SDS-PAGE Running Buffer: A Complete Guide to Composition, Preparation, and Troubleshooting
This comprehensive guide details the critical role of SDS-PAGE running buffer in successful protein electrophoresis.
SDS-PAGE Running Buffer: A Complete Guide to Composition, Preparation, and Troubleshooting
Abstract
This comprehensive guide details the critical role of SDS-PAGE running buffer in successful protein electrophoresis. Tailored for researchers and drug development professionals, it covers the foundational principles of Tris-Glycine-SDS buffer systems, provides step-by-step preparation protocols, and offers advanced troubleshooting for common issues like smeared bands and poor resolution. The article further explores methodological adaptations for specific applications and comparative analyses with alternative buffer systems, serving as an essential resource for ensuring accuracy and reproducibility in biomedical protein analysis.
The Science Behind SDS-PAGE Running Buffer: Principles and Components
The Role of Running Buffer in Protein Separation and Electrophoresis
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) is a foundational technique in biochemical research and drug development, enabling the separation of proteins based on their molecular weight. The efficacy of this technique is heavily dependent on the precise composition and preparation of the running buffer. This critical component not only facilitates the transport of proteins through the gel matrix but also maintains the denatured state of proteins and ensures a stable pH environment throughout the electrophoretic process. The running buffer, typically comprising Tris, glycine, and SDS, creates the ionic environment necessary for the discontinuous buffer system that underpins high-resolution protein separation [1] [2]. For researchers in protein chemistry and biotechnology, mastering the preparation and function of running buffer is essential for generating reproducible, reliable data in analytical and preparative applications.
Chemical Composition and Function of Running Buffer
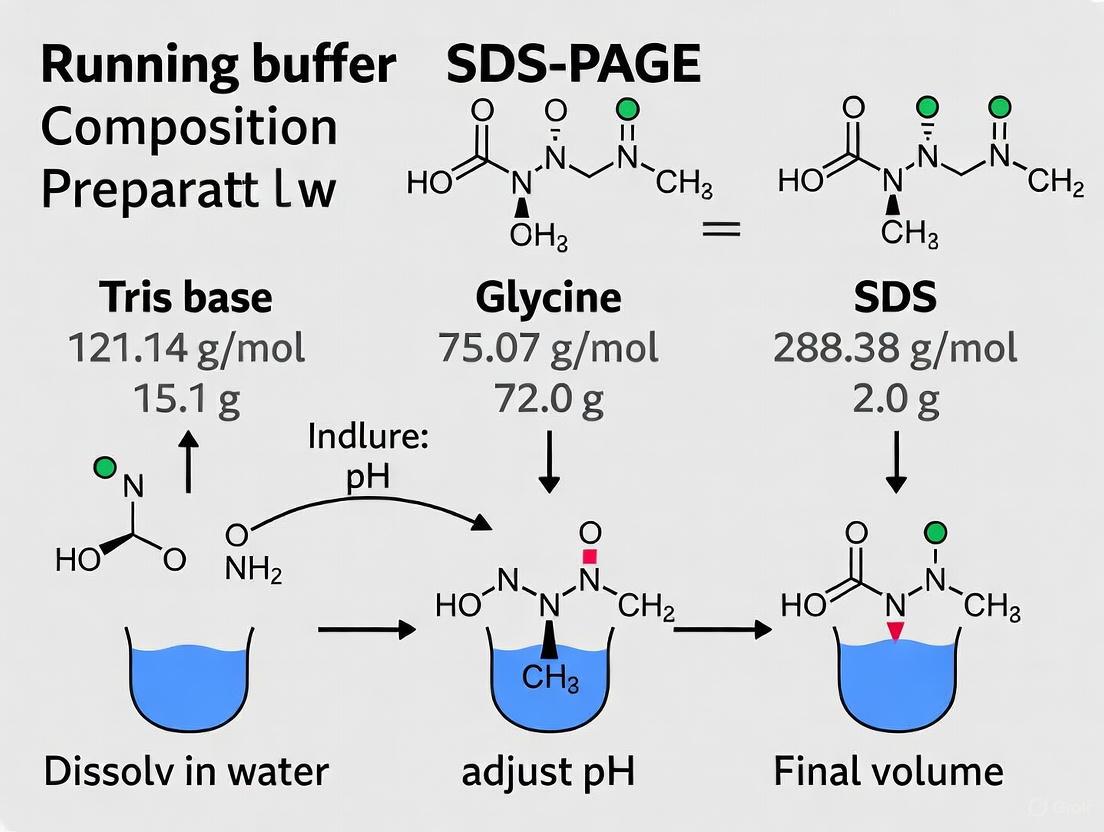
The standard SDS-PAGE running buffer is a ternary system whose components work in concert to create optimal separation conditions. Table 1 details the standard 10X concentrated recipe and the final working concentration of the 1X buffer.
Table 1: Standard 10X SDS-PAGE Running Buffer Recipe and Final Working Concentrations
| Component | Amount for 10X Buffer (per Liter) | Final 1X Concentration | Molecular Weight | Primary Function |
|---|---|---|---|---|
| Tris base | 30.285 g [3] | 25 mM [3] | 121.14 g/mol [3] | Maintains pH 8.3; charge carrier |
| Glycine | 144.4 g [3] | 192 mM [3] | 75.07 g/mol [3] | Leading ion in stacking gel; charge carrier |
| SDS | 10 g [3] | 0.1% (w/v) [3] | 288.38 g/mol [3] | Maintains protein denaturation and negative charge |
Each component in the running buffer fulfills a specific and critical role:
- Tris (tris(hydroxymethyl)aminomethane): This pH-buffering agent maintains the running buffer at pH 8.3. Its pKa of 8.1 makes it ideal for buffering in the slightly basic range required for SDS-PAGE [1]. It provides chloride ions (from Tris-HCl) that act as highly mobile leading ions in the stacking phase of electrophoresis [2].
- Glycine: An amino acid whose charge state is pH-dependent, glycine serves as the trailing ion in the discontinuous buffer system. At the pH 8.3 of the running buffer, glycine exists predominantly as a glycinate anion, carrying a negative charge [1]. Its ability to change charge states at the interface between the stacking and resolving gels is fundamental to the stacking phenomenon.
- SDS (Sodium Dodecyl Sulfate): This anionic detergent is included in the running buffer (typically at 0.1%) to ensure that proteins remain denatured and uniformly coated with a negative charge throughout their migration through the gel [1]. This sustains the primary principle of SDS-PAGE—that separation is based solely on molecular size rather than intrinsic charge.
The following diagram illustrates the coordinated mechanism of the running buffer's discontinuous system:
Diagram 1: Mechanism of the Discontinuous Buffer System in SDS-PAGE
As illustrated in Diagram 1, the running buffer's key mechanistic role is to create a discontinuous system that concentrates protein samples into sharp bands before they enter the resolving gel. When current is applied, glycinate ions from the running buffer (pH 8.3) enter the stacking gel (pH 6.8). At this lower pH, glycine loses charge and becomes predominantly a zwitterion, significantly reducing its electrophoretic mobility [1] [2]. Meanwhile, chloride ions from the Tris-HCl in the gel move rapidly toward the anode. This creates a narrow zone of high voltage gradient between the fast-moving chloride front (leading ions) and the slow-moving glycine zwitterions (trailing ions). Protein molecules, with mobilities intermediate between these two fronts, are compressed or "stacked" into extremely thin, sharp bands [2].
When this ion front reaches the resolving gel (pH 8.8), the environment changes dramatically. The higher pH causes the glycine zwitterions to regain their negative charge, transforming into fast-moving glycinate anions that overtake the proteins [1] [2]. The proteins, now deposited as sharp bands at the top of the resolving gel, are subjected to a uniform electric field and begin to separate based on their molecular size as they migrate through the sieving matrix of the resolving gel. This sophisticated mechanism, entirely dependent on the specific composition of the running buffer, is what enables the high-resolution separation that makes SDS-PAGE so powerful.
Detailed Experimental Protocols
Preparation of 10X Running Buffer
Principle: Preparing a 10X stock solution of running buffer ensures consistency across multiple experiments and saves preparation time. The correct molar ratios of Tris, glycine, and SDS are critical for maintaining proper ionic strength and buffer capacity during electrophoresis.
Materials:
- Tris base (Molecular Biology Grade)
- Glycine (Electrophoresis Grade)
- SDS (Sodium Dodecyl Sulfate, Electrophoresis Grade)
- Deionized water
- pH meter
- Magnetic stirrer and stir bar
- 1L graduated cylinder or volumetric flask
- Storage bottle
Procedure:
- Measure Components: Accurately weigh 30.285 g of Tris base, 144.4 g of glycine, and 10 g of SDS [3].
- Initial Dissolution: Add approximately 800 mL of deionized water to a 1L beaker or volumetric flask. Begin stirring with a magnetic stirrer.
- Sequential Addition: While stirring, add the Tris base to the water and allow it to dissolve completely. Next, add the glycine and stir until fully dissolved. Finally, carefully add the SDS powder, taking care to minimize dust formation.
- Final Volume: Once all components are completely dissolved, add deionized water to bring the final volume to 1L [3].
- pH Verification: Check the pH of the solution. The pH should be approximately 8.3 and typically requires no adjustment [4].
- Storage: Store the 10X running buffer at room temperature. For 1X working solution, dilute 100 mL of 10X stock with 900 mL deionized water before use [5].
Technical Notes:
- SDS can form suds when agitated vigorously; gentle stirring is sufficient for dissolution.
- If precipitate forms during storage, warm the solution to 37°C with mixing to redissolve.
- For optimal performance, the 1X working solution should be used fresh, though it can be stored at 4°C for up to one week [6].
SDS-PAGE Electrophoresis Procedure
Principle: SDS-PAGE separates protein mixtures based on molecular weight under denaturing conditions. The running buffer provides the ionic environment necessary for protein migration and maintenance of denaturation throughout the process.
Materials:
- Cast polyacrylamide gel (stacking and resolving layers)
- Prepared 1X SDS-PAGE running buffer
- Protein samples prepared in Laemmli buffer
- Pre-stained or unstained protein molecular weight markers
- Electrophoresis chamber and power supply
- Micropipettes and gel-loading tips
- Heating block (95°C)
- Microcentrifuge
Procedure:
- Gel Assembly: Place the polymerized gel into the electrophoresis chamber according to the manufacturer's instructions. If running a single gel, use a dummy cassette to balance the chamber [4].
- Buffer Addition: Fill the inner and outer chambers of the electrophoresis unit with 1X running buffer until the wells are completely submerged [5].
- Sample Preparation:
- For purified proteins: Mix 2.5 µL of 3X Laemmli buffer with 7.5 µL of protein sample [4].
- For cell lysates: Mix sample with an equal volume of 2X Laemmli buffer containing β-mercaptoethanol (e.g., 1 µL BME per 25 µL sample) [5].
- Heat samples at 95°C for 5 minutes to ensure complete denaturation [5] [4].
- Centrifuge heated samples at maximum speed (13,000 x g) for 3-5 minutes to pellet any insoluble debris [5] [4].
- Sample Loading: Using gel-loading tips, carefully load 5-35 µL of sample supernatant into the wells, avoiding disturbance of the pelleted debris. Include molecular weight markers in at least one well [5].
- Electrophoresis:
- Connect the power supply, ensuring the correct polarity (proteins migrate toward the anode).
- Run the gel at constant voltage: 80V until the dye front moves through the stacking gel and enters the resolving gel, then increase to 150-180V for the remainder of the run [5] [4].
- Continue electrophoresis until the bromophenol blue dye front reaches the bottom of the gel (typically 45-90 minutes) [5].
- Post-Electrophoresis Processing: Turn off the power supply, disconnect the electrodes, and carefully remove the gel from the cassette for subsequent staining or Western blotting.
Troubleshooting Tips:
- If protein bands appear smeared, ensure SDS is present in both the running buffer and sample buffer.
- If the dye front appears uneven, check for buffer leakage or improper gel seating in the chamber.
- If migration is unusually slow, verify that the running buffer was correctly diluted from the 10X stock.
Essential Research Reagent Solutions
Successful SDS-PAGE requires precisely formulated reagents beyond just the running buffer. Table 2 catalogues the essential components for protein electrophoresis, their specific functions, and standard formulations.
Table 2: Essential Reagent Solutions for SDS-PAGE Experiments
| Reagent | Composition | Function in SDS-PAGE |
|---|---|---|
| 10X Running Buffer | 25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3 [3] | Creates ionic environment for electrophoresis; maintains protein denaturation |
| Laemmli Sample Buffer | Tris-HCl (pH 6.8), SDS, glycerol, bromophenol blue, β-mercaptoethanol [1] [2] | Denatures proteins, adds visual tracking, provides density for well loading |
| 4X Resolving Gel Buffer | 1.5 M Tris-HCl, pH 8.8 [4] | Buffers resolving gel at high pH for optimal separation |
| 4X Stacking Gel Buffer | 0.5 M Tris-HCl, pH 6.8 [4] | Buffers stacking gel at lower pH for protein stacking |
| 30% Acrylamide/Bis Solution | 29:1 ratio acrylamide to bisacrylamide [4] | Forms the polyacrylamide gel matrix for size-based separation |
| Catalyst System | 10% ammonium persulfate (APS) and TEMED [1] [4] | Initiates and catalyzes acrylamide polymerization |
Troubleshooting and Optimization
Even with proper running buffer preparation, researchers may encounter challenges during SDS-PAGE. Common issues and their solutions include:
Precipitation in Running Buffer: If white precipitate forms in the running buffer, it may indicate SDS precipitation, often caused by the presence of potassium chloride (KCl) from samples. KCl causes SDS to precipitate as potassium dodecyl sulfate [2]. To prevent this, avoid high salt concentrations in samples or ensure all lanes contain similar salt concentrations to maintain even running conditions.
Optimizing Gel Concentration: The acrylamide percentage directly impacts resolution of different molecular weight ranges. Table 3 provides guidance for gel concentration selection based on target protein size.
Table 3: Optimization of Gel Percentage for Target Protein Sizes
| Protein Molecular Weight Range | Recommended Gel Concentration |
|---|---|
| 100-600 kDa | 4% [7] |
| 50-500 kDa | 7% [7] |
| 30-300 kDa | 10% [7] |
| 10-200 kDa | 12% [7] |
| 3-100 kDa | 15% [7] |
Artifactual Banding: If bands appear distorted or streaked, verify that the running buffer pH is correct (8.3) and that the buffer is not excessively reused. Running buffer becomes contaminated with protein fragments and chloride ions after each run, which can interfere with subsequent separations [6]. For optimal results, prepare fresh running buffer for each experiment.
Anomalous Migration: Some proteins may migrate at positions inconsistent with their known molecular weight. This can occur with heavily glycosylated proteins, membrane proteins, or proteins with unusual amino acid compositions that bind SDS differently [1] [2]. Including appropriate controls and using Western blotting for specific identification can help address this limitation.
The running buffer is far more than a simple conductive medium in SDS-PAGE; it is an active component of the sophisticated discontinuous buffer system that enables high-resolution protein separation. The precise coordination between Tris, glycine, and SDS at specific pH values creates the conditions necessary for both the initial stacking of proteins and their subsequent separation by molecular size in the resolving gel. For research and drug development professionals, a thorough understanding of running buffer composition, preparation, and function is fundamental to generating reliable, reproducible protein analysis data. By adhering to the detailed protocols and troubleshooting guidelines outlined in this application note, researchers can optimize their electrophoretic separations and avoid common pitfalls that compromise data quality.
The Laemmli buffer system, named after its formulator U.K. Laemmli, has been the foundational method for discontinuous sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) since 1970 [2]. This system enables the separation of denatured proteins based primarily on their molecular weight, forming a critical step in protein analysis for countless biochemical and biomedical research applications [8]. The running buffer, a crucial component of this system, creates the environment for electrophoretic migration. Its standard formulation consists of three key ingredients: Tris base, glycine, and SDS [9] [10]. The precise interplay of these components within a discontinuous pH system is what allows proteins to be first concentrated into a sharp band before being resolved in the separating gel. This application note deconstructs the formula of the standard Tris-Glycine-SDS running buffer, elucidating the function of each chemical constituent, providing optimized protocols, and contextualizing its use within modern drug development research.
Chemical Deconstruction and Function
The 10X Tris-Glycine-SDS running buffer is designed for convenience and is diluted to a 1X working solution for use. The final 1X composition in the electrode chamber is 0.025 M Tris, 0.192 M glycine, and 0.1% SDS, with a pH of 8.3 [9]. Each component plays a specific and critical role in the electrophoretic process.
Tris (C₄H₁₁NO₃): The Buffer Agent Tris (tris(hydroxymethyl)aminomethane) serves as the primary buffering agent in both the running buffer and the gels. Its pKa of approximately 8.1 makes it ideal for maintaining a stable pH within the physiological range (7-9) relevant for most biological samples [10]. In the discontinuous Laemmli system, Tris is used to create gels at two different pH levels: the stacking gel at pH 6.8 and the resolving gel at pH 8.8 [2]. The running buffer pH of 8.3 completes this discontinuous triad, establishing the conditions necessary for the stacking phenomenon.
Glycine (C₂H₅NO₂): The Trailing Ion Glycine is an amino acid whose charge state is profoundly pH-dependent, a property that is the cornerstone of the discontinuous buffer system [10]. In the running buffer at pH 8.3, glycine exists predominantly as a negatively charged glycinate anion [2]. However, when this anion enters the low-pH (6.8) environment of the stacking gel, its charge state shifts. At this pH, a significant proportion of glycine molecules become zwitterions, possessing both positive and negative charges and thus an overall neutral charge [10]. This change drastically reduces glycine's electrophoretic mobility, making it the "trailing ion" in the system.
SDS (Sodium Dodecyl Sulfate, C₁₂H₂₅NaO₄S): The Denaturant and Charge Uniformizer SDS is an anionic detergent that performs two essential functions. First, it binds to proteins via hydrophobic interactions, disrupting their secondary and tertiary non-covalent structures and causing them to unfold into linear chains [10]. Second, because SDS is negatively charged, it coats the proteins in a uniform negative charge cloud. This binding occurs at a nearly constant ratio of 1.4 g SDS per 1 g of protein, overwhelming the proteins' intrinsic charges and giving all proteins a similar charge-to-mass ratio [2]. This allows separation to be based almost entirely on molecular size rather than native charge.
Table 1: Composition of SDS-PAGE Running Buffer
| Component | Molecular Weight | 10X Concentration | 1X Working Concentration | Primary Function |
|---|---|---|---|---|
| Tris Base | 121.14 g/mol | 0.25 M | 0.025 M | Buffering agent; maintains pH 8.3 |
| Glycine | 75.07 g/mol | 1.92 M | 0.192 M | Trailing ion; enables stacking via pH-dependent charge |
| SDS | 288.38 g/mol | 1% (w/v) | 0.1% (w/v) | Denatures proteins and imparts uniform negative charge |
The Mechanism of Discontinuous Electrophoresis
The genius of the Laemmli system lies in the coordinated interplay of Tris, glycine, and SDS across different pH environments to concentrate samples into sharp bands before separation.
Diagram 1: Ionic dynamics in Laemmli's discontinuous SDS-PAGE system.
As illustrated in Diagram 1, when an electric current is applied:
- In the Stacking Gel (pH 6.8): The chloride ions (Cl⁻) from Tris-HCl in the gel become the highly mobile "leading ions." The glycinate ions from the running buffer enter the low-pH stacking gel and become neutral zwitterions, turning into the slow-moving "trailing ions" [10] [2]. The protein-SDS complexes, with a mobility intermediate between the two, are compressed into a extremely narrow zone between these fronts. This phenomenon, occurring in a low-acrylamide gel with little sieving, herds all proteins into a sharp band before they enter the resolving gel [2].
- In the Resolving Gel (pH 8.8): Upon reaching the high-pH resolving gel, the glycine zwitterions lose protons and become fast-moving glycinate anions once again. They overtake the protein-SDS complexes [10] [2]. The proteins, now deposited at the top of the resolving gel in a tight band and moving through a gel with smaller pores, begin to separate based solely on their molecular size, with smaller proteins migrating faster than larger ones [8].
Research Reagent Solutions
A successful SDS-PAGE experiment requires a suite of optimized reagents beyond the running buffer. The table below details the essential components.
Table 2: Essential Reagents for SDS-PAGE Analysis
| Reagent / Solution | Composition / Key Feature | Primary Function in the Workflow |
|---|---|---|
| SDS-PAGE Running Buffer (10X) | 0.25 M Tris, 1.92 M Glycine, 1% SDS, pH 8.3 [9] | Provides the conductive medium and ions for electrophoresis; SDS maintains protein denaturation. |
| 2X Laemmli Sample Buffer | 100 mM Tris-HCl (pH 6.8), 4% SDS, 0.2% Bromophenol Blue, 20% Glycerol; often includes 200 mM DTT or BME [11] | Denatures proteins, adds tracking dye, provides density for well loading, and reduces disulfide bonds. |
| Polyacrylamide Gel System | Stacking Gel: Lower acrylamide %, Tris-HCl, pH 6.8. Resolving Gel: Variable acrylamide %, Tris-HCl, pH 8.8 [10] | Creates a sieving matrix for size-based separation; discontinuous layers enable sample stacking. |
| Protein Stain (e.g., Coomassie) | Coomassie Brilliant Blue in methanol/acetic acid [8] | An anionic dye that binds proteins non-specifically, enabling visualization of separated bands. |
| Protein Molecular Weight Marker | Mixture of purified proteins of known molecular weights [8] | Allows for estimation of the apparent molecular weight of unknown proteins in the sample. |
Detailed Experimental Protocol
Preparation of 10X Tris-Glycine-SDS Running Buffer
Materials:
- Tris base (MW: 121.14 g/mol)
- Glycine (MW: 75.07 g/mol)
- SDS (Sodium Dodecyl Sulfate)
- Deionized water
- pH meter
- Magnetic stirrer and stir bar
- 1 L graduated cylinder and bottle for storage
Method:
- Measure out 800 mL of deionized water into a beaker on a magnetic stirrer.
- While stirring, add 30.28 g of Tris base (final concentration 0.25 M).
- Add 144.13 g of Glycine (final concentration 1.92 M).
- Add 10 g of SDS (final concentration 1% w/v). To avoid excessive foaming, sprinkle the SDS in slowly.
- Stir the solution until all components are completely dissolved. The mixture may be slightly cloudy due to SDS but will clear as it mixes.
- Carefully adjust the pH of the solution to 8.3 using HCl or NaOH if necessary. Note: The buffering capacity of Tris-glycine often results in a pH close to this target without adjustment.
- Transfer the solution to a 1 L graduated cylinder and add deionized water to bring the final volume to 1 L.
- Store the 10X buffer at room temperature. For use, dilute 100 mL of 10X buffer with 900 mL deionized water to make 1 L of 1X working solution [9] [12].
Standard SDS-PAGE Protocol
Materials:
- Prepared protein samples in 1X or 2X Laemmli sample buffer
- 1X Tris-Glycine-SDS Running Buffer
- Cast polyacrylamide gel (stacking and resolving)
- Electrophoresis chamber and power supply
- Heating block or water bath (95-100°C)
- Microcentrifuge
Method:
- Sample Preparation:
- Mix protein sample with an equal volume of 2X Laemmli sample buffer. If using a 5X buffer, adjust volumes accordingly.
- Heat the mixture at 95-100°C for 5-10 minutes to ensure complete denaturation [8].
- Briefly centrifuge (e.g., 12,000g for 30 seconds) to collect condensation and ensure the entire sample is at the bottom of the tube [8].
Gel Setup:
- Assemble the cast gel into the electrophoresis chamber.
- Fill the inner (upper) and outer (lower) chambers with freshly prepared 1X Tris-Glycine-SDS Running Buffer.
- Ensure the wells are fully submerged and remove any air bubbles trapped at the bottom of the gel.
Loading and Running:
- Load equal volumes of prepared samples and protein molecular weight marker into the wells.
- Secure the lid and connect the electrodes to the power supply.
- Run the gel at a constant voltage: 80-90 V until the dye front (bromophenol blue) has moved through the stacking gel and entered the resolving gel.
- Increase the voltage to 120-150 V and continue running until the dye front reaches the bottom of the gel [8].
Post-Electrophoresis:
- Turn off the power supply and disassemble the apparatus.
- Carefully pry the glass plates apart and remove the gel.
- Proceed with downstream applications such as Coomassie staining [8] or Western blotting for immunodetection.
