Somatic Hypermutation and Neutralization Breadth: Mechanisms, Measurement, and Therapeutic Implications
This comprehensive review examines the critical relationship between B cell receptor somatic hypermutation (SHM) and the development of neutralizing antibody breadth against rapidly evolving pathogens.
Somatic Hypermutation and Neutralization Breadth: Mechanisms, Measurement, and Therapeutic Implications
Abstract
This comprehensive review examines the critical relationship between B cell receptor somatic hypermutation (SHM) and the development of neutralizing antibody breadth against rapidly evolving pathogens. Drawing from recent advances in immunology and computational biology, we explore the fundamental mechanisms governing affinity maturation in germinal centers, methodological approaches for tracking BCR evolution, challenges in optimizing SHM for vaccine design, and comparative validation across pathogen systems including SARS-CoV-2, HIV, and influenza. For researchers and drug development professionals, this synthesis provides a framework for leveraging SHM dynamics to develop next-generation immunotherapies and broadly protective vaccines against antigenically diverse threats.
Germinal Center Dynamics: Where SHM Shapes Antibody Breadth
The generation of high-affinity antibodies and the development of broadly neutralizing antibodies (bnAbs) are central goals of vaccination strategies. For decades, the prevailing paradigm of affinity maturation has been one of stringent selection, where B cells with the highest affinity for a specific antigen are selectively favored within germinal centers (GCs). However, emerging evidence challenges this deterministic view, suggesting that GCs are more permissive environments than previously thought. This permissiveness allows B cells with a broader range of affinities to persist and mature, thereby promoting clonal diversity and enabling the rare emergence of bnAbs [1] [2]. This paradigm shift has profound implications for developing vaccines against rapidly evolving pathogens such as HIV, influenza, and SARS-CoV-2, where breadth of neutralization is often more valuable than affinity for a single epitope.
This review compares these competing paradigms of affinity maturation, examining the underlying experimental evidence and the advanced computational tools driving this conceptual evolution. We focus particularly on the critical relationship between B cell receptor somatic hypermutation and the development of neutralization breadth, providing researchers with a framework for evaluating and manipulating these processes in therapeutic development.
Germinal Center Dynamics: Reassessing Established Models
The Traditional Stringent Selection Paradigm
Germinal centers are transient microanatomical structures where the Darwinian evolution of antibody responses occurs. The classical model posits a highly coordinated process with spatial and functional segregation:
- Dark Zone (DZ): A site of rapid B cell proliferation and somatic hypermutation (SHM), where activation-induced cytidine deaminase (AID) introduces point mutations into immunoglobulin variable region genes at an exceptionally high rate [3] [4].
- Light Zone (LZ): Where B cells test their newly mutated B cell receptors (BCRs) against antigens displayed on follicular dendritic cells (FDCs). B cells that successfully acquire and present antigen receive survival signals from T follicular helper (Tfh) cells, enabling them to re-enter the DZ for further rounds of mutation [1] [2].
In this traditional death-limited selection model, competition for Tfh cell help is fierce, and only B cells with the highest antigen-binding affinity survive this selective bottleneck [1]. This model effectively explains how B cell responses can be progressively refined to achieve exceptionally high affinity against stable antigens.
Evidence for Permissive Selection in Germinal Centers
Recent research reveals a more complex picture, demonstrating that GC selection is not exclusively affinity-driven. Several key findings support this permissive selection paradigm:
Relaxed pMHCII Density Requirements: A critical experiment using MHCII haploinsufficient mice demonstrated that once GCs are established, B cells with half the normal peptide-MHCII (pMHCII) complex density compete equally with wild-type B cells in terms of persistence, mutation acquisition, and affinity maturation [5]. This indicates that the selection threshold in established GCs is sufficiently low to accommodate B cells with suboptimal antigen presentation capacity.
Birth-Limited Selection Model: An alternative to the death-limited model proposes that a B cell's ability to proliferate upon re-entering the DZ depends on signal strength received in the LZ, rather than strictly facing apoptosis [1] [2]. This model allows for a broader range of affinities to persist, as B cells receive varying opportunities to proliferate rather than facing binary live/die decisions.
Stochastic Elements in Fate Decisions: The mechanisms determining whether GC B cells differentiate into antibody-secreting plasma cells versus memory B cells remain incompletely understood, with evidence supporting roles for antigen distribution during cell division, temporal switches in GC reactions, and stochastic processes [1].
The following diagram illustrates the revised understanding of GC dynamics incorporating both stringent and permissive elements:
Revised Germinal Center Dynamics: This flowchart illustrates the contemporary understanding of GC reactions, incorporating both traditional stringent selection pathways and more recently recognized permissive elements that allow B cells with varying affinity levels to participate in the affinity maturation process.
Quantitative Comparison of Selection Paradigms
The following table summarizes key differences between the stringent and permissive selection paradigms, highlighting how this conceptual shift changes our understanding of GC function and its implications for antibody development.
Table 1: Comparison of Stringent versus Permissive Selection Paradigms in Affinity Maturation
| Feature | Stringent Selection Paradigm | Permissive Selection Paradigm |
|---|---|---|
| Selection Mechanism | Death-limited selection; binary live/die decisions based on affinity | Birth-limited selection; probabilistic survival and proliferation based on signal strength |
| GC Environment | Highly competitive; only highest-affinity B cells survive | Accommodating; allows B cells with broad affinity range to persist |
| Role of pMHCII Density | Critical determinant of positive selection; linear relationship with survival | Important but with lower threshold; halving pMHCII has minimal impact once GC established [5] |
| Clonal Diversity | Progressively narrowed as highest-affinity clones dominate | Maintained throughout GC response, allowing more clones to participate |
| Tfh Cell Help | Limited resource; determines which B cells survive | More widely available; modulates proliferation rate rather than determining survival |
| Outcome for bnAb Development | Unfavorable, as bnAb precursors often have lower initial affinity | Favorable, as allows rare bnAb precursors with suboptimal affinity to persist and mature |
Somatic Hypermutation: Mechanisms and Modeling
Molecular Mechanisms of SHM
Somatic hypermutation is the diversity-generating engine of affinity maturation, initiated by AID-mediated deamination of cytosine to uracil in DNA [3] [4]. This process occurs predominantly in the DZ of GCs and exhibits several key characteristics:
- Sequence Context Dependence: AID preferentially targets cytosine within specific DNA motifs, particularly the WRCY sequence context (W = A/T, R = A/G, Y = C/T) [4].
- DNA Repair Pathways Determine Mutation Outcomes: The initial U:G mismatch can be processed through multiple repair pathways:
- DNA replication leads to C→T and G→A transitions
- Base excision repair (BER) with error-prone polymerases introduces mutations at the original site
- Mismatch repair (MMR) creates broader mutation spectra around the original lesion [4]
- Transcriptional Coupling: SHM is strongly associated with transcription, with mutations beginning approximately 100 bp downstream of the transcription start site and peaking around 200 bp downstream [4].
Advanced Modeling of SHM Patterns
Traditional models of SHM were based on 5-mer sequence contexts, but newer "thrifty" models use convolutional neural networks on 3-mer embeddings to capture wider sequence contexts (up to 13-mers) with fewer parameters [6] [7]. These models reveal that:
- SHM patterns can be effectively modeled without position-specific effects when sufficient nucleotide context is included
- Models trained on out-of-frame sequences (minimally affected by selection) differ significantly from those trained on synonymous mutations
- The mutation process at each site is largely independent of mutations at other sites, though strongly dependent on local sequence context [6] [7]
Table 2: Key Computational Models for Predicting Somatic Hypermutation Patterns
| Model Type | Context Size | Key Features | Applications | Limitations |
|---|---|---|---|---|
| S5F 5-mer Model | 5 nucleotides | Independent mutation rate for each 5-mer motif | Predicting mutation probabilities for antibody maturation; established baseline | Limited context; exponential parameter growth with larger contexts |
| 7-mer Models | 7 nucleotides | Extended context with 3 flanking bases on each side | Improved accuracy for mutation hotspot prediction | High parameter count; requires substantial training data |
| "Thrifty" CNN Models | Up to 13 nucleotides | 3-mer embeddings with convolutional filters; parameter-efficient | Wide-context modeling with reduced parameters; slightly outperforms 5-mer models | Modest performance gains with modern machine learning; limited by available data [6] [7] |
Experimental Evidence Linking SHM to Neutralization Breadth
HIV Broadly Neutralizing Antibodies as a Case Study
HIV bnAbs provide compelling evidence for the relationship between extensive SHM and neutralization breadth. These antibodies typically show unusually high levels of SHM, with nucleotide sequences diverging 7-32% from their germline precursors [8]. For example:
- The VRC01 CD4 binding site bnAb shows approximately 30% mutation in its heavy chain variable region
- The PGT121-134 family of glycan-dependent bnAbs shows 17-23% divergence in heavy chain variable regions [8]
Strikingly, studies of inferred intermediate antibodies in the PGT121 lineage demonstrate that antibodies with approximately half the mutation level of mature bnAbs can still neutralize 40-80% of PGT121-sensitive viruses, though at reduced potency [8]. This suggests a correlation between SHM accumulation and neutralization breadth, while also indicating that moderately mutated intermediates may offer more tractable targets for vaccine design.
Experimental Approaches for Studying SHM-Breadth Relationships
Several key methodologies have been developed to interrogate the relationship between SHM and antibody function:
Deep Sequencing of B Cell Repertoires: High-throughput sequencing of BCR genes from antigen-specific B cells or memory B cell populations, followed by bioinformatic analysis to reconstruct lineage relationships and mutation trajectories [8] [9].
Inferred Intermediate Characterization: Phylogenetic reconstruction of antibody lineages to identify and synthesize putative intermediate antibodies, followed by functional characterization of their binding and neutralization properties [8].
Germline Reversion Studies: Systematic reversion of mutated positions in mature antibodies to their germline configurations to determine the functional contribution of specific mutations to breadth and potency [8].
The following diagram illustrates a representative experimental workflow for analyzing SHM and neutralization breadth:
SHM and Neutralization Breadth Analysis Workflow: This flowchart outlines a standard experimental approach for studying the relationship between somatic hypermutation and antibody function, combining next-generation sequencing with functional validation.
The Scientist's Toolkit: Key Research Reagents and Methods
Table 3: Essential Research Tools for Studying Affinity Maturation and SHM
| Tool/Reagent | Function/Application | Key Features |
|---|---|---|
| AID-Deficient Mice | Studying AID-specific functions in SHM and CSR | Complete absence of SHM and CSR; reveals AID-dependent processes [3] |
| Photoactivatable GFP Mice | Tracking B cell migration and fate decisions in GCs | Enables precise spatiotemporal monitoring of B cell movements between DZ and LZ [3] |
| Next-Generation Sequencing | Comprehensive antibody repertoire analysis | Enums enumeration of BCR diversity, SHM frequency, and clonal lineages [8] [9] |
| NetAM Python Package | Modeling SHM patterns with wide nucleotide context | Implements "thrifty" CNN models for predicting mutation probabilities [6] [7] |
| Luminex Bead Avidity Assays | Quantitative assessment of antibody-antigen binding strength | Enables high-throughput avidity measurement with variant antigens [5] |
| Single-Cell BCR Sequencing | Paired heavy and light chain sequence analysis | Preserves natural antibody pairings; enables recombinant expression of native antibodies |
The evolving understanding of affinity maturation—from a strictly stringent process to a more permissive and stochastic one—represents a significant paradigm shift in immunology. This revised framework better explains how the immune system balances the competing demands of affinity optimization and diversity maintenance, ultimately enabling the development of broadly protective antibodies against complex pathogens.
The permissive GC model provides a more optimistic outlook for vaccine development, suggesting that strategies designed to maintain GC persistence and clonal diversity may favor the emergence of bnAbs. Furthermore, advanced computational models of SHM continue to improve our ability to predict and manipulate antibody maturation pathways. As these tools become increasingly sophisticated and integrated with experimental validation, they offer promising avenues for rational vaccine design against challenging pathogens that have thus far evaded conventional vaccination approaches.
Germinal centers (GCs) are transient microanatomical structures that form in secondary lymphoid organs following exposure to T cell-dependent antigens [10]. Within these specialized microenvironments, B cells undergo an iterative process of somatic hypermutation (SHM) and selection that drives antibody affinity maturation, ultimately producing high-affinity plasma cells and memory B cells [10] [11]. The GC is functionally partitioned into two distinct compartments—the dark zone (DZ) and light zone (LZ)—that facilitate complementary roles in the antibody refinement process [10] [12]. Understanding the sophisticated coordination between DZ-based mutation and LZ-based selection is fundamental to research exploring the correlation between B cell receptor somatic hypermutation and the development of antibodies with broad neutralization capabilities.
Dark Zone and Light Zone: A Functional Comparison
The dark and light zones represent specialized microenvironments with distinct cellular compositions, functions, and molecular regulation that collectively drive affinity maturation.
Table 1: Core Functional Characteristics of Dark Zone and Light Zone
| Feature | Dark Zone (DZ) | Light Zone (LZ) |
|---|---|---|
| Primary Function | Somatic hypermutation (SHM) and rapid proliferation [12] [2] | Affinity-based selection and T cell help [12] [2] |
| Key Resident Cells | Proliferating B cells, CXCL12-expressing reticular cells [10] | B cells, Follicular Dendritic Cells (FDCs), T follicular helper (TFH) cells [10] [12] |
| Defining Molecular Regulators | BCL-6, CXCR4, FoxO1 (repressed) [10] [12] | BCL-6, CXCR5, CD40, c-Myc (induced upon selection) [10] [12] |
| B Cell Status | B cells degrade pre-SHM BCRs and undergo apoptosis if mutations are damaging [13] | B cells test newly expressed BCRs against antigen displayed on FDCs [12] [2] |
| Selection Pressure | --- | Based on BCR affinity and successful receipt of TFH cell help [12] |
Experimental Models and Methodologies for GC Studies
Research into GC dynamics relies on sophisticated model systems and precise tracking of B cell fate. The following workflow outlines a standard experimental approach for investigating the SHM-selection cycle.
Table 2: Key Experimental Models and Their Applications in GC Research
| Experimental Model / Reagent | Primary Function/Mechanism | Key Research Application |
|---|---|---|
| NP-OVA/KLH Conjugates | T cell-dependent model antigen; response dominated by VH186.2 B cells with traceable W33L affinity-enhancing mutation [14] | Tracking affinity maturation in response to a well-defined antigen [14] |
| H2b-mCherry Reporter Mice | Doxycycline-controlled histone reporter; dilution indicates division history [15] | Quantifying cell division dynamics and its correlation with SHM load in vivo [15] |
| DEC-205-Antibody Ag Delivery | Delivers antigen directly to endosomal compartments independently of BCR [12] | Manipulating and studying T cell help independent of BCR affinity [12] |
| scRNA-seq + BCR Sequencing | Couples whole-transcriptome data with B cell receptor sequence from single cells [15] [14] | Linking transcriptional states (e.g., metabolic programs) with clonal history and affinity [14] |
| Cox10fl/fl Aicda+/cre Mice | Enables B-cell-specific deletion of a protein essential for mitochondrial complex IV assembly [14] | Studying the role of OXPHOS metabolism in GC B cell selection and expansion [14] |
Molecular Mechanisms of SHM and Selection
The cyclic migration of B cells between the DZ and LZ is governed by a tightly regulated molecular program. The following diagram illustrates the core signaling and regulatory pathways active in each zone.
Evolving Paradigms in Light Zone Selection
The classical model of LZ selection posits a stringent, affinity-dependent process where B cells with higher-affinity BCRs acquire more antigen from FDCs, present more peptide-MHCII, and consequently outcompete lower-affinity neighbors for limited TFH cell help [12]. This interaction induces critical survival and proliferation signals, marked by the induction of the transcription factor c-Myc [12].
However, emerging evidence supports a more permissive selection model [12] [2]. This model suggests GCs initially select a wider range of B cells, including some with lower affinity, which are then outcompeted by higher-affinity clones over subsequent divisions rather than being immediately culled [12]. This permissiveness is thought to be crucial for maintaining clonal diversity, which is a prerequisite for the development of broadly neutralizing antibodies (bnAbs) that target variable pathogens [2].
Regulated Somatic Hypermutation in the Dark Zone
A groundbreaking 2025 study revealed that SHM is not a static process with a fixed mutation rate [15]. Using H2b-mCherry mice to track division history, researchers demonstrated that B cells receiving stronger TFH signals (typically higher-affinity clones) divide more frequently but paradoxically exhibit a lower mutation rate per division [15].
This affinity-dependent modulation of SHM protects high-affinity lineages from accumulating deleterious mutations during expansive proliferation, thereby enhancing the overall efficiency of affinity maturation [15]. This finding resolves the long-standing theoretical problem of "affinity backsliding," where the most expanded clones would otherwise risk generational degradation of affinity [15].
Metabolic Regulation of GC B Cell Fate
Cell fate decisions within the GC are underpinned by distinct metabolic programs. LZ B cells primarily utilize glycolysis, whereas DZ B cells undergoing rapid proliferation rely on oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) [14]. Research shows that high-affinity B cell clones preferentially upregulate OXPHOS, and its pharmacological enhancement promotes affinity maturation, highlighting metabolism as a critical regulator of selection [14].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Germinal Center Biology
| Reagent / Resource | Function in GC Research | Specific Example/Application |
|---|---|---|
| T cell-dependent model antigens | To induce synchronized, tractable GC responses in mice. | NP-OVA or NP-KLH; allows tracking of affinity-enhancing W33L mutation in VH186.2 B cells [14] |
| Cell fate and division trackers | To monitor proliferation history and lineage relationships in vivo. | H2b-mCherry/doxycycline system [15]; CFSE dye dilution |
| Tetramer-based reagents | To identify and isolate antigen-specific B cells via their BCR. | Fluorescently labeled antigen tetramers for flow cytometry |
| Recombinant cytokines & receptor ligands | To provide specific signals and probe pathways in vitro. | Recombinant CD40L to mimic TFH help; IL-4/IL-21 cytokines |
| Genetic models (Knockout/Cre-Lox) | To dissect gene function in a cell-type and time-specific manner. | B cell-specific knockout mice (e.g., Cox10fl/fl Aicda+/cre for metabolism studies [14]) |
| scRNA-seq platforms | To profile transcriptomes and pair them with BCR sequences at single-cell resolution. | 10X Genomics Chromium platform for clustering GC B cells and identifying zone-specific programs [15] [14] |
The germinal center microenvironment executes a sophisticated evolutionary algorithm where the dark zone functions as a mutation engine and the light zone as an affinity-based selection filter. The prevailing model of stringent selection is being refined by evidence of greater permissiveness and dynamic regulation of the SHM rate itself. These mechanistic insights into the correlation between BCR diversification and selection stringency are pivotal for rationally designing next-generation vaccines aimed at steering the immune system toward the production of broadly neutralizing antibodies against challenging pathogens like HIV and influenza.
The generation of broadly neutralizing antibodies (bnAbs) against rapidly evolving pathogens represents a paramount goal in modern immunology and vaccine development. This process is orchestrated within germinal centers (GCs), where the interplay of B cell receptor (BCR) signaling, T follicular helper (Tfh) cell help, and antigen capture mechanisms dictates the outcome of affinity maturation. Traditional models posit a straightforward selection for the highest-affinity B cells. However, emerging research reveals a more complex, regulated, and permissive system that balances affinity with breadth. This guide compares the key molecular drivers—BCR signaling, Tfh help, and antigen capture—synthesizing current experimental data to provide a foundational resource for researchers developing next-generation immunotherapies and vaccines.
Germinal centers (GCs) are transient, specialized microenvironments within secondary lymphoid organs where B cells undergo rapid proliferation, somatic hypermutation (SHM) of their immunoglobulin genes, and affinity-based selection [1] [2]. This process, known as affinity maturation, is fundamental to adaptive immunity. GCs are spatially organized into a dark zone (DZ), where B cells divide and mutate, and a light zone (LZ), where they test their newly mutated BCRs against antigen displayed on follicular dendritic cells (FDCs) and compete for help from Tfh cells [1] [15] [2]. The cyclic journey of B cells between these zones is the engine of antibody evolution.
The broader thesis of contemporary research is that the extent and nature of somatic hypermutation are directly correlated with the development of neutralization breadth [16] [17]. While high-affinity antibodies against a single antigen variant often accumulate numerous mutations, the elicitation of bnAbs—which can recognize a diverse set of pathogen variants—requires a GC reaction that permits sufficient clonal diversity and selects for B cells targeting conserved, vulnerable epitopes, even if their initial affinity is not maximal [1] [16]. The molecular drivers reviewed here are the core mechanisms regulating this delicate balance.
Comparative Analysis of Core Molecular Drivers
The following section provides a structured, data-driven comparison of the three primary molecular drivers in the GC reaction. The tables below summarize key characteristics, functions, and experimental evidence for each.
Table 1: Comparative Overview of BCR Signaling, Tfh Cell Help, and Antigen Capture
| Feature | BCR Signaling | Tfh Cell Help | Antigen Capture |
|---|---|---|---|
| Primary Function | Antigen internalization; Cell survival priming; Synergism with Tfh signals [18] | Licensing B cells for DZ re-entry; Determining division magnitude [15] [2] | Antigen acquisition from FDCs; Generation of pMHC for Tfh cell recognition [1] |
| Key Readouts | Phosphorylation of Syk, BTK; Calcium flux; Survival post-antigen engagement [18] | Expression of c-Myc in B cells; Quantity of CD40L and IL-21 [15] [2] | Amount of antigen internalized; pMHC-II density on B cell surface [1] [18] |
| Selection Model | Birth-limited (primes for survival/proliferation) [1] [18] | Death-limited & Birth-limited (determines survival & division cycles) [1] [15] | Pre-selection filter for Tfh help eligibility [1] |
| Impact on SHM | Ensures functional BCRs before SHM; Regulates GC B cell survival [18] | Modulates SHM rate; High help correlates with more divisions but lower mutation rate per division [15] | Determines which B cells get the chance to undergo further SHM cycles [1] |
Table 2: Experimental Evidence from Key Studies
| Driver Studied | Experimental Model/System | Key Finding | Quantitative Outcome |
|---|---|---|---|
| BCR Signaling | BTK drug-resistant mouse model; In vivo antigen presentation tracker [18] | BCR signaling is critical for LZ B cell survival and primes them to receive Tfh help. | B cells with inhibited BCR signaling showed significantly reduced survival in the LZ. |
| Tfh Cell Help & SHM Regulation | H2b-mCherry mouse model (NP-OVA immunization); scRNA-seq [15] | High-affinity B cells receiving strong Tfh help shorten cell cycle and reduce SHM rate per division. | B cells dividing ≥6 times had a 3-fold decrease in mutations per division, increasing progeny output from ~27 to ~41 cells [15]. |
| Antigen Capture & GC Permissiveness | Computational GC simulation; Probabilistic models [1] [2] | Permissive antigen capture, allowing a range of affinity B cells to persist, promotes clonal diversity and bnAb emergence. | Models show lower stringency in antigen capture leads to a broader B cell repertoire, a prerequisite for bnAbs [1]. |
| Affinity Maturation & Breadth | Human cohort study; Ad26.COV2.S vaccination [17] | Increased SHM over 8 months post-vaccination correlated with broader neutralizing antibody responses. | Highly mutated mAbs neutralized more SARS-CoV-2 variants than less mutated comparators [17]. |
Detailed Experimental Protocols
Understanding the methodologies behind the key findings is crucial for evaluating data and designing new experiments.
Protocol: Tracking B Cell Division and SHM In Vivo
This protocol is based on the seminal study investigating the relationship between Tfh help, cell division, and SHM rates [15].
- 1. Animal Model: Use transgenic mice expressing a doxycycline (DOX)-sensitive histone-2b-mCherry (H2b-mCherry) reporter.
- 2. Immunization: Immunize mice with a model antigen like NP-OVA or a SARS-CoV-2 vaccine.
- 3. Reporter Activation: On day ~12.5 post-immunization, administer DOX to turn off the mCherry reporter. From this point, cells that do not divide remain mCherryhigh, while the fluorescence dilutes with each successive division, creating mCherrylow populations.
- 4. Cell Sorting and Analysis: At a specific time point (e.g., 36 hours after DOX), isolate GC B cells from lymphoid organs. Sort populations based on mCherry intensity (mCherryhigh for low-division cells vs. mCherrylow for high-division cells).
- 5. Single-Cell RNA Sequencing (scRNA-seq): Perform scRNA-seq on sorted populations using a platform like 10X Genomics to obtain paired heavy- and light-chain sequences.
- 6. Data Analysis:
- Clonality Analysis: Reconstruct B cell clones from sequencing data to identify families of related cells.
- SHM Quantification: Calculate the number of nucleotide mutations in the variable regions of immunoglobulin genes for each cell.
- Affinity Assessment: For model antigens like NP, identify known affinity-enhancing mutations (e.g., W33L in IgHV1-72). Alternatively, use antigen-binding assays (e.g., NP-fluorophore binding) to correlate division history with affinity.
Protocol: Dissecting BCR Signaling in GC Selection
This protocol outlines the approach for defining the role of BCR signaling beyond antigen internalization [18].
- 1. Genetic Model: Generate a Bruton's tyrosine kinase (BTK) drug-resistant mouse model. This allows for selective pharmacological inhibition of endogenous BTK while the resistant BTK transgene maintains function in specific cells.
- 2. Antigen Presentation Tracker: Develop a traceable system for antigen binding and presentation. This can involve fluorescently tagged antigens or MHC-II reporters to track which B cells have successfully captured, processed, and presented antigen.
- 3. In Vivo Manipulation and Assessment: Treat immunized mice with a BTK inhibitor. This selectively disrupts BCR signaling without blocking antigen internalization via the BCR.
- 4. Flow Cytometry and Functional Assays: Analyze GC B cells by flow cytometry for markers of apoptosis (e.g., Annexin V), activation, and T cell priming. Compare the survival and functionality of B cells with inhibited vs. intact BCR signaling.
- 5. Key Readout: The critical measurement is whether B cells that successfully capture antigen but lack BCR signaling can survive in the LZ and receive Tfh help, compared to controls.
Signaling Pathways and Workflow Visualizations
The following diagrams, generated using DOT language, illustrate the core processes and relationships governing B cell fate in the germinal center.
Germinal Center B Cell Cycle
This diagram visualizes the cyclical journey of a B cell between the dark and light zones, highlighting the roles of the key molecular drivers.
