparticles, microparticles, and nanocapsules, where it can simultaneously quantify drug loading, encapsulation efficiency, and release kinetics [7] [28].
For pharmaceutical analysis in biological matrices or complex formulations, UFLC-DAD provides the necessary selectivity to resolve analytes from interfering compounds. The technique's enhanced sensitivity and lower detection limits make it suitable for impurity profiling and trace analysis where UV-Vis spectrophotometry lacks the required detectability [48] [47]. When method transferability and ruggedness are concerns, UFLC-DAD methods have demonstrated successful inter-laboratory transfer with consistent performance, a critical attribute for regulatory submissions [47].
Regulatory Considerations and Validation Requirements
Both UV-Vis spectrophotometric and UFLC-DAD methods require comprehensive validation according to International Conference on Harmonisation (ICH) guidelines before implementation in regulated environments. The validation parameters include specificity, linearity, accuracy, precision, range, robustness, and system suitability testing [48] [6]. For UV-Vis methods, specificity demonstration involves confirming the absence of interference from excipients at the analytical wavelength, typically through comparison with placebo formulations [6]. For UFLC-DAD methods, specificity requires baseline resolution of the analyte peak from known and potential impurities, supported by peak purity assessment using spectral comparison [48].
Stability-indicating methods must provide documented evidence that the analytical procedure is capable of reliably detecting and quantifying changes in API concentration while being unaffected by degradation products, excipients, or other potential impurities [46]. UFLC-DAD has become the benchmark technique for stability-indicating assays due to its superior separation power and peak purity assessment capability. The forced degradation studies required for these methods involve subjecting the drug substance to stress conditions including acid, base, oxidation, thermal, and photolytic degradation, then demonstrating that the method can resolve degradation products from the main peak [48] [47]. The validation data must establish that the method remains accurate and precise even in the presence of these degradation products.
Method transfer between laboratories requires additional demonstration of ruggedness, which evaluates method performance under different conditions including different instruments, analysts, and days [47]. UFLC-DAD methods have shown excellent transferability when system suitability parameters are properly defined and controlled. For both techniques, complete documentation of validation parameters and experimental data is essential for regulatory submissions, with the validation scope tailored to the method's intended application in quality control or stability assessment.
Overcoming Challenges: Troubleshooting and Enhancing Performance
In the field of pharmaceutical analysis, researchers routinely face two persistent challenges that compromise analytical accuracy: matrix interference and overlapping spectra. Matrix interference occurs when other components in a sample, such as excipients in formulations or endogenous substances in biological fluids, affect the accurate quantification of the target analyte. Overlapping spectra presents a different challenge, where the absorption profiles of multiple compounds in a mixture coincide, making individual quantification difficult without prior separation. These issues are particularly problematic in quality control laboratories and clinical settings where time and cost constraints are significant factors.
The persistence of these challenges has led to the development of various analytical strategies, primarily falling into two categories: mathematical spectrophotometric techniques and separation-based chromatographic methods. This article provides a comparative analysis of these approaches, focusing specifically on the performance of advanced spectrophotometric methods versus Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD). The evaluation is framed within the context of analytical efficiency, with experimental data drawn from recent pharmaceutical applications to provide practical insights for researchers and drug development professionals.
Comparative Analysis: Spectrophotometric vs. UFLC-DAD Methods
Performance Metrics and Applications
Table 1: Comparison of analytical techniques for managing overlapping spectra and matrix interference
| Analytical Technique | Typical Linear Range (μg/mL) | Limit of Detection (LOD) | Key Advantages | Reported Recovery (%) | Application Examples |
|---|---|---|---|---|---|
| Ratio Derivative Spectrophotometry | 1-15 [49] | 0.26-0.92 μg/mL [49] | Simple instrumentation, rapid analysis | 96-100 [7] | Remdesivir and Moxifloxacin in COVID-19 therapy [49] |
| Area Under Curve (AUC) Method | 2-30 [50] | Varies by compound | Effective for binary/ternary mixtures without separation | 98-101 [7] | Metronidazole, Diloxanide, Mebeverine HCl [50] |
| UFLC-DAD | 2-25 [7] | <1 μg/mL for most compounds | High sensitivity, excellent resolution | 98-101 [7] | Lychnopholide in nanocapsules [7], Amoxicillin and Diclofenac [51] |
| HPLC-DAD | 0.5-400 [27] | Varies by analyte | Good separation efficiency, wide linear range | 96-100 [27] | Analgin, Caffeine, Ergotamine [27] |
Experimental Protocols and Methodologies
Advanced Spectrophotometric Methods for Overlapping Spectra
For the analysis of remdesivir and moxifloxacin hydrochloride with overlapping spectra, six distinct spectrophotometric methods were developed and validated [49]. The ratio derivative method utilized Δλ = 4 nm and a scaling factor of 10, with measurements taken at 250 nm for remdesivir and 290 nm for moxifloxacin [49]. The ratio difference method calculated differences in peak amplitudes at 247 nm and 262 nm for remdesivir, and at 299 nm and 313 nm for moxifloxacin [49]. The mean centering method applied mathematical processing to ratio spectra followed by measurement at 247 nm for remdesivir and 299 nm for moxifloxacin [49].
For ternary mixtures such as metronidazole, diloxanide, and mebeverine HCl, the Area Under Curve (AUC) method employed mathematical processing where the total AUC across selected wavelength ranges equals the sum of individual AUCs of each component [50]. This approach utilizes Cramer's Rule and matrix calculations to resolve the mixture, with the relationship defined by the equation: Total AUCλ1-λ2 = AUCX + AUCY + AUCZ [50].
Table 2: Research reagent solutions for spectroscopic analysis
| Reagent/Material | Function in Analysis | Application Example |
|---|---|---|
| Methanol | Solvent for standard and sample preparation | Preparation of remdesivir and moxifloxacin stock solutions [49] |
| Phosphate Buffers | pH adjustment for mobile phase | HPLC analysis of amoxicillin and diclofenac [51] |
| C18 Reverse-Phase Columns | Stationary phase for compound separation | BDS Hypersil C18 for amoxicillin and diclofenac separation [51] |
| Trifluoroacetic Acid | Mobile phase modifier | Adjustment of pH to 2.8 in flavonoid glycosides analysis [52] |
| Ammonium Format Buffer | Mobile phase component for gradient elution | HPLC analysis of ternary mixture of analgin, caffeine, ergotamine [27] |
UFLC-DAD Methodology for Complex Matrices
The UFLC-DAD approach for analyzing lychnopholide in nanocapsules utilized an RP C18 column with isocratic elution of methanol-water (60:40 v/v) mobile phase at a flow rate of 0.8 mL/min and detection at 265 nm [7]. For amoxicillin and diclofenac analysis, a reversed-phase BDS Hypersil C18 column was employed with a mobile phase consisting of methanol:acetonitrile:water:orthophosphoric acid (60:30:9:1 by volume) at a flow rate of 1 mL/min with UV detection at 250 nm [51]. The retention times were 3.906 minutes for amoxicillin and 6.997 minutes for diclofenac, demonstrating effective separation of the compounds [51].
The Shimadzu Prominence UFLC system, which can endure pressures up to 66 MPa, typically includes a degasser, pump, autosampler, column oven, and DAD detector [17] [53]. This system achieves highly stable flow-rate performance across a broad range from micro to semi-preparative scale, making it suitable for various applications [53]. The system's sensitivity is enhanced by a flow cell volume of 10 μL, which minimizes band broadening and allows for high-resolution separation [53].
Strategic Approach Selection and Implementation
Decision Framework for Method Selection
The choice between advanced spectrophotometric methods and UFLC-DAD depends on several factors, including analysis time, equipment availability, sample complexity, and required sensitivity. For routine quality control of formulations without complex matrices, spectrophotometric methods offer advantages of rapid analysis, minimal solvent consumption, and lower operational costs [49] [50]. The greenness assessment of these methods using Eco-Scale, ComplexGAPI, and AGREE metrics further supports their environmental sustainability [49].
For analyses requiring high sensitivity in complex matrices such as biological samples, or when dealing with multiple components with severe spectral overlap, UFLC-DAD provides superior resolution and specificity [7] [51]. The diode array detection capability allows for peak purity assessment by comparing spectra at different points across the peak, providing an additional tool for managing matrix interference [53].
Implementation Workflows
Diagram 1: Analytical method selection workflow
The workflow begins with sample preparation, followed by initial spectroscopic assessment. Based on the degree of spectral overlap and matrix complexity, the appropriate analytical path is selected. For samples with no to mild overlap, mathematical spectrophotometric methods provide efficient quantification. For severe overlap or significant matrix effects, UFLC-DAD separation is recommended for accurate results.
Diagram 2: Technical approaches for overlapping spectra
Both advanced spectrophotometric methods and UFLC-DAD offer effective solutions for managing matrix interference and overlapping spectra, with distinct advantages for specific applications. Mathematical spectrophotometric techniques provide cost-effective, rapid analysis suitable for laboratories with limited resources and for applications where green chemistry principles are prioritized [49] [50]. UFLC-DAD offers superior resolution and sensitivity for complex matrices and multi-component systems, making it invaluable for research and development applications where highest data quality is essential [7] [51] [39].
The choice between these approaches should be guided by specific analytical requirements, available resources, and the complexity of the sample matrix. Future developments in both fields will likely focus on increasing automation, improving data processing algorithms, and enhancing green chemistry metrics to make analyses more efficient and environmentally friendly.
Ultra-Fast Liquid Chromatography coupled with Diode Array Detection (UFLC-DAD) represents a significant advancement in analytical technology, offering enhanced separation capabilities coupled with comprehensive spectral data collection. This technique has become indispensable in pharmaceutical analysis, quality control, and research, where precise quantification of complex mixtures is required [54]. The diode array detector (DAD) operates on principles distinct from single-wavelength UV-Vis detectors; it employs multiple photodiode arrays to capture absorption data across a wide spectrum of wavelengths simultaneously, enabling both quantitative analysis and compound identification through spectral matching [55]. This capability for peak purity assessment and library searching makes DAD particularly valuable when analyzing samples with potential co-eluting compounds that might otherwise go undetected with single-wavelength monitoring [55] [7].
Meanwhile, UV-Vis spectrophotometry remains a widely used technique in analytical laboratories, prized for its simplicity, cost-effectiveness, and rapid operational capabilities [1]. The technique measures component absorption at specific wavelengths in the ultraviolet or visible region, typically employing a deuterium discharge lamp (190-380 nm) for UV detection and potentially a tungsten lamp for extended visible range measurements [55]. However, spectrophotometry faces significant limitations when dealing with complex mixtures, as overlapping absorption bands can complicate quantitative analysis and reduce method specificity compared to chromatographic techniques [1]. This fundamental difference in capability between the two techniques frames the ongoing discussion in analytical science regarding their appropriate application, with UFLC-DAD offering superior separation power and identification capabilities, while spectrophotometry provides operational simplicity and economic advantages [1].
Critical UFLC-DAD Parameters and Optimization Strategies
Mobile Phase Composition and Gradient Optimization
The mobile phase composition and gradient profile constitute perhaps the most influential parameters in UFLC-DAD method development, directly impacting resolution, peak shape, and analysis time. A carefully designed binary gradient elution program proved essential for the satisfactory separation of orotic acid from endogenous species in milk samples, with the total run time for analysis including re-equilibration being 27 minutes [54]. Similarly, in the analysis of sweeteners, preservatives, and caffeine in sugar-free beverages, an optimized gradient employing acetonitrile and phosphate buffer (12.5 mM, pH = 3.3) enabled the simultaneous separation of seven target analytes in less than 9 minutes [56]. The gradient program progressed from 5% to 50% acetonitrile over 10 minutes, maintained for 5 minutes, then returned to initial conditions for column re-equilibration [56]. This approach demonstrates how strategic mobile phase programming can efficiently separate compounds with significantly different physicochemical properties within a short timeframe.
The pH of the mobile phase similarly requires careful optimization, as it can dramatically affect the ionization state of analytes and their interaction with the stationary phase. In the determination of vandetanib, different mobile phase compositions were evaluated, with one method utilizing methanol: ammonium acetate buffer (90:10 v/v) at pH values optimized for retention and peak shape [57]. Another approach employed 0.5% triethylamine with pH adjusted to 3.0, highlighting how both pH and additive selection can be leveraged to improve chromatographic performance [57]. These parameters must be optimized systematically, as they influence not only retention but also peak symmetry and overall method robustness.
Column Selection and Temperature Control
Column selection represents another critical parameter in UFLC-DAD optimization, with stationary phase chemistry, particle size, and dimensions all contributing to separation efficiency. The improved method for orotic acid determination utilized two Kinetex C18 columns (1.7 µm, 150 mm × 2.1 mm, i.d., 100 Å) fitted with a pre-column, which provided sufficient theoretical plates for satisfactory separation [54]. In comparative studies, the Luna Omega C18 column resulted in better separation of some tocopherol and tocotrienol components compared to the Kinetex C18 column, though neither could resolve β- and γ-forms regardless of the column or elution program used [38]. This highlights both the importance of column selection and its limitations for certain separation challenges.
Column temperature significantly influences retention times, efficiency, and reproducibility in UFLC-DAD analyses. Most methods employ controlled temperatures, typically between 30-40°C, to ensure retention time stability and optimal kinetics [54] [56]. The orotic acid method maintained the column at 35°C while acknowledging ambient temperature variations of 21-24°C [54], whereas the sweetener analysis set the column oven precisely to 30°C [56]. Temperature control becomes particularly important when transferring methods between laboratories or when seeking to enhance method robustness for routine analysis.