Applications in Research and Drug Development
The Tris-Glycine-SDS PAGE system is a versatile workhorse in life sciences. Its primary application is the analysis of protein purity and composition, where it can reveal the presence and relative abundance of contaminating proteins in a sample [8]. It is also indispensable for estimating the apparent molecular weight of an unknown protein by comparing its migration distance to that of a standard curve generated by a protein marker [8]. Furthermore, it serves as the critical first separation step for Western blotting (immunoblotting), enabling subsequent protein detection with specific antibodies for identification, quantification, and study of post-translational modifications [8]. In a clinical and diagnostic context, SDS-PAGE is used, for example, in confirmatory HIV testing to separate viral proteins before detection with patient sera [8].
Advancements and Alternative Buffer Systems
While the Tris-Glycine system remains the gold standard, it has limitations, including poor resolution of small proteins (<15 kDa) and relatively long run times, which can be a bottleneck in high-throughput drug development pipelines [13]. Recent research has focused on developing alternative buffer systems to overcome these challenges.
The Tris-Tricine-HEPES buffer is one such advancement. This novel running buffer formulation replaces glycine with a combination of Tricine and HEPES. This creates multiple ionic boundaries instead of two, which significantly improves the resolving power, particularly for low molecular weight proteins [13]. A key advantage is the ability to resolve a very wide molecular weight range (15–450 kDa) in a single 10% polyacrylamide gel, a feat difficult to achieve with the traditional system [13]. Additionally, this system allows for a substantially reduced running time without the excessive generation of Joule's heat that plagues Tris-Glycine buffers at higher voltages, making it highly suitable for accelerated research and development workflows [13].
How Buffer Chemistry Governs Current Flow and pH Maintenance
In the context of SDS-PAGE (Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis) research, the running buffer is not merely a passive medium but a dynamic component that governs the fundamental processes of current flow and pH maintenance. A buffer is a special solution that stops massive changes in pH levels, possessing both a specific buffer capacity (the amount of acid or base that can be added before pH changes significantly) and buffer range (the pH range where it effectively neutralizes added acids and bases) [14]. For SDS-PAGE, the running buffer—typically composed of Tris, glycine, and SDS at pH 8.3 [15]—creates a discontinuous system that is fundamental to achieving high-resolution protein separation. This application note details the underlying chemistry of this system and provides standardized protocols for its preparation and use in drug development and basic research settings.
Theoretical Foundations of Buffer Chemistry
Principles of pH Maintenance
The ability of a buffer to maintain a stable pH is explained by the equilibrium between a weak acid (HA) and its conjugate base (A⁻), as described by the Henderson-Hasselbalch equation [14] [16]:
[pH = pK_a + \log\dfrac{[A^-]}{[HA]}]
This equation demonstrates that the pH of a buffer solution depends on the pKa of the weak acid and the ratio of the concentrations of the conjugate base and acid. When this ratio is 1, the pH equals the pKa. The buffer is most effective, meaning it has the highest buffering capacity, when the pH is within approximately one unit of its pKa (pH range of pKa -1 to pK_a +1) [16]. The buffering capacity (β) can be empirically calculated as [16]:
[\beta = \frac{\Delta cb}{\Delta pH} = -\frac{\Delta ca}{\Delta pH}]
where Δca and Δcb represent changes in the molar concentration of acid or base, and ΔpH is the resulting change in pH.
Mechanism of Current Flow and Conductivity
In an SDS-PAGE apparatus, the application of an electric current (typically 100-150 V [17]) drives redox reactions that involve the water molecules in the running buffer. At the negatively charged cathode, reduction occurs, and hydrogen ions (H⁺) are converted to hydrogen gas (H₂). At the positively charged anode, oxidation occurs, and oxygen ions (O²⁻) are converted to oxygen gas (O₂) [15]. The observation of more bubbles at the cathode reflects the stoichiometry of water (H₂O), with two hydrogen atoms produced for every oxygen atom [15]. The Tris and glycine ions in the running buffer serve as charge carriers, facilitating current flow through the system. Their specific charge states, which change with the local pH, are critical for establishing the voltage gradients that drive protein stacking and separation.
The Discontinuous Buffer System in SDS-PAGE
The remarkable resolving power of SDS-PAGE hinges on its discontinuous buffer system, which features different pH values and ionic compositions in the stacking versus resolving gels and the running buffer [15].
Key Components and Their Roles
Table 1: Key Components of the SDS-PAGE Running Buffer System
| Component | Typical Concentration | Primary Function | Role in Discontinuous System |
|---|---|---|---|
| Tris Base | 25 mM (1x) [12] | Buffering agent (pK_a ≈ 8.1) [15] | Maintains pH at 8.3 in running buffer; provides Cl⁻ leading ions in stacking gel [15]. |
| Glycine | 192 mM (1x) [12] | Charge-carrying trailing ion | Exists as zwitterion (slow) in stacking gel (pH 6.8) and as glycinate (fast) in resolving gel (pH 8.8) [15]. |
| SDS (Sodium Dodecyl Sulfate) | 0.1% (1x) [12] | Ionic detergent | Coats proteins with uniform negative charge; maintains protein denaturation [18] [15]. |
The Stacking Mechanism Visualized
The following diagram illustrates the stepwise process of how the buffer chemistry creates a stacking effect, focusing on the change in glycine's ionic state.
The key to this mechanism is glycine's status as a zwitterion. Its charge is highly dependent on environmental pH [15]. In the running buffer (pH 8.3), glycine is predominantly a negatively charged glycinate anion and moves rapidly. Upon entering the low-pH (6.8) environment of the stacking gel, most glycine molecules become zwitterions, bearing both positive and negative charges and thus moving slowly. Chloride ions (Cl⁻) from Tris-HCl in the gel remain highly mobile. This creates a steep voltage gradient between the fast Cl⁻ front (leading ion) and the slow glycine zwitterion front (trailing ion). Proteins, with mobilities between these two fronts, are compressed ("stacked") into a narrow zone. When this zone reaches the resolving gel (pH 8.8), glycine regains its negative charge and speeds away, depositing the proteins as a tight band at the top of the resolving gel where size-based separation begins [15].
Research Reagent Solutions
The following table catalogues the essential materials required for preparing and running an SDS-PAGE experiment.
Table 2: Essential Reagents for SDS-PAGE Running Buffer and Related Preparations
| Reagent/Category | Specifications | Function in Experiment |
|---|---|---|
| Tris Base | MW: 121.14 g/mol [12] | Primary buffering agent; maintains pH in running buffer and gels [15]. |
| Glycine | MW: 75.07 g/mol [12] | Trailing ion in discontinuous buffer system; critical for protein stacking [15]. |
| SDS (Sodium Dodecyl Sulfate) | MW: 288.38 g/mol [12] | Ionic detergent that denatures proteins and confers uniform negative charge [18] [17]. |
| 10X Running Buffer Stock | 250 mM Tris, 1.92 M Glycine, 1% SDS [12] | Concentrated stock for convenient dilution to 1X working solution. |
| Acrylamide/Bis-Acrylamide | Ratio typically 37.5:1 or 29:1 [19] | Forms the cross-linked polyacrylamide gel matrix; pore size determines resolution [15]. |
| Ammonium Persulfate (APS) | 10% (w/v) solution in water | Initiator of acrylamide polymerization [15]. |
| TEMED | N,N,N',N'-Tetramethylethylenediamine | Catalyst that accelerates acrylamide polymerization by reacting with APS [15]. |
| Laemmli Sample Buffer | Contains Tris-HCl, SDS, glycerol, Bromophenol Blue, +/- BME/DTT [15] | Denatures proteins, adds density for loading, and provides visual tracking dye. |
| β-Mercaptoethanol (BME) or DTT | 0.55M final in sample buffer [17] | Reducing agents that break disulfide bonds for complete denaturation [18]. |
Detailed Experimental Protocols
Protocol 1: Preparation of 10X SDS-PAGE Running Buffer
This protocol describes the preparation of a 1-liter stock of 10X running buffer, which can be diluted to 1X for use [12].
Materials:
- Tris base (MW: 121.14 g/mol)
- Glycine (MW: 75.07 g/mol)
- SDS (MW: 288.38 g/mol)
- Distilled or deionized water
- pH meter
- 1 L graduated cylinder or volumetric flask
- Magnetic stirrer and stir bar
Method:
- Prepare Solution Base: Measure approximately 800 mL of distilled water and pour it into a suitable 1 L container.
- Dissolve Components: Add the following components to the water while stirring:
- 30.2 g of Tris base (Final concentration: 250 mM)
- 144.0 g of Glycine (Final concentration: 1.92 M)
- 10.0 g of SDS (Final concentration: 1% w/v)
- Adjust Volume: Continue stirring until all components are completely dissolved. Bring the final volume to 1.0 L with distilled water. The pH of the 10X solution should be approximately 8.3 and typically does not require adjustment.
- Storage: Store the 10X running buffer at room temperature. For use, dilute to 1X with distilled water (e.g., 50 mL of 10X buffer + 450 mL water for 500 mL of 1X buffer) [17].
Protocol 2: SDS-PAGE Electrophoresis Run
This protocol assumes a pre-cast polyacrylamide gel is being used.
Materials:
- Pre-cast SDS-PAGE gel
- Protein molecular weight standards
- Prepared protein samples in Laemmli buffer
- 1X SDS-PAGE Running Buffer
- Vertical electrophoresis unit and power supply
- Heating block or water bath (95°C)
- Microcentrifuge
Method:
- Gel Setup: Place the gel in the electrophoresis chamber and secure it according to the manufacturer's instructions.
- Buffer Addition: Fill the inner chamber of the gel unit completely with 1X running buffer. Pour the remaining 1X buffer into the outer chamber [17].
- Sample Denaturation: Ensure protein samples are mixed with an equal volume of 2X Laemmli buffer containing a reducing agent (e.g., BME to 0.55 M). Heat the samples at 95°C for 5 minutes to fully denature the proteins [18] [17]. Briefly centrifuge (3 minutes) to pellet any debris [17].
- Sample Loading: Using gel-loading tips for precision, load the denatured samples and molecular weight standards into the wells. Record the loading order.
- Electrophoresis Run: Place the lid on the chamber, connecting the electrodes correctly (black to cathode, red to anode). Set the power supply to a constant voltage of 100-150 V [18] [17]. Turn on the power. The Bromophenol Blue dye front should be visible moving through the gel.
- Run Completion: Allow the gel to run until the dye front has just migrated out of the bottom of the gel (typically 40-90 minutes). Immediately turn off the power supply [17].
- Analysis: Disconnect the apparatus, carefully open the gel cassette, and proceed with the desired detection method (e.g., Coomassie staining, Western blotting).
Optimization and Troubleshooting
Optimizing Separation Conditions
Table 3: Guidelines for Gel Concentration Selection Based on Protein Size
| Target Protein Molecular Weight Range | Recommended Gel Concentration |
|---|---|
| 100 - 600 kDa | 4% - 8% [17] [19] |
| 50 - 500 kDa | 7% [19] |
| 30 - 300 kDa | 10% [19] |
| 10 - 200 kDa | 12% [19] |
| 3 - 100 kDa | 15% [19] |
Addressing Common Issues
- Smiling Bands (Faster migration in outer lanes): Caused by uneven heat distribution. Ensure efficient heat transfer by completely filling the outer chamber with buffer and consider using a magnetic stirrer. Running at a lower current can also help maintain a constant temperature (10-20°C) [18].
- Poor Resolution or Smearing: Can result from incomplete protein denaturation. Ensure samples are heated to 95°C for 5 minutes in the presence of sufficient SDS and reducing agent [18]. Overloading the gel with too much protein can also cause smearing; for complex mixtures like cell lysates, load ≤20 µg per well for Coomassie staining [18].
- Unexpected Protein Migration: Post-translational modifications (e.g., glycosylation, phosphorylation) can alter SDS binding and thus protein mobility. Highly hydrophobic or intrinsic membrane proteins may also bind SDS differently and run at non-expected molecular weights [15].
Understanding Ionic Strength and Buffer Concentration (1X vs. 10X)
In SDS-polyacrylamide gel electrophoresis (SDS-PAGE), the running buffer is a critical component that serves two primary functions: it carries the electrical current necessary for electrophoretic separation and maintains a stable pH to ensure consistent protein migration [20]. The standard SDS-PAGE running buffer is a Tris-glycine-SDS system, which is commonly prepared as a 10X concentrated stock solution and diluted to 1X working concentration for use [12] [3]. The ionic strength of this buffer, determined by the concentration of ions in solution, directly impacts the efficiency of separation, the resolution of protein bands, and the heat generated during electrophoresis [21] [20]. Understanding the distinction between 1X and 10X concentrations and their effects on ionic strength is therefore fundamental for optimizing SDS-PAGE results, particularly in sensitive applications like proteomic analysis and drug development.
Theoretical Foundations: Ionic Strength and Buffer Concentration
The Role of Ionic Strength in Electrophoresis
Ionic strength refers to the total concentration of ions in a solution and is a measure of the intensity of the electric field in the solution. In SDS-PAGE, the ionic strength of the running buffer influences several key parameters [20]:
- Electrophoretic Mobility: The mobility of protein-SDS complexes is proportional to the potential gradient (voltage) and inversely proportional to resistance. Higher ionic strength increases the share of current carried by buffer ions, which can slow down sample migration.
- Heat Generation: High ionic strength buffers conduct electricity more efficiently, leading to increased current and substantial heat generation. This heat can cause diffusion of separation bands, resulting in poor resolution and potential protein denaturation [20].
- Resolution and Band Sharpness: Optimum ionic strength is necessary for achieving sharp protein bands. Excessively low ionic strength reduces overall current and resolution, while excessively high ionic strength generates heat that broadens protein bands [20].
The Chemistry of SDS-PAGE Running Buffer
The standard SDS-PAGE running buffer consists of three key components [3]:
- Tris Base (C₄H₁₁NO₃): Serves as the buffering agent to maintain stable pH throughout the electrophoresis run.
- Glycine (NH₂CH₂COOH): Functions as the leading ion in the discontinuous buffer system formulated by Laemmli [2].
- Sodium Dodecyl Sulfate (SDS) (C₁₂H₂₅O₄NaS): An anionic detergent that maintains the denatured state of proteins and provides uniform negative charge.
The 10X concentrated stock solution contains 0.25 M Tris base, 1.923 M glycine, and 1% (w/v) SDS [3]. When diluted to 1X working concentration, the buffer contains 25 mM Tris, 192 mM glycine, and 0.1% SDS. This specific formulation creates a discontinuous buffer system that enables both stacking and separation of proteins during electrophoresis [2].
Table 1: Composition of SDS-PAGE Running Buffer at 10X and 1X Concentrations
| Component | Molecular Weight | 10X Concentration | 1X Concentration |
|---|---|---|---|
| Tris base | 121.14 g/mol | 30.285 g/L (0.25 M) | 3.03 g/L (25 mM) |
| Glycine | 75.07 g/mol | 144.4 g/L (1.923 M) | 14.44 g/L (192 mM) |
| SDS | 288.38 g/mol | 10 g/L (1% w/v) | 1 g/L (0.1% w/v) |
The Discontinuous Buffer System Mechanism
The Laemmli buffer system utilizes a clever manipulation of pH and ionic strength to concentrate proteins into sharp bands before separation [2]. The key mechanism involves:
- Formation of a Moving Boundary: When current is applied, glycine ions in the running buffer (pH ~8.3) enter the stacking gel (pH 6.8) where they become protonated and lose much of their charge, slowing down dramatically.
- Creation of a High-Field Strength Zone: Highly mobile chloride ions from the Tris-HCl in the stacking gel move ahead quickly, creating a narrow zone of high electrical resistance and field strength.
- Protein Stacking: All protein-SDS complexes migrate rapidly in this high-field strength zone but cannot outrun the chloride front, resulting in concentration into a tight band.
- Separation in Resolving Gel: When this stack reaches the resolving gel (pH 8.8), the increased pH causes glycine to deprotonate, increasing its mobility. The proteins then separate by molecular weight in the uniform gel matrix [2].
Practical Protocols and Applications
Preparation of SDS-PAGE Running Buffer
Protocol: Preparation of 10X SDS-PAGE Running Buffer Stock Solution
Materials Required:
- Tris base (MW: 121.14 g/mol)
- Glycine (MW: 75.07 g/mol)
- Sodium dodecyl sulfate (SDS) (MW: 288.38 g/mol)
- Distilled water
- pH meter
- Magnetic stirrer and stir bar
- 1 L graduated cylinder or volumetric flask
- Storage bottle
Procedure:
- Measure approximately 800 mL of distilled water and pour into a clean 1 L beaker.
- Add 30.285 g of Tris base to the water while stirring [3].
- Add 144.4 g of glycine to the solution [3].
- Add 10 g of SDS powder to the mixture [3].
- Continue stirring until all components are completely dissolved.
- Transfer the solution to a 1 L volumetric flask and add distilled water to bring the final volume to 1 L.
- Store the 10X stock solution at room temperature. The solution is stable for several months.
Protocol: Dilution to 1X Working Concentration
Procedure:
- Measure the required volume of 10X stock solution based on the electrophoresis apparatus capacity (e.g., 50 mL for 500 mL of 1X buffer).
- Add the 10X concentrate to an appropriate container.
- Add distilled water to achieve a 1:10 dilution (e.g., add 450 mL water to 50 mL of 10X stock) [22].
- Mix thoroughly by stirring. The 1X working solution should be used immediately or stored for short periods at room temperature.
Optimization of Buffer Conditions for Specific Applications
Different protein separation challenges may require optimization of buffer concentration and ionic strength:
Table 2: Effects of Buffer Ionic Strength on SDS-PAGE Performance
| Ionic Strength Condition | Migration Rate | Band Resolution | Heat Generation | Recommended Applications |
|---|---|---|---|---|
| Standard 1X Buffer | Optimal for most proteins | High | Moderate | Routine protein separation, molecular weight determination |
| Higher than 1X Concentration | Slower migration | Decreased (band diffusion) | High | Not generally recommended |
| Lower than 1X Concentration | Faster, potentially erratic | Decreased (band smiling) | Low | Specialized applications requiring optimization |
Research by Zhao et al. demonstrated that buffer composition significantly affects separation efficiency, particularly for challenging proteins like phycoerythrins [21]. Their findings indicate that:
- Lower Tris concentration in the resolving gel can improve separation of similar molecular weight subunits [21].
- The ratio of SDS monomer to micelle, influenced by ionic strength, affects the amount of SDS bound to polypeptides and their electrophoretic mobility [21].
- For specific applications such as separation of phycoerythrin subunits, modified buffer conditions (lower SDS concentration, lower Tris concentration, higher pH) may be necessary [21].
The Scientist's Toolkit: Essential Reagents for SDS-PAGE
Table 3: Key Research Reagent Solutions for SDS-PAGE
| Reagent | Function | Application Notes |
|---|---|---|
| Tris-Glycine-SDS Running Buffer | Conducts current, maintains pH, provides SDS for protein charge | Use 1X concentration for standard runs; 10X stock for storage [12] [3] |
| Acrylamide/Bis-acrylamide | Forms cross-linked polyacrylamide gel matrix | Concentration determines pore size; varies from 8-16% for different protein size ranges [23] |
| Ammonium Persulfate (APS) | Polymerizing agent for polyacrylamide gels | Fresh preparation recommended as it degrades over 1-2 weeks [2] |
| TEMED | Catalyzes polymerization reaction | Promotes free radical production from APS; store refrigerated in dark [23] |
| SDS Sample Buffer | Denatures proteins, provides charge and density for loading | Typically contains SDS, glycerol, tracking dye, and reducing agent [22] |
| Molecular Weight Markers | Reference standards for size determination | Include pre-stained or unstained proteins of known molecular weights [23] |
Troubleshooting and Technical Considerations
Common Issues Related to Buffer Concentration and Ionic Strength
- Precipitated SDS: High concentrations of potassium chloride (KCl >200 mM) in samples can cause SDS to precipitate. Dilute samples or methanol-precipitate and resuspend in 1X sample buffer to avoid this issue [2].
- Buffer Depletion: Reusing running buffer multiple times can alter ionic strength due to electrolysis and pH changes, leading to inconsistent results. Fresh 1X buffer is recommended for each run.
- Heat Effects: High ionic strength increases current and heat generation, which can cause "band smiling" (curved migration patterns). Using a cooling system or reducing voltage can mitigate this problem.
- Altered Migration Patterns: As demonstrated in research on phycoerythrins, unusual protein characteristics may require optimization of standard buffer conditions, including adjustments to Tris concentration, pH, or SDS concentration [21].
Specialized Applications and Modified Buffer Systems
Recent research has explored the use of low SDS concentrations (0.1%) for specific applications where maintaining some protein structure or function is desirable [24]. These conditions represent an intermediate between negligible and extensive SDS binding, highlighting the potential for novel applications in decellularization and protein fractionation while preserving certain structural features [24].
The proper preparation and use of SDS-PAGE running buffer at appropriate concentrations (1X vs. 10X) is fundamental to successful protein separation. The ionic strength of the buffer significantly impacts electrophoretic mobility, resolution, and heat generation during the process. The standardized Tris-glycine-SDS system, when prepared correctly according to established protocols, provides reproducible results for most applications. However, researchers should be aware that specific protein separation challenges may require optimization of buffer conditions, including adjustments to Tris concentration, pH, or SDS content. Understanding these principles enables scientists to troubleshoot effectively and adapt methodology to meet specialized research needs in protein analysis and drug development.
Why the Buffer's pH is Critical and Should Not Be Adjusted
The discontinuous buffer system is a cornerstone of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), a foundational technique in proteomics and drug development. This application note elucidates the fundamental biochemical principles governing the Tris-Glycine running buffer pH, explaining why its prescribed value of pH 8.3 is critical and must not be empirically adjusted. Deviation from this specified pH disrupts the delicate balance of ionic mobilities, compromising the essential phenomena of sample stacking and subsequent size-based separation. Adherence to validated buffer preparation protocols is therefore a non-negotiable prerequisite for obtaining reproducible, high-resolution protein separation, ensuring data integrity in research and development workflows.
SDS-PAGE is the most widely used method for separating denatured proteins primarily by molecular weight [25]. The technique relies on a discontinuous buffer system—first described by Laemmli—that employs buffers of different pH and ionic composition in the gel and the electrode chambers [2]. This system is engineered to concentrate protein samples into extremely sharp bands before they enter the separating gel, a process known as stacking.
The running buffer, typically composed of Tris, glycine, and SDS, is a key component of this system [26]. Its specified pH of 8.3 is not arbitrary; it is meticulously calculated to control the charge states and electrophoretic mobilities of the glycine ions and the protein-SDS complexes throughout the electrophoresis process. Adjusting the pH of this buffer disrupts the precise interplay of leading and trailing ions, leading to poor resolution, band smearing, and unreliable migration, ultimately jeopardizing experimental results and downstream analyses.
The Scientific Principle: Ionic States and Moving Boundaries
The core mechanism of the discontinuous system hinges on the creation of a moving boundary of ions with different electrophoretic mobilities, all within a pH gradient.
The Key Players: Leading, Trailing, and Common Ions
The system functions via the coordinated movement of three ions [27]:
- Chloride (Cl⁻): The leading ion, supplied by Tris-HCl in the gel. It is highly mobile and forms a fast-moving front.
- Glycinate: The trailing ion, supplied by glycine in the running buffer. Its mobility is dynamically controlled by the local pH.
- Tris base (Tris⁺): The common cation present throughout the system, which does not participate directly in the moving boundary but maintains electrical neutrality.
The Critical Role of pH in Controlling Glycine's Charge
Glycine is an amino acid with two ionizable groups. Its net charge is profoundly dependent on the pH of its environment, which directly dictates its electrophoretic mobility [26] [2]:
- At running buffer pH (8.3): Glycine exists predominantly as a glycinate anion, carrying a significant negative charge and moderate mobility.
- At stacking gel pH (6.8): As glycinate ions enter the low-pH stacking gel, their amino group becomes protonated. They transition into zwitterions with a net charge close to zero, drastically reducing their mobility.
- At separating gel pH (8.8): Upon reaching the high-pH separating gel, glycine loses a proton and rapidly reverts to the highly mobile glycinate anion.
The following diagram illustrates this dynamic process and its effect on protein separation.
Diagram 1: The dynamic change in glycine's ionic state, controlled by local pH, is the engine of the discontinuous buffer system. It first creates a stacking effect, then releases proteins for separation.
Consequences of Improper Buffer pH Adjustment
Altering the pH of the running buffer from its specified value disrupts the entire separation mechanism. The following table summarizes the primary failure modes.