Tfh Help Regulates SHM and Division
This flowchart depicts the novel regulatory mechanism where Tfh cell help inversely couples cell division to the SHM rate, protecting high-affinity lineages.
The Scientist's Toolkit: Key Research Reagents
This section catalogs essential reagents and models used in the cited research, providing a resource for experimental design.
Table 3: Essential Research Reagents for Investigating GC Molecular Drivers
| Reagent / Model | Function / Application | Key Insight Enabled | Example Source |
|---|---|---|---|
| H2b-mCherry Reporter Mice | Tracks in vivo cell division history via fluorescent protein dilution. | Revealed that high-affinity B cells divide more but mutate less per division [15]. | [15] |
| BTK Drug-Resistant Mice | Enables selective inhibition of BCR signaling in vivo without affecting antigen uptake. | Demonstrated that BCR signaling is essential for LZ B cell survival and priming for Tfh help [18]. | [18] |
| In Vivo Antigen Presentation Tracker | Labels and tracks B cells that have bound, internalized, and presented specific antigen. | Allows direct correlation between antigen capture efficiency and subsequent B cell fate [18]. | [18] |
| Single-Cell BCR Sequencing (scRNA-seq) | Recovers paired heavy- and light-chain sequences from single B cells. | Enables reconstruction of clonal lineages, SHM tracking, and identification of affinity-enhancing mutations [15] [19]. | [15] [19] |
| Agent-Based GC Simulations | Computational models to test hypotheses about GC dynamics and selection rules. | Predicted that permissive selection and variable SHM rates enhance bnAb development [1] [15]. | [1] [15] |
The integrated function of BCR signaling, Tfh cell help, and antigen capture is not a simple linear pathway but a dynamic, regulated network that optimizes antibody responses. The prevailing model of pure affinity-based stringency is giving way to a more nuanced understanding of permissive selection, which is critical for the development of antibody breadth. The experimental data and tools summarized in this guide underscore that BCR signaling provides a survival primer, Tfh help quantitatively and qualitatively shapes the proliferation-SHM balance, and the efficiency of antigen capture initiates the entire selection cascade. For researchers aiming to design vaccines that elicit bnAbs against challenging pathogens like HIV, influenza, or future pandemic coronaviruses, the key lies in strategically manipulating these molecular drivers to guide the GC reaction toward favoring B cells with the potential for breadth, often by promoting sufficient clonal diversity and allowing for extended, but safeguarded, affinity maturation.
This guide examines the pivotal role of B Cell Receptor (BCR) Somatic Hypermutation (SHM) in broadening epitope recognition, moving beyond the conventional focus on affinity enhancement. We compare data from key vaccine and infection studies, detailing the experimental protocols that underpin this advanced understanding of neutralizing antibody development.
Somatic hypermutation is a cornerstone of adaptive immunity, traditionally credited for improving antibody affinity. Contemporary research reveals a more profound function: SHM systemically diversifies the BCR repertoire to recognize distinct and evolving epitopes. The comparative data below demonstrates that increased SHM correlates directly with enhanced neutralization breadth against heterologous viral variants, a critical consideration for vaccine design and therapeutic antibody development.
Quantitative Data Comparison
The following tables consolidate quantitative findings from recent studies, highlighting the correlation between SHM levels and the development of cross-reactive, broad-neutralizing antibodies.
Table 1: SHM and Neutralization Breadth in Vaccine Studies
| Study / Intervention | Cohort / Model | Time Post-Immunization | SHM Increase (Heavy Chain) | Neutralization Breadth Observation | Key Metrics |
|---|---|---|---|---|---|
| Ad26.COV2.S Vaccine (Phase 1/2a trial) [20] | SARS-CoV-2 naive individuals (n=20) | 8 months | Significant increase (p<0.0001) | Increased breadth to B.1.351 (Beta) & B.1.617.2 (Delta) | 2 to 3.2-fold increase in variant NT50; correlation between SHM & serum breadth (r=0.38-0.60) |
| SARS-CoV-2 mRNA/Model Antigen Immunization [15] | H2b-mCherry mice | 36 hours post DOX (Day 14) | Variable mutation rate model | High-affinity B cells underwent more divisions but mutated less per division | Proposed mechanism safeguards high-affinity lineages, enhancing affinity maturation outcomes |
Table 2: SHM and Antibody Function in Breakthrough Infection Studies
| Study / Context | Patient Cohort | B Cell Characteristics | Isotype & SHM Profile | Key Antibody Findings | Structural Features Linked to SHM |
|---|---|---|---|---|---|
| Delta Variant Breakthrough Infections [21] | Primarily vaccinated individuals (n=15, 13 vaccinated) | High percentage of switched memory B cells (90.2%) | Increased IgG1; Lower proportion of unmutated VH genes (6.03%) vs. naive infection (10.73%) | Isolation of mAbs cross-reactive to Omicron variants; mAbs from selected cells had avg. 11.88% SHM (nucleotide) | Unusual HCDR2 insertions and altered CDR residues introduced by SHM |
Experimental Protocols
To ensure reproducibility and provide clarity on the data generation process, here are the detailed methodologies from the cited cornerstone experiments.
Protocol 1: Tracking SHM and Serum Breadth Post-Vaccination
This protocol is adapted from the study of the Ad26.COV2.S vaccine [20].
- 1. Cohort & Sample Collection: Enroll SARS-CoV-2 naive individuals in a vaccine trial (e.g., Phase 1/2a). Collect peripheral blood samples at defined timepoints (e.g., 1, 3, and 8 months post-vaccination). Confirm absence of infection via longitudinal nucleocapsid serology.
- 2. Serum Neutralization Assay: Perform pseudovirus neutralization assays using ancestral and variant SARS-CoV-2 spikes (e.g., WA1/2020, B.1.351, B.1.617.2). Calculate the half-maximal inhibitory titer (NT50) for each serum sample against each variant.
- 3. B Cell Sorting: Isolate peripheral blood mononuclear cells (PBMCs) from donor samples. Use fluorescence-activated cell sorting (FACS) to single-cell sort Spike-specific memory B cells (e.g., using labeled Spike protein probes).
- 4. BCR Sequencing and SHM Analysis: From sorted B cells, amplify and sequence the variable regions of the immunoglobulin heavy (IgVH) and light (IgVL) chains via next-generation sequencing (NGS). Analyze sequences using tools like IMGT/HighV-QUEST to compare them to germline V(D)J sequences and calculate the number of nucleotide mutations per variable region.
- 5. Monoclonal Antibody (mAb) Production and Testing: Clone the variable region sequences from sorted B cells into antibody expression vectors to produce recombinant mAbs. Express and purify these mAbs, then test their binding (e.g., via BLI or ELISA) and neutralization breadth against a panel of viral variants.
Protocol 2: Interrogating SHM Regulation in Germinal Centers
This protocol is based on the murine study investigating variable SHM rates [15].
- 1. Animal Model and Immunization: Use H2b-mCherry reporter mice, where a histone-2b-mCherry fusion protein is expressed under a doxycycline (DOX)-sensitive promoter. Immunize mice with the antigen of interest (e.g., NP-OVA or SARS-CoV-2 vaccine).
- 2. Cell Division Tracking: Administer DOX at the peak of the germinal center response (e.g., day 12.5) to turn off the mCherry reporter. As cells divide, the mCherry signal dilutes. After a set period (e.g., 36 hours), analyze mCherry intensity via flow cytometry to identify B cells that have undergone low (mCherryhigh) versus high (mCherrylow) numbers of divisions.
- 3. Single-Cell RNA Sequencing (scRNA-seq): FACS sort GC B cells based on mCherry intensity. Perform scRNA-seq using a platform like 10X Chromium to obtain paired heavy- and light-chain sequences and transcriptome data from individual B cells.
- 4. Clonal Analysis and Affinity Assessment: Reconstruct B cell clonal families from the sequence data. Map affinity-enhancing mutations (e.g., W33L for NP-OVA) and antigen binding (e.g., via NP-fluorophore staining by flow cytometry) onto the phylogenetic trees of the clones to correlate division history, SHM, and affinity.
Key Mechanism Visualizations
SHM Optimization in Germinal Centers
The following diagram illustrates the proposed model where B cells receiving stronger T-cell help divide more but mutate less per division, protecting high-affinity lineages.
Structural Mechanisms of Broad Neutralization
This diagram shows how SHM-introduced structural changes, like CDR insertions, enable antibodies to recognize diverse epitopes across viral variants.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for BCR SHM and Breadth Research
| Reagent / Solution | Function in Research | Example Application in Context |
|---|---|---|
| Fluorescently Labeled Antigens | FACS-based sorting of antigen-specific B cells | Isolation of Spike-specific B cells from PBMCs for single-cell sequencing [20] [21]. |
| Single-Cell BCR Sequencing Kits (e.g., 10X Genomics 5') | High-throughput sequencing of paired BCR heavy and light chains from single cells | Profiling SHM levels and clonal relationships within B cell populations [15] [21]. |
| Pseudovirus Neutralization Assay Kits | Safe and scalable measurement of neutralizing antibody breadth against viral variants | Quantifying serum and mAb neutralization potency (NT50/IC50) against VoCs like Omicron [20] [21]. |
| Activation-Induced Cytidine Deaminase (AID) | Key enzyme for inducing SHM in in vitro maturation systems | Engineered expression in cell lines (e.g., HEK293) for directed evolution of antibodies [22]. |
| Germinal Center Reporter Mouse Models (e.g., H2b-mCherry) | In vivo tracking of B cell division and GC dynamics | Studying the link between cell division history, SHM rate, and antibody affinity [15]. |
| Bio-Layer Interferometry (BLI) / Surface Plasmon Resonance (SPR) | Label-free analysis of binding kinetics and affinity | Characterizing cross-reactivity of mAbs by testing binding to RBDs from different variants [21]. |
The adaptive immune system relies on the production of high-affinity antibodies for effective long-term protection against pathogens. Somatic hypermutation (SHM), a process whereby B cells accumulating mutations in their immunoglobulin variable region genes, serves as the fundamental mechanism underlying antibody affinity maturation. This process occurs within germinal centers and is critical for generating potent neutralizing antibodies against diverse pathogens, including HIV and SARS-CoV-2. Understanding the temporal dynamics of SHM across different immune challenges—natural infection versus vaccination—provides crucial insights for vaccine design and therapeutic antibody development. This guide systematically compares the kinetics of SHM in these distinct contexts, synthesizing experimental data from recent studies to elucidate how timing and quality of immune responses differ based on antigen exposure route.
Fundamental Principles of Somatic Hypermutation
Molecular Mechanisms of SHM
Somatic hypermutation is initiated by activation-induced cytidine deaminase (AID), which deaminates cytosine residues to uracils in single-stranded DNA during transcription. This process preferentially targets specific motifs, with recent research identifying AGCTNT as a novel and highly mutated AID hotspot [23]. The resulting U:G mismatches are then processed by either base excision repair (BER) or mismatch repair (MMR) pathways, leading to point mutations that can enhance, diminish, or not affect antibody affinity [23].
The mutation process is not random but exhibits predictable biases based on local sequence context. Advanced computational models now incorporate wider nucleotide contexts through "thrifty" parameter-efficient convolutional neural networks, enabling more accurate prediction of SHM patterns and probabilities [24]. These models reveal that SHM patterns are established independently of specific local nascent transcriptional features, suggesting that the process is guided primarily by sequence-specific targeting rather than transcriptional landscapes [25].
Germinal Center Dynamics and Affinity Maturation
Within germinal centers, B cells cycle between dark and light zones, undergoing repeated rounds of mutation and selection. Recent experimental evidence challenges the traditional view of a fixed SHM rate, suggesting instead that B cells expressing higher-affinity antibodies may divide more frequently but mutate less per division [15]. This regulated SHM mechanism serves to protect high-affinity lineages from accumulating deleterious mutations while allowing for expansive clonal bursts, thereby optimizing affinity maturation outcomes.
Table: Key Molecular Components of Somatic Hypermutation
| Component | Function | Experimental Detection Methods |
|---|---|---|
| Activation-induced cytidine deaminase (AID) | Initiates SHM through cytosine deamination | Immunoblotting, immunohistochemistry, AID-deficient models |
| Base excision repair (BER) pathway | Processes U:G mismatches, leading primarily to transitions | Ung-/- murine models [23] |
| Mismatch repair (MMR) pathway | Processes U:G mismatches, leading to transitions and transversions | Msh2-/- murine models [23] |
| RNA Polymerase II | Transcribes immunoglobulin genes, generating single-stranded DNA substrates | PRO-seq, PRO-cap, ChIP-seq [25] |
| SPT5 | Stalling factor that associates with AID | ChIP-seq, machine learning prediction models [23] |
SHM Kinetics in Natural Infection
HIV-1 Infection Patterns
In HIV-1 infection, the development of broadly neutralizing antibodies (bNAbs) requires extensive SHM accumulation over prolonged periods. Research on elite neutralizers reveals that neutralization breadth often results from complementary polyclonal responses rather than a single bNAb lineage [26]. These individuals develop multiple antibody lineages targeting distinct envelope epitopes—including the gp120-gp41 interface, CD4-binding site, silent face, and V3 region—with each lineage neutralizing different sets of HIV-1 viruses from various clades [26].
Longitudinal tracking of Env-specific B cells shows substantial genetic distances within antibody lineages arising from extensive SHM acquired during multiple years of infection [26]. The remarkable serum breadth and potency in these individuals emerges through complementary neutralizing mechanisms, including receptor binding site blockade, glycan binding, and trimer disassembly, achieved through distinct antibody lineages that collectively provide comprehensive coverage [26].
SARS-CoV-2 Infection Dynamics
SARS-CoV-2 infection induces a distinct SHM kinetic profile characterized by continuous maturation even after viral clearance. Studies demonstrate that SHM continues to accumulate between 6 and 12 months post-infection, with convalescent individuals showing ongoing clonal evolution and antibody gene mutation [27]. This prolonged maturation occurs despite relatively stable neutralizing antibody titers, suggesting qualitative improvements in the antibody repertoire.
Notably, infection stimulates robust antibody responses in infants and young children that are maintained for over 300 days, with SHM in V-genes accumulating progressively over 9 months [28]. The restricted SHM in SARS-homologous clonotypes early in infection suggests initial extrafollicular B cell maturation, followed by more traditional germinal center responses as time progresses [29].
Figure 1: SHM Kinetics During Natural Infection Timeline
SHM Kinetics in Vaccination
mRNA Vaccine Responses
SARS-CoV-2 mRNA vaccination induces a prolonged germinal center response that persists for at least six months, driving substantial SHM accumulation over time [30]. Research shows that spike-specific GC B cells increase their SHM frequency by approximately 3.5-fold within six months post-vaccination, with memory B cells and bone marrow plasma cells accumulating high SHM levels that correlate with enhanced antibody avidity and neutralizing capacity [30].
Notably, vaccination promotes SHM acquisition through germinal center-dependent responses, with distinct patterns of SHM targeting compared to natural infection [29]. The continuous evolution of the B cell receptor repertoire after vaccination demonstrates pronounced repertoire renewal and preferential targeting of specific codons within the VH domain, supporting ongoing affinity maturation within germinal centers [29].
Hybrid Immunity Patterns
Individuals with prior SARS-CoV-2 infection who subsequently receive mRNA vaccination exhibit enhanced SHM kinetics and antibody breadth. Studies reveal that vaccination in convalescent individuals boosts neutralizing titers by nearly 50-fold and expands cross-reactive memory B cell clones [27]. This "hybrid immunity" results in antibodies that are exceptionally resistant to SARS-CoV-2 RBD mutations found in variants of concern, with B cell clones expressing broad and potent antibodies being selectively retained in the repertoire and expanding markedly after vaccination [27].
The mechanism underlying these enhanced responses involves ongoing antibody somatic mutation and memory B cell clonal turnover, demonstrating that vaccination can leverage the established immune history from prior infection to generate superior protection [27].
Comparative Analysis of SHM Patterns
Temporal Kinetics Comparison
Direct comparison of SHM patterns reveals distinct temporal dynamics between infection and vaccination scenarios. The table below summarizes key quantitative differences in SHM accumulation and functional outcomes across these immune challenges.
Table: Comparative SHM Kinetics Across Infection and Vaccination
| Parameter | Natural Infection | Vaccination | Experimental Evidence |
|---|---|---|---|
| Onset of SHM | 1-2 weeks post-exposure | 1-2 weeks post-priming | Longitudinal BCR sequencing [28] [30] |
| Peak SHM accumulation | 6-12 months | 6 months | Single-cell BCR sequencing of GC B cells [27] [30] |
| SHM frequency in antigen-specific B cells | Progressive increase over 12+ months | 3.5-fold increase in 6 months | Flow cytometry with antigen probes [30] |
| Rate of clonal turnover | High, with 61% new clones at 12 months | Moderate, with expansion of persistent clones | Phylogenetic analysis of B cell clones [27] [30] |
| Neutralization breadth development | Gradual, over months to years | Rapid, within months after booster | Pseudovirus neutralization assays [26] [27] |
| Duration of GC reaction | Variable, typically weeks | Persistent, at least 6 months | Longitudinal lymph node fine needle aspirates [30] |
Qualitative Differences in SHM Patterns
Beyond kinetic differences, infection and vaccination drive distinct SHM characteristics. Natural infection often produces more heterogeneous SHM patterns with greater epitope diversity, as seen in HIV-1 elite neutralizers generating multiple antibody lineages targeting distinct envelope regions [26]. In contrast, vaccination typically focuses SHM on specific antigenic targets, such as the spike protein in SARS-CoV-2 vaccines, potentially leading to more focused antibody responses [30].
Infection with SARS-CoV-2 stimulates extrafollicular responses with limited SHM early in infection, followed by germinal center-driven maturation [29]. Vaccination, however, promotes immediate germinal center-dependent responses with more controlled SHM acquisition and pronounced repertoire renewal [29]. This fundamental difference in initial B cell activation pathways influences the subsequent maturation trajectory and ultimate antibody breadth.
Figure 2: SHM Pattern Comparison Across Immune Challenges
Experimental Methodologies for SHM Analysis
B Cell Receptor Repertoire Sequencing
Comprehensive SHM analysis relies on targeted next-generation sequencing of immunoglobulin variable regions from sorted B cell populations [29]. The standard workflow involves: (1) isolation of antigen-specific B cells using fluorescently labeled probes; (2) single-cell sorting and RNA extraction; (3) reverse transcription and PCR amplification of heavy and light chain variable regions; (4) high-throughput sequencing; and (5) bioinformatic analysis of SHM patterns using tools like Change-O and Immcantation [26] [29].
For temporal tracking of SHM kinetics, researchers employ longitudinal sampling of peripheral blood mononuclear cells (PBMCs) or lymph node specimens, with fine-needle aspirates allowing direct assessment of germinal center B cells [30]. SHM frequency is typically calculated as the number of nucleotide mutations per base pair in the variable region compared to the germline sequence, with normalization for sequence length.
Functional Validation of SHM Impact
Beyond sequencing, functional validation establishes the physiological relevance of SHM patterns. Key methodologies include:
- Pseudovirus neutralization assays to quantify antibody neutralization breadth and potency against diverse viral variants [26] [27]
- Surface plasmon resonance or biolayer interferometry to measure binding affinity and kinetics of recombinant monoclonal antibodies [30]
- Enzyme-linked immunosorbent assays to determine antibody avidity and cross-reactivity [27]
- Flow cytometry with antigen-specific probes to enumerate and characterize antigen-specific memory B cells [28] [30]
These functional assays directly correlate SHM patterns with antibody efficacy, providing critical insights for vaccine design and therapeutic antibody development.
Research Reagent Solutions Toolkit
Table: Essential Research Reagents for SHM Kinetics Studies
| Reagent/Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Antigen Probes | BG505 SOSIP, AMC009 SOSIP [26] | Isolation of antigen-specific B cells | Fluorescently labeled, stabilized trimeric proteins |
| Sequencing Standards | S5F 5-mer model [24] | SHM pattern analysis | Context-specific mutation rate references |
| SHM Modeling Tools | Thrifty wide-context models [24] | Prediction of SHM probabilities | Parameter-efficient convolutional networks |
| Cell Sorting Markers | CD19, CD20, CD27, CD38 | B cell subset isolation | Identification of memory, naive, and plasma B cells |
| AID Activity Reporters | Ung-/-Msh2-/- murine models [23] | AID targeting assessment | Detection of unprocessed AID deamination events |
Implications for Vaccine and Therapeutic Design
Understanding SHM temporal patterns has profound implications for rational vaccine design. The demonstration that vaccination can induce persistent germinal centers driving continuous affinity maturation for at least six months supports extended prime-boost intervals to maximize antibody quality [30]. Similarly, the superior breadth and potency of antibodies from hybrid immunity suggest strategic use of vaccination in convalescent individuals to enhance protection against variants [27].
For difficult targets like HIV, where extreme SHM is required for broad neutralization, vaccine strategies may need to explicitly guide B cell maturation along pathways observed in elite neutralizers [26]. The finding that multiple moderately broad antibodies can achieve comprehensive coverage through complementarity suggests an alternative approach to the elusive goal of eliciting single ultra-broad antibodies [26].
Future vaccine efforts may incorporate regulated SHM principles, potentially through kinetic control of germinal center reactions or selective modulation of mutation rates in high-affinity B cell lineages [15]. Such approaches could optimize the balance between antibody affinity and breadth, potentially overcoming current limitations in vaccine development against highly variable pathogens.
Tracking BCR Evolution: Computational and Single-Cell Approaches
Next-Generation Sequencing for BCR Repertoire Analysis
B cell receptor (BCR) repertoire sequencing using next-generation sequencing (NGS) has revolutionized our ability to study the adaptive immune system at unprecedented depth and scale. The BCR, or membrane-bound antibody, is composed of two heavy chains and two light chains, with the antigen specificity primarily determined by the complementary determining region 3 (HCDR3) within the variable region. This region exhibits extraordinary diversity due to V(D)J recombination, somatic hypermutation (SHM), and class-switch recombination [31]. High-throughput sequencing of BCR repertoires enables researchers to capture this diversity, providing critical insights into B-cell dynamics, immune responses, and the development of neutralizing antibodies.
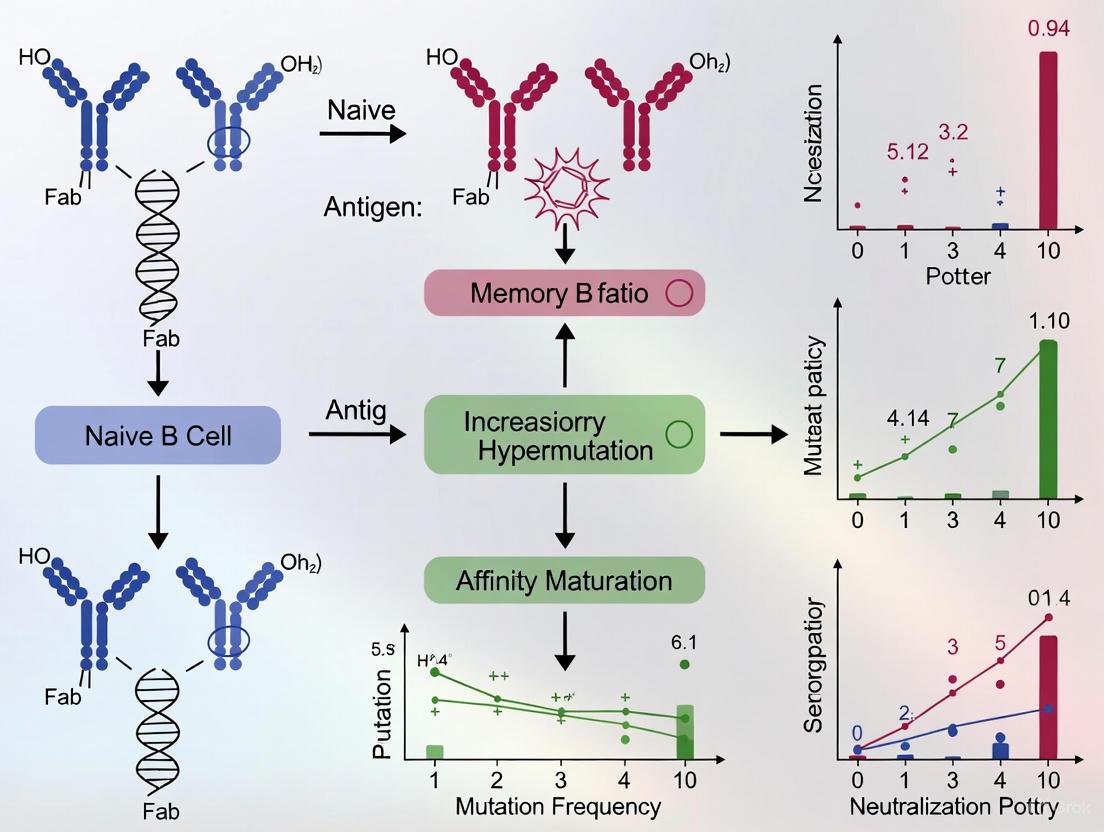
The correlation between B cell receptor somatic hypermutation and neutralization breadth represents a critical area of investigation in immunology. SHM introduces point mutations in the variable regions of BCRs during germinal center reactions, allowing for affinity maturation and the selection of B cells producing antibodies with superior antigen-binding capabilities [20]. Recent studies have demonstrated that increased SHM levels correlate strongly with enhanced neutralizing antibody breadth against viral variants, including SARS-CoV-2 and HIV [20] [21] [32]. This relationship underscores the importance of sophisticated NGS methodologies that can accurately capture and quantify SHM to advance vaccine development and therapeutic antibody discovery.
Comparative Analysis of NGS Platforms and Methods
Experimental Design Considerations
Template Selection represents a fundamental decision in BCR repertoire study design. Genomic DNA (gDNA) as a template captures both productive and non-productive BCR rearrangements, providing a comprehensive view of repertoire diversity and enabling precise clone quantification since each cell contributes a single template. However, gDNA-based approaches cannot inform on transcriptional activity or functional immune responses. In contrast, RNA templates reflect the actively expressed, functional repertoire but are less stable and susceptible to biases during extraction and reverse transcription. Complementary DNA (cDNA) synthesized from mRNA retains functional relevance while offering improved stability for experimental workflows [31].
