SDS-PAGE vs BN-PAGE: Choosing the Right Electrophoresis Method for Functional Protein Analysis
This article provides researchers, scientists, and drug development professionals with a comprehensive comparison of SDS-PAGE and Blue Native PAGE (BN-PAGE) for the analysis of functional protein properties.
SDS-PAGE vs BN-PAGE: Choosing the Right Electrophoresis Method for Functional Protein Analysis
Abstract
This article provides researchers, scientists, and drug development professionals with a comprehensive comparison of SDS-PAGE and Blue Native PAGE (BN-PAGE) for the analysis of functional protein properties. We explore the fundamental principles governing these techniques, with SDS-PAGE offering high-resolution separation by molecular weight but denaturing proteins, and BN-PAGE preserving native structures, enzymatic activities, and protein complexes at a cost to resolution. The scope includes detailed methodological protocols, troubleshooting advice for common challenges, and validation strategies through in-gel activity assays and two-dimensional electrophoresis. We also examine hybrid techniques like NSDS-PAGE that aim to balance resolution with functional retention, providing a practical guide for selecting the optimal method based on research objectives in biomedical and clinical applications.
Core Principles: Understanding How SDS-PAGE and BN-PAGE Work
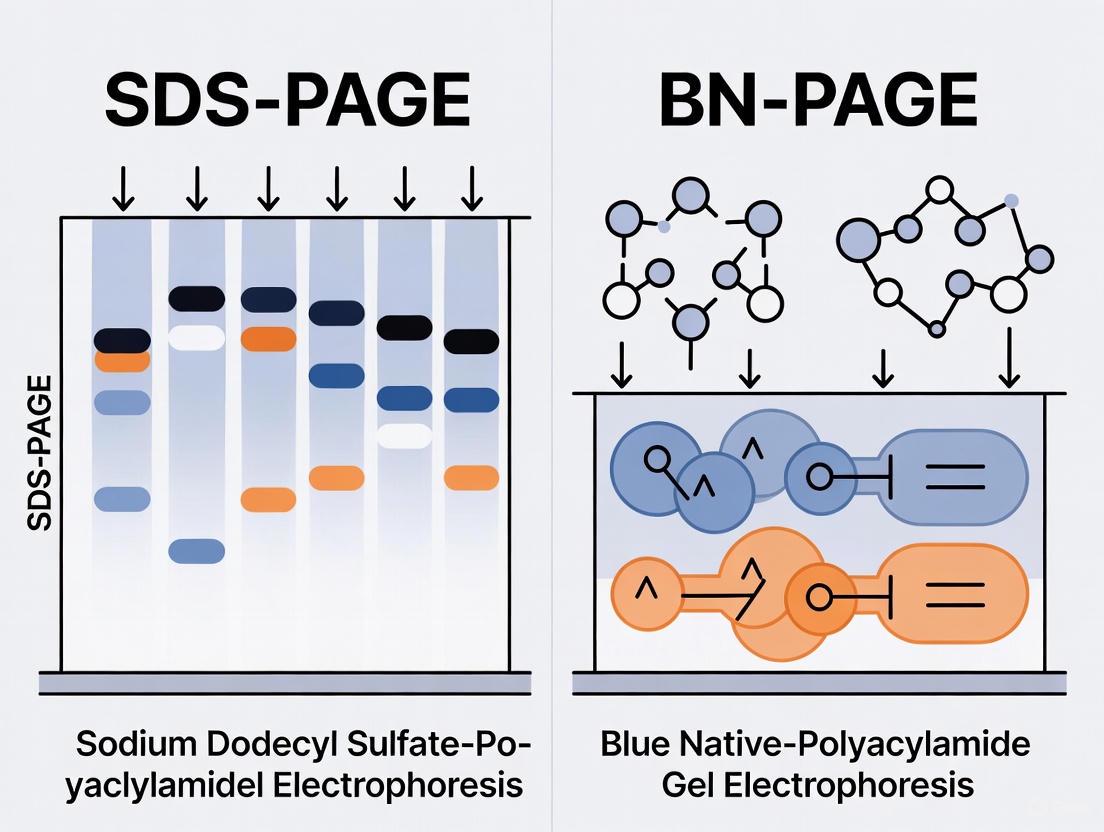
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stands as a cornerstone technique in biochemistry and molecular biology laboratories worldwide. Since its refinement by Ulrich Laemmli in 1970, this method has become indispensable for separating protein mixtures, determining molecular weights, and analyzing protein purity [1]. The technique's widespread adoption stems from its ability to provide high-resolution separation of complex protein mixtures with relatively simple instrumentation. However, the very mechanism that makes SDS-PAGE so effective for molecular weight-based separation—complete protein denaturation—also represents its most significant limitation for researchers interested in native protein properties.
This article examines the fundamental principles of SDS-PAGE that render it unsuitable for studying functional protein characteristics and compares it with alternative electrophoretic methods, particularly blue native PAGE (BN-PAGE), designed to preserve native protein properties. We will explore how the detergent-rich environment of SDS-PAGE systematically dismantles higher-order protein structures, destroying the very functional attributes that many researchers seek to investigate, and present emerging techniques that bridge the gap between resolution and native property retention.
Principles of SDS-PAGE and Protein Denaturation
Mechanism of SDS-Induced Denaturation
SDS-PAGE operates on the principle of complete protein denaturation to achieve separation based primarily on molecular weight. The anionic detergent SDS plays a pivotal role in this process by binding to proteins in a uniform ratio—approximately 1.4 g SDS per 1 g of protein [2]. This binding mechanism unfolds proteins by forming micellar structures where proteins coat the micelle surface, effectively disrupting hydrogen bonds, hydrophobic interactions, and other non-covalent forces that maintain secondary and tertiary structures [1].
The denaturation process masks the proteins' intrinsic charges and imposes a consistent negative charge-to-mass ratio across all polypeptides. Consequently, when subjected to an electric field within the polyacrylamide gel matrix, proteins migrate solely based on size, with smaller molecules moving more rapidly through the porous network [1]. This fundamental principle enables researchers to estimate molecular weight by comparing migration distances against standardized markers, but simultaneously eliminates any capacity to study native protein functions.
The Denaturation Protocol
Standard SDS-PAGE protocols incorporate multiple denaturation steps that collectively dismantle native protein structures. The process begins with sample preparation where proteins are mixed with SDS-containing buffer and heated at 70-100°C for 10 minutes [3] [1]. This heat treatment accelerates the denaturation process, ensuring complete unfolding of resistant protein domains. The running buffer additionally contains 0.1% SDS and EDTA (ethylenediaminetetraacetic acid), the latter serving as a chelating agent that strips essential metal ions from metalloproteins [3]. The combination of reducing agents (such as β-mercaptoethanol or dithiothreitol) further disrupts disulfide bonds, dismantling quaternary structures and ensuring complete dissociation into monomeric subunits.
Limitations for Functional Protein Analysis
Destruction of Functional Properties
The denaturing conditions of SDS-PAGE systematically destroy the structural features essential for protein function. Non-covalently bound cofactors, including metal ions critical for catalytic activity in metalloenzymes, are removed during electrophoresis [3]. Experimental evidence demonstrates that standard SDS-PAGE conditions result in approximately 74% loss of bound zinc ions from metalloproteins [3]. This metal stripping directly abolishes enzymatic activity in metalloenzymes, as confirmed by activity assays showing complete loss of function across nine model enzymes subjected to SDS-PAGE [3].
Protein-protein interactions maintained by non-covalent forces cannot survive SDS treatment, making it impossible to study oligomeric states or multiprotein complexes. Similarly, enzymatic activity is irreversibly lost due to structural unfolding and cofactor displacement. The technique also disrupts binding sites for ligands, substrates, and other interacting molecules, eliminating the possibility of studying these functionally critical interfaces. These limitations fundamentally restrict SDS-PAGE to applications focusing exclusively on covalent protein structure rather than biological function.
Comparative Functional Retention Across Electrophoresis Methods
Table 1: Quantitative Comparison of Functional Property Retention Across Electrophoretic Methods
| Functional Property | SDS-PAGE | BN-PAGE | NSDS-PAGE | 05SAR-PAGE |
|---|---|---|---|---|
| Metal Ion Retention | 26% [3] | >95% [3] | 98% [3] | Not Specified |
| Enzyme Activity Retention | 0/9 model enzymes [3] | 9/9 model enzymes [3] | 7/9 model enzymes [3] | Not Specified |
| Protein-Protein Interactions | Destroyed [4] | Preserved [5] [6] | Partially Preserved [3] | Preserved [4] |
| Resolution | High [3] | Low to Moderate [3] | High [3] | Moderate [4] |
| Molecular Weight Determination | Accurate [1] | Shape/Charge Dependent [4] | Accurate [3] | Shape/Charge Dependent [4] |
Alternative Methods for Native Protein Separation
Blue Native PAGE (BN-PAGE)
BN-PAGE represents a specialized electrophoretic technique specifically designed for separating protein complexes under non-denaturing conditions. First described by Schägger and von Jagow in 1991, this method employs Coomassie Blue G-250 dye, which binds to protein surfaces without disrupting tertiary or quaternary structures [5]. The dye imparts a negative charge proportional to protein size, enabling migration through polyacrylamide gels while preserving native properties [7] [5].
The BN-PAGE protocol involves solubilizing membrane protein complexes with mild detergents like n-dodecyl-β-D-maltopyranoside and staining with Coomassie dye before electrophoresis [5]. Unlike SDS-PAGE, no heating or reducing agents are used, and EDTA is excluded from buffers to prevent metal chelation [3]. This preservation of native structure allows BN-PAGE to maintain enzymatic activities, protein-protein interactions, and bound cofactors, making it particularly valuable for studying mitochondrial complexes, oxidative phosphorylation systems, and other multiprotein assemblies [5] [6].
Emerging Native Electrophoresis Techniques
Native SDS-PAGE (NSDS-PAGE)
NSDS-PAGE represents a hybrid approach that modifies standard SDS-PAGE conditions to balance resolution with native property retention. This method eliminates SDS and EDTA from sample buffers, omits the heating step, and reduces running buffer SDS concentration to 0.0375% [3]. These modifications dramatically increase zinc retention from 26% to 98% compared to standard SDS-PAGE and preserve activity in seven of nine model enzymes [3]. NSDS-PAGE thus offers a compelling compromise, maintaining high resolution while significantly improving functional preservation.
05SAR-PAGE
This recently developed technique utilizes the mild anionic detergent sarkosyl (sodium lauroyl sarcosinate) at low concentration (0.05% w/v) to minimize protein denaturation while enabling electrophoretic separation [4]. NMR studies confirm that 0.05% SAR has subtle effects on native protein structure, allowing identification of dimerization states and post-translational modifications that would be undetectable by SDS-PAGE [4]. The method has successfully demonstrated non-covalent dimerization of PhoBN and PhoRcp proteins and identification of phosphorylated or methylated protein states [4].
Comparative Experimental Workflows
Table 2: Key Buffer Compositions Across Electrophoretic Methods
| Method | Sample Buffer | Running Buffer | Critical Denaturing Components |
|---|---|---|---|
| SDS-PAGE | 2% LDS, 0.51 mM EDTA [3] | 0.1% SDS, 1 mM EDTA [3] | SDS/LDS, EDTA, heating |
| BN-PAGE | 50 mM BisTris, 50 mM NaCl, 10% glycerol [3] | 50 mM BisTris, 50 mM Tricine, 0.02% Coomassie [3] | None |
| NSDS-PAGE | 100 mM Tris HCl, 150 mM Tris Base, 10% glycerol [3] | 0.0375% SDS [3] | Trace SDS only |
| 05SAR-PAGE | Not specified | 0.05% sarkosyl [4] | Low sarkosyl concentration |
Method Selection and Applications
Choosing the Appropriate Electrophoresis Method
The selection of an electrophoretic method should align with specific research objectives. SDS-PAGE remains the optimal choice for applications requiring precise molecular weight determination, assessment of protein purity, or analysis of subunit composition under denaturing conditions [1]. Its high resolution and reproducibility make it ideal for routine analytical applications where native properties are not relevant.
BN-PAGE is particularly suited for investigating intact protein complexes, physiological protein-protein interactions, and mitochondrial complexes [5] [6]. The technique enables determination of native molecular weights and oligomeric states while maintaining enzymatic functionality, though with lower resolution than SDS-PAGE [3].
NSDS-PAGE offers an excellent compromise when both high resolution and partial functional retention are desired. This method is particularly valuable for metalloprotein studies where metal cofactor retention is essential [3]. 05SAR-PAGE provides specialized capabilities for analyzing weak protein-protein interactions, dimerization states, and post-translational modifications without disrupting native conformations [4].
Research Reagent Solutions
Table 3: Essential Reagents for Native Electrophoresis Studies
| Reagent | Function | Example Application |
|---|---|---|
| n-dodecyl-β-D-maltopyranoside | Mild detergent for solubilizing membrane proteins without denaturation [5] | BN-PAGE sample preparation [5] |
| Coomassie Blue G-250 | Charge-conferring dye for native electrophoresis [3] [5] | BN-PAGE and NSDS-PAGE [3] [5] |
| Sarkosyl (SAR) | Mild anionic detergent for minimal protein denaturation [4] | 05SAR-PAGE for dimerization studies [4] |
| 6-aminocaproic acid | Ionic compound for native buffer systems [5] | BN-PAGE gel and buffer formulations [5] |
| Protease Inhibitor Cocktails | Prevent protein degradation during native preparations [5] | All native electrophoresis methods [5] |
| PEPPI-MS Extraction Solution | Efficient protein recovery from gels for MS analysis [8] | Top-down proteomics after native PAGE [8] |
SDS-PAGE's denaturing nature provides excellent resolution for molecular weight-based separation while systematically eliminating the capacity to study functional protein properties. The technique's requirement for SDS and heating fundamentally disrupts non-covalent interactions, destroys enzymatic activity, and strips essential metal cofactors. For researchers investigating native protein properties, alternative methods like BN-PAGE, NSDS-PAGE, and 05SAR-PAGE offer varying balances of resolution and functional preservation. The selection of an appropriate electrophoretic method must therefore align with specific research objectives, recognizing that the denaturing principles that make SDS-PAGE effective for size-based separation simultaneously render it unsuitable for functional proteomics. As electrophoretic technologies continue to evolve, newly developed methods increasingly bridge the historical gap between resolution and native property preservation, expanding the analytical toolbox available to protein scientists.
In the field of proteomics and protein research, electrophoresis techniques represent fundamental tools for separating and analyzing complex protein mixtures. For decades, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) has served as the workhorse method for analytical protein separation, providing high-resolution separation based primarily on molecular mass. This denaturing technique unfolds proteins using an ionic detergent, imparting a uniform negative charge that enables separation through a polyacrylamide gel matrix [3] [9]. However, this very strength constitutes its fundamental limitation: the complete destruction of native protein structures, including higher-order complexes, enzymatic activity, and non-covalently bound cofactors such as metal ions [3].
To address this critical limitation, Blue Native PAGE (BN-PAGE) emerged as a complementary technique that preserves native protein structures and functions. First described by Schägger and von Jagow in 1991, BN-PAGE was specifically developed to study mitochondrial oxidative phosphorylation (OXPHOS) complexes but has since expanded to numerous applications across biological systems [10] [11] [12]. This guide provides a comprehensive comparison between these complementary techniques, focusing on their fundamental principles, methodological approaches, and applications in functional protein research.
Fundamental Principles: A Tale of Two Mechanisms
SDS-PAGE: The Denaturing Workhorse
SDS-PAGE operates on a straightforward denaturing principle. The anionic detergent sodium dodecyl sulfate (SDS) binds to proteins in a uniform ratio (approximately 1.4 g SDS per 1 g protein), effectively unfolding secondary and tertiary structures while imparting a consistent negative charge-to-mass ratio [9] [2]. This process eliminates intrinsic charge differences between proteins and dismantles non-covalent interactions, including protein-protein interactions and metal ion coordination. Consequently, separation occurs almost exclusively by molecular weight as proteins migrate through the polyacrylamide gel sieve toward the anode [3] [9]. While excellent for determining molecular weight, assessing purity, and initial expression analysis, SDS-PAGE irrevocably destroys all native functional properties during separation.
BN-PAGE: Preserving Native Architecture
BN-PAGE employs a fundamentally different mechanism designed to maintain proteins in their native, functional state. The technique utilizes mild non-ionic detergents such as n-dodecyl-β-d-maltoside or digitonin to gently solubilize membrane proteins without disrupting protein-protein interactions [13] [10] [11]. Instead of SDS, BN-PAGE employs the anionic dye Coomassie Brilliant Blue G-250, which binds hydrophobically to protein surfaces, providing the necessary charge for electrophoretic migration without causing denaturation [10] [11] [12]. This dye creates a negative charge shift that enables migration toward the anode while maintaining protein complexes intact [11] [14]. The resulting separation depends not only on molecular mass but also on native charge and three-dimensional structure, preserving enzymatic activity, subunit interactions, and cofactor binding throughout the separation process [10] [11].
Table 1: Fundamental Mechanism Comparison Between SDS-PAGE and BN-PAGE
| Parameter | SDS-PAGE | BN-PAGE |
|---|---|---|
| Detergent Type | Ionic (SDS) | Non-ionic (n-dodecyl-β-d-maltoside, digitonin) |
| Charge Agent | SDS itself | Coomassie Brilliant Blue G-250 |
| Protein State | Denatured, unfolded | Native, folded |
| Complex Preservation | Dissociates complexes | Maintains intact complexes |
| Separation Basis | Primarily molecular mass | Mass, charge, and 3D structure |
| Functional Retention | None | Enzymatic activity, cofactors, interactions |
Experimental Data: Quantitative Comparison of Performance
Direct comparative studies demonstrate significant functional differences between these separation techniques. Research examining zinc metalloproteins revealed striking disparities in metal retention: standard SDS-PAGE preserved only 26% of bound Zn²⁺ ions, while modified native conditions (approaching BN-PAGE principles) retained 98% of metal ions [3]. Similarly, enzymatic activity assays showed that seven of nine model enzymes, including four zinc-dependent proteins, maintained functionality after BN-PAGE separation, whereas all nine were completely inactivated by SDS-PAGE treatment [3].
The resolution capabilities of each technique also differ substantially. While SDS-PAGE typically provides superior resolution for individual polypeptide chains, BN-PAGE excels at resolving intact complexes ranging from ~100 kDa to several MDa [11] [14]. This makes BN-PAGE particularly valuable for studying large supercomplexes such as respiratory chain assemblies in mitochondria [10] [14] or photosystem megacomplexes in thylakoid membranes [14].
Table 2: Experimental Performance Comparison for Functional Protein Studies
| Performance Metric | SDS-PAGE | BN-PAGE |
|---|---|---|
| Metal Ion Retention | 26% (Zn²⁺) | 98% (Zn²⁺) |
| Enzyme Activity Preservation | 0/9 model enzymes | 7/9 model enzymes |
| Molecular Weight Resolution | Excellent for polypeptides (10-300 kDa) | Excellent for complexes (100 kDa - 10 MDa) |
| Membrane Protein Complexes | Dissociates into subunits | Maintains intact complexes and supercomplexes |
| Downstream Applications | Western blotting, mass spectrometry (denatured) | In-gel activity assays, native immunoblotting, 2D analysis |
Methodology: Step-by-Step Experimental Protocols
BN-PAGE Standard Protocol
Sample Preparation:
- Isolate mitochondria or membrane fractions from tissues or cells using differential centrifugation [10] [5].
- Resuspend sedimented mitochondria (0.4 mg) in 40 μL of solubilization buffer (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris, pH 7.0) [5].
- Add 7.5 μL of 10% n-dodecyl-β-d-maltoside (or digitonin for supercomplex preservation) [10] [14].
- Incubate on ice for 30 minutes with occasional mixing [5].
- Centrifuge at 20,000-72,000 × g for 30 minutes at 4°C to remove insoluble material [5] [12].
- Collect supernatant and add 2.5 μL of 5% Coomassie Blue G-250 solution in 0.5 M aminocaproic acid [5].
Gel Electrophoresis:
- Prepare a native gradient gel (typically 4-16% acrylamide) using a gradient mixer [10] [5].
- Load 5-20 μL of prepared sample per well [5].
- Conduct electrophoresis at 4°C using cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G-250, pH 7.0) and anode buffer (50 mM Bis-Tris, pH 7.0) [5] [12].
- Run at constant voltage (150 V for small gels) until the dye front approaches the gel bottom (typically 2-4 hours) [5] [15].
BN-PAGE Experimental Workflow
Critical Technical Considerations
Detergent Optimization: The choice and concentration of detergent significantly impact complex preservation. For fragile supercomplexes, digitonin often outperforms n-dodecyl-β-d-maltoside [10] [14]. Optimal detergent concentrations typically range between 0.5-2% (w/v) with detergent-to-protein ratios of 1:1 to 10:1 [13].
Gel System Selection: While gradient gels (4-16% or 3-12% acrylamide) provide optimal resolution across complex size ranges [10] [5], non-gradient highly porous gels (e.g., 8% acrylamide with 100:1 acrylamide:bis ratio) can also effectively separate complexes I-V while allowing simultaneous detection of additional proteins like Hsp60 polymers and dihydrolipoamide dehydrogenase [12].
In-Gel Activity Assays: Following BN-PAGE separation, complexes can be directly assayed for enzymatic activity through histochemical staining [10] [11] [12]:
- Complex I/DLDH: Incubate gel in 50 mM potassium phosphate buffer (pH 7.0) with 0.2 mg/mL NBT and 0.1 mg/mL NADH [12]
- Complex II: Stain with 0.5 M sodium succinate, 215 μM phenazine methosulfate, and 20 mg NBT in 5 mM Tris-HCl (pH 7.4) [12]
- Complex IV: Incubate with 50 mM sodium phosphate (pH 7.2), 20 mg 3,3'-diaminobenzidine tetrachloride, and 50 mg cytochrome c [12]
- Complex V: Detect ATP hydrolysis activity in 35 mM Tris, 270 mM glycine (pH 8.3), 14 mM MgCl₂, 0.2% Pb(NO₃)₂, and 8 mM ATP [12]
Research Reagent Solutions: Essential Materials for BN-PAGE
Table 3: Essential Reagents and Materials for BN-PAGE Experiments
| Reagent/Material | Function/Purpose | Example Specifications |
|---|---|---|
| n-dodecyl-β-d-maltoside | Mild non-ionic detergent for membrane protein solubilization | 10% solution in water, high purity [5] |
| Digitonin | Mild non-ionic detergent for supercomplex preservation | 1-2% (w/v) for fragile complexes [10] [14] |
| Coomassie Blue G-250 | Anionic dye for charge shift without denaturation | 5% solution in 0.5 M aminocaproic acid [5] |
| 6-Aminocaproic Acid | Zwitterionic salt for membrane protein solubilization | 0.75 M in Bis-Tris, pH 7.0 [5] [12] |
| Bis-Tris | Buffering agent for native conditions | 50 mM, pH 7.0 [5] |
| Protease Inhibitors | Prevent protein degradation during extraction | PMSF (1 mM), leupeptin (1 μg/mL), pepstatin (1 μg/mL) [5] |
| Acrylamide/Bis Solution | Gel matrix formation | 30-50% stock, varying crosslinking ratios [5] [12] |
| Gradient Former | Creating polyacrylamide gradients | For 6-13% or 4-16% linear gradients [5] |
Applications and Research Contexts
Mitochondrial Research
BN-PAGE has become indispensable in mitochondrial research, particularly for studying oxidative phosphorylation (OXPHOS) complexes. The technique enables analysis of individual complexes (I-V) and their organization into higher-order supercomplexes (respirasomes) [10]. This application proves particularly valuable for investigating mitochondrial disorders, as BN-PAGE can identify assembly defects in patient-derived fibroblasts, skeletal muscle biopsies, and cell models [10]. The method also supports dynamic studies of complex assembly pathways and the impact of genetic mutations on OXPHOS integrity [10] [11].
Membrane Protein Complexes
Beyond mitochondria, BN-PAGE facilitates the study of diverse membrane protein complexes, including thylakoid membrane complexes in plants [14], nuclear protein complexes [12], and multiprotein receptors in immunology [15]. The technique's capacity to preserve labile protein-lipid and protein-protein interactions enables researchers to characterize native complex composition, stoichiometry, and functional interactions under near-physiological conditions [13] [11].
Two-Dimensional Analysis
BN-PAGE frequently serves as the first dimension in two-dimensional separation systems, followed by denaturing SDS-PAGE in the second dimension [5] [15]. This approach combines the complex-preserving benefits of BN-PAGE with the high polypeptide resolution of SDS-PAGE, enabling comprehensive analysis of complex subunit composition, identification of novel complex components, and detection of post-translational modifications within native assemblies [15].
The choice between BN-PAGE and SDS-PAGE fundamentally depends on research objectives and the nature of the biological questions being addressed. SDS-PAGE remains the superior choice for applications requiring precise molecular weight determination, assessment of protein purity, expression analysis, and immunodetection of individual subunits without concern for native functionality. Its high resolution for denatured polypeptides and established protocols make it ideal for routine analytical applications.
Conversely, BN-PAGE provides unique capabilities for functional proteomics studies investigating native protein properties, including enzymatic activities, protein-protein interactions, complex assembly states, and metal cofactor retention. While requiring more optimization in detergent conditions and buffer systems, BN-PAGE offers unparalleled insights into the native architecture and functional organization of multiprotein complexes. For comprehensive analysis, many researchers employ both techniques within complementary experimental frameworks, leveraging their respective strengths to obtain both structural and functional information about their protein systems of interest.
In the field of protein separation science, the choice of electrophoretic technique profoundly influences the biological relevance of the results. For researchers investigating functional protein properties, the fundamental chemistry underlying Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Blue Native PAGE (BN-PAGE) dictates their experimental utility. This comparison guide examines the key chemical differences in detergents, dyes, and buffer systems between these techniques, providing researchers and drug development professionals with the experimental data necessary to select the optimal approach for preserving protein function while achieving high-resolution separation.
Core Chemical Principles and Composition
The fundamental separation mechanisms of SDS-PAGE and BN-PAGE diverge in their chemical treatment of protein samples, primarily through their use of distinct detergents, dyes, and buffer systems that dictate whether proteins remain native or denatured throughout electrophoresis.
Table 1: Core Chemical Compositions of SDS-PAGE and BN-PAGE
| Chemical Component | SDS-PAGE | BN-PAGE |
|---|---|---|
| Primary Detergent/Dye | Sodium Dodecyl Sulfate (SDS) [16] [17] | Coomassie Blue G-250 dye [18] |
| Chemical Function | Denatures proteins; confers uniform negative charge [16] [17] | Imparts negative charge while preserving native structure [18] |
| Sample Buffer Additives | Reducing agents (DTT, BME); EDTA [16] [3] | Glycerol; mild detergents (digitonin, dodecylmaltoside) [3] [18] |
| Running Buffer Additives | SDS; EDTA [3] | Coomassie Blue (cathode buffer) [3] |
| Key Chemical Omissions | None | No SDS; no reducing agents; no EDTA [16] |
| Protein State Post-Separation | Denatured and non-functional [16] | Native and often functional [16] [18] |
In SDS-PAGE, the anionic detergent Sodium Dodecyl Sulfate (SDS) is the primary chemical workhorse. SDS comprehensively denatures proteins by binding to hydrophobic regions, unfolding them into linear rods and masking their intrinsic charges [16] [17]. This creates a uniform negative charge-to-mass ratio, ensuring separation occurs almost exclusively by molecular weight [2]. Sample buffers typically include reducing agents like β-mercaptoethanol or DTT to break disulfide bonds, and EDTA to chelate metal ions, thereby stripping away any non-covalently bound cofactors [16] [3].
In contrast, BN-PAGE replaces the denaturing detergent with Coomassie Blue G-250 dye. This dye binds non-covalently to the surface of proteins, providing the necessary negative charge for electrophoretic migration without causing significant unfolding [18]. This fundamental chemical difference preserves the protein's tertiary and quaternary structure. The buffer systems in BN-PAGE avoid reducing agents and strong chelators like EDTA, and often include mild non-ionic detergents such as digitonin to solubilize membrane proteins while maintaining protein-protein interactions within complexes [3] [18].
Experimental Protocols and Methodologies
Standard SDS-PAGE Protocol
The following protocol, adapted from commercial systems, highlights the key chemical steps that ensure complete denaturation [3]:
- Sample Preparation: Mix protein sample with a 4X LDS (Lithium Dodecyl Sulfate) sample buffer. The final composition is typically 106 mM Tris-HCl, 141 mM Tris Base, 0.51 mM EDTA, 2% LDS, and 10% glycerol, pH 8.5 [3].
- Denaturation: Heat the sample mixture at 70°C for 10 minutes. This step, combined with LDS/SDS, ensures complete unfolding of proteins [16] [3].
- Electrophoresis: Load samples onto a polyacrylamide gel and run at room temperature with a running buffer containing 50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, and 0.1% SDS, pH 7.7 [3].
- Visualization: Resolved proteins are typically visualized using Coomassie staining or silver stain, but are irreversibly denatured and cannot be recovered for functional assays [16].
Standard BN-PAGE Protocol
This protocol is designed to maintain protein complexes in their native state [3] [18]:
- Sample Preparation: Mix protein sample with a 4X BN-PAGE sample buffer. A typical composition is 50 mM BisTris, 50 mM NaCl, 10% glycerol, and 0.001% Ponceau S, pH 7.2 [3]. Mild detergents are added to the lysate to solubilize complexes without disruption [18].
- No Heating: The sample is not heated prior to loading [16] [3].
- Electrophoresis: Load samples onto a native gradient gel (e.g., 4-16% Bis-Tris). Use separate cathode (50 mM BisTris, 50 mM Tricine, 0.02% Coomassie G-250, pH 6.8) and anode (50 mM BisTris, 50 mM Tricine, pH 6.8) running buffers [3]. Run at 4°C or with cooling to minimize heat-induced denaturation [16] [3].
- Visualization and Recovery: Proteins can be visualized with Coomassie stain. Crucially, proteins can often be eluted from the gel in their active, native form for downstream functional studies [16] [18].
Native SDS-PAGE (NSDS-PAGE): A Hybrid Approach
To address the limitations of both techniques, a modified method called Native SDS-PAGE (NSDS-PAGE) has been developed. This protocol aims to balance resolution with function retention [3]:
- Sample Preparation: Use a modified sample buffer (100 mM Tris-HCl, 150 mM Tris Base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5) that omits SDS and EDTA [3].
- No Denaturation: Omit the heating step [3].
- Electrophoresis: Use a standard polyacrylamide gel but a modified running buffer with a significantly reduced SDS concentration (0.0375% instead of 0.1%) and no EDTA [3].
Quantitative Performance Data and Functional Outcomes
The chemical differences between these methods lead to measurable disparities in their ability to preserve protein function, particularly for metalloproteins and enzymes.
Table 2: Quantitative Functional Outcomes Across PAGE Methods
| Performance Metric | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Enzyme Activity Retention | 0 out of 9 model enzymes active [3] | 9 out of 9 model enzymes active [3] | 7 out of 9 model enzymes active [3] |
| Metalloprotein Zinc Retention | 26% [3] | Not Reported | 98% [3] |
| Resolution of Complex Mixtures | High [3] [19] | Lower than SDS-PAGE [3] | High, comparable to SDS-PAGE [3] |
| Suitable Molecular Weight Range | 5 - 250 kDa [17] | 100 kDa - 10 MDa [18] | Not Reported |
Experimental data demonstrates that standard SDS-PAGE conditions result in near-total loss of protein function. A comparative study showed that none of the nine model enzymes tested retained activity after SDS-PAGE, and only 26% of zinc was retained in metalloproteins [3]. In stark contrast, BN-PAGE preserved the activity of all nine enzymes [3]. The hybrid NSDS-PAGE method achieved high-resolution separation comparable to SDS-PAGE while successfully preserving the activity for seven of the nine enzymes and retaining 98% of the zinc in metalloproteins [3].
These findings highlight a critical trade-off: the denaturing chemicals in SDS-PAGE enable excellent resolution based purely on polypeptide size, but at the cost of functional properties. The native chemicals in BN-PAGE preserve function but can compromise resolution, particularly for complex proteomic mixtures [3] [19].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these electrophoretic techniques requires careful selection of key reagents, each playing a specific chemical role.
Table 3: Essential Reagents for PAGE Techniques
| Reagent | Core Function | Application Notes |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Denatures proteins; imparts uniform charge [16] [17] | Critical for SDS-PAGE; omitted in BN-PAGE and reduced in NSDS-PAGE [16] [3] |
| Coomassie Blue G-250 | Imparts charge for migration in native systems [18] | Used in BN-PAGE cathode buffer; can be omitted for Colorless Native PAGE (CN-PAGE) [18] |
| Dithiothreitol (DTT) / β-Mercaptoethanol | Reduces disulfide bonds [16] | Used in SDS-PAGE sample buffer; typically omitted in native techniques [16] |
| Mild Detergents (Digitonin) | Solubilizes membrane proteins gently [18] | Used in BN-PAGE sample preparation to maintain protein complexes [18] |
| EDTA (Ethylenediaminetetraacetic acid) | Chelates metal ions [3] | Present in SDS-PAGE buffers; omitted in native methods to preserve metalloproteins [3] |
| Glycerol | Increases sample density; prevents diffusion [17] | Common component in sample buffers for both SDS-PAGE and BN-PAGE [3] |
Experimental Workflow and Decision Pathway
The following diagram illustrates the logical decision process for selecting an electrophoresis method based on research objectives and the corresponding experimental workflows.