Table 1: Consequences of deviating from the recommended running buffer pH.
| pH Deviation | Impact on Glycine State | Observed Experimental Defects |
|---|---|---|
| pH too high | Remains fully deprotonated; mobility remains too high, failing to act as an effective trailing ion. | Loss of stacking; diffuse and smeared bands from the start; poor resolution of protein bands [28]. |
| pH too low | Remains in or near zwitterionic state; mobility remains too low, acting as too strong a trailing ion. | Inefficient protein entry into the gel; distorted or wavy bands; extended run times; potential protein precipitation. |
Essential Reagents and Compositions for SDS-PAGE
A successful SDS-PAGE experiment relies on a system of carefully formulated reagents. The table below details the key components, with special emphasis on the critical, non-adjustable pH values.
Table 2: Key reagents for the Tris-Glycine SDS-PAGE system and their functions [27] [26] [12].
| Reagent | Standard Composition | Function | Critical pH & Rationale |
|---|---|---|---|
| SDS Running Buffer (10X) | 250 mM Tris, 1.92 M Glycine, 1% (w/v) SDS [12] | Conducts current; maintains pH for ion mobility; supplies trailing ion (glycine) and keeps proteins denatured. | pH 8.3 (not adjusted). Optimizes glycinate concentration to function as a trailing ion in the stacking gel (pH 6.8). |
| Resolving Gel Buffer | 1.5 M Tris-HCl | Sets the high-pH environment for size-based separation; supplies leading ions (Cl⁻). | pH ~8.8. Ensures glycinate gains high mobility, passing proteins and ending the stacking process. |
| Stacking Gel Buffer | 0.5 M Tris-HCl | Sets the low-pH environment that triggers glycine's charge shift to initiate stacking. | pH ~6.8. Maximizes the proportion of zwitterionic glycine, creating the slow-moving trailing ion front. |
| SDS Sample Buffer (2X) | Tris-HCl, SDS, Glycerol, Bromophenol Blue, ß-mercaptoethanol (or DTT) [27] [26] | Denatures proteins; provides negative charge; adds density for loading; includes reducing agent and tracking dye. | pH ~6.8. Matches the stacking gel pH to ensure proper protein mobility entering the stacking zone. |
Validated Experimental Protocol for SDS-PAGE
Preparation of Running Buffer
Principle: The running buffer must be prepared to the exact specifications without pH adjustment to ensure proper ionic dynamics [27] [12].
Materials:
- Tris base (MW: 121.14 g/mol)
- Glycine (MW: 75.07 g/mol)
- SDS (Sodium Dodecyl Sulfate)
- Deionized water
Method:
- To prepare 1 L of 10X Tris-Glycine-SDS Running Buffer:
- Add 30.0 g of Tris base and 144.0 g of glycine to approximately 800 mL of deionized water [12].
- Stir until completely dissolved.
- Add 10.0 g of SDS and stir until dissolved.
- Bring the final volume to 1 L with deionized water. Do not adjust the pH. The final pH should be approximately 8.3.
- Dilute the 10X stock to 1X with deionized water prior to use. For a standard mini-gel apparatus, you will require approximately 200 mL for the inner chamber and 600 mL for the outer chamber [27].
Gel Electrophoresis Procedure
Materials:
- Pre-cast or hand-cast Tris-Glycine polyacrylamide gel
- Prepared protein samples in 1X SDS sample buffer
- Protein molecular weight standard
- Power supply and electrophoresis unit
Method:
- Sample Preparation: Dilute protein samples in 1X SDS sample buffer. For denatured samples, heat at 70–100°C for 2-5 minutes [27]. Do not heat samples for native electrophoresis.
- Gel Assembly: Remove the gel cassette from its pouch, rinse with deionized water, and remove the comb. Rinse wells with 1X running buffer. Assemble the gel in the electrophoresis chamber according to the manufacturer's instructions [27].
- Loading: Fill the inner and outer chambers with 1X running buffer. Load equal volumes of prepared samples and molecular weight standards into the wells.
- Electrophoresis: Connect the apparatus to a power supply, ensuring correct polarity. Run the gel at a constant voltage of 125 V for approximately 90 minutes, or until the bromophenol blue tracking dye front reaches the bottom of the gel [27].
- Analysis: Upon completion, turn off the power, disassemble the apparatus, and carefully open the cassette to remove the gel for staining (e.g., Coomassie Blue) or western blotting.
Troubleshooting Common Issues Related to Buffer pH
Poor buffer preparation is a common source of experimental failure. The table below links symptoms to potential causes related to buffer integrity.
Table 3: Troubleshooting guide for common SDS-PAGE issues linked to buffer problems [28].
| Observed Problem | Potential Causes Related to Buffer | Solution |
|---|---|---|
| Smeared bands | - Incorrect running buffer pH.- Deteriorated or contaminated running buffer.- Buffer ion concentration too low. | - Prepare fresh running buffer from stock without pH adjustment.- Ensure proper dilution of 10X stock to 1X. |
| Poor resolution | - Buffer pH deviating from 8.3, disrupting stacking.- Incorrect gel buffer pH.- Extended run time generating excessive heat. | - Verify the pH of all stock buffers.- Check that the running buffer is not old or contaminated.- Ensure adequate cooling during the run. |
| Unusual band migration | - Running buffer prepared with wrong reagents or concentrations.- Sample contaminants affecting local pH or conductivity. | - Remake running buffer with correct reagents and concentrations.- Desalt or precipitate samples to remove interfering salts. |
Preparing and Using SDS-PAGE Running Buffer: A Step-by-Step Protocol
Within the framework of SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE) research, the running buffer constitutes a critical component of the discontinuous buffer system essential for successful protein separation. The Tris-Glycine-SDS running buffer facilitates the electrophoretic migration of proteins through the gel matrix, ensuring their denaturation and conferring a uniform negative charge via SDS binding. This application note delineates the standard preparation of a 10X Tris-Glycine-SDS Running Buffer, a staple reagent in biochemistry and molecular biology laboratories for the analysis of protein samples in drug development and basic research [29] [27]. The 10X concentrate offers storage convenience and is diluted to a 1X working concentration for use, which regulates the system at an approximate pH of 8.6, providing reproducible separation of a wide range of proteins into well-resolved bands [29].
Principles of the Tris-Glycine Discontinuous Buffer System
The Tris-Glycine-SDS running buffer is integral to the Laemmli system [27]. Its efficacy hinges on a discontinuous buffer system involving three key ions that establish a moving boundary within the gel, stacking proteins into a sharp line before they enter the separating gel.
- Chloride Ions: Supplied by the gel buffer, chloride acts as the leading ion due to its small size and high electrophoretic mobility toward the anode [27].
- Glycine Ions: supplied by the running buffer, glycine serves as the trailing ion. At the neutral pH of the stacking gel, glycine exists predominantly as a zwitterion with a net charge near zero, causing it to migrate slowly. This creates a mobility gradient between the fast chloride and slow glycine ions, effectively stacking the protein samples into a thin, sharp zone [27].
- Tris Base: As a common cation present in both the gel and running buffers, Tris base provides the necessary conductivity and buffering capacity throughout the electrophoretic process. The interplay of these ions in the charged environment establishes an operating pH of approximately 9.5 in the separation region of the gel, allowing glycine to become more fully ionized and mobilize behind the proteins, which are then separated based on size in the resolving gel [27].
The following diagram illustrates the orchestrated interplay of ions and proteins during the electrophoretic process.
Standard Recipe and Preparation
The Scientist's Toolkit: Essential Reagents for SDS-PAGE
The following table details the key components required to prepare one liter of 10X Tris-Glycine-SDS Running Buffer concentrate.
Table 1: Research Reagent Solutions for 10X Tris-Glycine-SDS Running Buffer
| Component | Chemical Formula / Description | Molecular Weight (g/mol) | Function in the Buffer System |
|---|---|---|---|
| Tris base | C₄H₁₁NO₃ [3] | 121.14 [3] | Provides the common cation and buffering capacity; essential for pH regulation and conductivity throughout the electrophoresis system [29] [27]. |
| Glycine | NH₂CH₂COOH [3] | 75.07 [3] | The trailing ion in the discontinuous system; its pH-dependent charge state is critical for protein stacking and separation [27]. |
| Sodium Dodecyl Sulfate (SDS) | C₁₂H₂₅O₄NaS [3] | 288.38 [3] | An anionic detergent that denatures proteins and confers a uniform negative charge, enabling separation primarily by molecular weight [30]. |
| Distilled Water | H₂O | 18.02 [29] | Solvent for preparing the buffer solution; ensures purity and absence of ions that might interfere with electrophoresis. |
Quantitative Formulation
The precise recipe for one liter of 10X Tris-Glycine-SDS Running Buffer is summarized in the table below. This formulation is consistent across multiple commercial and technical sources, ensuring reliability and reproducibility [3] [29] [31].
Table 2: Standard Recipe for 10X Tris-Glycine-SDS Running Buffer (1 L)
| Component | Amount | Final Concentration (10X) | CAS Number |
|---|---|---|---|
| Tris base | 30.285 g [3] | 0.25 M [3] [29] | 77-86-1 [3] [29] |
| Glycine | 144.4 g [3] | 1.92 M - 1.923 M [3] [29] | 56-40-6 [3] [29] |
| SDS | 10 g [3] [31] | 1% (w/v) [3] [31] | 151-21-3 [3] [29] |
Step-by-Step Preparation Protocol
Materials Required:
- High-precision analytical balance
- 1 L graduated cylinder or volumetric flask
- Magnetic stirrer and stir bar
- Appropriate container for storage
- Distilled or deionized water
- Weighing boats and spatula
Methodology:
- Prepare Base Solution: Measure approximately 800 mL of distilled water into a 1 L beaker or volumetric flask and begin stirring [12] [3].
- Dissolve Components: Sequentially add the weighed components to the water:
- Bring to Final Volume: Once all components are fully dissolved, add distilled water until the total volume reaches 1.0 L [12] [3].
- pH Verification: The pH of the 1X diluted working solution should be approximately 8.3 - 8.6 and typically does not require adjustment [29] [32] [33]. It is critical not to adjust the pH of the concentrated 10X solution [31].
- Storage: The 10X running buffer concentrate can be stored at room temperature. For the working solution, dilute the 10X concentrate ten-fold with deionized water prior to use [29] [32].
Application Protocol for SDS-PAGE Electrophoresis
Recommended Workflow for SDS-PAGE Using Tris-Glycine Buffer
The following protocol outlines the standard procedure for running a denaturing SDS-PAGE gel using the prepared Tris-Glycine-SDS Running Buffer.
Materials:
- Pre-cast or laboratory-cast Tris-Glycine polyacrylamide gel [27]
- Protein samples and molecular weight markers
- Vertical electrophoresis unit with power supply
- Tris-Glycine SDS Sample Buffer (2X) [27]
Experimental Procedure:
- Sample Preparation: Mix the protein sample with an equal volume of 2X Tris-Glycine SDS Sample Buffer. For reduced samples, add a reducing agent (e.g., DTT or β-mercaptoethanol) to a final concentration of 1X immediately before heating. Heat the samples at 85°C for 2-5 minutes to denature the proteins [27].
- Gel Assembly: Remove the pre-cast gel from its packaging and rinse the cassette with deionized water. Remove the comb and thoroughly rinse the sample wells with 1X Tris-Glycine SDS Running Buffer [27].
- Buffer Chamber Setup:
- Assemble the gel in the electrophoresis tank according to the manufacturer's instructions.
- Fill the inner (upper) and outer (lower) buffer chambers with the 1X Tris-Glycine SDS Running Buffer. Ensure the buffer level in the upper chamber covers the electrodes and submerges the sample wells [27].
- Sample Loading: Load the denatured samples and protein molecular weight markers into the designated wells.
- Electrophoresis Conditions: Connect the electrodes to the power supply, ensuring correct polarity. Run the gel at a constant voltage of 125 V. The expected current should start at 30-40 mA per gel and end at 8-12 mA. The run time is typically about 90 minutes, or until the bromophenol blue tracking dye front reaches the bottom of the gel [27].
- Post-Electrophoresis Analysis: After the run is complete, power off the supply, disassemble the apparatus, and carefully open the gel cassette. The gel can then be processed for staining (e.g., Coomassie Blue, silver stain) or for western blotting [30] [27].
Concluding Remarks
The precise preparation and application of 10X Tris-Glycine-SDS Running Buffer are foundational to achieving high-quality, reproducible results in SDS-PAGE. This buffer is a cornerstone of the discontinuous Laemmli system, enabling the precise separation of proteins based on molecular weight, which is a critical step in proteomic analysis, protein purity assessments, and biomarker discovery in drug development. Mastery of this fundamental reagent preparation ensures the integrity and reliability of electrophoretic data, forming the basis for subsequent analytical techniques and scientific conclusions.
Step-by-Step Guide from 10X Stock to 1X Working Solution
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is a fundamental technique in biochemistry and molecular biology for separating proteins based on their molecular weight. The running buffer is a critical component of this system, providing the ions necessary to conduct current and establish the conditions for protein separation. This protocol details the preparation of the standard 1X Tris-Glycine-SDS running buffer from a 10X concentrated stock, a routine but vital laboratory procedure. The 10X stock solution, often based on the original Laemmli method, allows for convenient storage and preparation of the working solution used in the electrophoresis tank [34] [35]. Framed within a broader research context on buffer composition, this guide ensures that scientists and drug development professionals can achieve highly reproducible and optimal protein separation, which is a cornerstone technique for downstream applications like Western blotting and protein characterization [36].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details the key reagents and materials required for the preparation and use of SDS-PAGE running buffer.
Table 1: Essential Reagents and Materials for SDS-PAGE Running Buffer Preparation
| Item | Function / Description |
|---|---|
| 10X SDS Running Buffer Stock | Concentrated solution containing Tris, Glycine, and SDS [34]. Provides the ionic components for the electrophoresis circuit and protein separation. |
| Tris Base | A buffering agent (pKa ~8.1) that maintains the pH required for the discontinuous buffer system to function [35]. |
| Glycine | An amino acid that acts as the trailing ion in the discontinuous buffer system. Its charge state changes with pH, which is crucial for protein stacking [35]. |
| Sodium Dodecyl Sulfate (SDS) | An anionic detergent that denatures proteins and confers a uniform negative charge, allowing separation based primarily on size [35]. |
| Distilled or Deionized Water | Solvent for diluting the 10X stock solution; its purity ensures no ions interfere with the electrophoretic process. |
| Measuring Cylinder / Volumetric Flask | For accurate measurement and dilution of the 10X stock solution to the final 1X working volume. |
Composition of the 10X Stock Solution
The 10X SDS-PAGE running buffer is a concentrated solution whose final chemical composition upon dilution to 1X is designed to match the classic Laemmli buffer system. The table below summarizes the precise concentrations of components in both the 10X stock and the resulting 1X working solution, based on consolidated data from commercial and academic protocols [34] [3] [37].
Table 2: Quantitative Composition of 10X and 1X SDS-PAGE Running Buffer
| Component | Molecular Weight (g/mol) | Amount per Liter of 10X Stock | Concentration in 10X Stock | Concentration in 1X Working Solution |
|---|---|---|---|---|
| Tris Base | 121.14 | 30.3 g | 0.25 M | 25 mM |
| Glycine | 75.07 | 144.0 g - 144.4 g | 1.92 M - 1.923 M | 192 mM - 250 mM * |
| SDS | 288.38 | 10.0 g | 1% (w/v) | 0.1% (w/v) |
Note: The stated concentration of glycine in the 1X working solution varies between sources. Most protocols, including the original Laemmli method, use 192 mM, though some commercial stocks are formulated to yield 250 mM [34] [38]. The pH of the 1X working solution is approximately 8.3 and typically does not require adjustment [37] [35].
Step-by-Step Protocol for 1X Working Solution Preparation
This section provides a detailed methodology for preparing the 1X SDS-PAGE running buffer from a 10X concentrate.
Materials and Equipment
- 10X SDS-PAGE Running Buffer stock solution [34]
- Distilled or deionized water
- A clean container or beaker (capacity should exceed the final volume needed)
- A graduated cylinder or serological pipettes for accurate volume measurement
- A magnetic stirrer and stir bar (optional, for mixing)
Procedure
- Determine the Required Volume: Calculate the total volume of 1X running buffer needed to fill the inner and outer chambers of your specific electrophoresis apparatus.
- Dilute the 10X Stock: For any desired final volume of 1X buffer, use a 1:10 dilution factor. This means one part of 10X stock is mixed with nine parts of water.
- Mix Thoroughly: Gently stir or swirl the solution to ensure it is homogenous. If the 10X stock was stored at low temperature and crystals formed, it should have been warmed gently (not exceeding 40°C) and mixed thoroughly before use to re-dissolve all components [34].
- Verification: The prepared 1X working solution should be clear. Its pH is expected to be ~8.3 and does not normally require verification if the 10X stock was prepared and stored correctly.
Scientific Workflow and Mechanism of Action
The prepared 1X running buffer is integral to the sophisticated discontinuous buffer system that underpins SDS-PAGE. The following diagram and explanation outline the workflow and the critical role of the running buffer's components.
Diagram 1: SDS-PAGE Running Buffer Workflow
The mechanism of action is a two-stage process driven by the running buffer's interaction with the gel's distinct layers:
In the Stacking Gel (pH 6.8): The running buffer, at pH 8.3, contains glycinate ions. Upon entering the low-pH stacking gel, these ions become predominantly zwitterions (neutral charge), drastically reducing their mobility. This creates a steep voltage gradient between the highly mobile chloride ions (from the Tris-HCl in the gel) and the slow-moving glycine. All proteins, with mobilities intermediate to these two fronts, are compressed into extremely sharp bands before entering the resolving gel [35].
In the Resolving Gel (pH 8.8): When the ion fronts hit the resolving gel at pH 8.8, glycine molecules lose protons and become rapidly moving negatively charged glycinate ions. They quickly overtake the protein bands, which are now deposited at the top of the resolving gel in a tight line. The proteins, coated with SDS and thus uniformly negatively charged, then migrate through the pores of the polyacrylamide matrix. Separation occurs solely based on molecular size, as smaller proteins navigate the web more easily than larger ones [35].
Tips for Optimal SDS-PAGE Separation
- Temperature Control: Maintain a constant temperature (between 10°C-20°C) during the run to prevent "smiling" effects where outer lanes migrate slower than center lanes. Efficient heat transfer can be achieved by ensuring the buffer chamber is full and using a magnetic stirrer [36].
- Running Parameters: Follow the manufacturer's recommendations for your gel apparatus. Typical conditions are 100-150 volts for 40-60 minutes. Running too long will cause low molecular weight proteins to run off the gel, while running too short will result in poor resolution [36].
- Sample Preparation: For complete denaturation, heat samples at 95°C for 5 minutes. The inclusion of reducing agents like DTT or β-mercaptoethanol in the sample buffer is critical for breaking disulfide bonds in complex protein structures [36].
Best Practices for Buffer Storage and Shelf-Life Management
This application note details the best practices for the storage and shelf-life management of electrophoresis running buffers, with a specific focus on the Tris-Glycine-SDS system central to SDS-PAGE research. Proper management of these reagents is not merely a matter of convenience but is a fundamental prerequisite for the reproducibility and reliability of experimental data in drug development and basic research. Inferior buffer quality, resulting from improper preparation or extended storage, directly compromises protein separation resolution, leading to smearing, distorted bands, and erroneous molecular weight determinations that can invalidate downstream analyses, including western blotting.
Running Buffer Composition and Standardization
The Tris-Glycine-SDS buffer system is a discontinuous buffer system, where the ionic composition and pH of the running buffer are distinct from those of the gel, serving to stack proteins into sharp bands before they enter the separating gel [39]. The standard 10X running buffer concentrate consists of three key components, each with a critical function.
Table 1: Composition of a Standard 10X Tris-Glycine-SDS Running Buffer
| Component | Molecular Weight (g/mol) | Final 1X Concentration | Primary Function in Electrophoresis |
|---|---|---|---|
| Tris Base | 121.14 | 25 mM | Provides the buffering capacity; the common cation in the system [27] [40] [39]. |
| Glycine | 75.07 | 192 mM | The trailing ion in the discontinuous buffer system; its charge state changes with pH to enable protein stacking [27] [39]. |
| SDS (Sodium Dodecyl Sulfate) | 288.38 | 0.1% | Anionic detergent that coats proteins with a uniform negative charge, allowing separation primarily by molecular weight [40] [39]. |
The standard protocol involves diluting the 10X concentrate to a 1X working solution with deionized water prior to use. The pH of the 1X running buffer should be approximately 8.3 [40] [39].
Storage Conditions and Shelf-Life Stability
Adherence to defined storage parameters is critical for maintaining buffer integrity and performance. The following recommendations synthesize manufacturer guidelines and empirical research.
Table 2: Buffer Storage Conditions and Shelf-Life
| Buffer Form | Storage Temperature | Expected Shelf-Life | Key Storage Considerations |
|---|---|---|---|
| 10X Concentrate | Room Temperature (+15°C to +25°C) | 6-12 months | Stable in a sealed container protected from light. |
| 1X Working Solution | +4°C | 1-4 weeks | Susceptible to microbial growth and pH drift over time. |
| Pre-Cast Gels | +4°C | 4-8 weeks from manufacture | Do not freeze. Performance is impaired by extended exposure to room temperature [27]. |
For critical experiments, it is considered best practice to prepare 1X running buffer fresh from a 10X stock. This minimizes the risk of oxidation, contamination, and pH shift. Furthermore, while some laboratories reuse running buffer for economy, this practice is not recommended for optimal results, as it leads to the depletion of buffering ions and SDS, potentially causing uneven migration and poor resolution [41].
- Novel Long-Term Storage Method for Gels: Beyond liquid buffers, a validated method for long-term storage of electrophoresed and stained gels involves placing the destained gel in a flexible polyethylene bag. The polyethylene sheets adhere air-tightly to the gel, creating a micro-saturated environment that prevents significant cracking, shrinking, or protein diffusion for several months at room temperature without any additional storage buffer [42].
Detailed Experimental Protocols
Protocol 1: Preparation of 10X Tris-Glycine-SDS Running Buffer
This protocol is for preparing one liter of 10X running buffer concentrate.
Research Reagent Solutions:
- Tris Base: (121.14 g/mol); provides the buffering milieu.
- Glycine: (75.07 g/mol); acts as the trailing ion.
- SDS: (288.38 g/mol); denatures proteins and confers negative charge.
- Deionized Water: solvent for all components.
Methodology:
- Prepare 800 mL of deionized water in a clean, graduated 1 L beaker or bottle.
- Weigh and add 30.3 g of Tris base to the water while stirring.
- Weigh and add 144.1 g of Glycine to the solution. Continue stirring until all powders are fully dissolved.
- Weigh and add 10 g of SDS to the solution. Stir gently to avoid excessive foaming until the SDS is completely dissolved.
- Top up the solution with deionized water to a final volume of 1 L.
- Verify that the pH of the 10X stock is approximately 8.3; typically, no adjustment is needed.
- Store the 10X concentrate in a sealed, labeled container at room temperature.
Protocol 2: Electrophoresis Using Pre-Cast Gels and Prepared Buffer
This protocol assumes the use of a standard mini-gel apparatus and pre-cast gels.
Methodology:
- Gel Equilibration: Remove the pre-cast gel from its packaging and rinse the cassette with deionized water. Remove the comb and thoroughly rinse the sample wells with 1X running buffer [27].
- Buffer Preparation: Dilute the 10X running buffer concentrate to a 1X working solution with deionized water. For a mini-gel cell, this typically requires ~800 mL for the inner and outer chambers combined [27].
- Apparatus Assembly: Assemble the gel cassette(s) in the electrophoresis chamber according to the manufacturer's instructions. Fill the inner (upper) and outer (lower) chambers with the 1X running buffer.
- Sample Loading: Load prepared protein samples and molecular weight markers into the wells.
- Electrophoresis Run: Connect the apparatus to a power supply and run at constant voltage (e.g., 125-150 V) until the dye front (bromophenol blue) reaches the bottom of the gel [27] [43].
- Post-Run Buffer Management: After the run, the 1X running buffer can be discarded. For non-critical runs, it may be reused once or twice if immediately returned to the storage bottle and labeled with a use-count, though fresh buffer is always recommended for optimal performance [41].
Workflow and Management Strategies
The following workflow diagram outlines the logical process for managing buffer preparation, use, and storage to ensure quality and reproducibility.