The choice between CDR3-only and full-length sequencing involves significant trade-offs. CDR3-focused approaches provide cost-effective profiling of the most variable and antigen-specific receptor region, enabling efficient clonotype analysis and diversity assessment with simplified bioinformatics. However, this approach limits functional interpretation as it lacks structural context from CDR1 and CDR2 regions that contribute to antigen recognition, and it cannot determine paired heavy and light chain associations. Full-length sequencing captures complete variable regions plus constant domains, enabling comprehensive analysis of receptor functionality, MHC-binding characteristics, and native chain pairing—critical for understanding antigen specificity and therapeutic antibody development [31].
Bulk versus single-cell sequencing approaches offer complementary advantages. Bulk sequencing pools nucleic acids from cell populations, providing a cost-effective overview of repertoire diversity suitable for large cohort studies. However, it averages clonal distributions and loses cellular resolution and chain pairing information. Single-cell sequencing preserves native heavy and light chain pairing at individual cell resolution, enabling direct linking of BCR sequences to clonal lineages and functional states, albeit at higher cost and computational complexity [33] [31].
Platform Performance Comparison
Table 1: Comparison of BCR Sequencing Methodologies
| Method | Read Length | Key Applications | Chain Pairing | Error Rate | Cost Efficiency |
|---|---|---|---|---|---|
| Short-read bulk | ~150-300bp | Large cohort diversity studies | Indirect statistical inference | Moderate | High |
| Long-read | >500bp | Full-length transcripts, isotypes, lineage trees | Direct for full transcripts | Higher, mitigated by consensus | Moderate |
| Single-cell multi-omics | Varies by platform | Paired chains with cell state/function | Direct native pairing | Low with UMIs | Lower |
Short-read bulk sequencing (e.g., Illumina) remains dominant for large cohort studies due to its cost-effectiveness and mature bioinformatics support. This approach works well for general diversity assessments but struggles with heavy mutation loads and provides only indirect chain pairing through statistical inference [33]. Long-read technologies (e.g., Nanopore, PacBio) capture near full-length V(D)J and constant regions in single reads, enabling complete characterization of isotype usage, splice variants, and clean lineage trees. The FLIRseq method, employing rolling-circle amplification plus nanopore sequencing, has demonstrated strong accuracy in profiling full-length immune receptor transcripts [33].
Single-cell multi-omics platforms represent the most advanced approach, simultaneously capturing natively paired heavy and light chains alongside transcriptomic, protein surface marker, or chromatin accessibility data from the same cell. Methods like CITE-seq (cellular indexing of transcriptomes and epitopes by sequencing) and TEA-seq (transcriptomics, epitopes, and accessibility sequencing) enable researchers to connect specific BCR sequences to cellular phenotypes, activation states, and functional capacities [33]. A 2024 study demonstrated that single-cell V(D)J sequencing could effectively capture B cells with elevated SHM rates, revealing clonal expansions of cross-reactive B cells in individuals with Delta variant breakthrough infections [21].
Bioinformatics Tools for BCR Repertoire Analysis
Table 2: Comparison of BCR Clonotyping Engines
| Tool | Speed (20M reads) | Sensitivity | False Positive Control | Reference Handling | Single-cell Support |
|---|---|---|---|---|---|
| MiXCR | <2 hours | High (especially with error-prone data) | Excellent (minimal false clones) | Built-in curated library, allele discovery | Yes, robust with low-read data |
| Immcantation | >10 hours | Moderate | Moderate (100-200x more clones than MiXCR) | TIgGER for allele inference, manual management | Limited |
| TRUST4 | Intermediate | Moderate | Poor (~20x more clones than MiXCR) | User-provided references only, no discovery | Yes, but lacks noise filters |
| Cell Ranger | Platform-optimized | Good with sufficient reads | Good | Custom references possible, limited to 10x | Native for 10x Genomics |
Accurate bioinformatics analysis is crucial for reliable BCR repertoire interpretation. MiXCR demonstrates superior processing speed, completing analysis of 20-million-read datasets in under 2 hours compared to over 10 hours for Immcantation. In sensitivity benchmarks, MiXCR maintains performance advantages particularly as sequencing error rates increase. Most notably, MiXCR excels at minimizing false positives—in monoclonal hybridoma datasets, it correctly identified minimal clones while TRUST4 reported approximately 20x more clones and Immcantation reported 100-200x more clones [34].
Reference library management significantly impacts analysis accuracy. Most tools depend on IMGT germline references, which have limitations including slow updates, population bias, and incomplete allele coverage. Using incomplete references causes cascading errors where germline polymorphisms may be misidentified as somatic mutations. MiXCR's built-in continuously updated library with automatic novel allele discovery (findAlleles) recovers 15-20% more productive sequences than static IMGT-only approaches in non-European populations, substantially reducing systematic bias [34].
For single-cell data, cell detection efficiency is paramount. Both MiXCR and Cell Ranger perform comparably with sufficient sequencing depth, but MiXCR maintains significantly higher detection rates with low-read data. When computationally downsampled to 50% of original reads, MiXCR's robust performance allows researchers to multiplex more samples per run, substantially reducing per-sample costs without sacrificing data quality [34].
Experimental Protocols for BCR Repertoire Studies
Sample Preparation and Library Construction
Cell Sorting and Sample Preparation: For antigen-specific BCR analysis, researchers typically sort memory B cells using fluorescently labeled antigens and surface markers. A standard protocol involves isolating peripheral blood mononuclear cells (PBMCs) from whole blood using Ficoll gradient centrifugation, followed by staining with fluorescent-conjugated antibodies against CD19, CD20, CD27, IgM, and IgA, along with labeled antigen (e.g., SARS-CoV-2 Spike protein). Antigen-binding CD19+CD20+CD27+IgM-IgA- memory B cells are then sorted using fluorescence-activated cell sorting (FACS) [32]. Total RNA is extracted using TRIzol followed by purification with RNeasy Mini Kit including on-column DNase digestion to remove genomic DNA contamination [35].
Library Construction Methods: Multiple library preparation approaches exist, each with distinct advantages:
Multiplex PCR: Uses multiple V-gene specific primers and a conserved C-region primer to amplify BCR transcripts. This method is efficient but may introduce amplification biases due to differential primer efficiencies across V-gene families [35].
5' RACE (Rapid Amplification of cDNA Ends): Employing SMARTer technology with a template-switching mechanism, 5' RACE captures complete variable regions without V-gene specific primers, reducing amplification bias. The protocol involves reverse transcription with a constant region primer, template switching using SMARTer oligonucleotides, and PCR amplification with primers complementary to the adapter sequences [35].
RNA-capture: This hybridization-based method uses biotinylated RNA baits to target BCR transcripts from total RNA libraries. The protocol involves mRNA isolation by polyA+ selection, cDNA synthesis, fragmentation, adapter ligation, and hybridization with target-specific baits (e.g., Agilent SureSelect) followed by streptavidin bead-based pulldown of target regions [35].
Each method demonstrates high correlation in IgHV gene usage frequencies, though read length significantly impacts captured repertoire structure. Full-length BCR sequences are most informative for comprehensive repertoire analysis as diversity outside the CDR provides valuable phylogenetic information [35].
Quality Control and Error Correction
Implementing rigorous error control measures is essential for accurate SHM quantification. Unique molecular identifiers (UMIs) are short random nucleotide sequences that label individual mRNA molecules before amplification, enabling bioinformatic consensus building to correct for PCR and sequencing errors. Two-strand consensus methods further reduce errors by requiring agreement between complementary strands [33].
The AIRR (Adaptive Immune Receptor Repertoire) Community has established reporting standards for BCR repertoire studies, including detailed sample metadata, library construction parameters, sequencing specifications, and analysis pipelines. Adherence to these guidelines ensures reproducibility and facilitates data sharing across research groups [33].
BCR Sequencing Workflow
Key Research Applications and Findings
SARS-CoV-2 Vaccine and Infection Studies
Research on B cell responses to SARS-CoV-2 vaccination and infection has provided compelling evidence for the relationship between SHM and neutralization breadth. A 2024 study of the Ad26.COV2.S COVID-19 vaccine demonstrated that serum neutralizing antibody breadth against variants including B.1.351 (Beta) and B.1.617.2 (Delta) increased significantly over 8 months post-vaccination without additional boosting or infection. Concurrently, SHM levels in spike-specific B cells increased substantially, with the median mutation rate in IgVH genes correlating directly with neutralization potency against variants (B.1.617.2: r=0.3827, p=0.0488; B.1.351: r=0.5952, p=0.0011) [20].
Monoclonal antibodies derived from highly mutated BCR sequences isolated 8 months post-vaccination demonstrated superior variant cross-neutralization compared to less mutated antibodies from earlier timepoints. These findings indicate that the Ad26.COV2.S vaccine induces prolonged germinal center reactions and affinity maturation, resulting in progressively broadened neutralization capacity [20].
Studies of Delta variant breakthrough infections revealed that memory B cells in these individuals exhibited elevated SHM rates compared to those from non-vaccinated individuals infected early in the pandemic. Only 6.03±0.74% of sorted B cells from vaccinated individuals with breakthrough infections expressed unmutated VH genes, compared to 10.73±1.26% in non-vaccinated individuals. Cross-reactive neutralizing antibodies isolated from these individuals, such as YB9-258 and YB13-292, featured unusual heavy chain CDR2 insertions and altered CDR residues putatively introduced by SHM, which directly contributed to their broad neutralization capacity against Omicron variants [21].
HIV Broadly Neutralizing Antibody Research
HIV research has provided fundamental insights into the relationship between extreme SHM and neutralization breadth. Analysis of HIV envelope-specific memory B cells from "controller" individuals who develop broad neutralization capacity revealed that IGHV and IGLV mutation frequencies directly correlated with serum neutralization breadth. The repertoire of the most mutated antibodies was dominated by a small number of large clones with evolutionary signatures suggesting they had reached peak affinity maturation [32].
Notably, this study demonstrated that BCR selection for extended SHM and clonal evolution can occur even in the setting of low plasma HIV antigenemia, challenging the previous paradigm that chronic high-level viremia is necessary for bnAb development. The most effective neutralizing antibodies in these controllers were characterized by exceptionally long CDRH3 regions and distinctive IGHV/IGL pairings, with IGHV1-69/IGKV3-20 combinations predominating in broad neutralizers [32].
SHM-Neutralization Relationship
HIV Immune Reconstitution Studies
BCR repertoire analysis has revealed distinctive signatures associated with incomplete immune reconstitution in people living with HIV (PLWH) despite antiretroviral therapy. Immunological non-responders (INRs) who fail to recover CD4+ T cell counts exhibit BCR repertoires characterized by longer HCDR3 regions and reduced usage of IGHV1-69, IGHJ2, and specific IGHV-IGHJ pairings compared to immune responders [36].
Notably, INRs carried HCDR3 sequences highly homologous to anti-HIV broadly neutralizing antibodies targeting the six-helix bundle (6HB) in envelope gp41, and their plasma exhibited increased reactivity to FPPR-N36, a peptide within 6HB. These findings suggest that BCR repertoire features and antibodies targeting specific HIV envelope regions may be associated with inadequate immune reconstitution, providing new perspectives for understanding B cell immunity in HIV infection [36].
Essential Research Reagents and Tools
Table 3: Research Reagent Solutions for BCR Repertoire Analysis
| Reagent/Tool | Function | Example Products |
|---|---|---|
| Cell Sorting Reagents | Isolation of antigen-specific B cells | Fluorescently-labeled antigens, anti-CD19, anti-CD20, anti-CD27 antibodies |
| Nucleic Acid Extraction Kits | High-quality RNA/DNA extraction | TRIzol, RNeasy Mini Kit, DNase digestion columns |
| Library Preparation Kits | BCR target enrichment and NGS library construction | SMARTer Pico cDNA Synthesis Kit, TruSight Rapid Capture, Ion AmpliSeq Library Kit 2.0 |
| Sequencing Platforms | High-throughput sequencing | Illumina MiSeq, NovaSeq; Oxford Nanopore; PacBio |
| Bioinformatics Tools | Data processing and analysis | MiXCR, Immcantation, TRUST4, Cell Ranger |
| Reference Materials | Method validation and standardization | Genome in a Bottle (GIAB) reference materials |
Next-generation sequencing technologies have transformed BCR repertoire analysis, enabling unprecedented resolution in studying B cell immunity and the relationship between somatic hypermutation and neutralization breadth. The continuing evolution of sequencing platforms, library preparation methods, and bioinformatics tools will further enhance our ability to decipher the complex dynamics of B cell responses, with significant implications for vaccine design, therapeutic antibody development, and understanding immune correlates of protection.
Single-Cell RNA-seq with Paired BCR Sequencing (Benisse Model)
Technology Comparison for scRNA-seq with Paired BCR Analysis
The integration of single-cell RNA sequencing (scRNA-seq) with B-cell receptor (BCR) sequencing enables researchers to simultaneously investigate the transcriptomic state and clonal history of individual B cells. This multi-modal approach is critical for studying the correlation between B cell receptor somatic hypermutation and neutralization breadth. The table below compares the leading technologies and computational methods in this field.
Table 1: Comparison of Single-Cell Multi-Omic Technologies for B Cell Analysis
| Technology/Method | Key Technology/Method Principle | BCR Sequence Recovery | Paired with Transcriptome | Key Applications in B Cell Research |
|---|---|---|---|---|
| Benisse Model [37] | BCR embedding graphical network informed by scRNA-seq; uses contrastive learning on CDR3H sequences. | Full-length heavy and light chain (from paired data) | Yes, integrates BCR sequences with cell transcriptomics | Revealing B-cell activation gradients; studying coupling between BCRs and gene expression in COVID-19 [37]. |
| B3E-Seq [38] | Probe-based enrichment of BCRs from 3'-barcoded scRNA-seq libraries (e.g., 10x 3' GEX, Seq-Well). | Full-length variable region | Yes, from 3'-barcoded libraries | Identifying convergent BCR responses to vaccination; profiling antigen-specific B cells [38]. |
| 10x Genomics Immune Profiling [39] | Droplet-based partitioning with gel beads containing barcoded oligos for 5' transcript and V(D)J capture. | Paired heavy and light chain V(D)J | Yes, with 5' gene expression | High-throughput paired BCR and transcriptome analysis; tracking clonal dynamics [39]. |
| BASIC [40] | Semi-de novo assembly of BCRs from full-length scRNA-seq data using anchor sequences. | Full-length heavy and light chain | Yes, from full-length scRNA-seq data | Assembling BCR sequences for functional testing; coupling gene expression with immune repertoire [40]. |
| Parse Biosciences Evercode BCR [41] | Combinatorial barcoding (split-pool); no specialized instrument required; fixation-compatible. | TCR and BCR repertoire | Yes, with whole transcriptome | Immune profiling from fixed samples; scalable time-course experiments [41]. |
Experimental Protocols for Key Methodologies
Protocol 1: B3E-Seq for Full-Length BCR Recovery from 3' scRNA-seq Libraries
The B3E-seq method addresses a significant limitation of widely used 3'-barcoded scRNA-seq platforms, which natively capture minimal coverage of the BCR variable region located at the 5' end of the transcript [38].
Workflow:
- Input Material: A portion of the 3'-barcoded Whole Transcriptome Amplification (WTA) product from platforms like 10x Genomics 3' GEX or Seq-Well.
- BCR Enrichment: The WTA product is subjected to a probe-based affinity capture using biotinylated oligonucleotides that target the constant regions of BCR heavy and light chain isotypes.
- Re-amplification: The enriched product is reamplified using the original Universal Primer Site (UPS).
- Primer Extension: The product is modified using a set of oligonucleotides comprising a shared 5' UPS (UPS2) linked to sequences specific for the leader (L) or framework 1 (FR1) region of BCR variable (V) segments.
- Library Construction: The primer extension product is amplified with primers containing sequencing adapters linked to regions specific for either UPS2 (5'-end) or the original UPS (3'-end).
- Sequencing: The amplicons are sequenced using two overlapping reads in opposite directions (5' to 3' via UPS2, and 3' to 5' via custom BCR constant region primers) and a third read to capture the cellular barcode and UMI [38].
Validation Data: In tests using human PBMCs, B3E-seq recovered full-length heavy chain sequences from 56.1-66.7% of B cells and paired heavy and light chain sequences from 42.2-52.2% of B cells, demonstrating its utility for profiling antigen-specific B cell responses [38].
Protocol 2: The Benisse Computational Analysis Pipeline
Benisse (BCR embedding graphical network informed by scRNA-seq) is a mathematical model that integrates high-dimensional BCR sequence data with single-cell gene expression data to infer functional relevance [37].
Workflow:
- BCR Sequence Embedding:
- Input: Amino acid sequences of the Complementarity-Determining Region 3 of the heavy chain (CDR3H).
- Encoding: Each amino acid is represented by five numeric Atchley factors, which capture biochemical properties.
- Dimension Reduction: Contrastive learning is applied to reduce the encoded matrix into a 20-dimensional numeric vector. This model learns an embedding space where similar CDR3H sequences are positioned close together [37].
- Integration with Gene Expression:
- Input: The 20-dimensional BCR embedding and the scRNA-seq gene expression matrix for the same single cells.
- Graph Learning: Benisse employs a sparse graph learning model to place BCR clonotypes (unique V/J gene and CDR3H) into a low-dimensional latent space supervised by the gene expression data. The model constructs a graph where BCRs with similar sequences and from cells with similar transcriptomic profiles are connected, forming "BCR networks" [37].
- Validation: The Benisse embedding was validated using LIBRA-seq data, showing a correlation of 0.616 between BCR sequence similarity and antigen specificity similarity. It also successfully reconstructed a known phylogenetic lineage of HIV antibodies, demonstrating a linear evolution pattern [37].
Visualizing Experimental and Analytical Workflows
B3E-Seq Wet-Lab Protocol for BCR Recovery
Diagram 1: B3E-seq wet-lab workflow for recovering full-length BCR sequences from 3' scRNA-seq libraries.
Benisse Computational Model for BCR-Expression Integration
Diagram 2: The Benisse computational model integrates BCR sequences and transcriptomic data.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful execution of single-cell BCR and transcriptome studies requires a suite of specialized reagents and computational tools. The following table details essential components for building a robust research pipeline.
Table 2: Key Reagent Solutions for scRNA-seq with Paired BCR Sequencing
| Item Name | Type | Primary Function in Workflow |
|---|---|---|
| 10x Barcoded Gel Bead Primers [42] | Wet-lab Reagent | Enable cell partitioning, mRNA capture, and incorporation of cell barcode/UMI during GEM generation in 10x platforms. |
| Biocytinylated BCR Constant Region Probes [38] | Wet-lab Reagent | Used in B3E-seq for targeted enrichment of BCR transcripts from complex WTA products. |
| V(D)J-specific Primers (UPS2-linked) [38] | Wet-lab Reagent | Enable primer extension for full-length variable region sequencing in enrichment-based methods. |
| Fixation/Permeabilization Reagents [41] | Wet-lab Reagent | Allow sample preservation for flexible experimental timing (e.g., Parse Evercode, 10x Flex). |
| Benisse Software [37] | Computational Tool | Provides a supervised model for integrating BCR sequence embeddings with single-cell gene expression data. |
| BASIC Software [40] | Computational Tool | Performs semi-de novo assembly of full-length BCR sequences from scRNA-seq data using anchor guidance. |
| Cell Ranger / V(D)J Pipeline [43] | Computational Tool | Processes raw sequencing data from 10x platforms to generate BCR contigs, annotate genes, and define clonotypes. |
| ScIB / Single-Cell Benchmarking [44] | Computational Tool | Evaluates the quality of data integration, helping researchers choose the best methods for their multi-omic data. |
Phylogenetic Reconstruction of Antibody Lineages
The adaptive immune system counters pathogens through a sophisticated process of antibody evolution. A cornerstone of this process is affinity maturation, which occurs in germinal centers and involves cycles of somatic hypermutation (SHM) in B cell receptor (BCR) genes and selection for mutants with higher antigen affinity [20] [15]. Phylogenetic reconstruction of antibody lineages is a powerful computational approach that traces this evolutionary history, mapping the relationships between clonally related B cells from a common ancestor to its diversified descendants [45] [46]. This guide objectively compares the performance of leading computational tools and experimental methods central to this field, framing the comparison within the critical research context of how somatic hypermutation correlates with the development of neutralization breadth—the ability of an antibody to neutralize diverse viral variants [20] [47] [48].
Core Concepts and Research Context
The Link Between SHM and Neutralization Breadth
Affinity maturation is not a simple, linear process. Research indicates that the rate of SHM itself may be regulated. A recent 2025 study proposed a model where B cells expressing higher-affinity antibodies undergo more cell divisions but experience a lower mutation rate per division, a mechanism that potentially protects high-affinity lineages from accumulating deleterious mutations and enhances the overall outcome of affinity maturation [15]. This refined understanding underscores the complexity embedded within the phylogenetic trees researchers aim to reconstruct.
The primary value of lineage reconstruction lies in its ability to illuminate the molecular pathways to potent antibody responses. Studies of both HIV-1 and SARS-CoV-2 have demonstrated a strong positive correlation between increased levels of SHM in antibody genes and greater breadth of neutralization across viral variants [20] [47]. For instance, in individuals vaccinated with the Ad26.COV2.S COVID-19 vaccine, the neutralizing antibody breadth against variants like Beta (B.1.351) and Delta (B.1.617.2) significantly increased over eight months. This expansion in breadth was correlated with a measurable rise in SHM, and highly mutated monoclonal antibodies derived from these lineages neutralized more variants than their less-mutated counterparts [20]. Similarly, the development of broadly neutralizing antibodies (bnAbs) against HIV-1 is characterized by extensive affinity maturation and is often driven by exposure to a series of related viral immunotypes that guide the antibody lineage toward breadth [47].
The Workflow of Antibody Lineage Analysis
The process of reconstructing antibody lineages involves a multi-step workflow, from wet-lab sequencing to computational inference, as illustrated below.
Comparative Analysis of Immunoinformatic Tools
The initial and critical step of annotating antibody sequences and assigning germline V(D)J genes directly impacts all downstream analyses, including phylogenetic accuracy.
Performance Benchmarking of Annotation Tools
Table 1: Benchmarking performance of immunoinformatic annotation tools using simulated and experimental high-throughput sequencing datasets [49].
| Tool | Alignment Accuracy (Mishit Frequency) | CDR3 Annotation Reproducibility | Speed (Sequences/Unit Time) | Germline Database |
|---|---|---|---|---|
| IMGT/HighV-QUEST | Intermediate | 4.3% - 77.6% (preprocessed data) | Intermediate | IMGT |
| IgBLAST | Highest (0.004 mishit frequency) | 4.3% - 77.6% (preprocessed data) | Not Specified | NCBI (IMGT, GenBank) |
| MiXCR | Lower (0.02 mishit frequency) | 4.3% - 77.6% (preprocessed data) | Fastest | User-defined (e.g., IMGT) |
Key Insights:
- Germline Database Inconsistency: A significant source of annotation discrepancy is the lack of a standardized germline database. One analysis found that only 40% (73/183) of human V, D, and J genes were shared between the reference germline sets used by different tools [49].
- Tool Selection Guidance: The choice of tool involves trade-offs. IgBLAST is superior for alignment accuracy, which is crucial for identifying true somatic mutations. MiXCR offers the best performance for processing large datasets quickly, while IMGT/HighV-QUEST is a widely recognized standard [49].
Protocols for Key Experiments
Protocol 1: In Silico Benchmarking of Annotation Tools [49]
- Dataset Generation: Use IgSimulator software to generate two types of in silico human antibody heavy-chain repertoires: a "diverse" repertoire (100,000 base sequences, 200,000 mutated sequences) and a "polarized" repertoire (20,000 base sequences, 100,000 mutated sequences).
- Tool Processing: Run the simulated datasets through the annotation tools (e.g., IMGT/HighV-QUEST, IgBLAST, MiXCR) using their default parameters.
- Accuracy Assessment: Compare the tool's output V, D, and J gene assignments and CDR3 annotations against the known input sequences. Calculate the frequency of gene "mishits" (incorrect assignments).
- Reproducibility & Speed: Measure the consistency of CDR3 annotation between tools and the computational time required to process the datasets.
Protocol 2: Tracing Antibody Lineage Evolution Post-Vaccination [20]
- Sample Collection & Sorting: Collect peripheral blood mononuclear cells (PBMCs) from vaccinated individuals at multiple timepoints (e.g., 1, 3, and 8 months post-vaccination). Use flow cytometry to sort single Spike-protein-specific memory B cells.
- BCR Sequencing: Perform single-cell reverse transcription and PCR to amplify the variable regions of the heavy (IgVH) and light (IgVL) chains. Sequence the amplicons using next-generation sequencing.
- SHM Quantification: Align the resulting sequences to human germline V(D)J genes using a tool like IMGT/HighV-QUEST. Calculate the level of SHM as the number of nucleotide changes per IgVH or IgVL gene.
- Lineage Analysis: Cluster sequences into clonal lineages. For selected lineages, recombinantly produce monoclonal antibodies (mAbs) from ancestors and descendants and test their neutralization breadth against a panel of viral variants.
Advanced Phylogenetic Inference and Lineage Reconstruction
Beyond initial annotation, specialized statistical methods are required to accurately infer phylogenetic relationships and the unmutated common ancestor within a lineage.
A Hierarchical Bayesian Framework
A advanced method for lineage reconstruction uses a hierarchical Bayesian model to account for the unique processes of B cell receptor generation [45]. The core of this method is Bayes' Theorem:
P(α | Q) = P(Q | α) P₀(α) / Σ P(Q | α') P₀(α')
Where:
- P(α | Q) is the posterior probability of the unmutated ancestor α given the observed clonally related sequences Q.
- P₀(α) is the prior probability of α, derived from the stochastic process of V(D)J recombination.
- P(Q | α) is the likelihood of observing the sequences Q given the ancestor α, based on the process of somatic hypermutation.
This framework systematically integrates the probabilities of V, D, and J gene segment selection, recombination point location, and N-nucleotide addition to compute a posterior distribution over possible ancestral sequences, providing a thorough accounting of the inherent uncertainty in the reconstruction [45].
Visualizing the Statistical Inference of an Unmutated Ancestor
The following diagram outlines the logical workflow of this statistical inference process.
Correlation of SHM with Neutralization Breadth: Experimental Data
The ultimate test of a reconstructed lineage is its ability to explain functional outcomes, such as neutralization breadth. The following table synthesizes quantitative evidence from key studies.