Diagram Title: Method Selection and Experimental Workflow
The chemical components of electrophoretic systems—detergents, dyes, and buffer additives—are not merely technical details but fundamental determinants of experimental outcomes. SDS-PAGE, employing the denaturing power of SDS, remains the gold standard for determining molecular weight and achieving high-resolution separation of polypeptides, but it obliterates functional protein properties. BN-PAGE, through the strategic use of Coomassie dye and mild buffers, preserves native structure and function at the potential cost of some resolving power. The emerging NSDS-PAGE method offers a promising compromise, modifying traditional SDS-PAGE chemistry to retain high resolution while dramatically improving the retention of metal cofactors and enzymatic activity. For researchers in drug development and functional proteomics, the informed selection of an electrophoretic method based on these chemical principles is crucial for generating biologically relevant data on protein complexes and their activities.
For researchers and drug development professionals, polyacrylamide gel electrophoresis (PAGE) serves as a fundamental tool for protein analysis. However, a fundamental trade-off has long persisted between resolution and functional preservation. On one hand, denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) provides high-resolution separation of complex protein mixtures based primarily on molecular mass, but it deliberately destroys higher-order structure and functional properties through detergent denaturation and heating [3]. On the other hand, blue native polyacrylamide gel electrophoresis (BN-PAGE) preserves native protein conformations, enzymatic activity, and protein-protein interactions, but achieves significantly lower resolution of proteomic mixtures and can complicate molecular weight determination [3] [20].
This methodological divide creates significant challenges for fields ranging from structural biology to drug development, where understanding both composition and function is critical. Recent methodological advances, particularly the introduction of native SDS-PAGE (NSDS-PAGE), seek to bridge this gap by modifying traditional SDS-PAGE conditions to maintain high resolution while preserving functional properties [3]. This comparison guide objectively evaluates these electrophoretic techniques based on experimental data, providing researchers with a framework for selecting appropriate methodologies based on their specific analytical needs.
Methodological Principles: Mechanism of Separation and Denaturation
SDS-PAGE: Denaturing Separation
Standard SDS-PAGE employs the anionic detergent sodium dodecyl sulfate (SDS) to denature protein samples, coupled with heating (typically 70-100°C) to ensure complete unfolding. The SDS binds to denatured proteins at a relatively constant ratio of approximately 1.4 g SDS per 1 g protein, imparting a uniform negative charge density that allows separation based primarily on molecular size through a polyacrylamide gel matrix [3]. The process includes EDTA (ethylenediaminetetraacetic acid) in buffers to chelate metal ions, effectively stripping them from metalloproteins. While this enables excellent resolution and mass estimation, it destroys tertiary and quaternary structures, enzymatic activity, and non-covalent protein-metal partnerships [3].
BN-PAGE: Native Separation
BN-PAGE utilizes the dye Coomassie Blue G-250, which confers negative charges to protein surfaces without causing significant denaturation. This approach preserves native protein conformations, oligomeric states, and interactions by maintaining proteins in their physiological conditions without detergents like SDS or heating steps [3] [20]. Separation depends on a complex combination of size, charge, and three-dimensional structure, which maintains functionality but results in lower resolution and more challenging molecular weight interpretation due to these multiple influencing factors [21]. BN-PAGE has proven particularly valuable for studying mitochondrial respiratory complexes, protein-protein interactions, and supramolecular structures [20].
NSDS-PAGE: Hybrid Approach
NSDS-PAGE represents a modified approach that reduces denaturing conditions while maintaining resolution. This method eliminates SDS and EDTA from sample buffers, omits the heating step, and reduces SDS concentration in running buffers to 0.0375% (compared to 0.1% in standard SDS-PAGE) [3]. The technique maintains the fundamental size-based separation mechanism of SDS-PAGE while significantly improving the retention of metal ions and functional properties, effectively creating a middle ground between traditional SDS-PAGE and BN-PAGE.
Table 1: Key Buffer Composition Differences Between Electrophoretic Methods
| Component | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| SDS | 0.1% in running buffer | None | 0.0375% in running buffer |
| EDTA | 1 mM in running buffer | None | None |
| Heating Step | 70°C for 10 minutes | None | None |
| Coomassie Dye | None | In cathode buffer (0.02%-0.002%) | In sample buffer (0.01875%) |
| Primary Separation Mechanism | Molecular mass | Size, charge, and 3D structure | Molecular mass |
Quantitative Comparison: Experimental Data on Functional Property Retention
Metal Ion Retention Capabilities
For metalloprotein research, the retention of bound metal ions during electrophoresis is crucial for maintaining structural integrity and function. Experimental comparisons using laser ablation-inductively coupled plasma-mass spectrometry and in-gel Zn-protein staining with fluorophore TSQ have demonstrated significant differences between methods:
Table 2: Zinc Retention in Model Metalloproteins Across Electrophoretic Methods
| Method | Zinc Retention | Experimental Model |
|---|---|---|
| SDS-PAGE | 26% | Pig kidney (LLC-PK1) cell proteome |
| BN-PAGE | >95% | Pig kidney (LLC-PK1) cell proteome |
| NSDS-PAGE | 98% | Pig kidney (LLC-PK1) cell proteome |
The dramatically improved metal retention in NSDS-PAGE (98%) compared to traditional SDS-PAGE (26%) highlights its potential for metalloprotein studies, approaching the near-complete preservation achieved with BN-PAGE while maintaining higher resolution [3].
Enzymatic Activity Preservation
Enzyme function requires proper folding and often intact cofactor binding sites. Studies with nine model enzymes, including four zinc-binding proteins (yeast alcohol dehydrogenase, bovine alkaline phosphatase, superoxide dismutase, and carbonic anhydrase), revealed stark contrasts in functional preservation:
Table 3: Enzymatic Activity Retention After Electrophoresis
| Method | Enzymes Active/Total Tested | Percentage Active |
|---|---|---|
| SDS-PAGE | 0/9 | 0% |
| BN-PAGE | 9/9 | 100% |
| NSDS-PAGE | 7/9 | 78% |
These findings demonstrate that while BN-PAGE completely preserves enzymatic activity, NSDS-PAGE maintains functionality for most enzymes (78%), representing a significant improvement over traditional SDS-PAGE, which denatures all enzymatic function [3].
Experimental Protocols: Detailed Methodologies
NSDS-PAGE Protocol
The following protocol for NSDS-PAGE is adapted from published methodology that successfully preserved zinc binding and enzymatic activity [3]:
Sample Preparation: Combine 7.5 μL of protein sample (5-25 μg protein) with 2.5 μL of 4X NSDS sample buffer (100 mM Tris HCl, 150 mM Tris base, 10% v/v glycerol, 0.0185% w/v Coomassie G-250, 0.00625% w/v Phenol Red, pH 8.5).
Gel Preparation: Use precast NuPAGE Novex 12% Bis-Tris 1.0 mm mini-gels. Pre-run at 200V for 30 minutes in double distilled H₂O to remove storage buffer and unpolymerized acrylamide.
Running Buffer: Prepare NSDS-PAGE running buffer (50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7).
Electrophoresis: Load samples and run at constant voltage (200V) for approximately 45 minutes at room temperature until the dye front reaches the end of the gel.
The critical modifications from standard SDS-PAGE include the elimination of EDTA, reduced SDS concentration, and omission of the heating step before loading, which collectively help preserve functional properties while maintaining resolution.
BN-PAGE Protocol for Protein Complexes
For studying intact protein complexes, the following BN-PAGE protocol has been successfully applied to mitochondrial respiratory complexes and other multi-protein assemblies [20]:
Mitochondrial Isolation: Isolate mitochondria from tissue samples using differential centrifugation followed by purification on 27%-45%-60% Percoll gradients.
Protein Solubilization: Solubilize mitochondrial proteins in digitonin using a NativePAGE Sample Prep Kit according to manufacturer instructions.
Sample Preparation: Mix 7.5 μL of protein sample with 2.5 μL of 4X BN-PAGE sample buffer (50 mM BisTris, 50 mM NaCl, 16 mM HCl, 10% Glycerol, 0.001% Ponceau S, pH 7.2).
Gel Electrophoresis: Load samples onto NativePAGE Novex 4-16% Bis-Tris 1.0 mm minigels. Run at constant voltage (150V) at room temperature using 1X solutions of Anode (50 mM BisTris, 50 mM Tricine, pH 6.8) and Cathode (50 mM BisTris, 50 mM Tricine, 0.02% Coomassie G-250, pH 6.8) Running Buffers.
Coomassie Transition: After 50 minutes of electrophoresis, replace the "dark cathode buffer" containing 0.02% Coomassie with "light cathode buffer" containing 0.002% Coomassie.
This protocol has been effectively used to resolve supercomplexes of mitochondrial respiratory chains and analyze their compositional differences between species with different thermogenic patterns [20].
Multimer-PAGE for Complex Stabilization
The Multimer-PAGE technique combines BN-PAGE with cross-linking and SDS-PAGE to stabilize and resolve native protein complexes from unmodified tissue lysates [21]:
Tissue Homogenization: Homogenize 20 mg of target tissue in 1 mL ice-cold BN-PAGE sample buffer with 30 strokes of a dounce homogenizer. Add detergent (2% digitonin) for membrane protein solubilization.
Partial Separation: Load homogenate onto BN polyacrylamide gel (3% T stacking, 6% T resolving layer) and electrophorese at 150V until dye progresses ~2 cm into resolving layer.
Cross-Linking: Excise gel strip containing migrated proteins, equilibrate in PBS, and incubate with 25 mM dithiobis(succinimidyl propionate) (DSP) for 30 minutes to covalently stabilize complexes.
Quenching: Treat with 0.375 M Tris-HCl, pH 8.8, containing 2% SDS to quench unreacted DSP.
Denaturing Separation: Cast cross-linked gel strip into SDS-PAGE gel for second dimension separation under denaturing conditions.
This method reduces nonspecific background cross-linking by partially separating proteins before stabilization, enabling better characterization of native protein complexes without requiring purification or pull-down assays [21].
Workflow Visualization: Integrated Structural Proteomics Approaches
The integration of PAGE separation with mass spectrometry has created powerful workflows for structural proteomics, particularly with the development of efficient protein recovery methods like PEPPI-MS [8].
Diagram 1: Integrated PAGE-MS Structural Proteomics Workflow. This workflow combines SDS-PAGE separation with efficient protein recovery using PEPPI-MS (Passively Eluting Proteins from Polyacrylamide Gels as Intact Species for MS) for subsequent structural analysis by mass spectrometry. The CBB (Coomassie Brilliant Blue) enhanced recovery step enables high protein recovery rates (mean 68% below 100 kDa) for comprehensive structural proteomics [8].
Advanced Applications: Method-Specific Research Applications
BN-PAGE for Mitochondrial Complex Analysis
BN-PAGE has proven particularly valuable for studying mitochondrial respiratory complexes and their organization into supercomplexes. Research comparing thermogenic plants (Symplocarpus renifolius and Arum maculatum) revealed that while constituents of respiratory complexes were generally similar, several mitochondrial components showed differential expression [20]. Notably, complex II in S. renifolius was detected as a 340 kDa product, suggesting an oligomeric or supramolecular structure in vivo that differed from conventional expectations. Alternative oxidase was detected as smear-like signals elongated on the first dimension with a peak at around 200 kDa in both species [20]. These findings demonstrate BN-PAGE's unique capability to resolve native complex variations that would be destroyed by denaturing methods.
NSDS-PAGE for Metalloprotein Studies
The preservation of metal ions in NSDS-PAGE makes it particularly suitable for metalloprotein research. In studies of zinc-binding proteins, NSDS-PAGE demonstrated near-complete zinc retention (98%) compared to minimal retention (26%) in standard SDS-PAGE [3]. This capability enables researchers to study metalloproteins without losing essential structural components, bridging a significant gap in conventional electrophoretic approaches. The method has been successfully applied to zinc-binding enzymes including alcohol dehydrogenase, alkaline phosphatase, superoxide dismutase, and carbonic anhydrase, with most maintaining activity after separation [3].
Two-Dimensional Approaches for Complex Analysis
Two-dimensional electrophoretic techniques combining BN-PAGE with SDS-PAGE provide powerful tools for analyzing protein complex composition:
Diagram 2: Two-Dimensional BN/SDS-PAGE Workflow for Protein Complex Analysis. This approach separates intact protein complexes in the first native dimension, followed by denaturing separation of constituent subunits in the second dimension, enabling comprehensive analysis of complex composition and organization [20] [6].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents for Electrophoretic Protein Separation
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| Digitoxin | Membrane protein solubilization | Critical for extracting membrane-bound complexes while maintaining native structure for BN-PAGE [20] |
| Coomassie Blue G-250 | Charge conferral dye | Imparts negative charge to protein surfaces in BN-PAGE without significant denaturation [3] |
| Dithiobis(succinimidyl propionate) (DSP) | Amine-reactive cross-linker | Stabilizes protein complexes in Multimer-PAGE; cell-permeable with cleavable disulfide bond [21] |
| PEPPI-MS Extraction Solution | Protein recovery from gels | 0.05% SDS/100 mM ammonium bicarbonate with CBB enables 68% mean protein recovery for MS analysis [8] |
| NativePAGE Sample Prep Kit | Native protein solubilization | Optimized for BN-PAGE applications; maintains protein complexes in their native state [20] |
| Tetrahydroxyborate-cross-linked agarose | Capillary gel matrix | Enables baseline hump-free SDS-CGE of therapeutic proteins; excellent for high MW proteins [22] |
| Propidium Iodide | Fluorescent detection | In-gel fluorescent dye for SDS-CGE; affects sieving properties and enables LIF detection [23] |
The choice between SDS-PAGE, BN-PAGE, and hybrid approaches like NSDS-PAGE depends primarily on research objectives and the specific protein properties of interest:
Choose SDS-PAGE when the primary need is high-resolution separation for molecular weight determination, purity assessment, or immunoblotting, and when functional preservation is not required.
Select BN-PAGE when studying native protein complexes, protein-protein interactions, enzymatic activity, or oligomeric states, particularly for membrane-bound complexes like mitochondrial respiratory chains.
Employ NSDS-PAGE when both high resolution and partial functional preservation are needed, particularly for metalloprotein studies or when maintaining some enzymatic activity is desirable.
Utilize integrated PAGE-MS workflows when comprehensive structural proteomics information is required, combining the separation power of electrophoresis with the analytical depth of mass spectrometry.
As electrophoretic methodologies continue to evolve, researchers now have an expanded toolkit for balancing the traditionally competing demands of resolution and functional preservation, enabling more sophisticated protein characterization across diverse research and development applications.
Historical Development and Evolution of Both Techniques
The analysis of protein complexes is fundamental to understanding cellular function. For decades, electrophoretic techniques have been crucial tools for separating and characterizing proteins. This guide examines the historical development and evolution of two principal methodologies: Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Blue Native-PAGE (BN-PAGE). While SDS-PAGE emerged as the standard for denaturing protein separation, BN-PAGE was developed specifically to study protein complexes under native conditions. The ongoing innovation in this field is illustrated by the more recent introduction of Native SDS-PAGE (NSDS-PAGE), which seeks to combine the high resolution of traditional SDS-PAGE with improved retention of native protein properties. This article objectively compares the performance of these techniques for researchers focused on retaining functional protein properties, including enzymatic activity and metal cofactors.
Historical Context and Technical Evolution
SDS-PAGE: The Denaturing Standard
SDS-PAGE became the cornerstone of protein analytical biochemistry following its introduction in the 1970s. The technique relies on the ionic detergent sodium dodecyl sulfate to denature proteins and impart a uniform negative charge, enabling separation primarily by molecular mass with high resolution. A crucial initial step involves heating proteins in the presence of SDS and EDTA, which denatures the protein structure and strips away non-covalently bound metal ions [3]. While this method revolutionized protein purity assessment and molecular weight determination, it systematically destroyed all native functional properties, including enzymatic activity and protein-metal interactions [3] [24].
BN-PAGE: Preserving Native Complexes
Blue Native-PAGE was developed by Schägger and von Jagow in 1991 specifically to address the limitations of denaturing methods [11] [5]. The technique was initially designed for studying mitochondrial respiratory chain complexes, which are embedded in the inner mitochondrial membrane [11]. The key innovation was replacing SDS with Coomassie Blue G250 (Serva Blue G) and mild detergents like dodecylmaltoside [11] [25]. The dye provides the necessary negative charge for electrophoretic mobility without causing complex dissociation, while mild detergents solubilize membranes without disrupting protein-protein interactions [11] [25]. This preservation of native structure enables the study of protein-protein interactions, oligomeric states, and supramolecular assemblies, or "supercomplexes" [25].
NSDS-PAGE: A Hybrid Approach
A more recent evolutionary step came with the development of Native SDS-PAGE (NSDS-PAGE), which modifies traditional SDS-PAGE conditions to retain some native properties without sacrificing resolution. This method eliminates the heating step, removes EDTA from sample buffers, and drastically reduces the SDS concentration in the running buffer from 0.1% to 0.0375% [3] [24]. These modifications result in dramatically improved retention of bound metal ions (Zn²⁺ retention increased from 26% to 98%) and enzymatic activity while maintaining high-resolution separation [24].
Table 1: Historical Development and Key Characteristics of PAGE Techniques
| Feature | SDS-PAGE (1970s) | BN-PAGE (1991) | NSDS-PAGE (2014) |
|---|---|---|---|
| Primary Developer | Laemmli et al. | Schägger & von Jagow | Petering et al. |
| Key Reagent | SDS (0.1-0.2%) | Coomassie Blue G250 + Mild Detergents | Reduced SDS (0.0375%) |
| Sample Preparation | Heating with SDS & EDTA | Cold Solubilization with Mild Detergents | No Heat, No EDTA |
| Separation Basis | Molecular Mass | Native Mass & Shape | Molecular Mass with Native Features |
| Protein State | Denatured | Native | Partially Native |
| Key Advantage | High Resolution | Preserves Complexes & Activity | High Resolution + Metal Retention |
Performance Comparison: Experimental Data
Direct comparison of these techniques reveals significant differences in their ability to preserve functional protein properties. Experimental data from model enzyme systems provides objective performance metrics.
Retention of Enzymatic Activity
Studies with nine model enzymes, including four zinc metalloproteins, demonstrate stark contrasts between techniques. When subjected to BN-PAGE, all nine enzymes retained their catalytic activity. Similarly, NSDS-PAGE preserved activity in seven of the nine enzymes. In contrast, standard SDS-PAGE denatured all nine enzymes, completely abolishing their activity [3] [24]. This indicates that BN-PAGE provides the most reliable conditions for preserving enzymatic function, while NSDS-PAGE offers a viable compromise when higher resolution is required.
Metal Cofactor Retention
For metalloproteins, the retention of bound metal ions is crucial for function. Research measuring zinc retention in proteomic samples found that standard SDS-PAGE retained only 26% of bound Zn²⁺, with EDTA in the buffer chelating and removing metals during electrophoresis [24]. In contrast, NSDS-PAGE (without EDTA and with reduced SDS) retained 98% of bound zinc ions [24]. Laser ablation-inductively coupled plasma-mass spectrometry and in-gel staining with the zinc-specific fluorophore TSQ confirmed these findings [3] [24]. BN-PAGE also demonstrates excellent metal retention, as it avoids denaturing conditions altogether.
Table 2: Quantitative Performance Comparison for Functional Property Retention
| Performance Metric | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Enzyme Activity Retention | 0/9 Model Enzymes [24] | 9/9 Model Enzymes [24] | 7/9 Model Enzymes [24] |
| Metal Ion Retention (Zn²⁺) | 26% [24] | High (Qualitative) [3] | 98% [24] |
| Resolution | High [3] | Moderate [3] | High [3] |
| Membrane Protein Complex Integrity | Dissociates Complexes [26] | Preserves Complexes & Supercomplexes [25] | Limited Data |
| Typical Run Time | ~45 minutes [3] | 90-95 minutes [3] | Similar to SDS-PAGE [3] |
Experimental Protocols and Methodologies
BN-PAGE Protocol for Mitochondrial Complexes
The standard BN-PAGE protocol for analyzing mitochondrial protein complexes involves specific steps to preserve native interactions [11] [5]:
- Sample Preparation: Isolate mitochondria and resuspend in aminocaproic acid buffer (0.75 M aminocaproic acid, 50 mM Bis-Tris, pH 7.0). Solubilize by adding 10% n-dodecyl-β-D-maltopyranoside (approximately 2g detergent/g protein) and incubate on ice for 30 minutes [5].
- Clarification: Centrifuge at 72,000 × g for 30 minutes to remove insoluble material [5].
- Dye Addition: Add Coomassie Blue G250 (5% solution in 0.5 M aminocaproic acid) to the supernatant [5].
- Gel Electrophoresis: Load samples onto a linear acrylamide gradient gel (e.g., 6-13%) [5]. Run with specific anode (50 mM Bis-Tris, pH 7.0) and cathode (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0) buffers at 150V for approximately 2 hours [5].
- Second Dimension (Optional): For subunit analysis, excise BN-PAGE lanes, soak in SDS buffer, and run on SDS-PAGE perpendicular to the first dimension [26] [5].
NSDS-PAGE Protocol
The NSDS-PAGE method modifies standard SDS-PAGE conditions as follows [3]:
- Sample Buffer: 100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5. Note the absence of SDS and EDTA [3].
- Sample Preparation: Mix protein sample with 4X NSDS sample buffer without heating [3].
- Running Buffer: 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7. Note the reduced SDS concentration (0.0375% vs. 0.1% in standard SDS-PAGE) and absence of EDTA [3].
- Electrophoresis: Use precast Bis-Tris gels and run at constant voltage (200V) for approximately 45 minutes [3].
Essential Research Reagent Solutions
Successful implementation of these electrophoretic techniques requires specific reagents optimized for preserving protein structure and function.
Table 3: Essential Research Reagents for Native Electrophoresis
| Reagent | Function | BN-PAGE Application | NSDS-PAGE Application |
|---|---|---|---|
| Coomassie Blue G250 | Imparts negative charge for electrophoretic mobility without denaturation | Critical component in cathode buffer and sample [11] [25] | Present in sample buffer (0.0185%) [3] |
| n-Dodecyl-β-D-Maltoside | Mild non-ionic detergent for solubilizing membranes | Releases protein complexes from mitochondrial membrane without dissociation [11] [5] | Not typically used |
| Digitonin | Mild detergent for labile complexes | Preserves supramolecular assemblies (supercomplexes) [25] | Not typically used |
| 6-Aminocaproic Acid | Zwitterionic salt | Aids complex extraction, prevents aggregation [11] [5] | Not used |
| Bis-Tris Buffer | Neutral pH buffer system | Maintains stable pH during native electrophoresis [5] | Used in gel matrix and running buffer [3] |
| Reduced SDS Concentration | Minimal denaturation | Not used | Critical modification (0.0375% in running buffer) [3] |
The historical evolution from SDS-PAGE to BN-PAGE and the more recent development of NSDS-PAGE represents a continuous effort to balance the competing demands of high resolution separation and preservation of native protein properties. SDS-PAGE remains the gold standard for determining protein purity and molecular weight when functional properties are not relevant. BN-PAGE excels in the analysis of intact protein complexes, protein-protein interactions, and membrane protein supercomplexes where maintaining native structure is paramount. NSDS-PAGE emerges as a promising hybrid technique, offering a unique combination of high resolution and retention of metal ions and enzymatic activity for many proteins. The choice between these techniques should be guided by the specific research question, prioritizing BN-PAGE for maximal functional preservation and NSDS-PAGE when both resolution and certain native properties must be maintained.
Practical Protocols: When and How to Apply Each Method
Step-by-Step BN-Page Protocol for Mitochondrial Complex Analysis
The choice of electrophoresis technique is pivotal in proteomics research, determining whether proteins are separated solely by mass or as intact, functional complexes. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), the longstanding industry standard, denatures proteins into uniform linear chains using an ionic detergent, enabling precise molecular weight separation but destroying native structure, enzymatic activity, and non-covalently bound cofactors [3]. In contrast, Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) preserves protein complexes in their native state through mild non-ionic detergents and Coomassie dye, allowing researchers to study intact complexes, their interactions, and biological activities [10] [5]. This comparison guide objectively evaluates both methodologies, focusing on their performance in mitochondrial complex analysis—a critical application in metabolic disease and drug development research.
Core Principles and Performance Comparison
Fundamental Separation Mechanisms
The foundational difference between these techniques lies in their treatment of protein structure:
SDS-PAGE Mechanism: Proteins are denatured by heating in a buffer containing the ionic detergent SDS and the reducing agent DTT, which unfolds secondary and tertiary structures. SDS binds uniformly along the polypeptide backbone, imparting a consistent negative charge-to-mass ratio. Separation occurs primarily by molecular mass as proteins migrate through a polyacrylamide gel matrix [3].
BN-PAGE Mechanism: Developed by Schägger and von Jagow in the 1990s, this technique solubilizes membranes with mild non-ionic detergents like n-dodecyl-β-D-maltoside while preserving protein-protein interactions. The anionic dye Coomassie Blue G-250 binds to hydrophobic protein surfaces, imposing a negative charge shift that drives electrophoretic migration without denaturation. Separation occurs by both size and native charge, maintaining enzymatic activity and subunit interactions [10] [5] [27].
Quantitative Performance Metrics
Table 1: Direct Performance Comparison Between SDS-PAGE, BN-PAGE, and NSDS-PAGE
| Performance Metric | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Structural Preservation | Denatures proteins; destroys quaternary structures [3] | Preserves native protein complexes and supercomplexes [10] [27] | Partial preservation; maintains some metal ions [3] |
| Metal Cofactor Retention | 26% Zn²⁺ retention in model systems [3] | High retention of metal cofactors [3] | 98% Zn²⁺ retention in model systems [3] |
| Enzymatic Activity Post-Electrophoresis | All 9 model enzymes denatured [3] | All 9 model enzymes remained active [3] | 7 of 9 model enzymes remained active [3] |
| Resolution Capability | High resolution separation by molecular mass [3] | Lower resolution for complex proteomic mixtures [3] | High resolution comparable to SDS-PAGE [3] |
| Typical Applications | Purity assessment, immunoblotting, molecular weight determination [3] | Protein-protein interactions, complex assembly analysis, in-gel activity assays [10] [27] | Metalloprotein analysis, functional studies requiring high resolution [3] |
Table 2: Mitochondrial Complex Analysis Capabilities in BN-PAGE
| Mitochondrial Complex | In-Gel Activity Staining | Supercomplex Formation | Key Applications in Research |
|---|---|---|---|
| Complex I | Well-established activity staining [10] [28] | Forms part of respirasomes (I+III₂+IVₙ) [27] | Assembly studies in mitochondrial disorders [10] [28] |
| Complex II | Reliable activity staining possible [10] [28] | Does not form supercomplexes [27] | Assessment of electron transport chain integrity [28] |
| Complex III | No direct in-gel activity stain available [10] | Forms dimers (III₂) and supercomplexes [27] | Assembly pathway analysis [10] |
| Complex IV | Moderate sensitivity activity staining [10] | Forms supercomplexes (I+III₂+IVₙ) [27] | Detection of Cox7a2l-dependent supercomplexes [27] |
| Complex V | Enhanced activity staining with protocol modifications [10] [28] | Forms dimers in curved cristae membranes [28] | ATP synthesis mechanism studies [10] |
Detailed BN-PAGE Protocol for Mitochondrial Complexes
Sample Preparation from Cultured Cells
Proper sample preparation is critical for preserving labile protein complexes during BN-PAGE:
Cell Harvesting: Grow A549, HEK293T, or fibroblast cells to 80-90% confluence. Dislodge cells by trypsinization, wash with cold PBS, and pellet by centrifugation at 680 × g [10] [28].
Mitochondrial Isolation (Recommended): Resuspend cell pellet in mitochondrial isolation buffer (70 mM sucrose, 230 mM mannitol, 15 mM MOPS pH 7.2, 1 mM EDTA). Homogenize with 40 strokes in a Wheaton glass homogenizer, keeping the probe submerged to avoid foaming. Centrifuge at 600 × g for 10 minutes at 4°C to remove debris. Transfer supernatant and centrifuge at 8,000 × g for 10 minutes to pellet mitochondria [27] [29].
Membrane Protein Solubilization: Resuspend mitochondrial pellet (0.4 mg) in 40 μL Buffer A (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris/HCl, pH 7.0) containing protease inhibitors (1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin). Add 7.5 μL of 10% n-dodecyl-β-D-maltoside, mix gently, and incubate on ice for 30 minutes [5] [29].
Clarification and Dye Addition: Centrifuge at 72,000 × g for 30 minutes at 4°C. Collect supernatant and add 2.5 μL of 5% Coomassie Blue G-250 in 0.5 M aminocaproic acid [5].
Gel Casting and Electrophoresis Conditions
Manual Gel Casting Protocol
While commercial precast gels are available (Thermo Fisher NativePAGE), manual casting provides greater flexibility and cost efficiency [10]:
Gel Solution Preparation: For a 10-gel casting chamber, prepare 38 mL of 6% acrylamide solution (7.6 mL 30% acrylamide/Bis solution, 19 mL 1 M aminocaproic acid pH 7.0, 1.9 mL 1 M Bis-Tris pH 7.0, 9 mL ddH₂O) and 32 mL of 13% acrylamide solution (14 mL 30% acrylamide/Bis, 16 mL 1 M aminocaproic acid pH 7.0, 1.6 mL 1 M Bis-Tris pH 7.0, 0.2 mL ddH₂O) [5].
Polymerization: Add 200 μL of 10% ammonium persulfate and 20 μL TEMED to each solution immediately before pouring. Use a gradient former to create linear 6-13% acrylamide gradients. Overlay with 50% isopropanol to ensure even polymerization [5].
Stacking Gel: After polymerization, prepare stacking gel (0.7 mL 30% acrylamide, 2.5 mL 1 M aminocaproic acid pH 7.0, 0.25 mL 1 M Bis-Tris pH 7.0, 1.6 mL ddH₂O, 40 μL 10% APS, 10 μL TEMED). Insert combs and allow to polymerize for 30 minutes [5].
Electrophoresis Conditions
Buffer Systems: Anode buffer (50 mM Bis-Tris, pH 7.0); Cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G-250, pH 7.0) [5] [29].
Running Conditions: Load 5-20 μL samples per well. Run at constant voltage (150 V) for approximately 2 hours or until the blue dye front approaches the gel bottom. For enhanced resolution, replace the cathode buffer with Coomassie-free version when the dye front reaches one-third of the gel length [5] [29].
Downstream Applications and Modifications
In-Gel Activity Staining
BN-PAGE uniquely enables direct enzymatic activity detection within the gel matrix:
Complex I: Incubate gel in 50 mM potassium phosphate (pH 7.0) containing 0.1 mg/mL NADH and 0.2 mg/mL nitro blue tetrazolium (NBT) at room temperature. Complex I activity produces purple formazan precipitates [29].
Complex IV: Stain with 5 mg 3,3'-diaminobenzidine tetrahydrochloride (DAB), 10 mg cytochrome c, and 225 mg sucrose in 15 mL 50 mM phosphate buffer (pH 7.2) [27].
Complex V: Enhanced sensitivity achieved through modified staining with 15 mM MgSO₄, 10 mM ATP, 0.2% Pb(NO₃)₂, and 175 mM Tris-HCl (pH 9.0) [10] [28].
Two-Dimensional BN/SDS-PAGE
For subunit resolution, excise BN-PAGE lanes and incubate in SDS denaturing buffer (2% SDS, 50 mM DTT, 62.5 mM Tris-HCl pH 6.8, 10% glycerol) for 20 minutes. Place strips onto SDS-PAGE gels (10-20% gradient) for second dimension separation. This approach successfully identified HNE-modified complex I subunits in diabetic kidney mitochondria [29].
Clear Native PAGE (CN-PAGE) Variation
CN-PAGE replaces Coomassie dye with mixed anionic/neutral detergents in the cathode buffer, eliminating dye interference with activity assays and spectral analysis. This variation is particularly valuable for fluorescent detection methods and sensitive enzymatic assays [10] [30].