The Scientist's Toolkit: Essential Reagents for SDS-PAGE
Table 3: Key Research Reagent Solutions for SDS-PAGE
| Reagent | Function | Critical Storage Parameters |
|---|---|---|
| 10X Running Buffer | Concentrate for preparing the electrophoresis mobile phase. | Store at room temperature; 6-12 month shelf-life in a sealed container. |
| Pre-Cast Gels | Acrylamide matrix for protein separation. | Store at +4°C; shelf-life of 4-8 weeks. Do not freeze [27]. |
| Ammonium Persulfate (APS) | Catalyst for polyacrylamide gel polymerization. | Aliquot and store at -20°C for long-term stability, or at +4°C for up to one month [41]. |
| Protein Samples | Analyte of interest, often in Laemmli buffer. | After denaturation, can be stored at -20°C for extended periods (up to a year) [41]. |
| Polyethylene Bags | For long-term storage of stained gels. | Room temperature; enables buffer-less gel storage for months [42]. |
Concluding Remarks
Robust and reproducible science in SDS-PAGE-based research is fundamentally linked to reagent quality. The implementation of disciplined buffer storage and shelf-life management protocols, as detailed in this application note, mitigates a significant and often overlooked source of experimental variance. By standardizing the preparation, use, and storage of electrophoresis buffers, researchers and drug development professionals can ensure the integrity of their protein separation data, thereby strengthening the foundation of their scientific conclusions.
Within the broader context of running buffer composition and preparation for SDS-PAGE research, the Tris-Tricine-SDS buffer system represents a significant advancement over traditional Tris-Glycine systems for resolving low molecular weight proteins and peptides. This application note details the fundamental principles, optimized protocols, and practical implementation of Tris-Tricine-SDS electrophoresis, enabling researchers to overcome the limitations of conventional Laemmli-based methods. By providing enhanced separation of proteins below 20 kDa, this alternative buffer system has become indispensable in proteomics, peptide characterization, and drug development workflows where analysis of small proteins is critical.
Principles of Tris-Tricine-SDS Electrophoresis
The Tris-Tricine-SDS buffer system was specifically developed to address a fundamental limitation of traditional Tris-Glycine SDS-PAGE: the poor resolution of low molecular weight proteins below 20 kDa [44]. In conventional Laemmli systems, the trailing glycine ion creates a zone of stacked dodecylsulfate (DS) micelles that causes mixing with smaller proteins, resulting in fuzzy bands and decreased resolution [44]. This interference particularly affects the fixing and staining of smaller proteins, making accurate analysis challenging.
Tris-Tricine-SDS electrophoresis overcomes these limitations through two key modifications: replacement of the trailing glycine ion with tricine and adjustment of the gel buffer to a lower pH [44]. The tricine ion, with its different electrophoretic mobility compared to glycine, enables more efficient stacking and destacking of low molecular weight proteins, resulting in superior separation of small peptides [44]. The system creates a distinct separation boundary where smaller proteins that would typically co-migrate with DS micelles in Tris-Glycine systems become well-resolved, yielding sharper bands and enhanced resolution in the 1-20 kDa range [44].
Recent advancements have further optimized this approach with the development of Tris-Tricine-HEPES composite buffers, which provide gradient-like simultaneous separation of both small (<10 kDa) and large (>400 kDa) proteins in single-percentage polyacrylamide gels [45] [13]. This innovative buffer composition creates multiple ionic boundaries throughout the gel (Chloride > Tricine > HEPES > protein ions), significantly improving the resolving power while reducing running times by minimizing excessive heat generation at higher voltages [13].
Comparative Analysis of SDS-PAGE Buffer Systems
Table 1: Quantitative comparison of SDS-PAGE buffer system compositions
| Component | Tris-Glycine (Laemmli) | Traditional Tris-Tricine | Advanced Tris-Tricine-HEPES |
|---|---|---|---|
| Leading Ion | Chloride | Chloride or Acetate | Chloride or Acetate |
| Trailing Ion | Glycine | Tricine | Tricine + HEPES |
| Tris Concentration | 0.25 M (10X) [31] | 1.2 M (20X) [31] | Varies (25-100 mM HEPES) [13] |
| Buffer Concentration | 1.92 M Glycine (10X) [31] | 0.8 M Tricine (20X) [31] | 100-200 mM Tricine [13] |
| SDS Concentration | 1% (10X) [31] | 2% (20X) [31] | 0.1% [13] |
| Typical pH Range | 8.6 [31] | 8.3-8.5 [46] [31] | 7.5-8.0 [13] |
| Optimal Separation Range | 10-200 kDa [13] | 2-20 kDa [44] | <10->400 kDa [45] [13] |
Table 2: Performance characteristics of different SDS-PAGE buffer systems
| Performance Parameter | Tris-Glycine | Tris-Tricine | Tris-Tricine-HEPES |
|---|---|---|---|
| Small Protein Resolution (<15 kDa) | Poor [13] | Excellent [44] | Superior [13] |
| Large Protein Resolution (>100 kDa) | Good | Limited [13] | Excellent [13] |
| Typical Running Time | 60-90 minutes | ~90 minutes [44] | ~35 minutes [13] |
| Optimal Voltage Range | 100-200V | 125V [44] | 150V + 200V [13] |
| Heat Generation at High Voltage | Excessive [13] | Moderate | Reduced [13] |
| Compatibility with Western Blotting | Good | Excellent [44] | Excellent [13] |
Experimental Protocols
Standard Tris-Tricine SDS-PAGE Protocol
Materials Required:
- Novex Tricine SDS Sample Buffer (2X) [44]
- Tricine SDS Running Buffer (10X) [44]
- NuPAGE Reducing Agent (10X) for reduced samples [44]
- Protein molecular weight markers appropriate for small proteins [44]
- Pre-cast Tricine gels or materials for casting custom gels [44]
Sample Preparation:
- For reduced samples, combine in a microcentrifuge tube: 4 μL protein sample, 5 μL Tricine SDS Sample Buffer (2X), and 1 μL NuPAGE Reducing Agent (10X) for a total volume of 10 μL [44].
- For non-reduced samples, combine: 5 μL protein sample, 5 μL Tricine SDS Sample Buffer (2X), and adjust to 10 μL with deionized water [44].
- Heat samples at 85°C for 2 minutes and load immediately onto the gel [44].
Gel Electrophoresis:
- Prepare 1X Tricine SDS running buffer by diluting 100 mL of 10X concentrate with 900 mL deionized water [44].
- Remove pre-cast gel from packaging, rinse with deionized water, and remove the comb carefully [44].
- Rinse sample wells three times with 1X Tricine SDS running buffer [44].
- Assemble gel apparatus according to manufacturer instructions (e.g., XCell SureLock Mini-Cell) [44].
- Load prepared samples and appropriate molecular weight markers into wells [44].
- Run gel at constant voltage of 125V for approximately 90 minutes, or until the phenol red tracking dye reaches the bottom of the gel [44].
- Expected current: start at 80 mA, ending at 40 mA for single gels [44].
Advanced Tris-Tricine-HEPES Protocol for Broad Range Separation
Running Buffer Preparation: Prepare a novel running buffer composed of Tris, Tricine, and HEPES (FRB - fast-running buffer) with systematic variation of HEPES concentrations (25, 50, 75, and 100 mM) to achieve a final pH in the range of 7.5-8.0 without pH adjustment [13].
Electrophoresis Conditions:
- Use pre-cast Tris-Acetate gels for optimal results with this buffer system [13].
- Run gels using "fast run" conditions: 150V for 15 minutes followed by 200V for 20 minutes (total running time: 35 minutes) [13].
- This protocol enables simultaneous resolution of proteins across an exceptionally broad molecular weight range (15-450 kDa) in a single 10% gel [13].
Tris-Tricine SDS-PAGE Experimental Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential reagents for Tris-Tricine SDS-PAGE research
| Reagent | Function | Specifications | Commercial Examples |
|---|---|---|---|
| Tris-Tricine-SDS Running Buffer | Cathode (upper) buffer for protein separation | 100 mM Tris, 100 mM Tricine, 0.1% SDS (1X) [46] | Sigma-Aldrich T1165 [46] |
| Tricine SDS Sample Buffer | Denatures and prepares proteins for electrophoresis | 2X concentration, contains SDS | Novex Tricine SDS Sample Buffer [44] |
| Reducing Agent | Reduces disulfide bonds for complete denaturation | 10X concentration (DTT or similar) | NuPAGE Reducing Agent [44] |
| Pre-cast Tricine Gels | Provides optimized gel matrix for separation | Various percentages, 10-well format | Novex Tricine Pre-Cast Gels [44] |
| High-Resolution Resolving Gel | Separates small proteins and peptides | 16.5% acrylamide, may include urea [47] [48] | Custom-cast gels [48] |
| Protein Molecular Weight Markers | Reference for protein size determination | Low molecular weight range (2.5-200 kDa) | Various suppliers |
Technical Notes and Troubleshooting
Critical Considerations:
- Never adjust the pH of commercial running buffers, as this compromises their buffering capacity and electrophoretic performance [31].
- Store pre-cast gels at +4°C and use within their shelf life (typically 4-8 weeks) to maintain optimal separation characteristics [44].
- Use gels immediately after removing from refrigeration, as extended exposure to room temperature seriously impairs performance [44].
- Always wear gloves when handling gels due to the presence of residual acrylamide monomer, a known neurotoxin [44].
Troubleshooting Common Issues:
- Faint shadow or "ghost" bands below expected protein bands: Caused by gel lifting from cassette due to expired gels or improper storage. Replace with fresh gels stored at appropriate temperature [44].
- Streaking of proteins: Results from sample overload, high salt concentration, or particulate contaminants. Reduce protein load, desalt samples, or clarify by centrifugation [44].
- Extended run times: Caused by overly dilute running buffer. Prepare fresh running buffer at correct dilution according to manufacturer instructions [44].
- Bands in outer lanes curving upwards ("smiling" effect): Often indicates use of expired gels or concentrated buffer. Use fresh gels and properly diluted buffers [44].
Applications in Research and Development
The Tris-Tricine-SDS buffer system has proven particularly valuable in pharmaceutical development and proteomics research, where analysis of low molecular weight proteins and peptides is essential. Its superior resolution in the 2-20 kDa range makes it ideal for characterizing therapeutic peptides, antibody fragments, and protein degradation products [44]. The system also facilitates direct sequencing of proteins after transfer to PVDF membranes, as tricine does not interfere with sequencing processes [44].
Recent innovations combining Tris-Tricine with HEPES have further expanded application possibilities by enabling simultaneous analysis of both small and large molecular weight species, significantly streamlining high-throughput workflows in drug discovery [13]. This advanced buffer formulation also reduces running times without excessive heat generation, addressing a critical bottleneck in proteomic studies requiring rapid turnaround [13].
The compatibility of Tris-Tricine separated proteins with downstream applications such as western blotting, mass spectrometry, and protein sequencing ensures its continued relevance in comprehensive protein characterization pipelines, particularly in the context of biomarker discovery and biopharmaceutical development.
Within the framework of a broader thesis on running buffer composition and preparation for SDS-PAGE research, this application note highlights the critical role of electrophoretic buffers in the specific context of food allergen analysis. The reliable detection and quality control of allergenic proteins in food products present significant challenges for the pharmaceutical and food safety industries. SDS-PAGE (Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis) serves as a foundational technique in this field, providing a robust method for separating complex protein mixtures from food extracts based on molecular weight [49] [50]. The consistency and reliability of these separations are fundamentally dependent on the precise composition and preparation of the running buffer, underscoring its importance in generating reproducible and analytically sound data.
Variability in raw materials, purification processes, and formulation techniques can lead to significant differences in the protein composition, potency, and biological activity of commercial allergen extracts [51]. These inconsistencies pose substantial risks, as poor-quality extracts can reduce the accuracy of diagnostic tests, potentially leading to false negatives and underestimation of allergic responses [51]. Consequently, rigorous quality assessment using techniques like SDS-PAGE is indispensable for ensuring the consistency, reliability, and safety of products used both in allergy diagnosis and therapy.
The Role of SDS-PAGE Running Buffer in Allergen Analysis
Fundamental Principles of SDS-PAGE Running Buffer
In SDS-PAGE, the running buffer creates the essential environment for protein separation. Its primary functions are to carry the electrical current through the gel system and to maintain a stable pH that ensures proper protein charge and mobility [49] [6]. The standard Tris-glycine discontinuous buffer system, pioneered by Laemmli, employs a running buffer typically composed of Tris base, glycine, and SDS at pH 8.3 [49] [27]. This specific pH is critical because it influences the charge states of the glycine ions and the proteins themselves, directly impacting the stacking and separation processes [49].
The running buffer works in concert with the gel buffers (stacking gel at pH 6.8 and resolving gel at pH 8.8) to create a discontinuous system that first concentrates protein samples into sharp bands before they enter the resolving gel [49]. During electrophoresis, the applied electric field causes the negatively charged proteins (due to SDS binding) to migrate toward the positively charged anode [49]. The glycine in the running buffer plays a pivotal role in this process; at pH 8.3, glycine exists predominantly as glycinate anions that become neutral zwitterions upon entering the lower pH stacking gel, creating a trailing ion front that herds proteins into a tight zone between the highly mobile chloride ions (leading ions) and the slower glycine zwitterions [49]. When this ion front reaches the higher pH resolving gel, glycine regains negative charge and speeds past the proteins, depositing them in a sharp band at the top of the resolving gel where size-based separation begins [49].
Quantitative Composition of Standard Running Buffers
Table 1: Standard formulation for 10X SDS-PAGE Running Buffer Concentrate
| Component | Molecular Weight (g/mol) | Final Concentration (10X) | Function in Separation |
|---|---|---|---|
| Tris base | 121.14 | 0.25 M | Maintains pH; provides common cation (Tris+) throughout the system [49] [27] |
| Glycine | 75.07 | 1.92 M | Primary anion in running buffer; acts as trailing ion in stacking phase [49] [27] |
| SDS (Sodium Dodecyl Sulfate) | 288.38 | 1% (w/v) | Denatures proteins; provides uniform negative charge [49] [50] |
For use, this 10X concentrate is typically diluted to 1X with distilled water, resulting in final working concentrations of 25 mM Tris, 192 mM glycine, and 0.1% SDS [12]. The pH of the 1X running buffer should be approximately 8.3 and generally does not require adjustment [27]. It is recommended to prepare fresh running buffer for each experiment, as reuse can lead to contamination with protein fragments, degraded SDS, and unreliable results due to altered pH and conductivity [6].
Experimental Protocol: Quality Control of Food Allergen Extracts via SDS-PAGE
Workflow for Allergen Extract Analysis
The following diagram illustrates the complete experimental workflow for analyzing food allergen extracts using SDS-PAGE, from sample preparation to final analysis.
Detailed Methodology
3.2.1 Sample Preparation
- Denaturation: Mix the protein extract from food sources (e.g., peanut, milk, egg) with an equal volume of 2X Laemmli sample buffer [50] [27]. A typical 2X sample buffer contains:
- Heating: Heat the mixture at 85°C for 2-5 minutes to ensure complete denaturation [27]. Do not heat samples if subsequent native (non-denaturing) PAGE is planned.
3.2.2 Gel Electrophoresis
- Buffer Preparation: Prepare 1X SDS running buffer by diluting the 10X concentrate (see Table 1) with distilled water. For a mini-gel system, approximately 1 liter of 1X buffer is required [27].
- Gel Setup: Assemble the electrophoresis unit. Load the prepared samples and appropriate molecular weight markers into the wells of a pre-cast or hand-cast polyacrylamide gel [27].
- Electrophoresis Run: Fill the upper and buffer chambers with the 1X running buffer. Connect the power supply and run the gel at constant voltage (e.g., 125 V for Tris-Glycine gels) until the dye front reaches the bottom of the gel (approximately 90 minutes) [27].
3.2.3 Post-Electrophoresis Processing and Analysis
- Gel Staining: After separation, proteins are visualized using stains such as Coomassie Blue (for general protein detection), silver stain (higher sensitivity), or fluorescent stains (broad dynamic range) [50].
- Band Analysis: The gel is imaged using a documentation system. Protein bands are identified by comparing their migration distance to that of the molecular weight standards [50].
- Quality Assessment: The protein banding pattern is analyzed for presence, intensity, and sharpness of expected allergen bands. Inconsistent patterns or smearing between different production batches of allergen extracts can indicate problems with purity, degradation, or inconsistent composition [51].
Advanced Applications and Complementary Techniques
Beyond SDS-PAGE: Biosensing for High-Throughput Quality Control
While SDS-PAGE is a cornerstone technique, recent advancements have introduced sophisticated biosensing technologies for massive screening of allergen extracts. These platforms, such as protein array technologies immobilized on polycarbonate chips, can assess the biological activity (IgE-binding capacity) of allergen extracts by incubating them with serum from allergic patients [51]. This provides functional quality control that goes beyond mere separation by size. Such high-throughput methods enable the effective comparison of manufacturers and offer a reliable alternative to traditional approaches like SDS-PAGE and ELISA [51]. Detection for these arrays can be achieved using cost-effective technologies, including smartphone imaging and DVD reader-based assays, enhancing accessibility for routine quality control in production processes [51].
Lateral Flow Assays for Rapid Allergen Detection
In industrial settings, lateral flow devices are widely used for rapid, qualitative detection of allergenic residues in food products and on processing equipment. These tests utilize specific antibodies and require specialized buffers, such as the Reveal 3-D Food Buffer, to facilitate the extraction and detection of low levels of allergens like almond, egg, peanut, and soy from complex food matrices [52]. This represents another critical application of buffer systems in the broader field of food allergen control.
Comparison of Buffer Systems in Protein Analysis
Table 2: Key differences between Running Buffer and Transfer Buffer in Western Blotting
| Characteristic | Running Buffer | Transfer Buffer |
|---|---|---|
| Primary Function | Facilitates protein separation based on size during SDS-PAGE [6] | Transfers separated proteins from gel to membrane for detection [6] |
| Typical Composition | Tris, Glycine, SDS [49] [12] | Tris, Glycine, Methanol (often 20%); may contain low SDS [6] |
| Key Components' Roles | SDS maintains protein denaturation and charge [49] | Methanol promotes protein adhesion to the membrane [6] |
| When It's Used | During gel electrophoresis separation [6] | During Western blotting (electrophoretic transfer) [6] |
The Scientist's Toolkit: Essential Reagents for Allergen Analysis
Table 3: Key research reagents for SDS-PAGE-based quality control of allergen extracts
| Reagent / Material | Function / Role in Experiment |
|---|---|
| SDS Running Buffer | Creates the conductive medium for electrophoresis; ensures proper pH and ion mobility for protein separation [49] [6] |
| Laemmli Sample Buffer | Denatures proteins, adds negative charge, and provides density for loading samples into wells [49] [50] |
| Pre-cast Polyacrylamide Gels | Provide a consistent, ready-to-use matrix with defined pore sizes for separating proteins by molecular weight [27] |
| Molecular Weight Markers | Allow estimation of the molecular weights of separated proteins in the sample [50] [27] |
| Reducing Agent (DTT or BME) | Breaks disulfide bonds in proteins to ensure complete unfolding and accurate size-based separation [49] [27] |
| Protein Stains (Coomassie, Silver) | Visualize separated protein bands on the gel after electrophoresis [50] |
| Polycarbonate Chips & Array Robots | Enable high-throughput, biosensor-based quality screening of allergen extracts' IgE reactivity [51] |
The critical role of running buffer in SDS-PAGE extends directly into the realm of food allergen detection and quality control, where precise and reproducible protein separation is non-negotiable. The standardized Tris-glycine-SDS running buffer system is fundamental to obtaining reliable protein profiles of allergen extracts, enabling scientists to assess purity, identify contaminants, and ensure batch-to-batch consistency. As the field advances, the integration of traditional electrophoretic methods with novel biosensing platforms promises even more robust and high-throughput approaches for safeguarding individuals with food allergies. A deep understanding of buffer composition and function remains the foundation upon which reliable analytical outcomes are built.
Troubleshooting SDS-PAGE: How Running Buffer Affects Gel Results
Smeared bands in SDS-PAGE are a common frustration that can compromise data integrity and obscure critical results in drug development. Within the broader context of running buffer composition and preparation, these smears are frequently a direct consequence of two key factors: excessive voltage during electrophoresis and improper running buffer formulation or condition. This application note provides a detailed, evidence-based protocol for diagnosing and resolving these issues to ensure reproducible, high-quality protein separation.
Systematic Troubleshooting for Smeared Bands
A systematic approach is essential for diagnosing the root cause of smearing. The following table outlines the primary culprits related to voltage and buffer, along with their confirmed solutions.
| Problem & Cause | Underlying Principle | Recommended Solution | Experimental Verification |
|---|---|---|---|
| Excessive Voltage [53] [54] [55] | High voltage causes Joule heating, leading to protein denaturation and uneven migration through the gel matrix. | Run the gel at a lower voltage (e.g., 80-150V) for a longer duration [53] [4] [56]. Use a constant current setting if available [55]. | Monitor buffer temperature; it should not feel warm to the touch. A cooler run will produce sharper bands. |
| Improper Running Buffer [53] [54] | Incorrect ion concentration/pH disrupts current flow and protein charge, while old/contaminated buffer can degrade. | Prepare fresh 1X running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH ~8.3) [57] [4] [6]. Do not reuse buffer from previous runs [6]. | Check buffer pH before use. Fresh buffer will resolve issues of unusually fast or slow migration [53] [54]. |
| High Salt Concentration in Sample [54] [55] | High salt increases local conductivity, distorting the electric field and causing band spreading and smearing. | Desalt samples via dialysis, desalting columns, or trichloroacetic acid (TCA) precipitation prior to loading [54]. | Compare a desalted sample versus a crude sample on the same gel; the desalted sample will show reduced smearing. |
| Overloaded Protein [54] [56] | Exceeding the gel's protein capacity overwhelms the resolving power, causing bands to merge and smear. | Reduce the total protein load per lane. Perform a protein concentration assay and run a loading series to determine the optimal amount. | A dilution series of the same sample will show that smearing decreases with lower protein concentration. |
Other Contributing Factors
While voltage and buffer are central, a complete diagnosis must also consider:
- Sample Degradation: Protease contamination can degrade proteins into a heterogenous mixture. Always use fresh protease inhibitors and keep samples on ice [55].
- Incomplete Denaturation: Ensure samples are properly mixed with SDS-containing Laemmli buffer and heated at 95-100°C for 5 minutes to achieve full denaturation [57] [54].
Experimental Protocol for Optimal SDS-PAGE
This detailed protocol is designed to preemptively avoid smearing by enforcing best practices for buffer preparation and gel running.
Running Buffer Preparation (1X Formula)
- Recipe: To prepare 1 liter of 1X running buffer from a 10X stock:
- Critical Step: Verify the pH of the diluted 1X buffer is approximately 8.3. No adjustment is typically needed [4] [6].
- Storage: For best results, prepare fresh 1X buffer for each run. If necessary, store at 4°C and use within a few days. Do not reuse buffer from previous electrophoresis runs [6].
Gel Electrophoresis Method
- Apparatus Setup: Assemble the gel electrophoresis unit according to the manufacturer's instructions. Pour the freshly prepared 1X running buffer into the inner and outer chambers, ensuring the wells are fully submerged [57].
- Sample Loading: Load prepared protein samples and molecular weight markers into the wells. Minimize the time between loading and starting the run to prevent sample diffusion from the wells [53].
- Voltage Parameters:
- Initial Run: Apply a constant voltage of 80V until the dye front has moved through the stacking gel and entered the resolving gel [4].
- Main Separation: Increase the voltage to a constant 150V for the remainder of the run [57]. For larger gels or to minimize heating, a lower voltage (e.g., 110-120V) for a longer time is acceptable [53] [56].
- Temperature Management: If the apparatus feels warm to the touch during the run, reduce the voltage further. For critical experiments, run the gel in a cold room or use a unit with a cooling module [53] [55].
- Run Completion: Stop electrophoresis immediately when the bromophenol blue dye front reaches approximately 1 cm from the bottom of the gel [53] [57].
Diagnostic Workflow and Reagent Solutions
Decision Pathway for Smeared Bands
The following diagnostic diagram outlines a step-by-step logical workflow to identify and correct the cause of smeared bands in your SDS-PAGE results.