Table 2: Correlation between somatic hypermutation (SHM) and neutralization breadth in antibody responses against SARS-CoV-2 and HIV-1.
| Study Context / Antibody | SHM Measurement | Neutralization Breadth & Potency | Experimental Evidence |
|---|---|---|---|
| Ad26.COV2.S Vaccine [20] | SHM in Spike-specific B cells increased significantly over 8 months (p<0.0001 for IgVH and IgVL). | Serum NAb breadth to Beta (B.1.351) & Delta (B.1.617.2) variants increased; highly mutated mAbs neutralized more variants. | Positive correlation between SHM level and variant NT50 (e.g., B.1.351 r=0.595, p=0.0011). |
| HIV-1 bnAb PGT145 [48] | N/A (Design focused) | 54% breadth (vs. 39% for WT) on 208-strain panel; >100-fold potency increase against some viruses. | In silico design (OSPREY) optimized side-chain interactions with variable epitope residues. |
| HIV-1 V2-apex bnAb Lineage [47] | Developed after superinfection; moderate SHM levels. | Acquired heterologous neutralization breadth within months of superinfection. | Breadth correlated with antibody's ability to neutralize diverse immunotypes within the patient. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key reagents and computational tools for antibody lineage reconstruction and analysis.
| Tool / Reagent | Function / Application | Example Use in Research |
|---|---|---|
| IMGT/HighV-QUEST | Web portal for annotation of Ig sequences against IMGT reference directory. | Standardized annotation of V(D)J genes, SHM, and CDR3 from NGS data [20] [49]. |
| IgBLAST | Command-line tool for aligning Ig sequences to germline gene databases. | High-accuracy alignment of antibody repertoire sequencing data [49]. |
| OSPREY | Computational protein design software. | In silico engineering of antibody variants for increased breadth and potency, as with HIV bnAbs [48]. |
| H2b-mCherry Reporter Mice | In vivo tracking of cell division via fluorescent protein dilution. | Experimental validation of linked cell division and SHM rates in germinal center B cells [15]. |
| Single-Cell BCR Sequencing | Paired heavy- and light-chain sequencing from individual B cells. | Essential for defining clonal lineages and generating accurate phylogenetic trees [20] [15]. |
| Karolinska Institutet Macaque Database (KIMDB) | Comprehensive database of macaque IG germline genes. | Critical for accurate VDJ gene assignment in macaque studies, a common model for vaccine research [50]. |
Phylogenetic reconstruction of antibody lineages has matured into an indispensable discipline for deconvoluting the complex process of affinity maturation. As the data demonstrates, the choice of computational tool—from initial annotation with IgBLAST or MiXCR to advanced phylogenetic inference with Bayesian methods—profoundly impacts the biological insights gained, particularly regarding the critical relationship between SHM and neutralization breadth. The emerging paradigm, supported by both computational and experimental evidence, is that breadth is not achieved by mere accumulation of mutations but through an optimized evolutionary path shaped by antigen exposure and potentially regulated SHM rates [20] [47] [15]. For drug development professionals, these methodologies provide a roadmap for rational antibody engineering, enabling the design of next-generation therapeutics and vaccines that can proactively guide the immune response toward broadly protective immunity.
Somatic hypermutation (SHM) serves as a critical mechanism in the adaptive immune system, refining antibody responses to achieve high-affinity binding and broad neutralization against rapidly evolving pathogens. This guide systematically compares how distinct SHM patterns correlate with structural and functional changes in epitope recognition. We synthesize experimental data from HIV-1, SARS-CoV-2, and other viral systems to objectively evaluate how SHM-induced modifications in antibody paratopes influence neutralization breadth and potency. The analysis is framed within the broader thesis that understanding these structural correlates is essential for guiding the development of vaccines and therapeutic antibodies, particularly against antigenically variable targets.
Affinity maturation in germinal centers optimizes antibody responses through somatic hypermutation—a process that introduces point mutations into immunoglobulin variable genes. For pathogens with high antigenic diversity, such as HIV-1 and SARS-CoV-2, broadly neutralizing antibodies (bnAbs) often exhibit unusually high levels of SHM, far exceeding typical vaccination responses [8]. These mutations occur in both complementarity-determining regions (CDRs) that directly contact antigen and framework regions (FWRs) that provide structural scaffolding, collectively enhancing epitope complementarity.
Research consistently demonstrates a positive correlation between SHM accumulation and the development of neutralization breadth and potency. For instance, phylogenetic modeling of the HIV-1 PGT121-134 bnAb lineage revealed that intermediate antibodies with approximately half the mutation level of mature bnAbs could still neutralize 40-80% of viruses, suggesting a viable pathway for vaccine design [8]. This guide examines the structural basis for these functional improvements by comparing experimental findings across multiple antibody lineages and epitope targets.
Quantitative Comparison of SHM Impact Across Antibody Lineages
Table 1: SHM Levels and Functional Outcomes in Characterized bnAbs
| Antibody / Lineage | Target / Virus | VH Mutation Frequency (%) | VL Mutation Frequency (%) | Neutralization Breadth | Key Structural Correlates |
|---|---|---|---|---|---|
| PGT121-134 [8] | HIV-1 V3-glycan | 17-23% | 11-28% | ~70-80% (74-virus panel) | Increased affinity for native Env over monomeric gp120; changes in glycan dependency |
| Inferred Intermediates [8] | HIV-1 V3-glycan | ~8-12% (estimated) | ~5-14% (estimated) | 40-80% (PGT121-sensitive viruses) | Moderate breadth with significantly lower SHM burden |
| VRC01-class [51] | HIV-1 CD4-binding site | Up to 30% | Up to 19% | >90% | CDRL1 truncation to avoid N276 glycan; pre-configured CDRH2 |
| 3D1 [52] | Pan-coronavirus HR1 | 14% (Heavy Chain) | Not specified | Multiple sarbecoviruses (excludes Omicron Q954H) | Recognition of β-turn fold in pre-hairpin intermediate; germline-encoded affinity |
| M15 [53] | Sarbecovirus S2 | Clonotype-enriched SHMs in light chain | Enhanced affinity for occluded epitope | Public antibody targeting conserved interface | Light-chain SHMs selectively enriched in clonotype |
Table 2: Experimental Techniques for Structural Correlation Studies
| Technique | Information Obtained | Application in SHM Studies | Key Limitations |
|---|---|---|---|
| X-ray Crystallography [54] | Atomic-resolution structures of antigen-antibody complexes | Direct visualization of paratope-epitope interfaces; conformational changes from SHM | Requires high-quality crystals; challenging for flexible complexes |
| Cryo-EM [55] | Near-atomic resolution of large complexes | Structure determination of antibody-Env trimer complexes | Lower resolution than crystallography; complex data processing |
| Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS) [51] | Local protein dynamics and flexibility | Measures SHM-induced changes in paratope structural dynamics | Indirect structural information; requires careful interpretation |
| Deep Mutational Scanning [54] | Comprehensive antigenic sequence determinants | Identifies escape mutations and critical epitope residues | Specialized library construction required |
| Benisse Model [37] | BCR embedding guided by single-cell gene expression | Correlates BCR sequence relationships with transcriptional state | Computational complexity; requires paired scRNA-seq+scBCR-seq data |
| SAAB+ Pipeline [56] | Structural annotation of BCR repertoires | High-throughput structural classification of CDR loops | Limited to CDR-H3 prediction; coverage varies by species |
Structural Mechanisms of SHM-Enhanced Epitope Recognition
Paratope Optimization Through Conformational Refinement
Somatic hypermutation enhances epitope recognition through several distinct structural mechanisms that improve shape complementarity. Studies of HIV-1 bnAbs reveal that SHM-induced stabilization and structural changes improve epitope complementarity while minimizing clashes with dynamic glycans on HIV-1 Env [51]. For VRC01-class antibodies, maturation involves CDRL1 truncation to avoid steric clashes with the N276 glycan on HIV-1 Env—a critical modification that enables access to the conserved CD4-binding site [51]. Framework mutations distant from the paratope can reorient CDR loops to improve antigen engagement, demonstrating that not all functionally important mutations occur in direct contact residues.
Hydrogen/deuterium-exchange mass spectrometry (HDX-MS) studies provide direct evidence that SHM modifies structural dynamics, with changes primarily observed at paratope peripheries adjacent to Env glycans [51]. This restriction of flexibility at the paratope periphery minimizes non-productive interactions with variable features while maintaining precise complementarity with conserved epitope elements.
Diagram 1: Structural and dynamic mechanisms linking SHM to improved antibody function. SHM induces both structural changes and dynamic modifications that collectively enhance epitope complementarity and reduce steric clashes with variable features, ultimately improving neutralization breadth and potency.
Epitope-Specific Maturation Patterns
Different epitope classes impose distinct constraints on antibody maturation, resulting in characteristic SHM patterns. Antibodies targeting glycan-dependent epitopes on HIV-1, such as the V3-glycan site recognized by PGT121-134, undergo mutations that alter glycan dependency and recognition over the course of affinity maturation [8]. These antibodies show a preference for native Env binding over monomeric gp120, suggesting selection for trimer-specific configurations during maturation.
For conserved cryptic epitopes, such as the HR1 domain targeted by coronavirus bnAb 3D1, SHM can refine recognition of transient intermediate states. The 3D1 antibody recognizes a β-turn fold comprising a 6-mer peptide that forms exclusively during a pre-hairpin transition state in membrane fusion [52]. Notably, the germline version of 3D1 retained binding affinity, suggesting that some bnAbs may originate from natural antibodies that undergo limited affinity maturation [52].
Experimental Approaches for Establishing Structural Correlates
Integrating BCR Sequencing with Transcriptional Profiling
Advanced single-cell technologies now enable correlated analysis of BCR sequences and transcriptional states. The Benisse model (BCR embedding graphical network informed by scRNA-seq) integrates BCR sequence information with single-cell gene expression data to reveal functional relationships within B-cell populations [37]. This approach demonstrates a positive correlation between BCR sequence similarity and transcriptional similarity, with B cells in the same clonotype showing more similar expression profiles than those from different clonotypes [37].
Application of Benisse to COVID-19 infection revealed stronger coupling between BCRs and B-cell gene expression during infection, and identified a directed pattern of continuous linear evolution in BCRs compared to the convergent evolution pattern of T-cell receptors [37]. This integrative approach moves beyond sequence-only analysis to connect SHM patterns with functional B-cell states.
Structural Annotation of BCR Repertoires
High-throughput structural annotation methods enable classification of SHM impacts across entire BCR repertoires. The SAAB+ pipeline rapidly annotates BCR sequences with structural information by mapping CDR loops to known structural templates [56]. This approach has revealed that B-cell types can be distinguished based solely on CDR structural properties, with antigen-unexperienced BCR repertoires using the highest number and diversity of CDR structures [56].
Notably, patterns of naïve repertoire paratope usage are highly conserved across individuals, while differentiated B-cells become more personalized in CDR structure usage [56]. This suggests that SHM drives individual-specific structural solutions to common antigenic challenges.
Diagram 2: Experimental workflow for establishing structural correlates of SHM. The integrated approach combines sequence analysis, structural annotation, and multi-modal data integration to connect SHM patterns with functional outcomes through structural mechanisms.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagent Solutions for SHM-Structural Correlation Studies
| Reagent / Platform | Specific Function | Application Context | Key Features |
|---|---|---|---|
| SAAB+ Pipeline [56] | Structural annotation of BCR repertoires | High-throughput structural classification of CDR loops in repertoire sequencing studies | Rapid processing (~4.5M sequences/day); CDR-H3 structural clustering; species-specific template usage |
| Benisse Model [37] | BCR embedding guided by single-cell gene expression | Integrating BCR sequencing with transcriptomic states from scRNA-seq data | Graph-based learning; correlates BCR similarity with transcriptional similarity; reveals B-cell activation gradients |
| HDX-MS Platform [51] | Measurement of local protein dynamics and flexibility | Characterizing SHM-induced changes in antibody paratope structural dynamics | Solution-phase measurements; localizes dynamics changes to specific regions; complements crystallographic data |
| ProtoArray Protein Microarray [57] | High-throughput antibody specificity profiling | Assessing auto- and polyreactivity of unmutated vs. mature antibodies | 9,374 human proteins; internal controls; quantitative comparison of binding profiles |
| AP205 VLP Library [58] | Epitope mapping via peptide display on virus-like particles | Defining minimal epitopes and assessing multivalency effects in antibody recognition | High immunogenicity; well-ordered multimerization; accessible C- and N-termini for epitope fusion |
| ImmuniTree [8] | Phylogenetic modeling of antibody lineage evolution | Reconstructing SHM pathways and identifying functional intermediates from deep sequencing data | Models antibody SHM specifically; identifies less-mutated intermediates with substantial breadth |
The structural correlates linking SHM patterns to epitope recognition reveal a sophisticated evolutionary process wherein antibodies are refined through mutation and selection to achieve optimal complementarity with their target epitopes. Key principles emerging from comparative studies include: (1) SHM optimizes both structural conformation and dynamics of paratopes; (2) different epitope classes impose distinct maturation pathways; and (3) moderate SHM levels can sometimes achieve substantial neutralization breadth, offering hope for vaccine development.
Future research directions should focus on leveraging these structural correlates to design vaccine immunogens that selectively expand B cells with favorable SHM patterns. The integration of structural biology, repertoire sequencing, and single-cell multimodal analysis represents a powerful approach to decipher the complex relationship between antibody sequence, structure, and function. As these methodologies continue to advance, researchers will gain unprecedented ability to guide antibody maturation toward predefined breadth and specificity targets, ultimately enabling development of next-generation vaccines and immunotherapies against challenging viral pathogens.
High-Throughput Antigen Specificity Mapping (LIBRA-seq)
Understanding the relationship between B cell receptor (BCR) somatic hypermutation and the development of neutralization breadth remains a central challenge in immunology and rational vaccine design. Traditional methods for connecting antibody sequence to antigen specificity have been limited by low throughput, typically allowing screening of only a few antigens simultaneously and requiring labor-intensive recombinant antibody expression and validation [59] [60]. The emergence of Linking B cell Receptor to Antigen specificity through sequencing (LIBRA-seq) represents a paradigm shift, enabling high-throughput mapping of paired heavy-/light-chain BCR sequences to their cognate antigen specificities through next-generation sequencing [59]. This technology transforms antibody-antigen interactions into sequence-detectable events by utilizing DNA-barcoded antigens, allowing simultaneous recovery of both BCR sequence and antigen specificity information from thousands of single B cells [59] [61]. Within the broader thesis of B cell receptor somatic hypermutation and neutralization breadth research, LIBRA-seq provides an unprecedented window into the functional consequences of affinity maturation, revealing how specific mutation patterns correlate with broad neutralization potential across diverse pathogen variants.
Fundamental Principles and Workflow
LIBRA-seq fundamentally transforms the problem of detecting physical protein-protein interactions into a sequencing-based readout through DNA barcoding. The core innovation involves conjugating recombinant antigens with unique DNA oligonucleotide barcodes, enabling the simultaneous assessment of BCR sequence and specificity within single-cell RNA sequencing workflows [59] [61]. When a BCR binds its cognate antigen, the associated DNA barcode is captured alongside the BCR mRNA during single-cell library preparation, creating a direct, sequence-based link between antigen specificity and BCR sequence [59].
The LIBRA-seq workflow comprises five key stages, as illustrated below:
The process begins with incubating a B cell population with a panel of DNA-barcoded antigens, all labeled with the same fluorophore to enable enrichment of antigen-binding cells [59] [61]. Antigen-positive B cells are isolated via fluorescence-activated cell sorting (FACS), followed by single-cell encapsulation using droplet microfluidics platforms such as 10x Genomics [61] [62]. Within each droplet, both cellular mRNA (including BCR transcripts) and bound antigen barcodes are tagged with a common cell barcode, preserving the relationship between each cell's BCR sequence and its antigen specificity [59]. Bioinformatic analysis then recovers paired heavy and light chain sequences and maps them to antigen specificities based on the associated barcodes, generating a LIBRA-seq score that quantifies binding strength for each BCR-antigen pair [59].
Key Technological Innovations
LIBRA-seq builds upon several key technological advances that enable its high-throughput capabilities:
DNA-Barcoded Antigens: Each antigen is conjugated with a unique DNA oligonucleotide containing a universal PCR handle, antigen-specific barcode region, and poly(A) tail, making it compatible with standard single-cell RNA-sequencing chemistries [59] [62].
Droplet Microfluidics: Commercial platforms like 10x Genomics enable simultaneous processing of thousands of single cells, dramatically increasing throughput compared to well-based approaches [62].
Multiplexed Antigen Screening: The theoretical number of antigens that can be screened simultaneously is limited only by the diversity of possible barcode sequences, enabling panels of dozens of antigens to be used in a single experiment [59] [60].
Integrated Bioinformatics: Specialized computational pipelines process the sequencing data to associate BCR sequences with antigen barcodes, calculate binding scores, and perform clonal analysis [62].
LIBRA-seq Variants and Technological Evolution
Advanced Methodologies for Enhanced Functionality
The core LIBRA-seq platform has evolved to incorporate additional functional dimensions that provide deeper insights into antibody characteristics. These advanced methodologies significantly enhance the ability to profile neutralizing antibodies without extensive downstream validation.
Table 1: LIBRA-seq Technological Variants and Applications
| Method Variant | Key Innovation | Research Application | Key Advantage |
|---|---|---|---|
| LIBRA-seq with Ligand Blocking [63] | Incorporates DNA-barcoded receptor ligands (e.g., ACE2) alongside antigens | Identification of neutralizing antibodies that block receptor engagement | Direct sequencing-based readout of neutralization potential; 67-86% success rate in identifying SARS-CoV-2 neutralizing antibodies |
| LIBRA-seq with Epitope Mapping [64] | Utilizes antigen panels with point mutations at critical residues | Residue-level epitope determination for thousands of B cells simultaneously | High-throughput mapping of antibodies to specific epitopes of interest (e.g., HIV-1 Env CD4-binding site) |
| Next-Generation Antigen Barcoding [62] | Optimized barcode designs and processing protocols | Improved recovery of rare antigen-specific B cells (e.g., bnAb precursors) | Enhanced sensitivity for detecting rare B cell populations; streamlined computational framework |
Visualizing LIBRA-seq with Ligand Blocking
LIBRA-seq with ligand blocking incorporates DNA-barcoded receptor proteins alongside viral antigens, enabling simultaneous detection of antigen binding and receptor blocking capability at sequencing scale [63]. The workflow and decision logic for this advanced variant can be visualized as follows:
This ligand-blocking approach enables direct identification of B cells expressing antibodies that simultaneously bind viral antigens and prevent receptor engagement—a key characteristic of potent neutralizing antibodies. During analysis, B cells with high LIBRA-seq scores for the target antigen but low scores for the ligand represent those producing antibodies capable of blocking the antigen-ligand interaction [63]. This method demonstrated remarkable efficiency in SARS-CoV-2 antibody discovery, with 67-86% of antibodies predicted to block ACE2 binding showing actual neutralizing activity in validation assays [63].
Performance Comparison: LIBRA-seq vs. Alternative Technologies
Direct Comparison of B Cell Profiling Technologies
LIBRA-seq occupies a unique position in the landscape of B cell profiling technologies, offering distinct advantages and limitations compared to established methods. The table below provides a comprehensive comparison of key technologies used to interrogate human B cell responses.
Table 2: Performance Comparison of B Cell Profiling Technologies
| Technology | BCR Sequence Recovery | Antigen Specificity | Throughput | Neutralization Function | Key Applications |
|---|---|---|---|---|---|
| LIBRA-seq [59] [65] | Yes (paired heavy/light) | Yes (multiplexed) | High (thousands of cells) | With ligand blocking variant | High-throughput antibody discovery, repertoire analysis |
| LIBRA-seq with Ligand Blocking [63] | Yes (paired heavy/light) | Yes (multiplexed) | High (thousands of cells) | Direct assessment | Neutralizing antibody discovery, vaccine development |
| Antigen-Specific Single-Cell Sorting [60] [65] | Yes (paired heavy/light) | Yes (limited) | Low (tens to hundreds) | Requires validation | Targeted antibody isolation, mAb development |
| CITE-seq [65] | Yes (with RNA-seq) | Limited (surface markers) | High (thousands of cells) | No | Phenotypic profiling, subset identification |
| CyTOF [65] | No | Limited (phenotypic) | High (thousands of cells) | No | Deep immunophenotyping, signaling analysis |
| Spec-Seq [65] | Yes (with RNA-seq) | No (requires validation) | Medium (hundreds of cells) | Requires validation | Transcriptome-BCR coupling, subset function |
Quantitative Performance Metrics
LIBRA-seq demonstrates significant advantages in key performance metrics relevant to B cell receptor research:
Throughput: LIBRA-seq routinely recovers thousands of antigen-specific B cells per experiment, with studies reporting 828-957 antigen-specific B cells in single experiments [63] and up to 2321 cells with BCR sequence and antigen mapping information in validation studies [59].
Efficiency in Neutralizing Antibody Discovery: LIBRA-seq with ligand blocking dramatically improves hit rates for neutralizing antibodies, with 67-86% of selected antibodies demonstrating neutralizing activity compared to traditional rates of 2-55% with conventional antigen bait approaches [63].
Multiplexing Capacity: LIBRA-seq panels have successfully incorporated 5-10 antigens simultaneously [59] [61], with the theoretical limit constrained mainly by antigen competition for BCR binding rather than barcode diversity [62].
Sensitivity for Rare B Cells: Optimized protocols enable isolation of extremely rare B cell populations, including precursors of broadly neutralizing antibody classes such as VRC01 and IOMA with frequencies below 0.01% [62].
Application Case Studies in Pathogen Research
HIV-1 Research: Uncovering Broadly Neutralizing Antibodies
LIBRA-seq has proven particularly valuable in HIV-1 research, where it has accelerated the discovery of broadly neutralizing antibodies (bNAbs) that target multiple viral strains. In a foundational study, researchers applied LIBRA-seq to peripheral blood mononuclear cells from two HIV-infected donors using a panel of HIV Env proteins and influenza hemagglutinin as a control antigen [59]. The approach successfully identified 29 BCRs clonally related to the VRC01-class bNAb lineage, with 86% showing high LIBRA-seq scores for HIV-1 antigens [59]. Subsequent recombinant expression and validation confirmed binding for selected antibodies, demonstrating LIBRA-seq's accuracy in predicting antigen specificity [59].
Notably, LIBRA-seq enabled the discovery of a novel broadly neutralizing HIV-1 antibody, 3602-870, which bound all five diverse HIV-1 Env antigens in the screening panel but not influenza antigens [61]. The LIBRA-seq scores showed high correlation with ELISA validation data, confirming the robustness of the sequencing-based specificity assessment [61]. This application highlights LIBRA-seq's power to rapidly identify cross-reactive B cells from complex polyclonal samples, providing crucial insights into how somatic hypermutation patterns correlate with neutralization breadth.
SARS-CoV-2 Response: Rapid Antibody Discovery During Pandemic
The COVID-19 pandemic showcased LIBRA-seq's utility in rapid response scenarios. Researchers applied LIBRA-seq with ligand blocking to B cells from convalescent COVID-19 donors, using SARS-CoV-2 spike protein and DNA-barcoded ACE2 as screening reagents [63]. This approach efficiently identified B cells producing antibodies that bound spike protein while blocking ACE2 interaction—a key characteristic of potent neutralizing antibodies.
The results demonstrated LIBRA-seq's exceptional efficiency: 57% of antibodies from experiment 1 and 67% from experiment 3 demonstrated ACE2 blocking via ELISA, with 86% and 67% respectively showing neutralizing activity in viral neutralization assays [63]. Notably, the ACE2 LIBRA-seq scores significantly correlated with reduction in ACE2 binding (Spearman r = -0.54, p = 0.017), validating the sequencing-based functional prediction [63]. This case study underscores how LIBRA-seq's integrated sequence-function approach can dramatically accelerate therapeutic antibody discovery during emerging outbreaks.
Coronavirus Cross-Reactivity and Pediatric Responses
LIBRA-seq has also illuminated aspects of B cell responses to coronaviruses across different populations. In one study, researchers used LIBRA-seq to characterize SARS-CoV-2-specific antibody repertoires in children aged 5 months to 18 years [66]. The analysis revealed that children generate antibodies with similar genetic features and public clonotypes to adults, yet these antibodies showed potent neutralization of diverse SARS-CoV-2 variants, including those resistant to most approved monoclonal antibody therapeutics [66].
In another application, LIBRA-seq identified cross-reactive antibodies capable of binding both SARS-CoV-2 and SARS-CoV spike proteins while blocking ACE2 interaction [63]. From 120 IgG+ B cells with high LIBRA-seq scores for both spike proteins, researchers identified a subset with low ACE2 scores; subsequent validation showed 88% exhibited the predicted cross-reactivity and 63% demonstrated strong ACE2-blocking ability [63]. These findings highlight LIBRA-seq's utility in profiling cross-reactive B cell responses, informing both therapeutic development and vaccine design strategies against related viral families.
The Scientist's Toolkit: Core Components for LIBRA-seq
Successful implementation of LIBRA-seq requires careful selection and quality control of key research reagents. The table below outlines essential components and their functional requirements.
Table 3: Essential Research Reagents for LIBRA-seq Implementation
| Reagent Category | Specific Examples | Functional Requirements | Quality Control Considerations |
|---|---|---|---|
| DNA-Barcoded Antigens | HIV-1 Env SOSIP trimers [59], SARS-CoV-2 Spike [63], Influenza HA [59] | Proper folding, specific barcode assignment, controlled labeling ratio | Antigenicity validation (ELISA, BLI), barcode uniqueness confirmation, functional assessment |
| Barcoded Ligands | ACE2 for SARS-CoV-2 studies [63] | Native binding conformation, specific barcode | Binding affinity validation, competition assay verification |
| Cell Sorting Reagents | Fluorophore conjugate (same for all antigens) [59], viability dyes, surface marker antibodies | Bright fluorophore (e.g., PE, Alexa Fluor), minimal spectral overlap | Titration to determine optimal staining concentration, validation with control cells |
| Single-Cell Library Prep | 10x Genomics 5' Gene Expression with Feature Barcoding [61] [62] | Feature barcoding compatibility, cell viability preservation | Cell number quantification, viability assessment (>90% recommended) |
| Positive Control Cells | Ramos B cell lines expressing known BCRs [59] | Defined specificity, stable BCR expression | Regular validation of antigen binding, monitoring of BCR expression levels |
| Bioinformatic Tools | LIBRA-seq analysis pipeline [62], scab for multi-omics [62] | Barcode demultiplexing, UMI counting, BCR assembly | Positive control analysis, cross-sample normalization |
LIBRA-seq represents a transformative methodology in the field of B cell biology, particularly for research investigating the relationship between somatic hypermutation and neutralization breadth. By simultaneously capturing BCR sequence and antigen specificity information from thousands of single cells, this technology enables unprecedented insights into how antibody diversity translates to functional breadth against diverse pathogens. The continued evolution of LIBRA-seq—through ligand blocking, epitope mapping, and optimized barcoding strategies—promises to further accelerate therapeutic antibody discovery and rational vaccine design. As the technology becomes more widely adopted and integrated with other single-cell modalities, it will continue to illuminate the complex relationship between BCR genetics, affinity maturation, and protective immunity, ultimately advancing our ability to harness the immune system against challenging pathogens.