Essential Research Reagent Solutions
Table 3: Key Reagents for BN-PAGE Experiments
| Reagent | Function | Concentration/Formula | Critical Notes |
|---|---|---|---|
| n-dodecyl-β-D-maltoside | Mild non-ionic detergent for membrane solubilization | 1% in extraction buffer [5] [29] | Preserves protein complexes; superior to SDS for native structure |
| Digitonin | Very mild detergent for supercomplex preservation | 2-8 g/g protein [10] [27] | Maintains respirasome integrity (I+III₂+IVₙ) |
| Coomassie Blue G-250 | Charge-shift dye for protein migration | 0.02% in cathode buffer [5] [27] | Binds hydrophobic surfaces; induces negative charge |
| 6-Aminocaproic Acid | Zwitterionic solubilization aid | 0.75 M in sample buffer [5] [29] | Zero net charge at pH 7.0; prevents aggregation |
| Bis-Tris Buffer | Primary buffering system | 50-75 mM, pH 7.0 [5] [29] | Compatible with native electrophoresis; non-reactive |
| Protease Inhibitor Cocktail | Prevents protein degradation | 1 mM PMSF, 1 μg/mL leupeptin/pepstatin [5] | Essential for preserving labile complexes |
Technical Considerations and Limitations
While BN-PAGE provides unparalleled capability for native complex analysis, researchers should acknowledge its limitations:
Comparative Insensitivity: Complex IV in-gel activity staining shows lower sensitivity compared to spectrophotometric assays [10].
No Direct Complex III Activity Stain: Complex III activity cannot be directly visualized in gels, requiring alternative assessment methods [10].
Interference Issues: Residual Coomassie dye can interfere with some downstream applications, making CN-PAGE the preferred option for these cases [10] [30].
Resolution Trade-offs: Although excellent for complex separation, BN-PAGE resolution for complex proteomic mixtures remains inferior to SDS-PAGE, though the development of NSDS-PAGE offers a promising compromise [3].
BN-PAGE represents a powerful alternative to SDS-PAGE when investigating functional protein properties in mitochondrial complexes and beyond. While SDS-PAGE remains the gold standard for molecular weight determination and purity assessment, BN-PAGE excels in preserving native protein interactions, enzymatic activities, and metal cofactors—critical dimensions in metabolic research and drug development. The choice between these techniques should be guided by research objectives: SDS-PAGE for denatured protein analysis, BN-PAGE for native complex functionality, and emerging hybrid methods like NSDS-PAGE for balancing resolution with functional preservation. As mitochondrial research continues to illuminate the pathogenesis of metabolic diseases, BN-PAGE stands as an indispensable tool for elucidating the structural and functional integrity of oxidative phosphorylation systems.
The journey of protein analysis, particularly in functional proteomics and drug development, is highly dependent on the initial steps of sample preparation. Among these, solubilization and detergent selection are not merely preliminary tasks but are critical determinants that dictate the success of downstream applications. Within the context of comparing SDS-PAGE and Blue Native (BN)-PAGE, these steps define the fundamental trade-off between high-resolution separation and the preservation of functional protein properties. SDS-PAGE, employing strong ionic detergents, provides excellent resolution based on molecular mass but completely denatures proteins, destroying their functional characteristics [16] [31]. In contrast, BN-PAGE utilizes mild non-ionic detergents to maintain proteins in their native, enzymatically active state, enabling the study of intact complexes, albeit sometimes at a cost to resolution [10] [25]. This guide objectively compares these methodologies, providing the experimental data and protocols necessary for researchers to make an informed choice based on their specific research goals, whether for molecular weight determination or for the functional analysis of protein complexes, ligands, and cofactors.
Core Principles: SDS-PAGE vs. BN-PAGE
Fundamental Separation Mechanisms
The core difference between these techniques lies in their treatment of the protein's native structure. SDS-PAGE relies on the powerful anionic detergent Sodium Dodecyl Sulfate (SDS). SDS comprehensively denatures proteins by binding to hydrophobic regions and breaking non-covalent bonds, effectively linearizing them. More importantly, it confers a uniform negative charge density, meaning the charge-to-mass ratio is nearly identical for all proteins. This eliminates separation based on intrinsic charge, making molecular weight the sole determinant of migration through the polyacrylamide gel [16] [31].
BN-PAGE, conversely, is designed to preserve the native state. It uses mild non-ionic detergents (e.g., Dodecyl-β-D-maltoside, Digitonin) for solubilization. These detergents disrupt the lipid membrane but do not disrupt protein-protein interactions or strip away bound cofactors. The negative charge required for electrophoretic migration is provided by the dye Coomassie Blue G-250, which binds hydrophobically to the protein complexes without causing denaturation [10] [25] [5]. Consequently, separation in BN-PAGE depends on a complex interplay of the protein complex's size, overall charge, and shape.
Comparative Technique Profiles
Table 1: Core Characteristics of SDS-PAGE and BN-PAGE.
| Feature | SDS-PAGE | BN-PAGE |
|---|---|---|
| Separation Basis | Molecular weight / mass [16] | Size, overall charge, and shape [16] |
| Protein State | Denatured / Unfolded [16] | Native / Folded [16] |
| Key Detergent | SDS (Ionic, strong) [31] | Dodecyl Maltoside, Digitonin (Non-ionic, mild) [10] [5] |
| Functional Retention | Function destroyed [16] [3] | Function retained (enzymatic activity, bound metals) [16] [3] |
| Primary Applications | Molecular weight determination, purity checks, protein expression analysis [16] | Study of protein complexes, protein-protein interactions, oligomeric state, in-gel activity assays [10] [25] |
Experimental Data and Performance Comparison
Quantitative Functional Retention
The theoretical preservation of function in BN-PAGE is strongly supported by empirical data. A modified approach called NSDS-PAGE (Native SDS-PAGE), which omits heating and reduces SDS concentration, demonstrates the profound impact of gentle solubilization. Research shows that this method can retain up to 98% of bound Zn²⁺ ions in metalloproteins, a stark contrast to the mere 26% retention in standard SDS-PAGE [3]. Furthermore, activity assays for a panel of nine model enzymes revealed that while all were denatured and inactivated by standard SDS-PAGE, seven retained their enzymatic activity following NSDS-PAGE, and all nine remained active after BN-PAGE [3]. This data provides quantitative backing for the superiority of native techniques in functional studies.
Impact on Complex Analysis and Crystallization
The choice of solubilization detergent within BN-PAGE itself can lead to different biological insights. For instance, using dodecyl maltoside for mitochondrial membrane solubilization typically reveals individual respiratory complexes (I-V). However, switching to the even milder detergent digitonin often preserves larger "supercomplexes" (e.g., respirasomes containing complexes I, III, and IV), supporting a "solid-state" model of respiratory chain organization [25]. This has direct implications for structural biology. A strong correlation has been observed between the monodispersity of a membrane protein sample as assessed by BN-PAGE and its propensity to form high-quality crystals. BN-PAGE thus serves as a valuable, economical tool for screening detergent conditions and protein constructs for crystallization trials [32].
Table 2: Comparative Performance in Functional and Structural Studies.
| Performance Metric | SDS-PAGE | BN-PAGE |
|---|---|---|
| Metal Cofactor Retention | Low (e.g., ~26% Zn²⁺) [3] | Very High (e.g., ~98% Zn²⁺) [3] |
| Enzymatic Activity Post-Run | Not retained [16] [3] | Retained (7/9 model enzymes in NSDS-PAGE, 9/9 in BN-PAGE) [3] |
| Reveals Supercomplexes | No | Yes, with specific detergents (e.g., Digitonin) [25] |
| Utility for Crystallization | Low, due to denaturation | High, correlates with monodispersity [32] |
| Resolution | High (separation by mass) | Good to High (dependent on complex stability) [3] |
Detailed Experimental Protocols
Protocol 1: Standard SDS-PAGE Sample Preparation
This protocol is optimized for complete denaturation and high-resolution separation by molecular weight [16] [31].
- Sample Lysate Preparation: Lyse cells or tissues in a buffer containing 2% (w/v) SDS, 62.5 mM Tris-HCl (pH 6.8), and a protease inhibitor cocktail. For solid tissues, mechanical homogenization is required.
- Denaturation and Reduction:
- Mix the protein sample with an equal volume of 2X SDS-PAGE sample buffer (125 mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 0.004% bromophenol blue).
- Add a reducing agent, typically β-mercaptoethanol (BME) or dithiothreitol (DTT), to a final concentration of 5-10%. This step reduces disulfide bonds.
- Heat the mixture at 95-100°C for 5-10 minutes to ensure complete denaturation.
- Clearing: Centrifuge the heated samples at >12,000 x g for 5 minutes to pellet any insoluble debris. The supernatant is now ready for loading onto the gel.
Protocol 2: BN-PAGE for Membrane Protein Complexes
This protocol, based on Schägger's method, is designed for the isolation of native membrane protein complexes and supercomplexes [10] [5].
- Isolation of Mitochondria/Membranes: Begin with a purified membrane fraction (e.g., isolated mitochondria from tissue or cell culture) to reduce sample complexity.
- Solubilization:
- Resuspend the membrane pellet in ice-cold solubilization buffer (e.g., 750 mM 6-aminocaproic acid, 50 mM Bis-Tris, pH 7.0) containing protease inhibitors.
- Add a non-ionic detergent. Common choices include:
- n-Dodecyl-β-D-maltoside (DDM) at 1-2% (w/v) for solubilizing individual complexes.
- Digitonin at a detergent-to-protein ratio of 2-4 g/g for preserving supercomplexes.
- Mix gently and incubate on ice for 30 minutes. Avoid vigorous shaking to prevent foaming and complex disruption.
- Clarification: Centrifuge the solubilized mixture at high speed (e.g., 100,000 x g for 30-45 minutes at 4°C) to remove non-solubilized material and large aggregates.
- Loading Preparation: To the clarified supernatant, add Coomassie Blue G-250 from a 5% (w/v) stock to a final concentration of approximately 0.25-0.5%. This provides the necessary charge for electrophoresis. The sample is now ready for loading onto a native gradient gel.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Solubilization and Electrophoresis.
| Reagent/Material | Function / Purpose | Example Usage & Notes |
|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | Strong ionic detergent; denatures proteins and imparts uniform negative charge [31]. | Core component of SDS-PAGE sample and running buffers. |
| Dodecyl-β-D-Maltoside (DDM) | Mild non-ionic detergent; solubilizes membranes while preserving protein-protein interactions [10] [5]. | Workhorse detergent for BN-PAGE, typically used at 1-2%. |
| Digitonin | Very mild, non-ionic detergent derived from plants; gentler than DDM [25]. | Used in BN-PAGE to preserve labile supercomplexes (e.g., respiratory chain respirasomes). |
| Coomassie Blue G-250 | Anionic dye; binds to protein surfaces, providing charge for electrophoresis without denaturation [10] [5]. | Added to BN-PAGE samples and cathode buffer. Critical for native separation. |
| β-Mercaptoethanol (BME) / DTT | Reducing agents; break disulfide bonds between cysteine residues [16]. | Essential for complete denaturation in SDS-PAGE. Omitted in BN-PAGE. |
| 6-Aminocaproic Acid | Zwitterionic salt; provides ionic strength and helps inhibit proteases during solubilization [10]. | Key component of BN-PAGE solubilization and gel buffers. |
| Bis-Tris | Buffer compound; provides stable pH environment for native electrophoresis (typical pH ~7.0) [5]. | Preferred over Tris-glycine in BN-PAGE for better performance at neutral pH. |
Workflow and Decision Pathway
The following diagram illustrates the critical decision points in the sample preparation workflow and the divergent paths leading to either a denaturing (SDS-PAGE) or native (BN-PAGE) analysis.
The choice between SDS-PAGE and BN-PAGE is fundamentally a choice between maximizing resolution and preserving biological function. SDS-PAGE, with its robust denaturing protocol, remains the unmatched gold standard for determining molecular weight and assessing sample purity. However, this power comes at the cost of destroying the very functional properties that define a protein's biological role. BN-PAGE, through its careful use of mild non-ionic detergents and a charge-providing dye, forgoes a degree of resolution to maintain the native state of proteins, enabling the study of intact complexes, their interactions, and their activities.
The experimental data is clear: techniques that prioritize native solubilization result in dramatically higher retention of metal cofactors and enzymatic function. For researchers in drug development and functional proteomics, where understanding protein interactions and activity is paramount, BN-PAGE and its variants offer an essential toolkit. The critical steps of solubilization and detergent selection are, therefore, not just technical details but foundational decisions that directly shape the validity and scope of scientific discovery.
BN-PAGE for Studying Respiratory Chain Supercomplexes
The study of mitochondrial respiratory chain complexes represents a cornerstone of metabolic and biochemical research, with profound implications for understanding cellular energy production, neurodegenerative diseases, and aging. These complexes—notably Complex I (CI), Complex III (CIII), Complex IV (CIV), and ATP synthase (CV)—exist both as individual entities and as stable assemblies called supercomplexes or respirasomes [27]. The structural and functional organization of these complexes has been a matter of intense scientific debate for more than six decades, primarily between two historical models: the "fluid state" model, where individual OXPHOS complexes diffuse freely in the mitochondrial inner membrane, and the "solid state" model, which proposes that OXPHOS complexes are organized in rigid higher-order assemblies [27]. It is now generally accepted that both organizations coexist in what is termed the "dynamic aggregate" or "plasticity" model [27].
Traditional protein separation techniques like SDS-PAGE denature proteins, destroying the very supercomplex structures researchers aim to study. Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) has emerged as an indispensable tool that overcomes this limitation by preserving native protein interactions while providing high-resolution separation. This technique has become particularly crucial for investigating the stoichiometric combinations of respiratory complexes and their functional implications in health and disease [33]. The following analysis compares BN-PAGE directly with SDS-PAGE and related techniques, providing experimental data and detailed methodologies to guide researchers in selecting the optimal approach for studying respiratory chain supercomplexes.
Technical Comparison: BN-PAGE Versus SDS-PAGE and Related Methods
Fundamental Separation Principles and Applications
Table 1: Core Differences Between BN-PAGE and SDS-PAGE Separation Techniques
| Parameter | BN-PAGE | SDS-PAGE | Native SDS-PAGE (NSDS-PAGE) |
|---|---|---|---|
| Separation Basis | Size, charge, and shape [16] | Molecular weight only [16] | Molecular weight with partial native state preservation [3] |
| Protein State | Native conformation, folded [16] | Denatured, linearized [16] | Intermediate native state preservation [3] |
| Detergent/Dye | Coomassie Blue G-250 [27] [5] | SDS (ionic detergent) [16] | Reduced SDS (0.0375%) + Coomassie [3] |
| Sample Preparation | Not heated [16] | Heated (70-95°C) [16] | Not heated [3] |
| Functional Retention | Proteins retain function [16] | Proteins lose function [16] | Partial function retention (7/9 enzymes active) [3] |
| Metal Cofactor Retention | Preserved | Lost | High retention (98% Zn²⁺) [3] |
| Primary Applications | Studying structure, subunit composition, supercomplexes [16] | Molecular weight determination, protein expression [16] | High-resolution separation with partial native function |
The fundamental distinction between these techniques lies in their treatment of protein structures. SDS-PAGE employs the ionic detergent sodium dodecyl sulfate (SDS) and heating to denature proteins, linearizing them and masking their native charge [16]. While excellent for determining molecular weight and assessing protein purity, this approach destroys higher-order structures like supercomplexes. In contrast, BN-PAGE uses the non-ionic dye Coomassie Blue G-250, which binds to proteins without disrupting their tertiary or quaternary structure, imparting a negative charge that facilitates electrophoretic separation while preserving native interactions [27] [5].
An intermediate approach, Native SDS-PAGE (NSDS-PAGE), reduces SDS concentration (0.0375% versus 0.1% in standard SDS-PAGE) and eliminates heating and EDTA from sample preparation [3]. This modification significantly improves native property retention—achieving 98% zinc retention compared to 26% with standard SDS-PAGE—while maintaining higher resolution than BN-PAGE [3]. Seven of nine model enzymes, including four zinc metalloproteins, retained activity after NSDS-PAGE separation, whereas all were denatured during standard SDS-PAGE [3].
Diagram 1: Separation technique outcomes and applications. BN-PAGE uniquely preserves functional protein complexes essential for respiratory chain studies.
Quantitative Performance Comparison
Table 2: Experimental Performance Metrics Across Electrophoresis Methods
| Performance Metric | BN-PAGE | SDS-PAGE | NSDS-PAGE | Experimental Context |
|---|---|---|---|---|
| Metal Retention (Zn²⁺) | Preserved (not quantified) | 26% | 98% | Pig kidney epithelial cell proteome [3] |
| Enzyme Activity Retention | 9/9 enzymes active | 0/9 enzymes active | 7/9 enzymes active | Model enzymes including Zn-proteins [3] |
| Resolution | Moderate | High | High | Separation of complex protein mixtures [3] |
| Supercomplex Preservation | Yes, including respirasomes [27] | No | No | Mitochondrial respiratory complexes [27] |
| Typical Run Time | 3-4 hours (mini-gel) [34] | ~45 minutes [3] | ~45 minutes [3] | Standard mini-gel systems |
The experimental data demonstrates that BN-PAGE uniquely preserves supercomplex structures and maintains full enzymatic activity, while NSDS-PAGE offers a compelling compromise with high metal retention and mostly preserved function while maintaining the resolution familiar to SDS-PAGE users [3]. The critical advantage of BN-PAGE for respiratory chain research is its ability to maintain the structural integrity of respirasomes—the functional assemblies of CI, CIII₂, and CIV—which is impossible with any denaturing method [27].
BN-PAGE Experimental Protocol for Respiratory Chain Supercomplexes
Mitochondria Isolation and Sample Preparation
The initial and most critical step for successful supercomplex analysis is the isolation of intact mitochondria under conditions that preserve native protein interactions [27] [35].
Protocol for Mitochondria Isolation from Mouse Tissues:
- Tissue Homogenization: Sacrifice the mouse and immediately excise approximately 30 mg of liver tissue. Rinse briefly with ice-cold isolation buffer (IB: 250 mM sucrose, 1 mM EGTA, 5 mM HEPES, pH 7.4 with protease inhibitors). Place tissue in a Wheaton glass homogenizer with 2 mL ice-cold IB [27].
- Homogenization: Homogenize at 1500 rpm with 20 strokes, keeping the homogenizer probe submerged to avoid foaming [27].
- Differential Centrifugation: Transfer homogenate to a 2 mL tube and centrifuge at 600 × g for 10 min at 4°C to remove cell debris. Transfer supernatant to a new tube and centrifuge at 6,800-10,000 × g for 10 min to pellet mitochondria [27] [35].
- Mitochondrial Lysis: Resuspend mitochondrial pellet in mitochondrial buffer (0.75 M aminocaproic acid, 50 mM Bis-Tris, pH 7.0) with protease inhibitors. Add lauryl maltoside to 1% final concentration (or digitonin for alternative protocols) and incubate on ice for 30 min [5] [35].
- Clarification: Centrifuge at 72,000 × g for 30 min (or 16,000 × g for small volumes). Collect supernatant containing solubilized mitochondrial complexes [5].
- Sample Preparation: Add Coomassie blue G-250 to the supernatant (final concentration 0.5-1%) [5].
Critical Considerations:
- Protease inhibitors are essential throughout the process to prevent protein degradation [5] [35].
- Mild detergents like lauryl maltoside or digitonin effectively solubilize membranes while preserving protein interactions [35].
- Rapid processing at 4°C maintains complex integrity [27].
BN-PAGE Electrophoresis and Detection
Gel Preparation and Electrophoresis:
- Gradient Gel Casting: Prepare a linear acrylamide gradient gel (typically 4-16% or 6-13%) using a gradient mixer. The 4% solution contains 4% acrylamide, 0.75 M aminocaproic acid, 50 mM Bis-Tris (pH 7.0), while the 15% solution contains 15% acrylamide with the same buffer components [34] [5]. Add APS and TEMED immediately before pouring.
- Stacking Gel: After polymerization of the separating gel, pour a 3.2-4% stacking gel containing the same buffer system [34].
- Sample Loading: Load 5-20 μL of prepared sample into wells. For reference, include native molecular weight markers [5].
- Electrophoresis Conditions: Run at 150 V for approximately 2 hours using cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G-250, pH 7.0) and anode buffer (50 mM Bis-Tris, pH 7.0) until the blue dye front approaches the gel bottom [5]. Maintain temperature at 4°C throughout.
Detection Methods:
- In-Gel Activity Staining: Following electrophoresis, specific complexes can be visualized by activity assays. For example, Complex IV activity can be detected using cytochrome c and diaminobenzidine [27].
- Immunoblotting: Transfer proteins to PVDF membrane (150 mA for 1.5 hours) and probe with antibodies against specific complex subunits (e.g., NDUFS3 for CI, UQCRC2 for CIII, COX4 for CIV) [5] [35].
- Two-Dimensional Analysis: For subunit resolution, excise BN-PAGE lanes, incubate in SDS buffer, and run perpendicularly on SDS-PAGE [34].
Diagram 2: BN-PAGE workflow for supercomplex analysis. The process maintains native protein interactions from sample preparation through detection.
Key Research Applications and Findings
Genetic Regulation of Supercomplex Assembly
BN-PAGE has been instrumental in identifying genetic factors controlling supercomplex formation. Research has revealed that Cox7a2l (SCAFI) plays a primary role in supercomplex assembly, particularly in forming structures containing multiple copies of CIV [27]. Mouse strains with functional Cox7a2l (e.g., DBA, CBA, 129) form five distinct supercomplexes, including I+III₂+IV₂ (SC 3) and I+III₂+IV₃ (SC 4), while Cox7a2l-negative strains (e.g., C57BL/6, BALB/c) form only three of the five supercomplexes [27]. This assembly mechanism exhibits tissue specificity—certain supercomplexes can form in heart tissue even without Cox7a2l, though to a much lesser extent [27].
The organization of complexes into supercomplexes may offer structural and functional advantages, including prevention of destabilization and degradation, enhanced electron transport efficiency, substrate channeling, and reduced electron or proton leakages [27]. This structural interdependence has major biomedical implications, as assembly defects in one complex often induce pleiotropic effects on others in mitochondrial diseases [27].
Investigating the Essentiality of Respirasomes
A groundbreaking 2023 study employed BN-PAGE to challenge conventional wisdom about respirasome essentiality. Researchers generated knockin mice with profoundly decreased respirasome levels (homozygous Uqcrc1delEED) but normal individual complex levels [36]. Surprisingly, these mutant mice displayed:
- Preserved respiratory chain capacity
- Normal exercise performance
- Apparently normal physiology despite dramatically reduced CI-CIII₂-CIV supercomplexes [36]
This finding contradicts the long-standing assumption that respirasomes are indispensable for efficient oxidative phosphorylation, suggesting instead that high respirasome levels may serve alternate functions such as regulating protein stability and preventing age-associated aggregation [36].
Disease Modeling and Diagnostic Applications
BN-PAGE enables the investigation of mitochondrial dysfunction mechanisms in human diseases. The technique has revealed that structural alterations primarily affecting one complex often induce pleiotropic effects on others [27]. For instance:
- Pathogenic mutations in CIII subunits lead to combined deficiencies of CI and CIV [27]
- Mutations in CIV subunits may cause secondary CI deficiencies [27]
- Mutations affecting CI-specific genes can produce combined CI and CIII, or CI and CIV deficiencies [27]
These cascade effects likely result from impacts on supercomplex formation, which normally stabilizes individual complexes [27]. BN-PAGE thus provides critical insights for understanding disease pathogenesis beyond what SDS-PAGE can offer.
Essential Research Reagents and Materials
Table 3: Key Reagents for BN-PAGE Analysis of Respiratory Supercomplexes
| Reagent/Category | Specific Examples | Function and Importance | Technical Notes |
|---|---|---|---|
| Detergents | Lauryl maltoside, Digitonin [35] | Membrane solubilization while preserving native interactions | Concentration critical (typically 1%); digitonin for plasma membrane permeabilization [35] |
| Protease Inhibitors | PMSF, Leupeptin, Pepstatin [5] | Prevent protein degradation during isolation | Essential for preserving complex integrity; use cocktails for broad protection [5] |
| Dyes | Coomassie Blue G-250 (not R-250) [35] | Imparts charge shift for electrophoretic mobility | Binds hydrophobic regions without denaturing complexes [27] |
| Buffers | Aminocaproic acid, Bis-Tris, Tricine [5] [35] | Maintain pH and native conditions | Aminocaproic acid improves complex stability and resolution [35] |
| Antibodies | Subunit-specific: NDUFS3 (CI), UQCRC2 (CIII), COX4 (CIV) [35] | Detection of specific complexes after transfer | Must recognize native epitopes; sequential incubation possible [35] |
| Electrophoresis | Gradient gels (4-16%), PVDF membranes [5] | Separation and transfer of complexes | PVDF preferred over nitrocellulose for better protein retention [5] |
BN-PAGE represents a uniquely powerful technique for investigating mitochondrial respiratory chain supercomplexes, offering capabilities unmatched by denaturing methods like SDS-PAGE. The preservation of native protein interactions enables researchers to study supercomplex assembly, stability, and functional relationships under near-physiological conditions. Recent findings demonstrating that respirasomes may be dispensable for basic oxidative phosphorylation highlight the ongoing importance of this technique for challenging and refining fundamental biological concepts [36].
For researchers studying mitochondrial function, disease mechanisms, or protein complex dynamics, BN-PAGE provides critical insights that would be impossible to obtain through denaturing methods. While requiring more specialized protocols than SDS-PAGE, its ability to maintain functional protein interactions makes it indispensable for modern mitochondrial research. As techniques continue to evolve, including modifications like NSDS-PAGE that bridge the gap between denaturing and native approaches, the toolbox for studying respiratory supercomplexes will continue to expand, offering ever more sophisticated approaches to understanding cellular energy production.
Two-Dimensional BN/SDS-PAGE for Comprehensive Complex Analysis
In the study of proteomes, a fundamental challenge lies in the choice between achieving high-resolution separation of individual proteins and preserving their native, functional states. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) has long been the workhorse for analytical protein separation, providing excellent resolution by denaturing proteins and separating them primarily by molecular mass [3] [1]. However, this denaturation comes at a significant cost: the destruction of functional properties, including enzymatic activity, protein-protein interactions, and non-covalently bound cofactors such as metal ions [3] [24]. In contrast, Blue Native PAGE (BN-PAGE) preserves protein complexes in their native state, maintaining these functional properties, but at the expense of protein resolving power [3] [25].
Two-dimensional BN/SDS-PAGE emerges as a powerful hybrid technique that leverages the strengths of both methods. This comprehensive approach combines the native complex separation of BN-PAGE in the first dimension with the high-resolution subunit separation of SDS-PAGE in the second dimension, enabling researchers to analyze multi-protein complexes with unprecedented detail [29] [37]. The technique has become indispensable for investigating respiratory chains [25], mitochondrial complexes [29], and protein interaction networks in various biological systems.
Fundamental Principles: How 2D BN/SDS-PAGE Works
First Dimension: Blue Native PAGE
BN-PAGE operates on the principle of separating intact protein complexes under native conditions. Unlike SDS-PAGE, which uses ionic detergents to denature proteins and impart uniform charge, BN-PAGE employs mild non-ionic detergents for solubilization and the dye Coomassie Blue G250 to provide negative charges [25]. The Coomassie dye binds to protein complexes without disrupting their structure, enabling separation based on both size and charge while maintaining enzymatic activity and protein-protein interactions [5] [25].
The critical aspect of BN-PAGE is the careful selection of detergents for solubilizing membrane protein complexes. Suitable detergents must be mild enough to disrupt lipid-lipid interactions without disturbing protein-protein interactions. Commonly used detergents include n-dodecylmaltoside, Triton X-100, and digitonin, with the specific choice significantly impacting the results and biological interpretations [25]. For instance, different detergents can reveal either individual respiratory complexes or larger supercomplexes, supporting different models of electron transport chain organization [25].
Second Dimension: SDS-PAGE
Following BN-PAGE separation, the second dimension employs denaturing SDS-PAGE to dissociate the complexes into their constituent polypeptides. Gel strips from the first dimension are equilibrated in SDS-containing buffer, then applied to an SDS-polyacrylamide gel for separation based on molecular mass [29]. This orthogonal separation approach allows researchers to correlate intact complexes with their subunit composition, providing a comprehensive view of the protein complexome.
Visualizing the Process
The following diagram illustrates the complete workflow of the 2D BN/SDS-PAGE technique:
Quantitative Comparison of Electrophoretic Methods
The choice between electrophoresis methods involves significant trade-offs between resolution, native property retention, and analytical applications. The table below provides a comprehensive comparison of key performance metrics across different PAGE techniques:
Table 1: Quantitative Comparison of Electrophoresis Methods for Protein Analysis
| Parameter | SDS-PAGE | BN-PAGE | NSDS-PAGE | 2D BN/SDS-PAGE |
|---|---|---|---|---|
| Protein Resolution | High resolution based on molecular mass [3] | Lower resolution of complexes [3] | High resolution similar to SDS-PAGE [3] | Highest resolution (complexes + subunits) [29] |
| Native Property Retention | Denatures proteins; destroys functional properties [3] | Preserves native structure and function [3] [25] | Retains some native properties [3] | Combines native and denaturing separation [29] |
| Metal Ion Retention | 26% Zn²⁺ retention [3] [24] | High metal retention [3] | 98% Zn²⁺ retention [3] [24] | Context-dependent (1D: high, 2D: low) [29] |
| Enzyme Activity Retention | 0/9 model enzymes active [3] | 9/9 model enzymes active [3] | 7/9 model enzymes active [3] | 1D gel: activity possible; 2D gel: lost [29] |
| Complex Analysis | Not possible (denaturing) [3] | Excellent for intact complexes [5] [25] | Limited complex analysis [3] | Excellent for complex composition [29] [37] |
| Typical Applications | Molecular weight determination, purity assessment [1] | Studying protein-protein interactions, supercomplexes [25] | Metalloprotein analysis, some functional studies [3] | Comprehensive complexome analysis, PTM studies [29] |
Experimental Protocols: Implementing 2D BN/SDS-PAGE
Sample Preparation for BN-PAGE
Proper sample preparation is critical for successful native electrophoresis. For mitochondrial complexes, the following protocol has been established:
- Isolation and Solubilization: Resuspend 0.4 mg of sedimented mitochondria in 40 μL of buffer containing 0.75 M aminocaproic acid and 50 mM Bis-Tris (pH 7.0). Add 7.5 μL of 10% n-dodecyl-β-D-maltopyranoside and mix thoroughly [5].
- Extraction: Incubate on ice for 30 minutes, then centrifuge at 72,000 × g for 30 minutes. Collect the supernatant containing solubilized complexes [5].
- Staining: Add 2.5 μL of 5% Coomassie Blue G250 solution in 0.5 M aminocaproic acid to the supernatant [5].
- Protease Inhibition: Include protease inhibitors (e.g., 1 mM PMSF, 1 μg/mL leupeptin, and 1 μg/mL pepstatin) to prevent protein degradation [5].
For whole cellular lysates, dialysis may be necessary to make samples compatible with BN-PAGE conditions [37].
First Dimension: BN-PAGE Electrophoresis
The first dimension separation employs native gradient gels:
- Gel Preparation: Prepare a linear acrylamide gradient gel (typically 4-16% or 6-13%) using a gradient former. The gel buffer should contain 500 mM aminocaproic acid and 50 mM Bis-Tris (pH 7.0) [5] [29].
- Electrophoresis Conditions: Load 5-20 μL samples and run at 100-150 V initially. For enhanced resolution, start with cathode buffer containing 0.02% Coomassie Blue, then switch to cathode buffer without dye when the front reaches one-third of the gel [29].
- Complex Visualization: After electrophoresis, complexes can be visualized by Coomassie staining, activity staining, or prepared for second dimension separation [29].
Second Dimension: SDS-PAGE Separation
The transition to the denaturing second dimension requires careful equilibration:
- Gel Strip Equilibration: Excise BN-PAGE lanes and equilibrate in buffer containing 5% 2-mercaptoethanol, 62.5 mM Tris-HCl (pH 6.8), 2% SDS, and 10% glycerol for 20 minutes [29].
- Gel Assembly: Place the equilibrated strip onto an SDS-polyacrylamide gel (typically 10-20% acrylamide). A wider gel may be necessary to accommodate the first dimension strip [5].
- Electrophoresis: Perform SDS-PAGE according to standard protocols, typically at 100-150 V until the dye front reaches the bottom [29].
Detection and Analysis
Following two-dimensional separation, multiple detection approaches can be employed:
- Protein Staining: Use Coomassie Brilliant Blue for abundant complexes or silver staining for enhanced sensitivity [29].
- Western Blotting: Transfer proteins to PVDF membranes for immunodetection with specific antibodies [5] [29].
- Activity Staining: For first-dimension gels, specific enzyme activities can be detected using appropriate substrates [29].
- Mass Spectrometry: Excise protein spots of interest for identification by mass spectrometric peptide sequencing [29].