The Scientist's Toolkit: Essential Research Reagents
The quality and proper use of core reagents are fundamental to preventing smearing and achieving optimal protein separation.
| Reagent / Material | Function & Importance in Preventing Smearing |
|---|---|
| Tris-Glycine-SDS Running Buffer [6] [58] | Maintains optimal pH and conductivity. Fresh buffer ensures correct ion concentration for uniform charge and migration, preventing blurry bands. |
| High-Purity Acrylamide/Bis-Acrylamide [56] | Forms the polyacrylamide gel matrix. Properly mixed and degassed solutions ensure a uniform pore size for consistent sieving of proteins. |
| Laemmli Sample Buffer (with SDS & BME) [4] [58] | Denatures proteins and coats them with negative charge. SDS linearizes proteins, while β-mercaptoethanol (BME) reduces disulfide bonds, preventing aggregation. |
| Ammonium Persulfate (APS) & TEMED [4] | Catalyzes the polymerization of acrylamide. Fresh reagents are critical for forming a consistent gel without irregularities that can distort bands. |
| Precision Voltage Power Supply [53] [55] | Provides stable electrical conditions. Running at optimized, lower voltages minimizes Joule heating, a primary cause of band smiling and smearing. |
Key Takeaways for Reproducible Results
Achieving sharp, well-resolved bands in SDS-PAGE is critical for accurate analysis in research and drug development. The most effective strategy for eliminating smearing is often a combination of using freshly prepared running buffer and optimizing electrophoresis conditions to minimize heat generation. By adhering to the detailed protocols and systematic troubleshooting guide provided, researchers can consistently produce high-quality gels, ensuring reliable data for their scientific conclusions.
Within the framework of a broader thesis on running buffer composition and preparation for SDS-PAGE research, this application note addresses a critical yet frequently overlooked variable: buffer integrity. The Tris-glycine discontinuous buffer system, foundational to SDS-PAGE, relies on precise ionic conditions and pH to concentrate and resolve protein samples effectively [59]. When running buffers are improperly formulated, stored beyond their shelf life, or experience pH drift, the resulting poor band separation compromises data quality and reproducibility. This document provides researchers and drug development professionals with detailed protocols and analytical data to diagnose, troubleshoot, and prevent buffer-related resolution failures, thereby enhancing the reliability of protein analysis in downstream applications like western blotting.
The Critical Role of Running Buffer in SDS-PAGE
SDS-PAGE separates proteins primarily by molecular weight through a sophisticated discontinuous buffer system that involves both a stacking gel and a resolving gel [59]. The running buffer, typically containing Tris, glycine, and SDS, is not merely a conductive medium but an active participant in establishing the ionic conditions necessary for sharp band separation [27].
The fundamental mechanism hinges on the creation of moving ionic boundaries. In the standard Tris-Glycine system, chloride ions (Cl⁻) from the gel buffer act as highly mobile leading ions, while glycine ions from the running buffer serve as trailing ions [59] [27]. At the pH of the stacking gel, glycine exists predominantly in a zwitterionic state with a low electrophoretic mobility, creating a narrow, high-voltage gradient that forces protein samples into a tight stack between the chloride and glycine fronts [59]. When this ionic front reaches the higher pH of the resolving gel, glycine ions become fully negatively charged and overtake the proteins, which then separate according to size as they migrate through the polyacrylamide matrix [59] [27]. Any deviation in buffer composition, concentration, or pH disrupts this delicate balance of mobilities, leading to diffuse bands, poor resolution, and unreliable molecular weight determination.
Troubleshooting Buffer-Related Band Separation Issues
The following table summarizes common symptoms, their probable buffer-related causes, and corrective actions for poor band separation.
Table 1: Troubleshooting Guide for Poor Band Separation in SDS-PAGE
| Symptom | Probable Buffer-Related Cause | Corrective Action |
|---|---|---|
| Smeared Bands | Improper running buffer concentration (too diluted); excessive voltage causing buffer heating [60] [61]. | Prepare running buffer at correct salt concentration; reduce voltage and extend run time [60]. |
| Poor Resolution (Unclear/Overlapping Bands) | Improper running buffer ion concentration or pH, disrupting current flow and pH maintenance [60]. | Remake running buffer to ensure correct ion concentration and pH (8.3 for Tris-Glycine) [60] [61]. |
| 'Smiling' Bands (Curved bands) | Excessive heat generation from high voltage, causing uneven gel temperature [60] [61]. | Reduce voltage; run gel in a cold room or use a cooling apparatus [60]. |
| Vertical Streaking | High salt contamination in sample, increasing local conductivity and disrupting the voltage gradient [61]. | Desalt samples using dialysis or desalting columns before loading [61]. |
| Inconsistent Migration Between Runs | Use of old or degraded buffer; pH drift over time [61]. | Always prepare fresh running buffer or monitor pH/conductivity of stored buffers; avoid reuse [61]. |
The logical relationships between buffer status, the resulting problems in the electrophoresis process, and the final gel outcomes are mapped in the workflow below.
Experimental Protocol for Buffer Preparation and Evaluation
Standard Tris-Glycine SDS Running Buffer Preparation
This protocol details the preparation of the traditional Laemmli running buffer, suitable for separating proteins within the 10-200 kDa range [27].
Materials:
- Tris Base (MW = 121.14 g/mol)
- Glycine (MW = 75.07 g/mol)
- SDS (Sodium Dodecyl Sulfate)
- Deionized Water
- pH Meter
Method:
- To prepare 1X running buffer, add the following to deionized water to a final volume of 1 liter:
- 2.5 mM Tris: 0.3 g
- 192 mM Glycine: 14.4 g
- 0.1% (w/v) SDS: 1 g
- Stir until all components are completely dissolved. No pH adjustment is typically required, as the components should yield a pH of approximately 8.3 [27].
- Verify the pH using a calibrated pH meter. If necessary, adjust with HCl or NaOH.
- The 1X buffer can be stored at 4°C for up to one week. For longer-term storage, prepare a 10X stock solution without SDS, which is stable for months. Dilute the stock and add the appropriate amount of SDS fresh before use [61].
Fast-Running Tris-Tricine-HEPES (FRB) Buffer Protocol
For high-throughput applications, a novel running buffer utilizing Tris, Tricine, and HEPES offers significantly reduced run times and superior resolution across a wider molecular weight range (15-450 kDa) in a single 10% gel [13].
Materials:
- Tris Base
- Tricine (N-[Tris(hydroxymethyl)methyl]glycine)
- HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- SDS
- Deionized Water
Method:
- The specific molarities for an optimized FRB are defined in the primary literature. A representative formulation involves combining Tris, Tricine, and HEPES in a specific ratio to achieve a final pH in the range of 7.5–8.0 without further adjustment [13].
- Add 0.1% (w/v) SDS to the buffer solution.
- Electrophoresis Conditions: Run gels at 150 V for 15 minutes, followed by 200 V for 20 minutes (total run time: 35 minutes) [13].
Table 2: Quantitative Comparison of Standard and Fast-Running Buffer Systems
| Parameter | Tris-Glycine-SDS (Standard) | Tris-Tricine-HEPES (FRB) |
|---|---|---|
| Running Buffer Composition | 25 mM Tris, 192 mM Glycine, 0.1% SDS, pH 8.3 [27] | Tris, Tricine, HEPES (specific molarities as per literature), 0.1% SDS, pH ~7.5-8.0 [13] |
| Effective Separation Range | ~10-200 kDa [13] | 15-450 kDa in a single 10% gel [13] |
| Typical Running Conditions | 125-150 V constant, 60-90 minutes [27] [60] | 150 V for 15 min, then 200 V for 20 min (Total: 35 min) [13] |
| Key Advantages | Low cost; well-established protocol [13] | Fast separation; wide molecular weight range; reduced heat generation [13] |
| Key Limitations | Poor resolution of small proteins (<15 kDa); long running times; excessive heat at high voltages [13] | Requires optimization; non-standard buffer components [13] |
The Scientist's Toolkit: Essential Reagents for SDS-PAGE Buffer Studies
Table 3: Key Research Reagents and Materials for SDS-PAGE Buffer Experiments
| Item | Function/Application |
|---|---|
| Tris Base | Primary buffering agent in gels and running buffers; maintains pH in the 7-9 range critical for electrophoresis [59] [27]. |
| Glycine | Trailing ion in the discontinuous buffer system; its charge state changes with pH, enabling protein stacking and separation [59] [27]. |
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent that denatures proteins and confers a uniform negative charge, allowing separation by size rather than native charge [59]. |
| Tricine | Alternative trailing ion; improves resolution of low molecular weight proteins (<15 kDa) [13]. |
| HEPES | Zwitterionic buffer used in novel running buffer formulations to create multiple ionic boundaries, enhancing resolution over a wide mass range [13]. |
| Precast Gels (e.g., Tris-Glycine) | Ensure consistency and reproducibility when comparing buffer performance, eliminating variability from gel casting [27]. |
| Protein Molecular Weight Marker | Essential control for monitoring separation efficiency and accurately determining protein sizes under different buffer conditions [27]. |
| β-Mercaptoethanol or DTT | Reducing agents added to sample buffer to break disulfide bonds, ensuring complete protein denaturation [59] [27]. |
The integrity of the running buffer is a non-negotiable factor for achieving optimal band separation in SDS-PAGE. Researchers must be vigilant in the fresh preparation and proper storage of buffers to prevent the resolution issues detailed in this note. Furthermore, the exploration of novel buffer systems, such as Tris-Tricine-HEPES, presents a promising avenue for overcoming the inherent limitations of traditional Tris-Glycine buffers, particularly for high-throughput and wide-mass-range applications in modern drug development. By adhering to rigorous buffer management protocols and considering advanced formulations, scientists can ensure the generation of high-quality, reproducible protein data.
Preventing the 'Smiling Band' Effect and Managing Gel Heat
The 'smiling band' effect, characterized by curved protein bands that dip towards the edges of the gel, is a common artifact in SDS-PAGE that compromises data quality and interpretation. This phenomenon is directly caused by uneven heat distribution across the polyacrylamide gel during electrophoresis [62] [63]. When an electric current flows through the gel, resistance generates heat. If this heat is not dissipated uniformly, the outer lanes, which are often in closer contact with the cooler glass plates, can become warmer than the center lanes. This temperature gradient causes proteins in the warmer regions to migrate faster, resulting in the characteristic upward-curving "smile" shape [62]. Effectively managing this joule heat is therefore critical not only for preventing smiling bands but also for ensuring consistent, high-resolution protein separation, which forms the foundation for reliable analysis in drug development and research contexts.
Understanding the Root Cause: Joule Heating in SDS-PAGE
The fundamental principle of SDS-PAGE relies on the motion of charged proteins through a polyacrylamide matrix under an electric field. The running buffer, typically a Tris-Glycine-SDS system, provides the ions necessary to conduct current [27] [64]. The relationship between power (P), voltage (V), and current (I) is given by P = V × I. This power is dissipated as joule heat within the gel matrix. Excessive or uneven heat causes localized expansion of the gel pores, reducing their sieving effect and allowing proteins in those regions to migrate faster and with less resolution [62] [63]. Furthermore, high heat can destabilize the buffer system and even lead to protein degradation or aggregation. The traditional Tris-Glycine buffer system is particularly prone to these issues at higher voltages, which are often used to reduce run times in high-throughput applications [13]. Thus, the composition and pH of the running buffer, along with the physical setup of the electrophoresis apparatus, are key factors in heat management.
Systematic Troubleshooting and Prevention Strategies
A multi-faceted approach is required to effectively eliminate the smiling band effect. The strategies below are listed in order of practicality and effectiveness.
Voltage and Current Optimization: Running the gel at a lower constant voltage is the most straightforward corrective action. While a common practice is to run mini-gels at 150-200V, reducing the voltage to 125V or lower significantly minimizes heat generation [27] [62]. If time is a constraint, a two-step protocol can be used: an initial low-voltage step (e.g., 80V) to allow proteins to stack, followed by a higher-voltage step (e.g., 150V) for the separation phase, monitoring the current to ensure it does not exceed 30-40 mA per gel at the start [27].
Active and Passive Cooling Methods: Implementing efficient cooling is paramount.
- External Cooling: Performing the electrophoresis run in a cold room (4°C) is a highly effective method [62].
- Apparatus Cooling: If a cold room is not available, placing ice packs directly in the outer buffer chamber of the gel tank can help maintain a low temperature [62].
- Buffer Stirring: For systems equipped with a magnetic stirrer, continuously stirring the cathode buffer ensures even heat distribution and prevents the formation of local hot spots [65].
Apparatus and Buffer Considerations:
- Ensure the outer buffer chamber is completely filled with running buffer to maximize heat capacity and transfer [65].
- Avoid reusing running buffer, as its ionic composition can change, potentially altering conductivity and heat generation. Always use freshly prepared buffer for critical experiments [63].
Table 1: Troubleshooting Guide for the 'Smiling Band' Effect
| Problem Indicator | Primary Cause | Corrective Action | Preventive Measure |
|---|---|---|---|
| Severe smiling in outer lanes | Excessive voltage leading to overheating | Reduce voltage; run gel in cold room or with ice packs [62] [63] | Adopt a standard voltage of 125V for Tris-Glycine gels [27] |
| Mild smiling across entire gel | Uneven heat distribution | Use a magnetic stirrer in the outer buffer chamber [65] | Ensure gel apparatus is on a level surface and outer chamber is full [65] |
| Smiling accompanied by smeared bands | Combined effect of high heat and voltage | Lower voltage and ensure sample is properly denatured [62] [63] | Follow recommended sample prep protocols (heating at 85-95°C for 5 min) [27] [65] |
Advanced Solution: Alternative Buffer Systems
Recent research has identified alternative buffer systems that are inherently less prone to heat-related issues and can provide superior resolution, especially for specific protein types.
The Tris-Acetate buffer system is particularly advantageous for separating large proteins (>150 kDa), such as monoclonal antibodies. Its operating pH is 7.0, significantly lower than the pH 9.5 operating environment of Tris-Glycine systems [66]. This lower pH reduces gel-induced protein modifications and results in sharper bands, higher resolution, and a more accurate determination of molecular weight for complex molecules like IgG1 and IgG2 [66].
Another innovative development is the Tris-Tricine-HEPES (FRB - Fast-Running Buffer) system. This formulation creates multiple ionic boundaries (Chloride > Tricine > HEPES > protein ions), which enhances resolving power. A key benefit is the ability to perform "fast runs" (e.g., 150 V for 15 min followed by 200 V for 20 min) without the excessive heat generation that plagues the Tris-Glycine system, thereby intrinsically preventing the smiling effect [13]. This system also allows for the gradient-like separation of a very wide molecular weight range (15–450 kDa) on a single 10% gel [13].
Table 2: Comparison of SDS-PAGE Running Buffer Systems
| Parameter | Tris-Glycine | Tris-Acetate | Tris-Tricine-HEPES (FRB) |
|---|---|---|---|
| Standard Composition | 25 mM Tris, 192 mM Glycine, 0.1% SDS [13] | Tris-Acetate, 0.1% SDS [66] | Tris, Tricine, HEPES, 0.1% SDS [13] |
| Operating pH | ~9.5 [27] [66] | ~7.0 [66] | 7.5 - 8.0 [13] |
| Optimal Protein Size Range | 6 - 200 kDa [27] | Best for >150 kDa [66] | 15 - 450 kDa [13] |
| Primary Advantage | Low cost, widely established | Superior for large proteins (e.g., mAbs) [66] | Fast run time, superior resolution of low & high MW proteins [13] |
| Heat Generation | High at increased voltages | Lower | Significantly reduced, allowing faster runs [13] |
Integrated Experimental Protocol for Heat-Managed SDS-PAGE
The following detailed protocol is designed to systematically prevent smiling bands and manage gel heat, incorporating both standard and advanced methods.
Materials and Reagent Solutions
Table 3: Research Reagent Solutions for SDS-PAGE
| Reagent / Solution | Function / Purpose | Key Consideration |
|---|---|---|
| Pre-cast Tris-Glycine Gel | Matrix for protein separation based on size. | Store at +4°C; use before expiration. Separating range typically 6-200 kDa [27]. |
| Tris-Glycine SDS Running Buffer (10X) | Conducts current, maintains pH for separation. | Dilute to 1X; prepare fresh or store properly to avoid pH drift [27] [63]. |
| SDS Sample Buffer (2X) with Reducing Agent | Denatures proteins, confers uniform negative charge. | For reduced samples, add DTT (final 50 mM) or β-ME (final 2.5%) before heating [27] [65]. |
| Protein Molecular Weight Marker | Allows monitoring of run and protein size estimation. | Load in at least one well to monitor electrophoresis progress and band curvature [27] [62]. |
Step-by-Step Procedure
Sample Preparation:
- Mix protein sample with an equal volume of 2X SDS sample buffer.
- For reduced samples, add DTT to a final concentration of 50 mM or β-mercaptoethanol to 2.5% [27] [65].
- Denature the samples by heating at 85-95°C for 5 minutes [63] [65].
- Centrifuge at maximum speed for 2-3 minutes to pellet any insoluble material.
Gel Apparatus Setup:
- Remove a pre-cast gel from its pouch and rinse the cassette with deionized water.
- Remove the comb and rinse the wells thoroughly with 1X running buffer.
- Assemble the gel cassette into the electrophoresis chamber according to the manufacturer's instructions. Fill the inner (upper) and outer (lower) chambers with the appropriate volume of freshly prepared 1X running buffer [27] [63].
Loading and Run Conditions:
- Load samples and molecular weight markers into the wells. Do not leave wells empty; if necessary, load a control sample or sample buffer to prevent the "edge effect" which can distort neighboring lanes [62].
- Start electrophoresis immediately after loading to prevent samples from diffusing out of the wells [62] [65].
- Apply the power. For standard Tris-Glycine mini-gels, the recommended condition is constant voltage of 125 V for approximately 90 minutes, or until the dye front reaches the bottom of the gel [27].
Integrated Heat Management During the Run:
- Standard Method (Tris-Glycine): If the run is performed at room temperature, set the voltage to 125 V. If smiling is observed, reduce the voltage to 100 V or lower, accepting a longer run time [62].
- Advanced Cooling Method: Place the entire electrophoresis apparatus in a cold room (4°C) or equip it with a built-in cooling module. Alternatively, place ice packs around the outer buffer chamber [62].
- Alternative Buffer Method: Replace the traditional Tris-Glycine buffer with a Tris-Tricine-HEPES (FRB) system. This allows for a faster run (e.g., 150 V for 15 min, then 200 V for 20 min) with significantly less heat production [13].
The following workflow diagram summarizes the logical decision process for selecting the optimal strategy to prevent smiling bands:
The 'smiling band' effect is a readily manageable artifact of SDS-PAGE. A thorough understanding of its root cause—uneven joule heating—empowers researchers to select the most effective mitigation strategy. For routine analyses using Tris-Glycine buffers, simple adjustments like reducing voltage and implementing external cooling are highly effective. For more demanding applications, particularly those involving a wide range of protein sizes or requiring high throughput, switching to advanced buffer systems like Tris-Acetate or Tris-Tricine-HEPES offers a robust solution by fundamentally altering the electrophoretic conditions to minimize heat-related issues. By integrating these protocols, researchers can ensure the generation of high-quality, reproducible protein data that is critical for rigorous scientific research and drug development.
Addressing Edge Effects and Distorted Peripheral Lanes
Edge effects, manifesting as distorted or tailing protein bands in the peripheral lanes of SDS-PAGE gels, represent a common artifact that compromises data reliability in protein analysis. This application note examines the underlying causes of this phenomenon within the critical context of running buffer composition and preparation. We provide detailed protocols for preventing and mitigating edge effects through optimized buffer systems and electrophoretic practices, enabling researchers to achieve superior band resolution and reproducibility for drug development and basic research applications.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) is a cornerstone technique in biochemistry and molecular biology, enabling the separation of proteins based on their molecular weight [67]. The technique relies on a discontinuous buffer system involving stacking and resolving gels with different pH and porosity, coupled with a running buffer that creates moving ionic boundaries to sharpen and separate protein bands [68] [13].
A frequent challenge in this system is the "edge effect," where protein bands in the outermost lanes (left and right) appear distorted, curved, or show tailing compared to the sharp, straight bands in the central lanes [69] [70]. This artifact compromises the accuracy of molecular weight determination and quantitative analysis. As running buffer composition is fundamental to establishing a uniform electric field and consistent ionic fronts across the entire gel, its role in mitigating edge effects is paramount. This note details the causes and solutions, with a specific focus on buffer-related parameters.
Mechanisms and Causes of Edge Effects
The primary cause of edge effects is an inhomogeneous electric field across the width of the gel. When the outermost wells are left empty, the path of least electrical resistance is along the edges of the gel. This results in a stronger electric field in the peripheral regions compared to the center, causing proteins in the edge lanes to migrate faster and unevenly, leading to distorted or "smiling" bands [69]. The fundamental principle is that electric current takes the path of least resistance, and without a sample in every well, this path is altered.
Another contributing factor, particularly in older Weber-Osborn-type SDS-PAGE systems, is a phenomenon known as "edge tailing," where protein bands exhibit tailing at both ends. This specific distortion has been linked to buffer and gel matrix interactions and can be eliminated by modifications to the sample-well gel composition [70].
Preventive Strategies and Reagent Solutions
A multi-faceted approach is most effective for preventing edge effects. The strategies below can be implemented individually or in combination.
Core Experimental Practices
The most straightforward and highly recommended practice is to avoid leaving any wells empty [69]. If all wells are not filled with experimental samples, the peripheral wells should be loaded with a control protein, such as a protein ladder or a non-critical protein stock from the laboratory [69]. This ensures a uniform distribution of ions and current flow across the entire gel face, preventing the field distortion that causes edge effects.
Furthermore, ensuring the gel apparatus is properly assembled, with no loose contacts, and that the gel is fully submerged under a consistent depth of running buffer (typically 3-5 mm) helps maintain a stable and uniform electric field [71].
Running Buffer Composition and Novel Formulations
The standard Tris-Glycine-SDS running buffer, while widely used, has known limitations, including long run times and poor resolution of small proteins, which can be exacerbated at higher voltages due to excessive heat generation [13]. Heat is a known contributor to band distortion, including the "smiling" effect where bands in the center migrate faster than those on the edges [69] [71].
Recent advancements propose novel running buffer formulations to address these issues. For instance, a Tris-Tricine-HEPES (FRB) buffer system has been shown to provide superior separation across a wide molecular weight range (15–450 kDa) in a single 10% gel while significantly reducing the running time without generating excessive Joule's heat [13]. This reduced heat production inherently minimizes one of the root causes of band distortion.
Table 1: Comparison of Common SDS-PAGE Running Buffer Systems
| Buffer System | Typical Composition | Optimal Separation Range | Advantages | Disadvantages |
|---|---|---|---|---|
| Tris-Glycine (Laemmli) [13] | 25 mM Tris, 192 mM Glycine, 0.1% SDS, pH 8.3 | 10 - 200 kDa | Low cost, well-established protocol. | Long run times, excessive heat at high voltage, poor resolution of small proteins (<15 kDa). |
| Tris-Tricine [13] | Tris, Tricine, SDS | 1 - 100 kDa | Excellent resolution of low molecular weight proteins. | Cannot resolve small and large proteins simultaneously; long run times. |
| Tris-Tricine-HEPES (FRB) [13] | Tris, Tricine, HEPES, SDS | 15 - 450 kDa | Wide-range separation in one gel; fast run time; reduced heat generation. | Less common, requires formulation. |
Gel Matrix Modifications
A simple yet effective modification to the gel itself can eliminate edge tailing. Research demonstrates that supplementing the sample-well gel (the stacking gel) with glycerol at a concentration of 10-15% (v/v) makes protein bands as sharp and straight as those achieved in Laemmli-type SDS-PAGE [70]. The glycerol likely alters the viscosity and pore dynamics at the critical point of sample entry, standardizing migration across all lanes.
Detailed Experimental Protocols
Protocol 1: Standard SDS-PAGE with Edge Effect Prevention
Research Reagent Solutions:
- Running Buffer (Tris-Glycine-SDS): 25 mM Tris base, 192 mM glycine, 0.1% (w/v) SDS, pH ~8.3. Do not adjust pH with acid or base.
- 30% Acrylamide/Bis Solution: 29.2% acrylamide, 0.8% bis-acrylamide.
- Resolving Gel Buffer: 1.5 M Tris-HCl, pH 8.8.
- Stacking Gel Buffer: 0.5 M Tris-HCl, pH 6.8.
- 10% Ammonium Persulfate (APS): Prepare fresh weekly.
- TEMED
- Protein Ladder/Control Protein: For loading into empty wells.
Methodology:
- Gel Casting: Prepare the resolving gel according to the desired percentage (e.g., 10% for 10-250 kDa proteins). Pour between glass plates, overlay with isopropanol or water for a flat interface, and allow to polymerize completely. Prepare the stacking gel (5% acrylamide) and pour over the polymerized resolving gel, immediately inserting a clean comb.
- Sample Preparation: Mix protein samples with Laemmli buffer, heat at 95-100°C for 5 minutes (or 75°C to avoid Asp-Pro bond cleavage for sensitive proteins [72]), and centrifuge briefly to pellet insoluble material.
- Loading Strategy: Critical Step: Load protein samples into the wells. Load protein ladder or control protein into any unused wells, especially the left-most and right-most wells. Do not leave any well empty [69].