Vaccine Design Challenges: Steering SHM Toward Broad Neutralization
The process of somatic hypermutation (SHM) is a cornerstone of adaptive immunity, driving the evolution of antibodies with enhanced pathogen-fighting capabilities. During affinity maturation within germinal centers, B cells undergo iterative cycles of mutation and selection, ultimately producing antibodies with superior affinity and specificity. A critical area of modern immunology research focuses on the relationship between the level of SHM and the development of neutralization breadth—the ability of antibodies to recognize diverse pathogen variants. This guide examines the experimental evidence underlying the SHM-breadth tradeoff, comparing data across different pathogens and vaccination strategies to inform vaccine design and therapeutic antibody development.
Quantitative Evidence: Correlating SHM with Neutralization Breadth
Substantial clinical and experimental evidence demonstrates that increased somatic hypermutation is frequently associated with enhanced antibody breadth across multiple pathogen systems. The following table summarizes key findings from recent studies.
Table 1: Documented Correlations Between SHM and Neutralization Breadth
| Pathogen / Condition | SHM Measurement | Breadth Assessment | Key Correlation Finding | Reference |
|---|---|---|---|---|
| SARS-CoV-2 (Ad26.COV.2S vaccine) | Nucleotide changes in IgVH & IgVL over 8 months | Serum neutralization of variants (B.1.351, B.1.617.2) | Significant positive correlation (B.1.351: r=0.60, p=0.0011; B.1.617.2: r=0.38, p=0.049) [20] | [20] |
| SARS-CoV-2 (mRNA boost) | Amino acid mutation count in mAbs from memory B cells | Neutralization IC50 against variants (e.g., Omicron) | Antibodies from 3rd dose (more mutated) showed significantly increased potency and breadth versus 2nd dose [67] | [67] |
| HIV (Controllers) | IGHV and IGLV mutation frequency in Env-specific MBCs | Serum neutralization % of tier 2/3 virus panel | Frequency of genomic mutations directly correlated with serum neutralization breadth [32] | [32] |
| SARS-CoV-2 (Repeated Exposure) | SHM level in cross-reactive B cell clonotypes | Neutralization of SARS-CoV-2 variants & sarbecoviruses | IGHV3-74 mAbs developing cross-sarbecovirus neutralization were isolated after immune recalls, implying SHM role [68] | [68] |
Experimental Protocols for SHM-Breadth Analysis
Researchers employ several sophisticated methodologies to quantify SHM and link it to functional antibody breadth.
Longitudinal Tracking of SHM and Serum Neutralization
This protocol, used in studies of vaccines like Ad26.COV.2S, tracks the natural evolution of the antibody response over time [20].
- B Cell Isolation and Sequencing: At multiple time points post-vaccination (e.g., 1, 3, and 8 months), peripheral blood mononuclear cells (PBMCs) are collected. Spike-specific memory B cells are sorted using labeled antigens (e.g., Spike protein or RBD). Single-cell RNA sequencing is performed to obtain paired heavy- and light-chain variable region sequences [20].
- SHM Quantification: The obtained antibody sequences are aligned to germline human BCR genes using databases like IMGT. The number of nucleotide changes per variable region (IgVH and IgVL) from the germline sequence is calculated, providing a measure of SHM [20].
- Serum Neutralization Assay: A pseudovirus neutralization assay is employed. Serum samples collected concurrently with PBMCs are tested against a panel of pseudoviruses expressing spikes from different viral variants (e.g., ancestral, Beta, Delta). The half-maximal neutralization titer (NT50) is calculated for each variant [20].
- Correlation Analysis: Statistical analysis (e.g., Pearson correlation) is performed to relate the median SHM level in individuals to their serum NT50 values against variants, establishing a population-level SHM-breadth relationship [20].
Functional Validation of Clonally-Evolved Antibodies
This approach moves beyond correlation to directly test the functional impact of mutations isolated from evolved B cells.
- Monoclonal Antibody (mAb) Generation: Representative antibody sequences from sorted B cells (e.g., from pre- and post-boost time points) are recombinantly expressed as monoclonal antibodies [67].
- High-Throughput Neutralization Screening: The panel of mAbs is screened for neutralization potency (IC50) and breadth against a wide spectrum of viral variants using standardized pseudovirus assays [67].
- Comparative Potency Analysis: The neutralization profiles of mAbs derived from less-mutated (e.g., after primary vaccination) and highly-mutated (e.g., after boost or chronic infection) B cells are compared. This directly tests whether antibodies accumulating more SHM possess superior breadth and potency [67] [32].
Computational Modeling of Affinity Maturation
Computational models provide a theoretical framework to understand SHM dynamics and optimize vaccination protocols.
- Agent-Based Simulation: These models simulate the germinal center reaction. Virtual B cells, each with a unique BCR, undergo cycles of proliferation, mutation (with set probabilities for silent, deleterious, or enhancing mutations), and affinity-based selection based on interaction with antigens and T follicular helper cells [69] [15].
- Protocol Optimization: Researchers can simulate different vaccination strategies, such as sequential immunization with variant proteins versus mixture immunization. The model outputs, like the average breadth of evolved antibodies, can identify protocols that best guide SHM toward bnAb development [69].
- Modeling SHM Regulation: More recent models incorporate hypotheses like affinity-dependent mutation rates, where B cells receiving stronger T cell help divide more but mutate less per division. These models are tested against experimental data from mouse immunization studies to validate proposed mechanisms [15].
The workflow for the first two experimental protocols can be visualized as follows:
Mechanisms Underlying the SHM-Breadth Tradeoff
The positive relationship between SHM and breadth is not accidental but is driven by specific immunological mechanisms.
Targeting Conserved Epitopes
Broadly neutralizing antibodies often target cryptic, conserved regions of viral proteins (e.g., the CD4 binding site on HIV). These epitopes can be difficult to access sterically or are shielded by variable loops and glycans. Achieving high-affinity binding to these sites frequently requires numerous mutations to refine the antibody paratope, enabling it to navigate around variable regions and make precise contacts with conserved residues [69]. Studies of mRNA vaccine boosts found that newly developing B cell clones after the third dose differed from persisting clones by targeting more conserved regions of the RBD, a process enabled by ongoing SHM [67].
Sequential Antigen Exposure
Natural infection with highly mutable pathogens or sequential vaccination with variant antigens presents the immune system with a series of related but distinct selection pressures. This "conflicting selection" favors B cells whose binding to the antigen is dominated by interactions with the shared, conserved parts of the protein. Sequential immunization drives the immune system further from its steady state in an optimal fashion, allowing B cells to become bnAbs via diverse evolutionary paths [69].
Regulation of SHM to Preserve High-Affinity Lineages
Emerging evidence suggests that the germinal center response may be optimized to protect high-affinity lineages from the detrimental effects of random mutation. A proposed model of regulated SHM suggests that B cells expressing high-affinity antibodies, which receive strong T cell help, may undergo more cell divisions but with a lower mutation rate per division. This strategy allows for expansive clonal growth of the fittest B cells without accumulating as many deleterious mutations, thereby enhancing the overall efficiency of affinity maturation [15].
The Scientist's Toolkit: Essential Research Reagents and Models
Investigating the SHM-breadth relationship requires a suite of specialized reagents, computational tools, and model systems.
Table 2: Key Research Tools for SHM and Breadth Analysis
| Tool / Reagent | Primary Function | Application in SHM-Breadth Research |
|---|---|---|
| Fluorescently-Labeled Antigens | Flow cytometric sorting of antigen-specific B cells | Isolation of pathogen-specific (e.g., SARS-CoV-2 Spike, HIV Env) memory B cells for subsequent sequencing and analysis [20] [32] |
| Single-Cell BCR Sequencing | High-throughput sequencing of paired B cell heavy and light chains | Provides the raw sequence data necessary for quantifying SHM load and tracking clonal lineages [20] [15] |
| Pseudovirus Neutralization Assay | Safe measurement of neutralizing antibody potency | Benchmarking serum neutralization breadth or testing the breadth of individual mAbs against a panel of viral variants [20] [67] |
| H2B-mCherry Reporter Mice | Experimental tracking of cell division history in vivo | Studying the relationship between number of B cell divisions, acquired mutations, and affinity in germinal centers [15] |
| Agent-Based GC Models | Computational simulation of affinity maturation | Theoretical exploration of vaccination protocols (e.g., sequential immunization) to optimize for bnAb development [69] [15] |
| SHM Modeling Software (e.g., netam) | Probabilistic modeling of mutation biases | Predicting the likelihood of specific amino acid changes during SHM, relevant for vaccine design [70] [24] |
The collective evidence from studies of HIV, SARS-CoV-2, and other pathogens solidly supports a model where cumulative somatic hypermutation is a critical driver for the acquisition of neutralizing antibody breadth. While the optimal "level" of SHM is context-dependent, the consensus is that extended affinity maturation, often fueled by repeated or sequential antigen exposure, is necessary for B cells to refine their paratopes to target functionally constrained, conserved epitopes effectively. Future research, leveraging the sophisticated tools and protocols outlined here, will focus on designing vaccine regimens that actively steer this evolution to efficiently generate protective, broad antibodies against the world's most challenging mutable pathogens.
A central challenge in modern vaccinology lies in overcoming B cell immunodominance—the phenomenon where antibody responses preferentially target specific, often highly variable epitopes on complex protein antigens. This asymmetric targeting presents a fundamental barrier to eliciting broadly neutralizing antibodies (bnAbs), which typically target functionally conserved epitopes but constitute only a minor fraction of the overall repertoire. These rare bnAb clonotypes are often immunologically subdominant, outcompeted by B cells targeting immunodominant, variable epitopes that do not confer broad protection. Understanding the correlation between B cell receptor somatic hypermutation and neutralization breadth requires sophisticated analytical frameworks to dissect the complex evolutionary pathways leading to bnAb development. This guide compares the leading experimental and computational approaches designed to overcome immunodominance hierarchies, providing researchers with objective performance comparisons and detailed methodologies to advance next-generation vaccine design.
Theoretical Framework: Linking SHM Patterns to Neutralization Breadth
Somatic Hypermutation as a Driver of Antibody Breadth
Somatic hypermutation (SHM) represents the diversity-generating process in antibody affinity maturation, occurring at rates approximately 10⁶-fold higher than background somatic mutation rates. The mutation biases are predictable from local sequence context, with recent models expanding beyond traditional 5-mer contexts to wider nucleotide frameworks using parameter-efficient convolutional neural networks. These "thrifty" models demonstrate that SHM patterns contain predictive information about the subsequent selection forces that drive antibody evolution toward breadth. Advanced probabilistic models of SHM are now essential for analyzing rare mutations, understanding selective forces guiding affinity maturation, and computing models of natural selection on antibodies [71].
Analytical Challenges in Clonal Family Identification
Accurately identifying B cell clonal families from next-generation sequencing data remains challenging, as members of a B cell clone share the same initial V(D)J rearrangement but accumulate SHMs during affinity maturation. Traditional methods focused primarily on junction region similarity, but newer approaches like SCOPer (Spectral Clustering for clOne Partitioning) leverage shared SHMs in V and J segments alongside junction sequences to improve sensitivity and specificity for identifying clonally related sequences [72]. The definition of clonal relationships significantly impacts diversity measurements, with different grouping methods leading to substantially different clonal definitions and diversity quantifications [73].
Table: Comparison of Clonal Identification Methods
| Method | Key Features | Advantages | Limitations |
|---|---|---|---|
| Germline Alignment-Based | Uses reference V/J gene alignments with CDR3 similarity | High accuracy with complete sequence information | Prone to alignment errors with short reads |
| Spectral Clustering (SCOPer) | Combines junction similarity with shared SHMs | Improved sensitivity/specificity; accounts for mutation patterns | Higher computational complexity |
| Alignment-Free NLP Techniques | Uses natural language processing independent of gene alignments | Suitable for shorter sequencing reads; alignment-free | May overlook evolutionary relationships |
Comparative Analysis of Immunogen Design Strategies
Multimeric display strategies leverage the repetitive presentation of viral antigens to increase BCR crosslinking and amplify humoral responses. Ferritin-based nanoparticles displaying trimeric influenza HA antigens elicit higher antigen-specific titers with increased breadth and protection compared to recombinant trimeric HAs, demonstrating a shift toward cross-reactive and subdominant responses. Similarly, SpyTag-SpyCatcher technology enables spontaneous covalent linkage of antigens to nanoparticle scaffolds, creating an easily modifiable 'plug and display' approach for presenting diverse antigens [74]. Despite significant benefits, these approaches introduce additional epitopes that may expand interclonal competition and potentially skew immunodominance hierarchies away from subdominant epitopes of interest.
Table: Multimeric Display Platforms for Immunogen Design
| Platform | Mechanism | Applications | Impact on Subdominant Responses |
|---|---|---|---|
| Ferritin Nanoparticles | Genetic fusion at three-fold symmetry axis | Influenza HA, RSV F, HIV Env | Increases stem-directed titers and breadth |
| SpyTag-SpyCatcher | Covalent isopeptide bond formation | Betacoronavirus RBDs, influenza HAs | Enables heterologous antigen display |
| Synthetic Liposomes | Mixed antigen presentation on lipid bilayers | Various viral glycoproteins | Amplifies serum titers relative to recombinant protein |
| Virus-Like Particles | Native viral structure mimicry | Hepatitis B, flock house virus | Presents small, linear peptides effectively |
Restricting Off-Target Responses
Epitope removal represents a direct approach to modulating immunodominance by physically eliminating undesired epitopes, thereby reducing the pool of competing B cells. Domain-based constructs such as 'mini-HAs' (headless, stem-only domains) and 'engineered outer domain' (eOD) Env constructs maintain target epitope integrity while minimizing off-target epitopes [74]. Complementary to this approach, steric occlusion uses immune complexes between antibodies and antigens to mask epitopes, with recent refinements employing single-chain variable fragments (scFvs) or covalently stabilized complexes through chemical crosslinking. This shielding biases antibody responses toward desired epitopes by physically preventing BCR engagement with off-target regions.
Rational Antibody Discovery Platforms
Rational Antibody Discovery platforms systematically identify optimal target epitopes and immunoengineer immune responses that result in the majority of produced antibodies hitting desired epitopes. These platforms overcome immunodominance by conditionally modifying B cells during immunization to direct optimal antibody responses to epitopes of interest determined with AI-aided algorithms [75]. This approach represents a significant departure from classical methods, where the majority of antibodies produced miss optimal epitopes due to immunodominance hierarchies.
Experimental Protocols and Methodologies
Spectral Clustering for B Cell Clonal Partitioning
The SCOPer method employs independent distance functions that capture junction similarity and shared mutations, combining these in a spectral clustering framework to infer BCR clonal relationships [72].
Protocol Details:
- Sequence Preprocessing: Annotate V(D)J genes using IMGT/HighV-QUEST or similar tools
- Distance Calculation: Compute two independent distance matrices:
- Junction region distance based on nucleotide similarity
- Shared mutation distance based on pattern of SHMs in V and J segments
- Spectral Clustering: Apply spectral clustering to the combined distance matrix to partition sequences into clonal families
- Validation: Use known lineage relationships from simulations or experimental data to optimize parameters
Performance Metrics: Using both simulated and experimental data, this model improves both sensitivity and specificity for identifying B cell clones compared to junction-only methods.
High-Throughput Single-Cell BCR Sequencing
Massively parallel single-cell B-cell receptor sequencing (scBCR-seq) enables rapid discovery of diverse antigen-reactive antibodies by obtaining accurately paired full-length variable regions from individual B cells [76].
Experimental Workflow:
- Cell Encapsulation: Isolate individual B cells using emulsion-based encapsulation
- cDNA Generation: Generate 5' barcoded cDNA from thousands of individual B cells in parallel
- Targeted Amplification: Amplify VH and VL regions using custom primers while retaining cell barcode
- Library Preparation: Shear 5' barcoded cDNAs and convert into sequencing-ready libraries
- Bioinformatic Analysis:
- Cell detection and de novo contig assembly
- Variable domain annotation and pairing of full-length VH and VL sequences
- Filter for open reading frames encoding entire variable regions
Quality Control: Require minimum read support (10 for VH, 100 for VL) and one dominant VH and VL contig (≥80% read support). This approach has demonstrated 99% antigen-reactivity confirmation when expressed clones are tested [76].
scBCR-seq Experimental Workflow: This diagram illustrates the key steps in massively parallel single-cell B-cell receptor sequencing, from cell isolation to final paired BCR sequences.
Thrifty Wide-Context SHM Modeling
Recent advances in somatic hypermutation modeling use convolutional neural networks to develop "thrifty" models with wide nucleotide context yet fewer parameters than traditional 5-mer models [71].
Method Implementation:
- Data Preparation: Process out-of-frame BCR sequences to minimize selective pressure confounding
- Phylogenetic Reconstruction: Cluster sequences into clonal families and infer ancestral sequences
- Parent-Child Pairing: Split phylogenetic trees into parent-child sequence pairs for mutation analysis
- Model Architecture:
- Map each 3-mer into an embedding space with trainable parameters
- Apply convolutions to capture wider context without exponential parameter growth
- Model mutation process as Exponential waiting time with rate λᵢ per site
- Training: Optimize models to predict probability of observed SHM in child sequences relative to parents
Applications: These models enable prediction of amino acid change probabilities in affinity maturation, crucial for understanding prospects of selecting mutations that lead to bnAbs in reverse vaccinology approaches.
The Scientist's Toolkit: Essential Research Reagents
Table: Key Reagent Solutions for B Cell Immunodominance Research
| Reagent/Platform | Function | Application Context |
|---|---|---|
| SCOPer R Package | Spectral clustering for clonal partitioning | Identifies B cell clones using junction similarity and shared SHMs [72] |
| Netam Python Package | SHM modeling with wide context | Predicts somatic hypermutation probabilities using thrifty models [71] |
| scBCR-seq Platform | Single-cell paired VH-VL sequencing | Obtains full-length variable regions from individual B cells at scale [76] |
| Ferritin Nanoparticles | Multimeric antigen display | Presents trimeric antigens with precise geometry for subdominant epitope focusing [74] |
| SpyTag-SpyCatcher System | Covalent antigen coupling | Enables 'plug-and-display' antigen attachment to nanoparticle scaffolds [74] |
| Rational Antibody Discovery Platform | Epitope-focused antibody discovery | Immunoengineers responses toward predetermined conserved epitopes [75] |
Pathway to bnAb Development: A Conceptual Framework
bnAb Development Pathway: This conceptual diagram outlines the critical steps in the development of broadly neutralizing antibodies, highlighting key bottlenecks where immunogen design strategies can intervene to promote rare clonotypes.
Overcoming immunodominance to promote rare bnAb clonotypes requires an integrated approach combining structural biology, protein engineering, and advanced computational analysis of B cell repertoire dynamics. The most promising strategies include epitope-focused immunogens that direct responses toward conserved regions, multimeric display systems that enhance subdominant epitope immunogenicity, and rational antibody discovery platforms that bypass natural immunodominance hierarchies. Critical to these efforts are advanced analytical methods for clonal lineage tracking and SHM pattern analysis, which enable researchers to reconstruct the evolutionary paths leading to breadth and identify the rare mutational patterns that confer neutralization breadth. As these technologies mature, the systematic navigation of immunodominance landscapes will become increasingly central to vaccine development for rapidly evolving pathogens such as HIV, influenza, and betacoronaviruses.
Age-Related Limitations in SHM Efficiency and Repertoire Diversity
Somatic hypermutation (SHM) is a fundamental process in adaptive immunity, driving the affinity maturation of B cell receptors (BCRs) and antibodies within germinal centers. This programmed mechanism introduces point mutations into the variable regions of immunoglobulin genes, enabling B cells to generate high-affinity antibody receptors crucial for effective pathogen neutralization [77]. The efficiency of SHM and the resulting diversity of the B cell repertoire are not static throughout life; they undergo significant changes from early development through advanced age. Understanding these age-related trajectories is essential for elucidating the molecular underpinnings of immunosenescence and its impact on vaccine efficacy and infection control, particularly within research focused on correlating BCR somatic hypermutation with the development of neutralization breadth [77] [78]. This guide synthesizes current experimental data and methodologies to objectively compare SHM performance across age groups, providing a resource for researchers and drug development professionals.
Quantitative Comparison of SHM Across the Lifespan
Early-Life Development of SHM
The SHM machinery becomes active shortly after birth, with efficiency increasing rapidly during the first two years of life. This period represents a critical window for the maturation of the adaptive immune system.
Table 1: SHM Development in Early Life
| Age Group | Mean SHM Level (%) | Key Findings | Experimental Method |
|---|---|---|---|
| Neonates (Cord Blood) | Low (Baseline) | Foundation of the memory B cell compartment is established. | Igκ-REHMA [77] |
| First 2 Years | Rapid increase to ~68% | SHM level rises steeply, reflecting immune maturation and expansion of memory B cell subsets. | Igκ-REHMA [77] |
| Childhood (>2 years) | 68% (Range: 38-89%) | SHM levels stabilize, correlating with a relatively stable memory B cell population. | Igκ-REHMA [77] |
Adult Aging and Sex-Biased Senescence of SHM
In adulthood, SHM efficiency plateaus before exhibiting signs of senescence in older age. Notably, recent research reveals that this decline is not uniform and shows significant sexual dimorphism.
Table 2: SHM Alterations in Older Age
| Subject Group | Key SHM Alteration | Impact on BCR Phenotype | Experimental Method |
|---|---|---|---|
| Older Males | ↓ Frequency of Phase II (MMR/BER)-induced mutations; ↓ Expression of MMR genes (e.g., MSH2, MSH6). | Altered immunoglobulin amino acid composition; potential for reduced affinity and breadth. | AIRR-seq [78] |
| Older Females | SHM mutability hierarchies and Phase II mutation frequency are largely maintained. | Relative preservation of SHM quality and resulting BCR diversity compared to age-matched males. | AIRR-seq [78] |
| CVID Patients | Lower SHM (Mean: 31%, Range: 6-77%) | Serves as a pathological benchmark for impaired SHM, often accompanied by diminished memory B cells. | Igκ-REHMA [77] |
Detailed Experimental Protocols for SHM Assessment
The Igκ-Restriction Enzyme Hot-Spot Mutation Assay (Igκ-REHMA)
The Igκ-REHMA protocol provides a quantitative estimate of the overall SHM level in a peripheral blood B cell population [77].
Workflow:
- RNA Isolation & cDNA Synthesis: Total RNA is extracted from peripheral blood mononuclear cells (PBMCs) or sorted B cell subsets, followed by reverse transcription to generate cDNA.
- Fluorescent PCR Amplification: A nested or standard PCR is performed using HEX-coupled Vκ3-20U forward primer and 6-carboxyfluorescein (FAM)-coupled Vκ3-20 L reverse primer. This amplifies the rearranged Vκ3-20 gene segments.
- Restriction Enzyme Digestion: The PCR products are digested with the restriction enzymes DdeI and Fnu4HI. These enzymes specifically cut at unmutated hot-spot motifs within the amplified sequence.
- Capillary Electrophoresis: The digested fragments are separated by size using a capillary sequencer (e.g., ABI3130XL).
- Data Analysis: Unmutated gene products are visualized as short (106/109-bp) FAM-coupled fragments. Mutated gene products, which resist digestion due to mutations in the enzyme recognition site, are visualized as a long (244-bp) FAM-coupled fragment. The SHM level is estimated as the percentage of the 244-bp fragment within the total FAM-coupled products [77].
Igκ-REHMA Workflow: This diagram illustrates the key steps for estimating SHM levels using the Igκ-REHMA protocol, from sample processing to data analysis.
Adaptive Immune Receptor Repertoire Sequencing (AIRR-seq)
AIRR-seq provides a high-resolution, in-depth view of SHM patterns, allowing for the analysis of mutation targeting and repair pathways [78].
Workflow:
- Sample Preparation: Genomic DNA or cDNA is obtained from B cell populations (e.g., sorted memory B cells or PBMCs).
- Library Construction: Ig gene loci (e.g., IGHV, IGKV) are amplified using multiplexed primers in a bias-controlled manner. Sequencing adapters and barcodes are added for high-throughput sequencing.
- High-Throughput Sequencing: Libraries are sequenced on platforms like Illumina to generate millions of paired-end reads.
- Bioinformatic Processing:
- Quality Control & Assembly: Raw reads are quality-filtered and assembled using tools like pRESTO.
- Germline Assignment: V(D)J gene segments are assigned, and the germline sequence is inferred using IMGT/HighV-QUEST or IgBLAST.
- Clonotype Grouping: Sequences are partitioned into clonal groups based on shared IGHV, IGHJ genes, and junction length using the Change-O toolkit.
- Mutation Analysis: Mutations are identified by comparing each sequence to its inferred germline. Advanced tools like shazam in R are used to build 5-mer targeting models and analyze substitution patterns to distinguish Phase Ia, Ib, and Phase II mutations [78] [79].