Research Reagent Solutions for 2D BN/SDS-PAGE
Successful implementation of 2D BN/SDS-PAGE requires specific reagents optimized for native protein separation. The following table details essential components and their functions:
Table 2: Essential Research Reagents for 2D BN/SDS-PAGE
| Reagent Category | Specific Examples | Function and Importance |
|---|---|---|
| Detergents | n-Dodecyl-β-D-maltoside, Triton X-100, Digitonin [5] [25] | Solubilize membrane protein complexes while maintaining native interactions; choice affects complex stability and supercomplex formation [25] |
| Dyes | Coomassie Blue G250, Serva Blue G [5] [29] | Impart negative charge to proteins for electrophoretic mobility in native state; critical for BN-PAGE separation principle [25] |
| Protease Inhibitors | PMSF, Leupeptin, Pepstatin [5] | Prevent protein degradation during sample preparation and electrophoresis; essential for preserving labile complexes [5] |
| Buffers | Aminocaproic acid, Bis-Tris, Tricine [5] [29] | Maintain optimal pH and ionic conditions; aminocaproic acid improves solubilization of membrane complexes [5] |
| Electrophoresis Reagents | Acrylamide/Bis solution, APS, TEMED [5] | Form the polyacrylamide gel matrix; gradient gels enhance separation range for complexes of different sizes [5] |
| Detection Reagents | Coomassie R-250, Silver Nitrate, Nitro Blue Tetrazolium (NBT) [29] | Visualize proteins and enzyme activities; NBT used for complex I activity staining [29] |
Applications and Case Studies in Protein Complex Analysis
Identification of HNE-Modified Mitochondrial Complex I Subunits
The 2D BN/SDS-PAGE technique has proven invaluable for studying post-translational modifications in specific protein complexes. Wu et al. applied this method to identify subunits of mitochondrial complex I modified by 4-hydroxynonenal (HNE), a lipid peroxidation product [29]. Complex I was first separated by BN-PAGE, visualized, then further resolved by SDS-PAGE. HNE-modified proteins were detected by Western blotting with anti-HNE antibodies, and specific modified bands were excised for identification by mass spectrometry. This approach successfully identified two complex I subunits with enhanced HNE modifications in diabetic kidney mitochondria, providing insights into diabetes-related mitochondrial dysfunction [29].
Analysis of Multi-Protein Complexes from Whole Cellular Lysates
Camacho-Carvajal et al. demonstrated that 2D BN/SDS-PAGE could be applied to whole cellular lysates, significantly expanding its utility beyond purified organellar fractions [37]. Through dialysis to make samples compatible with BN-PAGE conditions, they visualized different multi-protein complexes including forms of the eukaryotic proteasome. They studied complex dynamics after gamma interferon stimulation and developed an antibody shift assay to detect protein-protein interactions directly in BN-PAGE. The method successfully identified defined protein complexes of various proteins including the tumor suppressor p53 and c-Myc, highlighting its potential for functional proteomics and interaction network studies [37].
Plant Proteomics and Supercomplex Analysis
In plant sciences, 2D BN/SDS-PAGE has revealed remarkable insights into the organization of respiratory and photosynthetic complexes. The technique has been particularly valuable in demonstrating the existence of supercomplexes—stable associations between different respiratory complexes—especially when using digitonin for solubilization [25]. This finding has led to a paradigm shift from the "liquid state model" of individually diffusing complexes to a "solid state model" of defined supercomplex arrangements, with significant implications for our understanding of metabolic efficiency and regulation in plants [25].
Technical Considerations and Optimization Strategies
Detergent Selection and Optimization
The choice of detergent profoundly impacts the results of BN-PAGE and subsequent two-dimensional analysis. Researchers must empirically determine the optimal detergent and detergent-to-protein ratio for their specific protein complexes of interest [25]. n-Dodecylmaltoside typically solubilizes individual complexes, while digitonin often preserves supercomplex associations. Comparative testing of different detergents is essential, as the optimal choice depends on the membrane lipid composition and the specific protein complexes being studied [25].
Gel System Variations
Different gel configurations can be employed based on research needs:
- Non-gradient vs. Gradient Gels: While non-gradient BN-PAGE (e.g., 4-5% acrylamide) works well for specific applications, gradient gels (typically 4-16% or 6-13%) generally provide superior separation across a broader molecular mass range [5] [29].
- Acrylamide Concentration: The appropriate acrylamide concentration depends on the size of the complexes being separated. Lower percentages (3-5%) better separate larger complexes, while higher percentages (13-16%) improve resolution of smaller complexes [25].
Troubleshooting Common Issues
Several challenges may arise during 2D BN/SDS-PAGE experiments:
- Smearing or Distorted Bands: Often results from inappropriate salt concentrations or incomplete solubilization. Buffer exchange through dialysis or dilution may be necessary [25] [37].
- Poor Complex Stability: Some soluble complexes may be sensitive to Coomassie Blue exposure. In such cases, adding minimal dye to the sample and relying mainly on the cathode buffer dye may improve results [25].
- Vertical Streaking in 2D Gels: Can indicate incomplete equilibration or aggregation. Optimizing equilibration time and ensuring proper reduction of disulfide bonds can mitigate this issue [29].
Two-dimensional BN/SDS-PAGE represents a powerful integration of complementary separation techniques, offering researchers unprecedented capability to analyze protein complexes in comprehensive detail. By preserving native interactions in the first dimension while achieving high-resolution subunit separation in the second, this method bridges the critical gap between functional proteomics and structural analysis.
The continued refinement of this technique—including optimized detergent systems, improved sensitivity, and enhanced compatibility with downstream mass spectrometric analysis—ensures its enduring relevance in an era increasingly dominated by gel-free proteomic methods. For researchers investigating protein complexes, particularly those involved in metabolic pathways, signal transduction, and cellular stress responses, 2D BN/SDS-PAGE remains an indispensable tool that provides unique insights unobtainable through either native or denaturing approaches alone.
As proteomics continues to evolve toward more integrative and functional analyses, the dual perspective offered by 2D BN/SDS-PAGE—simultaneously capturing the holistic picture of intact complexes and the detailed composition of their subunits—positions it as a cornerstone methodology for deciphering the complex protein networks that underlie cellular function and dysfunction.
For researchers investigating complex biological mixtures like venoms or characterizing multisubunit enzymes, selecting the appropriate electrophoretic technique is a critical first step that dictates the functional information obtainable from an experiment. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Blue Native-PAGE (BN-PAGE) represent two fundamentally different approaches to protein separation. SDS-PAGE, the workhorse of molecular weight-based separation, denatures proteins into their constituent polypeptides, providing high resolution of individual protein chains but destroying higher-order structures and often abolishing biological activity [3]. In contrast, BN-PAGE separates protein complexes under native conditions, preserving their quaternary structures, enzymatic activities, and non-covalently bound cofactors [38] [30]. This guide provides a detailed objective comparison of these techniques, focusing on their applications in venom proteomics and multisubunit enzyme characterization, to help researchers select the optimal method for their functional protein studies.
Technical Principles and Separation Mechanisms
The fundamental difference between these techniques lies in their sample preparation and separation mechanisms, which directly determine the type of information they yield.
SDS-PAGE relies on the strong anionic detergent SDS, which denatures proteins by disrupting non-covalent interactions and confers a uniform negative charge proportional to molecular mass. This process dismantles protein complexes into individual subunits and generally destroys enzymatic activity, as it eliminates the three-dimensional structure required for function [3] [2]. The resulting separation is based almost exclusively on polypeptide chain molecular mass.
BN-PAGE utilizes the anionic dye Coomassie Brilliant Blue G-250, which binds to the surface of native proteins without causing significant denaturation. The dye provides a charge shift that facilitates electrophoretic migration toward the anode while maintaining protein-protein interactions, quaternary structures, and biological activity. Separation occurs primarily by protein complex size and shape rather than subunit molecular weight [38] [3]. This preservation of native state enables subsequent functional analyses, including in-gel activity assays.
Table 1: Fundamental Separation Principles of SDS-PAGE and BN-PAGE
| Parameter | SDS-PAGE | BN-PAGE |
|---|---|---|
| Sample Condition | Denaturing (SDS + heat) | Native (Coomassie dye) |
| Charge Source | SDS micelles | Coomassie Brilliant Blue G-250 |
| Separation Basis | Polypeptide molecular mass | Native complex size/shape |
| Complex Preservation | No (disassembles complexes) | Yes (maintains quaternary structure) |
| Enzymatic Activity | Typically destroyed | Often retained |
| Typical Applications | Molecular weight determination, purity assessment | Protein-protein interactions, oligomeric state analysis, in-gel activity assays |
Performance Comparison in Venom Proteomics Applications
Snake venoms represent exceptionally complex mixtures of proteins and peptides, many of which form functionally important complexes. Research applications in venom characterization benefit significantly from techniques that preserve these native structures.
Revealing Protein Complexes in Venoms
BN-PAGE has demonstrated unique capabilities for visualizing protein complexes in snake venoms that remain undetected by conventional SDS-PAGE. A groundbreaking study analyzing Bothrops atrox, B. erythromelas, and B. jararaca venoms revealed numerous native protein complexes that maintained their collagenolytic and esterase activities after separation, enabling direct correlation of specific complexes with biological function [38]. Similarly, research on Cape Cobra (Naja nivea) venom provided the first evidence of covalent quaternary structures in this species, including complexes that likely enhance venom potency through synergistic interactions between components [39].
Comparative analysis using two-dimensional BN/SDS-PAGE (first dimension BN-PAGE followed by second dimension denaturing SDS-PAGE) has proven particularly powerful. This approach separates intact complexes in the first dimension, then dissociates them into subunits in the second dimension, creating a map that links native complexes to their constituent proteins [6] [38]. The technique has been successfully applied to study complex dynamics in response to cellular stimuli and to detect protein-protein interactions through antibody shift assays in BN-PAGE [6].
Functional Retention for Enzymatic Characterization
The preservation of enzymatic activity following BN-PAGE separation enables direct functional characterization through zymography techniques. Venom metalloproteinases and serine proteinases from Bothrops species maintained their specific proteolytic activities after BN-PAGE separation, allowing researchers to directly link specific protein bands to biological function [38]. This functional retention is particularly valuable for identifying the active forms of enzymes that require specific quaternary structures for activity.
Table 2: Application-Based Technique Selection for Venom Proteomics
| Research Objective | Recommended Technique | Key Advantages | Experimental Evidence |
|---|---|---|---|
| Mapping protein complexes | BN-PAGE or BN/SDS-PAGE | Preserves non-covalent interactions; reveals quaternary structure | Identification of SVMP and SVSP complexes in Bothrops venoms [38] |
| Detecting covalent complexes | Non-reducing SDS-PAGE + reducing SDS-PAGE | Identifies disulfide-linked complexes by comparing migration patterns | First evidence of covalent complexes in Naja nivea venom [39] |
| Molecular weight determination | SDS-PAGE | High resolution separation by polypeptide size | Standard proteomic characterization of venom components [40] |
| In-gel activity assessment | BN-PAGE zymography | Retains enzymatic function after separation | Collagenolytic and amidolytic activities in native venom complexes [38] |
Performance Comparison in Multisubunit Enzyme Characterization
Many biologically important enzymes function as multisubunit complexes, and understanding their structure-function relationships requires techniques that preserve these native assemblies.
Analyzing Oligomeric State and Activity Relationships
BN-PAGE provides unique insights into the relationship between oligomeric state and enzymatic function. A sophisticated application for medium-chain acyl-CoA dehydrogenase (MCAD) demonstrated how high-resolution clear native PAGE (hrCN-PAGE), a variant of BN-PAGE, could separate active tetramers from inactive lower molecular weight forms generated by pathogenic variants [30]. This separation enabled researchers to directly correlate specific oligomeric states with enzymatic activity, revealing how disease-associated mutations disrupt quaternary structure and impair function.
The in-gel activity assay for MCAD coupled enzyme separation with activity detection using a colorimetric system. After electrophoretic separation, gels were incubated with the physiological substrate octanoyl-CoA and nitro blue tetrazolium chloride, which forms an insoluble purple precipitate upon reduction [30]. This approach showed linear correlation between protein amount, FAD content, and enzymatic activity, demonstrating the quantitative potential of native electrophoresis for functional enzymology.
Metal Cofactor Retention and Enzymatic Activity Preservation
The preservation of non-covalently bound metal cofactors represents a significant advantage of native electrophoresis techniques. Comparative studies have demonstrated that BN-PAGE and related native methods preserve zinc ions in metalloproteins, with one study showing 98% metal retention compared to substantial metal loss during standard SDS-PAGE [3]. This metal preservation is crucial for maintaining the activity of metalloenzymes, including many venom metalloproteinases.
When seven model enzymes, including four zinc metalloproteins, were subjected to NSDS-PAGE (a modified native approach), most retained enzymatic activity, whereas all were inactivated by standard denaturing SDS-PAGE [3]. This functional preservation enables researchers to directly link specific protein bands to enzymatic activity without the need for complex renaturation procedures.
Experimental Protocols for Core Applications
BN/SDS-PAGE for Venom Protein Complex Analysis
Protocol adapted from Bothrops venom characterization [38]:
Sample Preparation: Dissolve 200 µg of lyophilized venom in native sample buffer (50 mM BisTris, 50 mM NaCl, 10% glycerol, 0.001% Ponceau S, pH 7.2). Do not heat the sample.
First Dimension (BN-PAGE):
- Prepare a 4-16% gradient polyacrylamide gel.
- Load samples and electrophorese at 150V for 90-95 minutes using cathode buffer (50 mM BisTris, 50 mM Tricine, 0.02% Coomassie G-250, pH 6.8) and anode buffer (50 mM BisTris, 50 mM Tricine, pH 6.8).
- Continue electrophoresis until the dye front reaches the gel bottom.
Lane Excision and Equilibration:
- Carefully excise individual lanes from the BN-PAGE gel.
- Incubate lanes in SDS sample buffer (2% SDS, 66 mM Tris-HCl, pH 7.0) for 30 minutes with gentle agitation.
Second Dimension (SDS-PAGE):
- Place the equilibrated BN-PAGE lane horizontally atop a denaturing SDS-polyacrylamide gel.
- Seal with agarose solution.
- Perform standard SDS-PAGE separation at 200V for approximately 45 minutes.
Analysis:
- Proteins can be visualized with Coomassie staining, transferred for Western blotting, or used for in-gel activity assays.
In-Gel Activity Assay for Multisubunit Enzymes
Protocol adapted from MCAD characterization [30]:
Native Electrophoresis:
- Separate 1-10 µg of enzyme sample using high-resolution clear native PAGE (4-16% gradient gel).
- Use mild conditions without Coomassie in the cathode buffer to preserve enzymatic activity.
Activity Staining:
- Incubate the gel in reaction buffer containing 100 mM Tris-HCl (pH 8.0), 0.5-1.0 mM octanoyl-CoA (substrate), and 0.2-0.5 mg/mL nitro blue tetrazolium.
- Include 0.1 mM phenazine methosulfate as electron coupler for some applications.
- Incubate at 37°C in the dark with gentle agitation.
Reaction Monitoring:
- Monitor formation of purple diformazan precipitate indicating enzymatic activity.
- Typically appears within 10-15 minutes for active enzyme complexes.
- Stop reaction by rinsing with distilled water.
Quantification:
- Capture gel images and perform densitometric analysis using appropriate software.
- Compare band intensities to standards for semi-quantitative assessment.
Experimental Workflows and Decision Pathways
The following diagram illustrates the key decision pathways for selecting and implementing the most appropriate electrophoretic method based on research objectives:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Electrophoretic Techniques
| Reagent/Equipment | Function/Purpose | Technique | Key Considerations |
|---|---|---|---|
| Coomassie G-250 | Binds protein surfaces, provides charge shift for migration | BN-PAGE | Critical for native separation; different from G-250 used in staining [38] [3] |
| SDS (Sodium Dodecyl Sulfate) | Denatures proteins, confers uniform charge | SDS-PAGE | Disassembles complexes; destroys most enzymatic activity [3] [2] |
| Bis-Tris Gels | Matrix for protein separation | Both | Preferred for BN-PAGE due to neutral pH; minimizes protein modification [3] |
| NativeMark Standards | Molecular weight standards for native conditions | BN-PAGE | Essential for estimating native complex sizes [3] |
| Nitro Blue Tetrazolium | Electron acceptor in activity assays | In-gel activity | Forms insoluble purple formazan upon reduction [30] |
| Protease Inhibitors | Prevent protein degradation during separation | Both | Particularly important for native techniques preserving enzymatic activity |
| Substrate Cocktails | Enzyme-specific activity detection | Zymography | Must be compatible with gel matrix and detection method [38] [30] |
The choice between SDS-PAGE and BN-PAGE represents a fundamental strategic decision in experimental design for venom proteomics and enzyme characterization. SDS-PAGE remains the gold standard for determining polypeptide molecular weights and assessing sample purity with high resolution [2]. However, BN-PAGE and related native techniques provide unique capabilities for studying functionally relevant protein complexes, preserving enzymatic activity, and maintaining metal cofactors [38] [3] [30].
For comprehensive analyses, two-dimensional approaches combining both techniques offer the most powerful solution, enabling researchers to map native complexes to their constituent subunits while retaining functional information [6] [38]. As venom research increasingly focuses on understanding synergistic interactions between components and characterizing complex biological activities, native electrophoretic techniques will continue to provide critical insights not obtainable through denaturing methods alone.
In-Gel Activity Staining for Functional Validation After BN-PAGE
In the study of proteins, the choice of electrophoretic technique dictates the biological insights you can uncover. While SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) has been a workhorse for separating proteins by molecular weight, its denaturing nature destroys higher-order structure and function [16] [17]. BN-PAGE (Blue-Native PAGE) emerged to fill this critical gap, enabling the separation of native protein complexes and, crucially, their subsequent functional validation through in-gel activity staining [41] [42]. This guide objectively compares these techniques, providing the experimental data and protocols necessary to select the optimal method for research focused on retaining functional protein properties.
The fundamental difference lies in the state of the separated proteins. SDS-PAGE employs a denaturing anionic detergent, linearizing proteins and masking their native charge. In contrast, BN-PAGE uses mild non-ionic detergents for solubilization and the dye Coomassie Blue G-250 to impart charge, preserving protein complexes in their native, active conformations [42] [25]. This preservation is the cornerstone of functional validation directly within the gel matrix.
Technique Comparison: Separation Philosophy and Capabilities
The choice between SDS-PAGE, BN-PAGE, and its variants dictates the scope of your functional analysis. The table below summarizes their core operational differences.
Table 1: Fundamental Differences Between SDS-PAGE, BN-PAGE, and CN-PAGE
| Criteria | SDS-PAGE | BN-PAGE | CN-PAGE (Clear-Native PAGE) |
|---|---|---|---|
| Separation Principle | Molecular weight only [16] [17] | Size, charge, and native shape [16] | Size, charge, and native shape [28] |
| Gel Condition | Denaturing [16] [17] | Non-denaturing [16] | Non-denaturing [28] |
| Key Reagents | SDS, reducing agents (e.g., DTT/BME) [16] | Mild detergents (e.g., DDM), Coomassie G-250 [42] | Mild detergents, mixed micelles (replaces Coomassie) [28] |
| Sample Preparation | Heating required [16] | No heating [16] | No heating [28] |
| Protein State | Denatured, linearized [16] [17] | Native, folded, active complexes [41] [42] | Native, folded, active complexes [28] |
| Functional Recovery | Not possible; function is destroyed [16] [17] | Proteins retain function; can be recovered [16] [43] | Proteins retain function; can be recovered [28] |
| Primary Applications | Molecular weight determination, purity checks [16] | Study of protein complexes, supercomplexes, in-gel activity [41] | In-gel activity staining without dye interference [28] [30] |
For researchers targeting functional properties, the data is clear: BN-PAGE and CN-PAGE are the only viable options. The ability of BN-PAGE to resolve intact complexes is exemplified by its pivotal role in discovering mitochondrial respiratory supercomplexes (respirasomes). The detergent used during solubilization is a critical parameter; digitonin, milder than dodecylmaltoside, preserves these fragile supercomplexes for analysis [28] [42].
Table 2: Functional Retention: Comparative Experimental Data
| Experimental Metric | SDS-PAGE | BN-PAGE | CN-PAGE / NSDS-PAGE |
|---|---|---|---|
| Enzyme Activity Retention | 0 out of 9 model enzymes active [3] | 9 out of 9 model enzymes active [3] | 7 out of 9 model enzymes active [3] |
| Metal Cofactor Retention | Low (e.g., 26% for Zn²⁺) [3] | High [3] | High (e.g., 98% for Zn²⁺) [3] |
| Complex V In-Gel Staining | Not applicable | Sensitive, with enhanced protocols [41] | Highly sensitive, no dye interference [28] |
| Respiratory Supercomplex Analysis | Not possible | Excellent with digitonin solubilization [28] [42] | Excellent with digitonin solubilization [28] |
Experimental Protocols for In-Gel Activity Staining
The following protocols, adapted from validated methodologies, provide a roadmap for functional validation after BN-PAGE.
Core BN-PAGE Methodology
Sample Preparation (Mitochondrial Complexes)
- Solubilization: Solubilize mitochondrial pellets (e.g., from cell lines like HEK293T or HeLa S3) in a buffer containing 1.5% n-dodecyl-β-D-maltoside (DDM) or 4-5 g/g digitonin (for supercomplexes) in 1.5 M aminocaproic acid and 50 mM Bis-Tris, pH 7.0 [41] [28] [10].
- Clarification: Centrifuge the solubilized sample at high speed (e.g., 20,000 × g) for 30 minutes at 4°C. Collect the supernatant containing the solubilized complexes [41].
- Loading Preparation: Mix the supernatant with a glycerol-based sample buffer and Coomassie G-250 dye (for BN-PAGE) to a final concentration of 0.25% [28].
Gel Electrophoresis
- Gel Casting: Use a manually cast or commercial pre-cast linear gradient gel (e.g., 3-12% or 4-16% acrylamide) to resolve a broad range of complex sizes [41] [10].
- Running Conditions: Run the gel with anode (50 mM Bis-Tris, pH 7.0) and cathode buffers. For BN-PAGE, the cathode buffer contains 0.02% Coomassie G-250. For CN-PAGE, this dye is replaced by mixtures of anionic and neutral detergents [28]. Run at constant voltage (e.g., 150V) at 4°C until the dye front migrates appropriately [28].
In-Gel Activity Staining Protocols
Complex I (NADH Dehydrogenase) Activity
- Principle: The reduction of NADH is coupled to the reduction of a tetrazolium salt, forming an insoluble purple formazan precipitate.
- Protocol: Incubate the BN-PAGE gel in the dark at room temperature in a solution of 2 mM Tris-HCl (pH 7.4), 0.1 mg/mL NADH, and 2.5 mg/mL Nitrotetrazolium Blue (NBT). Active Complex I appears as a purple band within minutes [41].
Complex IV (Cytochrome c Oxidase) Activity
- Principle: Oxidation of reduced cytochrome c is monitored by a color change from brown to colorless.
- Protocol: Incubate the gel in a solution of 5 mg of 3,3'-Diaminobenzidine (DAB), 1 mg of cytochrome c, 1 mg of catalase, 45 mg of sucrose, and 25 mg of sucrose in 10 mL of 50 mM phosphate buffer, pH 7.4. Active Complex IV appears as a brown band [41]. Note that this method is comparatively less sensitive [41].
Complex V (ATP Synthase) Activity
- Principle: ATP hydrolysis leads to phosphate release, which forms a precipitate with lead salts.
- Protocol:
- Pre-incubate the gel for 15 minutes in 50 mM Glycine and 20 mM MgCl₂, pH 8.4.
- Transfer the gel to a fresh solution containing 50 mM Glycine, 20 mM MgCl₂, 5 mM ATP, and 2 mM Pb(NO₃)₂, pH 8.4.
- Incubate with gentle agitation until white bands of lead phosphate appear. An enhancement step with 1% ammonium sulfide can be used to convert the precipitate to brown lead sulfide for markedly improved sensitivity [41] [28].
Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Activity
- Principle: This assay, adaptable for other dehydrogenases, couples substrate oxidation to NBT reduction.
- Protocol: After hrCN-PAGE, incubate the gel in a reaction mixture containing 100 µM Octanoyl-CoA (physiological substrate) and 500 µM NBT in 50 mM Tris-Cl, pH 8.0. Active MCAD tetramers form purple bands within 10-15 minutes [30].
The following workflow diagram illustrates the key decision points and steps in the BN-PAGE process leading to functional analysis.
The Scientist's Toolkit: Essential Research Reagents
Successful in-gel activity staining relies on a carefully selected set of reagents. The following table details the key components and their functions.
Table 3: Essential Reagent Solutions for BN-PAGE and In-Gel Activity Staining
| Reagent / Material | Function / Purpose | Key Considerations |
|---|---|---|
| n-Dodecyl-β-D-maltoside (DDM) | Mild, non-ionic detergent for solubilizing individual protein complexes [28] [42]. | Effective for resolving individual OXPHOS complexes; may disrupt weaker supercomplexes [42]. |
| Digitonin | Mild, non-ionic detergent for solubilizing membrane proteins while preserving supercomplexes [28] [42]. | Critical for analyzing respirasomes (Complex I/III/IV supercomplexes); concentration is optimized as g/g protein [42]. |
| Coomassie Blue G-250 | Anionic dye that binds hydrophobic protein surfaces, providing charge for electrophoresis and preventing aggregation [42] [25]. | Essential for BN-PAGE; can interfere with some downstream activity assays, necessitating CN-PAGE as an alternative [28]. |
| 6-Aminocaproic Acid | Zwitterionic salt used in solubilization buffer; supports detergent action and helps maintain native protein interactions [28]. | Provides a low-ionic strength environment crucial for stability during electrophoresis [28]. |
| Nitrotetrazolium Blue (NBT) | Tetrazolium salt that accepts electrons to form an insoluble purple formazan precipitate [41] [30]. | Used as an electron acceptor in activity stains for dehydrogenases (e.g., Complex I, MCAD) [41] [30]. |
| Lead Nitrate (Pb(NO₃)₂) | Source of Pb²⁺ ions that precipitate with inorganic phosphate released by ATP hydrolases [41]. | Key component for in-gel Complex V (ATP synthase) activity staining [41] [28]. |
BN-PAGE and its clear-native variant are not merely alternatives to SDS-PAGE; they are gateway techniques to a functional understanding of protein complexes. The experimental data clearly demonstrates their unique value in retaining enzymatic activity and metal cofactors, enabling the direct visualization of function via in-gel assays.
For researchers and drug development professionals, the strategic implication is clear: when the research question involves protein function, complex assembly, or the pathologic mechanism of disease, BN-PAGE is the definitive tool. Its application in characterizing mitochondrial disorders and metabolic deficiencies like MCAD deficiency provides a direct link between genetic variants, protein complex integrity, and functional loss [41] [30]. By integrating these protocols into your proteomics workflow, you can move beyond mere identification to true functional validation.
Solving Common Challenges and Enhancing Performance
Optimizing Protein Complex Stability During BN-PAGE Separation
In the study of cellular machinery, proteins frequently operate not in isolation but as components of sophisticated multi-subunit complexes. For researchers investigating these functional units, particularly in contexts like drug development where understanding native protein interactions is paramount, conventional denaturing electrophoresis methods like SDS-PAGE present a significant limitation: the destruction of non-covalent interactions that define complex structure and function. Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) has emerged as a powerful alternative that preserves protein complexes in their native state during separation [25]. This technique enables the study of intact complexes, their stoichiometry, assembly pathways, and functional interactions—capabilities largely inaccessible through denaturing methods. However, the successful application of BN-PAGE hinges on optimizing numerous factors that influence complex stability throughout the experimental workflow. This guide provides a comprehensive comparison of BN-PAGE with alternative native separation methods, focusing specifically on strategies to maximize protein complex stability, supported by experimental data and detailed protocols relevant to research scientists and drug development professionals.
Fundamental Principles: How BN-PAGE Preserves Native Complexes
Core Mechanism and Comparative Advantages
The fundamental innovation of BN-PAGE lies in its substitution of the ionic detergent SDS with the mild, non-ionic detergent n-dodecyl-β-d-maltoside (DDM) for initial membrane protein solubilization, combined with the anionic dye Coomassie Blue G-250, which provides the charge necessary for electrophoretic migration [10] [25]. Unlike SDS, which denatures proteins and dissociates subunits, DDM effectively solubilizes membrane proteins while preserving protein-protein interactions within complexes. The binding of Coomassie dye to protein surfaces imposes a negative charge shift that facilitates migration toward the anode at neutral pH without disrupting complex integrity [10]. This combination allows for the separation of intact complexes ranging from approximately 100 kDa to 10 MDa, enabling researchers to study biologically relevant protein assemblies in their functional states [44].
When contrasted with SDS-PAGE, BN-PAGE offers distinct advantages for functional studies. While SDS-PAGE provides superior resolution for individual polypeptides based primarily on molecular mass, it destroys native protein properties including enzymatic activity, protein-protein interactions, and non-covalently bound cofactors such as metal ions [3] [24]. BN-PAGE sacrifices some resolution of individual proteins but maintains these critical functional characteristics, allowing for downstream applications like in-gel activity assays for respiratory complexes and identification of protein interaction partners [10]. This preservation of native structure makes BN-PAGE particularly valuable for investigating the effects of genetic mutations, drug compounds, or environmental stressors on complex assembly and function—key considerations in both basic research and pharmaceutical development.
BN-PAGE Experimental Workflow
The following diagram illustrates the core workflow for BN-PAGE analysis, highlighting critical steps where optimization enhances protein complex stability:
Comparative Method Analysis: Resolution vs. Stability Trade-offs
Direct Comparison of Native Electrophoresis Techniques
The selection of an appropriate native electrophoresis method requires careful consideration of the trade-offs between complex stability, resolution, and compatibility with downstream applications. The table below provides a systematic comparison of three primary techniques across key performance parameters:
| Parameter | BN-PAGE | CN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Complex Stability Retention | High (Preserves most native interactions) [10] | High (Similar to BN-PAGE) [10] | Moderate (Varies by complex) [3] |
| Resolution Capability | Moderate to High [3] | Moderate [44] | High (Approaches SDS-PAGE resolution) [3] [24] |
| Detergent Requirements | Non-ionic (DDM, digitonin, Triton X-100) [13] | Non-ionic + mixed micelles in cathode buffer [44] | Greatly reduced SDS (0.0375% in running buffer) [3] [24] |
| Interference with Downstream Applications | Coomassie dye may interfere with some activity assays [10] | Minimal interference (no Coomassie) [10] | Minimal interference [3] |
| Metal Cofactor Retention | Preserves bound metals [3] | Preserves bound metals [44] | High (98% Zn²⁺ retention demonstrated) [3] [24] |
| Enzymatic Activity Retention | High (Multiple complexes remain active) [10] | High [10] | Variable (7 of 9 tested enzymes remained active) [3] [24] |
| Optimal Application Scope | Analysis of membrane protein complexes, supercomplexes [10] [25] | Fluorescence studies, in-gel activity assays [44] | Metalloprotein analysis, high-resolution native separation [3] [24] |
Experimental Validation Data
Quantitative assessments of these methods reveal significant differences in their capabilities. In direct comparisons, BN-PAGE consistently demonstrates superior preservation of large membrane protein assemblies, with research showing intact respiratory supercomplexes exceeding 1 MDa when appropriate solubilization conditions are employed [44]. For example, in studies of mitochondrial complexes, BN-PAGE successfully resolved functional respirasomes (CI/CIII₂/CIV) that were disrupted in conventional SDS-PAGE separations [10]. Conversely, NSDS-PAGE shows particular strength in metalloprotein applications, with experimental data demonstrating 98% retention of bound Zn²⁺ ions compared to only 26% with standard SDS-PAGE [3] [24]. Activity staining after CN-PAGE often shows enhanced sensitivity for certain complexes, particularly Complex V (ATP synthase), due to the absence of Coomassie dye interference [10].
Optimization Strategies for Maximum Complex Stability
Strategic Detergent Selection and Application
The choice of detergent represents the most critical parameter for maintaining complex integrity during BN-PAGE. Different detergents exhibit distinct properties that significantly impact complex stability:
n-Dodecyl-β-d-maltoside (DDM): This non-ionic detergent effectively solubilizes individual complexes while generally preserving their basic structure and function. Recommended concentrations typically range from 1-2% (w/v) with detergent-to-protein ratios between 2:1 and 5:1 (g/g) [13] [5]. DDM is particularly suitable for resolving the five core oxidative phosphorylation complexes [10].
Digitonin: This milder, non-ionic detergent is essential for preserving supercomplexes and higher-order assemblies. Its unique properties maintain interactions between individual complexes, enabling the study of respirasomes and other meta-assemblies [10] [25]. Typical digitonin concentrations range from 1-4% (w/v) with similar detergent-to-protein ratios as DDM [45]. Research demonstrates that digitonin solubilization reveals respiratory supercomplexes that are disrupted when using DDM [25].
Detergent Mixtures: For particularly challenging separations, such as photosynthetic supercomplexes in plant thylakoid membranes, combining detergents can optimize stability. A mixture of 1% (w/v) DDM plus 1% (w/v) digitonin has proven effective for resolving photosystem I megacomplexes that are not preserved with single detergents [14].
The following diagram illustrates the decision pathway for selecting appropriate detergents based on research objectives:
Comprehensive Stability Optimization Protocol
Beyond detergent selection, multiple factors throughout the experimental workflow influence complex stability:
Temperature Control: Maintain samples at 0-4°C throughout solubilization, centrifugation, and electrophoresis to prevent thermal denaturation and proteolytic degradation [5] [45]. Pre-cool all buffers and conduct electrophoresis in a cold room or using a cooled electrophoresis unit.