Detection Wavelength and Spectral Parameters
The DAD detector provides unique advantages through its ability to monitor multiple wavelengths simultaneously and collect full spectral data for each eluting peak. Wavelength selection should be based on the maximum absorption of target analytes, as demonstrated in the orotic acid method which used 278 nm [54], the lychnopholide analysis at 265 nm [7], and the vandetanib quantification at 341 nm [57]. For methods analyzing multiple compounds with different optimal wavelengths, the time program function can be employed to measure each component at its maximum absorption wavelength during the analysis [55].
The DAD's ability to record complete spectra enables valuable applications beyond simple quantification, including peak purity assessment and library searching for compound identification [55]. In the monitoring of sulfamethoxazole degradation, detection using DAD allowed better correspondence between species monitored spectrophotometrically with those analyzed chromatographically [58]. The spectral resolution and collection rate should be optimized based on the analysis requirements, balancing data richness with file size considerations, especially in methods with short run times and narrow peaks.
Table 1: Optimized UFLC-DAD Parameters from Representative Applications
| Analysis Target | Column Type | Mobile Phase | Gradient/Flow | Temperature | Detection Wavelength | Analysis Time |
|---|---|---|---|---|---|---|
| Orotic acid in milk [54] | Kinetex C18 (1.7 µm, 150 mm × 2.1 mm) | Binary gradient (specifics not detailed) | 27 min total run time | 35°C | 278 nm | 27 min |
| Sweeteners, preservatives, caffeine [56] | Kromasil C18 (5 µm, 150 mm × 4.6 mm) | Acetonitrile/Phosphate buffer (12.5 mM, pH 3.3) | 0-10 min: 5-50% A; 10-15 min: 50% A; 15-16 min: 50-5% A | 30°C | 200-380 nm (multiple wavelengths) | <9 min |
| Tocopherols, tocotrienols [38] | Luna Omega C18 vs. Kinetex C18 | Binary gradient programs | Not specified | Not specified | 195 nm, 278 nm, and 205 nm | Not specified |
| Lychnopholide [7] | RP C18 | Methanol-water (60:40 v/v) | Isocratic, 0.8 mL/min | Not specified | 265 nm | Not specified |
| Vandetanib [57] | C18 Atlantis | Acetonitrile/0.5% triethylamine (pH 3.0) | Gradient, 1.0 mL/min | Ambient | 341 nm | Not specified |
Experimental Protocols for Method Optimization
Systematic Approach to Method Development
A structured methodology for UFLC-DAD optimization begins with sample preparation tailored to the matrix, followed by systematic evaluation of chromatographic parameters. For biological samples like milk, protein precipitation is often necessary, as demonstrated in the orotic acid method where samples were treated with acetonitrile (1:1, v/v) and centrifuged at 4°C before dilution and injection [54]. For pharmaceutical formulations such as tablets containing metoprolol tartrate, extraction typically involves dissolving the active component in appropriate solvents, with protection from light to prevent degradation [1]. The sample preparation protocol must be optimized to minimize matrix effects while maintaining analyte stability throughout the analysis.
Initial scoping experiments should evaluate columns with different stationary phases and dimensions, followed by mobile phase composition screening. The optimal solvent for oil samples in tocopherol analysis was found to be propan-2-ol, which provided suitable extraction efficiency without requiring saponification [38]. For chromatographic separation, method development should systematically assess organic modifier type (acetonitrile vs. methanol), buffer composition and pH, and gradient profile. The simultaneous determination of four tyrosine kinase inhibitors achieved complete elution of analytes within 2.8-3.8 minutes using a reversed-phase column with methanol (65%, v/v) and 0.1% formic acid aqueous solution (35%, v/v) [57], demonstrating how mobile phase optimization can dramatically reduce analysis time.
Workflow Visualization
The following diagram illustrates the systematic workflow for optimizing UFLC-DAD methods:
Figure 1: Systematic UFLC-DAD Method Optimization Workflow
Performance Evaluation and Validation
Following method development, systematic validation is essential to demonstrate reliability and fitness for purpose. Validation parameters should include specificity, linearity, accuracy, precision, limits of detection and quantification, and robustness [1]. For the orotic acid method, validation included assessment of recoveries (96.7-105.3%), inter-and intra-assay coefficient of variation (below 1.3%), and detection/quantification limits (0.04 ng and 0.12 ng, respectively) [54]. Similarly, the method for sweeteners in beverages demonstrated excellent linearity (R² ≥ 0.9995), satisfactory accuracy (recovery values between 94.1-99.2%), and repeatability (RSD ≤ 2.49%) [56].
System suitability tests verify adequate performance of the HPLC system before analysis, evaluating parameters such as peak retention time, capacity factor (k′), selectivity (α), resolution (R), and peak asymmetry (As) [56]. Acceptance criteria typically include k' ≥ 1, α > 1, R ≥ 1.5, and As between 0.8-1.2 [56]. For the sweetener analysis method, these parameters were evaluated using three replicates of a mixture standard solution at 20 mg/L concentration [56]. Robustness testing examines method resilience to deliberate variations in parameters such as mobile phase pH, temperature, and flow rate, ensuring the method remains reliable under normal operational variations.
Comparative Analysis: UFLC-DAD vs. UV-Spectrophotometry
Performance Metrics Comparison
When comparing UFLC-DAD and UV-spectrophotometric methods, distinct differences emerge across multiple performance parameters that inform their appropriate application. A comprehensive comparison study of metoprolol tartrate (MET) determination demonstrated that while both techniques showed excellent linearity (r² > 0.999), UFLC-DAD provided significantly lower limits of detection (0.014 µg/mL) compared to spectrophotometry (0.060 µg/mL) [28]. This enhanced sensitivity makes UFLC-DAD particularly valuable for trace analysis and compounds with weak chromophores. Similarly, in the analysis of lychnopholide in nanocapsules, HPLC-DAD provided a wider linear range (2-25 µg/mL) compared to UV-spectrophotometry (5-40 µg/mL), along with superior sensitivity for release kinetic studies [7].
Specificity represents perhaps the most significant differentiator between the techniques. UFLC-DAD provides two dimensions of identification - retention time and spectral matching - enabling reliable quantification even in complex matrices [55] [59]. As demonstrated in the absorption-based quality control of Traditional Chinese Medicine Preparations, the combination of chromatographic separation with DAD detection allowed specific quantification of baicalein, wogonin, paeonol and emodin despite the massive numbers of ingredients in the formulation [59]. In contrast, UV-spectrophotometry struggles with specificity in mixtures due to overlapping absorption bands, making accurate quantification challenging without prior separation [1].
Table 2: Comparative Performance Metrics of UFLC-DAD vs. UV-Spectrophotometry
| Parameter | UFLC-DAD | UV-Spectrophotometry | References |
|---|---|---|---|
| Linearity | R² ≥ 0.999 for metoprolol tartrate, lychnopholide | R² > 0.999 for metoprolol tartrate | [7] [28] |
| LOD | 0.014 µg/mL for methotrexate, 0.04 ng for orotic acid | 0.060 µg/mL for methotrexate | [54] [28] |
| Specificity | High (retention time + spectrum) | Limited in mixtures | [55] [59] [1] |
| Analysis Time | Longer (9-27 min) but multi-component | Rapid but single component | [54] [56] |
| Multi-component Analysis | Excellent (simultaneous) | Limited without separation | [59] [56] |
| Equipment Cost | High | Low | [1] |
| Operational Complexity | High | Low | [1] |
| Environmental Impact | Higher solvent consumption | Lower solvent consumption | [1] |
Analytical Applications and Limitations
The choice between UFLC-DAD and spectrophotometry often depends on the specific analytical requirements, sample complexity, and available resources. UFLC-DAD excels in applications requiring precise quantification of multiple analytes in complex matrices, as demonstrated in the simultaneous determination of sweeteners, preservatives, and caffeine in beverages [56], and the analysis of orotic acid in milk with interfering endogenous compounds [54]. The technique's ability to provide spectral confirmation of peak identity makes it invaluable for method development and troubleshooting, where unknown peaks may appear in chromatograms [55].
UV-spectrophotometry remains a valuable technique for routine quality control of single-component pharmaceuticals, raw material identification, and applications where high throughput and operational simplicity are prioritized over specificity [1]. The MET study concluded that quality control of tablets could be effectively monitored using UV spectrophotometry rather than UFLC, offering a substantially more cost-effective and environmentally friendly alternative [1]. However, this approach is only viable when the formulation matrix doesn't interfere with quantification and when analyzing single components without need for separation.
Essential Research Reagent Solutions
Successful implementation of UFLC-DAD methods requires specific reagents and materials optimized for chromatographic performance. The following table details key research reagents and their functions in method development and application:
Table 3: Essential Research Reagents for UFLC-DAD Method Development
| Reagent/Material | Function/Application | Examples from Literature |
|---|---|---|
| Kinetex C18 Columns | Core separation media for reversed-phase chromatography | 1.7 µm, 150 mm × 2.1 mm used for orotic acid separation [54] |
| Acetonitrile (HPLC grade) | Primary organic modifier in mobile phase | Used in binary gradients with aqueous buffers [54] [56] |
| Methanol (HPLC grade) | Alternative organic modifier | Used in isocratic elution for lychnopholide (60:40 with water) [7] |
| Ammonium acetate buffer | Mobile phase additive for pH control | Used in vandetanib analysis (90:10 with methanol) [57] |
| Phosphate buffer | Aqueous mobile phase component | 12.5 mM, pH 3.3 used in sweetener analysis [56] |
| Formic acid | Mobile phase acidifier | 0.1% aqueous formic acid used in gradient elution [59] |
| Acetonitrile (protein precipitation) | Sample preparation solvent | 1:1 (v/v) with milk samples for deproteinization [54] |
| Propan-2-ol | Alternative solvent for lipid-soluble analytes | Optimal solvent for tocopherol/tocotrienol extraction from oils [38] |
| Trifluoroacetic anhydride | Derivatization reagent | Esterification of hydroxyl groups of tocols for improved separation [38] |
| α-tocopheryl acetate | Internal standard for fat-soluble vitamins | Used in tocopherol/tocotrienol quantification [38] |
UFLC-DAD technology represents a powerful analytical tool when method requirements include separation of complex mixtures, specific identification, and precise quantification of multiple components. Through systematic optimization of critical parameters including mobile phase composition, gradient profile, column selection, temperature, and detection wavelengths, analysts can achieve remarkable improvements in resolution, peak shape, and analysis time. The experimental protocols and comparative data presented provide a roadmap for method development that balances separation efficiency with practical considerations of analysis time and resource utilization.
While UV-spectrophotometry offers advantages in simplicity, cost, and environmental impact [1], its limitations in specificity make it unsuitable for complex matrices or multi-analyte determinations. The choice between these techniques should be guided by the specific analytical problem, with UFLC-DAD providing the necessary separation power and confirmation capabilities for challenging applications, and spectrophotometry serving well for simpler, single-component analyses. As analytical science continues to evolve, the fundamental principles of separation science and detection optimization outlined here will remain relevant for developing reliable, efficient methods in pharmaceutical analysis and quality control.
Sensitivity Enhancement Strategies for Both Techniques
In the field of analytical chemistry, the accurate and sensitive detection of chemical compounds is paramount across various applications, including pharmaceutical development, environmental monitoring, and food safety. Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) and UV-Visible Spectrophotometry represent two cornerstone techniques for compound separation and quantification. UFLC-DAD combines high-resolution chromatographic separation with multi-wavelength detection, enabling the analysis of complex mixtures. UV-Visible Spectrophotometry, while generally simpler and more cost-effective, provides direct quantification of analytes based on their light absorption properties. Within pharmaceutical analysis and drug development, the sensitivity of these methods directly impacts their ability to detect low concentrations of active ingredients, impurities, or degradation products, thereby influencing drug quality, safety, and efficacy. This guide provides a comparative analysis of sensitivity enhancement strategies for both techniques, framed within experimental contexts and supported by quantitative data, to aid researchers in selecting and optimizing appropriate methodologies.
Comparative Sensitivity Performance Data
The following tables summarize key performance metrics for UFLC-DAD and UV-Visible Spectrophotometry, based on validated experimental methods from recent literature.
Table 1: Sensitivity and Linear Range Comparison for Pharmaceutical Compounds
| Analytical Technique | Compound(s) Analyzed | Linear Range | Limit of Detection (LOD) | Limit of Quantification (LOQ) | Citation |
|---|---|---|---|---|---|
| HPLC-DAD | Analgin | 50-400 µg/mL | Not Specified | Not Specified | [27] |
| Caffeine | 25-200 µg/mL | Not Specified | Not Specified | [27] | |
| Ergotamine | 0.5-10 µg/mL | Not Specified | Not Specified | [27] | |
| Spectrophotometry | Analgin | 10-35 µg/mL | Not Specified | Not Specified | [27] |
| (DDRD & RDW) | Caffeine | 2-30 µg/mL | Not Specified | Not Specified | [27] |
| Ergotamine | 10-70 µg/mL | Not Specified | Not Specified | [27] | |
| HPLC-DAD | Lychnopholide | 2-25 µg/mL | Not Specified | Not Specified | [7] |
| UV-Spectrophotometry | Lychnopholide | 5-40 µg/mL | Not Specified | Not Specified | [7] |
| HPLC-DAD | Polyphenols (CA, TF, FL) | Nanogram level | At nanogram per milliliter (ng/mL) level | At nanogram per milliliter (ng/mL) level | [25] |
Table 2: Method Validation Parameters Exemplifying Sensitivity and Reliability
| Validation Parameter | HPLC-DAD for Lychnopholide [7] | UV-Spectrophotometry for Lychnopholide [7] | HPLC-DAD for Polyphenols [25] |
|---|---|---|---|
| Accuracy | 98-101% | 96-100% | 98-102% |
| Precision (RSD) | Intra-day and inter-day RSD values were low | Intra-day and inter-day RSD values were low | Repeatability < 2%; Intermediate Precision < 5% |
| Linearity (R²) | > 0.999 | Not Specified | Satisfactory |
Detailed Experimental Protocols for Sensitivity Enhancement
Protocol 1: Enhanced Sensitivity for a Ternary Drug Mixture using HPLC-DAD and Spectrophotometry
This protocol outlines the simultaneous determination of analgin, caffeine, and ergotamine in a market formulation (Amigraine) for migraine treatment [27].