AIRR-seq Analysis Pipeline: This diagram outlines the computational workflow for processing AIRR-seq data to analyze SHM patterns at single-nucleotide resolution.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for SHM and Repertoire Research
| Reagent / Solution | Function in Experimental Protocol |
|---|---|
| Vκ3-20 Primers (HEX/FAM) | Fluorescently-labeled primers for specific amplification of a common Ig light chain rearrangement in the Igκ-REHMA assay [77]. |
| Restriction Enzymes (DdeI, Fnu4HI) | Digest unmutated PCR products at specific hot-spot motifs to distinguish mutated from unmutated sequences in Igκ-REHMA [77]. |
| Multiplex Ig Primers | Amplify a broad spectrum of V(D)J rearrangements from complex samples for comprehensive AIRR-seq library generation [78] [79]. |
| pRESTO Pipeline | A suite of computational tools for pre-processing raw immune repertoire sequencing data, including quality control, filtering, and assembly [78]. |
| IMGT/HighV-QUEST | The international standard for annotating Ig sequences, assigning V/D/J genes, and identifying mutations [78] [79]. |
| Change-O & shazam | Bioinformatics toolkits for advanced repertoire analysis, including clonal lineage assignment and quantitative profiling of SHM targeting [78]. |
Molecular Mechanisms and Aging-Related SHM Dysregulation
SHM is initiated by Activation-Induced Cytidine Deaminase (AID), which deaminates cytosine to uracil in Ig genes. The subsequent DNA repair processes are categorized into two phases. Phase I mutations occur at the original C/G base pairs: Phase Ia (C-to-T transitions via replication) and Phase Ib (mutations at C/G from base excision repair). Phase II mutations result from error-prone mismatch repair, introducing mutations at A/T bases and neighboring nucleotides [78]. Aging, particularly in males, is associated with a documented decline in the expression of key Mismatch Repair (MMR) genes like MSH2 and MSH6. This molecular shift leads to the observed reduction in Phase II mutations, altering the quality and potential functionality of the antibody repertoire [78].
SHM Mechanism & Age-Related Decline: This diagram shows the core phases of somatic hypermutation and highlights the specific Phase II pathway that is impaired with age, particularly in males.
Immunogen Design Strategies to Guide Desired Mutation Pathways
The correlation between B cell receptor (BCR) somatic hypermutation (SHM) and the development of neutralization breadth represents a foundational principle in modern vaccinology. Somatic hypermutation is a programmed process introducing point mutations into the variable regions of immunoglobulin genes at rates approximately 10^6-fold higher than background mutation rates, serving as the primary engine for antibody affinity maturation [80]. While traditional vaccine development often relied on empirical approaches, a new paradigm has emerged wherein immunogens are strategically designed to guide B cell lineages along specific mutation pathways that culminate in broadly neutralizing antibodies (bNAbs) against challenging pathogens like HIV-1 and SARS-CoV-2 [16] [81] [82].
The fundamental challenge in eliciting bNAbs through vaccination lies in the extensive somatic mutation typically required for broad neutralization. For instance, VRC01-class antibodies against HIV-1 can contain up to ~40% mutations from their germline-encoded sequences [82]. Furthermore, recent research has revealed that the relationship between SHM and affinity maturation is more sophisticated than previously recognized. A 2025 study demonstrated that B cells producing high-affinity antibodies can actually reduce their mutation rates per cell division, thereby protecting beneficial BCR configurations from the accumulation of deleterious mutations during clonal expansion [15]. This nuanced understanding of SHM regulation provides critical insights for designing immunogens that can steer B cell responses toward desired outcomes. The strategic design of immunogens to guide specific mutation pathways represents the cutting edge of reverse vaccinology, enabling researchers to overcome the natural immune evasion tactics employed by rapidly mutating viruses [16] [83].
Comparative Analysis of Immunogen Design Strategies
Strategic Approaches and Experimental Evidence
Immunogen design strategies have evolved from simple antigen presentation to sophisticated engineering approaches that actively guide B cell receptor evolution. The table below compares three prominent strategies, their molecular mechanisms, and supporting experimental data.
Table 1: Comparison of Immunogen Design Strategies for Guiding Mutation Pathways
| Strategy | Molecular Mechanism | Key Experimental Findings | Pathogen Model | Reported Outcomes |
|---|---|---|---|---|
| Germline Targeting | Engages unmutated precursor B cells using engineered immunogens that bind germline-encoded BCRs [82]. | Priming with germline-targeting Env protein (426c.Mod.Core) resulted in greater expansion of VRC01-class B cells compared to non-Env prime [82]. | HIV-1 | Env-Env prime-boost: Higher frequency of on-target B cells, larger GC responses, higher antibody titers [82]. |
| Sequential Immunization | Uses a series of distinct immunogens to recapitulate the natural evolution of bNAbs, guiding B cells through desired mutation pathways [16]. | In murine models, sequential immunization drove accumulation of specific mutations (e.g., in CDRL1) to accommodate conserved glycans [82]. | HIV-1 | Aims to drive maturation of neutralizing breadth by selecting for B cells with increasing neutralization capacity [16] [82]. |
| Structure-Guided Design | Leverages atomic-level structural data of antibody-antigen complexes to design immunogens focusing responses on conserved epitopes [16]. | Analysis of bnAb-Env complexes identified mechanisms of viral escape (e.g., G446S in SARS-CoV-2 RBD introduces steric clash) [16]. | SARS-CoV-2, HIV-1 | Enables targeting of conserved epitopes (e.g., Class 3 RBD epitopes) despite surrounding viral diversity [16]. |
Quantitative Assessment of Strategy Performance
The efficacy of these immunogen strategies can be quantitatively assessed through their impact on B cell recruitment, mutational landscapes, and functional antibody outputs. The following table synthesizes key performance metrics from recent studies.
Table 2: Performance Metrics of Immunogen Design Strategies
| Strategy | B Cell Recruitment & Expansion | SHM Profile Guidance | Neutralization Breadth | Limitations & Challenges |
|---|---|---|---|---|
| Germline Targeting | Effectively activates rare precursor B cells (e.g., VRC01-class) [82]. | Non-Env immunogens (e.g., ai-mAb) can drive off-track mutations away from Env recognition [82]. | Sequential Env boosting required to achieve breadth; priming with non-Env immunogens disfavors boosting [82]. | Precursor frequency and competition with off-target B cells limit effectiveness [82]. |
| Sequential Immunization | Can expand B cell lineages through multiple rounds of selection. | Can guide specific, pre-defined mutations (e.g., CDRL1 shortening for glycan accommodation) [82]. | Designed to progressively increase breadth; success demonstrated in animal models [16]. | Requires precise epitope-specific steering; complex vaccine regimens. |
| Structure-Guided Design | Focuses recruitment on B cells targeting sub-dominant, conserved epitopes. | Models predict context-dependent mutation rates; high-affinity B cells may mutate less per division [15]. | Targets conserved vulnerable sites; can elicit antibodies against multiple viral clades [16]. | Must overcome immunodominance of variable epitopes; can be circumvented by new escape mutations. |
Experimental Protocols for Evaluating Immunogen Efficacy
Analyzing SHM Patterns and Clonal Lineages
Comprehensive analysis of somatic hypermutation patterns is essential for evaluating how immunogens guide B cell mutation pathways. The following protocol outlines key methodological approaches:
Single-Cell BCR Sequencing: Isolate antigen-specific B cells from germinal centers or memory compartments using fluorescently labeled antigen probes or sorting for specific B cell markers (e.g., CD19+CD38+ for germinal center B cells). Perform single-cell RNA sequencing using platforms like 10X Chromium to obtain paired heavy- and light-chain sequences [15]. This approach enables the reconstruction of clonal lineages and identification of shared mutations across related B cells.
Lineage Tree Reconstruction: Utilize computational tools (e.g., Immcantation framework, SCOPer) to align BCR sequences to germline V(D)J genes, identify somatic mutations, and reconstruct phylogenetic trees [84] [15]. These trees visualize the mutational pathways selected by immunogens, revealing patterns of convergent mutation and clonal expansion.
SHM Rate Quantification: Compare mutation frequencies in different B cell populations. For example, in the 2025 Nature study, researchers sorted germinal center B cells based on cell division history (using a H2b-mCherry reporter system) and quantified mutations via sequencing, finding that B cells undergoing more divisions had enriched affinity-enhancing mutations despite potentially lower mutation rates per division [15].
In Vivo Assessment of B Cell Responses
In vivo models provide critical insights into how immunogens direct B cell fate decisions and functional outcomes. The following protocol details a representative adoptive transfer approach:
Adoptive Transfer Model: Isolate B cells expressing the BCR of interest (e.g., inferred germline version of a bNAb) from donor mice. Transfer these cells (e.g., 500,000 cells) into congenic wild-type recipient mice that can be distinguished by CD45 allelic variants (CD45.1 vs. CD45.2) [82]. This model allows tracking of rare, target B cell populations in a physiologically competitive environment.
Immunization and Immune Monitoring: Immunize recipient mice with test immunogens formulated with adjuvants (e.g., SMNP nanoparticle adjuvant). At designated time points post-immunization (e.g., day 14), analyze serum for antigen-specific antibody titers by ELISA and monitor transferred B cell populations in spleen and lymph nodes using flow cytometry [82].
Germinal Center Analysis: Isolate germinal center B cells (identified by markers such as B220+GL7+Fas+) for downstream analysis including BCR sequencing to quantify SHM patterns, or use them in antigen-binding assays to assess affinity maturation [15] [82]. This approach can directly measure how immunogens influence the selection of specific mutational pathways.
Visualizing Key Concepts and Workflows
SHM Regulation in High-Affinity B Cells
The following diagram illustrates the recently discovered mechanism where high-affinity B cells modulate their mutation rates to preserve beneficial BCR configurations during clonal expansion.
Diagram 1: Affinity-Dependent SHM Regulation
Sequential Immunization Workflow
This diagram outlines the sequential immunization strategy used to guide B cell lineages toward broadly neutralizing antibodies through structured antigen exposure.
Diagram 2: Sequential Immunization Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful research in immunogen design requires specialized reagents and tools for evaluating B cell responses and mutation pathways. The following table catalogues essential research solutions used in the cited studies.
Table 3: Essential Research Reagents for SHM and Immunogen Studies
| Research Reagent/Tool | Function & Application | Example Use Case |
|---|---|---|
| H2b-mCherry Reporter Mice | Tracks cell division history in vivo; doxycycline turns off reporter, enabling measurement of divisions via fluorescence dilution [15]. | Quantifying division-dependent SHM rates in GC B cells after immunization [15]. |
| Adoptive Transfer Models (CD45 congenic) | Enables tracking of rare, defined B cell populations (e.g., bNAb precursors) at physiological frequencies in wild-type hosts [82]. | Testing germline-targeting immunogens for their ability to recruit and expand specific B cell lineages [82]. |
| Single-Cell BCR Sequencing (10X Chromium) | Obtains paired heavy- and light-chain sequences from individual B cells, enabling clonal lineage reconstruction and SHM analysis [15]. | Profiling clonality and mutation patterns in GC B cell subpopulations [15]. |
| SCOPer (Spectral Clustering) | Computational tool for identifying B cell clones from repertoire data by integrating junction similarity and shared SHMs [84]. | Improving sensitivity and specificity of clonal family identification in AIRR-seq data [84]. |
| Germline-Targeting Immunogens (e.g., 426c.Mod.Core, eOD-GT8) | Engineered antigens designed to bind and activate unmutated precursor B cells with specific BCR characteristics [82]. | Priming rare VRC01-class B cells for subsequent boosting with native-like Env immunogens [82]. |
| CATNAP/CAByN Database & Tools | Public repository and analysis platform for antibody neutralization data; allows definition of numerical criteria for bnAbs [81]. | Systematically identifying antibodies meeting specific potency and breadth thresholds across viral panels [81]. |
Prime-Boost Regimens to Maximize Affinity Maturation Duration
The duration and quality of affinity maturation, the process by which B cells evolve to produce antibodies with increasing affinity for a target antigen, are critical determinants of vaccine efficacy. This process occurs within germinal centers (GCs), dynamic microenvironments where B cells undergo cycles of somatic hypermutation (SHM) and selection. The strategic design of prime-boost vaccination regimens—varying the platform, timing, and antigen—can profoundly influence GC dynamics, thereby shaping the breadth and longevity of the resulting B cell receptor (BCR) repertoire. Within the context of broader research on the correlation between BCR somatic hypermutation and neutralization breadth, optimizing these regimens is paramount for developing countermeasures against rapidly evolving pathogens such as SARS-CoV-2 and HIV. This guide objectively compares the performance of different prime-boost strategies, supported by experimental data, to inform researchers and drug development professionals.
Mechanisms of Affinity Maturation in Germinal Centers
Affinity maturation is a complex evolutionary process primarily orchestrated within GCs, which are divided into a dark zone and a light zone. In the dark zone, B cells proliferate rapidly and undergo SHM, introducing random mutations into their antibody genes. These B cells then migrate to the light zone, where they are tested for affinity against antigens displayed on follicular dendritic cells (FDCs). B cells that successfully bind antigen present it to T follicular helper (Tfh) cells; the quality of this interaction determines whether the B cell receives survival signals, re-enters the dark zone for further rounds of mutation, or exits the GC as a long-lived plasma cell or memory B cell [2].
Traditional models posited that GC selection was highly stringent, favoring only the highest-affinity B cell clones. However, emerging evidence suggests GCs are more permissive, allowing a broader range of affinities to persist. This permissiveness promotes clonal diversity, which is a crucial substrate for the eventual development of broadly neutralizing antibodies (bnAbs) that can target variable pathogen epitopes [2]. The following diagram illustrates the key dynamics of a GC reaction.
Comparative Analysis of Prime-Boost Vaccination Strategies
The structure and duration of GC reactions are not fixed; they can be modulated by vaccine design. Key variables include the interval between prime and boost, the specific vaccine platforms used (homologous vs. heterologous), and the inherent properties of the platform itself. The table below summarizes the impact of different prime-boost strategies on GC dynamics and affinity maturation, based on recent preclinical and clinical findings.
Table 1: Impact of Prime-Boost Strategies on Affinity Maturation and Immune Outcomes
| Prime-Boost Strategy | Impact on GC & B Cell Responses | Key Experimental Findings | Pathogen/Vaccine Model |
|---|---|---|---|
| Extended Prime-Boost Interval [85] | ↑ Germinal Center B cells↑ Antigen-specific Antibody-Secreting Cells→ Unchanged serum IgG & Tfh cell induction | A long (18-week) interval vs. short (4-week) interval elicited higher numbers of GC-B cells and H56-specific antibody-secreting cells [85]. | H56 tuberculosis antigen + Alum (Mouse) |
| Heterologous: Viral Vector Prime / Protein Boost [86] | ↑ Binding antibody titers→ Potentiated humoral immunity↑ Bone marrow plasma cells | Ad5 prime/Protein boost resulted in a 3.9 to 36-fold superior binding antibody titers vs. homologous Ad5/Ad5. Similar patterns observed with poxvirus (VV) and vesicular stomatitis virus (VSV) vectors [86]. | SARS-CoV-2, LCMV (Mouse) |
| Heterologous: Inactivated Virus Prime / Viral Vector Boost [87] | ↑ Spike-specific antibodies↑ Th1-biased immunityRobust CD4+ & CD8+ T-cell responses | VLA2001 (inactivated) prime / Prime-2-CoV (Orf virus vector) boost induced stronger antibody and T-cell responses vs. homologous regimens [87]. | SARS-CoV-2 (Mouse) |
| Homologous mRNA / mRNA [88] | ↑ Clonal continuity between GC reactions→ Broader neutralizing antibody coverage of variants | mRNA/mRNA promoted higher clonal continuity of B cell clones between primary and secondary GC reactions compared to rAd/mRNA, associated with more durable GC responses [88]. | SARS-CoV-2 (Mouse) |
| Heterologous: Adenovirus Prime / mRNA Boost [89] | ↑ Neutralizing antibody titers↑ CD8+ T cell responses→ Fibroblast-driven chemokine responses | Heterologous (Ad/mRNA) vaccination elicited higher nAb titers and stronger CD8+ T cell responses against variants vs. homologous regimens. Single-cell RNA-seq showed amplified innate immune activation upon boost [89]. | SARS-CoV-2 (Mouse) |
The data from these studies collectively indicate that while heterologous regimens often enhance the magnitude and breadth of antibody responses, homologous mRNA vaccination excels at promoting clonal continuity in GCs. This continuity allows the same B cell lineage to undergo repeated rounds of SHM across multiple GC reactions, which is a potential pathway for developing bnAbs [88]. The following diagram synthesizes the experimental workflow commonly used to arrive at these conclusions.
Diagram Title: Prime-Boost Regimen Evaluation Workflow
Detailed Experimental Protocols for Key Findings
To enable replication and critical evaluation, this section details the methodologies from pivotal studies cited in this guide.
- Immunization: C57BL/6 mice were primed subcutaneously at the base of the tail with a vaccine formulation containing the H56 tuberculosis antigen (2 µg/mouse) combined with aluminum hydroxide (alum) adjuvant.
- Boosting: Mice were boosted subcutaneously with a lower dose of H56 antigen (0.5 µg/mouse) at either a short (4 weeks) or a long (18 weeks) interval after priming.
- Sample Collection: Blood, draining lymph nodes (sub iliac, medial, external), and spleens were collected at multiple time points post-priming and post-boosting.
- Immune Monitoring:
- Serology: Serum antigen-specific IgG responses were measured by ELISA.
- Flow Cytometry: Cell samples from draining lymph nodes were stained with fluorescent antibodies (e.g., anti-CD45R/B220, anti-GL-7, anti-CD95, anti-CD3) to identify and quantify germinal center B cells, T follicular helper cells, and antibody-secreting cells.
- Priming: C57BL/6 mice were primed intramuscularly with an adenovirus serotype 5 (Ad5) vector vaccine expressing a target antigen (e.g., SARS-CoV-2 spike or LCMV glycoprotein).
- Boosting: Approximately four weeks later, mice were boosted either homologously with the same Ad5 vector or heterologously with a purified protein vaccine of the same antigen.
- Immune Monitoring:
- Humoral Response: Binding antibody titers were measured in blood by ELISA. Neutralizing antibody titers were assessed using virus-specific assays (e.g., pseudovirus or live virus neutralization).
- Cellular Response: Antigen-specific CD8+ T cells were quantified in the blood via intracellular cytokine staining or MHC-multimer staining.
- Plasma Cells: Bone marrow was analyzed by flow cytometry to quantify long-lived antibody-secreting plasma cells.
- Immunization: Mice were vaccinated with homologous (mRNA/mRNA) or heterologous (rAd/mRNA) prime-boost regimens.
- GC B Cell Analysis: Germinal center B cells specific for the SARS-CoV-2 antigen were characterized.
- B Cell Fate-Mapping: Advanced techniques were employed to track the participation of individual B cell clones in the primary and secondary GC reactions following prime and boost immunizations.
- Assessment: Clonal continuity was defined as the recurrence of the same B cell clone in the secondary GC reaction that was initially identified in the primary GC reaction. This was correlated with the durability of the GC response and the breadth of neutralizing antibodies against viral variants.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and their applications for studying prime-boost regimens and affinity maturation, as utilized in the cited research.
Table 2: Key Research Reagents for Prime-Boost and GC Studies
| Research Reagent / Solution | Primary Function in Experimental Context | Example Application |
|---|---|---|
| Alum Adjuvant [85] | Th2-skewing vaccine adjuvant; promotes antigen uptake and presentation. | Used in model vaccine formulations with protein antigens to boost immunogenicity and study the effect of prime-boost intervals [85]. |
| CpG 1018 [87] | Toll-like receptor 9 (TLR9) agonist; promotes Th1-biased immune responses. | Combined with alum in inactivated vaccines (e.g., VLA2001) to achieve a more balanced humoral and cellular immune profile [87]. |
| Fluorophore-conjugated Antibodies for Flow Cytometry [85] | Multiparametric identification and quantification of specific immune cell populations. | Staining for surface markers (e.g., CD45R/B220, GL-7, CD95, CD3, CD19) to identify GC B cells, Tfh cells, and plasma cells from lymphoid tissues [85]. |
| ORFV-based Vector (e.g., Prime-2-CoV) [87] | Viral vector platform; delivers encoded antigens and induces strong T-cell immunity with short-lived vector-specific immunity, allowing effective re-immunization. | Used in heterologous prime-boost studies to assess synergy with inactivated virus vaccines and its potent Th1-polarizing capacity [87]. |
| MHC Multimers [86] | Ex vivo detection and isolation of antigen-specific T cells by T cell receptor (TCR) recognition. | Quantifying the population of antigen-specific CD8+ T cells induced by viral vector vaccines in blood or lymphoid organs [86]. |
| Single-cell RNA-sequencing (scRNA-seq) [89] | High-resolution profiling of transcriptomes from individual cells; reveals cellular heterogeneity and activation states. | Used to analyze immune cell infiltration and stromal activation at the vaccination site, providing mechanistic insights into heterologous regimen efficacy [89]. |
The pursuit of vaccine regimens that maximize the duration and quality of affinity maturation is a cornerstone of modern immunology. Experimental evidence clearly demonstrates that there is no single optimal strategy; rather, the choice of prime-boost regimen must be tailored to specific immunological goals. Extended intervals between doses can enhance GC B cell responses, while heterologous regimens, particularly those combining viral vectors with mRNA or protein subunits, frequently outperform homologous schedules in eliciting potent and broad antibody titers. However, homologous mRNA vaccination shows a unique capacity to foster clonal continuity in GCs, a feature that may be critical for guiding B cell evolution toward breadth against highly mutable pathogens. For researchers, the decision matrix involves balancing the desired immune outcome (e.g., peak antibody titer, T cell response, or bnAb development) with practical considerations, leveraging the distinct immunological footprints left by each vaccine platform.
Cross-Pathogen Evidence: SHM Role in HIV, SARS-CoV-2, and Influenza
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants with mutated spike proteins has posed significant challenges to vaccine efficacy. Among the deployed vaccines, Ad26.COV2.S (Jcovden), a recombinant, replication-incompetent adenovirus serotype 26 vector vaccine encoding a prefusion-stabilized spike protein, has demonstrated a unique capacity to elicit broadening neutralizing antibody responses over time, even without additional boosting. This distinctive immunological trajectory is increasingly linked to the process of B cell somatic hypermutation within germinal centers. This case study provides a comprehensive analysis of Ad26.COV2.S-induced antibody maturation, directly comparing its performance against mRNA-based alternatives (BNT162b2 and mRNA-1273) and situating these findings within the broader research on how SHM governs neutralization breadth against diverse SARS-CoV-2 variants.
Comparative Vaccine Performance and Immunological Profile
Key Immunological Metrics Across Vaccine Platforms
Table 1: Comparative immunogenicity of COVID-19 vaccines over time
| Vaccine Parameter | Ad26.COV2.S | BNT162b2 | mRNA-1273 |
|---|---|---|---|
| Dosing Schedule | Single dose | Two-dose series | Two-dose series |
| Peak WT RBD GMT (BAU/mL) | 2-3 Log10 [90] | 3-5 Log10 [90] | 3-5 Log10 [90] |
| 6-Month WT RBD GMT | Moderate decline [90] | Significant decline [90] | Most durable [90] |
| Neutralization Fold-Change vs. B.1.351 (Beta) | 5.0-fold reduction [91] | Not reported | Not reported |
| Neutralization Fold-Change vs. P.1 (Gamma) | 3.3-fold reduction [91] | Not reported | Not reported |
| T Cell Response Durability | Maintained at 6 months [90] | Declined at 6 months [90] | Declined at 6 months [90] |
| SHM Increase (8 months) | Significant [92] [17] | Not reported | Not reported |
Variant-Specific Neutralization Breadth
A defining characteristic of the Ad26.COV2.S vaccine is its capacity to elicit expanding neutralization breadth against variants of concern over time, even in the absence of boosting. Research demonstrates that serum neutralizing antibody responses increase in coverage of variants including B.1.351 (Beta) and B.1.617.2 (Delta) through 8 months post-vaccination without additional boosting or infection [92]. This phenomenon is not merely a maintenance of initial responses but represents a qualitative improvement in antibody functionality.
In mouse models, this effect was observed to persist long-term, with significant expansion of antibody breadth and durability of humoral responses over 15 months following a single Ad26.COV2.S immunization [93]. Evaluation of bone marrow and spleen tissues revealed that Ad26.COV2.S-immunized mice contained spike-specific antibody-secreting cells, providing a mechanistic basis for the persistent antibody production [93].
Somatic Hypermutation and Affinity Maturation
SHM as the Mechanism for Broadening Neutralization
The observed increase in neutralization breadth following Ad26.COV2.S vaccination is mediated by affinity maturation, a process driven by somatic hypermutation in germinal centers. Research examining human recipients of Ad26.COV2.S revealed that the level of somatic hypermutation, measured by nucleotide changes in the VDJ region of the heavy and light antibody chains, increased in spike-specific B cells over 8 months post-vaccination [92] [17].
Critical evidence supporting the functional significance of SHM comes from monoclonal antibody studies. When researchers compared monoclonal antibodies derived from these sequences, they found that highly mutated mAbs neutralized more SARS-CoV-2 variants than less mutated comparators [92] [17]. This direct correlation between mutation load and neutralization breadth provides compelling evidence that the prolonged affinity maturation process is responsible for the expanding variant coverage observed with Ad26.COV2.S.
Theoretical Framework of Regulated Somatic Hypermutation
Recent research has illuminated how regulated SHM enhances antibody affinity maturation. The conventional understanding posited that SHM occurs at a fixed rate per B cell division, potentially leading high-affinity B cells to accumulate deleterious mutations through excessive divisions. However, emerging evidence suggests an optimized system where mutation rates are variable and dependent on B cell affinity [15].
Table 2: Key experimental models for SHM research
| Experimental System | Utility in SHM Research | Key Findings |
|---|---|---|
| H2b-mCherry Reporter Mice | Tracks GC B cell division in vivo [15] | High-affinity B cells divide more but mutate less per division |
| NP-OVA Immunization Model | Characterizes affinity-enhancing mutations [15] | Cells with more divisions enriched for affinity-enhancing mutations |
| Agent-Based Computational Models | Simulates GC dynamics and mutation probabilities [15] | Affinity-dependent pmut facilitates establishment of high-affinity B cells |
| Single-cell BCR Sequencing | Resolves clonal families and mutation history [15] | Decreasing pmut produces larger populations of identical high-affinity B cells |
In this refined model, B cells expressing higher-affinity antibodies receive stronger T follicular helper cell signals, leading to more cell divisions but with a reduced mutation probability per division (pmut). This mechanism safeguards high-affinity lineages from accumulating deleterious mutations while allowing expansive clonal proliferation [15]. This paradigm aligns with observations from Ad26.COV2.S vaccination, where prolonged germinal center activity may facilitate such optimized affinity maturation.