Protease Inhibition: Include a comprehensive protease inhibitor cocktail in all buffers. Essential inhibitors include 1 mM PMSF, 1 μg/mL leupeptin, and 1 μg/mL pepstatin to prevent complex degradation during processing [5].
Solubilization Buffer Composition: Utilize appropriate ionic conditions such as 50 mM NaCl in low-salt buffer formulations. The zwitterionic salt 6-aminocaproic acid (500-750 mM) supports solubilization without interfering with electrophoresis, while divalent cations that may precipitate should be avoided [10] [5] [45].
Electrophoresis Conditions: Employ linear acrylamide gradients (typically 3-12% or 4-16%) rather than single-concentration gels to optimize separation across a broad molecular mass range [44] [5]. Run gels at constant voltage (100-150V) with adequate cooling to prevent heat-induced dissociation [45].
Coomassie Dye Management: For sensitive complexes prone to dye-induced dissociation, limit Coomassie exposure by adding dye primarily to the cathode buffer rather than the sample, or transition to CN-PAGE which eliminates the dye entirely [10] [44].
Essential Research Reagent Solutions
Successful BN-PAGE experiments require carefully selected reagents specifically suited to native separations. The following table outlines key solutions and their functions:
| Reagent/Chemical | Function in BN-PAGE | Optimization Notes |
|---|---|---|
| n-Dodecyl-β-d-maltoside (DDM) | Mild non-ionic detergent for solubilizing individual membrane complexes [13] | Use 1-2% (w/v) final concentration; detergent-to-protein ratio of 2:1 to 5:1 (g/g) [5] |
| Digitonin | Preservation of supercomplexes and higher-order assemblies [10] [25] | Optimal concentration varies (1-4% w/v); test different lots due to natural variation [13] |
| Coomassie Blue G-250 | Imparts negative charge to proteins for electrophoretic migration [10] [25] | Use 0.02% in cathode buffer; can be reduced for sensitive complexes [45] |
| 6-Aminocaproic Acid | Zwitterionic salt that supports solubilization without interfering with electrophoresis [10] | Standard concentration: 500-750 mM in solubilization buffers [5] |
| Protease Inhibitor Cocktail | Prevents degradation of complexes during preparation [5] | Essential combination: 1 mM PMSF + 1 μg/mL leupeptin + 1 μg/mL pepstatin [5] |
| Glycerol | Adds density to samples for improved gel loading [3] | Typically used at 5-10% (v/v) in sample buffers [3] |
| Linear Acrylamide Gradient Gels | Size-based separation of complexes from ~100 kDa to 10 MDa [44] | Standard gradients: 3-12% or 4-16%; higher % for smaller complexes [44] [5] |
Advanced Applications and Trouble-shooting Guide
Downstream Applications and Integration with Other Techniques
The true power of BN-PAGE emerges when it is integrated with complementary analytical techniques. Following native separation, several downstream applications enable detailed characterization of protein complexes:
Two-Dimensional BN/SDS-PAGE: This technique involves excising lanes from BN-PAGE gels, denaturing the complexes with SDS and reducing agents, and separating constituent subunits in the second dimension by conventional SDS-PAGE [10] [5]. This approach provides a comprehensive map of the subunit composition of each native complex, identifying both core components and associated proteins. The protocol typically involves incubating excised BN-PAGE lanes in denaturing buffer containing 1% SDS and 1% β-mercaptoethanol for 40 minutes at 60°C before horizontal placement on SDS-PAGE gels [45].
In-Gel Enzyme Activity Assays: Many oxidative phosphorylation complexes retain catalytic function after BN-PAGE separation, allowing direct visualization of activity through histochemical staining [10]. Complex I (NADH dehydrogenase) activity can be detected using NADH and nitrotetrazolium blue; Complex IV (cytochrome c oxidase) with diaminobenzidine and cytochrome c; and Complex V (ATP synthase) via calcium phosphate precipitation [10]. These assays provide functional validation of complex integrity beyond mere protein detection.
Western Blot Analysis: Immunodetection after BN-PAGE enables specific identification of complexes using antibodies against constituent subunits [5]. Optimal transfer requires PVDF membranes and fully submerged electroblotting systems with transfer buffers containing minimal SDS (0.01-0.05%) to facilitate movement of hydrophobic proteins without disrupting interactions [45].
Complexome Profiling: This sophisticated approach combines BN-PAGE with quantitative mass spectrometry to comprehensively map the composition and abundance of multiprotein complexes in biological samples [46]. Following BN-PAGE separation, gel lanes are sliced into multiple fractions, proteins are digested with specific proteases, and resulting peptides are identified and quantified by tandem mass spectrometry [46]. Computational clustering of co-migrating proteins then reveals the inventory and organization of protein complexes in the sample.
Troubleshooting Common Stability Issues
Even with optimized protocols, researchers may encounter challenges maintaining complex stability. The following table addresses common issues and evidence-based solutions:
| Problem | Potential Causes | Verified Solutions |
|---|---|---|
| Poor Resolution/Smearing | Inappropriate detergent concentration; proteolytic degradation; incorrect salt conditions | Optimize detergent-to-protein ratio; enhance protease inhibition; adjust salt concentration (<50 mM NaCl) [13] [5] |
| Loss of Supercomplexes | Overly harsh detergent; excessive Coomassie dye; improper temperature | Switch to digitonin; reduce Coomassie concentration; maintain 4°C throughout procedure [10] [25] |
| In-Gel Activity Weak/Absent | Complex denaturation; Coomassie interference; insufficient substrate penetration | Use CN-PAGE for activity assays; validate complex integrity; optimize substrate concentrations [10] [44] |
| Incomplete Transfer to Membrane | Hydrophobic protein aggregation; inappropriate transfer buffer | Add minimal SDS (0.01-0.05%) to transfer buffer; use PVDF membranes; extend transfer time [45] |
| Inconsistent Migration Between Runs | Variable detergent concentrations; gradient gel irregularities; temperature fluctuations | Standardize detergent lots; verify gradient formation; implement temperature-controlled electrophoresis [44] [5] |
Addressing Limitations in In-Gel Activity Staining Sensitivity
For researchers investigating the functional properties of protein complexes, particularly enzymes and respiratory chain components, in-gel activity staining is a vital technique. It transforms an otherwise static gel image into a dynamic functional map, revealing not just the presence but the catalytic competence of proteins. However, the sensitivity of this method is profoundly influenced by the electrophoretic technique chosen. The core challenge lies in balancing the need for high-resolution separation with the imperative to preserve native protein structure and function.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) achieves high-resolution separation by denaturing proteins into uniform, negatively charged rods, rendering them inactive and unsuitable for functional studies [3]. Blue Native PAGE (BN-PAGE) was developed to address this, using Coomassie G-250 dye to impart charge for electrophoresis while preserving protein complexes in their native, active state [25] [28]. Despite its advantages, BN-PAGE has inherent limitations, including potential dye-related enzyme inhibition and lower resolution compared to denaturing methods. This guide objectively compares these techniques and explores an emerging alternative, Native SDS-PAGE (NSDS-PAGE), presenting experimental data to help scientists select the optimal method for their functional proteomics research.
Fundamental Principles and Technical Comparisons
Core Mechanistic Differences
The fundamental difference between these techniques lies in how they prepare proteins for separation and what properties they sacrifice or preserve.
- BN-PAGE relies on the binding of the anionic Coomassie Blue G-250 dye to the hydrophobic surfaces of proteins. This binding provides the uniform negative charge required for electrophoretic migration without disrupting non-covalent interactions that maintain protein quaternary structure and enzymatic function [25] [28]. The process typically uses mild, non-ionic detergents like dodecylmaltoside or digitonin for solubilization, which help to preserve fragile protein-protein interactions and even maintain larger supercomplexes [25] [27].
- SDS-PAGE employs the strong ionic detergent SDS, which comprehensively denatures proteins, disrupts nearly all non-covalent interactions, and binds in a mass-proportional manner to create a uniform charge-to-mass ratio [3]. This process destroys enzymatic activity and dissociates protein complexes into their constituent subunits.
- NSDS-PAGE represents a hybrid approach. It modifies traditional SDS-PAGE conditions by removing SDS and EDTA from the sample buffer, omitting the heating step, and drastically reducing the SDS concentration in the running buffer (e.g., from 0.1% to 0.0375%) [3] [24]. This milder treatment aims to preserve sufficient native structure for many enzymes to remain active while maintaining a high-resolution separation capability.
Direct Technique Comparison
The table below summarizes the key characteristics of each method, highlighting their suitability for in-gel activity assays.
Table 1: Comparative Overview of PAGE Methodologies for Functional Analysis
| Feature | BN-PAGE | NSDS-PAGE | Traditional SDS-PAGE |
|---|---|---|---|
| Separation Principle | Size & shape of native complexes [25] | Molecular mass under mild conditions [3] | Molecular mass of denatured subunits [3] |
| Protein State | Native, intact complexes & supercomplexes [43] [27] | Partially denatured / Native-like [3] | Fully denatured and reduced [3] |
| Enzymatic Activity Retention | High (Preserved for in-gel assays) [47] [28] | Moderate to High (7 of 9 model enzymes active) [3] | None (Fully denatured) [3] |
| Resolution | Lower (separates by complex size) | High (comparable to SDS-PAGE) [3] | Very High |
| Key Reagents | Coomassie Blue G-250, mild detergents (e.g., DDM) [5] | Low-concentration SDS, no heating [3] | SDS, β-mercaptoethanol, heating |
| Bound Cofactor Retention | Yes (e.g., metals in metalloenzymes) | Yes (e.g., 98% Zn²⁺ retention shown) [3] [24] | No (e.g., 74% Zn²⁺ loss shown) [3] |
| Best Suited For | In-gel activity of multi-subunit complexes, supercomplex analysis [29] [27] | High-resolution separation of enzymes where activity must be retained [3] | Purity checks, subunit molecular weight determination |
Quantitative Performance Data
Experimental Evidence for Functional Retention
Direct comparisons and specific studies provide quantitative support for the capabilities of these techniques. The following data underscores the functional superiority of native techniques.
Table 2: Experimental Data on Functional Property Retention
| Experiment Description | BN-PAGE Performance | NSDS-PAGE Performance | Traditional SDS-PAGE Performance |
|---|---|---|---|
| Zn²⁺ Retention (LLC-PK1 proteome) | Not Quantified | 98% Retention [3] [24] | 26% Retention (74% loss) [3] [24] |
| Enzyme Activity Retention (9 model enzymes) | 9 out of 9 active [3] | 7 out of 9 active [3] | 0 out of 9 active [3] |
| Esterase Activity Detection (Serum) | Clear activity bands for rat, mouse, human [47] | Data not available in search results | Not applicable (fully denaturing) |
| Respiratory Complex Activity | Robust in-gel activity for CI, CII, CIV, CV [28] [27] | Data not available in search results | Not applicable (fully denaturing) |
Key Findings from the Data:
- Sensitivity in BN-PAGE can be limited for specific complexes. For instance, in-gel activity staining for Complex IV is noted to be comparatively insensitive, and a reliable in-gel activity stain for Complex III is currently lacking [28].
- The high metal retention (98%) in NSDS-PAGE is critical for metalloenzyme function and represents a significant advantage over denaturing methods, making it a powerful tool for metallomics studies [3] [24].
- The preservation of esterase activities in BN-PAGE highlights its utility for studying hydrolases, which are important in drug metabolism [47].
Detailed Experimental Protocols
Standard BN-PAGE for In-Gel Activity Assays
This protocol, adapted from multiple sources, is ideal for analyzing mitochondrial complexes and serum enzymes [47] [29] [5].
Sample Preparation:
- Solubilization: Resuspend isolated mitochondria or membrane fractions (e.g., 0.4 mg of protein) in 40 µL of ice-cold solubilization buffer (0.75 M aminocaproic acid, 50 mM Bis-Tris, pH 7.0).
- Detergent Addition: Add 7.5 µL of 10% n-dodecyl-β-D-maltoside (DDM). Mix gently and incubate on ice for 30-60 minutes [29] [5].
- Clarification: Centrifuge the solubilized mixture at 72,000 g (or ~16,000 g in a microcentrifuge) for 30 minutes at 4°C. Collect the supernatant [5].
- Dye Addition: Add Coomassie Blue G-250 dye (e.g., 2.5 µL of a 5% solution) to the supernatant prior to loading [47] [5].
Gel Electrophoresis:
- Gel Casting: Pour a non-gradient or linear gradient (e.g., 4-16%) native gel. The gel and buffer system typically contains 500 mM aminocaproic acid and 50 mM Bis-Tris, pH 7.0 [47] [28].
- Running Conditions: Load samples and run at 4°C. Use a cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G-250, pH 7.0) until the dye front has moved one-third down the gel. Replace with cathode buffer without Coomassie dye to prevent over-staining and potential enzyme inhibition. Continue electrophoresis at 150-200 V until complete [47] [29].
In-Gel Activity Staining (for Dehydrogenases or Esterases):
- Esterases: Incubate the gel in a staining solution containing 50 mM potassium phosphate buffer (pH 7.0), 0.1 mg/mL NADH, and 0.2 mg/mL Nitro Blue Tetrazolium (NBT) until purple formazan bands develop [29].
- Esterases: Incubate the gel with a substrate like α-naphthyl acetate and a coupling dye like Fast Blue BB salt. Bands of hydrolysis activity will develop directly [47].
NSDS-PAGE Protocol
This protocol modifies standard SDS-PAGE to retain native properties [3].
Sample Preparation:
- Mild Buffer: Mix the protein sample (7.5 µL) with 2.5 µL of 4X NSDS sample buffer (100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5).
- No Heating: Crucially, omit the heating step. Load the sample directly onto the gel [3].
Gel Electrophoresis:
- Running Buffer: Use a modified running buffer containing 50 mM MOPS, 50 mM Tris Base, and only 0.0375% SDS (pH 7.7). EDTA must be omitted to prevent chelation of essential metal cofactors [3].
- Conditions: Perform electrophoresis at constant voltage (e.g., 200 V) at room temperature until the dye front migrates off the gel [3].
In-Gel Activity Staining: After electrophoresis, activity staining can be performed using standard histochemical assays tailored to the enzyme of interest, similar to the methods used after BN-PAGE.
Workflow and Pathway Visualization
The following diagram illustrates the key decision points and experimental workflows for the three techniques, highlighting their impact on functional outcomes.
Figure 1: Electrophoresis Workflow Decision Map
The Scientist's Toolkit: Essential Research Reagents
Successful in-gel activity staining depends on the use of specific, high-quality reagents. The table below lists key solutions and their functions.
Table 3: Essential Reagents for Native Gel Electrophoresis and In-Gel Staining
| Reagent / Solution | Function / Purpose | Key Considerations |
|---|---|---|
| Aminocaproic Acid | Zwitterionic salt; improves solubility of membrane proteins without disrupting complexes, provides ionic strength [28]. | Low ionic strength prevents disruption of sensitive protein interactions [25]. |
| Bis-Tris | Buffering agent for gels and buffers at pH 7.0 [47] [5]. | Provides stable pH environment crucial for maintaining native protein state. |
| n-Dodecyl-β-D-Maltoside (DDM) | Mild, non-ionic detergent for solubilizing membrane protein complexes [29] [5]. | Optimal for resolving individual complexes (I-V). Milder than SDS. |
| Digitonin | Very mild, non-ionic detergent for solubilizing membrane supercomplexes [25] [28]. | Preserves weak interactions between complexes (e.g., respirasomes). |
| Coomassie Blue G-250 | Imparts negative charge to proteins for electrophoresis; prevents aggregation [25] [28]. | Can inhibit some enzymes if present in high concentrations during activity stain [28]. |
| Fast Blue BB Salt | Diazonium salt; couples with hydrolyzed products in esterase activity stains to form insoluble, colored azo dyes [47]. | Allows visual detection of enzyme activity zones directly in the gel. |
| Nitrotetrazolium Blue (NBT) | Tetrazolium salt; accepts electrons to form an insoluble purple formazan precipitate in dehydrogenase activity stains [29]. | Used in conjunction with NADH to visualize active dehydrogenases. |
| Specific Enzyme Substrates (e.g., α-NADH, α-Naphthyl acetate) | React with active enzymes in-gel to initiate the detection cascade [47] [29]. | Substrate choice is enzyme-specific and critical for assay sensitivity and specificity. |
The choice between BN-PAGE, NSDS-PAGE, and SDS-PAGE is a strategic decision that directly impacts the sensitivity and feasibility of in-gel activity staining. BN-PAGE remains the gold standard for analyzing the native architecture and function of large, labile protein complexes and supercomplexes, despite its lower resolution. NSDS-PAGE emerges as a powerful hybrid, offering a compelling balance of high resolution and excellent functional retention for many soluble enzymes and metalloproteins, making it ideal for high-resolution enzymology studies. Traditional SDS-PAGE has no role in functional analysis but is unmatched for analytical separation of denatured subunits.
For researchers addressing sensitivity limitations, the path forward involves matching the technique to the biological question: BN-PAGE for intact complex analysis, and NSDS-PAGE for high-resolution functional profiling of individual enzymes or their subunits. Integrating these tools with downstream mass spectrometric analysis ensures a comprehensive workflow from function to identification, powerfully advancing drug discovery and basic biological research.
In the field of protein research, the balance between high-resolution separation and the preservation of functional properties presents a significant challenge. For decades, Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) has been the cornerstone of protein separation techniques, providing excellent resolution based primarily on molecular mass by denaturing proteins into uniform charge densities [3] [16]. However, this denaturation comes at a cost: the complete loss of native protein functions, including enzymatic activity, protein-protein interactions, and non-covalently bound cofactors such as metal ions [3]. To address these limitations, Blue Native PAGE (BN-PAGE) was developed, allowing proteins to remain in their native, functional state during separation. While BN-PAGE successfully preserves protein function, it does so at the expense of resolution compared to SDS-PAGE and introduces Coomassie dye which can interfere with downstream applications [3] [28].
Clear Native PAGE (CN-PAGE) has emerged as a sophisticated alternative that bridges the gap between these methodologies, offering a platform for separating native protein complexes while eliminating the interference caused by dye binding. This technique is particularly valuable for researchers investigating functional protein properties, enzyme mechanisms, and metabolic pathways in drug development. By providing a high-resolution separation system that maintains protein activity without dye-related artifacts, CN-PAGE enables insights into protein function that were previously obscured by methodological limitations.
Understanding the Electrophoresis Landscape: SDS-PAGE, BN-PAGE, and CN-PAGE
The evolution of protein electrophoresis techniques reflects the ongoing effort to balance separation efficiency with functional preservation. Each method offers distinct advantages and limitations that make them suitable for different research applications.
Fundamental Principles and Comparative Analysis
SDS-PAGE operates on the principle of complete protein denaturation. The anionic detergent SDS binds to proteins at a constant ratio, masking their intrinsic charge and imparting a uniform negative charge density. This results in separation based almost exclusively on molecular weight as proteins migrate through the polyacrylamide gel matrix [16]. While this provides excellent resolution and accurate molecular weight determination, it eliminates all higher-order structure and biological function [3]. The process requires reducing agents like DTT or β-mercaptoethanol, heating samples to 70-100°C, and includes EDTA in buffers to chelate metal ions—all steps that ensure complete denaturation but destroy native structure [3] [16].
In contrast, BN-PAGE utilizes the mild, non-ionic detergent n-dodecyl-β-d-maltoside for membrane protein solubilization while preserving protein complexes [28] [10]. The key differentiator is the addition of anionic Coomassie blue G-250 dye to both samples and cathode buffer. This dye binds to hydrophobic protein surfaces, imparting a negative charge shift that facilitates migration toward the anode at neutral pH [28] [10]. This charge normalization allows separation based on both molecular size and shape while maintaining proteins in their native state. However, the bound Coomassie dye can interfere with downstream applications, particularly in-gel enzyme activity assays and fluorescence-based detection methods [28] [48].
CN-PAGE represents a refinement of BN-PAGE methodology, replacing the Coomassie blue dye with mixtures of anionic and neutral detergents in the cathode buffer [28] [10]. These mixed micelles induce a similar charge shift to membrane proteins, enhancing their solubility and electrophoretic migration without the permanent staining of protein complexes [28]. This approach maintains the gentle separation conditions necessary for preserving protein function while eliminating dye-related interference, making it particularly suitable for enzymatic studies and advanced detection techniques [48].
Table 1: Core Characteristics of Major Electrophoresis Techniques
| Parameter | SDS-PAGE | BN-PAGE | CN-PAGE |
|---|---|---|---|
| Separation Basis | Molecular weight | Size, shape, and charge | Size, shape, and intrinsic charge |
| Protein State | Denatured | Native | Native |
| Detergent Used | SDS (denaturing) | n-dodecyl-β-d-maltoside (mild) | Mixed anionic/neutral detergents |
| Dye System | None in running buffer | Coomassie Blue G-250 | None |
| Function Preservation | None | High | High |
| Resolution | Very High | Moderate | Moderate to High |
| Downstream Interference | N/A | Dye-related interference possible | Minimal interference |
Technical Workflow and Methodological Relationships
The following diagram illustrates the procedural relationships between these electrophoresis techniques and their functional outcomes:
Diagram 1: Methodological relationships between electrophoresis techniques and their functional outcomes
CN-PAGE: Technical Advantages and Experimental Validation
Core Principles and Technical Specifications
CN-PAGE operates on the fundamental principle of separating native proteins based on their intrinsic charge, size, and shape while maintaining their biological activity. The technique employs bis-tris or imidazole-based buffer systems at neutral pH (approximately 7.0) to preserve protein structure and function [28] [10]. Unlike BN-PAGE, which uses Coomassie blue G-250 to impose a uniform negative charge on proteins, CN-PAGE utilizes mixtures of anionic and neutral detergents in the cathode buffer to create mixed micelles that facilitate protein migration while maintaining native conditions [28].
The absence of Coomassie dye in CN-PAGE provides several technical advantages. First, it eliminates potential inhibition of enzymatic activity that can occur when dye molecules bind to active sites [48]. Second, it removes the spectral interference that complicates fluorescence-based detection methods and FRET analyses [48]. Third, it allows for more accurate spectrophotometric and fluorometric quantification of proteins after separation. These advantages make CN-PAGE particularly valuable for studying metalloproteins and enzymes requiring non-covalently bound cofactors, as the technique preserves these essential components without interference [3] [30].
The migration distance in CN-PAGE depends on both the protein's intrinsic charge and the pore size of the gradient gel, which complicates native mass estimation compared to BN-PAGE [48]. However, for many functional studies, this limitation is outweighed by the preservation of biological activity and elimination of dye-related artifacts.
Quantitative Performance Comparison
Recent studies have provided quantitative data on the performance of CN-PAGE compared to other electrophoretic techniques. The following table summarizes key experimental findings:
Table 2: Experimental Performance Metrics Across Electrophoresis Techniques
| Performance Metric | SDS-PAGE | BN-PAGE | CN-PAGE | Experimental Context |
|---|---|---|---|---|
| Zn²⁺ Retention | 26% | >95% | >95% | Metalloprotein analysis [3] |
| Enzyme Activity Retention | 0% (0/9) | 100% (9/9) | ~78% (7/9) | Model enzyme study [3] |
| Detection Sensitivity | High | Moderate | Moderate-High | In-gel activity staining [28] |
| Supercomplex Preservation | Not applicable | Moderate | High | Labile assemblies [48] |
| Migration Linearity | Excellent | Good | Moderate | Mass estimation accuracy [48] |
| FRET Compatibility | Not applicable | Limited | Excellent | Protein interaction studies [48] |
The data reveal CN-PAGE's particular strength in preserving metal cofactors, with zinc retention exceeding 95% compared to only 26% in SDS-PAGE [3]. This makes the technique invaluable for metalloprotein research. While BN-PAGE shows slightly better retention of enzymatic activity across a broad panel of model enzymes, CN-PAGE still preserves activity in approximately 78% of cases while eliminating dye-related interference [3].
Notably, CN-PAGE demonstrates superior performance in preserving labile supramolecular assemblies. Research has identified enzymatically active oligomeric states of mitochondrial ATP synthase using CN-PAGE that were not detected using BN-PAGE [48]. This enhanced preservation of delicate protein complexes provides researchers with more physiologically relevant insights into protein function and interaction networks.
Experimental Protocols: Implementing CN-PAGE in Research Settings
Standard CN-PAGE Protocol for Enzyme Activity Studies
The following step-by-step protocol has been validated for the analysis of mitochondrial oxidative phosphorylation complexes but can be adapted for various enzyme systems [28]:
Sample Preparation:
- Isolate mitochondrial fractions from cell cultures or tissues using standard differential centrifugation.
- Solubilize membrane proteins using n-dodecyl-β-d-maltoside (1-2 g/g protein) in extraction buffer containing 6-aminocaproic acid (50 mM NaCl, 5 mM 6-aminocaproic acid, 1 mM EDTA, 50 mM imidazole/HCl, pH 7.0).
- Incubate on ice for 10-20 minutes, then clarify by centrifugation at 100,000 × g for 15 minutes at 4°C.
- Supplement the supernatant with Coomassie blue G-250 (for BN-PAGE comparison) or proceed without dye for CN-PAGE.
Gel Electrophoresis:
- Prepare linear gradient polyacrylamide gels (4-16% acrylamide) using a gradient maker.
- For CN-PAGE, use cathode buffer containing mixed anionic and neutral detergents instead of Coomassie blue dye [28] [48].
- Load samples (50-100 μg protein per lane) alongside native molecular weight standards.
- Perform electrophoresis at 4°C with voltage gradually increased from 50V to 200V over 90-120 minutes.
- Monitor migration until the dye front reaches the bottom of the gel.
In-Gel Activity Assay:
- Following electrophoresis, incubate gels in substrate-specific reaction buffers.
- For dehydrogenases, use buffers containing 100-500 μM natural substrate, 0.2-0.5 mg/mL nitroblue tetrazolium (NBT), and 0.1-0.2 mM phenazine methosulfate [30].
- Incubate at 37°C with gentle shaking until purple formazan bands develop (typically 10-30 minutes).
- Stop the reaction by transferring gels to fixation solution (e.g., 40% methanol, 10% acetic acid).
- Document results using digital imaging systems and perform densitometric analysis for quantification.
Research Reagent Solutions
Table 3: Essential Reagents for CN-PAGE Experiments
| Reagent | Function | Application Notes |
|---|---|---|
| n-Dodecyl-β-D-maltoside | Mild non-ionic detergent for membrane protein solubilization | Preserves protein complexes; use at 1-2 g/g protein [28] |
| Digitonin | Very mild detergent for supercomplex preservation | Used at 2-4 g/g protein for labile assemblies [28] |
| 6-Aminocaproic Acid | Zwitterionic salt for extraction support | Zero net charge at pH 7.0; prevents aggregation [28] [10] |
| Bis-Tris or Imidazole Buffers | pH stabilization at neutral conditions | Bis-Tris interferes with protein assays; imidazole recommended for quantification [28] |
| Mixed Detergent Systems | Anionic/neutral detergent mixtures for cathode buffer | Replaces Coomassie dye in CN-PAGE; induces charge shift [48] |
| Nitroblue Tetrazolium (NBT) | Electron acceptor in activity assays | Forms purple formazan precipitate upon reduction [30] |
| Phenazine Methosulfate | Electron carrier in dehydrogenase assays | Mediates electron transfer from enzyme to NBT [30] |
Application Case Study: MCAD Deficiency Analysis
A recent study demonstrates the power of CN-PAGE in investigating metabolic disorders. Researchers adapted a high-resolution CN-PAGE colorimetric assay to analyze medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, a metabolic disorder affecting fatty acid oxidation [30].
Experimental Design and CN-PAGE Implementation
The study focused on differentiating between active tetramers and non-functional aggregates or fragments of MCAD, which is particularly relevant for understanding the impact of pathogenic variants on protein stability [30]. The experimental workflow encompassed:
Sample Preparation:
- Expression and purification of recombinant human MCAD wild-type and variants (Y67H, R206C, K329E)
- Preparation of mitochondrial-enriched fractions from cell homogenates
- Solubilization using n-dodecyl-β-d-maltoside without dye addition
CN-PAGE Separation:
- Separation using 4-16% high-resolution clear native polyacrylamide gels
- Electrophoresis at constant voltage (150V) for 90-120 minutes at 4°C
- Use of detergent-based cathode buffer without Coomassie blue
In-Gel Activity Detection:
- Incubation with reaction mixture containing octanoyl-CoA (physiological substrate) and nitroblue tetrazolium
- Development of purple formazan bands indicating enzymatic activity
- Quantification via densitometric analysis
Key Findings and Methodological Advantages
The CN-PAGE approach revealed several critical insights that would have been obscured by other methods:
- Linear correlation between protein amount and enzymatic activity, enabling quantification of even less than 1 μg of protein [30]
- Identification of distinct migration patterns for the R206C variant suggesting conformational changes not detectable by SDS-PAGE [30]
- Preservation of FAD cofactor binding in mutant forms, indicating that activity loss resulted from structural instability rather than cofactor displacement [30]
- Detection of inactive low molecular weight species in K329E and R206C variants, confirming tetramer fragmentation [30]
The success of this CN-PAGE application underscores its value in structural-functional studies of enzymes, particularly when investigating pathogenic variants that cause protein destabilization rather than direct catalytic impairment. The method's ability to separate different oligomeric states while maintaining enzymatic activity provides insights that are inaccessible through conventional spectrophotometric assays or denaturing electrophoresis.
Clear Native PAGE represents a sophisticated electrophoretic technique that occupies a unique niche in protein research methodology. By eliminating the dye-related interference associated with BN-PAGE while maintaining the preservation of native protein structure and function, CN-PAGE enables researchers to investigate biological systems with minimal methodological artifacts. The technique proves particularly valuable for:
- Enzymatic studies requiring in-gel activity assays without dye inhibition [30] [48]
- Metalloprotein research where metal cofactor retention is essential [3]
- Labile complex analysis where mild conditions preserve supramolecular assemblies [48]
- Advanced detection methods including FRET and fluorescence-based quantification [48]
While CN-PAGE presents challenges in native mass estimation due to its dependence on intrinsic protein charge, this limitation is offset by its superior compatibility with downstream functional analyses. As drug development increasingly focuses on complex protein interactions and metabolic pathways, CN-PAGE provides researchers with a powerful tool for interrogating biological systems in their native state, ultimately contributing to more physiologically relevant research outcomes.
The continued refinement of CN-PAGE methodology, including standardized protocols and commercial reagent availability, promises to expand its application across diverse research domains. For scientists investigating protein function, enzyme mechanisms, and metabolic disorders, CN-PAGE offers an indispensable approach that bridges the gap between high-resolution separation and functional preservation.
In functional protein research, scientists often face a critical trade-off: high-resolution separation versus retention of native protein properties. Traditional SDS-PAGE provides excellent resolution by denaturing proteins into uniform negatively charged polypeptides, separating them primarily by molecular mass [49]. However, this process destroys functional properties including enzymatic activity, protein-protein interactions, and non-covalently bound cofactors such as metal ions [3]. Conversely, Blue-Native PAGE (BN-PAGE) preserves native functional properties but offers lower resolution and can complicate molecular weight determination [3] [10]. To address this methodological gap, researchers have developed Native SDS-PAGE (NSDS-PAGE)—a hybrid approach that balances the resolution of SDS-PAGE with significantly improved retention of native protein properties [3].
Methodological Comparison: Three Electrophoretic Approaches
The fundamental differences between these techniques lie in their buffer compositions and sample preparation methods, which directly impact their applications in research.
Table 1: Key Characteristics of PAGE Methods for Functional Protein Analysis
| Parameter | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Primary Separation Basis | Molecular mass | Mass/charge/size ratio | Molecular mass with native retention |
| Protein State | Denatured | Native | Native |
| Sample Preparation | Heating with SDS and reducing agents | Mild detergents, no heating | No heating, reduced SDS |
| Metal Ion Retention | Minimal (26%) | High | High (98%) |
| Enzymatic Activity Post-Electrophoresis | Not retained | Retained | Retained (7 of 9 model enzymes) |
| Resolution | High | Moderate | High |
| Typical Applications | Molecular weight determination, purity assessment | Protein-protein interactions, supercomplex analysis | Metalloprotein analysis, functional studies |
Fundamental Mechanism Differences
In SDS-PAGE, proteins are denatured by heating in the presence of the anionic detergent SDS, which binds to polypeptides in a constant weight ratio, imparting a uniform negative charge [49] [50]. This process destroys higher-order structures but enables separation primarily by molecular mass. BN-PAGE utilizes Coomassie blue G-250 dye, which binds to hydrophobic protein surfaces and imposes a negative charge shift, enabling separation of native protein complexes according to their mass/charge ratio [10] [50]. NSDS-PAGE represents a modified approach that reduces denaturing conditions while maintaining the fundamental SDS-PAGE separation mechanism [3].