- 1. Objective: To develop and validate sensitive HPLC-DAD and novel spectrophotometric methods for a ternary mixture.
- 2. HPLC-DAD Method:
- Chromatography: An Inertsil-C8 column was used with a gradient elution of acetonitrile and ammonium format buffer (pH 4.2).
- Detection: Ultraviolet detection for analgin and caffeine was at λ = 280 nm and 254 nm, respectively. A key sensitivity enhancement strategy was the use of fluorometric detection for ergotamine at λexc = 310 nm and λemm = 360 nm, leveraging its native fluorescence for lower detection limits.
- Sensitivity Outcome: The method achieved a wide linear range for ergotamine (0.5-10 µg/mL), demonstrating high sensitivity for this component [27].
- 3. Spectrophotometric Methods (DDRD & RDW):
- Strategy: To overcome spectral overlapping in the mixture, which can reduce sensitivity and accuracy, two advanced mathematical techniques were employed.
- Double Divisor Ratio Spectra Derivative (DDRD): This method was used to determine ergotamine at 355 nm and caffeine at 268 nm via their derivative spectra.
- Ratio Dual Wavelength (RDW): This method utilized amplitude differences for the determination of caffeine and analgin.
- Sensitivity Outcome: These computational approaches enhanced the effective sensitivity and selectivity of the spectrophotometric method without requiring physical separation, allowing quantification of the drugs in their pharmaceutical formulation [27].
Protocol 2: Optimization for Polyphenol Quantification in Complex Plant Extracts using HPLC-DAD
This protocol focuses on enhancing the sensitivity and efficiency of extracting and quantifying polyphenols from Cecropia species leaves [25].
- 1. Objective: To optimize the extraction and HPLC-DAD quantification of chlorogenic acid (CA), total flavonoids (TF), and flavonolignans (FL).
- 2. Sensitivity Enhancement via Extraction Optimization:
- Design of Experiments (DoE): A Fractional Factorial Design (FFD) screened seven factors (e.g., methanol fraction, temperature, extraction time). A subsequent Central Composite Design (CCD) was used for response surface optimization.
- Optimal Conditions: The key factors for maximizing extraction yield (and thus detection sensitivity) were identified as methanol fraction (70-80% v/v for FL, 55-72% for CA) and extraction temperature. This systematic optimization ensured the highest possible amount of analyte was presented for analysis [25].
- 3. HPLC-DAD Analysis:
- Chromatography: Reversed-phase chromatography was employed.
- Quantification Strategy: For compounds without primary standards (e.g., flavonolignans), a secondary standard (vitexin) was used for quantification, a common strategy to enable sensitivity measurements for a wider range of compounds.
- Sensitivity Outcome: The fully validated method demonstrated excellent sensitivity, with LODs and LOQs at the nanogram per milliliter (ng/mL) level, and high accuracy (98-102%) [25].
Signaling Pathways, Workflows, and Logical Diagrams
The following diagrams illustrate the core logical workflows for the described sensitivity enhancement strategies.
Enhancing Spectrophotometric Sensitivity via Mathematical Resolution
Systematic Sensitivity Enhancement in HPLC-DAD Analysis
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Sensitivity-Optimized Analysis
| Item | Function in Analysis | Example from Context |
|---|---|---|
| C8 or C18 Chromatography Columns | Stationary phase for reversed-phase separation of analytes based on hydrophobicity. | Inertsil-C8 column for separating analgin, caffeine, ergotamine [27]. |
| Buffers (e.g., Ammonium Formate) | Control mobile phase pH to improve peak shape and separation reproducibility. | Ammonium format buffer (pH 4.2) used in ternary mixture analysis [27]. |
| HPLC-grade Organic Solvents | Act as the mobile phase for eluting analytes from the column. Acetonitrile and methanol are most common. | Acetonitrile used in gradient elution for ternary mixture and polyphenol analysis [27] [25]. |
| Fluorescence Detector | Provides highly sensitive and selective detection for native fluorescent compounds or those derivatized with fluorescent tags. | Fluorometric detection for ergotamine at λexc=310 nm/λemm=360 nm [27]. |
| Design of Experiments (DoE) Software | Statistically plans and analyzes experiments to efficiently optimize multiple variables for maximum response (e.g., extraction yield). | Used to optimize methanol fraction and temperature for polyphenol extraction [25]. |
| Chemical Standards | High-purity reference materials essential for method development, calibration, and validation. | Chlorogenic acid, rutin, and vitexin used as standards for polyphenol quantification [25]. |
UFLC-DAD and Spectrophotometry are both powerful techniques whose sensitivity can be significantly enhanced through targeted strategies. UFLC-DAD generally provides superior sensitivity and selectivity for complex mixtures, with enhancements achieved through advanced detection modes (like fluorescence) and systematic optimization of extraction and separation parameters. UV-Visible Spectrophotometry, while having a inherently higher detection limit, can effectively analyze mixtures through mathematical resolution techniques (like DDRD and RDW) that circumvent the need for physical separation, offering a cost-effective and "green" alternative [27]. The choice between techniques depends on the specific application, required detection limits, complexity of the sample matrix, and available resources. The continuous development of optimization protocols and data processing algorithms ensures that both techniques remain indispensable tools in the analytical scientist's arsenal.
In the field of analytical chemistry, the reliability of data is paramount. Robustness testing represents a critical validation parameter that measures a method's capacity to remain unaffected by small, deliberate variations in method parameters. It provides an indication of the method's reliability during normal usage and is an essential component of method validation according to guidelines established by the International Conference on Harmonisation (ICH). This comparative guide examines the resilience of two prominent analytical techniques: traditional UV-Vis spectrophotometry and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD).
Within the context of a broader thesis on comparative analysis of spectroscopic methods, this assessment provides experimental data and performance metrics to guide researchers, scientists, and drug development professionals in selecting appropriate methodologies for their specific applications. The fundamental differences between these techniques lie in their separation capabilities and detection mechanisms. While UV-Vis spectrophotometry offers simplicity and rapid analysis of bulk properties, UFLC-DAD provides superior resolution for complex mixtures through combined chromatographic separation and multi-wavelength detection.
Theoretical Foundations and Technical Principles
UV-Vis Spectrophotometry
Ultraviolet-Visible (UV-Vis) spectrophotometry is an analytical technique that measures the absorption of ultraviolet and visible light by molecules in a solution. The fundamental principle operates on Beer-Lambert's law, which states that absorbance is proportional to the concentration of the absorbing species and the path length of the sample. When molecules contain chromophore groups that can absorb specific wavelengths of light, electronic transitions occur, resulting in characteristic absorption spectra [60]. This method is widely applicable for compounds possessing chromophores, including many pharmaceuticals, natural products, and biochemical substances.
Traditional UV-Vis spectrophotometers typically utilize a single wavelength or scanning monochromator system with a cuvette-based sample introduction. The technique provides several advantages including operational simplicity, rapid analysis time, and minimal solvent consumption. However, its primary limitation lies in its lack of separation capability, making it susceptible to interference from coexisting absorbing substances in complex matrices [61] [6].
UFLC-DAD Technology
Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD) represents an advanced analytical platform that combines high-efficiency chromatographic separation with full-spectrum ultraviolet-visible detection. The system employs high-pressure pumping systems and specialized column chemistry to achieve rapid separation of complex mixtures, typically in minutes rather than the longer run times associated with conventional HPLC [40].
The Diode Array Detection component represents a significant advancement over single-wavelength detectors. Unlike conventional detectors that measure absorbance at a fixed wavelength, a DAD simultaneously captures spectral data across a range of wavelengths (typically 190-800 nm) by utilizing an array of photodiodes. This enables the collection of complete UV-Vis spectra for each eluting compound during the chromatographic run, providing both quantitative and qualitative information [25] [62]. The resulting three-dimensional data (time-wavelength-absorbance) facilitates peak purity assessment and method optimization.
Experimental Protocols for Robustness Assessment
Standard Robustness Testing Framework
Robustness testing follows a systematic approach where method parameters are deliberately varied within a realistic operating range, and the impact on method performance is quantitatively assessed. The standard protocol involves:
Parameter Selection: Identify critical method parameters that may vary during routine application, including mobile phase composition, pH, flow rate, column temperature, detection wavelength, and sample preparation variables.
Experimental Design: Implement structured variation of parameters using fractional factorial or central composite designs to efficiently evaluate multiple factors with minimal experiments.
Response Monitoring: Quantify the impact of variations on critical method attributes including retention time, peak area, resolution, tailing factor, and theoretical plates for chromatographic methods, or absorbance maxima and bandwidth for spectrophotometric methods.
Acceptance Criteria: Establish predefined acceptance limits for each response metric based on method requirements, typically requiring less than 2-5% variation for quantitative methods [25].
UFLC-DAD Robustness Assessment Protocol
For UFLC-DAD methods, robustness testing typically evaluates the impact of variations in chromatographic conditions on separation efficiency and detection stability. A representative protocol adapted from the analysis of Jatropha isabellei demonstrates the approach [40]:
Chromatographic Conditions:
- Column temperature variations (±2°C)
- Mobile phase composition (±2-5% organic modifier)
- Flow rate variations (±0.1 mL/min)
- pH of aqueous phase (±0.1 units)
- Detection wavelength variations (±3 nm)
Sample Preparation:
- Extraction time variations (±10%)
- Solvent composition variations (±5%)
- Extraction temperature variations (±5°C)
Assessment Metrics:
- Retention time stability (%RSD)
- Peak area reproducibility (%RSD)
- Resolution between critical peak pairs
- Tailing factor consistency
- Spectral purity match thresholds
UV-Vis Spectrophotometry Robustness Protocol
For UV-Vis methods, robustness testing focuses on parameters affecting absorbance measurement and sample preparation. A protocol adapted from fluoxetine and vildagliptin analysis illustrates the approach [61] [6]:
Instrumental Parameters:
- Wavelength accuracy (±1-2 nm)
- Slit width variations (±20%)
- Stray light threshold testing
- Cell path length verification
- Baseline drift assessment
Sample Preparation Variables:
- Solvent composition variations (±5%)
- Extraction time variations (±10%)
- Temperature variations (±5°C)
- pH variations (±0.2 units for ionizable compounds)
- Filter compatibility testing
Assessment Metrics:
- Absorbance precision (%RSD)
- Calibration curve linearity (R²)
- Limit of detection and quantification
- Recovery percentages
- Method specificity against interferences
Comparative Performance Data
Quantitative Method Performance Metrics
Table 1: Comparative Validation Parameters of Spectrophotometric and UFLC-DAD Methods
| Validation Parameter | UV-Vis Spectrophotometry (Typical Range) | UFLC-DAD (Typical Range) | Key Implications |
|---|---|---|---|
| Linearity (R²) | 0.997-0.999 [61] [6] | >0.999 [40] [28] | Superior quantitative reliability with UFLC-DAD |
| Precision (%RSD) | 0.06-1.26% [61] [6] | <1-2% [40] [25] | Comparable precision between techniques |
| Detection Limit | 0.951 μg/mL (Vildagliptin) [6] | 0.014 μg/mL (Methotrexate) [28] | 10-100x better sensitivity with UFLC-DAD |
| Analysis Time | 1-5 minutes | 10-60 minutes [40] [25] | UV-Vis offers faster single-component analysis |
| Specificity | Low to moderate [24] | High [40] [25] | UFLC-DAD superior for complex mixtures |
| Recovery (%) | 98-101% [6] | 89.5-105.5% [28] | Comparable accuracy when properly validated |
Robustness Testing Outcomes
Table 2: Documented Robustness Responses to Parameter Variations
| Parameter Variation | UV-Vis Spectrophotometry Impact | UFLC-DAD Impact | Regulatory Significance |
|---|---|---|---|
| Wavelength Variation (±3 nm) | Significant absorbance changes (5-15%) [62] | Minimal area variation (<2%) [25] | Critical for UV-Vis; mitigated by DAD spectral collection |
| Mobile Phase Composition (±2%) | Not applicable | Retention time shifts (1-3%) [40] | Method-specific tolerance limits required |
| Flow Rate Variation (±0.1 mL/min) | Not applicable | Retention time shifts (2-4%) [40] | Well-characterized and predictable effect |
| pH Variation (±0.1 units) | Significant for ionizable compounds [6] | Retention and resolution effects [25] | Critical for both techniques with pH-sensitive analytes |
| Temperature Variation (±2°C) | Minimal effect (<1%) | Retention time shifts (1-2%) [40] | UFLC-DAD more temperature-sensitive |
Methodological Workflows and Signaling Pathways
The fundamental differences in operational principles between UV-Vis spectrophotometry and UFLC-DAD can be visualized through their methodological workflows:
Robustness Testing Decision Pathway
Implementing a systematic approach to robustness testing requires careful consideration of methodological characteristics and application requirements:
Essential Research Reagent Solutions
The experimental protocols for robustness assessment require specific reagents and materials to ensure reliable and reproducible results:
Table 3: Essential Research Reagents and Materials for Robustness Testing
| Reagent/Material | Function in Robustness Testing | Technical Specifications | Quality Considerations |
|---|---|---|---|
| HPLC-Grade Solvents | Mobile phase preparation; sample extraction | Low UV absorbance; high purity | HPLC-grade with low particulate matter |
| Buffer Salts | pH control in mobile phase and samples | Analytical grade with specified pH tolerance | Certified reference materials for accuracy |
| Reference Standards | Method calibration and quantification | Certified purity >95% | Pharmacopeial standards when available |
| Chromatographic Columns | Stationary phase for separation | Specified dimensions and particle size | Multiple columns from different lots |
| Volumetric Glassware | Precise solution preparation | Class A tolerance | Certified and periodically calibrated |
| pH Meters | Mobile phase and solution pH adjustment | Accuracy ±0.01 units | Regular calibration with standard buffers |
| Filter Membranes | Sample clarification | Specified pore size and composition | Compatibility testing to avoid adsorption |
Based on the comparative experimental data and robustness assessment protocols presented, several key conclusions emerge for researchers and method development professionals:
Application-Specific Method Selection: UV-Vis spectrophotometry demonstrates adequate robustness for simple analytical applications involving single-component analysis in uncomplicated matrices, offering advantages in speed, cost, and operational simplicity. Conversely, UFLC-DAD provides superior performance for complex mixtures and demanding regulatory applications, with enhanced specificity, sensitivity, and reliability despite more extensive robustness testing requirements.