Experimental Protocols and Methodologies
Key Assays for Evaluating Vaccine-Induced Immunity
Table 3: Essential methodologies for vaccine immunogenicity assessment
| Methodology | Application | Key Output Parameters |
|---|---|---|
| Lentiviral Pseudovirus Neutralization Assay [91] [93] | Quantifies neutralizing antibody titers against specific variants | ID50 values (50% inhibitory dilution) |
| Enzyme-Linked Immunosorbent Assay (ELISA) [91] [94] | Measures binding antibodies to spike/RBD | Geometric mean titers (GMT) in BAU/mL |
| Electrochemiluminescence Assay (ECLA) [91] | Quantifies binding antibodies to variant spike/RBD | Relative light units (RLU) |
| Enzyme-Linked Immunospot (ELISPOT) [91] [90] | Detects antigen-specific T cell responses | Spot-forming units (SFU) per PBMCs |
| Intracellular Cytokine Staining (ICS) [91] | Characterizes T cell functionality and phenotype | Cytokine-positive CD4+/CD8+ T cell percentages |
| Single-Cell BCR Sequencing [92] [15] | Profiles somatic hypermutation in B cell clones | Nucleotide changes in VDJ regions, SHM frequency |
Detailed Protocol: Assessing SHM and Neutralization Breadth
Objective: To quantify somatic hypermutation in spike-specific B cells and correlate with neutralization breadth following Ad26.COV2.S vaccination [92].
Sample Collection:
- Collect peripheral blood mononuclear cells from vaccinated individuals at multiple timepoints (e.g., 1, 3, 6, and 8 months post-vaccination)
- Isulate B cells using negative selection or antigen-specific sorting with spike protein probes
BCR Sequencing and SHM Analysis:
- Perform single-cell RNA sequencing on sorted B cells using 10X Chromium platform
- Amplify and sequence immunoglobulin heavy and light chain variable regions
- Align sequences to germline references and quantify nucleotide substitutions
- Calculate SHM frequency as mutations per base pair in VDJ regions
Monoclonal Antibody Generation and Characterization:
- Express recombinant monoclonal antibodies from sequenced B cells
- Evaluate neutralization capacity against a panel of SARS-CoV-2 variants using pseudovirus neutralization assays
- Compare neutralization breadth between highly mutated and less mutated antibodies
Statistical Analysis:
- Correlate SHM load with neutralization breadth using linear regression models
- Track temporal evolution of SHM and neutralization capacity
Signaling Pathways and Germinal Center Dynamics
Figure 1: Germinal center optimization for high-affinity B cell selection. The process illustrates how T follicular helper cell help received in the light zone determines both division potential and somatic hypermutation rates in the dark zone, creating an optimized system where high-affinity B cells undergo expanded division with reduced mutation probability per division to minimize accumulation of deleterious mutations [15].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key research reagents for investigating vaccine-induced B cell responses
| Research Reagent | Application | Function/Purpose |
|---|---|---|
| Prefusion-Stabilized Spike Protein [91] | Vaccine immunogen; B cell sorting | Maintains native antigen conformation for optimal immunity |
| Spike-Specific Peptide Megapools [90] | T cell ELISPOT assays | Detect spike-specific cellular immune responses |
| Fluorophore-Conjugated Spike Probes [15] | Antigen-specific B cell sorting | Identify and isolate spike-reactive B cells |
| Lentiviral Pseudovirus Panel [91] [93] | Neutralization assays | Evaluate antibody neutralization against variants |
| Anti-Human Ig Antibodies [94] | ELISA, flow cytometry | Detect and quantify antigen-specific antibodies |
| Single-Cell BCR Amplification Primers [92] [15] | BCR sequencing | Amplify immunoglobulin variable regions for SHM analysis |
| Recombinant Antibody Expression Systems [92] | mAb characterization | Produce monoclonal antibodies from sequenced B cells |
Discussion and Research Implications
The unique temporal pattern of immune maturation following Ad26.COV2.S vaccination, characterized by delayed but broadening neutralization capacity, provides valuable insights for vaccine design against rapidly evolving pathogens. The demonstrated correlation between somatic hypermutation and neutralization breadth underscores the importance of vaccine platforms that sustain germinal center reactions over extended periods.
This case study reveals that Ad26.COV2.S, while eliciting lower initial antibody titers compared to mRNA vaccines, induces a more prolonged affinity maturation process that enhances variant cross-reactivity over time. This characteristic may be particularly valuable in regions where booster access is limited or against pathogens where breadth outweighs peak titer as a protective correlate.
Future vaccine development may leverage these insights by designing immunization strategies that specifically optimize germinal center persistence and regulated SHM. Combining platform advantages through heterologous prime-boost regimens represents a promising direction, as suggested by studies showing that ORFV-S prime followed by Ad26.COV2.S boost elicited superior antibody titers and cellular responses compared to homologous regimens [95]. The continued investigation of how vaccine platforms shape B cell maturation will be crucial for addressing future pandemic threats and improving vaccines against highly variable pathogens like HIV and influenza.
Broadly neutralizing antibodies (bnAbs) against HIV-1 represent a critical goal for vaccine development, as they can neutralize diverse viral strains by targeting conserved regions of the envelope glycoprotein (Env). A defining characteristic of most HIV bnAbs is their exceptionally high level of somatic hypermutation (SHM), a process occurring in germinal centers where B cells accumulate mutations in their variable genes to achieve superior antigen-binding affinity. The correlation between extensive SHM and the development of neutralization breadth is well-established but presents a significant challenge for vaccine design, as current strategies have been unable to recapitulate the natural maturation pathways that yield these antibodies.
This guide systematically compares the SHM requirements across different classes of HIV bnAbs, providing experimental data and methodologies central to ongoing research. Understanding these requirements is essential for developing targeted immunization strategies capable of eliciting protective antibody responses.
Quantitative Comparison of SHM Across bnAb Classes
The extent of SHM varies considerably among different bnAb classes, influenced by their target epitope and the structural constraints of the HIV Env glycan shield. The table below summarizes key characteristics of major bnAb classes.
Table 1: Somatic Hypermutation and Genetic Features of Major HIV bnAb Classes
| bnAb Class (Binding Region) | % Heavy Chain SHM (Mean ± SE) | Average CDRH3 Length (aa) | % of Antibodies | Common Germline Genes |
|---|---|---|---|---|
| gp120-gp41 Interface | 25.0 ± 1.9 | 16 ± 1.7 | 1% | IGHV1-69, IGHV4-4 |
| Silent Face | 25.0 ± 3.3 | 21 ± 1.3 | 1% | Data Limited |
| CD4 Binding Site (CD4bs) | 17.0 ± 1.2 | 16 ± 0.3 | 25% | IGHV1-2*02, IGHV1-46, IGHV3-30 |
| Glycan-Dependent (V3) | 18.0 ± 0.9 | 23 ± 0.9 | 12% | IGHV4-39, IGHV5-51 |
| V1/V2 Apex | 13.0 ± 0.5 | 27 ± 0.8 | 18% | IGHV1-69, IGHV2-5 |
| MPER | 13.0 ± 1.0 | 20 ± 0.6 | 5% | IGHV1-69, IGHV3-15 |
| Fusion Peptide | 15.0 ± 1.3 | 14 ± 0.9 | 9% | IGHV1-3, IGHV1-18 |
Source: Data compiled from CATNAP database analysis [96] and recent studies [32] [97].
Key observations from the data include:
- gp120-gp41 interface-targeting bnAbs require the highest levels of SHM, with a mean of 25% mutation in the heavy chain variable region [96].
- CD4bs bnAbs, one of the most prevalent classes, also show high SHM (~17%) and typically use a restricted set of germline genes like IGHV1-2*02 [97].
- V1/V2 Apex bnAbs, such as PCT64 and PG9, often forgo extreme SHM but instead rely on exceptionally long heavy chain complementarity-determining region 3 (HCDR3) loops (averaging 27 amino acids) to penetrate the Env glycan shield [98].
- A direct correlation exists between SHM frequency and serum neutralization breadth in HIV-infected individuals, underscoring the critical role of mutation accumulation in achieving breadth [32].
Experimental Evidence and Supporting Data
Case Study: The FD22 CD4bs bnAb with Extreme SHM
The recently isolated FD22 antibody exemplifies the extreme SHM requirement for some CD4bs bnAbs. Isolated from an elite neutralizer, FD22 exhibits 37% SHM in its heavy chain (derived from IGHV3-30) and a long 20-amino-acid CDRH3. This antibody neutralized 82% of a 145-virus panel (Geometric Mean IC₅₀ = 0.27 µg/mL), a breadth and potency comparable to the well-known bnAb VRC01 [97].
Table 2: Neutralization Performance of High-SHM bnAbs
| Antibody | bnAb Class | Heavy Chain SHM | Neutralization Breadth | Geometric Mean IC₅₀ (µg/mL) | Key Feature |
|---|---|---|---|---|---|
| FD22 | CD4bs | 37% | 82% (119/145 viruses) | 0.27 | Uses rare IGHV3-30; robust ADCC |
| VRC01 | CD4bs | ~30-35% | 88% (128/145 viruses) | 0.25 | Classic IGHV1-2*02-derived |
| BG18 | V3-glycan | High (Data not specified) | Elite neutralization | Data not specified | Isolated from an HIV controller [32] |
| PCT64-lineage | V1/V2 Apex | Relatively low | Priming achieved in models | N/A | Depends on long HCDR3 >24 aa [98] |
SHM and Viral Control: Evidence from HIV Controllers
Research on HIV controllers (individuals who control viral load without therapy) provides evidence that high SHM can develop even with low-level antigen exposure. One study found that the B-cell repertoires of top neutralizers were dominated by a small number of large, highly mutated B-cell clones. The frequency of these mutations was directly correlated with the serum's neutralization breadth, suggesting these clones had undergone extensive affinity maturation to achieve peak affinity [32].
Essential Experimental Protocols in bnAb Research
B-cell Receptor Repertoire Analysis
Purpose: To characterize the immunoglobulin gene repertoire, clonal expansions, and SHM levels in antigen-specific B cells from vaccinated or infected individuals [32].
Detailed Workflow:
- Cell Sorting: HIV-1 Envelope-specific memory B cells (CD19+CD20+IgM-IgA-) are isolated from Peripheral Blood Mononuclear Cells (PBMCs) using Fluorescence-Activated Cell Sorting (FACS).
- Single-Cell Sequencing: Natively paired heavy- and light-chain B-cell receptor (BCR) transcripts are amplified from single B cells using immune repertoire capture or similar techniques.
- High-Throughput Sequencing: Amplified products are sequenced via next-generation sequencing (NGS) platforms (e.g., Illumina MiSeq).
- Bioinformatic Analysis:
- Annotation: Sequences are annotated using tools like IMGT/HighV-QUEST to assign V(D)J genes and identify mutations.
- SHM Calculation: The level of SHM is calculated as the percentage of nucleotide mutations from the inferred germline sequence.
- Clonality Analysis: Clonal families are identified based on shared V(D)J genes and identical or highly similar HCDR3 amino acid sequences.
Germline-Targeting Immunogen Evaluation
Purpose: To test engineered immunogens designed to prime and expand rare B-cell precursors with specific genetic features (e.g., long HCDR3s), guiding them toward bnAb development [98].
Detailed Workflow:
- Immunogen Design: Create immunogens like ApexGT6, engineered with specific mutations to enhance affinity for the inferred germline precursors of target bnAbs (e.g., PCT64, PG9).
- Animal Immunization: Administer the immunogen to model organisms (e.g., rhesus macaques) via adjuvanted protein or mRNA-LNP platforms.
- Immune Response Monitoring:
- ELISPOT/Sorting: Isolate antigen-specific B cells.
- BCR Sequencing: Sequence the BCRs of responding cells to identify elicited antibodies with desired features (e.g., long HCDR3s, specific motifs like DDY).
- Structural Analysis: Use cryo-electron microscopy (cryo-EM) to determine structures of elicited antibodies in complex with Env, confirming binding mode similarity to mature bnAbs.
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 3: Essential Reagents for HIV bnAb and SHM Research
| Reagent / Solution | Primary Function | Application in Research |
|---|---|---|
| Engineered Env Trimers (e.g., ApexGT5/6) | Germline-targeting priming immunogen | Designed to bind and activate rare bnAb-precursor B cells with specific genetic features [98]. |
| CATNAP / CAByN Database | Public neutralization & sequence database | Analyzing bnAb neutralization breadth/potency and defining numerical criteria for bnAbs [96]. |
| IMGT/HighV-QUEST | Immunoglobulin sequence annotation | Standardized analysis of V(D)J gene usage, SHM, and CDR3 characterization from NGS data [99] [32]. |
| Pseudovirus Panels (Tier 2/3) | Standardized neutralization assay | High-throughput quantification of antibody neutralization breadth and potency against diverse global HIV strains [96] [97]. |
| Anti-human IgG (Fc-specific) | B-cell sorting & detection | Isolation of antigen-specific memory B cells (e.g., CD19+CD20+IgM-IgA-) for single-cell cloning and repertoire sequencing [32]. |
The development of HIV bnAbs is intrinsically linked to extreme somatic hypermutation, which enables the antibody flexibility and affinity required to neutralize the diverse and shielded HIV Env trimer. While the specific SHM requirements vary by epitope class, the consistent correlation between high mutation levels and neutralization breadth presents a formidable barrier for vaccine design. Current research, utilizing sophisticated germline-targeting immunogens and deep BCR repertoire analysis, is focused on understanding and mimicking these natural maturation pathways. Success in this endeavor will depend on strategically designed vaccination regimens that can selectively prime and expand rare B-cell precursors and guide them through the complex evolutionary paths necessitated by extreme SHM.
Somatic hypermutation (SHM) of B cell receptors (BCRs) serves as a critical mechanism for broadening antibody neutralization capacity against influenza viruses. This review synthesizes recent findings demonstrating that sustained germinal center (GC) reactions following pandemic or novel influenza exposure correlate with increased SHM loads and enhanced antibody breadth. In contrast, repeated seasonal vaccination often elicits more constrained SHM patterns, though under specific conditions of homologous boosting, can gradually promote cross-reactive responses. Structural biology and longitudinal immunological tracking reveal that persistent GC activity facilitates the accumulation of critical mutations in BCR complementarity-determining regions (CDRs), enabling recognition of conserved epitopes across divergent viral strains. These findings establish a direct correlation between SHM patterns induced by different exposure contexts and the development of broadly neutralizing antibodies, providing a framework for rational vaccine design.
The adaptive immune system's capacity to generate high-affinity, broadly protective antibodies against influenza viruses hinges on the process of SHM within GCs. During T cell-dependent immune responses, B cells undergo clonal expansion and affinity maturation, whereby point mutations are introduced into the variable regions of immunoglobulin genes at a rate approximately 10³-fold higher than the background mutation rate in somatic cells [100]. These mutations, followed by selective pressure, can enhance antibody affinity and potentially broaden neutralization capacity against heterologous strains.
The pattern and extent of SHM differ significantly depending on the nature of immune exposure. Seasonal influenza vaccines, typically reformulated annually to match circulating strains, often recall pre-existing memory B cells with limited diversification. In contrast, pandemic or novel influenza virus exposure can initiate de novo GC responses that persist for extended periods, allowing for iterative rounds of mutation and selection. This review systematically compares SHM patterns arising from these distinct exposure contexts and explores their implications for developing universal influenza vaccines.
SHM in Seasonal Influenza Vaccination Responses
Typical SHM Patterns in Seasonal Vaccination
Seasonal influenza vaccination in previously exposed individuals typically stimulates a memory recall response characterized by moderate SHM levels. Studies tracking GC B cell responses after seasonal inactivated influenza vaccination in humans found that antigen-specific GC B cells persist for at least 13 weeks in some individuals [101]. In these persistent GCs, monoclonal antibodies (mAbs) derived from later time points exhibit enhanced binding affinity and breadth to influenza hemagglutinin (HA) antigens compared to earlier clonotypes [101].
Analysis of B cells from blood samples reveals distinct selection pressures across antibody regions. The framework regions (FWRs) experience strong purifying selection on both short and long timescales, preserving structural integrity. Conversely, complementarity determining regions (CDRs) experience a combination of purifying and antigen-driven positive selection, resulting in net positive selection over time [100]. This differential selection pressure optimizes antigen binding while maintaining antibody stability.
Impact of Repeated Homologous Vaccination
Contrary to conventional understanding, recent evidence suggests that repeated vaccination with homologous influenza HA can gradually broaden antibody responses. In a longitudinal cohort receiving the same H1N1 2009 pandemic vaccine strain over four consecutive years, researchers observed that annual vaccination progressively enhanced receptor-blocking antibodies (HAI) to highly unmatched H1N1 strains [102] [103]. This broadening occurred gradually over the 4-year vaccination period, particularly in individuals devoid of initial memory recall against historical viruses [102].
Computational modeling suggests this phenomenon arises from epitope masking dynamics in GCs. After booster vaccinations, pre-existing high-affinity antibodies bind immunodominant epitopes, allowing previously subdominant epitopes to engage naïve B cells [102]. This "antigenic masking" promotes diversification of the antibody response against conserved, less immunogenic epitopes over multiple exposures.
Age-Related Differences in SHM
Aging significantly impacts SHM patterns in response to seasonal vaccination. Single-cell profiling of B cell responses to influenza vaccination reveals that older adults (over age 65) exhibit higher pre-vaccination SHM frequencies and increased abundance of activated B cells compared to young adults [104]. However, following vaccination, young adults mount a more clonally expanded response with a higher proportion of plasmablasts [104]. These quantitative and qualitative differences in SHM and B cell composition may contribute to the reduced vaccine effectiveness observed in elderly populations.
Table 1: SHM Patterns in Seasonal Influenza Vaccination
| Parameter | Typical Response | Repeated Homologous Vaccination | Age-Related Differences |
|---|---|---|---|
| SHM Frequency | Moderate increase [101] | Progressive accumulation over years [102] | Higher baseline in older adults [104] |
| Neutralization Breadth | Strain-specific initially, broadening with persistent GCs [101] | Gradual broadening to unmatched strains over 4 years [102] | Reduced adaptability in older adults [104] |
| GC Persistence | Up to 13 weeks in responders [101] | Enhanced by repeated antigen exposure [102] | Not reported |
| BCR Selection | Positive selection in CDRs, purifying in FWRs [100] | Diversification to subdominant epitopes [102] | Altered gene expression in activated B cells [104] |
SHM in Pandemic Influenza Exposure Responses
Enhanced GC Persistence and SHM in Novel Exposure
Pandemic or novel influenza virus exposure often elicits more robust and persistent GC reactions compared to seasonal vaccination. Studies of H5N1 vaccination reveal that GC-independent memory B cells generated during primary responses to novel antigens provide templates for mutation and selection in secondary GCs upon re-exposure [105]. These early MBCs can undergo further BCR diversification when encountering variant strains, building high-affinity repertoires for novel epitopes.
Notably, research on humans vaccinated with monovalent H5N1 vaccine 15 years prior demonstrates that long-lived memory B cells can persist and generate potent, broadly cross-neutralizing mAbs upon rechallenge. Isolated mAbs exhibited an average of 5% heavy-chain SHM and neutralized diverse H5 clades, including contemporary 2.3.4.4b H5N1 viruses [106]. Structural analyses revealed these mAbs target conserved epitopes within the HA globular head, with their potency and breadth directly correlated with their SHM profiles.
Structural Correlates of Broad Neutralization
Cryo-electron microscopy structures of broadly neutralizing mAbs in complex with HA reveal how specific SHM-acquired substitutions enable recognition of conserved epitopes across divergent viral strains. For H5Nx viruses, mAbs targeting regions surrounding the receptor-binding site (RBS), upper lateral regions, and the base of the HA head demonstrate varying levels of neutralizing potency and breadth [106]. The most potent mAbs (e.g., 310-12D03 and 310-7D11) neutralized currently circulating H5N1 strains with exceptional potency (IC80 of 0.01 µg mL⁻¹ or lower) [106].
The altered binding footprints of these mAbs, achieved through SHM, allow them to circumvent variable regions and engage conserved structural elements essential for viral entry. This structural adaptation represents a key mechanism by which SHM expands neutralization breadth against pandemic threats.
Table 2: SHM Patterns in Pandemic Influenza Exposure
| Parameter | Primary Response | Memory Recall After 15 Years | Structural Correlates |
|---|---|---|---|
| SHM Frequency | Extensive in persistent GCs [101] | ~5% heavy chain SHM in cross-reactive mAbs [106] | Critical mutations in CDRs [106] |
| Neutralization Breadth | Develops over weeks in GCs [101] | Broad cross-reactivity across H5 clades [106] | Altered binding footprints to conserved epitopes [101] [106] |
| GC Persistence | Sustained for months [101] | Long-lived memory pools [106] | Not reported |
| BCR Selection | Diversification against novel epitopes [105] | Selection for conserved epitope recognition [106] | Positive selection for RBS engagement [106] |
Experimental Protocols for SHM Analysis
Longitudinal Tracking of Antigen-Specific B Cells
Objective: To track the maturation of antigen-specific B cell clones in persistent GCs over time.
- Sample Collection: Serial fine needle aspirates (FNA) of draining lymph nodes collected at 0, 1, 2, 4, 9, 13, 17, and 26 weeks post-vaccination [101].
- Cell Sorting: Flow cytometry identification of total GC B cells (CD19+ IgDlo CD20+ CD38int) and HA-binding GC B cells using biotinylated HA probes [101].
- Single-Cell RNA Sequencing: scRNA-seq analysis on pooled MBC-enriched and whole peripheral blood mononuclear cells (PBMCs), FNA of lymph nodes, and bone marrow plasma cell-enriched aspirates [101].
- Lineage Tracing: Reconstruction of B cell lineage trees to identify trunk (shared) and canopy (subset-specific) mutations [100].
- mAb Characterization: Recombinant expression of isolated antibodies for binding affinity and neutralization potency assays against diverse viral strains [106].
Structural Biology Approaches for Epitope Mapping
Objective: To determine the structural basis of broad neutralization mediated by SHM.
- Antibody Isolation: Single-cell sorting of HA-specific memory B cells from vaccinated or convalescent individuals [106].
- mAb Production: Recombinant expression and purification of monoclonal antibodies.
- Complex Formation: Incubation of mAbs with recombinant HA proteins.
- Cryo-EM Grid Preparation: Vitrification of mAb-HA complexes.
- Data Collection & Processing: High-resolution imaging and 3D reconstruction.
- Structural Analysis: Identification of binding interfaces and mutation effects.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for SHM and Neutralization Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| HA Probes | Biotinylated HA from A/Michigan/45/2015 H1N1, A/Singapore/INFIMH-16-0019/2016 H3N2, B/Colorado/06/2017 [101] | Flow cytometry identification of antigen-specific B cells |
| Cell Isolation Tools | Anti-CD19, anti-IgD, anti-CD20, anti-CD38 antibodies [101] | Fluorescence-activated cell sorting of GC B cell populations |
| Single-Cell Platforms | 10x Genomics Chromium [104] | Parallel scRNA-seq and BCR sequencing |
| Expression Systems | HEK293 or ExpiCHO cells [106] | Recombinant mAb production for functional characterization |
| Neutralization Assays | Microneutralization, HA inhibition (HAI), focus reduction assays [107] [106] | Quantification of antibody potency and breadth |
| Structural Biology | Cryo-EM, negative stain EM [106] | High-resolution epitope mapping |
The patterns of somatic hypermutation in influenza responses exhibit distinct characteristics depending on exposure context. Seasonal vaccination typically recruits pre-existing memory B cells with moderate SHM levels, though persistent GC reactions and repeated homologous vaccination can gradually enhance breadth through epitope masking and diversification. In contrast, pandemic or novel virus exposure generates more extensive SHM through sustained GC reactions, creating BCR repertoires capable of recognizing conserved epitopes across divergent strains. The structural characterization of broadly neutralizing antibodies reveals how SHM fine-tunes paratopes to engage conserved viral epitopes, providing a blueprint for next-generation vaccine design. These insights underscore the importance of promoting sustained GC reactions and targeting subdominant conserved epitopes to elicit broadly protective immunity against both seasonal and pandemic influenza threats.
Breakthrough infections, occurring in individuals despite previous vaccination or recovery, have become a focal point in infectious disease immunology. For SARS-CoV-2, the Delta and Omicron variants have demonstrated a remarkable capacity to cause such infections. This review examines the hypothesis that these breakthrough events act as powerful drivers of B cell receptor evolution, accelerating somatic hypermutation (SHM) beyond what vaccination or primary infection alone achieves. SHM constitutes a critical mechanism of antibody affinity maturation within germinal centers, whereby point mutations in immunoglobulin variable regions generate B cell receptors with potentially enhanced antigen binding capabilities. The resulting antibody diversity directly influences the neutralization breadth achieved—the ability to recognize and counteract diverse viral variants. Within the context of an evolving pathogen, understanding how different antigen exposure scenarios shape B cell responses through SHM provides crucial insights for next-generation vaccine design against rapidly mutating viruses.
Comparative SHM Dynamics: Delta vs. Omicron Breakthroughs
SHM Elevation in Delta Breakthrough Infections
Infection with the SARS-CoV-2 Delta variant in previously vaccinated individuals triggers a recall of pre-existing memory B cells, leading to a rapidly matured antibody response. Single-cell sequencing of B cells from individuals with Delta breakthrough infections reveals a significant elevation in SHM levels compared to responses from primary infection in immunologically naive individuals.
Table 1: Somatic Hypermutation in Delta Variant Breakthrough Infections
| Study Cohort / Comparison Group | Average VH Gene SHM Rate (Nucleotide Level) | Key B Cell Phenotype Observations | Neutralization Breadth Outcome |
|---|---|---|---|
| Delta Breakthrough Infection (Vaccinated) [21] | 11.88% (average for candidate mAbs) | Rapid induction of isotype-switched B cells; significant proportion of IGHG1 expression; recall of memory B cells (e.g., IGHV3-53, IGHV3-66) | 63 of 117 mAbs bound Delta RBD/S1; 13 of 22 cross-reactive mAbs bound Omicron BA.1 RBD |
| Non-Vaccinated, Wild-Type Infected (Comparison) [21] | Lower proportion of unmutated VH genes (10.73%) vs. Delta breakthroughs (6.03%) | Predominantly germline-like antibodies with lower SHM from acute activation of naive B cells | Limited neutralization breadth against antigenically distant variants |
| Cross-Reactive mAbs from Delta Breakthroughs [21] | 9.63% (average for 63 binding mAbs) | Enriched in a specialized switched memory B-cell subpopulation (C6 cluster); high clonal diversity | Potent cross-neutralization of WT, Beta, Delta; YB9-258 & YB13-292 also neutralized Omicron BA.1 |
The data indicate that Delta breakthrough infections promote an antibody repertoire characterized by increased SHM, which is directly associated with the generation of antibodies possessing superior cross-reactive neutralization capabilities, including activity against the highly mutated Omicron BA.1 variant [21].