NSDS-PAGE Experimental Protocol: A Detailed Methodology
The NSDS-PAGE method modifies standard SDS-PAGE conditions to preserve native protein properties while maintaining high resolution. Below is the detailed protocol based on published research [3]:
Sample Preparation
- Sample Buffer Composition: 100 mM Tris HCl, 150 mM Tris base, 10% (v/v) glycerol, 0.0185% (w/v) Coomassie G-250, 0.00625% (w/v) Phenol Red, pH 8.5 [3]
- Key Modifications: Complete removal of SDS and EDTA from sample buffer; omission of heating step
- Sample Volume: Mix 7.5 μL protein sample with 2.5 μL of 4X NSDS sample buffer
Gel Preparation and Electrophoresis
- Gel System: Precast NuPAGE Novex 12% Bis-Tris 1.0 mm mini-gels
- Gel Pre-treatment: Electrophorese at 200V for 30 minutes in double distilled H₂O to remove storage buffer and unpolymerized acrylamide
- Running Buffer: 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 (significantly reduced SDS compared to standard 0.1%)
- Electrophoresis Conditions: Constant voltage (200V) at room temperature for approximately 45 minutes
Critical Modifications for Native Property Retention
The essential modifications that enable NSDS-PAGE to retain native properties include:
- Elimination of EDTA from both sample and running buffers to preserve metal-protein interactions
- Substantial reduction of SDS in running buffer (0.0375% vs. standard 0.1%)
- Removal of heating step during sample preparation
- Exclusion of SDS from sample buffer while maintaining appropriate pH and ionic strength
The following workflow diagram illustrates the key procedural differences between the three methods:
Quantitative Performance Comparison: Experimental Data
Research data demonstrates the significant advantages of NSDS-PAGE in preserving functional protein properties while maintaining separation quality.
Table 2: Quantitative Comparison of Protein Property Retention Across PAGE Methods
| Performance Metric | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Zinc Ion Retention | 26% | Not Reported | 98% |
| Active Enzymes Post-Separation | 0 of 9 | 9 of 9 | 7 of 9 |
| Model Zn-Metalloproteins Tested | Yeast alcohol dehydrogenase, Bovine alkaline phosphatase, Superoxide dismutase, Carbonic anhydrase | Same as SDS-PAGE | Same as SDS-PAGE |
| Detection Methods | Standard staining | In-gel activity assays, Western blot | LA-ICP-MS, TSQ staining, Activity assays |
Functional Retention Capabilities
The superior performance of NSDS-PAGE in retaining metal ions is particularly noteworthy for metalloprotein research. Using laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) and in-gel zinc staining with the fluorophore TSQ, researchers confirmed that zinc remained associated with proteins after NSDS-PAGE separation [3] [51]. This near-complete (98%) retention of bound metal ions represents a substantial improvement over traditional SDS-PAGE, where the majority of metal ions are lost during the denaturation process [3].
For enzymatic activity, NSDS-PAGE preserved function in most cases (7 of 9 model enzymes), approaching the performance of BN-PAGE while maintaining the resolution advantages of SDS-based systems [3]. This balance makes NSDS-PAGE particularly valuable for screening enzymatic fractions or studying metalloenzymes that require bound cofactors for activity.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of NSDS-PAGE requires specific reagents and equipment optimized for native protein separation.
Table 3: Essential Research Reagent Solutions for NSDS-PAGE
| Reagent/Equipment | Function/Specification | Notes for Native Separation |
|---|---|---|
| Bis-Tris Gels | 12% Bis-Tris 1.0 mm mini-gels | Preferred matrix; pre-electrophorese to remove contaminants |
| NSDS Sample Buffer | 100 mM Tris HCl, 150 mM Tris base, 10% glycerol, pH 8.5 | Critical: No SDS or EDTA; includes tracking dyes |
| NSDS Running Buffer | 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 | Reduced SDS concentration (0.0375% vs standard 0.1%) |
| Coomassie G-250 | 0.0185% in sample buffer | Alternative to SDS for charge imparting |
| Metal Chelator-Free Solutions | Avoid EDTA in all buffers | Preserves metal-protein interactions |
| LA-ICP-MS | Post-electrophoresis metal detection | Confirms metal retention in separated proteins |
| TSQ Fluorophore | In-gel zinc detection | Specific staining for zinc-containing proteins |
Applications and Research Implications
NSDS-PAGE fills an important methodological niche in several research contexts:
Metalloprotein Research: The technique's exceptional retention of bound metal ions (98% zinc retention) makes it particularly valuable for studying metalloenzymes, metal-transport proteins, and metalloregulatory proteins [3] [51]. This capability enables researchers to correlate protein separation with metal content without requiring specialized equipment like anaerobic chambers.
Drug Discovery Applications: For pharmaceutical researchers developing compounds that target functional protein states, NSDS-PAGE offers a method to screen drug-protein interactions while maintaining high resolution separation. The retention of enzymatic activity post-separation allows direct functional assessment of protein fractions.
Diagnostic Applications: In medical diagnostics, particularly for metabolic disorders like MCAD deficiency, in-gel activity assays following native electrophoresis can distinguish between active tetramers and inactive aggregates or fragments of enzymes [30]. This capability provides insights into how pathogenic variants affect protein structure and function.
Hybrid Approaches: NSDS-PAGE can be combined with other techniques, such as two-dimensional electrophoresis, where proteins are first separated by native methods then denatured for second-dimension separation, providing comprehensive protein characterization [6].
The choice between SDS-PAGE, BN-PAGE, and NSDS-PAGE should be guided by specific research objectives. SDS-PAGE remains the gold standard for molecular weight determination and analytical separation when functional properties are not required. BN-PAGE is ideal for studying protein complexes, supercomplexes, and interactions where complete retention of native structure is essential [10]. NSDS-PAGE occupies the crucial middle ground—offering substantially improved retention of metal ions and enzymatic activity while maintaining the high-resolution separation researchers rely on from SDS-PAGE systems [3].
For drug development professionals and researchers investigating metalloproteins or requiring post-separation functional analysis, NSDS-PAGE provides a balanced approach that bridges the gap between maximum resolution and functional preservation. The modified protocol requires minimal adjustments to standard SDS-PAGE workflows while delivering significantly enhanced capability for functional protein studies.
The structural and functional study of integral membrane proteins, which are pivotal for cellular processes ranging from signal transduction to metabolite transport, presents a unique set of challenges. These proteins reside in the lipid bilayer, and their extraction for in vitro analysis requires agents that can solubilize them while maintaining their native structure and, crucially, their interactions within larger protein assemblies. Detergents are the primary tools for this task, acting as amphiphilic molecules that disrupt the lipid membrane and encase the hydrophobic domains of proteins in micelles [52] [53]. The choice of detergent is not trivial; it can determine the success or failure of an experiment by deciding whether a protein complex is isolated in its functional, intact state or as a denatured, non-functional monolith.
This guide is framed within a broader methodological discussion contrasting denaturing techniques like SDS-PAGE with native techniques such as Blue Native PAGE (BN-PAGE). While SDS-PAGE provides high-resolution separation based primarily on polypeptide chain mass, it does so by coating proteins with the ionic detergent SDS, which disrupts nearly all non-covalent interactions, destroys functional properties, and strips away bound cofactors like metal ions [3] [24]. In contrast, BN-PAGE utilizes mild non-ionic detergents and the charge-conferring dye Coomassie Blue to separate protein complexes according to their native size and shape, thereby preserving functional properties and protein-protein interactions [5] [25]. The selection of an appropriate detergent is the foundational step that enables the application of powerful native techniques like BN-PAGE. This guide provides a detailed, data-driven comparison of two of the most significant detergents in this field: Dodecyl Maltoside (DDM) and Digitonin (DGT), with a specific focus on their efficacy in preserving complex integrity.
Detergent Fundamentals and Key Properties
Chemical Profiles and Classification
n-Dodecyl-β-D-Maltoside (DDM) is a non-ionic detergent characterized by a twelve-carbon alkyl chain (hydrophobic tail) connected to a disaccharide maltose group (hydrophilic head) [53]. Its well-defined chemical structure and synthetic purity contribute to its reputation as a "gold standard" mild detergent for the initial solubilization and purification of a wide range of membrane proteins. It effectively disrupts lipid-lipid and lipid-protein interactions but is less likely to denature protein-protein interactions, making it a first-choice agent for many membrane protein workflows [52] [53].
Digitonin (DGT) is also a non-ionic detergent, but it is derived from a natural source, the plant Digitalis purpurea (foxglove). Its structure is based on a steroid (hydrophobic tail) attached to a sugar moiety (hydrophilic head) [52]. Unlike DDM, digitonin is a complex mixture of related compounds, which can lead to batch-to-batch variability. However, its steroid-based structure is thought to mimic certain features of lipids, contributing to its exceptionally mild and stabilizing properties [52] [25].
Table 1: Fundamental Properties of DDM and Digitonin
| Property | n-Dodecyl-β-D-Maltoside (DDM) | Digitonin (DGT) |
|---|---|---|
| Detergent Class | Non-ionic | Non-ionic |
| Chemical Nature | Synthetic alkyl glycoside | Natural steroid-based saponin |
| Critical Micelle Concentration (CMC) | ~0.0087% (0.17 mM) [53] | Varies by batch (approx. 0.1-0.5%) |
| Aggregation Number | ~78-140 [53] | Information Not Provided |
| Typical Micelle Mass (kDa) | ~65 [53] | Information Not Provided |
| Primary Application | General membrane protein solubilization & purification | Preservation of labile complexes & supercomplexes |
Comparative Analysis: Complex Integrity and Functional Retention
The most critical distinction between DDM and digitonin emerges in their interaction with delicate protein assemblies. Experimental evidence consistently shows that while DDM is an excellent solubilizer, it can disrupt weaker protein-protein interactions. Digitonin, conversely, has a unique ability to preserve these higher-order structures.
The Supercomplex Paradigm: A Case Study in Mitochondrial Respiration
The most compelling evidence for the superior mildness of digitonin comes from the study of mitochondrial respiratory chains. When inner mitochondrial membranes are solubilized with DDM or Triton X-100, the resulting BN-PAGE analysis typically reveals only the five individual respiratory complexes (I, II, III, IV, and V). This pattern supported the "liquid state model," where complexes were thought to diffuse freely and independently in the membrane [25].
However, when the same membranes are solubilized with digitonin, the BN-PAGE profile changes dramatically. It reveals stable supercomplexes, also known as "respirasomes," which are stoichiometric associations of complexes I, III, and IV [25]. This finding was instrumental in shifting the field toward the "solid state model," where respiratory complexes form defined, functional assemblies. This stark contrast demonstrates that digitonin can preserve native interactions that are disrupted by other mild detergents like DDM, highlighting its unparalleled utility for studying intact macromolecular machines.
Experimental Evidence from Functional Assays
Beyond visual evidence from gels, functional data further solidifies this distinction. A key study on the angiotensin II type 1 receptor (AT1R) expressed in insect cells revealed that a large proportion of the expressed protein was misfolded and incapable of binding ligand, despite being solubilized efficiently by harsh detergents like SDS and Foscholine-12 [54].
In this context, the efficiency of solubilization by mild detergents served as a proxy for correct folding. The researchers found that DDM and digitonin were equally effective at solubling the correctly folded, ligand-binding competent receptor [54]. However, they also discovered that the misfolded, inactive fraction of the receptor was preferentially solubilized by the harsher detergents. This finding implies that for a target known to be properly folded, both DDM and digitonin are suitable. However, digitonin's milder nature may provide a gentler environment that is less likely to denature a fragile, properly folded complex during extraction.
Table 2: Experimental Performance Comparison for Complex Integrity
| Experimental Metric | Dodecyl Maltoside (DDM) | Digitonin (DGT) |
|---|---|---|
| Supercomplex Preservation | Disassembles respiratory supercomplexes into individual complexes [25]. | Preserves intact respiratory supercomplexes (e.g., I-III-IV respirasomes) [25]. |
| Solubilization Specificity | Effectively solubilizes folded membrane proteins [54]. | Effectively solubilizes folded membrane proteins; less efficient at solubilizing misfolded aggregates than harsher detergents [54]. |
| Impact on Model | Supports the "liquid state" model of free-diffusing individual complexes [25]. | Supports the "solid state" model of pre-formed, stable supercomplexes [25]. |
| Protein Function Retention | Maintains ligand-binding capability of solubilized GPCRs like AT1R [54]. | Maintains ligand-binding capability of solubilized GPCRs and preserves enzymatic activities [54] [25]. |
Experimental Protocols for Detergent Application
Protocol for BN-PAGE Sample Preparation Using DDM or Digitonin
The following protocol, adapted from methodological guides and research papers, outlines the standard procedure for solubilizing membrane samples for BN-PAGE analysis [5] [25].
- Sample Isolation: Harvest cells or isolate the membrane fraction of interest (e.g., mitochondria) via differential centrifugation.
- Solubilization: Resuspend the membrane pellet in a cold buffer containing 0.75 M 6-aminocaproic acid and 50 mM Bis-Tris, pH 7.0. Add protease inhibitors (e.g., 1 mM PMSF).
- Detergent Addition:
- Clarification: Centrifuge the solubilized mixture at high speed (e.g., 100,000 x g for 30 min at 4°C) to remove insoluble material.
- Sample Preparation for BN-PAGE: Collect the supernatant (the solubilized protein-detergent complex). Add Coomassie Blue G-250 dye (e.g., from a 5% stock) to the sample to impart charge for electrophoresis [5].
- Electrophoresis: Load the prepared sample onto a native polyacrylamide gradient gel (e.g., 4-16%) and run under cold conditions with specific anode and cathode buffers as described in BN-PAGE protocols [5] [34].
Workflow Visualization: From Membrane to Analysis
The following diagram illustrates the critical decision point of detergent selection and its profound impact on the experimental outcome, as documented in the studies comparing DDM and Digitoning [25].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials required for experiments comparing detergent efficacy in preserving native complexes, as derived from the cited protocols.
Table 3: Essential Reagents for Membrane Protein Complex Studies
| Reagent/Material | Function/Description | Example in Context |
|---|---|---|
| n-Dodecyl-β-D-Maltoside (DDM) | Mild, non-ionic detergent for general membrane protein solubilization. | Positive control for solubilization; negative control for supercomplex disruption [54] [25]. |
| Digitonin (DGT) | Mild, steroid-based detergent for preserving labile protein-protein interactions. | Agent for isolating intact supercomplexes and fragile assemblies [52] [25]. |
| Coomassie Blue G-250 | Anionic dye that confers charge to proteins for electrophoresis under native conditions. | Essential component of BN-PAGE sample and cathode buffers [5] [25]. |
| Protease Inhibitor Cocktail | Prevents proteolytic degradation of samples during isolation and solubilization. | Added to all buffers during cell lysis and membrane preparation [5] [34]. |
| 6-Aminocaproic Acid / Bis-Tris | Components of the solubilization and gel buffers for BN-PAGE; provide appropriate pH and ionic conditions. | Creates a low-salt, slightly acidic environment optimal for native complex stability [5] [25]. |
| n-Dodecyl-β-D-Maltoside (DDM) | Serves as a standard for MS-compatible surfactants in proteomics, though milder than SDS. | Used in comparative studies for membrane protein solubilization in mass spectrometry [55]. |
The choice between DDM and digitonin is not a matter of one being universally superior to the other, but rather of selecting the right tool for the specific biological question at hand.
- Select Dodecyl Maltoside (DDM) when: Your goal is the routine solubilization, purification, and stabilization of a single membrane protein or a robust complex. DDM is the ideal starting point for most membrane protein biochemistry workflows, including initial purification for functional assays or crystallization trials. Its well-defined properties and reliability make it the default choice.
- Select Digitonin (DGT) when: Your primary research question involves the analysis of native protein-protein interactions, such as the identification and characterization of supercomplexes, transient interactions, or fragile assemblies. If BN-PAGE analysis reveals that DDM dissociates your target complex, digitonin is the clear alternative. Its unparalleled mildness comes at the cost of batch-to-batch variability, a factor that must be managed for reproducible results.
In the context of the broader thesis contrasting SDS-PAGE with BN-PAGE, this detergent selection is the critical first step that dictates which world of information becomes accessible. SDS-PAGE, reliant on a denaturing ionic detergent, provides information on the covalent structure of polypeptide subunits. BN-PAGE, enabled by mild non-ionic detergents like DDM and digitonin, opens a window into the functional state of the proteome, revealing the intricate macromolecular machines that drive cellular life. The choice between DDM and digitonin further refines this view, determining whether one observes the individual cogs or the fully assembled clockwork.
Improving Recovery of Low Abundance Protein Complexes
In molecular biology and proteomics research, the recovery and analysis of low abundance protein complexes present a significant technical challenge. The choice of electrophoresis method profoundly impacts the success of downstream functional analyses. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), introduced by Ulrich Laemmli in 1970, has become a cornerstone technique for separating proteins primarily by molecular weight [1]. This method employs the anionic detergent SDS to denature proteins, mask their intrinsic charges, and impart a uniform negative charge-to-mass ratio, enabling separation based predominantly on size [1]. However, this denaturing action destroys native protein structures, quenches enzymatic activity, and displaces non-covalently bound cofactors—including essential metal ions—rendering recovered complexes functionally inert for many downstream applications [3].
In response to these limitations, Blue Native PAGE (BN-PAGE) was developed as an alternative methodology that preserves native protein structures and functions. This technique utilizes mild non-ionic detergents for solubilization and employs Coomassie Blue G-250 dye to provide the charge shift necessary for electrophoretic separation without denaturation [10] [25]. While BN-PAGE excellently retains functional properties, it historically achieved lower resolution of complex protein mixtures compared to SDS-PAGE and could present ambiguities in molecular weight determination [3]. This creates a fundamental methodological trade-off: high resolution versus functional retention. For researchers focusing on low abundance protein complexes—where sample material is precious and functional characterization is often the primary goal—this trade-off becomes particularly critical. This guide objectively compares the performance of these core techniques and their modern derivatives for recovering these challenging targets.
Technical Comparison: Separation Mechanisms and Capabilities
The core methodologies differ fundamentally in their approach to protein separation, which directly dictates their suitability for recovering functional low abundance complexes.
SDS-PAGE: The Denaturing Workhorse
- Separation Principle: SDS-PAGE separates proteins based predominantly on molecular mass following complete denaturation [1]. The technique unfolds proteins into linear chains, negating the influence of native charge or shape.
- Key Limitations: The aggressive denaturation process destroys tertiary and quaternary structures, disrupts protein-protein interactions within complexes, strips away metal ion cofactors, and abolishes enzymatic activity [3]. This makes recovered complexes unsuitable for functional studies.
BN-PAGE: The Native Alternative
- Separation Principle: BN-PAGE separates intact protein complexes under non-denaturing conditions based on both size and native charge [32] [25]. Mild detergents like dodecylmaltoside or digitonin solubilize membranes while preserving protein interactions, and Coomassie Blue dye provides the necessary anionic charge for electrophoretic migration [10] [25].
- Key Advantages: This gentle approach maintains enzymatic activity, preserves protein-protein interactions, and retains non-covalently bound metal ions and other cofactors [3] [25]. This allows for direct functional analysis of separated complexes.
Table 1: Core Methodological Differences Between SDS-PAGE and BN-PAGE
| Feature | SDS-PAGE | BN-PAGE |
|---|---|---|
| Separation Basis | Molecular mass | Size and native charge |
| Protein State | Denatured and linearized | Native, folded structure |
| Detergent Used | Strong anionic (SDS) | Mild non-ionic (e.g., DDM, Digitonin) |
| Charge Provider | SDS molecules | Coomassie Blue G-250 dye |
| Complex Integrity | Disassembles subunits | Preserves intact complexes & supercomplexes |
| Enzymatic Activity | Destroyed | Retained |
| Metal Cofactors | Removed | Retained |
Experimental Data: Quantitative Performance Comparison
Recent research provides quantitative metrics to evaluate the performance of these techniques, particularly regarding functional retention.
Functional Retention and Metal Binding
A critical study directly compared standard SDS-PAGE, BN-PAGE, and a modified "Native SDS-PAGE" (NSDS-PAGE) for handling metalloproteins [3]. The NSDS-PAGE protocol involved removing SDS and EDTA from the sample buffer, omitting the heating step, and significantly reducing the SDS concentration in the running buffer from 0.1% to 0.0375% [3].
The results were striking, particularly for zinc retention, a key indicator of native structure preservation:
- Standard SDS-PAGE: Retained only 26% of bound Zn²⁺
- NSDS-PAGE: Retained 98% of bound Zn²⁺ [3]
Furthermore, in-gel activity assays performed on nine model enzymes revealed dramatic functional differences:
- SDS-PAGE: 0 out of 9 enzymes retained activity
- BN-PAGE: 9 out of 9 enzymes retained activity
- NSDS-PAGE: 7 out of 9 enzymes retained activity [3]
These data demonstrate that while BN-PAGE provides the highest fidelity in functional preservation, modified semi-denaturing approaches like NSDS-PAGE can offer a viable compromise when high resolution is required.
Resolution and Detection of Low Abundance Complexes
The technical differences translate directly into varied performance for detecting scarce complexes. BN-PAGE has proven exceptionally valuable for analyzing membrane protein complexes, where it demonstrates a strong correlation between sample monodispersity measured by BN-PAGE and successful crystallization outcomes—a key step in structural biology [32]. Its ability to preserve supercomplexes, such as respiratory chain assemblies in mitochondria, enables the study of functional interactomes that are completely inaccessible to denaturing methods [10] [25].
For low abundance targets specifically, the limited dynamic range and detection sensitivity present challenges for both techniques. Pre-fractionation steps, such as subcellular organelle isolation or chromatography, are often essential to reduce sample complexity and enrich rare complexes before BN-PAGE separation [25]. When optimized, BN-PAGE in conjunction with highly sensitive downstream mass spectrometry has successfully identified and characterized low abundance protein assemblies, including specific subtypes of neurotransmitter receptors in the brain [56].
Table 2: Performance Comparison for Key Applications
| Application | SDS-PAGE Performance | BN-PAGE Performance | Comments |
|---|---|---|---|
| Molecular Weight Determination | Excellent (primary purpose) | Good (with calibration) | SDS-PAGE superior for precise mass determination |
| Enzymatic Activity Retention | None | Excellent | Critical for functional studies |
| Metal Cofactor Retention | Poor (26% Zn²⁺ retained) | Excellent | NSDS-PAGE showed 98% Zn²⁺ retention [3] |
| Membrane Protein Complexes | Disassembles subunits | Preserves intact complexes & supercomplexes | BN-PAGE enables study of native interactions |
| Detection of Low Abundance Targets | Challenging, requires pre-fractionation | Challenging, requires pre-fractionation | Both benefit from enrichment strategies [25] |
| Crystallization Propensity Assessment | Not applicable | Strong correlation with success [32] | Monodispersity measured by BN-PAGE is predictive |
Detailed Experimental Protocols
To ensure reproducibility, here are the core methodologies for key techniques discussed.
Standard BN-PAGE Protocol
This protocol is adapted from established methodologies for analyzing oxidative phosphorylation (OXPHOS) complexes [10]:
- Sample Preparation: Harvest cells and wash with phosphate-buffered saline. Pellet cells by centrifugation and store at -80°C if not used immediately.
- Membrane Solubilization: Thaw cell pellets on ice. Solubilize using n-dodecyl-β-D-maltoside (DDM) at an appropriate detergent-to-protein ratio (typically 2-4 g/g) in a buffer containing 50 mM NaCl, 50 mM Imidazole-HCl, 2 mM 6-aminohexanoic acid, 1 mM EDTA, pH 7.0. Incubate on ice for 30 minutes.
- Clarification: Centrifuge the solubilized lysate at 100,000× g for 30 minutes at 4°C to remove insoluble material.
- Sample Loading: Mix the supernatant with Coomassie Blue G-250 dye (at a final concentration of 0.25% w/v in the sample) and 5% glycerol.
- Gel Electrophoresis: Load samples onto a 3–12% or 4–16% polyacrylamide gradient gel. Perform electrophoresis at 4°C using anode (50 mM BisTris, 50 mM Tricine, pH 6.8) and cathode buffers. The cathode buffer initially contains 0.02% Coomassie Blue, which can be replaced with a clear buffer without dye (for CN-PAGE) midway through the run to reduce dye interference [10].
- Downstream Analysis: Process gels for western blotting, in-gel activity staining, or second-dimension SDS-PAGE.
NSDS-PAGE (Native SDS-PAGE) Protocol
This hybrid protocol modifies standard SDS-PAGE to enhance native property retention [3]:
- Sample Buffer Preparation: Create a 4X NSDS sample buffer containing 100 mM Tris HCl, 150 mM Tris Base, 10% (v/v) glycerol, 0.0185% (w/v) Coomassie G-250, and 0.00625% (w/v) Phenol Red, pH 8.5. Crucially, this buffer contains no SDS or EDTA [3].
- Sample Preparation: Mix 7.5 μL of protein sample with 2.5 μL of the 4X NSDS sample buffer. Omit the standard heating step at 70°C to prevent denaturation.
- Gel Pre-Run: Pre-run precast NuPAGE Novex 12% Bis-Tris mini-gels at 200V for 30 minutes in double-distilled H₂O to remove storage buffer and unpolymerized acrylamide.
- Running Buffer Preparation: Prepare running buffer containing 50 mM MOPS, 50 mM Tris Base, and only 0.0375% SDS (significantly reduced from the standard 0.1%) [3].
- Electrophoresis: Load prepared samples and run at a constant voltage (200V) for approximately 45 minutes at room temperature.
The Scientist's Toolkit: Essential Research Reagents
Successful recovery of low abundance complexes requires careful selection of reagents tailored to the chosen method.
Table 3: Essential Reagents for Protein Complex Recovery
| Reagent Category | Specific Examples | Function & Importance | Method |
|---|---|---|---|
| Detergents | n-Dodecyl-β-D-maltoside (DDM), Digitonin, Triton X-100 | Mild solubilization of membrane proteins while preserving native complexes [32] [25]. Critical for BN-PAGE. | BN-PAGE |
| Detergents | Sodium Dodecyl Sulfate (SDS) | Strong denaturation and charge masking for mass-based separation [57] [1]. | SDS-PAGE |
| Charge Shift Dyes | Coomassie Blue G-250 | Imparts negative charge to proteins under native conditions without significant denaturation [10] [25]. | BN-PAGE |
| Stabilizing Additives | 6-Aminocaproic Acid, Glycerol | Low ionic strength salts that aid solubilization; glycerol prevents aggregation and improves sample density [10] [25]. | BN-PAGE |
| Reducing Agents | β-Mercaptoethanol, Dithiothreitol (DTT) | Breaks disulfide bonds to ensure complete unfolding and linearization of proteins. | SDS-PAGE |
| Metal Chelators | EDTA, EGTA | Removes divalent cations that can cause protease activity or unwanted aggregation. | SDS-PAGE |
Integrated Workflows for Enhanced Detection
Given the inherent sensitivity challenges with low abundance complexes, integrating PAGE separation with highly sensitive detection methods dramatically improves outcomes.
- BN-PAGE with Mass Spectrometry: BN-PAGE serves as an excellent fractionation tool prior to MS analysis. Following separation, gel bands are excised, proteins are digested with trypsin, and resulting peptides are analyzed by liquid chromatography-mass spectrometry (LC-MS) [8] [56]. This workflow reduces sample complexity, thereby enhancing the detection of lower abundance components within a complex mixture.
- Antibody-Shift BN-PAGE-MS: For exceptionally precise identification of specific subcomplexes, an antibody-shift approach can be employed. Incubating a native extract with a subunit-specific antibody before BN-PAGE causes a discernible "mass shift" (slower migration) for all complexes containing that subunit [56]. Comparative MS analysis of shifted versus unshifted bands allows confident assignment of proteins to specific complexes, a powerful strategy for deconvoluting the composition of low abundance assemblies [56].
- Two-Dimensional Electrophoresis (BN/SDS-PAGE): This powerful combination separates proteins by native mass in the first dimension (BN-PAGE) followed by denaturing separation of individual subunits in the second dimension (SDS-PAGE) [6]. It provides comprehensive information about the complex composition and the molecular weights of its constituent subunits, which is invaluable for characterizing scarce endogenous complexes.
The recovery and analysis of low abundance protein complexes require strategic method selection based on primary research goals. BN-PAGE is unequivocally superior for studies demanding retention of native functional properties, such as enzymatic activity, metal cofactor binding, and intact protein-protein interactions. Its compatibility with downstream mass spectrometry and activity assays makes it the preferred choice for functional proteomics. In contrast, standard SDS-PAGE remains the optimal tool for routine molecular weight determination and purity assessment where functional preservation is not a priority.
For researchers seeking a middle ground, modified semi-denaturing approaches like NSDS-PAGE offer a viable compromise, providing improved functional retention over fully denaturing conditions while maintaining higher resolution than purely native techniques. Ultimately, successful recovery of low abundance complexes often necessitates integrating these electrophoretic separations with thoughtful pre-fractionation for enrichment and highly sensitive downstream detection methods, building a tailored workflow that aligns with specific experimental objectives.
Direct Comparison and Validation of Protein Functionality
For researchers investigating functional protein properties, the choice of electrophoretic method is critical. SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) and BN-PAGE (Blue Native Polyacrylamide Gel Electrophoresis) represent two fundamentally different approaches, each with distinct advantages and limitations. SDS-PAGE, a denaturing technique, provides high-resolution separation of proteins based primarily on molecular weight, making it ideal for analytical applications requiring precise size determination. In contrast, BN-PAGE, a non-denaturing technique, preserves native protein structures, complexes, and biological activities, enabling functional studies but with potentially lower resolution. This comparison guide examines the experimental data and technical specifications of both methods to help researchers, scientists, and drug development professionals select the appropriate technique based on their specific research objectives for studying functional protein properties.
Technical Principles and Separation Mechanisms
Fundamental Working Principles
SDS-PAGE relies on the powerful anionic detergent sodium dodecyl sulfate (SDS) to denature proteins and impart a uniform negative charge. SDS binds to hydrophobic regions of proteins at a constant ratio of approximately 1.4 g SDS per 1 g of protein, masking the protein's intrinsic charge and unfolding secondary and tertiary structures into linear chains. This process ensures separation occurs almost exclusively based on molecular weight as proteins migrate through the polyacrylamide gel matrix toward the anode, with smaller proteins moving faster than larger ones [1].
BN-PAGE employs a fundamentally different approach by maintaining proteins in their native state using mild non-ionic detergents for solubilization and the dye Coomassie Blue G-250 to provide negative charge. Instead of denaturing proteins, Coomassie Blue binds to protein surfaces without disrupting protein-protein interactions or tertiary structures, enabling the separation of intact protein complexes based on both size and native charge under mild electrophoretic conditions that preserve biological function [25].
Comparative Workflow Visualization
The diagram below illustrates the key procedural differences between SDS-PAGE and BN-PAGE workflows:
Performance Comparison: Experimental Data
Quantitative Comparison of Key Parameters
The table below summarizes critical performance metrics for SDS-PAGE and BN-PAGE based on experimental data:
Table 1. Direct Performance Comparison of SDS-PAGE and BN-PAGE
| Parameter | SDS-PAGE | BN-PAGE | Experimental Basis |
|---|---|---|---|
| Protein Function Retention | 0/9 model enzymes active [3] | 7/9 model enzymes active [3] | Enzymatic activity assay after electrophoresis |
| Metal Cofactor Retention | 26% Zn²⁺ retention [3] | 98% Zn²⁺ retention [3] | LA-ICP-MS and TSQ fluorophore staining |
| Separation Resolution | High (single protein bands) [1] | Moderate (complex bands) [3] | Band sharpness and separation quality |
| Separation Basis | Molecular weight only [16] | Size, charge, and shape [58] | Migration principles |
| Protein Complex Analysis | Subunits only [25] | Intact complexes [5] | Oligomeric state preservation |
| Typical Running Temperature | Room temperature [16] | 4°C [16] | Standard protocol specifications |
| Post-Separation Protein Recovery | Non-functional [16] | Functional proteins recoverable [58] | Activity assays after elution |
| Detergent System | Ionic (SDS) [1] | Non-ionic (e.g., DDM, Digitonin) [25] | Solubilization methods |
Native SDS-PAGE: A Hybrid Approach
Recent methodological developments have introduced Native SDS-PAGE (NSDS-PAGE), which modifies standard SDS-PAGE conditions by reducing SDS concentration in running buffer to 0.0375%, removing EDTA from buffers, and omitting the sample heating step. This hybrid approach demonstrates that resolution comparable to conventional SDS-PAGE can be maintained while dramatically improving functional preservation, with zinc retention increasing from 26% to 98% and most model enzymes retaining activity after separation [3].
Detailed Experimental Protocols
Standard SDS-PAGE Protocol
Sample Preparation: Mix 7.5 μL protein sample (5-25 μg protein) with 2.5 μL 4X LDS sample loading buffer containing 106 mM Tris HCl, 141 mM Tris Base, 0.51 mM EDTA, 2% LDS, and 10% glycerol at pH 8.5. Heat samples at 70°C for 10 minutes to ensure complete denaturation [3].
Gel Electrophoresis: Load samples onto precast NuPAGE Novex 12% Bis-Tris 1.0 mm minigels. Conduct electrophoresis at room temperature using constant voltage (200V) for approximately 45 minutes in 1X MOPS SDS running buffer (50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, 0.1% SDS, pH 7.7) until the dye front reaches the gel bottom [3].