- Electrophoresis: Assemble the gel unit, fill the tank with Tris-Glycine-SDS running buffer, and connect to a power supply. Run at a constant voltage (e.g., 150V for a mini-gel). To minimize heat-induced smiling, running the gel at a lower voltage for a longer time or in a cold room is effective [69]. Stop the run when the dye front reaches the bottom of the gel.
Protocol 2: Advanced Method Using Glycerol-Modified Stacking Gel
This protocol directly addresses the edge tailing artifact [70].
Research Reagent Solutions:
- All reagents from Protocol 1, plus:
- Molecular Biology Grade Glycerol
Methodology:
- Casting the Resolving Gel: Follow standard procedure as in Protocol 1.
- Casting the Glycerol-Modified Stacking Gel: Prepare the stacking gel solution as usual (5% acrylamide, Tris-HCl pH 6.8), but supplement it with glycerol to a final concentration of 10-15% (v/v) before adding APS and TEMED. Pour and polymerize as normal.
- Sample Loading and Electrophoresis: Follow the steps in Protocol 1, emphasizing the importance of loading all wells to prevent the primary edge effect, which this method complements.
The following workflow summarizes the core strategies for addressing lane distortion:
Logical workflow for addressing lane distortion in SDS-PAGE.
Protocol 3: Rapid, High-Resolution SDS-PAGE with FRB Buffer
This protocol leverages a novel running buffer for fast, high-quality results with minimal heat [13].
Research Reagent Solutions:
- Fast-Running Buffer (FRB): The exact molarities may require optimization, but the buffer consists of Tris, Tricine, and HEPES. The composite pH should fall within 7.5–8.0 without adjustment [13].
- Tris-Acetate Gels: The protocol is optimized with Tris-acetate gels, which use acetate as the leading ion.
Methodology:
- Gel Casting: Cast polyacrylamide gels (e.g., 10%) using Tris-acetate buffers for the resolving and stacking gels.
- Buffer Preparation: Prepare the FRB running buffer according to the optimized formulation.
- Electrophoresis: Load the gel and run with FRB. The run conditions can be significantly shortened, for example, 150 V for 15 min followed by 200 V for 20 min (total 35 min) [13].
- Downstream Processing: The gel is compatible with standard downstream applications like Western blotting.
Table 2: Summary of Key Preventive Methods for Edge Effects
| Method | Principle | Key Advantage | Consideration |
|---|---|---|---|
| Load All Wells [69] | Ensures uniform electric field distribution. | Simple, no protocol modification. | Requires spare protein for control wells. |
| Glycerol-Modified Stacking Gel [70] | Alters gel matrix to standardize band entry. | Directly targets edge tailing artifact. | Requires modification to standard gel recipe. |
| Novel FRB Buffer System [13] | Reduces heat and improves resolution. | Faster run times, wide separation range. | Requires preparation of non-standard buffer. |
| Reduced Voltage / Cooling [69] | Minimizes heat generation. | Easy to implement. | Increases total run time. |
Edge effects and distorted peripheral lanes are not inevitable artifacts of SDS-PAGE. They are manageable phenomena rooted in the electrophoretic conditions, particularly the running buffer system and sample loading strategy. By understanding the underlying causes—primarily electric field inhomogeneity and heat generation—researchers can employ targeted strategies. These include the fundamental practice of loading all wells, the specific biochemical modification of adding glycerol to the stacking gel, and the adoption of advanced, thermally efficient buffer systems like Tris-Tricine-HEPES. Implementing these protocols will significantly enhance the quality, reliability, and reproducibility of protein electrophoretic data, which is critical for rigorous scientific research and drug development.
In the realm of protein biochemistry, SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) stands as a fundamental technique for separating proteins by molecular weight. While the composition and preparation of the running buffer are critical foundational elements, the parameters governing the application of electricity during the electrophoretic run are equally pivotal for obtaining high-quality, reproducible results. The conditions of voltage, current, and power are not merely settings on an instrument; they are interconnected variables that directly influence the resolution of protein bands, the duration of the experiment, and the structural integrity of the gel matrix itself. This application note details the principles and practical protocols for optimizing these electrophoretic parameters, with particular emphasis on thermal management, to achieve superior separation outcomes within the established framework of Tris-Glycine-SDS running buffer systems.
The Interplay of Electrical Parameters in SDS-PAGE
The relationship between voltage (V, in volts), current (I, in amperes), and power (P, in watts) during electrophoresis is defined by fundamental physical laws. Ohm's Law (V = I × R) describes how voltage and current are related through the resistance (R) of the system [73] [74]. The power generated, which manifests as heat, is the product of voltage and current (P = I × V) [73]. This production of Joule heating is a critical consideration, as it can significantly impact the electrophoresis process.
- Heat as a Double-Edged Sword: A moderate amount of heat can be beneficial by assisting in protein denaturation [73]. However, excessive heat causes the acrylamide gel to expand, which can lead to uneven migration of proteins (often observed as "smiling" bands), distorted bands, or even render the gel unusable for subsequent analysis like Western blotting [73] [74]. Managing this heat is therefore a primary objective when selecting electrical settings.
Operational Modes: Constant Current, Voltage, and Power
Most modern power supplies allow the user to maintain one electrical parameter constant, while the other two fluctuate in response to changing conditions within the gel, such as evolving buffer ion concentrations.
Table 1: Comparison of SDS-PAGE Operational Modes
| Operational Mode | Key Characteristics | Advantages | Disadvantages |
|---|---|---|---|
| Constant Current [73] [74] | Migration rate is constant; voltage increases as resistance increases. | Predictable run time; sharper bands due to faster runs. | Risk of overheating; "smiling" bands from gel warping. |
| Constant Voltage [73] [27] [74] | Current decreases as resistance increases; heat production diminishes over time. | Safer (less risk of boiling); multiple chambers can run from one power pack. | Longer run times; potential for diffuse bands. |
| Constant Power [73] | A product of V and I; the power supply adjusts both to keep P constant. | Limits heat production. | Unpredictable sample migration; longer run times. |
Experimental Protocol for Optimized SDS-PAGE
This protocol is designed for a standard mini-gel format and utilizes the widely adopted Tris-Glycine discontinuous buffer system.
Reagent and Buffer Preparation
10X SDS Running Buffer [3]
- Recipe: 30.285 g Tris base, 144.4 g Glycine, 10 g SDS. Add components to 0.8 L of distilled water, mix to dissolve, and bring the final volume to 1 L. The final 1X working concentration is 25 mM Tris, 192 mM Glycine, and 0.1% SDS [75].
- Function: Tris provides the common cation, Glycine serves as the trailing ion in the discontinuous system, and SDS ensures proteins remain denatured and charged during electrophoresis [76].
Sample Preparation Buffer (2X Laemmli Buffer) [2] [75]
- Composition: 4% SDS, 20% glycerol, 0.004% bromophenol blue, 100 mM Tris-HCl (pH 6.8). Add 10% β-mercaptoethanol or 100 mM DTT fresh before use.
- Procedure: Mix protein sample with an equal volume of 2X buffer. Heat at 95°C for 5 minutes, then cool briefly on ice and centrifuge before loading [75].
Electrophoresis Procedure and Parameter Optimization
- Gel Casting & Assembly: Cast separating and stacking gels according to standard recipes [75]. Assemble the electrophoresis chamber, ensuring a tight seal. Add sufficient 1X running buffer to the inner and outer chambers.
- Load Samples: Load prepared samples and a molecular weight marker into the wells.
- Initial Stacking Phase: Apply a constant voltage of 50-80 V [73] [75]. This low voltage allows proteins to stack into sharp bands as they move through the low-percentage stacking gel. This step typically takes about 30 minutes, until the dye front enters the resolving gel.
- Separation Phase: Once proteins enter the resolving gel, increase to a higher voltage. A general guideline is 5-15 V per centimeter of gel [73] [74]. For a standard mini-gel, this often translates to 120-150 V constant voltage [27] [75]. Run until the bromophenol blue dye front reaches the bottom of the gel (approximately 60-90 minutes).
- Thermal Management: For runs at higher voltages or using constant current, active cooling is recommended. This can be achieved by:
- Performing the run in a 4°C cold room.
- Using an ice bath or a circulating water cooler designed for the electrophoresis apparatus [73] [74].
- Important: If cooling in a refrigerator, keep the power pack at room temperature to prevent condensation damage, and only run the electrode leads into the cooled environment [74].
The following workflow diagram illustrates the logical decision process for parameter selection and optimization as described in the protocol.
The Scientist's Toolkit: Essential Reagents for SDS-PAGE
Table 2: Key Research Reagent Solutions for SDS-PAGE
| Reagent / Solution | Core Function |
|---|---|
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent that denatures proteins and imparts a uniform negative charge, allowing separation by size rather than native charge [76] [2]. |
| Tris-Glycine Running Buffer | Provides the ionic environment and pH (8.3) for electrophoresis; glycine's charge state is key to the discontinuous stacking mechanism [76] [3]. |
| Acrylamide/Bis-Acrylamide | Monomer and cross-linker that polymerize to form a porous gel matrix, sieving proteins during separation [75]. |
| APS & TEMED | Catalyst (Ammonium Persulfate) and stabilizer (Tetramethylethylenediamine) that initiate and accelerate acrylamide polymerization [75]. |
| Laemmli Sample Buffer | Denatures proteins, provides tracking dye (bromophenol blue), and adds density (glycerol) for sample loading [76] [2]. |
| β-Mercaptoethanol or DTT | Reducing agents that break disulfide bonds, ensuring complete protein unfolding [2] [75]. |
The optimization of voltage, current, and cooling is a critical determinant of success in SDS-PAGE. While constant voltage offers a robust and safer approach suitable for most routine applications, constant current can provide sharper bands and predictable timing when paired with rigorous temperature control. The selection of the optimal parameters is not a one-size-fits-all decision but should be guided by the specific experimental goals, gel format, and equipment. By understanding the scientific principles underlying these electrical parameters and implementing the detailed protocols outlined herein, researchers can consistently achieve high-resolution protein separation, thereby enhancing the reliability and quality of their data in downstream analyses.
Beyond Standard Protocols: Validating and Comparing Buffer Performance
Techniques for Verifying Running Buffer Quality and Performance
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) is a foundational technique in molecular biology for the separation of denatured proteins based on their molecular weight [8]. The running buffer, an essential component of this system, creates the environment through which proteins migrate under an electric field. The most common running buffer, known as Laemmli buffer, consists of Tris, glycine, and SDS [77]. Its primary functions are to maintain stable pH during electrophoresis, provide ions necessary for conductivity, and ensure proteins remain denatured and uniformly charged. Verification of running buffer quality is crucial for obtaining reproducible, high-resolution protein separation, as subtle deviations in composition or pH can significantly impact electrophoretic results and downstream analyses such as western blotting.
The running buffer operates within a discontinuous buffer system where the stacking gel (pH ~6.8) and resolving gel (pH ~8.8) have different pH values and pore sizes [77]. Glycine, a key component, exists in different ionic forms depending on pH. In the stacking gel at pH 6.8, glycine is predominantly a zwitterion with minimal charge, moving slowly. In contrast, at the running buffer pH of 8.3 and in the resolving gel at pH 8.8, glycine becomes negatively charged (glycinate anion) and moves rapidly [77]. This creates a critical ion front that stacks proteins into sharp bands before they enter the resolving gel for separation based on size.
Composition and Standard Preparation
Standard Tris-Glycine-SDS Running Buffer
The conventional running buffer for SDS-PAGE is a Tris-Glycine-SDS system. The standard 10X stock solution can be prepared as follows [3]:
Table 1: Standard 10X SDS-PAGE Running Buffer Recipe
| Component | Amount per Liter | Final Concentration (10X) | Molecular Weight | Role in Buffer System |
|---|---|---|---|---|
| Tris base | 30.285 g | 0.25 M | 121.14 g/mol | pH buffer |
| Glycine | 144.4 g | 1.923 M | 75.07 g/mol | Leading ion |
| SDS | 10 g | 1% (w/v) | 288.38 g/mol | Protein charge modifier |
Preparation Protocol:
- Prepare 800 mL of distilled water in a suitable container [12].
- Add Tris base and stir until completely dissolved [3].
- Add glycine and mix thoroughly until dissolved [3].
- Add SDS slowly while stirring to prevent clumping [3].
- Bring the final volume to 1 L with distilled water [12].
- The 10X stock is typically diluted to 1X with distilled water before use [12].
Alternative Buffer Formulations
While Tris-Glycine-SDS remains the standard, alternative formulations have been developed to address specific limitations. A Tris-Tricine-HEPES buffer system has been shown to provide superior separation across a wider molecular weight range (15-450 kDa) in a single 10% gel while significantly reducing running time [13]. This novel running buffer creates multiple ionic boundaries instead of two, theoretically improving resolving power. For specialized applications requiring retention of native protein properties including enzymatic activity and bound metal ions, a Native SDS-PAGE (NSDS-PAGE) running buffer with reduced SDS concentration (0.0375%) can be employed [78].
Table 2: Alternative Running Buffer Formulations
| Buffer Type | Composition | Separation Range | Running Conditions | Key Advantages |
|---|---|---|---|---|
| Tris-Tricine-HEPES [13] | Tris, Tricine, HEPES | 15-450 kDa | 150V for 15 min, then 200V for 20 min (35 min total) | Wide separation range, reduced run time |
| NSDS-PAGE [78] | 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS | Similar to SDS-PAGE | 200V for 30-45 min | Retains protein function, preserves metal cofactors |
| Standard SDS-PAGE [3] | 25 mM Tris, 192 mM Glycine, 0.1% SDS | 10-200 kDa | Typically 90V-150V for 60-90 min | Well-established, low cost |
Assessment Techniques and Quality Control
Visual Performance Indicators
Several visual indicators during electrophoresis can reveal running buffer quality and performance:
- Bubble Formation: Proper bubble production at both electrodes confirms adequate current flow and buffer conductivity [77]. The cathode (negative) typically produces more bubbles (hydrogen gas) than the anode (positive, oxygen gas) due to water electrolysis [77].
- Dye Front Migration: A sharp, straight bromophenol blue dye front indicates proper ion mobility and buffer function [8]. A wavy or diffuse dye front suggests buffer degradation, improper formulation, or electrode issues.
- Heat Generation: Moderate warmth is expected, but excessive heat indicates high current resistance, potentially from buffer depletion or incorrect dilution.
Electrophoretic Performance Metrics
Systematic assessment of protein separation quality provides quantitative measures of buffer performance:
- Band Sharpness: Well-functioning buffer produces tight, crisp protein bands. Band smearing may indicate insufficient buffering capacity or SDS concentration.
- Resolution: Calculate resolution between adjacent protein standards to quantify separation efficiency. Optimal buffer maintains resolution R ≥ 1.5 between close molecular weight standards.
- Migration Linearity: The plot of log(MW) versus migration distance should be linear for standard proteins under optimal buffer conditions.
- Reproducibility: Consistent migration patterns and band intensities across multiple gels run with the same batch of buffer indicate quality and stability.
Buffer Stability and Storage Assessment
Running buffer quality degrades over time due to electrochemical reactions during electrophoresis and chemical decomposition. Implement these quality control measures:
- pH Monitoring: Regularly check 1X running buffer pH (should be ~8.3 for Tris-Glycine-SDS). Significant deviation (>0.3 pH units) indicates buffer exhaustion.
- SDS Precipitation: Visual inspection for SDS crystallization, particularly at cooler temperatures, suggests saturation issues or impurity presence.
- Performance Comparison: Always retain a aliquot of fresh buffer for side-by-side comparison with used or stored buffer.
- Microbial Contamination: Cloudiness or particulate matter in stored buffer indicates contamination requiring disposal.
Experimental Protocols for Verification
Side-by-Side Buffer Comparison Protocol
This protocol systematically evaluates buffer performance by comparing new and test buffers under identical conditions.
Materials:
- Pre-cast or freshly cast SDS-PAGE gels (consistent percentage) [8]
- Protein molecular weight standard
- Test samples (known complex protein mixture)
- Freshly prepared running buffer (control)
- Test running buffer (experimental)
- Electrophoresis apparatus and power supply [8]
Procedure:
- Prepare two identical gels in the same electrophoresis apparatus if possible, or run sequentially under identical conditions.
- Load identical protein samples and standards on both gels.
- Run gels simultaneously at constant voltage (e.g., 150V) until dye front reaches bottom.
- Process gels identically for staining and destaining [8].
- Compare:
- Migration distance of standards
- Band sharpness of sample proteins
- Overall resolution and background staining
- Document results with gel imaging system [8].
Interpretation: Significant differences in migration, band patterns, or resolution indicate compromised buffer quality. Buffer should be replaced if performance degradation exceeds 10% based on migration rates or resolution metrics.
Buffer Re-use Validation Protocol
This protocol determines how many times running buffer can be reused without significant performance degradation.
Materials:
- Freshly prepared running buffer
- Multiple identical gels and sample sets
- Materials for protein quantification (e.g., spectrophotometer)
Procedure:
- Prepare fresh running buffer and note initial pH.
- Run first gel, noting run time and voltage requirements.
- Collect and store used buffer in sealed container, noting usage number.
- Repeat process for multiple runs (typically 3-5) using same buffer.
- After each run:
- Measure and record buffer pH
- Document voltage and current patterns
- Assess gel resolution and band quality
- Compare all gels side-by-side after staining.
Acceptance Criteria: Buffer should be replaced when: (1) pH changes by >0.5 units from initial; (2) run time increases by >15% at constant voltage; (3) visible band smearing or distortion occurs; (4) excessive bubble formation decreases.
Advanced Verification Techniques
Western Blot Transfer Efficiency Assessment
Running buffer quality directly impacts downstream applications like western blotting. This protocol assesses buffer performance through transfer efficiency.
Procedure:
- Run duplicate SDS-PAGE gels with identical samples.
- Transfer one gel to membrane using standard transfer protocols.
- Compare post-transfer gel (stained with Coomassie) with non-transferred gel to assess residual proteins.
- Probe membrane for housekeeping proteins to quantify transfer efficiency.
Interpretation: Poor transfer efficiency may indicate issues with running buffer affecting protein state or gel structure, particularly if band patterns differ between pre- and post-transfer gels.
Enzymatic Activity Assay for NSDS-PAGE
For native SDS-PAGE applications, buffer quality verification includes assessment of retained protein function.
Procedure:
- Separate known enzymes (e.g., alcohol dehydrogenase, alkaline phosphatase) using NSDS-PAGE conditions [78].
- After electrophoresis, incubate gel in appropriate substrate solution.
- Compare activity bands with protein staining patterns.
- Quantify retained activity relative to non-electrophoresed controls.
Interpretation: Successful NSDS-PAGE running buffer should preserve ≥70% of enzymatic activity for most model enzymes while maintaining adequate resolution [78].
Implementation and Documentation
Research Reagent Solutions
Table 3: Essential Materials for Running Buffer Quality Assessment
| Reagent/Equipment | Function/Purpose | Specification Guidelines |
|---|---|---|
| Tris base | Primary buffering component | ≥99.9% purity, molecular biology grade |
| Glycine | Leading ion in electrophoresis | ≥99% purity, electrophoresis grade |
| SDS | Protein denaturation and charge uniformity | ≥98.5% purity, suitable for electrophoresis |
| Precast gels or acrylamide/bis-acrylamide | Separation matrix | Consistent lot-to-lot performance verified |
| Protein molecular weight standards | Migration reference | Broad range (10-250 kDa), prestained options available |
| Electrophoresis system | Separation platform | Consistent voltage/current delivery capability |
| Power supply | Electrical field application | Constant voltage capability, safety features |
| pH meter | Buffer quality verification | Regular calibration at appropriate pH values |
Experimental Workflow
The following diagram illustrates the complete workflow for running buffer quality verification:
Buffer Quality Assessment Workflow
Robust verification of running buffer quality is essential for reproducible, high-quality protein separation in SDS-PAGE. The techniques outlined here provide comprehensive assessment from basic visual inspection to advanced functional assays. Implementation of these verification protocols ensures experimental reliability and identifies buffer-related issues before they compromise valuable samples. As electrophoresis technology evolves with new buffer formulations like Tris-Tricine-HEPES for expanded separation ranges and NSDS-PAGE for native protein applications, corresponding verification methods must adapt to address their specific performance metrics. Regular quality control of running buffers represents a small investment in time and resources that pays substantial dividends in experimental consistency and data quality.
Within the framework of a broader thesis on running buffer composition for SDS-PAGE research, this application note provides a detailed comparative analysis of the Laemmli (Tris-Glycine) and Tris-Tricine buffer systems. The critical role of electrophoresis in proteomics and drug development necessitates a deep understanding of how buffer composition influences protein separation efficacy. The choice between these two predominant buffer systems dictates the successful resolution and analysis of proteins across different molecular weight ranges, particularly for low-mass polypeptides and complex biological samples. This document provides a structured comparison of their operational principles, detailed protocols for their application, and data-driven guidance to inform method selection for specific research objectives in protein characterization.
Fundamental Principles and Comparative Mechanics
The Laemmli (Tris-Glycine) system, the long-standing standard for SDS-PAGE, employs a discontinuous buffer system to achieve sharp protein bands. Its mechanism relies on the formation of ionic boundaries between leading chloride ions (from the gel buffer) and trailing glycinate ions (from the running buffer) [13]. In the stacking gel (pH ~6.8), glycine exists predominantly as a zwitterion with low electrophoretic mobility, creating a tight protein stack between the fast-moving chloride and slow-moving glycine. Upon entering the resolving gel (pH ~8.8), the increased pH causes glycine to become more fully deprotonated, increasing its mobility so it overtakes the proteins. The proteins, now in a gel with smaller pores and without the trailing ion boundary, separate primarily by molecular mass [79]. However, a major limitation of this system is its poor resolution of proteins smaller than 15 kDa, as these small peptides tend to co-migrate with the diffuse trailing ion front [13].
In contrast, the Tris-Tricine buffer system, developed by Schägger and von Jagow, replaces glycine with tricine as the trailing ion [80]. Tricine has a higher electrophoretic mobility (pK 8.15) compared to glycine (pK 9.6) [80] [13]. This key difference means that in the resolving gel, tricine remains behind the proteins, allowing even very small polypeptides (1-100 kDa) to be resolved effectively before the buffer front [80]. The system is particularly renowned for its superior performance in separating low molecular weight proteins and peptides in the 1 to 20 kDa range, which are often poorly resolved or diffuse in Tris-Glycine systems [80].
Recent advancements have led to the development of a novel Tris-Tricine-HEPES (FRB) running buffer. This system creates multiple ionic boundaries (Chloride > Tricine > HEPES > protein ions), which further enhances the resolving power. It enables the gradient-like simultaneous separation of a very wide molecular weight range (15–450 kDa) in a single-percentage polyacrylamide gel and significantly reduces the total running time to approximately 35 minutes without excessive heat generation [13].
The following diagram illustrates the decision-making workflow for selecting the appropriate buffer system based on research goals.
Comparative Data and Application Specifications
The operational characteristics and optimal application ranges of the Laemmli (Tris-Glycine) and Tris-Tricine buffer systems are quantitatively distinct. The table below summarizes the key parameters for direct comparison, aiding in informed method selection.
Table 1: Direct comparison of Laemmli (Tris-Glycine) and Tris-Tricine buffer systems
| Parameter | Laemmli (Tris-Glycine) System | Tris-Tricine System | Tris-Tricine-HEPES (FRB) System |
|---|---|---|---|
| Effective Separation Range | 20-200 kDa [80] [79] | 1-100 kDa [80] | 15-450 kDa [13] |
| Resolution of Small Proteins (<20 kDa) | Poor, bands are diffuse [80] [13] | Excellent [80] | Excellent [13] |
| Standard Running Buffer Composition (10X) | 0.25 M Tris, 1.92 M Glycine, 1% SDS [31] | 1.2 M Tris, 0.8 M Tricine, 2% SDS (20X) [31] | 25-100 mM Tris, Tricine, HEPES; 0.1% SDS [13] |
| Typical Running Time | ~1 hour or more [13] | 4-16 hours [80] | ~35 minutes [13] |
| Compatibility with High Voltage | Limited due to excessive Joule's heat [13] | Requires low voltage and cooling [80] | Yes, designed for fast run conditions [13] |
| Primary Trailing Ion | Glycinate (pK 9.6) [13] | Tricinate (pK 8.15) [80] [13] | Tricinate and HEPES [13] |
Detailed Experimental Protocols
Protocol for Laemmli (Tris-Glycine) SDS-PAGE
This protocol is adapted for a standard mini-gel format.