Affinity Maturation Following Omicron BA.1 Breakthrough
Omicron BA.1 breakthrough infections in vaccinated individuals drive further affinity maturation of the pre-existing, cross-reactive B cell repertoire. Longitudinal tracking of mRNA-vaccinated individuals for six months post-BA.1 infection demonstrates that the antibody response continues to evolve, increasing its specificity for Omicron at the population level.
Table 2: Somatic Hypermutation and Antibody Evolution in Omicron BA.1 Breakthrough Infections
| Parameter | Early Timepoint (∼1 Month Post-Infection) | Late Timepoint (5-6 Months Post-Infection) | Implication |
|---|---|---|---|
| Serum Neutralizing Antibodies [108] | High peak titers; cross-reactive, but preferential WT neutralization | Modest decline (2-4 fold); maintained breadth across variants | Durable, broad humoral immunity |
| SHM in Cross-Reactive B Cells [108] | Already elevated from vaccination | Accumulation of additional somatic mutations | Continued affinity maturation over time |
| Antibody Binding Preference [108] | Dominated by clones with preferential WT binding | Shift towards enhanced BA.1 recognition at the expense of WT binding | B cell memory evolves toward the breakthrough variant |
| B Cell Clonality [108] | High proportion of clones shared with vaccine-induced response | High clonal diversity; 4-30% shared with early timepoint | Persistent evolution of pre-existing B cell lineages |
This evolution is characterized by a shift in antibody binding preference, where B cell clones initially targeting the ancestral (Wuhan-1) strain accumulate further mutations that refine their ability to recognize the Omicron BA.1 variant, a process the authors describe as the original Wuhan antigenic sin driving "convergent antibody maturation" [109].
Key Experimental Methodologies for SHM Analysis
Investigating SHM in the context of breakthrough infections relies on a suite of advanced immunological and bioinformatic techniques.
Single-Cell B Cell Receptor Sequencing
The foundational protocol for profiling the B cell immune repertoire at single-cell resolution involves isolating antigen-specific memory B cells for sequencing [21] [108].
- Cell Sorting and Isolation: Peripheral blood mononuclear cells (PBMCs) are stained with fluorescently labeled probes, such as SARS-CoV-2 Spike protein, S1 subunit, or Receptor Binding Domain (RBD) tetramers (e.g., WT and BA.1). Fluorescence-Activated Cell Sorting (FACS) is used to isolate single CD19+ CD27+ memory B cells that bind the target antigen [21] [108].
- Single-Cell V(D)J Sequencing: The sorted single B cells are processed using platforms like the 10X Genomics Chromium system with kits such as the Next GEM Single Cell 5' Reagent Kit. This technology captures full-length, natively paired heavy- and light-chain variable region transcripts [21].
- Bioinformatic Analysis: The raw sequencing data is processed through pipelines like Cell Ranger to assemble V(D)J sequences. Subsequent analysis with specialized toolkits (e.g., Immcantation framework, SHazaM for SHM analysis, SCOPer for clonal grouping) is performed. The sequences are aligned to germline human immunoglobulin gene databases (e.g., IMGT) using tools like IMGT/HighV-QUEST to precisely quantify nucleotide substitutions and calculate SHM rates [43] [21].
Functional Characterization of Monoclonal Antibodies
To link SHM to antibody function, sequenced B cell receptors are recombinantly expressed and tested.
- Recombinant Antibody Expression: Heavy- and light-chain variable region genes from sorted B cells are cloned into IgG1 expression vectors. These plasmids are co-transfected into mammalian cell lines (e.g., ExpiCHO cells) for antibody production, followed by purification via Protein A or Protein G affinity chromatography [43].
- Antigen Binding Assessment: Techniques like Bio-Layer Interferometry (BLI) and Surface Plasmon Resonance (SPR) are used to quantify binding affinity and kinetics of monoclonal antibodies to various variant RBDs. These methods provide precise measurements of the functional outcome of SHM [43] [21].
- Virus Neutralization Assays: The gold-standard functional test involves pseudovirus neutralization assays (e.g., using murine leukemia virus or lentivirus backbones) or authentic virus neutralization. Antibodies are serially diluted and tested for their ability to inhibit virus infection of susceptible cells, determining the half-maximal inhibitory concentration (IC50 or ID50) against a panel of viral variants [20] [21] [108].
Visualizing B Cell Evolution and SHM Analysis
The following diagrams illustrate the core concepts of B cell evolution post-breakthrough infection and the experimental workflow for SHM analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for SHM and B Cell Research
| Research Reagent | Function in Experimental Protocol |
|---|---|
| Fluorescently Labeled RBD/S1 Tetramers [21] [108] | Critical probes for identifying and isolating antigen-specific memory B cells via FACS. |
| 10X Genomics Single Cell 5' Kit [43] [21] | Enables high-throughput capture of natively paired BCR sequences from single cells. |
| ExpiCHO or Expi293F Cell Lines [43] | Mammalian expression systems for high-yield production of recombinant monoclonal antibodies. |
| Protein A/G Affinity Resins [43] | For purification of recombinant IgG antibodies from culture supernatants. |
| MLV or Lentivirus Pseudovirus Systems [20] [108] | Safe, BSL-2 compatible platforms for measuring antibody neutralization potency and breadth. |
| IMGT/HighV-QUEST Database [20] [21] | The international reference for immunoglobulin gene alignment and SHM analysis. |
Delta and Omicron breakthrough infections serve as powerful natural immunization events that profoundly accelerate the process of antibody somatic hypermutation. The consistent finding across multiple studies is that these infections recall pre-existing cross-reactive B cell memory and drive them through additional rounds of affinity maturation, resulting in a quantitatively and qualitatively enhanced antibody repertoire. The elevated SHM rates documented in these contexts are directly correlated with the generation of antibodies possessing remarkable neutralization breadth, capable of countering diverse SARS-CoV-2 variants, including Omicron sublineages. This convergent evolution, guided by antigenic sin, suggests that strategic vaccination with antigenically distant variants or polyvalent vaccines could mimic this process, potentially leading to robust, pre-emptive protection against future viral escapes.
Somatic hypermutation (SHM) serves as a critical mechanism in adaptive immunity, driving the affinity maturation of antibodies in germinal centers (GCs) to combat diverse pathogens. However, the level of SHM required to elicit effective, broad-spectrum antibodies varies significantly across different pathogen classes. This comparative analysis examines the SHM thresholds associated with broadly neutralizing antibodies (bnAbs) against key viral pathogens, including HIV-1, SARS-CoV-2, and influenza. We synthesize quantitative data on SHM percentages, neutralization breadth, and potency to establish pathogen-specific patterns, providing a framework for rational vaccine design and therapeutic antibody development. Understanding these differential requirements is fundamental to advancing B cell receptor somatic hypermutation and neutralization breadth research.
SHM Patterns Across Key Viral Pathogens
Table 1: Comparative SHM Requirements for Broadly Neutralizing Antibodies Across Pathogen Classes
| Pathogen | Antibody Class/Target | Average SHM % (Heavy Chain) | Neutralization Breadth | Potency (IC₅₀) | Key References |
|---|---|---|---|---|---|
| HIV-1 | CD4-binding site (CD4bs) | 17 ± 1.2% | Multi-clade, >68% (elite bnAbs) | < 0.06 µg/mL (elite) | [96] [110] |
| HIV-1 | Glycan shield (V1/V2) | 13 ± 0.5% | Multi-clade | Varies by variant | [96] |
| HIV-1 | gp120-gp41 interface | 25 ± 1.9% | Limited data | Limited data | [96] |
| SARS-CoV-2 | VH1-58 public clonotype | Low (mutations often dispensable for early variants) | Cross-neutralization of early VOCs | High against Wu01, Alpha, Beta, Delta | [111] |
| SARS-CoV-2 | Omicron-specific | Critical for BA.1/BA.2 neutralization | Effective against Omicron sublineages | Restored via specific mutations | [111] |
| Influenza | Hemagglutinin (HA) stalk | Variable; can evolve from non-binding precursors | Broad group coverage | Model-dependent | [112] |
The data reveals a clear spectrum of SHM dependency. HIV-1 bnAbs consistently require high levels of SHM, particularly those targeting conserved but challenging epitopes like the CD4bs and the gp120-gp41 interface [96]. In contrast, effective SARS-CoV-2 neutralizing antibodies can originate from public clonotypes like VH1-58 with minimal initial SHM, though specific mutations become crucial for neutralizing escape variants like Omicron [111]. Furthermore, research on influenza and model antigens demonstrates that SHM can generate de novo antigen recognition, a process termed "affinity birth," suggesting that pre-existing high affinity is not an absolute prerequisite for GC entry and diversification [112].
Experimental Protocols for SHM Functional Analysis
SHM Reversion and Functional Characterization
A cornerstone methodology for defining the functional role of specific mutations is the reversion of SHMs to their germline counterparts, followed by binding and neutralization assays.
Detailed Protocol:
- Antibody Selection & Sequencing: Select bnAbs of interest (e.g., IOMA-class for HIV-1 CD4bs, VH1-58 for SARS-CoV-2). Obtain their variable region sequences [110] [111].
- Germline Inference: Use bioinformatic tools (e.g., IgBLAST, part of the Immcantation framework) to infer the unmutated common ancestor (germline) sequence [113].
- Library Generation: Generate a library of antibody variants using site-directed mutagenesis. This includes:
- Revertants: Variants where each SHM is individually reverted to germline.
- Minimally mutated variants (e.g., IOMAmin): Combinations of the fewest SHMs required to retain breadth [110].
- Recombinant Expression: Transfect plasmids encoding heavy and light chains of each variant into mammalian expression systems (e.g., ExpiCHO cells). Purify antibodies using Protein A or G affinity chromatography [43] [110].
- Functional Assays:
- Binding Affinity: Use Surface Plasmon Resonance (SPR) to characterize kinetics (Kon, Koff, KD) against recombinant antigen (e.g., HIV-1 Env trimer, SARS-CoV-2 RBD) [43].
- Neutralization Potency/Breadth: Utilize live virus or pseudovirus neutralization assays (e.g., PRNT) against a panel of viral variants (e.g., multi-clade HIV-1 pseudoviruses, SARS-CoV-2 VOCs). Calculate IC₅₀/IC₈₀ values [96] [114].
- Structural Analysis: For key variants (e.g., IOMAmin), determine high-resolution structures (e.g., via cryo-EM) complexed with antigen to interpret how SHMs enable binding and neutralization [110].
In Vitro Viral Escape Profiling
This protocol assesses how SHM shapes the potential for viral escape, a critical factor for antibody durability.
Detailed Protocol:
- Antibody Preparation: Express and purify wild-type and SHM-reverted monoclonal antibodies [43].
- Escape Selection: Incubate a replication-competent recombinant VSV expressing the target viral protein (e.g., SARS-CoV-2 spike) with sub-neutralizing concentrations of the antibody in cell culture. Perform serial passages to allow for escape mutant outgrowth [43].
- Variant Sequencing: Sequence the viral antigen gene (e.g., spike) from the breakthrough virus pools to identify escape mutations.
- Hotspot Mapping: Compare the escape profiles selected by wild-type versus SHM-reverted antibodies to identify how somatic mutations alter the "Achilles heel" of the antibody and suppress specific escape pathways [43].
Conceptual Framework of SHM in Antibody Evolution
The following diagram illustrates the fundamental pathways through which SHM contributes to antibody evolution against diverse pathogens, highlighting key concepts like affinity maturation, affinity birth, and bystander mutation.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Reagents and Tools for SHM and Neutralization Breadth Research
| Category | Reagent / Tool | Specific Function / Application | Example / Source |
|---|---|---|---|
| Bioinformatics | pRESTO/Change-O Suite | Processing BCR repertoire sequencing data; error correction, V(D)J assignment. | [113] |
| Immcantation Framework | Comprehensive pipeline for adaptive immune repertoire analysis. | [43] | |
| CAByN (Choose Antibodies by Neutralization) | Web-based tool to define numerical criteria for bnAbs based on neutralization data. | Los Alamos HIV Database [96] | |
| Neutralization Assays | Plaque-Reduction Neutralization Test (PRNT) | Gold-standard live virus neutralization assay to quantify antibody potency. | [114] |
| Pseudovirus Neutralization Assay | High-throughput, safer method for assessing neutralization breadth against envelope-pseudotyped viruses. | CATNAP Database [96] | |
| Binding & Affinity | Surface Plasmon Resonance (SPR) | Label-free kinetic analysis of antibody-antigen interactions (Kon, Koff, KD). | [43] |
| Structural Biology | Cryo-Electron Microscopy (cryo-EM) | High-resolution structural determination of antibody-antigen complexes. | [110] |
| In Vivo Models | Bone Marrow Chimeric (BMC) Mice | Study SHM and "affinity birth" in a competitive, polyclonal B cell environment. | [112] |
| Syrian Hamster Model | In vivo evaluation of antibody-mediated protection and escape variant selection for SARS-CoV-2. | [43] |
This comparative guide delineates a clear spectrum of SHM thresholds required for broad neutralization across major pathogen classes. HIV-1 represents a high-threshold paradigm, where elite neutralization demands extensive SHM, particularly for antibodies targeting conserved epitopes. In contrast, the response to SARS-CoV-2 demonstrates that public clonotypes with low SHM can achieve notable breadth, though specific mutations are critical for combating antigenically drifted variants. The emerging concept of "affinity birth" reveals that GCs can generate novel specificities beyond the primary repertoire, a mechanism with profound implications for vaccine design. These findings underscore that rational immunogen design must be tailored to the specific SHM landscape and evolutionary pathways characteristic of each pathogen.
Conclusion
The collective evidence establishes somatic hypermutation as a central determinant of neutralizing antibody breadth, with implications spanning fundamental immunology to clinical translation. Key insights reveal that permissive germinal center reactions maintaining clonal diversity, sustained affinity maturation timelines, and strategic immunogen design are critical for eliciting broadly neutralizing antibodies. Future directions should focus on developing vaccines that specifically guide SHM toward conserved epitopes, computational models predicting optimal mutation pathways, and therapeutic interventions overcoming age-related declines in SHM efficiency. For biomedical research, leveraging these principles promises accelerated development of broadly protective countermeasures against current and emerging viral threats.
References
- 1. A paradigm shift in simulating affinity maturation to elicit ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12259674/]
- 2. A paradigm shift in simulating affinity maturation to elicit ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1627674/full]
- 3. Regulation of AID, the B-cell genome mutator - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3553278/]
- 4. The off-target effects of AID in carcinogenesis [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1221528/full]
- 5. Germinal center entry not selection of B cells is controlled ... [https://www.nature.com/articles/s41467-018-03382-x]
- 6. Thrifty wide-context models of B cell receptor somatic ... [https://elifesciences.org/reviewed-preprints/105471v2]
- 7. Thrifty wide-context models of B cell receptor somatic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12396816/]
- 8. The Effects of Somatic Hypermutation on Neutralization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3836729/]
- 9. Different Somatic Hypermutation Levels among Antibody ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2017.00389/full]
- 10. Dynamics of B cells in germinal centres - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4399774/]
- 11. Review The unique biology of germinal center B cells [https://www.sciencedirect.com/science/article/pii/S1074761321003010]
- 12. Positive Selection in the Light Zone of Germinal Centers [https://pmc.ncbi.nlm.nih.gov/articles/PMC8044421/]
- 13. Germinal Center B Cells Replace Their Antigen Receptors ... [https://www.sciencedirect.com/science/article/pii/S1074761318303881]
- 14. A new perspective is needed for positive selection of ... [https://www.nature.com/articles/s41423-021-00823-4]
- 15. Regulated somatic hypermutation enhances antibody ... [https://www.nature.com/articles/s41586-025-08728-2]
- 16. Optimizing the breadth of SARS-CoV-2-neutralizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12285597/]
- 17. Article B cell somatic hypermutation following COVID-19 ... [https://www.sciencedirect.com/science/article/pii/S2589004224009386]
- 18. B cell receptor signaling in germinal centers prolongs ... [https://www.sciencedirect.com/science/article/pii/S1074761323000778]
- 19. Activation and maturation of antigen-specific B cells in ... [https://www.nature.com/articles/s41423-025-01285-8]
- 20. B cell somatic hypermutation following COVID-19 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11035370/]
- 21. Somatically hypermutated antibodies isolated from SARS ... [https://www.nature.com/articles/s41467-023-36761-0]
- 22. The Use of Somatic Hypermutation for the Affinity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6497467/]
- 23. A broad atlas of somatic hypermutation allows prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5839764/]
- 24. Thrifty wide-context models of B cell receptor somatic ... [https://elifesciences.org/articles/105471]
- 25. Somatic hypermutation patterns in immunoglobulin ... [https://elifesciences.org/reviewed-preprints/106566]
- 26. Complementary antibody lineages achieve neutralization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9714913/]
- 27. Naturally enhanced neutralizing breadth against SARS ... [https://www.nature.com/articles/s41586-021-03696-9]
- 28. Systems biological assessment of the temporal dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9915811/]
- 29. Imprints of somatic hypermutation on B-cell receptor ... [https://pubmed.ncbi.nlm.nih.gov/40489958/]
- 30. Germinal centre-driven maturation of B cell response to mRNA vaccination | Nature [https://www.nature.com/articles/s41586-022-04527-1]
- 31. Comprehensive Analysis of TCR and BCR Repertoires [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-024-00034-z]
- 32. Distinct clonal evolution of B-cells in HIV controllers with ... [https://elifesciences.org/articles/62648]
- 33. Immune Repertoire Sequencing Trends: TCR & BCR Insights [https://www.cd-genomics.com/biomedical-ngs/emerging-trends-in-immune-repertoire-sequencing-technologies.html]
- 34. A researcher's guide to TCR/BCR repertoire analysis [https://blog.platforma.bio/p/a-researchers-guide-to-tcrbcr-repertoire]
- 35. a comparison of B-cell receptor sequencing methods - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4243823/]
- 36. B cell receptor repertoires identified by next-generation ... [https://www.sciencedirect.com/science/article/abs/pii/S152166162500169X]
- 37. Interpreting the B-cell receptor repertoire with single- ... [https://www.nature.com/articles/s42256-022-00492-6]
- 38. Full-length single-cell BCR sequencing paired with RNA ... [https://www.nature.com/articles/s42003-024-06823-0]
- 39. Review B cell phylogenetics in the single cell era [https://www.sciencedirect.com/science/article/abs/pii/S1471490623002582]
- 40. BASIC: BCR assembly from single cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5408917/]
- 41. Parse Biosciences: Scalable Single Cell Sequencing [https://www.parsebiosciences.com/]
- 42. How does single cell RNA-seq work [https://www.10xgenomics.com/blog/how-does-single-cell-rna-seq-work]
- 43. Somatic hypermutation shapes the viral escape profile of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12148588/]
- 44. Best practices for single-cell analysis across modalities [https://pmc.ncbi.nlm.nih.gov/articles/PMC10066026/]
- 45. Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901458/]
- 46. Lineage Reconstruction of In Vitro Identified Antigen-Specific Autoreactive B Cells from Adaptive Immune Receptor Repertoires - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9820449/]
- 47. A pathway to HIV-1 neutralization breadth - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4769374/]
- 48. Improved HIV-1 neutralization breadth and potency of V2- ... [https://www.sciencedirect.com/science/article/pii/S2211124723007222]
- 49. Benchmarking immunoinformatic tools for the analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7075533/]
- 50. VDJ Gene Usage in IgM Repertoires of Rhesus and ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.815680/full]
- 51. Somatic Hypermutation-Induced Changes in the Structure and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5250619/]
- 52. A broadly neutralizing antibody recognizes a unique ... [https://www.nature.com/articles/s41467-025-63101-1]
- 53. Clonotype-enriched somatic hypermutations drive affinity ... [https://www.sciencedirect.com/science/article/pii/S2211124725008939]
- 54. An overview of methods for the structural and functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8637828/]
- 55. Complementary antibody lineages achieve neutralization ... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1010945]
- 56. Structural diversity of B-cell receptor repertoires along ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7048297/]
- 57. The process of somatic hypermutation increases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7927892/]
- 58. Structural basis of epitope recognition by anti-alpha- ... [https://www.nature.com/articles/s41531-024-00822-y]
- 59. High-throughput mapping of B-cell receptor sequences to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7158953/]
- 60. Advances in antibody discovery from human BCR repertoires [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2022.1044975/full]
- 61. LIBRA-seq: Accelerating antibody discovery [https://pipebio.com/blog/libra-seq-accelerating-antibody-discovery]
- 62. Efficient isolation of rare B cells using next-generation ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2022.962945/full]
- 63. Efficient discovery of SARS-CoV-2-neutralizing antibodies ... [https://www.nature.com/articles/s41587-022-01232-2]
- 64. High-Throughput B Cell Epitope Determination by Next ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.855772/full]
- 65. Bridging the B Cell Gap: Novel Technologies to Study ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8310089/]
- 66. SARS-CoV-2 antibodies from children exhibit broad ... [https://www.sciencedirect.com/science/article/pii/S2666379123004445]
- 67. Increased memory B cell potency and breadth after a ... [https://www.nature.com/articles/s41586-022-04778-y]
- 68. A rare B cell clonotype imprinted by ancestral SARS-CoV-2 ... [https://www.sciencedirect.com/science/article/pii/S2211124725007351]
- 69. Optimizing immunization protocols to elicit broadly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7443869/]
- 70. Thrifty wide-context models of B cell receptor somatic ... [https://elifesciences.org/reviewed-preprints/105471v1]
- 71. Thrifty wide-context models of B cell receptor somatic hypermutation [https://elifesciences.org/reviewed-preprints/105471]
- 72. Somatic hypermutation analysis for improved identification of B cell clonal families from next-generation sequencing data - PubMed [https://pubmed.ncbi.nlm.nih.gov/32574157/]
- 73. Exploring the impact of clonal definition on B-cell diversity [https://pmc.ncbi.nlm.nih.gov/articles/PMC10150052/]
- 74. Protein engineering strategies for rational immunogen design [https://pmc.ncbi.nlm.nih.gov/articles/PMC8683408/]
- 75. Antibody Platform | Hummingbird Bioscience [https://hummingbirdbioscience.com/antibody-platform/]
- 76. Massively parallel single-cell B-cell receptor sequencing ... [https://www.nature.com/articles/s42003-019-0551-y]
- 77. Levels of somatic hypermutations in B cell receptors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4233388/]
- 78. Sex-biased aging effects on immunoglobulin somatic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8582005/]
- 79. A bioinformatic framework for immune repertoire diversity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4489130/]
- 80. Somatic Hypermutation - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/somatic-hypermutation]
- 81. Defining criteria for broadly neutralizing HIV antibodies [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1624020/full]
- 82. Priming VRC01-precursor B cells with non-envelope ... [https://www.nature.com/articles/s41541-025-01235-5]
- 83. Immunological Strategies for Enhancing Viral ... [https://www.mdpi.com/2673-8449/5/3/21]
- 84. Somatic hypermutation analysis for improved identification of ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007977]
- 85. Short or Long Interval between Priming and Boosting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8003773/]
- 86. A viral vector prime and protein boost vaccine regimen elicits ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12579541/]
- 87. Heterologous prime-boost vaccination with VLA2001 and an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12488559/]
- 88. mRNA prime-boost vaccination promotes clonal continuity ... [https://www.sciencedirect.com/science/article/pii/S1525001625006380]
- 89. Heterologous prime-boost vaccination drives stromal ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1597417/full]
- 90. Comparative analysis of human immune responses ... [https://www.nature.com/articles/s41541-022-00504-x]
- 91. Immunogenicity of Ad26.COV2.S vaccine against SARS- ... [https://www.nature.com/articles/s41586-021-03681-2]
- 92. B cell somatic hypermutation following COVID-19 vaccination ... [https://pubmed.ncbi.nlm.nih.gov/38655202/]
- 93. Durability and expansion of neutralizing antibody breadth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8866515/]
- 94. Differences in SARS-CoV-2-Specific Antibody Responses ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9196038/]
- 95. Heterologous ORFV–Ad26 vaccination broadens antibody ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1715442/full]
- 96. Defining criteria for broadly neutralizing HIV antibodies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12310620/]
- 97. A potent and broad CD4 binding site neutralizing antibody ... [https://www.nature.com/articles/s41421-025-00808-x]
- 98. HIV broadly neutralizing antibody precursors to the Apex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12453070/]
- 99. Imprints of somatic hypermutation on B-cell receptor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12148301/]
- 100. The mutation patterns in B-cell immunoglobulin receptors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4528419/]
- 101. Maturation of germinal center B cells after influenza virus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11211068/]
- 102. Repeated vaccination with homologous influenza ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614894/]
- 103. Repeated vaccination with homologous influenza ... [https://elifesciences.org/articles/107042]
- 104. High-throughput single-cell profiling of B cell responses ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10564424/]
- 105. B cell memory: from generation to reactivation [https://www.nature.com/articles/s41420-024-01889-5]
- 106. Cross-neutralizing and potent human monoclonal ... [https://www.nature.com/articles/s41564-025-02137-x]
- 107. Antibody responses against influenza A decline with ... [https://www.nature.com/articles/s41541-024-01057-x]
- 108. Evolution of antibody immunity following Omicron BA.1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10180619/]
- 109. Antigenic sin and multiple breakthrough infections drive ... [https://www.sciencedirect.com/science/article/pii/S2211124724009963]
- 110. Article Mapping essential somatic hypermutations in a CD4 ... [https://www.sciencedirect.com/science/article/pii/S221112472500484X]
- 111. Somatic hypermutation introduces bystander mutations that ... [https://www.sciencedirect.com/science/article/pii/S1074761323004855]
- 112. Somatic hypermutation generates antibody specificities ... [https://www.sciencedirect.com/science/article/abs/pii/S1074761325001773]
- 113. Practical guidelines for B-cell receptor repertoire sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4654805/]
- 114. Neutralization capacity of antibodies elicited through ... [https://www.nature.com/articles/s41467-022-31556-1]