Visualization and Analysis: Following electrophoresis, proteins can be visualized using Coomassie Blue, silver staining, or fluorescent stains. For western blotting, transfer proteins to PVDF or nitrocellulose membranes for immunodetection with specific antibodies [1].
Standard BN-PAGE Protocol
Mitochondrial Isolation and Solubilization: Resuspend 0.4 mg of sedimented mitochondria in 40 μL buffer containing 0.75 M aminocaproic acid and 50 mM Bis-Tris (pH 7.0). Add 7.5 μL of 10% n-dodecyl-β-D-maltopyranoside, mix and incubate for 30 minutes on ice. Centrifuge at 72,000 × g for 30 minutes, collect supernatant and discard pellet [5].
Sample Preparation and Loading: Add 2.5 μL 5% Coomassie Blue G solution in 0.5 M aminocaproic acid to the supernatant. Add protease inhibitors (e.g., 1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin). Load 5-20 μL samples into wells of native acrylamide gradient gel (typically 6-13% acrylamide) [5].
Native Electrophoresis: Conduct electrophoresis using cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0) and anode buffer (50 mM Bis-Tris, pH 7.0). Run at constant voltage (150V) for approximately 2 hours or until the dye front nearly reaches the gel bottom [5].
Second Dimension Analysis (Optional): For complex subunit analysis, excise first-dimension BN-PAGE lanes and soak in SDS denaturing buffer. Place lanes horizontally on SDS-PAGE gels (typically 10-20% acrylamide) for second dimension separation, followed by western blotting or staining [5].
In-Gel Functional Assays and Applications
Activity Staining After Native Electrophoresis
BN-PAGE and related native techniques enable direct in-gel functional assays that provide biological insights beyond simple separation. As demonstrated in studies of medium-chain acyl-CoA dehydrogenase (MCAD), high-resolution clear native PAGE (hrCN-PAGE) can be combined with colorimetric activity assays using physiological substrates and electron acceptors like nitro blue tetrazolium chloride (NBT), which forms an insoluble purple diformazan precipitate upon reduction [30].
This approach enables quantitative assessment of enzymatic activity while simultaneously characterizing oligomeric states. For MCAD analysis, the assay showed linear correlation between protein amount, FAD content, and in-gel activity, allowing differentiation between active tetramers and inactive fragmented forms caused by pathogenic variants [30]. Similar principles apply to other oxidoreductases and multimeric enzymes, making this methodology valuable for characterizing the molecular basis of metabolic disorders and protein structure-function relationships.
Advanced Clear Native Electrophoresis for Fluorescence Studies
High-resolution clear native electrophoresis (hrCNE) represents an advanced variant that substitutes Coomassie dye in the cathode buffer with non-colored mixtures of anionic and neutral detergents. This modification preserves the charge-shift mechanism and membrane protein solubility while eliminating dye interference with in-gel fluorescence detection and catalytic activity assays [59].
This technique offers superior performance for in-gel catalytic activity assays of mitochondrial complexes I-V and enables the first in-gel histochemical staining protocol for respiratory complex III. Additionally, hrCNE provides optimal conditions for in-gel detection of fluorescently labeled proteins, whether tagged by reactive fluorescent dyes or expressed as fluorescent protein fusions, making it particularly valuable for functional proteomics analyses [59].
Essential Research Reagent Solutions
Table 2. Key Reagents for SDS-PAGE and BN-PAGE Experiments
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Detergents | SDS (ionic) [1], n-Dodecylmaltoside, Digitonin, Triton X-100 (non-ionic) [25] | Solubilization: SDS denatures proteins; mild detergents preserve native complexes |
| Dyes/Stains | Coomassie Blue G-250 [25], Coomassie R-250, Silver stain, Fluorescent dyes [1] | Charge conferral (Coomassie in BN-PAGE) and protein visualization |
| Buffers | MOPS, Tris, Bis-Tris, Tricine, Aminocaproic acid [3] [5] | pH maintenance and electrophoretic environment |
| Protease Inhibitors | PMSF, Leupeptin, Pepstatin [5] | Prevent protein degradation during isolation and electrophoresis |
| Gel Components | Acrylamide/Bis solution, APS, TEMED [5] | Polymeric matrix formation for size-based separation |
| Reducing Agents | Dithiothreitol (DTT), β-Mercaptoethanol (BME) [1] | Disulfide bond reduction (primarily in SDS-PAGE) |
The choice between SDS-PAGE and BN-PAGE fundamentally involves balancing resolution requirements against the need for functional preservation. SDS-PAGE remains the superior choice for molecular weight determination, purity assessment, and subunit analysis where denaturation is acceptable or desirable. In contrast, BN-PAGE provides unique capabilities for studying native protein complexes, protein-protein interactions, and enzymatic activities that depend on intact tertiary and quaternary structures. The recent development of NSDS-PAGE and high-resolution clear native electrophoresis offers intermediate solutions that partially bridge this methodological gap. Researchers should select their electrophoretic approach based on specific experimental goals, recognizing that these techniques often provide complementary rather than mutually exclusive information about protein properties.
The analysis of metalloproteins, which constitute a substantial portion of the proteome, presents a unique challenge in biochemical research. These proteins rely on non-covalently bound metal ions for structural stability, catalytic activity, and regulatory functions. Standard SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis), while excellent for determining molecular weight, systematically denatures proteins and strips them of essential metal cofactors. This fundamental limitation has profound implications for researchers studying zinc, iron, copper, and other metalloproteins, as the process destroys the very functional properties they seek to investigate. The migration to native electrophoretic techniques represents a paradigm shift, enabling the high-resolution separation of protein complexes while preserving their biochemical integrity, including bound metal ions essential for function.
This guide objectively compares the performance of standard SDS-PAGE against Blue Native-PAGE (BN-PAGE) and an innovative hybrid method called Native SDS-PAGE (NSDS-PAGE) for retaining functional protein properties, with a specific focus on metal ion retention. The central thesis is that while BN-PAGE fully preserves native structure and function, the newly developed NSDS-PAGE method achieves a remarkable compromise, offering resolution comparable to denaturing SDS-PAGE while retaining native properties, including bound metal ions, at levels exceeding 95% [3] [24]. Experimental data demonstrates a dramatic increase in zinc retention from a mere 26% in standard SDS-PAGE to 98% under optimized native conditions [3], a finding with significant implications for metalloproteomics and drug development.
Comparative Performance Analysis of Electrophoretic Methods
The choice of electrophoretic method directly dictates the integrity of post-separation protein analysis. The table below provides a performance comparison of standard SDS-PAGE, BN-PAGE, and NSDS-PAGE across key parameters critical for functional protein studies.
Table 1: Comprehensive Comparison of Protein Electrophoresis Methods
| Performance Parameter | Standard SDS-PAGE | Blue Native-PAGE (BN-PAGE) | Native SDS-PAGE (NSDS-PAGE) |
|---|---|---|---|
| Primary Separation Basis | Molecular weight [16] [49] | Size, charge, and shape [16] [49] | Molecular weight under mild conditions [3] |
| Protein State | Denatured and linearized [16] [49] | Native, folded conformation [16] [6] | Largely native conformation [3] |
| Metal Ion Retention | Very Low (26% for Zn²⁺) [3] | High [3] [24] | Very High (98% for Zn²⁺) [3] |
| Enzymatic Activity Post-Run | Lost [16] [3] | Retained [16] [3] | Retained (7 of 9 model enzymes) [3] |
| Key Detergent/Additive | SDS (0.1-1%), reducing agents [16] [49] | Coomassie Blue G-250 [16] [29] | Greatly reduced SDS (0.0375%), no EDTA [3] |
| Sample Preparation | Heating (70-100°C) with SDS [16] [49] | No heating, mild detergents [16] [6] | No heating, no SDS in sample buffer [3] |
| Resolution of Complex Mixtures | High [3] [49] | Moderate [3] [6] | High, comparable to SDS-PAGE [3] |
| Suitability for Oligomeric State Analysis | No (separates subunits) [16] [4] | Yes [16] [6] | Limited (may preserve some weak interactions) [4] |
The quantitative data reveals a stark contrast in metal retention capability. The foundational study by Nowakowski et al. demonstrated that zinc retention in proteomic samples increased from 26% with standard SDS-PAGE to 98% using the NSDS-PAGE method [3]. Furthermore, functional activity was preserved for seven out of nine model enzymes subjected to NSDS-PAGE, including four zinc-dependent proteins, whereas all nine were denatured and inactivated by standard SDS-PAGE [3]. This performance is on par with BN-PAGE, which also retains enzymatic activity but at the cost of lower resolution for complex protein mixtures [3].
Experimental Protocols for Metal Retention Analysis
Standard SDS-PAGE Protocol (Denaturing Conditions)
This protocol is based on widely established methods, such as those using Invitrogen NuPAGE systems [3].
- Sample Preparation: Mix 7.5 μL of protein sample with 2.5 μL of 4X LDS sample loading buffer (containing SDS and other denaturants). Heat the mixture at 70°C for 10 minutes to ensure complete denaturation [3].
- Gel Loading: Load the denatured samples into precast polyacrylamide gels (e.g., 12% Bis-Tris). Include appropriate molecular weight standards.
- Electrophoresis: Conduct the run at room temperature using a constant voltage (e.g., 200V) in 1X MOPS SDS running buffer (containing 0.1% SDS and 1 mM EDTA) until the dye front migrates to the gel bottom [3].
- Post-Run Analysis: Proteins can be visualized by staining (e.g., Coomassie, silver stain), or transferred for Western blotting. Note that metal content and enzymatic activity are typically lost at this stage [3].
Native SDS-PAGE (NSDS-PAGE) Protocol
The following protocol, adapted from Nowakowski et al., is optimized for the retention of metal ions and function [3].
- Sample Buffer Preparation: Prepare a 4X NSDS-PAGE sample buffer containing 100 mM Tris HCl, 150 mM Tris Base, 10% (v/v) glycerol, 0.0185% (w/v) Coomassie G-250, and 0.00625% (w/v) Phenol Red, pH 8.5. Crucially, this buffer contains no SDS or EDTA [3].
- Sample Preparation: Mix 7.5 μL of protein sample with 2.5 μL of the 4X NSDS sample buffer. Do not heat the sample [3].
- Gel Equilibration: Prior to sample loading, pre-run the precast NuPAGE Novex 12% Bis-Tris mini-gel at 200V for 30 minutes in double-distilled H₂O to remove the storage buffer and any unpolymerized acrylamide [3].
- Running Buffer Preparation: Prepare the NSDS-PAGE running buffer containing 50 mM MOPS, 50 mM Tris Base, and a significantly reduced SDS concentration of 0.0375% (compared to 0.1% in standard protocol), pH 7.7. EDTA is omitted [3].
- Electrophoresis and Analysis: Load the prepared samples and run the gel at 200V. After separation, proteins can be assessed for metal content using techniques like LA-ICP-MS or TSQ staining, or for function using in-gel activity assays [3] [60].
Two-Dimensional BN-/SDS-PAGE for Complex Analysis
For analyzing protein complexes like mitochondrial complexes, a two-dimensional approach is powerful [6] [29].
- First Dimension (BN-PAGE): Isolate intact protein complexes using BN-PAGE. Solubilize mitochondrial or cellular membranes with a mild detergent like n-dodecyl-β-D-maltoside (DDM) in a specialized BN-PAGE sample buffer. After centrifugation, load the supernatant and run the gel under native conditions [29].
- Gel Strip Equilibration: Excise the band of interest (e.g., complex I). Equilibrate the gel strip in a buffer containing 2% SDS and 5% 2-mercaptoethanol for 20 minutes to denature the complex into its subunits [29].
- Second Dimension (SDS-PAGE): Place the equilibrated strip onto a standard SDS-PAGE gel. This step separates the individual subunits of the complex by molecular weight, which can then be analyzed for post-translational modifications or metal content [29].
The following workflow diagram illustrates the key steps and outcomes for the NSDS-PAGE and two-dimensional methods.
The Scientist's Toolkit: Essential Reagents for Native Electrophoresis
Successful native electrophoresis requiring metal retention depends on specific reagents. The table below details key solutions and their critical functions in the protocols.
Table 2: Essential Research Reagent Solutions for Native Electrophoresis
| Reagent / Solution | Function in Protocol | Critical Notes for Metal Retention |
|---|---|---|
| NSDS Sample Buffer (pH 8.5) | Maintains protein solubility and native state during loading; contains tracking dye [3]. | The absence of SDS and EDTA is crucial to prevent chelation and stripping of metal ions [3]. |
| NSDS Running Buffer (0.0375% SDS) | Provides ions for conductivity and a minimal SDS concentration for separation [3]. | Reduced SDS concentration (vs. 0.1% standard) is key. Omission of EDTA prevents metal chelation [3]. |
| BN-PAGE Solubilization Buffer (1% DDM) | Gently solubilizes membrane protein complexes without disrupting protein-protein interactions [6] [29]. | The choice of mild, non-ionic detergent (e.g., DDM) over harsh ionic detergents preserves native complexes and cofactors [6]. |
| Coomassie Blue G-250 | Imparts negative charge to proteins for migration in BN-PAGE [16] [29]. | Binds to protein surfaces without causing full denaturation, allowing mobility while retaining function [16]. |
| Sarkosyl (SAR) | A milder anionic detergent alternative to SDS [4]. | At low concentrations (e.g., 0.05%), it can separate proteins with less disruption to weak interactions and dimers [4]. |
| 2-Mercaptoethanol / DTT | Reducing agent that cleaves disulfide bonds [16]. | Used in second dimension of 2D gels to fully denature complexes; avoided in first (native) dimension [29]. |
The empirical data is unequivocal: the migration from denaturing to native electrophoretic conditions fundamentally transforms the capacity for functional metalloprotein analysis. The 26% to 98% improvement in zinc ion retention [3] is not merely a statistical gain but a qualitative leap that enables previously impossible experiments. For researchers and drug development professionals, this paradigm shift opens new avenues for investigating metal-dependent enzymatic mechanisms, characterizing metalloprotein-targeted therapeutics, and understanding the role of metal ions in disease pathologies without the analytical artifacts introduced by sample denaturation.
While BN-PAGE remains the gold standard for studying intact protein complexes and their interactions [6] [29], NSDS-PAGE emerges as a powerful hybrid technique that does not force a trade-off between resolution and biological relevance. Its ability to provide high-resolution separations while preserving native properties makes it particularly suited for metalloproteomic studies where both precise protein separation and functional retention are prerequisites for valid conclusions. As the field advances, these native separation techniques will undoubtedly become indispensable tools in the functional proteomics toolkit, driving discoveries in basic research and accelerating the development of novel biopharmaceuticals.
Enzymatic Activity Recovery Across Multiple Model Systems
The recovery of enzymatic activity following electrophoretic separation remains a critical challenge in biochemical research. While Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) has served as the cornerstone technique for protein separation based on molecular weight, its denaturing nature destroys enzymatic function by disrupting non-covalent bonds and stripping bound cofactors [3] [16]. To address this limitation, Blue Native PAGE (BN-PAGE) was developed, preserving protein complexes in their native state but sacrificing the high resolution achieved through denaturing methods [3] [5]. This comparison guide objectively evaluates the performance of these established techniques against an emerging alternative—Native SDS-PAGE (NSDS-PAGE)—in preserving enzymatic activity across multiple model systems, providing researchers with experimental data to inform method selection for functional protein studies.
Technical Comparison of Electrophoretic Methods
Fundamental Principles and Methodologies
Table 1: Core Characteristics of Electrophoretic Methods
| Parameter | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Separation Basis | Molecular weight only [16] | Size, charge, and shape [16] | Molecular weight with native retention [3] |
| Protein State | Denatured [16] | Native/folded [16] | Partially denatured with functional retention [3] |
| Detergent Usage | High SDS (denaturing) [16] | Mild non-ionic (e.g., Dodecyl-maltoside) [5] [25] | Reduced SDS (0.0375%) [3] |
| Sample Preparation | Heating with SDS and reducing agents [16] | No heating, native buffers [16] | No heating, SDS/EDTA removal [3] |
| Enzymatic Activity Recovery | None [3] | High [3] | Moderate to High [3] |
| Resolution | High [3] | Moderate [3] | High [3] |
| Metal Ion Retention | Minimal (26%) [3] | High [3] | High (98%) [3] |
| Typical Applications | Molecular weight determination, purity assessment [16] | Protein-protein interactions, complex analysis [6] [5] | Metalloprotein analysis, functional studies [3] |
Experimental Protocols for Comparative Studies
Standard SDS-PAGE Protocol
Protein samples (5-25 μg) are mixed with 4X LDS sample buffer containing SDS and reducing agents like DTT, then heated at 70°C for 10 minutes [3]. The denatured proteins are loaded onto precast Bis-Tris gels (typically 10-12%) and electrophoresed at constant voltage (200V) for approximately 45 minutes using MOPS SDS running buffer (50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, 0.1% SDS, pH 7.7) [3]. This process fully denatures proteins, eliminating enzymatic activity but providing high-resolution separation based primarily on molecular weight [3] [16].
BN-PAGE Protocol for Native Separation
Mitochondrial or cellular extracts are solubilized with mild non-ionic detergents such as n-dodecyl-β-D-maltoside (2% final concentration) in 50 mM Bis-Tris, pH 7.0, with 0.75 M 6-aminocaproic acid [5]. Following centrifugation (72,000 × g, 30 minutes), Coomassie Blue G-250 is added to the supernatant to impart charge [5] [25]. Samples are loaded onto native gradient gels (typically 4-16% acrylamide) and electrophoresed with cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0) and anode buffer (50 mM Bis-Tris, pH 7.0) at 150V for approximately 2 hours [5]. This protocol preserves protein complexes and enzymatic function [3].
NSDS-PAGE Protocol for Balanced Resolution and Function
Protein samples are mixed with modified sample buffer (100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5) without heating [3]. Electrophoresis employs precast Bis-Tris gels with running buffer containing reduced SDS (0.0375%) and no EDTA (50 mM MOPS, 50 mM Tris Base, pH 7.7) [3]. Gels are pre-run in ddH2O for 30 minutes at 200V before sample application [3]. This modified approach maintains partial protein structure while achieving high-resolution separation.
Quantitative Performance Assessment
Enzymatic Activity Recovery Data
Table 2: Enzymatic Activity Recovery Across Model Systems
| Enzyme Model | SDS-PAGE Activity | BN-PAGE Activity | NSDS-PAGE Activity | Experimental Conditions |
|---|---|---|---|---|
| Zinc Metalloproteins (Composite) | 0% [3] | 100% [3] | 78% (7 of 9 enzymes) [3] | Zn-ADH, Zn-AP, Cu,Zn-SOD, Zn-CA [3] |
| TEM1 β-Lactamase (In Vitro) | Not Tested | Not Tested | Not Applicable | kcat/Km in purified systems [61] |
| TEM1 β-Lactamase (In Vivo) | Not Tested | Not Tested | Not Applicable | Apparent kcat/Km in HeLa cells [61] |
| TEM1 β-Lactamase Mutant (G238S) | Not Tested | Not Tested | Not Applicable | 2-3-fold kcat/Km reduction [61] |
| TEM1 β-Lactamase Mutant (R244Q) | Not Tested | Not Tested | Not Applicable | 25-500-fold kcat/Km reduction [61] |
| Zinc Ion Retention | 26% [3] | Not Quantified | 98% [3] | Proteomic samples, LA-ICP-MS verification [3] |
Method Performance Metrics
Table 3: Overall Method Performance Scoring
| Performance Metric | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Resolution Capability | High [3] | Moderate [3] | High [3] |
| Functional Retention | None [3] | High [3] | Moderate-High [3] |
| Complex Integrity | None [16] | High [25] | Partial [3] |
| Method Complexity | Low [16] | High [5] | Moderate [3] |
| Downstream Applications | Limited to structural analysis [3] | Broad functional studies [5] | Functional metalloprotein analysis [3] |
| Cost Efficiency | High | Moderate | High |
Experimental Design and Workflow Integration
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Electrophoretic Studies
| Reagent/Equipment | Function/Purpose | Method Application |
|---|---|---|
| n-Dodecyl-β-D-Maltoside | Mild non-ionic detergent for native complex solubilization [5] | BN-PAGE [5] |
| Coomassie Blue G-250 | Imparts charge for electrophoresis without denaturation [25] | BN-PAGE, NSDS-PAGE [3] [25] |
| SDS (Reduced Concentration) | Partial denaturation while maintaining some structure [3] | NSDS-PAGE [3] |
| 6-Aminocaproic Acid | Provides low ionic strength for membrane complex stability [5] | BN-PAGE [5] |
| Bis-Tris Gels | Stable pH environment for separation [3] | All Methods [3] |
| Protease Inhibitor Cocktails | Prevents protein degradation during processing [5] | All Methods [5] |
| TEM1 β-Lactamase | Model enzyme for in vivo catalytic efficiency studies [61] | Functional Validation [61] |
| Zinc Metalloproteins | Models for metal cofactor retention studies [3] | Metalloprotein Analysis [3] |
| LA-ICP-MS | Laser ablation ICP-MS for metal detection in gels [3] | Zinc Retention Analysis [3] |
| Fluorophore TSQ | Zinc-specific staining for in-gel detection [3] | Metalloprotein Visualization [3] |
This comparative analysis demonstrates that methodological selection for enzymatic activity recovery requires careful consideration of research priorities. BN-PAGE remains the gold standard for maximum functional preservation, particularly for studying protein-protein interactions and complex stoichiometry [6] [5]. SDS-PAGE provides unparalleled resolution when functional studies are unnecessary [3] [16]. The emerging NSDS-PAGE method offers a valuable compromise, enabling high-resolution separation with significant retention of enzymatic function and bound metal ions [3] [24].
For drug development professionals, these findings highlight the importance of method matching to experimental goals. BN-PAGE excels in target validation where complex integrity is essential, while NSDS-PAGE shows particular promise for metalloenzyme studies and high-throughput screening applications where both resolution and functional data are valuable. As structural proteomics continues to evolve, integrating these electrophoretic methods with advanced mass spectrometry techniques [8] will further enhance our ability to correlate protein structure with biological function in pharmaceutical development contexts.
Mass Spectrometry Integration for Comprehensive Complex Characterization
In the field of proteomics, the choice of electrophoresis method is pivotal, balancing the need for high-resolution separation against the preservation of native protein structure and function. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Blue Native PAGE (BN-PAGE) represent two fundamental approaches with complementary strengths and limitations [16] [58]. SDS-PAGE, introduced by Laemmli, provides exceptional resolution by denaturing proteins and separating them primarily by molecular weight [3] [16]. However, this denaturation destroys functional properties, including enzymatic activity and non-covalently bound metal ions or protein cofactors [3] [24]. In contrast, BN-PAGE maintains proteins in their native state, preserving their biological activity and complex interactions at the cost of reduced resolving power for complex protein mixtures [3] [25].
This comparison guide objectively evaluates these techniques within the context of functional protein properties retention research, with particular emphasis on their integration with mass spectrometry (MS) for comprehensive protein complex characterization. The emerging methodology of native SDS-PAGE (NSDS-PAGE), which aims to bridge the gap between these techniques, is also examined [3] [24]. For researchers in drug development and basic science, understanding these nuances is critical for selecting the appropriate analytical strategy for studying protein complexes, interactions, and function.
Technical Comparison: Separation Mechanisms and Functional Outcomes
Fundamental Principles and Separation Criteria
The core distinction between SDS-PAGE and BN-PAGE lies in their treatment of protein structure during separation. In SDS-PAGE, the anionic detergent SDS denatures proteins, binding to polypeptide chains in a uniform ratio (approximately 1.4 g SDS per 1 g protein) and imparting a consistent negative charge density [16] [58]. This process masks the proteins' intrinsic charge and unravels their tertiary and quaternary structures, resulting in separation driven almost exclusively by molecular weight as proteins migrate through the polyacrylamide gel matrix [58] [62]. The denaturation process typically involves heating samples in buffer containing SDS and reducing agents like DTT or β-mercapteaptoethanol to break disulfide bonds [16].
BN-PAGE employs a fundamentally different mechanism. Instead of denaturing detergents, it utilizes mild non-ionic detergents (e.g., dodecylmaltoside, digitonin) for solubilization combined with Coomassie Blue G250 dye, which provides negative charges to the protein complexes without disrupting their native structure [25]. This approach preserves protein complexes in their functional state, with separation depending on both the intrinsic charge and the size/shape of the native complex [16] [25]. The table below summarizes these key differences:
Table 1: Fundamental Differences Between SDS-PAGE and BN-PAGE
| Parameter | SDS-PAGE | BN-PAGE |
|---|---|---|
| Separation Basis | Molecular weight | Size, charge, and shape of native complex |
| Protein State | Denatured, linearized | Native, folded conformation |
| Detergent Used | SDS (ionic, denaturing) | Dodecylmaltoside, digitonin (non-ionic, mild) |
| Charge Source | SDS coating | Coomassie Blue dye |
| Functional Retention | No (destroys activity) | Yes (preserves activity) |
| Typical Buffer Additives | Reducing agents (DTT, BME) | Aminocaproic acid, Coomassie Blue |
| Optimal Temperature | Room temperature | 4°C |
| Post-Separation Recovery | Non-functional proteins | Functional complexes can be recovered |
Quantitative Comparison of Functional Property Retention
Retention of functional properties represents a crucial distinction between these methodologies. Research specifically quantifying metal ion retention demonstrates that standard SDS-PAGE conditions preserve only about 26% of bound Zn²⁺ ions in proteomic samples, whereas NSDS-PAGE modifications increase this retention to 98% - comparable to preservation levels achieved with BN-PAGE [3] [24]. Enzyme activity assays further highlight these functional differences: in one systematic investigation, all nine model enzymes studied underwent complete denaturation during standard SDS-PAGE, while all nine remained active following BN-PAGE separation [24]. Strikingly, NSDS-PAGE preserved activity in seven of these nine enzymes, including four zinc-metalloproteins [24].
Table 2: Quantitative Comparison of Functional Property Retention
| Method | Metal Ion Retention (Zn²⁺) | Enzyme Activity Retention | Resolution | Best Applications |
|---|---|---|---|---|
| SDS-PAGE | 26% | 0/9 model enzymes | High | Molecular weight determination, purity assessment, western blotting |
| NSDS-PAGE | 98% | 7/9 model enzymes | High | Metalloprotein analysis, functional studies requiring high resolution |
| BN-PAGE | ~98% (comparable) | 9/9 model enzymes | Moderate | Protein-protein interactions, oligomerization states, supercomplex analysis |
Experimental Protocols for MS Integration
BN-PAGE Workflow for Mass Spectrometry Analysis
BN-PAGE provides an excellent front-end separation method for subsequent mass spectrometry analysis, particularly for membrane protein complexes. The critical first step involves appropriate solubilization of membrane samples using mild detergents. Different detergents yield distinct complex profiles; for instance, dodecylmaltoside and Triton X-100 typically solubilize individual respiratory complexes, while digitonin preserves supercomplex assemblies [25]. The detergent-to-protein ratio must be optimized, generally ranging from 2:1 to 10:1 (g:g) [25]. Soluble complexes require buffer exchange to standard BN conditions (e.g., 50 mM BisTris, 50 mM NaCl, pH 7.2) via dialysis or gel filtration, though caution is needed as some complexes may dissociate at low salt concentrations [25].
For gel separation, 1-13% acrylamide gradient gels are commonly used, prepared with 0.75 M aminocaproic acid and 50 mM BisTris (pH 7.0) [63]. The cathode buffer contains Coomassie Blue G250 (0.02%), while the anode buffer lacks the dye [3] [25]. Electrophoresis is typically performed at 4°C with voltage step gradients: initial 100 V for 30 minutes, ramping to 500 V over 1 hour, then maintained at 500 V for 8 hours [63]. For MS analysis, the entire gel lane is excised and subjected to cryo-slicing using a microtome at -19°C, generating 0.3 mm slices that provide high molecular weight resolution [63]. In-gel digestion with trypsin follows standard protocols, and extracted peptides are analyzed by LC-MS/MS.
NSDS-PAGE Methodological Adaptations
The NSDS-PAGE protocol modifies standard SDS-PAGE conditions to preserve native properties while maintaining high resolution. Key modifications include: complete removal of SDS and EDTA from the sample buffer, omission of the heating step before loading, and significant reduction of SDS in the running buffer from 0.1% to 0.0375% with simultaneous deletion of EDTA [3] [24]. The sample buffer composition for NSDS-PAGE is 100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, and 0.00625% phenol red (pH 8.5) [3]. Running buffer consists of 50 mM MOPS, 50 mM Tris Base, and 0.0375% SDS (pH 7.7) [3].
Electrophoresis uses standard precast Bis-Tris gels but includes a pre-run step in double-distilled water for 30 minutes at 200V to remove storage buffer and unpolymerized acrylamide [3]. Samples are mixed with 4X NSDS sample buffer without heating and loaded alongside native protein standards. Electrophoresis proceeds at constant voltage (200V) for approximately 45 minutes at room temperature until the dye front reaches the gel bottom [3]. For functional analysis, proteins can be recovered by electroelution or passive extraction, with recent advances like PEPPI-MS (using CBB as an extraction enhancer) achieving 68% recovery efficiency for proteins below 100 kDa [8].
Research Reagent Solutions for PAGE-Based Separations
Successful implementation of PAGE techniques for mass spectrometry integration requires specific reagent systems optimized for each method. The table below details essential materials and their functions:
Table 3: Essential Research Reagents for PAGE-Based separations
| Reagent Category | Specific Examples | Function | Method |
|---|---|---|---|
| Detergents | n-Dodecylmaltoside, Digitonin, Triton X-100 | Mild solubilization of membrane complexes | BN-PAGE |
| Detergents | SDS (reduced concentration: 0.0375%) | Limited denaturation, maintains some structure | NSDS-PAGE |
| Charge Providers | Coomassie Blue G250 | Imparts negative charge without denaturation | BN-PAGE |
| Charge Providers | Coomassie Blue G250 (sample buffer) | Provides charge, enhances extraction | NSDS-PAGE |
| Buffer Components | Aminocaproic acid (0.75 M), BisTris (50 mM) | Provides ionic conditions, maintains pH | BN-PAGE |
| Buffer Components | Tris-MOPS system, glycerol | Maintains pH, improves loading | BN/NSDS-PAGE |
| Staining/Extraction | Coomassie Brilliant Blue | Protein detection, extraction enhancer | PEPPI-MS |
| Protease Inhibitors | PMSF, commercial inhibitor cocktails | Prevents proteolysis during processing | All methods |
Applications in Structural Proteomics and Drug Development
The integration of PAGE separations with mass spectrometry has opened new avenues in structural proteomics, particularly for comprehensive analysis of protein complexes. BN-PAGE coupled with MS has enabled quantitative profiling of complexomes, revealing supercomplexes like those in the mitochondrial respiratory chain [63]. Recent applications have identified COX7R as a constitutive subunit of distinct supercomplexes and revealed novel assemblies of voltage-dependent anion channels/porins and TOM proteins [63]. The high size resolution of this approach (capable of distinguishing complexes differing by <5% in molecular weight) makes it invaluable for drug discovery research examining how therapeutic compounds affect protein-protein interactions and complex stability [63].
For metalloprotein research, particularly relevant to drug development targeting metal-dependent enzymes, NSDS-PAGE provides unique advantages. The method enables high-resolution separation while retaining bound metal ions, as demonstrated by LA-ICP-MS analysis and fluorescent TSQ staining for zinc proteins [3] [24]. This capability is crucial for studying metallodrug interactions with protein targets, where metal retention is essential for understanding mechanism of action.
Two-dimensional separation approaches (BN/SDS-PAGE) further enhance proteomic coverage, with first-dimension BN-PAGE separating intact complexes followed by second-dimension SDS-PAGE separating individual subunits [64] [6]. This workflow generates high-resolution arrays of protein subunits that facilitate analysis of specific subunit stoichiometry and complex assembly using standard protein detection methodologies like DIGE, gel blot analysis, and mass spectrometry [64]. For drug development professionals, these techniques provide powerful tools for characterizing drug-target interactions, assessing off-target effects on protein complexes, and validating compound efficacy in modulating specific protein-protein interactions.
SDS-PAGE and BN-PAGE offer complementary approaches for protein separation with distinct advantages for different research objectives. SDS-PAGE remains the method of choice for high-resolution analytical separation based on molecular weight, while BN-PAGE excels in preserving native protein complexes and their functional interactions. NSDS-PAGE represents a promising intermediate approach, maintaining high resolution while preserving substantial functional characteristics, particularly bound metal ions. The integration of these electrophoretic techniques with advanced mass spectrometry methods, particularly through improved gel extraction and fractionation workflows like PEPPI-MS and cryo-slicing BN-MS, continues to expand our capability for comprehensive complex characterization. For researchers and drug development professionals, understanding these techniques' capabilities and limitations enables informed selection of the most appropriate methodology for specific protein analysis requirements in functional proteomics and structural biology.