- Gel Formulation: A 10% resolving gel is commonly used. Prepare the resolving gel by mixing 7.5 mL of 40% acrylamide solution, 3.9 mL of 1% bisacrylamide solution, 7.5 mL of 1.5 M Tris-HCl (pH 8.8), 0.3 mL of 10% SDS, and water to 30 mL. Catalyze polymerization with 0.3 mL of 10% ammonium persulfate (APS) and 0.03 mL of TEMED. Pour the gel and overlay with water or ethanol. Once polymerized, prepare the stacking gel (4-5% acrylamide) in Tris-HCl (pH 6.8) and pour over the resolving gel [79].
- Sample Preparation: Dilute protein samples 1:1 with 2X Laemmli sample buffer, which consists of 100 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 0.2% bromophenol blue, and 200 mM dithiothreitol (DTT) (added fresh). Heat the samples at 90-100°C for 5 minutes to fully denature the proteins [11] [79].
- Electrophoresis Conditions: Dilute the 10X running buffer to 1X (final: 25 mM Tris, 192 mM glycine, 0.1% SDS). Load samples into the wells. Apply a constant voltage of 120-150 V for approximately 60-90 minutes, or until the dye front reaches the bottom of the gel [79].
Protocol for Tris-Tricine SDS-PAGE
This protocol is ideal for separating low molecular weight proteins and peptides.
- Gel Formulation: The gel system often includes three layers: separating, spacer, and stacking gels. For a 16.5% separating gel for peptides <10 kDa, combine 10 mL of Acrylamide Solution B (49.5% T, 6% C), 10 mL of gel buffer (3M Tris, 3% SDS, pH 8.45), 4 g of glycerol, and bring to 30 mL with water. Add APS and TEMED to polymerize. A spacer gel (10% acrylamide) and a stacking gel (4% acrylamide) are then cast on top [80].
- Sample Preparation: Use a Tricine-compatible sample buffer: 50 mM Tris (pH 8.0), 12% glycerol, 4% SDS, 0.01% Coomassie Blue G-250, and 2% 2-mercaptoethanol (for reducing conditions). Heat samples at 40°C for 30 minutes or 90°C for 5 minutes [80].
- Electrophoresis Conditions: Use a discontinuous buffer system. The cathode (upper) buffer is 0.1 M Tris, 1 M Tricine, 0.1% SDS (pH ~8.25). The anode (lower) buffer is 0.2 M Tris (pH ~9.0). It is critical to run the gel at low voltage initially (30 V for 1 hour) before increasing to 180 V. The total run time is longer (4-16 hours), and the gel apparatus should be kept cool [80].
Protocol for Novel Tris-Tricine-HEPES (FRB) SDS-PAGE
This modern protocol allows for rapid, high-resolution separation of a broad molecular weight range.
- Gel Formulation: A single-percentage resolving gel (e.g., 10%) is cast using a 30% Acrylamide/Bis Solution (37.5:1 ratio) in Tris buffer, followed by a standard 5% stacking gel [13].
- Running Buffer Preparation: The fast-running buffer (FRB) consists of a mixture of Tris, Tricine, and HEPES. The optimal concentration can be systematically varied (e.g., 25, 50, 75, 100 mM HEPES) to achieve a final pH in the range of 7.5–8.0 without adjustment [13].
- Electrophoresis Conditions: This system is designed for speed. Run the gel at 150 V for 15 minutes, followed by 200 V for 20 minutes, for a total running time of just 35 minutes [13].
The following workflow diagram outlines the key procedural steps for the Tris-Tricine-HEPES protocol, highlighting its streamlined nature.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of SDS-PAGE experiments requires specific, high-quality reagents. The following table lists key materials and their critical functions.
Table 2: Essential research reagents for SDS-PAGE buffer systems
| Reagent/Material | Function/Purpose |
|---|---|
| Tris (Hydroxymethyl)aminomethane | Primary buffering agent; maintains pH in both gel and running buffers [80] [31]. |
| Glycine | Trailing ion in the Laemmli buffer system; establishes the ionic boundary for protein stacking [13] [31]. |
| Tricine (N-[Tris(Hydroxymethyl)methyl]glycine) | Higher mobility trailing ion in Tris-Tricine systems; essential for resolving small proteins [80] [13] [31]. |
| HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) | Zwitterionic buffer component in novel FRB system; creates multiple ionic boundaries for enhanced resolution [13]. |
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent that denatures proteins and confers a uniform negative charge, allowing separation by mass [79]. |
| Acrylamide/Bis-Acrylamide | Forms the cross-linked polyacrylamide gel matrix that acts as a molecular sieve for separating proteins [80] [79]. |
| Ammonium Persulfate (APS) & TEMED | Catalytic system for initiating and accelerating the free-radical polymerization of acrylamide gels [79]. |
| Dithiothreitol (DTT) or 2-Mercaptoethanol | Reducing agents added to sample buffer to break disulfide bonds and fully denature proteins [11] [80]. |
The Laemmli (Tris-Glycine) and Tris-Tricine buffer systems serve complementary yet distinct roles in the protein separation laboratory. The Laemmli system remains the robust, cost-effective gold standard for routine separation of proteins in the 20-200 kDa range. In contrast, the Tris-Tricine system is the unequivocal method of choice for high-resolution analysis of low molecular weight proteins and peptides below 20 kDa. The emergence of novel composite buffers, such as the Tris-Tricine-HEPES system, signifies a step change in SDS-PAGE technology, offering unprecedented speed and a vastly expanded effective separation range in a single gel. This evolution in running buffer composition directly empowers researchers in drug development and proteomics, enabling more efficient and comprehensive protein characterization.
Correlating Buffer Conditions with Protein Extraction Method Efficacy
In proteomic research, the efficacy of protein extraction methodologies is intrinsically linked to the buffer conditions used during subsequent separation by SDS-PAGE. The recovery, denaturation, and electrophoretic mobility of proteins are governed by a complex interplay between extraction reagents and running buffer composition. While numerous studies have optimized extraction protocols, the correlation between these methods and the electrophoretic buffer system remains inadequately characterized. This application note systematically evaluates how buffer conditions influence the detection and resolution of proteins obtained through different extraction techniques, providing researchers with a validated framework for optimizing proteomic workflows from cell lysis to gel separation. Understanding these correlations is paramount for obtaining reproducible, high-quality results in drug development and basic research applications.
Comparative Efficacy of Protein Extraction Methods
Systematic Evaluation of Extraction Protocols
Recent systematic comparisons of protein extraction methodologies reveal significant variability in protein recovery efficiency, particularly when applied to bacteria with differing cell wall structures. In a 2025 study evaluating four distinct protocols using both data-dependent (DDA) and data-independent (DIA) acquisition modes, the SDT-B-U/S method—combining thermal denaturation with ultrasonication—demonstrated superior performance for both Gram-negative (Escherichia coli) and Gram-positive (Staphylococcus aureus) bacteria [81].
Table 1: Protein and Peptide Identification Across Extraction Methods [81]
| Extraction Method | Total Peptides (E. coli) | Total Proteins (E. coli) | Total Peptides (S. aureus) | Total Proteins (S. aureus) | Technical Replicate Correlation (R²) |
|---|---|---|---|---|---|
| SDT-B (Boiling) | 14,220 | 1,532 | 8,407 | 986 | 0.87 |
| SDT-U/S (Ultrasonication) | 15,893 | 1,798 | 9,226 | 1,103 | 0.89 |
| SDT-B-U/S (Combined) | 16,560 | 1,942 | 10,575 | 1,288 | 0.92 |
| SDT-LNG-U/S (Liquid N₂) | 13,885 | 1,487 | 7,893 | 912 | 0.84 |
The enhanced performance of the combined thermal and mechanical disruption method (SDT-B-U/S) highlights the importance of employing complementary lysis mechanisms. This protocol achieved a 19.2% increase in peptide recovery from S. aureus compared to boiling alone and a 12.7% increase compared to liquid nitrogen grinding [81]. The method proved particularly effective for membrane protein recovery, successfully identifying challenging targets such as OmpC, and demonstrated enhanced extraction of proteins within specific molecular weight ranges (20–30 kDa for E. coli; 10–40 kDa for S. aureus) [81].
Gram-Class Specific Considerations
The correlation between extraction efficacy and bacterial cell structure necessitates gram-class specific methodological considerations:
- Gram-negative bacteria: The outer membrane presents the primary barrier, making it more susceptible to SDS-based disruption combined with thermal denaturation [81].
- Gram-positive bacteria: The thicker peptidoglycan layer requires more rigorous disruption methods, with ultrasonication alone yielding only one-third the protein compared to E. coli under identical conditions [81].
The SDT lysis buffer composition (4% SDS, 100 mM DTT, 100 mM Tris-HCl, pH 7.6) proved effective across extraction methods, with SDS playing the dual role of membrane disruption and protein denaturation [81].
SDS-PAGE Buffer Systems and Electrophoretic Separation
Traditional and Advanced Buffer Formulations
The discontinuous buffer system fundamental to SDS-PAGE relies on ionic boundaries to concentrate and separate proteins based on molecular weight. The traditional Laemmli system (Tris-Glycine, pH 8.3) has served as the benchmark for decades, yet newer formulations address specific limitations in resolution and separation time [13] [82].
Table 2: Comparison of SDS-PAGE Running Buffer Systems
| Buffer System | Leading Ion | Trailing Ion | Effective Separation Range | Run Time | Optimal Applications |
|---|---|---|---|---|---|
| Tris-Glycine (Laemmli) | Chloride | Glycinate | 10-200 kDa | 90 minutes | Standard protein separation; broad molecular weight ranges |
| Tris-Tricine | Chloride | Tricine | 1-100 kDa | 3-5 hours | Small proteins and peptides (<15 kDa) |
| Tris-Tricine-HEPES (FRB) | Acetate/Chloride | Tricine/HEPES | 15-450 kDa | 35 minutes | High-throughput applications; simultaneous large and small protein separation |
The fundamental mechanism involves creation of a voltage gradient between highly mobile leading ions (chloride) and slower trailing ions (glycinate or Tricine). At pH 8.3 in the running buffer, glycine exists primarily as zwitterions with minimal net charge, migrating slowly through the stacking gel (pH 6.8). Upon entering the resolving gel (pH 8.8), glycine molecules become predominantly negatively charged, overtaking proteins and depositing them in a tight band at the top of the resolving gel [82]. The Tris-Tricine-HEPES buffer creates multiple ionic boundaries instead of two, significantly improving resolving power while reducing Joule's heat generation [13].
Buffer Preparation Protocols
Traditional Tris-Glycine SDS Running Buffer (10X) [12] [27]
- Tris base: 30.3 g (250 mM final concentration)
- Glycine: 144.1 g (1.92 M final concentration)
- SDS: 10 g (1% final concentration)
- Add distilled water to 1 L total volume
- Adjust to pH 8.3 if necessary
- Dilute to 1X with distilled water before use
Fast-Running Buffer (FRB) Formulation [13]
- Tris base: 250 mM
- Tricine: 50 mM
- HEPES: 75 mM
- SDS: 0.1%
- No pH adjustment required (final pH approximately 7.5-8.0)
- Use with Tris-Acetate gels for optimal results
Integrated Experimental Workflows
Optimal Protein Extraction and Separation Workflow
The following workflow diagram illustrates the integrated process from protein extraction to SDS-PAGE separation, highlighting critical buffer-dependent steps:
Correlated Efficacy: Extraction Methods and Buffer Compatibility
Different protein extraction methods yield lysates with varying compatibility with SDS-PAGE buffer systems. The table below summarizes optimal pairings based on extraction efficiency and downstream separation quality:
Table 3: Correlation Between Extraction Methods and Buffer Conditions
| Extraction Method | Optimal Running Buffer | Recommended Gel Percentage | Key Advantages | Limitations |
|---|---|---|---|---|
| SDT-B-U/S (Recommended) | Tris-Tricine-HEPES (FRB) | 10% homogeneous gel | Superior membrane protein recovery; enhanced reproducibility; compatible with wide MW range | Slightly longer preparation time |
| SDT-B (Boiling) | Tris-Glycine | 8-12% gradient | Rapid processing; minimal equipment requirements | Poor efficiency for Gram-positive bacteria; limited membrane protein recovery |
| SDT-U/S (Ultrasonication) | Tris-Tricine-HEPES (FRB) | 10-12% homogeneous gel | Effective for heat-sensitive proteins; moderate efficiency for both Gram classes | Heat generation may denature thermolabile proteins |
| SDT-LNG-U/S (Liquid N₂) | Tris-Tricine | 12-15% for small proteins | Maximum disruption efficiency for tough cell walls | Time-consuming; requires specialized handling |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Integrated Protein Extraction and Electrophoresis
| Reagent/Solution | Composition | Function in Workflow | Considerations |
|---|---|---|---|
| SDT Lysis Buffer | 4% SDS, 100 mM DTT, 100 mM Tris-HCl (pH 7.6) | Cell membrane disruption; protein denaturation; reduction of disulfide bonds | Critical for efficient extraction; must be fresh prepared for optimal reduction |
| Laemmli Sample Buffer | Tris-HCl, SDS, glycerol, β-mercaptoethanol or DTT, Bromophenol Blue | Protein denaturation; sample visualization; well loading density | β-mercaptoethanol or DTT concentration affects reduction efficiency; glycerol prevents diffusion from wells |
| Tris-Glycine Running Buffer | 25 mM Tris, 192 mM glycine, 0.1% SDS (pH 8.3) | Electrophoretic separation; maintaining protein denaturation; establishing pH gradient | Reuse not recommended due to protein fragment contamination and SDS degradation [6] |
| Tris-Tricine-HEPES FRB | 250 mM Tris, 50 mM Tricine, 75 mM HEPES, 0.1% SDS | Rapid, high-resolution separation; wide molecular weight range compatibility | Optimal with Tris-Acetate gels; no pH adjustment required [13] |
| Polyacrylamide Gel | Acrylamide/bis-acrylamide, Tris-HCl, SDS, TEMED, ammonium persulfate | Molecular sieving matrix for size-based separation | Percentage determines resolution range; incomplete polymerization causes poor resolution [83] |
Troubleshooting Common Issues
Integration-Specific Challenges
Several common electrophoretic anomalies can be traced to suboptimal correlations between extraction methods and buffer conditions:
- Band smearing: Often results from incomplete denaturation during extraction or insufficient SDS in running buffer [54] [83]. Remedy by ensuring proper boiling duration (5 minutes at 98°C) and using fresh running buffer.
- Poor band resolution: Can indicate mismatch between protein size range and gel percentage, or excessive protein loading [54]. For extraction methods yielding broad MW distributions (e.g., SDT-B-U/S), gradient gels (4-20%) often provide superior resolution.
- Horizontal band distortion: Frequently caused by high salt concentration in extraction buffers [54]. Desalting through acetone precipitation or dialysis before electrophoresis is recommended.
- Missing bands: May indicate protein degradation during extraction or electrophoresis [54]. Maintain protease inhibitors during extraction and ensure gel apparatus cooling during runs.
The correlation between protein extraction methodologies and SDS-PAGE buffer conditions significantly influences proteomic analysis outcomes. The SDT-B-U/S extraction method combined with advanced buffer systems such as Tris-Tricine-HEPES FRB demonstrates superior performance for comprehensive proteome coverage, particularly for challenging samples including membrane proteins and organisms with robust cell walls. Researchers should select extraction and separation conditions as an integrated system rather than independent workflow steps, paying particular attention to gram-class specific requirements, molecular weight ranges of interest, and downstream application needs. This systematic approach ensures optimal protein recovery, resolution, and reproducibility—foundational requirements for rigorous scientific research and drug development.
The Impact of Buffer Integrity on Downstream Applications like Western Blotting
The integrity of running buffers is a foundational, yet frequently overlooked, factor determining the success of SDS-PAGE and the reliability of subsequent western blot data. The Tris-Glycine-SDS buffer system is central to the discontinuous gel electrophoresis technique, facilitating the separation of proteins based on molecular weight [84]. Any deviation in the composition, pH, or concentration of this buffer directly compromises protein separation, transfer efficiency, and ultimately, the accuracy of protein detection and quantification [85] [86]. This application note details the critical role of buffer integrity within the context of SDS-PAGE research, providing validated protocols and troubleshooting guides to ensure robust and reproducible western blot results.
The Science of SDS-PAGE Running Buffer
Composition and Function
The standard SDS-PAGE running buffer is a Tris-Glycine-SDS system. Each component plays a specific and vital role in the electrophoretic separation of proteins [84].
- Tris (trimethylaminomethane): Acts as the buffering agent. Its pKa of 8.1 makes it ideal for maintaining a stable pH throughout the electrophoresis process, which is crucial for consistent protein migration [84].
- Glycine: An amino acid that serves as the trailing ion in the discontinuous buffer system. Its charge state is pH-dependent, which is the key mechanism behind the stacking effect that concentrates proteins into sharp bands before they enter the resolving gel [84].
- SDS (Sodium Dodecyl Sulfate): An anionic detergent that uniformly coats denatured proteins, imparting a net negative charge proportional to their mass. This masks the proteins' inherent charges, ensuring separation is based primarily on molecular size [85] [84].
The delicate interplay of these components, particularly the ionic fronts of chloride and glycine, creates a stacking effect that concentrates protein samples into sharp lines before they enter the resolving gel, which is fundamental to achieving high-resolution separation [84].
Consequences of Buffer Compromise
The use of compromised running buffer—through improper preparation, contamination, or repeated use—leads to a range of experimental artifacts that degrade data quality.
Table 1: Common Issues Arising from Compromised Running Buffer
| Issue | Probable Cause | Impact on Downstream Western Blot |
|---|---|---|
| Poor Protein Separation [85] | Incorrect pH, outdated buffer, incorrect Tris/Glycine concentrations | Diffuse or smeared bands; inaccurate molecular weight estimation; difficulty in quantifying specific bands. |
| "Smiling" or "Bulging" Bands [85] | Buffer made incorrectly, leading to excessive heat during electrophoresis | Uneven protein migration across the gel; compromised densitometry and quantitative analysis. |
| Skewed Migration of Proteins | Low-quality or degraded SDS, leading to inconsistent protein coating | Proteins do not migrate according to true molecular weight; potential for misinterpretation. |
| High Background & Poor Transfer [87] | Microbial growth or contaminants in stored buffer | Increased non-specific antibody binding; high background noise on the western blot membrane. |
Protocols for Buffer Preparation and Quality Control
Standard SDS Running Buffer Formulation
A standard 10X running buffer stock solution can be prepared as follows and stored at 4°C [12] [88]. Dilute to 1X working concentration with deionized water prior to use.
Table 2: Standard Recipe for 10X SDS-PAGE Running Buffer
| Component | Molecular Weight | Final Concentration (10X) | Mass per Liter |
|---|---|---|---|
| Tris Base | 121.14 g/mol | 250 mM | 30.29 g |
| Glycine | 75.07 g/mol | 1.92 M | 144.13 g |
| SDS | 288.38 g/mol | 1% (w/v) | 10 g |
Quality Control and Optimization Measures
To ensure buffer integrity, implement the following quality control protocols:
- pH Verification: The pH of the 1X working solution must be 8.3 [88]. Always check the pH of a diluted aliquot; concentrating a 10X solution will give an inaccurate reading. Adjust if necessary with HCl or NaOH.
- Visual Inspection: Discard any buffer that shows signs of precipitation or microbial contamination.
- Single-Use Policy: For optimal and reproducible results, prepare fresh 1X working solution for each electrophoresis run. If re-use is necessary, limit it to a maximum of two runs and store at 4°C, noting a potential increase in background noise [89].
- Electrophoresis Conditions: For a standard mini-gel, run at a constant voltage of 125V for approximately 90 minutes, or until the dye front reaches the bottom of the gel [27].
The following workflow outlines the key steps from buffer preparation to analysis, highlighting critical control points.
The Researcher's Toolkit: Essential Reagents and Materials
Table 3: Essential Reagents for SDS-PAGE and Western Blotting
| Reagent | Function | Key Considerations |
|---|---|---|
| Tris-Glycine-SDS Running Buffer [84] [88] | Facilitates electrophoretic separation of proteins by size. | Must be prepared to exact specifications (25 mM Tris, 192 mM Glycine, 0.1% SDS, pH 8.3). |
| Precast or Handcast Polyacrylamide Gels [85] | Matrix for protein separation. Pore size determines resolution range. | Choose gel percentage based on target protein size (e.g., 8% for >50kDa, 15% for <50kDa). |
| Protein Molecular Weight Markers [85] | Calibrate gel and estimate size of unknown proteins. | Prestained markers allow real-time tracking. Unstained markers offer higher accuracy. |
| Sample Loading Buffer (Laemmli Buffer) [84] | Denatures proteins and provides dye for visualization. | Contains SDS, reducing agents (DTT/BME), glycerol, and tracking dye. |
| Transfer Buffer [86] | Medium for electrophoretic transfer of proteins from gel to membrane. | Composition (e.g., addition of methanol) affects efficiency, especially for high or low MW proteins. |
| PVDF or Nitrocellulose Membrane [86] [90] | Solid support for immobilizing transferred proteins for antibody probing. | PVDF offers higher protein retention and durability for reprobing. |
Troubleshooting Guide
Below is a structured guide to diagnosing and resolving common problems linked to buffer integrity.
Problem: Smeared Bands Across Lanes
- Cause: Insufficient SDS in the running buffer or sample, leading to incomplete protein denaturation and charge masking [85] [87].
- Solution: Prepare fresh running buffer with the correct 0.1% SDS concentration. Ensure sample buffer contains adequate SDS and fresh reducing agent, and boil samples for 5 minutes at 100°C [85].
Problem: "Smiling" or "Bulging" Bands
Problem: Unusual Banding Patterns or Poor Resolution
Problem: Slow Migration of Proteins
Buffer integrity is not a minor technical detail but a fundamental pillar of reproducible and high-quality SDS-PAGE and western blotting. Adherence to precise preparation protocols, coupled with rigorous quality control, ensures optimal protein separation and reliable data. By recognizing the critical impact of the Tris-Glycine-SDS system on downstream applications, researchers can significantly reduce experimental artifacts, enhance the validity of their findings, and accelerate the drug development process.
Running buffers form the foundational liquid environment that enables protein separation in both conventional Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and emerging microfluidic "lab-on-a-chip" technologies. While the fundamental chemistry of Tris-Glycine-SDS buffers remains consistent, their implementation in microfluidic systems presents unique challenges and opportunities for advancement. The core function of running buffers in SDS-PAGE is to maintain stable pH and provide ions necessary for conducting current while ensuring proteins remain uniformly coated with SDS for separation by molecular weight. Traditional discontinuous buffer systems, such as the Laemmli buffer (0.25 M Tris, 1.92 M Glycine, 1% SDS, pH ~8.6), have served as the gold standard for decades [31] [3]. However, the migration of this technique to microfluidic platforms necessitates significant reformulation and re-engineering of these established buffer systems to accommodate drastically reduced volumes, different material interfaces, and advanced functionality.
The emergence of microfluidic SDS electrophoresis represents a paradigm shift in protein analysis, offering dramatic reductions in analysis time (from hours to minutes) and sample/reagent consumption (typically less than 0.5 mL total volume per chip for 10 samples) compared to traditional methods [91]. This transition is driven by the broader life science industry's push toward automation, miniaturization, and high-throughput workflows [92]. Within this context, running buffers are evolving from simple conductive solutions into multifunctional components that actively participate in achieving high-resolution separations within the constraints of microfluidic architectures. This application note explores the evolving role of running buffers in these emerging systems, providing detailed protocols and analytical frameworks for researchers adapting SDS-PAGE methodologies to microfluidic platforms.
Comparative Analysis: Running Buffers in Traditional vs. Microfluidic Systems
Traditional SDS-PAGE Running Buffers
Traditional SDS-PAGE relies on robust, high-capacity buffer systems designed for macroscopic gel tanks. The standardized formulations for these systems are well-established, with precise concentrations optimized for decades of laboratory use.
Table 1: Standardized Buffer Formulations for Traditional SDS-PAGE
| Buffer System | Component | Final Concentration (1X) | 10X Stock Concentration | Amount per Liter (10X) | pH |
|---|---|---|---|---|---|
| Tris-Glycine-SDS (Laemmli) [31] [3] | Tris base | 25 mM | 0.25 M | 30 g | ~8.6 (unadjusted) |
| Glycine | 192 mM | 1.92 M | 144 g | ||
| SDS | 0.1% | 1% | 10 g | ||
| Tris-Tricine-SDS [31] | Tris base | 60 mM | 1.2 M | 145 g | ~8.5 (unadjusted) |
| Tricine | 40 mM | 0.8 M | 143 g | ||
| SDS | 0.1% | 2% | 20 g |
These traditional buffers are characterized by their relatively high ionic strength and volume consumption (typically several liters per run), which provides stable buffering capacity but generates significant heat requiring dissipation through bulky tank designs [91]. The Tris-Glycine-SDS system creates a discontinuous buffer system that stacks proteins into sharp bands before separation, a principle that must be preserved in microfluidic adaptations.