Robustness Testing Intensity: The scope and intensity of robustness testing should be proportional to method complexity and application criticality. While UV-Vis methods require focused testing on wavelength accuracy and sample preparation variables, UFLC-DAD methods demand comprehensive assessment of chromatographic parameters and detection stability.
Regulatory Compliance Considerations: For pharmaceutical applications requiring strict regulatory compliance, UFLC-DAD methods generally provide the necessary specificity and reliability, with robustness testing protocols aligning with ICH Q2(R1) guidelines. The documented resilience of UFLC-DAD to parameter variations, coupled with its built-in spectral verification capabilities, makes it particularly suitable for methods requiring transfer between laboratories or instruments.
The selection between these analytical approaches ultimately depends on the specific analytical problem, available resources, and data quality requirements. Proper robustness assessment during method development ensures reliable performance throughout the method lifecycle, regardless of the chosen technique.
Ensuring Reliability: Method Validation, Comparative Analysis, and Green Assessment
Analytical method validation is the fundamental process that guarantees the quality, reliability, and consistency of chemical measurements, confirming that an analytical procedure is suitable for its intended purpose. For researchers, scientists, and drug development professionals, understanding validation parameters is critical for ensuring that generated data meets regulatory standards and scientific rigor. Validation provides assurance that methods will consistently produce accurate results when applied to test samples, forming the bedrock of pharmaceutical analysis, food chemistry, and environmental monitoring. Before their introduction into routine use, and whenever method conditions change, analytical procedures require thorough validation to demonstrate their capabilities [29].
This guide objectively compares the performance of two prevalent analytical techniques—classical spectrophotometric methods and advanced Ultra-Fast Liquid Chromatography-Diode Array Detection (UFLC-DAD) systems—across key validation parameters. Spectrophotometry, based on the Beer-Lambert law which states that absorbance is proportional to concentration, represents a well-established, cost-effective approach for quantitative analysis [29]. In contrast, UFLC-DAD provides a high-resolution separation-based technique that combines rapid chromatographic separation with full spectral confirmation, offering enhanced specificity for complex mixtures. By examining experimental data across standardized validation parameters, this analysis provides a framework for selecting the appropriate methodology based on specific application requirements within the drug development pipeline.
Core Validation Parameters Explained
The validation of analytical methods rests upon several interconnected parameters that collectively define method performance and reliability. According to International Conference on Harmonisation (ICH) guidelines, these parameters establish that a method produces results that are consistent, predictable, and accurate for its intended purpose [29] [63].
Linearity refers to the ability of the method to obtain test results that are directly proportional to analyte concentration within a given range. It is demonstrated by preparing and analyzing a series of standard solutions at varying concentrations and establishing a calibration curve through statistical analysis. The correlation coefficient (r) or coefficient of determination (r²) quantifies the strength of this relationship, with values approaching 1.0 indicating excellent linearity [64] [63].
Limit of Detection (LOD) and Limit of Quantification (LOQ) define the sensitivity of a method. The LOD represents the lowest concentration of an analyte that can be reliably detected but not necessarily quantified, while the LOQ is the lowest concentration that can be quantitatively determined with acceptable precision and accuracy. These parameters are crucial for methods intended to detect trace impurities or measure drugs at low concentrations in biological matrices [65].
Precision expresses the closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample under prescribed conditions. It is typically investigated at three levels: repeatability (intra-day precision), intermediate precision (inter-day precision involving different days, analysts, or equipment), and reproducibility (between laboratories). Precision is expressed as relative standard deviation (RSD%) with lower values indicating better reliability [64] [27].
Accuracy reflects the closeness of agreement between the test result and an accepted reference value (true value). It demonstrates that a method is free from systematic error and is typically determined by spiking a known amount of analyte into a sample matrix (recovery study) or comparing results with those from a validated reference method. Recovery percentages close to 100% indicate high accuracy [7] [63].
Table 1: Summary of Key Validation Parameters and Their Definitions
| Parameter | Definition | Common Expression |
|---|---|---|
| Linearity | The ability to obtain results directly proportional to analyte concentration | Correlation coefficient (r), coefficient of determination (r²) |
| LOD | Lowest concentration that can be detected but not necessarily quantified | Signal-to-noise ratio of 3:1 |
| LOQ | Lowest concentration that can be quantified with acceptable accuracy and precision | Signal-to-noise ratio of 10:1 |
| Precision | Closeness of agreement between a series of measurements | Relative Standard Deviation (RSD%) |
| Accuracy | Closeness of agreement to the true or accepted reference value | Percent Recovery (%) |
Comparative Experimental Data: Spectrophotometry vs. UFLC-DAD
Direct comparison of experimental data from published studies reveals distinct performance characteristics between spectrophotometric and UFLC-DAD methods across different applications. The following structured data demonstrates how each technique performs when subjected to identical validation criteria.
Table 2: Comparison of Validation Parameters for Spectrophotometric Methods Across Different Applications
| Application | Linearity Range | Correlation Coefficient (r) | LOD | LOQ | Precision (RSD%) | Accuracy (% Recovery) |
|---|---|---|---|---|---|---|
| Total Polyphenol Content [64] | Not specified | High determination coefficients | Reasonably low | Reasonably low | Not specified | Not specified |
| Lychnopholide in Nanocapsules [7] | 5-40 μg/mL | >0.999 | Not specified | Not specified | Low RSD values (intra-day & inter-day) | 96-100% |
| Chloroaluminum Phthalocyanine [63] | 0.50-3.00 μg/mL | r=0.9992 | 0.09 μg/mL | 0.27 μg/mL | 0.58% to 4.80% | 98.9% to 102.7% |
| Analgin, Caffeine, Ergotamine [27] | 10-70 μg/mL | Not specified | Not specified | Not specified | Not specified | Not specified |
Table 3: Comparison of Validation Parameters for UFLC-DAD Methods Across Different Applications
| Application | Linearity Range | Correlation Coefficient (r) | LOD | LOQ | Precision (RSD%) | Accuracy (% Recovery) |
|---|---|---|---|---|---|---|
| Lychnopholide in Nanocapsules [7] | 2-25 μg/mL | >0.999 | Not specified | Not specified | Low RSD values (intra-day & inter-day) | 98-101% |
| Analgin, Caffeine, Ergotamine [27] | 0.5-10 μg/mL (ergotamine) | Not specified | Not specified | Not specified | Not specified | Not specified |
| Carbonyl Compounds in Soybean Oil [66] | Not specified | Not specified | High sensitivity demonstrated | High sensitivity demonstrated | Good precision | High accuracy |
| Bioactive Flavonoids from Cassia [67] | Not specified | Not specified | Demonstrated | Demonstrated | ≤2.0% intra-day, <4.0% inter-day | Not specified |
Detailed Methodologies and Experimental Protocols
Spectrophotometric Method for Total Polyphenol and Flavonoid Content
The Folin-Ciocalteu method for determining total polyphenol content and the aluminum chloride (AlCl₃) method for total flavonoid content represent well-established spectrophotometric protocols. The validation process for these methods utilized groups of polyphenol standards including phenolic acids (gallic, p-coumaric, caffeic, and chlorogenic acids), flavan-3-ols [(+)-catechin and procyanidins B1 and B2], flavonols (quercetin and quercetin-3-rutinoside), and dihydrochalcones (phloretin and phloretin-2′-glucoside). During method validation, all parameters fell within acceptable ranges with high determination coefficients, reasonably low LODs and LOQs, and high slopes in the calibration curves for both methods. The exception was observed for phloretin and phloretin-2′-glucoside, which exhibited low slopes in the calibration curves specifically for the AlCl₃ method. When applied to real samples, these validated methods successfully determined significant differences in polyphenol content across various wines (Plavac, Graševina, and Vranac) and juices (blueberry, strawberry, and blackcurrant) [64].
UFLC-DAD Method for Carbonyl Compounds in Soybean Oil
The development and validation of a UFLC-DAD-ESI-MS method for determining toxic carbonyl compounds (CCs) in thermally stressed soybean oil involved a sophisticated approach. The methodology centered on derivatization with 2,4-dinitrophenylhydrazine (2,4-DNPH), selected for its simultaneous reaction with aldehydes, ketones and carboxylic acids, fast reaction kinetics at room temperature, and high derivative stability. The critical extraction optimization phase compared solvents including acetonitrile, methanol, tetrahydrofuran, and dichloromethane, with acetonitrile demonstrating superior extraction capacity based on the sum of peak areas (as illustrated in Figure 1 of the original study). The validated method was applied to soybean oil samples continuously heated at 180°C for different time intervals. The UFLC-DAD-ESI-MS analysis successfully identified concerning carbonyl compounds—particularly the toxic compounds acrolein, 4-hydroxy-2-nonenal (HNE), and 4-hydroxy-2-hexenal (HHE)—with demonstrated good selectivity, precision, high sensitivity, and accuracy. This method proved effective for monitoring the formation of harmful degradation products during oil heating [66].
Comparative Study of HPLC-DAD and UV-Spectrophotometry for Lychnopholide
A direct comparison of HPLC-DAD and UV-spectrophotometry methods for quantifying lychnopholide (LYC) in poly-ε-caprolactone nanocapsules provides valuable insights into the relative strengths of each technique. The HPLC-DAD method employed an RP C18 column with isocratic elution using methanol-water (60:40 v/v) mobile phase at 0.8 mL/min flow rate and detection at 265 nm. In parallel, a UV-spectrophotometric method was developed for the same analyte. Both methods were thoroughly validated according to ICH guidelines, demonstrating excellent linearity (r² > 0.999) within their respective ranges: 2-25 μg/mL for HPLC-DAD and 5-40 μg/mL for spectrophotometry. Both methods exhibited satisfactory intra-day and inter-day precision with low RSD values and accuracy ranging from 98-101% for HPLC-DAD and 96-100% for UV-spectrophotometry. While both techniques were suitable for determining drug loading and encapsulation efficiency, only the HPLC-DAD method offered sufficient sensitivity for studying LYC release/dissolution in sink conditions, revealing that 100% of pure LYC dissolved after 24 hours compared to only 60% release from nanocapsules [7].
Diagram 1: Analytical Method Selection Workflow
Essential Research Reagent Solutions
The execution of validated analytical methods depends critically on appropriate selection of reagents and materials. The following table summarizes key research reagent solutions and their functions in both spectrophotometric and chromatographic analyses.
Table 4: Essential Research Reagents and Their Functions in Analytical Method Development
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Folin-Ciocalteu Reagent | Oxidation-reduction indicator for phenolic compounds | Total polyphenol content determination via spectrophotometry [64] |
| Aluminum Chloride (AlCl₃) | Complexing agent for flavonoid detection | Total flavonoid content determination via spectrophotometry [64] |
| 2,4-Dinitrophenylhydrazine (2,4-DNPH) | Derivatization reagent for carbonyl compounds | Analysis of aldehydes and ketones in oxidized oils via UFLC-DAD [66] |
| C18 Reverse-Phase Columns | Stationary phase for compound separation | UFLC separation of flavonoids, carbonyl hydrazones, and pharmaceuticals [66] [67] [39] |
| Methanol and Acetonitrile | HPLC-grade mobile phase components | Solvent system for chromatographic separations [66] [7] [39] |
| Poly-ε-caprolactone | Polymer for nanocarrier formulations | Nanocapsule material for drug delivery studies [7] |
The comparative analysis of spectrophotometric and UFLC-DAD methods reveals a clear performance-simplicity tradeoff that should guide analytical method selection. Spectrophotometry offers adequate performance for simpler analyses with advantages of cost-effectiveness, operational simplicity, and rapid implementation. UFLC-DAD delivers superior capabilities for complex mixtures and trace analysis, providing enhanced specificity, sensitivity, and the ability to resolve multiple components simultaneously.