Validation Through Western Blotting and Immunodetection After Native PAGE
In the study of functional protein properties, the choice of electrophoresis method is paramount. While Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) denatures proteins, separating them primarily by molecular weight, Native PAGE techniques, particularly Blue Native PAGE (BN-PAGE), preserve protein complexes in their functional state [16]. This capability makes Native PAGE indispensable for researching protein-protein interactions, oligomeric states, and enzymatic activity [25]. However, the true potential of this separation is only realized through effective immunodetection via Western blotting, a process that presents unique technical challenges compared to standard SDS-PAGE workflows. This guide objectively compares the performance of Western blotting after Native PAGE against traditional SDS-PAGE, providing researchers with the experimental data and protocols necessary to optimize functional protein studies in drug development and basic research.
The fundamental distinction lies in what each method preserves: SDS-PAGE provides high-resolution separation of denatured polypeptides by molecular weight, whereas BN-PAGE separates intact protein complexes by both size and charge under non-denaturing conditions [16]. This preservation of native structure comes with complexities in separation and transfer that must be addressed for successful immunodetection. As research increasingly focuses on protein complexes rather than individual subunits, mastering Western blotting after Native PAGE becomes essential for accurate functional analysis.
Technical Comparison: Western Blotting After Native PAGE vs. SDS-PAGE
Fundamental Methodological Differences
The processes of Western blotting after Native PAGE and SDS-PAGE differ significantly in their core principles and technical execution, each with distinct implications for the resulting data.
SDS-PAGE to Western Blot employs denaturing conditions throughout the workflow. Proteins are first denatured with SDS and reducing agents, then separated by molecular weight in a polyacrylamide gel [16]. The uniform negative charge imparted by SDS means migration distance depends almost entirely on polypeptide chain length. After separation, proteins are transferred to a membrane for immunodetection with antibodies. While this method offers excellent resolution and straightforward molecular weight determination, it destroys native protein structure, complexes, and function [3].
Native PAGE to Western Blot maintains proteins in their native state during separation. In BN-PAGE, the anionic dye Coomassie Blue G250 provides the necessary charge for electrophoretic migration instead of SDS, while mild non-ionic detergents like dodecylmaltoside or digitonin solubilize membrane proteins without disrupting complex integrity [5] [25]. This preservation allows separation of functional complexes, but introduces complexities in transfer and immunodetection, as larger complexes must migrate to the membrane and remain accessible to antibodies without disassembling.
The diagram below illustrates the key decision points for researchers selecting the appropriate method based on their experimental goals:
Performance Comparison: Quantitative Experimental Data
The choice between Native PAGE and SDS-PAGE significantly impacts experimental outcomes, particularly when studying metal-binding proteins, enzymatic activity, and protein complexes. The following table summarizes key performance differences based on experimental data:
| Performance Metric | SDS-PAGE/Western Blot | Native PAGE/Western Blot |
|---|---|---|
| Protein Function Retention | Enzymatic activity destroyed in all tested enzymes [3] | 7 of 9 model enzymes retained activity; all 9 active in BN-PAGE [3] |
| Metal Cofactor Retention | 26% Zn²⁺ retention in metalloproteins [3] | 98% Zn²⁺ retention with modified conditions [3] |
| Resolution Capability | High resolution of complex protein mixtures [3] | Lower resolution than BN-PAGE for some complexes [48] |
| Molecular Weight Determination | Direct determination possible [16] | Indirect estimation; depends on protein charge and size [48] |
| Detection of Supramolecular Structures | Cannot detect labile supramolecular assemblies [48] | Retains labile assemblies when combined with mild detergents like digitonin [48] |
| Sensitivity | Moderate (ng/mL range) [65] | Moderate (ng/mL range); potential interference from dye [65] |
| Quantitative Capability | Semi-quantitative [65] | Semi-quantitative [65] |
Beyond these metrics, Clear Native PAGE (CN-PAGE) offers particular advantages for specific applications. Research demonstrates that CN-PAGE can retain enzymatically active oligomeric states of mitochondrial ATP synthase that were not detected using BN-PAGE [48]. This makes CN-PAGE particularly valuable when Coomassie dye interferes with downstream techniques like catalytic activity measurements or fluorescence resonance energy transfer (FRET) analyses [48].
Advantages and Limitations for Research Applications
Each method offers distinct advantages and suffers from specific limitations that researchers must consider when designing experiments:
SDS-PAGE/Western Blot Advantages:
- High resolution separation of complex protein mixtures by molecular weight [3]
- Direct molecular weight determination possible [16]
- Standardized, widely-used protocol with extensive literature support [66]
- Excellent for immunodetection of individual protein subunits
SDS-PAGE/Western Blot Limitations:
- Complete destruction of protein function and enzymatic activity [3]
- Poor retention of metal cofactors in metalloproteins [3]
- Inability to study protein complexes or oligomeric states in their native form
- Cannot detect labile supramolecular assemblies [48]
Native PAGE/Western Blot Advantages:
- Preservation of enzymatic activity and protein function [3]
- Excellent retention of metal cofactors in metalloproteins [3]
- Ability to study protein-protein interactions and complex stoichiometry
- Detection of supramolecular structures and labile assemblies [48]
Native PAGE/Western Blot Limitations:
- More complex protocol with optimization required for different complexes [25]
- Potential interference from Coomassie dye in downstream applications [48]
- Indirect molecular weight estimation affected by protein intrinsic charge [48]
- Lower resolution for complex protein mixtures compared to SDS-PAGE [3]
Experimental Protocols and Methodologies
BN-PAGE and Western Blot Protocol for Protein Complex Analysis
This detailed protocol enables researchers to separate and immunodetection protein complexes in their native state, particularly suited for mitochondrial complexes and other multisubunit enzymes [5].
Stage 1: Sample Preparation
- Resuspend 0.4 mg of sedimented mitochondria in 40 μL of Buffer A (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris/HCl, pH 7.0) containing protease inhibitors (1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin) [5].
- Add 7.5 μL of 10% n-dodecyl-β-D-maltopyranoside (lauryl maltoside).
- Mix and incubate for 30 minutes on ice.
- Centrifuge at 72,000 × g for 30 minutes at 4°C (a bench-top microcentrifuge at 16,000 × g can be used but is not ideal).
- Collect supernatant and discard pellet.
- Add 2.5 μL of 5% Coomassie Blue G solution in 0.5 M aminocaproic acid to the supernatant.
Stage 2: Native Gel Electrophoresis (First Dimension)
- Prepare a linear gradient native gel (typically 6-13% acrylamide) using a gradient former:
- For 6% acrylamide: 7.6 mL 30% acrylamide, 9 mL dd water, 19 mL 1 M aminocaproic acid (pH 7.0), 1.9 mL 1 M Bis-Tris (pH 7.0), 200 μL 10% APS, 20 μL TEMED.
- For 13% acrylamide: 14 mL 30% acrylamide, 0.2 mL dd water, 16 mL 1 M aminocaproic acid (pH 7.0), 1.6 mL 1 M Bis-Tris (pH 7.0), 200 μL 10% APS, 20 μL TEMED [5].
- Pour gels and overlay with 50% isopropanol to ensure even polymerization.
- Once set, remove isopropanol and add stacking gel (0.7 mL 30% acrylamide, 1.6 mL dd water, 0.25 mL 1 M Bis-Tris pH 7.0, 2.5 mL 1 M aminocaproic acid pH 7.0, 40 μL 10% APS, 10 μL TEMED).
- Load samples (5-20 μL) into wells and run at 150V for approximately 2 hours using anode (50 mM Bis-Tris, pH 7.0) and cathode (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0) buffers until the dye front nearly reaches the bottom [5].
Stage 3: Second Dimension Electrophoresis (Optional)
- Cut each gel lane from the first dimension and soak in SDS denaturing buffer (10% glycerol, 2% SDS, 50 mM Tris pH 6.8, 0.002% Bromophenol blue, 50 mM DTT).
- Turn each lane 90° and load onto an SDS-PAGE gel (10-20% acrylamide).
- Perform standard SDS-PAGE for subunit analysis [5].
Stage 4: Electroblotting and Immunodetection
- Soak gel in transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol, 0.1% SDS) for 30 minutes.
- Transfer to PVDF membrane (recommended over nitrocellulose) at 150 mA for 1.5 hours using fully submerged system.
- Block membrane with PBS containing 5% non-fat milk powder.
- Proceed with standard immunodetection using primary and secondary antibodies [5].
The workflow below visualizes this comprehensive process from sample preparation to detection:
Critical Optimization Strategies for Native PAGE Western Blotting
Successful Western blotting after Native PAGE requires careful optimization at several critical points:
Detergent Selection and Optimization: The choice of detergent significantly impacts complex preservation. Different detergents yield different results:
- n-Dodecylmaltoside and Triton X-100 typically solubilize individual respiratory complexes [25].
- Digitonin preserves supramolecular structures (supercomplexes) that are disrupted by other detergents [25].
- Empirical testing of detergent-to-protein ratios is essential for optimal results [25].
Membrane Selection: PVDF membranes are strongly recommended over nitrocellulose for BN-PAGE Western blotting due to their superior protein binding capacity, physical strength, and chemical stability [5]. Nitrocellulose becomes brittle when dry and has lower protein retention, especially for smaller proteins [66].
Antibody Validation: Antibodies must be rigorously validated for Western blotting applications. Specificity should be confirmed using genetic controls (knockout validation), independent-epitope strategies, and multiple cell lines [67]. Batch-to-batch variation, particularly with polyclonal antibodies, can significantly impact reproducibility. Recombinant antibodies offer superior consistency between batches [67].
Transfer Conditions: Standard transfer conditions may require modification for native complexes. Larger complexes transfer less efficiently and may require extended transfer times or different buffer compositions. Complete transfer is indicated by the movement of the blue dye front to the membrane [5].
Essential Research Reagent Solutions
Successful Western blotting after Native PAGE requires specific reagents optimized for native protein work. The following table details essential materials and their functions:
| Reagent/Category | Specific Examples | Function in Native PAGE/Western Blot |
|---|---|---|
| Mild Detergents | n-Dodecylmaltoside, Digitonin, Triton X-100 | Solubilize membrane proteins while preserving complex integrity [25] |
| Specialty Dyes | Coomassie Blue G250 | Imparts negative charge for electrophoresis without denaturation [5] |
| Protease Inhibitors | PMSF, Leupeptin, Pepstatin | Prevent protein degradation during sample preparation [5] |
| Membranes | PVDF (Immobilon) | Superior protein binding for immunodetection of native complexes [5] |
| Validated Antibodies | Recombinant monoclonal antibodies | Ensure specificity and reduce batch-to-batch variability [67] |
| Electrophoresis Buffers | Aminocaproic acid, Bis-Tris, Tricine | Maintain native pH conditions and complex stability [5] |
Western blotting after Native PAGE provides unparalleled capability for studying functional protein properties in their native state, complementing the high-resolution separation of denatured proteins offered by SDS-PAGE. The experimental data presented demonstrates that BN-PAGE retains 98% of metal cofactors in metalloproteins and preserves enzymatic activity in 7 of 9 tested enzymes, compared to complete functional destruction in SDS-PAGE [3]. This capability comes with increased methodological complexity, requiring optimization of detergent conditions, transfer parameters, and antibody validation.
For researchers studying protein-protein interactions, oligomeric states, enzymatic function, or metal cofactor binding, Native PAGE with Western blotting offers essential insights unobtainable through denaturing methods. The protocols and optimization strategies provided here enable systematic implementation of this powerful technique. When paired with SDS-PAGE analysis of subunit composition, Native PAGE Western blotting provides a comprehensive approach to protein characterization that bridges the gap between structural information and functional understanding, ultimately advancing drug development and basic biological research.
The mitochondrial oxidative phosphorylation (OXPHOS) system is a fundamental component of cellular energy conversion, comprising five multi-subunit complexes (CI-CV) embedded in the inner mitochondrial membrane [68] [69]. These complexes work in concert to conduct cellular respiration and generate the bulk of cellular ATP. The proper biogenesis and assembly of these complexes is a sophisticated process requiring the coordinated expression of genes from both nuclear and mitochondrial DNA, followed by the precise incorporation of protein subunits and essential cofactors [68] [70]. Defects in this process represent a major cause of mitochondrial disorders, which are among the most frequent inborn errors of metabolism with an estimated prevalence of up to 1 in 5,000 live births [68] [69].
This case study focuses on the pivotal role of electrophoretic techniques, particularly Blue-Native PAGE (BN-PAGE), in diagnosing and researching these disorders. When the structural integrity of OXPHOS complexes is compromised by mutations in structural subunits or assembly factors, it disrupts the entire energy production system, leading to a spectrum of severe clinical manifestations, often affecting tissues with high energy demands like brain, muscle, and heart [68] [70]. Consequently, methods that can directly probe the assembly status and native functionality of these complexes are indispensable tools in both clinical and research settings.
Analytical Challenge: Preserving Functional Protein Properties in Electrophoresis
Traditional protein analysis often relies on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), a denaturing method that provides high-resolution separation based on polypeptide molecular mass. However, for studying OXPHOS complexes, this method has a critical limitation: it destroys native protein structures and functional properties. The SDS detergent denatures proteins, dissociates non-covalently bound subunits, and strips away essential cofactors such as metal ions, thereby obliterating enzymatic activity and disrupting protein-protein interactions within multi-subunit complexes [3]. This makes SDS-PAGE unsuitable for assessing whether a mutation affects the proper assembly of a holocomplex or for detecting the presence of enzymatically active enzymes.
The challenge, therefore, is to employ electrophoretic techniques that preserve the native state of proteins. This allows researchers to move beyond merely detecting the presence of individual protein subunits and toward evaluating the integrity and function of the entire macromolecular machine. As we will demonstrate, the choice between denaturing and native electrophoresis is not trivial; it fundamentally determines the type of biological questions one can answer, especially in the context of complex assembly defects in mitochondrial disorders.
Methodological Comparison: SDS-PAGE vs. BN-PAGE
To address the limitations of SDS-PAGE for native protein analysis, researchers developed Blue-Native PAGE (BN-PAGE). The table below provides a direct comparison of these two core techniques, highlighting their fundamental differences in application and outcome for the study of OXPHOS complexes.
Table 1: Core Methodological Comparison: SDS-PAGE vs. BN-PAGE
| Feature | SDS-PAGE (Denaturing) | BN-PAGE (Native) |
|---|---|---|
| Principle | Separation based on polypeptide molecular mass. | Separation based on native mass and shape of protein complexes. |
| Key Reagent | Ionic detergent SDS for uniform charge and denaturation. | Mild detergents (e.g., DDM, digitonin) and Coomassie G-250 dye for charge shift. |
| Protein State | Denatured; subunits dissociated. | Native; complexes remain intact and functional. |
| Functional Retention | Destroys enzymatic activity and cofactors. | Retains enzymatic activity and bound metal ions. |
| Key Application | Analyzing subunit composition, purity, and molecular weight. | Analyzing complex assembly, stability, supercomplex formation, and in-gel activity. |
| Resolution of OXPHOS | Separates individual subunits; complexes are not intact. | Resolves intact individual complexes (CI-CV) and supercomplexes. |
BN-PAGE operates on a fundamentally different principle. Instead of a denaturing ionic detergent, it uses mild non-ionic detergents like n-dodecyl-β-D-maltoside (DDM) or digitonin to gently solubilize membrane proteins without dissociating them from their native partners [25] [11]. The anionic dye Coomassie Blue G-250 is then added, which binds to the hydrophobic surfaces of the proteins, imparting a negative charge that allows them to migrate toward the anode during electrophoresis. This dye also helps to keep the solubilized complexes in solution, preventing aggregation [28] [25]. This elegant combination allows for the high-resolution separation of intact, enzymatically active protein complexes according to their native molecular mass.
The following diagram illustrates the fundamental workflow differences between these two techniques and the distinct information they yield regarding OXPHOS complexes.
A key advantage of using the milder detergent digitonin is its ability to preserve even higher-order structures known as respiratory supercomplexes (or respirasomes), which are stoichiometric associations of CI, CIII, and CIV [27] [25]. The ability to detect these supercomplexes is crucial, as their improper formation can be a pathological mechanism in mitochondrial diseases, even when individual complexes appear normal [27].
Experimental Data: Quantitative Comparison of Technique Performance
The theoretical advantages of native electrophoresis are borne out by robust experimental data. The following tables summarize key quantitative findings from the literature, demonstrating the superior performance of native techniques for preserving functional protein properties.
Table 2: Metal Retention and Enzymatic Activity After Electrophoresis
| Analysis Type | SDS-PAGE Performance | BN-PAGE / NSDS-PAGE Performance |
|---|---|---|
| Zinc (Zn²⁺) Retention | 26% retention in proteomic samples [3]. | 98% retention with Native SDS-PAGE [3]. |
| Enzymatic Activity | All nine model enzymes, including four Zn²⁺ proteins, were denatured and inactive [3]. | Seven of nine model enzymes retained activity after NSDS-PAGE; all nine were active after BN-PAGE [3]. |
| In-Gel Activity Assays | Not possible due to denaturation. | Possible for Complexes I, II, IV, and V, allowing direct functional assessment [28]. |
Table 3: Resolution of OXPHOS Complexes and Supercomplexes
| Electrophoresis Method | Detergent Used | Complexes Resolved | Supercomplexes Resolved |
|---|---|---|---|
| SDS-PAGE | SDS | No intact complexes; only denatured subunits. | None. |
| BN-PAGE | n-Dodecyl-β-D-maltoside (DDM) | Individual, intact complexes CI-CV [28] [71]. | Limited or none. |
| BN-PAGE | Digitonin | Individual, intact complexes CI-CV. | Yes (e.g., I+III₂+IV, I+III₂+IV₂) [27] [25]. |
The data in Table 2 clearly show that BN-PAGE and its close relative, Native SDS-PAGE (a modified version of SDS-PAGE with reduced detergent and no heating), are vastly superior for preserving metal cofactors and enzymatic function. This functional retention is the cornerstone for performing in-gel activity assays, a direct method to determine if a correctly assembled complex is also catalytically competent [28].
Furthermore, as indicated in Table 3, the choice of detergent during sample preparation for BN-PAGE is critical. While DDM effectively solubilizes individual OXPHOS complexes, the even milder detergent digitonin is often necessary to preserve the fragile supercomplex assemblies. This highlights a key strategic consideration for experimental design based on the research question [27] [25].
Detailed Experimental Protocols
BN-PAGE for OXPHOS Complex Analysis
The following is a consolidated protocol for analyzing OXPHOS complexes via BN-PAGE, adapted from established methodologies [27] [28].
- Step 1: Mitochondria Isolation. Tissues (e.g., ~30 mg liver) or cultured cells are homogenized in cold isolation buffer (e.g., 250 mM sucrose, 10 mM HEPES, 1 mM EGTA, pH 7.4). The homogenate is centrifuged at low speed (600 × g) to remove cell debris and nuclei, followed by a high-speed centrifugation (7,000 - 12,000 × g) to pellet the mitochondrial fraction [27].
- Step 2: Membrane Solubilization. The mitochondrial pellet is resuspended and solubilized using a mild non-ionic detergent. For individual complex analysis, n-dodecyl-β-D-maltoside (DDM) is used at a detergent-to-protein ratio of 2-4 g/g. For supercomplex analysis, digitonin is used at a higher ratio of 4-8 g/g. Solubilization is performed in the presence of a zwitterionic salt like 1-2 mM 6-aminocaproic acid to support the process. The mixture is incubated on ice for 10-30 minutes and then centrifuged at high speed (20,000 × g) to remove insoluble material [28] [25].
- Step 3: Sample Preparation and Gel Electrophoresis. Coomassie Blue G-250 dye is added to the supernatant (at a 1:4 dye-to-detergent ratio). The sample is then loaded onto a native polyacrylamide gel (typically a 3-12% or 4-16% gradient mini-gel for optimal resolution). Cathode buffer (with Coomassie dye) and anode buffer (without dye) are used. Electrophoresis is performed at a constant voltage (e.g., 100V for stacking, 150-200V for resolving) at 4°C until the dye front migrates to the bottom of the gel [28].
- Step 4: Downstream Applications.
- In-Gel Activity (IGA) Staining: The gel is incubated in specific substrate solutions to visualize active complexes. For example, Complex I activity is detected by NADH dehydrogenase activity using nitrotetrazolium blue (NBT), and Complex IV activity is detected by cytochrome c oxidase staining [28] [71].
- Western Blotting: Proteins are transferred to a membrane for immunodetection with antibodies against specific OXPHOS subunits to assess assembly.
- Two-Dimensional BN/SDS-PAGE: A lane from the BN-PAGE gel is excised, incubated in SDS buffer, and placed horizontally on top of an SDS-PAGE gel for a second dimension of separation, revealing the subunit composition of each native complex [6] [28].
A Modified Approach: Native SDS-PAGE (NSDS-PAGE)
A relevant methodological innovation is Native SDS-PAGE (NSDS-PAGE), which modifies standard SDS-PAGE conditions to better preserve native properties. The key modifications are [3]:
- Sample Buffer: Removal of SDS and EDTA, and omission of the heating step.
- Running Buffer: Reduction of SDS concentration from 0.1% to 0.0375% and deletion of EDTA. This method offers a compromise, providing high-resolution separation closer to that of SDS-PAGE while significantly improving the retention of metal ions and enzymatic activity compared to fully denaturing conditions [3].
The Scientist's Toolkit: Essential Reagents for BN-PAGE
Success in native electrophoresis depends critically on the choice of reagents. The following table details the essential materials and their specific functions in the analysis of OXPHOS complexes.
Table 4: Research Reagent Solutions for BN-PAGE Analysis of OXPHOS Complexes
| Reagent / Kit | Function / Role in Experiment |
|---|---|
| n-Dodecyl-β-D-maltoside (DDM) | Mild, non-ionic detergent for solubilizing individual OXPHOS complexes while preserving their integrity [25] [11]. |
| Digitonin | Mild, non-ionic detergent used for solubilizing mitochondrial membranes while preserving respiratory supercomplexes [27] [25]. |
| Coomassie Blue G-250 | Anionic dye that binds to protein complexes, providing a negative charge for electrophoresis and preventing aggregation [28] [11]. |
| 6-Aminocaproic Acid | Zwitterionic salt used in extraction buffers to support solubilization without interfering with electrophoresis [28] [11]. |
| Acrylamide Bis-Tris Gels | Gel matrix for native electrophoresis; Bis-Tris system at pH ~7.0 helps maintain protein stability [3] [28]. |
| Nitrotetrazolium Blue (NBT) | Substrate used in in-gel activity staining to detect NADH dehydrogenase activity of Complex I [28] [71]. |
| Cytochrome c (reduced) | Substrate used in in-gel activity staining to detect cytochrome c oxidase activity of Complex IV [28]. |
This case study underscores a critical principle in protein biochemistry: the analytical method must be matched to the biological question. For diagnosing and researching mitochondrial disorders characterized by OXPHOS complex deficiencies, BN-PAGE is an indispensable tool. Its unparalleled ability to separate intact, functional protein complexes and supercomplexes provides direct insight into the assembly and catalytic competence of the mitochondrial respiratory system—information that is completely lost in denaturing SDS-PAGE.
The quantitative data clearly demonstrates BN-PAGE's superiority in preserving metal cofactors and enzymatic activity. When combined with strategic detergent selection for solubilization and followed by powerful downstream applications like in-gel activity assays or 2D electrophoresis, BN-PAGE offers a comprehensive platform for unraveling the molecular pathology of mitochondrial diseases. It enables researchers to distinguish between defects in protein stability, complex assembly, and catalytic function, thereby guiding the development of targeted diagnostic and therapeutic strategies. For any investigation into the functional integrity of the OXPHOS system, BN-PAGE remains the electrophoretic technique of choice.
Conclusion
The choice between SDS-PAGE and BN-PAGE represents a fundamental trade-off between resolution and functional preservation. While SDS-PAGE remains invaluable for determining molecular weight and protein expression, BN-PAGE and its variants provide unique insights into native protein complexes, enzymatic activities, and metal cofactor retention that are essential for understanding biological function. The development of hybrid techniques like NSDS-PAGE and CN-PAGE offers promising middle ground for specific applications. For biomedical and clinical research, particularly in studying mitochondrial disorders, metabolic diseases, and protein interaction networks, BN-PAGE enables investigation of complex assembly pathways and pathological mechanisms that would be invisible to denaturing methods. Future directions include improved sensitivity for detecting low-abundance complexes, standardized protocols for clinical diagnostics, and enhanced integration with structural mass spectrometry techniques for comprehensive structural proteomics.
References
- 1. SDS-PAGE guide: Sample preparation to analysis [https://www.abcam.com/en-us/knowledge-center/western-blot/sds-page-guide]
- 2. Comparing SDS-PAGE and CE-SDS for Antibody Purity ... [https://www.bioprocessintl.com/qa-qc/comparing-sds-page-and-ce-sds-for-antibody-purity-analysis]
- 3. Native SDS-PAGE: High Resolution Electrophoretic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4517606/]
- 4. 05SAR-PAGE: Separation of protein dimerization and ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267019314497]
- 5. Blue native electrophoresis protocol [https://www.abcam.com/en-us/technical-resources/protocols/blue-native-electrophoresis]
- 6. Two-dimensional Blue Native/SDS Gel Electrophoresis of ... [https://www.sciencedirect.com/science/article/pii/S1535947620334228]
- 7. Mass Estimation of Native Proteins by Blue ... [https://www.sciencedirect.com/science/article/pii/S1535947620345199]
- 8. In-depth structural proteomics integrating mass ... [https://www.frontiersin.org/journals/analytical-science/articles/10.3389/frans.2022.1107183/full]
- 9. : Unlocking Sds Isolation Techniques | MedShun Protein [https://medshun.com/article/what-is-the-function-of-sds-during-protein-isolation]
- 10. Validation of blue- and clear-native polyacrylamide gel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445495/]
- 11. Blue Native electrophoresis to study mitochondrial and ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202302000385]
- 12. Resolving mitochondrial protein complexes using non- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2795571/]
- 13. Solubilization of membrane protein complexes for blue ... [https://www.sciencedirect.com/science/article/abs/pii/S1874391908000729]
- 14. Qualitative and quantitative evaluation of thylakoid complexes ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-022-00858-2]
- 15. Video: Blue Native Polyacrylamide Gel Electrophoresis BN - PAGE for... [https://www.jove.com/v/2164/blue-native-polyacrylamide-gel-electrophoresis-bn-page-for-analysis]
- 16. Differences between SDS PAGE and Native PAGE [https://www.geeksforgeeks.org/biology/differences-between-sds-page-and-native-page/]
- 17. SDS PAGE vs Native PAGE [https://byjus.com/biology/difference-between-sds-page-and-native-page/]
- 18. Principles of Blue Native-PAGE [https://blog.benchsci.com/principles-of-blue-native-page]
- 19. Comparison of the performance of 1D SDS-PAGE with ... [https://pubmed.ncbi.nlm.nih.gov/30599360/]
- 20. Different molecular bases underlie the mitochondrial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3385843/]
- 21. Multimer-PAGE: A Method for Capturing and Resolving ... [https://www.jove.com/t/55341/multimer-page-method-for-capturing-resolving-protein-complexes]
- 22. Rapid baseline hump-free analysis of therapeutic proteins ... [https://www.sciencedirect.com/science/article/pii/S0003267025005410]
- 23. Effect of the operational parameters on the electromigration ... [https://www.sciencedirect.com/science/article/pii/S0039914025000876]
- 24. Native SDS-PAGE: high resolution electrophoretic ... [https://pubs.rsc.org/en/content/articlelanding/2014/mt/c4mt00033a]
- 25. Blue-native PAGE in plants: a tool in analysis of protein ... [https://plantmethods.biomedcentral.com/articles/10.1186/1746-4811-1-11]
- 26. Advantages and Disadvantages of 2D Blue Native/SDS- ... [https://www.mtoz-biolabs.com/advantages-and-sisadvantages-of-2d-blue-native-sds-page-protein-complex-analysis.html]
- 27. Analysis of mitochondrial respiratory chain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4823378/]
- 28. and clear-native polyacrylamide gel electrophoresis protocols ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0332065]
- 29. Two dimensional blue native/SDS-PAGE to identify ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2015.00098/full]
- 30. High-resolution native electrophoresis in-gel activity assay ... [https://www.nature.com/articles/s41598-025-24684-3]
- 31. Detergents and their role in successful SDS-PAGE ... [https://info.gbiosciences.com/blog/detergents-and-their-role-in-successful-sds-page-electrophoresis]
- 32. The use of blue native PAGE in the evaluation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2648668/]
- 33. Protocol for the Analysis of Yeast and Human ... [https://www.sciencedirect.com/science/article/pii/S2666166720300769]
- 34. Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3339838/]
- 35. Analysis of Mitochondrial Respiratory Chain Complexes in ... [https://www.jove.com/t/59269/analysis-mitochondrial-respiratory-chain-complexes-cultured-human]
- 36. Preserved respiratory chain capacity and physiology in ... [https://www.sciencedirect.com/science/article/pii/S1550413123002899]
- 37. Two-dimensional Blue native/SDS gel electrophoresis of ... [https://pubmed.ncbi.nlm.nih.gov/14665681/]
- 38. Two-Dimensional Blue Native/SDS Polyacrylamide Gel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9611221/]
- 39. Proteomic Investigation of Cape Cobra (Naja nivea ... [https://www.mdpi.com/2072-6651/16/2/63?]
- 40. Comparative compositional and functional analyses of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0222206]
- 41. Validation of blue- and clear-native polyacrylamide gel ... [https://pubmed.ncbi.nlm.nih.gov/40966204/]
- 42. Blue-native PAGE in plants: a tool in analysis of protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1308860/]
- 43. Deciphering metabolic networks by blue native ... [https://www.sciencedirect.com/science/article/pii/S221296851500015X]
- 44. Mass Estimation of Native Proteins by Blue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2953912/]
- 45. Bench Tips: Blue Native-PAGE [https://blog.benchsci.com/bench-tips-blue-native-page]
- 46. Complexome Profiling—Exploring Mitochondrial Protein ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.796128/full]
- 47. Nongradient blue native gel analysis of serum proteins and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3034814/]
- 48. Advantages and limitations of clear-native PAGE [https://pubmed.ncbi.nlm.nih.gov/16220535/]
- 49. Overview of Protein Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 50. Gel electrophoresis of proteins - Wikipedia [https://en.wikipedia.org/wiki/Gel_electrophoresis_of_proteins]
- 51. [PDF] Native SDS-PAGE: high resolution electrophoretic separation of proteins with retention of native properties including bound metal ions. | Semantic Scholar [https://www.semanticscholar.org/paper/Native-SDS-PAGE:-high-resolution-electrophoretic-of-Nowakowski-Wobig/31919f2dee6d0a1da25134cbeeeca6f05060110a]
- 52. Detergents and alternatives in cryo-EM studies of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9828306/]
- 53. An Overview of the Top Ten Detergents Used for ... [https://www.mdpi.com/2073-4352/7/7/197]
- 54. Quality Control in Eukaryotic Membrane Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4271737/]
- 55. A New Mass Spectrometry-compatible Degradable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4384424/]
- 56. Blue Native PAGE–Antibody Shift in Conjunction with Mass ... [https://www.mdpi.com/1422-0067/24/8/7632]
- 57. SDS protein interactions [https://www.sciencedirect.com/science/article/abs/pii/S0301462225001462]
- 58. How Is Native PAGE Different from SDS-PAGE? [https://synapse.patsnap.com/article/how-is-native-page-different-from-sds-page]
- 59. High Resolution Clear Native Electrophoresis for In-gel ... [https://www.sciencedirect.com/science/article/pii/S153594762031690X]
- 60. high resolution electrophoretic separation of proteins with ... [https://www.semanticscholar.org/paper/Native-SDS-PAGE%3A-high-resolution-electrophoretic-of-Nowakowski-Wobig/31919f2dee6d0a1da25134cbeeeca6f05060110a]
- 61. Quantifying enzyme activity in living cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5612114/]
- 62. What's the Difference Between SDS-PAGE and Native ... [https://synapse.patsnap.com/article/whatE28099s-the-difference-between-sds-page-and-native-page]
- 63. Cryo-slicing Blue Native-Mass Spectrometry (csBN-MS), a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4739680/]
- 64. How to analyze protein complexes by 2D blue native SDS- ... [https://pubmed.ncbi.nlm.nih.gov/17893852/]
- 65. ELISA vs. Western Blot: Choosing the Best Immunoassay ... [https://www.metwarebio.com/elisa-vs-western-blot-comparison/]
- 66. Western blotting [https://www.sciencedirect.com/science/article/pii/S1046202306000065?via%3Dihub]
- 67. Antibody validation for Western blot: By the user, for the user [https://pmc.ncbi.nlm.nih.gov/articles/PMC6983856/]
- 68. Human diseases associated with defects in assembly of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6056716/]
- 69. Mitochondrial disorders of the OXPHOS system [https://pubmed.ncbi.nlm.nih.gov/33159691/]
- 70. Mitochondrial Diseases Part I: Mouse models of OXPHOS ... [https://www.sciencedirect.com/science/article/pii/S1567724915000203]
- 71. Resolving mitochondrial protein complexes using ... [https://www.sciencedirect.com/science/article/abs/pii/S000326970900219X]