For drug development professionals, this comparison underscores the importance of aligning method selection with application requirements. Routine quality control of simple formulations may efficiently employ spectrophotometric methods, while development of complex drug delivery systems or analysis of degraded products necessitates the enhanced capabilities of UFLC-DAD. The experimental data presented provides a framework for evidence-based method selection, ensuring that validation parameters meet the intended analytical needs while optimizing resource utilization in pharmaceutical research and development.
Diagram 2: Comparative Strengths and Limitations of Analytical Techniques
The accurate quantification of active pharmaceutical ingredients (APIs) is a cornerstone of pharmaceutical analysis, impacting every stage from formulation development to quality control and pharmacokinetic studies. Metoprolol tartrate, a cardioselective β1-adrenergic receptor blocker widely used in treating hypertension and angina, serves as an excellent model compound for methodological comparisons in analytical science. Within the broader context of comparative analysis of spectrophotometric versus UFLC-DAD methods research, this examination provides a concrete case study contrasting these fundamentally different approaches. The choice between these techniques represents a classic trade-off between simplicity, cost, and analysis speed against specificity, sensitivity, and comprehensive data resolution. This analysis objectively compares the performance of a kinetic spectrophotometric method and liquid chromatographic methods, including UFLC-DAD, for the determination of metoprolol tartrate, providing researchers and drug development professionals with critical experimental data and performance metrics to inform their analytical strategies.
Experimental Protocols and Methodologies
Kinetic Spectrophotometric Method
The kinetic spectrophotometric method offers a straightforward, cost-effective approach for quantifying metoprolol tartrate, particularly suited for routine quality control in pharmaceutical formulations. The protocol is based on the oxidative coupling reaction of metoprolol with alkaline potassium permanganate [68] [69].
Detailed Experimental Protocol:
- Reagent Preparation: A 0.015 M potassium permanganate solution is prepared freshly in doubly distilled water. A 0.60 M sodium hydroxide solution is similarly prepared. The standard test solution of metoprolol tartrate (0.01%) is made in doubly distilled water [69].
- Sample Preparation: Aliquots of 0.1-0.6 ml of the 0.01% metoprolol tartrate standard solution are pipetted into a series of 10 ml volumetric flasks. To each flask, 2.0 ml of 0.60 M NaOH is added, followed by 2.0 ml of 0.015 M potassium permanganate. The solutions are then diluted to volume with doubly distilled water maintained at 25±1°C [69].
- Analysis: The reaction is monitored spectrophotometrically by measuring the change in absorbance at 610 nm as a function of time. Two calibration approaches are utilized: the initial rate method and the fixed-time method (at 15.0 minutes) [68].
- Calibration: The calibration graphs are linear in the concentration range of 1.46 × 10^(-6) to 8.76 × 10^(-6) M (equivalent to 10.0-60.0 μg per 10 ml). The linear regression equations are reported as log(rate) = 3.634 + 0.999 log C for the initial-rate method and A = 6.300 × 10^(-4) + 6.491 × 10^(-2) C for the fixed-time method [68].
Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD)
While the search results do not contain a specific UFLC-DAD method for metoprolol tartrate, they do provide detailed methodology for closely related LC-DAD and LC-MS/MS techniques from which principles and parameters for UFLC-DAD can be inferred. UFLC represents an advancement of these methods with improved speed and resolution.
Inferred UFLC-DAD Protocol Based on Related Chromatographic Methods:
- Chromatographic Conditions: Separation is typically achieved using a reversed-phase C18 column (e.g., 150 × 2.1 mm ID, 5 μm). The mobile phase often consists of a mixture of methanol or acetonitrile and water, sometimes modified with acids like formic acid (0.2%) to improve peak shape [70]. For simultaneous determination with other compounds like atenolol and phenol red, a gradient elution method is necessary to achieve adequate separation [71].
- Detection: Detection is performed using a diode array detector, allowing for simultaneous monitoring at multiple wavelengths. For metoprolol, detection is typically in the UV range, though specific wavelengths for UFLC-DAD are not provided in the search results. Related HPLC-DAD methods for other compounds use wavelengths such as 265 nm [7].
- Sample Preparation: Biological samples such as plasma require protein precipitation prior to analysis. A simple protein precipitation method using a four-fold volume of methanol has been demonstrated to be effective for metoprolol in plasma samples, simplifying the process compared to more complex liquid-liquid extraction methods [70].
LC-MS/MS Method
Although not directly UFLC-DAD, the LC-MS/MS method represents a highly sensitive and specific complementary technique, particularly valuable for pharmacokinetic studies.
Detailed LC-MS/MS Protocol:
- Chromatographic Conditions: An Ultimate XB-C18 column (150 × 2.1 mm ID, 5 μm) is used with an isocratic mobile phase of methanol-water containing 0.2% formic acid (65:35, v/v) at a flow rate of 0.2 mL/min [70].
- Mass Spectrometric Detection: Analysis is performed using electrospray ionization (ESI) in positive ion mode. Monitoring ions are m/z 268.1→115.6 for metoprolol and m/z 373.1→150.2 for the internal standard (hydroxypioglitazone) [70].
- Sample Preparation: Plasma samples are prepared using protein precipitation with methanol, providing a simple and efficient sample clean-up procedure [70].
Figure 1: Experimental workflow comparison for metoprolol tartrate quantification methods.
Comparative Method Performance Data
Quantitative Performance Metrics
Table 1: Direct comparison of key validation parameters for metoprolol tartrate quantification methods
| Performance Parameter | Kinetic Spectrophotometry [68] | LC-MS/MS [70] | RP-HPLC (with ATN & PR) [71] |
|---|---|---|---|
| Linear Range | 1.46×10^(-6)-8.76×10^(-6) M (10.0-60.0 μg/10 ml) | 3.03-416.35 ng/mL | Not fully specified |
| Limit of Detection | 0.013 μg/mL (initial rate) | Not specified (but LLOQ: 3.03 ng/mL) | Not specified |
| Limit of Quantification | 0.040 μg/mL (initial rate) | 3.03 ng/mL | Not specified |
| Precision (RSD) | Not fully specified (stated as statistically acceptable) | Intra-day: 2.54-10.65%Inter-day: 5.01-8.51% | Not specified |
| Accuracy | 96-100% (recovery studies) | 95.20-99.96% | Not specified |
| Analysis Time | ~15 minutes (fixed-time method) | ~3.0 minutes per sample | Not specified |
Application-Based Performance
Table 2: Application suitability and comparative advantages of each quantification method
| Application Context | Kinetic Spectrophotometry | Liquid Chromatography Methods |
|---|---|---|
| Pharmaceutical Formulations | Excellent for routine quality control of bulk drug and simple formulations [68] | Suitable but potentially over-qualified; best for complex formulations |
| Biological Samples | Not suitable due to lack of specificity | Excellent for plasma, serum, and CSF samples [70] [72] |
| Simultaneous Compound Analysis | Not feasible | Ideal for multi-component analysis (e.g., with atenolol, phenol red) [71] |
| Permeability Studies | Not suitable | Well-suited for intestinal perfusion and permeability studies [71] |
| Pharmacokinetic Studies | Not suitable | Gold standard for concentration monitoring in biological fluids [70] |
| Equipment Requirements | Basic UV-Vis spectrophotometer | Advanced instrumentation (HPLC/UFLC, MS) |
| Method Development Time | Relatively short | Can be extensive, particularly for MS methods |
| Sample Throughput | Moderate | High, especially with automated systems |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key research reagents and materials for metoprolol tartrate quantification
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| Potassium Permanganate | Oxidizing agent for spectrophotometric derivatization | 0.015 M in doubly distilled water, prepared fresh [69] |
| Sodium Hydroxide | Provides alkaline medium for reaction | 0.60 M solution in doubly distilled water [69] |
| Methanol (HPLC Grade) | Mobile phase component; protein precipitation solvent | HPLC grade; used for plasma protein precipitation [70] [71] |
| Formic Acid | Mobile phase modifier to improve peak shape | 0.2% in mobile phase for LC-MS/MS [70] |
| C18 Chromatographic Column | Stationary phase for chromatographic separation | e.g., Ultimate XB-C18 (150 × 2.1 mm ID, 5 μm) [70] |
| Ammonia Solution | Alkaline agent for derivatization in alternative methods | Used in Cu(II) dithiocarbamate complex formation [73] |
| Carbon Disulfide | Derivatization reagent for AAS methods | Reacts with secondary amino group of metoprolol [73] |
| Copper Chloride | Complexing agent for indirect AAS determination | Forms Cu(II) dithiocarbamate complex [73] |
Discussion of Comparative Advantages and Limitations
Sensitivity and Specificity Considerations
The kinetic spectrophotometric method demonstrates adequate sensitivity for pharmaceutical dosage forms with a quantification limit of 0.04 μg/mL, making it suitable for quality control of tablet formulations [68]. However, it lacks the exceptional sensitivity of chromatographic methods, with LC-MS/MS achieving a lower limit of quantification of 3.03 ng/mL in biological matrices [70]. This thousand-fold difference in sensitivity highlights the fundamental limitation of spectrophotometry for pharmacokinetic studies where low plasma concentrations must be measured.
Regarding specificity, the spectrophotometric method relies on a chemical reaction between metoprolol and alkaline permanganate. While selective for the oxidizable functional groups in metoprolol, it may be susceptible to interference from other reducing agents or excipients in complex formulations. In contrast, chromatographic methods provide superior specificity through physical separation prior to detection. The LC-MS/MS method, in particular, offers unmatched specificity through mass transition monitoring (m/z 268.1→115.6), effectively eliminating potential interference from metabolites or matrix components [70].
Application to Different Sample Matrices
The applicability of each method varies significantly based on sample matrix complexity. For simple pharmaceutical formulations such as immediate-release tablets, the kinetic spectrophotometric method provides adequate performance with considerable advantages in simplicity and cost-effectiveness [68]. The method has been successfully applied to commercial formulations including Betaloc, Metapro, and Metolar with statistical comparison showing no significant difference in accuracy and precision compared to reference methods [68].
For biological matrices such as plasma, serum, or cerebrospinal fluid (CSF), chromatographic methods are unequivocally superior. The LC-MS/MS method has been successfully validated for metoprolol determination in beagle dog plasma [70], while similar methods have been applied to simultaneous human serum and CSF samples, revealing important neuropharmacokinetic properties of metoprolol [72]. These studies demonstrated that metoprolol achieves significant penetration into the CSF (approximately 43% of total serum concentrations), findings that would be difficult to obtain with less specific analytical techniques [72].
Analysis Time and Throughput Considerations
The fixed-time spectrophotometric method requires approximately 15 minutes for color development before measurement [68], while the LC-MS/MS method completes the analysis of each sample within 3.0 minutes [70]. However, when considering batch processing, spectrophotometry may offer higher throughput for simple formulations as multiple samples can be prepared and measured simultaneously. For complex samples requiring separation, chromatographic methods provide superior overall efficiency despite longer individual run times.
The development time also differs significantly between methods. The spectrophotometric approach requires minimal method development once established, while chromatographic methods, particularly those involving mass spectrometric detection, demand extensive validation for each new matrix or application. However, the initial development investment in chromatographic methods pays dividends in application flexibility and data quality.
Figure 2: Decision pathway for selecting appropriate metoprolol tartrate quantification methods based on research requirements.
This direct comparative analysis demonstrates that both spectrophotometric and chromatographic methods have distinct roles in the quantification of metoprolol tartrate, with the optimal choice heavily dependent on the specific research or quality control context. The kinetic spectrophotometric method provides a cost-effective, straightforward solution for routine analysis of pharmaceutical formulations, offering adequate sensitivity and precision for quality control applications. In contrast, UFLC-DAD and related chromatographic methods deliver superior specificity, sensitivity, and application flexibility, making them indispensable for complex matrices, pharmacokinetic studies, and situations requiring multi-component analysis.
The data presented in this comparison provides researchers and pharmaceutical scientists with a clear framework for method selection based on their specific needs, balancing analytical performance requirements with practical considerations of cost, time, and equipment availability. As analytical technologies continue to evolve, the fundamental principles highlighted in this case study—specificity versus simplicity, sensitivity versus speed—will remain central to methodological decision-making in pharmaceutical analysis.
In the development and validation of analytical methods, such as in the comparative analysis of spectrophotometric versus UFLC-DAD methods, establishing method equivalence is paramount. Statistical tests provide the objective framework needed to determine whether different methods yield comparable results. The t-test and Analysis of Variance (ANOVA) are two fundamental inferential statistics used for this purpose, each with specific applications in analytical method comparison [74] [75]. These tests help researchers and drug development professionals make data-driven decisions about method selection, validation, and implementation.
The core principle behind both tests is to determine whether observed differences in measurements between methods are statistically significant or merely due to random variation. Proper selection between these tests depends on the experimental design and the number of methods being compared. While both tests share common assumptions—including normal distribution of data, homogeneity of variances, and independence of observations—their application differs significantly based on the number of groups being compared [74].
Fundamental Concepts: t-test and ANOVA
Core Definitions and Applications
- t-test: A statistical test used to determine if there is a significant difference between the means of two groups. It is ideal for direct comparisons between two analytical methods [74]. For example, it would be appropriate for comparing a novel spectrophotometric method against an established UFLC-DAD method for a single analyte.
- ANOVA (Analysis of Variance): A statistical method used to compare means across three or more groups. It determines whether at least one group mean differs significantly from the others [74]. This would be used when comparing multiple modifications of a method, different laboratories using the same method, or multiple experimental conditions simultaneously.
Key Differences Between t-test and ANOVA
Table 1: Comparison between t-test and ANOVA
| Feature | t-test | ANOVA |
|---|---|---|
| Purpose | Compares means between two groups | Compares means across three or more groups |
| Number of Groups | Two groups only | Three or more groups |
| Test Statistic | t-statistic | F-statistic |
| Post-hoc Testing | Not required | Required to identify which specific groups differ |
| Experimental Design | Simple comparisons | Complex experimental designs |
| Example Application | Comparing two analytical methods for one analyte | Comparing multiple analytical methods or multiple laboratories |
Statistical Workflow and Decision Pathways
The following diagram illustrates the statistical decision process for method comparison studies, from experimental design to interpretation:
Experimental Design and Protocol Considerations
Sample Preparation and Measurement
Proper experimental design is crucial for valid statistical comparison of analytical methods. Studies typically involve multiple replicate measurements of the same samples using each method being compared. For example, in a comparison of spectrophotometric and UFLC-DAD methods for vitamin C quantification, researchers prepared a standard vitamin C solution and applied both methods to measure concentration repeatedly [15]. Similarly, in comparing methods for riociguat quantification in tablets, multiple tablets were analyzed using each method to account for variability [76].
The number of replicates should be sufficient to provide statistical power, typically with a minimum of 3-6 replicates per method, though more may be required for methods with high variability. Samples should represent the entire concentration range of interest, and measurements should be randomized to avoid systematic bias.
Data Collection and Assumption Checking
Before performing statistical tests, researchers must verify that data meet the necessary assumptions:
- Normality: The dependent variable (e.g., concentration measurement) should be approximately normally distributed within each method group. This can be assessed using normality tests (e.g., Shapiro-Wilk) or graphical methods (Q-Q plots).
- Homogeneity of variances: The variability of measurements should be similar across methods. Tests like Levene's test can evaluate this assumption.
- Independence: Measurements should be independent of each other, meaning the result of one measurement does not influence another.
If assumptions are violated, data transformations or non-parametric alternatives may be necessary.
Case Studies in Analytical Method Comparison
Vitamin C Quantification in Fruit Juices
A 2017 study directly compared titrimetric and spectrophotometric methods for quantifying vitamin C in industrialized orange and pineapple juices [15]. The researchers evaluated both methods using a standard vitamin C solution to establish linearity, precision, accuracy, and limits of detection and quantification.
Table 2: Comparison of analytical methods for vitamin C quantification
| Parameter | Titrimetric Method | Spectrophotometric Method |
|---|---|---|
| Interference | Affected by juice color (error >15%) | Minimal color interference |
| Sensitivity (LOD) | Not specified | 0.002 mg/mL |
| Sensitivity (LOQ) | Not specified | 0.010 mg/mL |
| Accuracy Error | >15% | <5% |
| Precision | Precise with standard solution | Precise with standard solution |
The spectrophotometric method demonstrated superior performance with greater sensitivity and accuracy, while the titrimetric method was significantly affected by sample color. Statistical comparison would be appropriate using a paired t-test to directly compare the concentration values obtained by each method for the same samples.
Simultaneous Drug Quantification in Formulations
A 2023 study developed and validated both HPLC-DAD and spectrophotometric methods for simultaneous determination of a ternary mixture of analgin, caffeine, and ergotamine in pharmaceutical formulations [27]. The researchers applied the methods over concentration ranges of 50-400, 25-200, and 0.5-10 μg/mL for HPLC-DAD and 10-35, 2-30, and 10-70 μg/mL for spectrophotometric methods for analgin, caffeine, and ergotamine, respectively.
The results demonstrated that both methods successfully determined the cited drugs in their pharmaceutical formulation. Statistical comparison with reported methods showed no significant difference, establishing their equivalence for this application. In such multi-analyte studies, ANOVA would be appropriate for comparing results across methods for each analyte, followed by post-hoc tests if significant differences are detected.
Riociguat Estimation in Tablets
A 2020 comparison of spectrophotometric and spectrodensitometric techniques for estimating riociguat in tablets developed and validated three analytical methods [76]. The limits of detection and quantification were found to be 0.240 and 0.728 μg/mL for spectrofluorimetric, 0.193 and 0.586 μg/mL for UV-spectrophotometric, and 3.35 and 10.15 ng/band for HPTLC method, respectively.
Statistical comparison was performed using ANOVA and paired t-test, showing no significant difference between the three methods. The successful application of these methods for content uniformity testing demonstrates how statistical comparison validates method equivalence for pharmaceutical quality control.
Multiple Comparison Procedures
Post-hoc Testing Following ANOVA
When ANOVA identifies significant differences between multiple methods, post-hoc tests determine which specific methods differ. The choice of post-hoc test depends on the research question and error tolerance:
- Tukey's Test: Preferred when comparing all experimental groups against all control groups, especially with unequal group sizes. It controls the familywise error rate and is conservative, reducing Type I errors (false positives) [77].
- Newman-Keuls Test: More powerful than Tukey's but with higher Type I error risk. Appropriate when detecting small differences is crucial, such as in preliminary method development [77].
- Bonferroni Correction: Adjusts significance level based on the number of comparisons. Ideal when testing a predetermined, limited set of comparisons [77].
- Scheffé Method: Tests all possible contrasts (simple and complex). Most appropriate when all comparisons are planned and sample sizes are equal [77].
Application in Analytical Method Comparison
The following diagram illustrates the multiple comparison decision process after obtaining a significant ANOVA result:
Essential Research Reagent Solutions
Successful method comparison studies require specific reagents and materials tailored to the analytical techniques being evaluated. The following table details key research reagent solutions used in the cited comparative studies:
Table 3: Essential research reagents for analytical method comparison studies
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| 2,6-Dichlorophenolindophenol | Titrimetric reagent for vitamin C quantification | Reduction-based measurement of ascorbic acid in titrimetric method [15] |
| Cuproine Complex | Spectrophotometric reagent for vitamin C | Detection through cupric ion reduction in spectrophotometric method [15] |
| C18 Reverse Phase Columns | Stationary phase for chromatographic separation | HPLC separation of ternary mixture of analgin, caffeine, and ergotamine [27] |
| 2,4-Dinitrophenylhydrazine | Derivatization agent for carbonyl compounds | HPLC-DAD detection of malondialdehyde after derivatization [26] |
| Thiobarbituric Acid | Colorimetric reagent for malondialdehyde | TBA-test for malondialdehyde detection using spectrophotometry [26] |
| Mobile Phase Buffers | Liquid phase for chromatographic separation | Gradient elution with ammonium format buffer (pH 4.2) for HPLC analysis [27] |
Statistical comparison using t-tests and ANOVA provides the scientific rigor necessary for establishing equivalence between analytical methods in pharmaceutical research and development. The case studies presented demonstrate successful application of these statistical approaches across various method comparison scenarios, from simple two-method comparisons to complex multi-analyte determinations.
When designing method comparison studies, researchers should consider the number of methods being compared, the required statistical power, and the balance between Type I and Type II error risks. Proper application of these statistical tools, coupled with appropriate experimental design and validation parameters, ensures robust conclusions regarding method equivalence, ultimately supporting quality control in drug development and manufacturing.
The pharmaceutical industry is increasingly mandated to evaluate and minimize the environmental impact of its processes and products, driven by the axiom that "we cannot manage what we cannot measure" [78]. Green Analytical Chemistry (GAC) has emerged as a fundamental discipline since the early 2000s, focusing on reducing the hazardous effects of analytical chemicals on both environmental and analyst safety [79]. Within this framework, specialized assessment tools have been developed to quantitatively evaluate the environmental friendliness, or "greenness," of analytical methods. These tools provide structured approaches to compare methodologies, guide development of more sustainable practices, and validate claims about ecological impact [80].
The evolution of assessment frameworks has progressed to include White Analytical Chemistry (WAC), which expands upon GAC principles by incorporating analytical quality and practical effectiveness alongside environmental metrics [79] [81]. This holistic approach recognizes that a truly excellent method must balance analytical performance (red principles), environmental impact (green principles), and practical efficiency (blue principles) [81]. Among the various assessment tools available, the Analytical GREEnness (AGREE) metric and Green Analytical Procedure Index (GAPI) have gained significant traction for their comprehensive and user-friendly approaches to evaluating method sustainability [82].
Available Metrics and Their Applications
Several assessment tools have been introduced for evaluating the greenness of analytical methods, each with unique advantages, disadvantages, and assessment protocols [82]. The four primary tools include:
- National Environmental Methods Index (NEMI): A simple tool that provides a basic pictogram but offers limited differentiation between methods [82].
- Eco-Scale Assessment (ESA): Provides a numerical score out of 100, with higher scores indicating greener methods [82].
- Green Analytical Procedure Index (GAPI): Offers a detailed pictogram that evaluates multiple aspects of the analytical process [82] [81].
- Analytical GREEnness (AGREE) metric: Provides a comprehensive 0-1 score and incorporates the 12 principles of green analytical chemistry [82].
A comparative study of these tools revealed that NEMI was the least effective in providing detailed information, as multiple methods often received identical pictograms. In contrast, AGREE, GAPI, and ESA provided more reliable and differentiated assessments, with AGREE particularly noted for its automation capabilities and ability to highlight the weakest points in analytical techniques [82].
AGREE and GAPI: A Detailed Comparison
AGREE (Analytical GREEnness metric) is a recently developed tool that evaluates methods based on all 12 principles of Green Analytical Chemistry. It provides an overall score between 0 and 1, with scores closer to 1 indicating greener methods [79] [82]. The tool generates a circular pictogram divided into 12 sections, each corresponding to one principle, with colors ranging from green (ideal) to red (undesirable). Key advantages of AGREE include its simplicity, automation capabilities, and comprehensive coverage of GAC principles [82].
GAPI (Green Analytical Procedure Index) employs a pictogram consisting of five pentagrams corresponding to different stages of the analytical process: sampling, sample preparation, reagents, instrumentation, and method type [81]. Each pentagram contains several subcategories evaluated using a three-color system: green for low environmental impact, yellow for medium impact, and red for high impact [81]. While more complex than NEMI and ESA, GAPI provides a comprehensive visual representation of a method's environmental performance across its entire workflow [82].
Table 1: Comparison of AGREE and GAPI Greenness Assessment Tools
| Feature | AGREE | GAPI |
|---|---|---|
| Assessment Basis | 12 principles of GAC | 5 stages of analytical process |
| Scoring System | 0-1 scale (1 = ideal) | Three-color system (green, yellow, red) |
| Output Format | Circular pictogram with sections | Series of pentagrams with subcategories |
| Key Strength | Automation, comprehensive GAC coverage | Detailed process-stage evaluation |
| Primary Limitation | Less detailed at individual process stages | More complex pictogram |
| Whiteness Assessment | Can be integrated with RGB model | Not inherently designed for whiteness |
Greenness Metrics in Analytical Method Comparison
Application to Spectrophotometric vs. Chromatographic Methods
Greenness metrics provide objective criteria for comparing different analytical techniques, such as spectrophotometric methods versus Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD). A comparative study of 16 chromatographic methods for hyoscine N-butyl bromide demonstrated how greenness assessment tools can objectively differentiate between methodologies [82]. The study found that AGREE, GAPI, and ESA provided reliable numerical assessments that effectively highlighted differences in environmental impact between methods [82].
In practical applications, spectrophotometric methods often demonstrate advantages in greenness due to their typically simpler instrumentation, reduced solvent consumption, and lower energy requirements. For instance, a study on vitamin C quantification found that a spectrophotometric method based on reduction of cupric ions offered superior sensitivity and accuracy compared to traditional titrimetric approaches, while also being less affected by sample color interference [15]. Similarly, a comparison of HPLC-DAD and UV-spectrophotometry for determination of lychnopholide in nanocapsules found both methods suitable for pharmaceutical analysis, with the spectrophotometric method demonstrating adequate linearity and accuracy for quality control applications [7].
Case Studies in Pharmaceutical Analysis
Recent pharmaceutical applications demonstrate the practical implementation of greenness metrics. A sustainable voltammetric method for difluprednate estimation in the presence of its alkaline degradation product was comprehensively evaluated using AGREE and GAPI [79]. The method utilized ethanol as a greener alternative to more hazardous solvents and employed a glassy carbon electrode with gold nanoparticles, demonstrating how electrochemical methods can offer environmentally friendly alternatives to traditional chromatographic techniques [79].
Another study developed a validated RP-HPLC method for cinnarizine estimation that explicitly compared its greenness and whiteness profiles against three reported methods using AES, AGREE, GAPI, RGB, and BAGI metrics [81]. The optimized method utilized methanol and 0.1% v/v orthophosphoric acid in a 95:05 ratio as the mobile phase, specifically selected for its improved environmental profile compared to acetonitrile-based systems [81]. This approach highlights how greenness metrics can guide method development toward more sustainable outcomes without compromising analytical performance.
Table 2: Greenness Comparison of Analytical Methods from Case Studies
| Method Application | Technique | AGREE Score | GAPI Assessment | Key Green Features |
|---|---|---|---|---|
| Difluprednate Estimation [79] | Voltammetry | 0.81 | Predominantly green | Ethanol solvent, minimal reagent use |
| Cinnarizine Analysis [81] | RP-HPLC | 0.78 | Mixed green/yellow | Methanol-based mobile phase, low flow rate |
| Hyoscine N-butyl Bromide [82] | Various HPLC | 0.42-0.76 | Variable | Method-dependent, showing improvement potential |
| Lychnopholide Determination [7] | HPLC-DAD vs. UV-Vis | Not reported | Not reported | Simpler UV-Vis method potentially greener |
AGREE and GAPI Assessment Methodologies
AGREE Assessment Protocol
The AGREE metric evaluates analytical methods against the 12 principles of green analytical chemistry, which include aspects such as miniaturization, sample preparation, solvent and reagent choice, energy consumption, waste generation, and operator safety [79]. The assessment is typically performed using dedicated software that calculates scores based on input parameters related to each principle. The output includes an overall score (0-1) and a circular pictogram where each section's color corresponds to the performance for that principle.
The AGREE calculator assigns different weights to each principle based on its relative importance to greenness, providing a balanced evaluation that highlights both strengths and areas for improvement. This automated approach reduces subjectivity and enables direct comparison between different methods [82]. The clear visualization helps researchers quickly identify which aspects of their method contribute most significantly to environmental impact and guides optimization efforts toward the most beneficial modifications.
GAPI Assessment Protocol
GAPI assessment involves a more detailed, step-by-step evaluation of the analytical process across five key areas [81]:
- Sample collection and preservation
- Sample preparation and transportation
- Reagent and solvent use
- Instrumentation and device operation
- Method type and final waste treatment
Each area contains multiple subcategories that are individually assessed using the three-color system. For example, in the reagent and solvent section, evaluations consider toxicity, volume, recyclability, and safety aspects. The instrumentation section evaluates energy consumption, occupational hazards, and throughput [81]. The complete assessment generates a visual profile of the method's environmental impact that clearly shows which stages contribute most significantly to its overall footprint.
The following diagram illustrates the workflow for conducting greenness assessments using AGREE and GAPI:
The Scientist's Toolkit: Essential Research Reagents and Materials
The implementation of green analytical methods requires careful selection of reagents and materials to minimize environmental impact while maintaining analytical performance. The following table outlines key research reagent solutions and their functions in sustainable method development:
Table 3: Essential Research Reagent Solutions for Green Analytical Methods
| Reagent/Material | Function | Green Considerations | Example Applications |
|---|---|---|---|
| Methanol | HPLC mobile phase component | Less toxic alternative to acetonitrile; biodegradable | RP-HPLC analysis of cinnarizine [81] |
| Ethanol | Solvent for extraction and analysis | Renewable source; lower toxicity than methanol | Voltammetric analysis of difluprednate [79] |
| Tetrabutylammonium tetrafluoroborate | Supporting electrolyte in voltammetry | Enables non-aqueous systems; recyclable | Electrochemical methods [79] |
| Gold Nanoparticles | Electrode modifier | Enhance sensitivity; reduce reagent consumption | Modified glassy carbon electrode [79] |
| Orthophosphoric Acid | Mobile phase modifier | Replace more hazardous ion-pairing reagents | HPLC method development [81] |
| Biodegradable Polymers | Drug delivery matrices | Reduce environmental persistence | Microparticle drug carriers [28] |
The adoption of greenness metrics represents a paradigm shift in analytical chemistry, moving beyond traditional performance criteria to include environmental impact as a key validation parameter. AGREE and GAPI have emerged as sophisticated tools that provide comprehensive, standardized assessments of method greenness, enabling objective comparison between techniques such as spectrophotometry and UFLC-DAD. The case studies examined demonstrate that these metrics are not merely theoretical exercises but practical tools that drive meaningful improvements in sustainable method development.
As the pharmaceutical industry faces increasing pressure to minimize its environmental footprint, the integration of greenness assessment into routine method validation represents a critical step forward. The expanding framework of White Analytical Chemistry, which balances analytical, practical, and environmental factors, promises to further refine how we define excellence in analytical science. By adopting these tools and principles, researchers and drug development professionals can contribute to a more sustainable future while maintaining the high-quality standards required for pharmaceutical analysis.
The selection of an appropriate analytical method is a critical decision in pharmaceutical research and drug development. This guide provides an objective comparison between traditional spectrophotometric methods and Ultra-Fast Liquid Chromatography with Diode Array Detection (UFLC-DAD), two prominent techniques with distinct performance characteristics and operational cost structures. By examining direct experimental comparisons and validated applications across diverse analytical scenarios, this analysis aims to equip scientists with the data necessary to make informed, cost-effective decisions that align with their specific research objectives and operational constraints.
Analytical Performance Comparison
The fundamental differences between spectrophotometry and UFLC-DAD translate into distinct performance profiles that directly impact their suitability for various applications. The table below summarizes key performance parameters based on experimental data from comparative studies:
Table 1: Direct Performance Comparison of Spectrophotometric and UFLC-DAD Methods
| Performance Parameter | Spectrophotometric Methods | UFLC-DAD Methods | Comparative Experimental Evidence |
|---|---|---|---|
| Specificity | Lower; susceptible to matrix interference in complex samples [26] [15] | Higher; separation step reduces interference [26] [40] | MDA detection in milk: Spectrophotometry required careful clean-up; UFLC-DAD provided reliable results with different clean-up procedures [26]. |
| Sensitivity | Good for standard assays; LOD for Vitamin C: 0.002 mg/mL [15] | Generally superior; suitable for trace analysis in plasma [40] [21] | Letrozole analysis: UV-spectrophotometry linear range 0.25–20.0 μg/mL [83]. MK-4 in plasma via UFLC-DAD: linear range 0.374–6 μg/mL [21]. |
| Linear Range | Wide linear ranges demonstrated (e.g., 0.25–20.0 μg/mL for Letrozole) [83] | Well-defined linear ranges, though possibly narrower [21] | Ternary drug mixture: HPLC-DAD covered 50-400 μg/mL (Analgin), while spectrophotometry covered 10-35 μg/mL [27]. |
| Precision & Reproducibility | High reproducibility and precision reported [26] [83] | High precision and validated reproducibility [40] [21] | MDA detection: Both methods showed "high reproducibility and precision" [26]. Letrozole analysis: RSD <1% for multiple spectrophotometric methods [83]. |
| Multi-Component Analysis | Limited; requires derivative techniques or chemical separation [27] [83] | Excellent; inherent capability for simultaneous multi-analyte detection [27] [21] | Ternary Mixture (Analgin, Caffeine, Ergotamine): Spectrophotometry required complex DDRD and RDW methods; HPLC-DAD achieved direct separation and detection [27]. |
Detailed Experimental Protocols
To illustrate the practical implementation of each technique, the following section outlines specific methodologies from published research.
Protocol 1: Third-Derivative Spectrophotometry for Malondialdehyde (MDA) Detection
This protocol, adapted from a study comparing methods for detecting lipid oxidation markers in infant formula, highlights the steps to enhance specificity in complex matrices [26].
- Sample Preparation: Milk samples (infant formula, human, cow milk) are freeze-dried and stored frozen until analysis. The thiobarbituric acid (TBA) test is employed, where MDA reacts with TBA to form a colored adduct [26].
- Derivatization and Measurement: One milliliter of the sample extract is mixed with 1.5 mL of 5% aqueous trichloroacetic acid (TCA) and 1.5 mL of 0.8% aqueous TBA. The resulting solution is measured using a UV spectrophotometer [26].
- Data Analysis: The absorbance spectrum is processed to obtain the third-derivative spectrum. This mathematical transformation helps resolve the MDA-TBA complex peak from interfering absorbing substances, improving the method's specificity without physical separation [26].
- Key Application Note: The study found that applying the correct clean-up procedure was crucial for achieving quantitative recovery yields and results that showed good agreement with the HPLC-DAD method [26].
Protocol 2: UFLC-DAD for Quantification of Jatrophone in Plant Material
This protocol, used for quantifying a bioactive diterpene in Jatropha isabellei, exemplifies the application of UFLC-DAD in natural product analysis [40].
- Sample Preparation and Extraction: The dried, powdered underground parts of the plant are macerated with 70% ethanol. The ethanol extract is then partitioned with dichloromethane to obtain a fraction enriched with non-polar compounds like jatrophone [40].
- Chromatographic Separation:
- Column: C18 column.
- Mobile Phase: Specific gradient or isocratic elution with solvents like acetonitrile and water or buffer.
- Flow Rate and Run Time: Typical flow rate of 1 mL/min with a run time optimized for separation (e.g., 10-15 minutes).
- Detection: A DAD detector scans over a wavelength range (e.g., 190-600 nm). Jatrophone is quantified at its specific absorption wavelength (e.g., 269 nm as used for MK-4) [21]. The DAD allows for peak purity assessment by comparing spectra across the peak.
- Validation: The method is validated for linearity, precision (intra-day and inter-day %RSD <10%), and accuracy (% recovery within acceptable limits) [40].
Operational Cost Analysis
Beyond analytical performance, the financial and operational implications of each method are critical for laboratory planning and sustainability.
Table 2: Comprehensive Cost and Operational Factor Analysis
| Cost Factor | Spectrophotometry | UFLC-DAD |
|---|---|---|
| Instrument Capital Cost | Low | High |
| Maintenance & Consumables | Low (cuvettes, standard chemicals) | High (HPLC-grade solvents, columns, costly replacement parts) |
| Operational Skill Level | Lower; standard technical training often sufficient | Higher; requires specialized training in chromatography and data software |
| Analysis Speed & Throughput | Very High (rapid measurements, no separation) | Moderate to Slow (time per sample includes column separation) |
| Sample Preparation Cost | Can be high if extensive clean-up is needed to achieve specificity [26] | Can be complex, but the separation step often reduces need for exhaustive pre-cleaning |
| Solvent Consumption & Waste Disposal | Low | High (continuous solvent flow generates waste) |
| Data Versatility | Limited to spectral and concentration data | Rich data including retention time, spectral confirmation, and peak purity |
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and materials central to the experiments discussed in this guide, along with their critical functions.
Table 3: Key Reagents and Materials for Spectrophotometric and UFLC-DAD Analyses
| Reagent/Material | Function in Analysis | Example Application |
|---|---|---|
| Thiobarbituric Acid (TBA) | Reacts with malondialdehyde (MDA) to form a pink chromophore for spectrophotometric detection. | Marker for lipid oxidation in milk and infant formula [26]. |
| 2,4-Dinitrophenylhydrazine (DNPH) | Derivatizing agent for carbonyl compounds like MDA, allowing for subsequent HPLC analysis. | Alternative derivatization method for HPLC-DAD detection of MDA [26]. |
| C18 Chromatography Column | Stationary phase for reverse-phase chromatography; separates compounds based on hydrophobicity. | Used in UFLC-DAD for separating jatrophone [40], menaquinone-4 [21], and drug mixtures [27]. |
| HPLC-Grade Solvents (Acetonitrile, Methanol) | High-purity mobile phase components to ensure low UV background and reproducible chromatography. | Essential for all UFLC-DAD applications to prevent system damage and baseline noise [27] [40] [21]. |
| Dichloromethane | Medium-polarity organic solvent for liquid-liquid extraction of non-polar compounds. | Used to obtain a terpene-enriched fraction from Jatropha isabellei [40]. |
The choice between spectrophotometry and UFLC-DAD is not a matter of superiority, but of context. The following diagram illustrates the recommended decision-making workflow to guide method selection based on project-specific goals and constraints.
In conclusion, spectrophotometric methods offer a compelling solution for high-throughput, cost-sensitive analyses where target analytes are well-defined and matrix effects are manageable. In contrast, UFLC-DAD provides an indispensable tool for method development, regulatory submission, and complex analyses requiring high specificity and reliable quantification of multiple components. A strategic approach, often employing spectrophotometry for rapid screening and UFLC-DAD for confirmatory analysis, represents the most effective path to balancing analytical performance with operational expense in modern drug development and research.
Conclusion
The choice between spectrophotometry and UFLC-DAD is not a matter of superiority but of context. Spectrophotometry offers a cost-effective, simple, and environmentally friendlier solution for routine analysis of single components where high specificity is not paramount. In contrast, UFLC-DAD provides the necessary selectivity, sensitivity, and robustness for complex matrices, stability-indicating methods, and multi-analyte determination. The validation framework and greenness metrics provide a scientifically sound and sustainable approach to method selection. Future directions in pharmaceutical analysis will likely involve further miniaturization, automation, and the development of even greener chromatographic solvents, guided by the comprehensive comparative principles outlined in this article.
References
- 1. Comparative analysis and validation of analytical techniques for quantification active component in pharmaceuticals: Green approach - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0019452224000530]
- 2. Study of UV And HPLC Comparative for Estimation of Drug Methods [https://www.ijsrtjournal.com/article/Comparative+Study+of+UV+And+HPLC+Methods+for+Estimation+of+Drug]
- 3. in Spectrophotometric Analysis: Principles... Methods Pharmaceutical [https://juniperpublishers.com/ijesnr/IJESNR.MS.ID.556391.php]
- 4. Development and Validation of a UPLC Method for Rapid and Simultaneous Analysis of Proton Pump Inhibitors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4309820/]
- 5. Cutting-edge assays for mirabegron and tadalafil combo therapy for... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-025-01497-z]
- 6. Analytical Method Development and Validation of UV-visible Spectrophotometric Method for the Estimation of Vildagliptin in Gastric Medium - PubMed [https://pubmed.ncbi.nlm.nih.gov/32746479/]
- 7. HPLC-DAD and UV-spectrophotometry for the determination of lychnopholide in nanocapsule dosage form: validation and application to release kinetic study - PubMed [https://pubmed.ncbi.nlm.nih.gov/23247030/]
- 8. A UPLC/DAD method for simultaneous determination of empagliflozin and three related substances in spiked human plasma - PubMed [https://pubmed.ncbi.nlm.nih.gov/31384830/]
- 9. Method validation for determination of amino acids in feed by UPLC | Accreditation and Quality Assurance [https://link.springer.com/article/10.1007/s00769-017-1281-9]
- 10. : Uses, Spectrophotometry & Applications | Danaher Life... Advantages [https://lifesciences.danaher.com/us/en/library/spectrophotometry.html]
- 11. Shining a Light on Spectrophotometry : Principles , Practices and... [https://sperdirect.com/blogs/news/shining-a-light-on-spectrophotometry-principles-practices-and-real-world-applications]
- 12. What is the Spectrophotometer ? Working, Uses, and Basics Principle [https://www.prestogroup.com/blog/what-is-the-spectrophotometer-principle-working-uses-and-basics/]
- 13. ACTTR Inc. - What Are The Advantages And Disadvantages of An UV-Vis Spectrophotometer? [https://www.acttr.com/en/en-faq/en-faq-uv-vis/134-en-faq-uv-vis-advantage-disadvantage.html]
- 14. Analytical Method Development and Validation of Filgrastim by UV and... [https://www.scirp.org/journal/paperinformation?paperid=112677]
- 15. Comparison between titrimetric and spectrophotometric methods for quantification of vitamin C - PubMed [https://pubmed.ncbi.nlm.nih.gov/28159298/]
- 16. Ultrafast Liquid Chromatography - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/ultrafast-liquid-chromatography]
- 17. Prominence Ultra Fast Liquid Chromatograph (UFLC XR) - Shimadzu Europa Analytical Instruments - PDF Catalogs | Technical Documentation [https://pdf.medicalexpo.com/pdf/shimadzu-europa-analytical-instruments/prominence-ultra-fast-liquid-chromatograph-uflc-xr/77760-82775.html]
- 18. Selective Detection with UV Spectroscopy in Gas Chromatography | LCGC International [https://www.chromatographyonline.com/view/selective-detection-with-uv-spectroscopy-in-gas-chromatography]
- 19. The Stability-Indicating Ultra High- Performance ... Liquid [https://www.mdpi.com/1424-8247/18/1/124]
- 20. The Stability-Indicating Ultra High- Performance ... Liquid [https://pubmed.ncbi.nlm.nih.gov/39861185/]
- 21. (PDF) An UFLC - DAD Method for the Quantification of... [https://www.academia.edu/112516405/An_UFLC_DAD_Method_for_the_Quantification_of_Menaquinone_4_in_Spiked_Rabbit_Plasma]
- 22. High-Performance Liquid Chromatography Market to Reach USD 7.74 Billion by 2032, Driven by Rising Demand for Precise Analytical Testing and Advancing Lab Technologies | SNS Insider [https://www.globenewswire.com/news-release/2025/11/19/3190839/0/en/High-Performance-Liquid-Chromatography-Market-to-Reach-USD-7-74-Billion-by-2032-Driven-by-Rising-Demand-for-Precise-Analytical-Testing-and-Advancing-Lab-Technologies-SNS-Insider.html]
- 23. High-Performance Liquid Chromatography Market Size & Share - Industry Growth | 2032 [https://www.skyquestt.com/report/high-performance-liquid-chromatography-market]
- 24. (PDF) Measurement of Capsaicinoids in Chiltepin Hot Pepper... [https://www.academia.edu/24269370/Measurement_of_Capsaicinoids_in_Chiltepin_Hot_Pepper_A_Comparison_Study_between_Spectrophotometric_Method_and_High_Performance_Liquid_Chromatography_Analysis]
- 25. Ultrasound-assisted extraction optimization and validation of an HPLC-DAD method for the quantification of polyphenols in leaf extracts of Cecropia species | Scientific Reports [https://www.nature.com/articles/s41598-018-37607-2]
- 26. PPT - Comparison between third derivative spectrophotometric ... [https://www.slideserve.com/Sunaina/comparison-between-third-derivative-spectrophotometric-method-and-hplc-dad-method-in-detection-of-malondialdehyde-in-inf]
- 27. Validated HPLC- DAD and Spectrophotometric for... Methods [https://pubmed.ncbi.nlm.nih.gov/36326356/]
- 28. (PDF) HPLC- DAD and UV-Vis Spectrophotometric for... Methods [https://www.academia.edu/129711648/HPLC_DAD_and_UV_Vis_Spectrophotometric_Methods_for_Methotrexate_Assay_in_Different_Biodegradable_Microparticles]
- 29. Uv-visible spectrophotometric development and validation of... method [https://ijpsr.com/bft-article/uv-visible-spectrophotometric-method-development-and-validation-of-assay-of-paracetamol-tablet-formulation/?view=fulltext]
- 30. of UV spectrophotometry and high Comparison liquid... performance [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658086/]
- 31. Genetic-algorithm-based wavelength in multicomponent... selection [https://pubmed.ncbi.nlm.nih.gov/18968913/]
- 32. Simultaneous spectrophotometric determination of drug components from their dosage formulations - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1386142521013962]
- 33. approach for deconvolving overlapped Spectrophotometric of... spectra [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-025-01565-4]
- 34. Novel two wavelength spectrophotometric methods for simultaneous determination of binary mixtures with severely overlapping spectra - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1386142514015819]
- 35. Study of RP-HPLC and UV Comparative ... Spectrophotometric [https://pmc.ncbi.nlm.nih.gov/articles/PMC3021696/]
- 36. Different spectrophotometric methods for simultaneous quantitation... [https://www.nature.com/articles/s41598-023-50097-1?error=cookies_not_supported&code=b8aa0e18-737e-4295-a88c-46fc277eecec]
- 37. of high-efficient pre- Optimization sample treatments and... column [https://pubmed.ncbi.nlm.nih.gov/37939419/]
- 38. Research Assistant - Bohrium | AI for Science with Global Scientists [https://www.bohrium.com/paper-details/optimization-of-high-efficient-pre-column-sample-treatments-and-c18-uflc-method-for-selective-quantification-of-selected-chemical-forms-of-tocopherol-and-tocotrienol-in-diverse-foods/949074592251510834-1325]
- 39. Ultra Fast Liquid Chromatography - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/chemistry/ultra-fast-liquid-chromatography]
- 40. Antinociceptive and anti-inflammatory activities of the Jatropha isabellei... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6130469/]
- 41. Comparison of optimization algorithms for automated method development of gradient profiles - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S0021967324009993]
- 42. Overview of Methods and Considerations for Handling Complex Samples | LCGC International [https://www.chromatographyonline.com/view/overview-methods-and-considerations-handling-complex-samples]
- 43. Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7412464/]
- 44. Matrix removal in state of the art sample preparation methods for serum by charged aerosol detection and metabolomics-based LC-MS - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S000326701630232X]
- 45. Ultraviolet Detectors: Perspectives, Principles, and Practices | LCGC International [https://www.chromatographyonline.com/view/ultraviolet-detectors-perspectives-principles-and-practices]
- 46. Choices of chromatographic methods as stability indicating assays for pharmaceutical products: A review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8027279/]
- 47. - Stability LC-MS/MS and LC- indicating for robust... DAD methods [https://pubmed.ncbi.nlm.nih.gov/32858342/]
- 48. Simultaneous stability - indicating for the determination of... method [https://ijpsr.com/bft-article/simultaneous-stability-indicating-method-for-the-determination-of-abacavir-dolutegravir-and-lamivudine-by-rp-hplc/?view=fulltext]
- 49. Selective six spectrophotometric methods for determination of... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-025-01607-x]
- 50. Three New Methods for Resolving Ternary Mixture with Overlapping ... [https://www.jstage.jst.go.jp/article/cpb/65/6/65_c17-00132/_html/-char/en]
- 51. (PDF) Application of HPLC- DAD and spectrophotometric continuous... [https://www.academia.edu/36869525/Application_of_HPLC_DAD_and_spectrophotometric_continuous_wavelet_transform_methods_for_simultaneous_determination_of_amoxicillin_and_diclofenac_in_their_pure_and_capsule_dosage_forms]
- 52. ultraviolet uv spectrophotometry: Topics by Science.gov [https://www.science.gov/topicpages/u/ultraviolet+uv+spectrophotometry]
- 53. Shimadzu Prominence 20 HPLC UFLC System Including Software [https://innovtrendlab.com/product/shimadzu-prominence-20-hplc-uflc-system-including-software-shimadzu-lc-solutions/]
- 54. The Improved Method for Determination of Orotic Acid in Milk by... [https://pubmed.ncbi.nlm.nih.gov/34827928/]
- 55. 7. Principle and Feature of Various... : Hitachi High-Tech Corporation [https://www.hitachi-hightech.com/global/en/knowledge/analytical-systems/hplc/hplc-basics/course7.html]
- 56. Optimization and validation of HPLC–DAD method for simultaneous analysis of sweeteners, preservatives, and caffeine in sugar-free beverages | European Food Research and Technology [https://link.springer.com/article/10.1007/s00217-023-04328-4]
- 57. High-Performance Liquid Chromatography With Diode-Array Detection - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/medicine-and-dentistry/high-performance-liquid-chromatography-with-diode-array-detection]
- 58. - UV absorption Vis and LC- spectrophotometry -MS-ESI(+)-ESI... DAD [https://pubmed.ncbi.nlm.nih.gov/37573101/]
- 59. Developing an Absorption–Based Quality Control Method for... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6274194/]
- 60. Ultraviolet Spectrophotometry - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ultraviolet-spectrophotometry]
- 61. Development and validation of an uv- spectrophotometric method for... [https://ijpsr.com/bft-article/development-and-validation-of-an-uv-spectrophotometric-method-for-the-estimation-of-fluoxetine-in-pure-and-tablet-dosage-forms/?view=fulltext]
- 62. Peak area for DAD vs Spectrophotometric detector... | Forum [https://www.chromforum.org/viewtopic.php?t=97694]
- 63. (PDF) Validated spectrophotometric and spectrofluorimetric methods ... [https://www.academia.edu/73791625/Validated_spectrophotometric_and_spectrofluorimetric_methods_for_determination_of_chloroaluminum_phthalocyanine_in_nanocarriers]
- 64. of Validation for the Determination of... Spectrophotometric Methods [https://pubmed.ncbi.nlm.nih.gov/28730980/]
- 65. 9 LoD and LoQ – Validation of liquid chromatography mass... [https://sisu.ut.ee/lcms_method_validation/9-lod-and-loq/]
- 66. Development, validation and application of an UFLC-DAD-ESI-MS method for determination of carbonyl compounds in soybean oil during continuous heating - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0308814616314200]
- 67. (PDF) Method development, optimization and validation of RP-UFLC method for bioactive flavonoids from Cassia auriculata [https://www.academia.edu/96856323/Method_development_optimization_and_validation_of_RP_UFLC_method_for_bioactive_flavonoids_from_Cassia_auriculata]
- 68. Validated kinetic spectrophotometric for the determination of... method [https://pubmed.ncbi.nlm.nih.gov/16079525/]
- 69. (PDF) Validated Kinetic Spectrophotometric for the... Method [https://www.academia.edu/108609231/Validated_Kinetic_Spectrophotometric_Method_for_the_Determination_of_Metoprolol_Tartrate_in_Pharmaceutical_Formulations]
- 70. Rapid and Sensitive LC-MS/MS Method for the Determination of Metoprolol in Beagle Dog Plasma with a Simple Protein Precipitation Treatment and Its Pharmacokinetic Applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6268535/]
- 71. Development of a RP-HPLC method for simultaneous determination of atenolol, metoprolol tartrate and phenol red for in-situ rat intestinal perfusion studies - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1570023224001685]
- 72. Quantification of Bisoprolol and Metoprolol in Simultaneous Human Serum and Cerebrospinal Fluid Samples - PubMed [https://pubmed.ncbi.nlm.nih.gov/28930747/]
- 73. AAS and spectrophotometric for the... | AVESİS methods [https://avesis.yildiz.edu.tr/yayin/e35b5755-6f98-41da-bca1-0c1649c74e14/aas-and-spectrophotometric-methods-for-the-determination-metoprolol-tartrate-in-tablets]
- 74. Difference between t - test and ANOVA - GeeksforGeeks [https://www.geeksforgeeks.org/data-science/difference-between-t-test-and-anova/]
- 75. - t & test ( ANOVA of Analysis ) | RayBiotech | RayBiotech Variance [https://www.raybiotech.com/t-test-anova]
- 76. study of Comparative and spectrodensitometric... spectrophotometric [https://link.springer.com/article/10.1007/s00764-020-00050-z]
- 77. Multiple comparison in analysis - Biochemia Medica testing ANOVA [https://www.biochemia-medica.com/en/journal/21/3/10.11613/BM.2011.029]
- 78. Green metrics in pharmaceutical development - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2452223621001206]
- 79. Greenness and whiteness assessment of a sustainable voltammetric method for difluprednate estimation in the presence of its alkaline degradation product | Scientific Reports [https://www.nature.com/articles/s41598-024-61712-0]
- 80. Evaluating the "greenness" of chemical processes and products in the pharmaceutical industry--a green metrics primer - PubMed [https://pubmed.ncbi.nlm.nih.gov/22076593/]
- 81. A validated RP-HPLC method for the estimation of Cinnarizine in tablet... [https://fjps.springeropen.com/articles/10.1186/s43094-025-00888-2]
- 82. study of four Comparative assessment tools for selection... greenness [https://pubmed.ncbi.nlm.nih.gov/33404016/]
- 83. Spectrophotometric methods for the determination of letrozole in bulk and pharmaceutical dosage forms - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3255416/]