Microfluidic System Requirements and Constraints
Microfluidic SDS electrophoresis introduces distinct constraints that directly impact buffer formulation and implementation. The Agilent Protein 80 kit, for instance, can separate proteins ranging from ~5 to 80 kDa within a miniaturized format [91]. Key considerations for buffers in these systems include:
- Volume Minimization: Chip-based systems consume less than 5% of the buffer volume required for traditional gel tanks [91].
- Heat Dissipation: High surface-to-volume ratios in microchannels enable efficient heat transfer but require careful buffer conductivity management to prevent Joule heating effects.
- Material Compatibility: Buffer chemistry must be compatible with polymer chip materials (e.g., PDMS, PMMA) without causing degradation or leaching.
- Interfacial Effects: Surface tension and electroosmotic flow become significant factors at microscale dimensions.
- Multifunctionality: Advanced systems utilize buffers that serve multiple purposes, including as reagent containers for on-chip SDS-protein complexation [93].
Table 2: Performance Comparison of Traditional vs. Microfluidic SDS-PAGE Systems
| Parameter | Traditional SDS-PAGE | Microfluidic SDS Electrophoresis | Implications for Running Buffers |
|---|---|---|---|
| Separation Time | 1-2 hours | < 30 minutes for 10 samples | Higher conductivity buffers possible |
| Sample Volume | 10-20 µL | Significantly reduced | Buffer protein compatibility at high concentrations |
| Buffer Consumption | Several liters | < 0.5 mL for 10 samples [91] | Concentrated stocks, reduced waste |
| Resolution | High for most milk proteins [91] | Comparable for major milk proteins (α-LA, β-LG, caseins) [91] | Requires optimized discontinuous systems |
| Automation Potential | Low to moderate | High with integrated designs | Compatibility with automated dispensing |
Advanced Microfluidic Applications and Protocols
Microfluidic 2-D PAGE with Discontinuous Buffers
Innovative microfluidic systems have been developed that integrate two-dimensional separation techniques, combining isoelectric focusing (IEF) with SDS-PAGE in a single chip platform. These systems employ in situ photopolymerized polyacrylamide (PAAm) gels that serve multiple functions beyond mere separation matrices [93]. Discrete polyacrylamide gel plugs act as:
- Separation media for high-resolution electrophoresis
- On-chip reagent containers holding defined quantities of SDS
- Isolation elements separating different on-chip media (anolyte, catholyte, sample/ampholyte)
- Buffer discontinuity enablers for sample band sharpening during SDS-PAGE
This multifunctional approach more than doubles the resolving power compared to single-dimension microfluidic separation [93]. The diagram below illustrates the workflow and functional components of such an integrated 2-D microfluidic system.
Protocol: Microfluidic SDS Electrophoresis with Integrated Buffer Systems
Objective: Perform protein separation using a microfluidic chip with multifunctional gel plugs and discontinuous buffers for enhanced resolution.
Materials:
- Microfluidic 2-D PAGE chip with angled IEF channel and backbiasing transfer channels [93]
- Multifunctional in situ photopolymerized polyacrylamide gels
- Protein samples (e.g., E. coli cell lysate)
- Anolyte (20 mM phosphoric acid)
- Catholyte (20 mM sodium hydroxide)
- SDS running buffer (modified Tris-Glycine with additives)
- Chip priming station
- High-voltage power supply
- Microfluidic chip analyzer with detection system
Methodology:
Chip Preparation and Priming
- Inspect microfluidic channels for defects or blockage.
- Prime separation channels with SDS running buffer using manufacturer-specified pressure and time parameters.
- Verify proper fluid movement through all channels, including backbiasing transfer channels.
Sample Preparation and Loading
- Dilute protein sample to appropriate concentration (typically 1-2 μg/μL) in IEF-compatible buffer.
- Load 1-5 μL of prepared sample into sample reservoir.
- Add anolyte and catholyte to respective reservoirs according to chip design.
First Dimension: Isoelectric Focusing
- Apply focusing voltage gradient according to manufacturer specifications (typically 100-1000 V/cm).
- Monitor current stabilization to determine focusing completion (typically 1-5 minutes).
- Utilize angled IEF channel design to minimize sample tailing effects [93].
Interdimensional Transfer
- Activate backbiasing channels to achieve uniform interdimensional sample transfer.
- Engage multifunctional gel plugs to release SDS for on-chip protein complexation.
- Establish buffer discontinuity for sample band sharpening prior to second dimension.
Second Dimension: SDS-PAGE Separation
- Apply separation voltage (typically 200-500 V) across SDS-PAGE dimension.
- Utilize discontinuous buffer system embedded in gel plugs for high-resolution separation.
- Monitor separation progress through integrated detection system.
Data Collection and Analysis
- Collect real-time electrophoregram data during separation.
- Calculate protein molecular weights based on migration time against standard curves.
- Perform quantification through integrated software algorithms.
Technical Notes:
- The 20-channel chip design demonstrates improved separation resolution compared to 10-channel configurations [93].
- Never adjust the pH of running buffers in microfluidic systems as this alters ionic characteristics critical for performance [31].
- Standard curves for major proteins (α-lactalbumin, β-lactoglobulin) show linear response with both microfluidic chip and traditional SDS-PAGE techniques [91].
The Scientist's Toolkit: Essential Reagents for Microfluidic Electrophoresis
Successful implementation of microfluidic SDS electrophoresis requires specialized reagents optimized for microscale separations. The following table details essential research reagent solutions and their specific functions in these advanced systems.
Table 3: Research Reagent Solutions for Microfluidic SDS Electrophoresis
| Reagent Solution | Composition | Function in Microfluidic Systems | Key Considerations |
|---|---|---|---|
| High-Efficiency SDS Running Buffer | Tris-Glycine with optimized conductivity | Provides stable electrophoretic field with minimal Joule heating | Pre-mixed, pH-stabilized formulations preferred for reproducibility [94] |
| Multifunctional Gel Matrix | In situ photopolymerized PAAm with embedded reagents | Serves as separation medium and reagent storage container [93] | Must maintain stability during chip storage and operation |
| Chip-Compatible Protein Ladders | Fluorescently-labeled standards (5-250 kDa) | Enables molecular weight calibration in absence of visible bands | Ultra-wide range markers essential for diverse applications |
| Microfluidic Surface Modifier | Polymeric surfactants and coating agents | Controls electroosmotic flow and prevents protein adsorption | Critical for maintaining separation efficiency in polymer chips |
| Integrated Staining Solution | Fluorescent dyes in optimized buffer | Enables in-chip protein detection without destaining | Must be compatible with chip materials and detection optics |
| Buffer Additive Kit | Conductivity modifiers, viscosity enhancers | Fine-tunes separation characteristics for specific applications | Allows customization without reformulating core buffer |
Future Outlook and Commercial Landscape
The SDS-PAGE electrophoresis buffer market, valued at $94.5 million in 2025 with a projected CAGR of 3.2% through 2033, reflects ongoing innovation in buffer formulations for emerging technologies [94]. Key future directions for running buffers in microfluidic systems include:
Innovation Trends:
- Pre-mixed, Ready-to-Use Formulations: Development of pre-aliquoted, pH-stabilized buffers specifically designed for gradient gels in microfluidic applications [92].
- Enhanced Conductivity Buffers: New formulations with improved conductivity enable faster electrophoresis runs without excessive heat generation [94].
- Specialized Application Buffers: Emerging buffers tailored for specific applications such as high-resolution separation of membrane proteins or improved transfer efficiency for microfluidic Western blotting [94].
- Additive-Enhanced Formulations: Incorporation of additives that reduce heat generation during electrophoresis and improve separation sharpness [94].
Market Dynamics: The competitive landscape features established life science leaders including Thermo Fisher Scientific, Bio-Rad Laboratories, and Merck KGaA, alongside specialized manufacturers developing innovative buffer solutions for microfluidic platforms [94]. These companies are expanding their portfolios to include buffers optimized for the unique requirements of chip-based electrophoresis, including compatibility with automated fluid handling systems and enhanced stability for long-term storage.
Technical Advancements: Future microfluidic systems will likely incorporate running buffers with dynamically adjustable properties, enabled by on-chip mixing of multiple buffer components to optimize separation conditions in real-time. The integration of microfluidic SDS electrophoresis with downstream analysis techniques such as mass spectrometry will further drive innovation in buffer formulations that facilitate seamless workflow transitions while maintaining protein integrity and compatibility with ionization processes.
Running buffers, once considered merely supportive components in traditional SDS-PAGE, have evolved into sophisticated, multifunctional solutions that enable high-performance protein separations in emerging microfluidic systems. The successful adaptation of Tris-Glycine-SDS buffer principles to microfluidic platforms requires careful consideration of volume constraints, material compatibility, and heat management while preserving the discontinuous buffer characteristics essential for high-resolution separations. As microfluidic technology continues to advance toward greater integration, automation, and miniaturization, running buffer formulations must similarly evolve to meet the demanding requirements of these next-generation protein analysis platforms. The protocols and analyses presented in this application note provide researchers with the foundational knowledge required to leverage these advanced buffer systems in their microfluidic SDS electrophoresis workflows.
Conclusion
The SDS-PAGE running buffer is far more than a simple salt solution; it is a foundational component that dictates the success of protein separation. Its precise composition ensures proper current flow, maintains critical pH, and keeps proteins denatured. Mastering its preparation and understanding its role in troubleshooting common issues like smeared or poorly resolved bands is non-negotiable for obtaining publication-quality data. As protein analysis evolves with technologies like lab-on-chip systems, the principles of robust buffer design remain paramount. For researchers in drug development and clinical fields, where reproducibility is key, a deep understanding of the running buffer directly translates to more reliable results in protein characterization, purity assessment, and biomarker validation, ultimately accelerating biomedical discovery.
References
- 1. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOopDKjWuX1L8K8u9ugh_Y-RBK5qisoVnWtOpnDc0vDJqz6_Eduoq]
- 2. SDS-PAGE - Mullins Lab - [https://mullinslab.ucsf.edu/sds-page/]
- 3. SDS-PAGE SDS Running Buffer (10x) [https://www.novoprolabs.com/tools/buffer-preparations-and-recipes/sds-page-sds-running-buffer-10x]
- 4. SDS-PAGE V.2 [https://www.protocols.io/view/sds-page-chqut5ww.html]
- 5. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOor2r38V895umJhug50APfR8nF7UZHYW-I795mev4rLMTaqAEspG]
- 6. What's the Difference between Running and Transfer Buffer? [https://azurebiosystems.com/blog/electrophoresis-101-the-difference-between-running-and-transfer-buffer/]
- 7. SDS-PAGE Optimization for Western Blot [https://www.bosterbio.com/protocol-and-troubleshooting/western-blotting-optimization/sds-page?srsltid=AfmBOoo_HD5zdAdsFbfzurkvOotAhP8-0XMQYDdtp8Ngseq3reyUsvKP]
- 8. SDS-PAGE Protocol [https://neutab.creative-biolabs.com/sds-page-analysis.htm]
- 9. 10X Tris-Glycine-SDS Buffer, 1 L-S1008 [https://www.ezbioresearch.com/10X-Tris-Glycine-SDS-Buffer-1-L_p_289.html]
- 10. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOooTeKPdRtvYb_sF87qInyeEL_RLwM56hxTW0glteMmHC-OrE5np]
- 11. Novex™ Tris-Glycine SDS Sample Buffer (2X) - FAQs [https://www.thermofisher.com/store/v3/products/faqs/LC2676]
- 12. SDS-PAGE SDS Running Buffer (10x) Preparation and ... [https://www.aatbio.com/resources/buffer-preparations-and-recipes/sds-page-10x-sds-running-buffer]
- 13. The gradient-like separation and reduced running time with ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269722002457]
- 14. How Does A Buffer Maintain pH? [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Acids_and_Bases/Buffers/How_Does_A_Buffer_Maintain_Ph]
- 15. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoomLsFXlzinaIe5ooNTXIsXgxtlWpXSwWcmfi55Z2UjB2mwdj1F]
- 16. Buffers, buffering capacity, oxidation and reduction, ... [https://www.wikilectures.eu/w/Buffers,_buffering_capacity,_oxidation_and_reduction,_electrode_processes_(1.LF_UK,_GM)]
- 17. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOor_GiT3Wv5NpXBsErN5lkZM4WxRVEi25GoHmn2l5Ok3L8r8i7z-]
- 18. Tips for Optimal SDS-PAGE Separation [https://www.rockland.com/resources/tips-for-optimal-sds-page-separation/?srsltid=AfmBOorxNj_f7MSxx6qAOXadxBL5WHHYVLSB_yXdDfhEWmI5Ft7I9Vub]
- 19. SDS-PAGE Optimization for Western Blot [https://www.bosterbio.com/protocol-and-troubleshooting/western-blotting-optimization/sds-page?srsltid=AfmBOop28yt7uEtwt9VQ6Rk2ragMiJe8JtH_m5_5321M6Zm-G2fCPxu6]
- 20. Electrophoresis [https://pubmed.ncbi.nlm.nih.gov/36251838/]
- 21. Influence of ionic strength, pH, and SDS concentration on ... [https://pubmed.ncbi.nlm.nih.gov/20195791/]
- 22. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOoqXt6NrpJ_QmhN6_PR0bqTEli-cdIxciiKoLU7_9Dflj8uvzR67]
- 23. Overview of Protein Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 24. SDS protein interactions [https://www.sciencedirect.com/science/article/abs/pii/S0301462225001462]
- 25. Overview of Protein Electrophoresis [https://www.thermofisher.com/id/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 26. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoo42BUZUzPBjNpG91lR1hYcpZdlFGY0fkVne9fpZ1tS_rJrZvbO]
- 27. One-Dimensional SDS and Non-Denaturing Gel ... [https://www.thermofisher.com/us/en/home/references/protocols/proteins-expression-isolation-and-analysis/sds-page-protocol/one-dimensional-sds-and-non-denaturing-gel-electrophoresis.html]
- 28. Troubleshooting SDS-PAGE Gel Running Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-gel-running-issues?srsltid=AfmBOoq6z52uyWTeM0Sc8MQBDcp9-PNCTLTpKMs7IatevkqCt1ZiMLZt]
- 29. Tris-Glycine-SDS Running Buffer (10X) [https://www.bostonbioproducts.com/products/tris-glycine-sds-running-buffer-10x-bp-150]
- 30. Introduction to SDS-PAGE - Separation of Proteins Based ... [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/protein-biology/gel-electrophoresis/sds-page?srsltid=AfmBOoo0cnJt1mPofFVcxrPhX7Vw_nw94XAYdKjQE2d7kpze-E5Lj290]
- 31. Running buffer preparation › SDS PAGE ... [https://www.serva.de/enDE/611_Information_Center_Electrophoresis_SDS_PAGE_Running_buffer_preparation.html]
- 32. Tris-Glycine SDS Running Buffer (10X) #4050 [https://www.cellsignal.com/products/buffers-dyes/tris-glycine-sds-running-buffer-10x/4050?srsltid=AfmBOoqs5LycbEQ76jUoMAQJc0IjrBy9vy7scilqIz84v93M1AlS--gd]
- 33. 10X Tris-Glycine-SDS Buffer Concentrate [https://www.ibisci.com/products/10x-tris-glycine-sds-buffer-concentrate]
- 34. ROTIPHORESE® 10 - x , 1 l, glass | Gel Buffer SDS PAGE Solutions [https://www.carlroth.com/com/en/gel-buffer-solutions/rotiphorese10x-sds-page/p/3060.1]
- 35. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoqePeqmLFMhXllK-pn47d5tBBbartfgYs0Aop11GKkGih82kiqb]
- 36. Tips for Optimal SDS-PAGE Separation [https://www.rockland.com/resources/tips-for-optimal-sds-page-separation/?srsltid=AfmBOopiQRx534gwJOy8c4ZWxtnL2nvJpqpPHLh_bU3YJvoIiPd0yBtV]
- 37. Preparation of SDS - PAGE Running Buffer ( 10 ) - Laboratory Notes x [https://www.laboratorynotes.com/preparation-of-sds-page-running-buffer-10x/]
- 38. Solutions and dilutions: working with stock solutions [https://www.ruf.rice.edu/~bioslabs/methods/solutions/stocks.htm]
- 39. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOooZYO29NAV3ovP6g_FjZEFrRxfJWqx-pigrH58Zz2qqcf8qFFh4]
- 40. Buffers and stock solutions for western blot [https://www.abcam.com/en-us/technical-resources/guides/western-blot-guide/buffers-and-stock-solutions]
- 41. SDS-PAGE Tips and Tricks to Save You Time [https://bitesizebio.com/28798/sds-page-tips-tricks-save-time/]
- 42. Long-term, buffer-less, wet gel storage in non-sealed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7310580/]
- 43. Tips for Optimal SDS-PAGE Separation [https://www.rockland.com/resources/tips-for-optimal-sds-page-separation/?srsltid=AfmBOoroFLHP7gGkuF_ovHqBp42Sk4687quBLj4VJ-Rjnb9eEKSThKUk]
- 44. One-Dimensional SDS Gel Electrophoresis of Peptides ... [https://www.thermofisher.com/us/en/home/references/protocols/proteins-expression-isolation-and-analysis/sds-page-protocol/one-dimensional-sds-gel-electrophoresis-of-peptides-and-small-proteins-with-pre-cast-gels.html]
- 45. The gradient-like separation and reduced running time with ... [https://pubmed.ncbi.nlm.nih.gov/35738440/]
- 46. Tris-Tricine-SDS Buffer 10× Concentrate [https://www.sigmaaldrich.com/US/en/product/sigma/t1165?srsltid=AfmBOorGWPwJQmWiW-wXvTC6WDv3gK-gqlmJb1kYntJpWasYzE2eq6NM]
- 47. Tris-Tricine SDS-PAGE and Immunoblotting [https://bio-protocol.org/exchange/minidetail?id=4117310&type=30]
- 48. Tris-Tricine SDS-PAGE Gel Protocol | PDF [https://www.scribd.com/document/295359266/16-5-Tris-Tricine-Gel]
- 49. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoqiEgWOLWinkIb92P1O9rwVGUA0KV9klaH-gTwddOgZIwRf9T6Y]
- 50. SDS-PAGE guide: Sample preparation to analysis [https://www.abcam.com/en-us/knowledge-center/western-blot/sds-page-guide]
- 51. Massive Screening of Food Extracts for Quality Assessment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11674539/]
- 52. Reveal® 3-D Food Buffer | Food Safety [https://www.neogen.com/en/categories/allergens/reveal-3d-food-buffer/]
- 53. Troubleshooting SDS-PAGE Gel Running Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-gel-running-issues?srsltid=AfmBOoqYdcA_grSAE4O7A9r9Ij5KdYpJYjciuyjVZbiL41BblUuGeeM4]
- 54. Troubleshooting Sodium Dodecyl Sulfate- Polyacrylamide ... [https://www.hycultbiotech.com/sds-page/troubleshooting/]
- 55. Troubleshooting Common Electrophoresis Problems and ... [https://www.labx.com/resources/troubleshooting-common-electrophoresis-problems-and-artifacts/5539]
- 56. What to do about Bendy Bands - SDS-PAGE [https://azurebiosystems.com/blog/bendy-bands-and-what-to-do-with-them/]
- 57. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOor62MXilFRsyTNBde--oMzRHOMaeVeGD6RRHv_qz_bn4LeeilkD]
- 58. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoraCIZ2pdlW2_uKDuzCGy3VJIQVjORvjlH69mUoKwWO7rZL-5uL]
- 59. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoogESl09Y3c_vWoSfI_lWt-HHQwAIPFVniFu7nHcHFnj2QSj5v4]
- 60. Troubleshooting SDS-PAGE Gel Running Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-gel-running-issues?srsltid=AfmBOorpzG4xj7qnduqY8YPaUm9vno-z0ZWW7Xnni22iOuJRhrywq3fV]
- 61. Guide to SDS-PAGE: Tips for Better Results with NuSep ... [https://nusep.us/pages/guide-to-sds-page-tips-for-better-results-with-nusep-precast-gels?srsltid=AfmBOoofLNIdX9VvF_TjPh6QDnl-BhaDOPJdSarTqpQUS56FLWcFrZPz]
- 62. Troubleshooting SDS-PAGE Gel Running Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-gel-running-issues?srsltid=AfmBOop5ORh9lWyqbAIo4OrwDxHotEHfQAL-yg5nYlD_qLNBAI16tTSw]
- 63. Guide to SDS-PAGE: Tips for Better Results with NuSep ... [https://nusep.us/pages/guide-to-sds-page-tips-for-better-results-with-nusep-precast-gels?srsltid=AfmBOopCm5qTnAkPlnyxQzdHeyGtHuBU8cGKsXob0L_YdpeJpXn-rG96]
- 64. Protein Electrophoresis Using SDS-PAGE: A Detailed ... [https://www.goldbio.com/blogs/articles/protein-electrophoresis-sds-page-overview?srsltid=AfmBOoraty6bBYbg3ywIq0oZ_Qe2MBdJRXEOSLyEnRyrkzrk6Uu0onNu]
- 65. Tips for Optimal SDS-PAGE Separation [https://www.rockland.com/resources/tips-for-optimal-sds-page-separation/?srsltid=AfmBOoqEJzeOzzO7DpLVNDgjxaCD4HucS5O73ZTsW70iG9ZHY-yC4dYg]
- 66. Improving SDS-PAGE method for monoclonal antibodies [https://www.sciencedirect.com/science/article/abs/pii/S1046592821000280]
- 67. SDS-PAGE: A Cornerstone of Protein Analysis in Modern ... [https://www.metwarebio.com/sds-page-protein-analysis/]
- 68. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoo41eN3rMHJdS2FVABoQG1x2Ws4Jj6LLhbuVHvWXepoOVhSQkaH]
- 69. Troubleshooting SDS-PAGE Gel Running Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-gel-running-issues?srsltid=AfmBOoo-UpoQaEIYzxbrahW84tXVhzZ6EzHIjfdLNi7YZwjequBWoDFg]
- 70. Elimination of Protein Band "Edge Tailing" in Weber ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269783714090]
- 71. Eight Tips to Improve Gel Electrophoresis Results [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/8-DNA-ladder-tips.html]
- 72. Common artifacts and mistakes made in electrophoresis [https://pmc.ncbi.nlm.nih.gov/articles/PMC7295095/]
- 73. How current, voltage, and power settings affect SDS-PAGE [https://www.biossusa.com/blogs/news/how-current-voltage-and-power-settings-affect-sds-page?srsltid=AfmBOoo7u6K5qlZU-RM5WgQ--SFjreZoHRjB6aX06E_9Ye4EBlKMoXLj]
- 74. Constant Current or Voltage in SDS-PAGE [https://bitesizebio.com/51744/constant-current-or-voltage-in-sds-page/]
- 75. How Does SDS-PAGE Work? A Comprehensive Guide [https://www.metwarebio.com/how-sds-page-works-guide/]
- 76. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOorMu_rvEz7LVFSdh10FDkSlPwxnitIt3jkeAoQUxFPhV8i4idGO]
- 77. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOopvNppcEK_wV71EJQUmsK10KPKyx7Qkypb2W3eyMQFVyczhs0a8]
- 78. Native SDS-PAGE: High Resolution Electrophoretic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4517606/]
- 79. Overview of Protein Electrophoresis [https://www.thermofisher.com/tw/zt/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 80. Tris Tricine SDS PAGE: What is it and how to PERFORM it? [https://info.gbiosciences.com/blog/tris-tricine-sds-page-what-is-it-and-how-to-perfrom-it]
- 81. Evaluation of protein extraction methodologies on bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12312612/]
- 82. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoqzZknaWSdF3wWnQmJREC1TJvyHCwlD3sSaM-b7CHDPNop08p1o]
- 83. 6 Best Tips for Troubleshooting Poor Band Separation on ... [https://azurebiosystems.com/blog/6-tips-for-troubleshooting-poor-band-separation-on-sds-page-gels/]
- 84. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOopnZdyQ6qt8aLDr7zTr5ZA2d7xLFRcDmIwhVooXl6C-4HttOIjk]
- 85. Western blotting guide: Part 2, Protein separation by SDS- ... [https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blotting-guide-part-2/]
- 86. An overview of technical considerations for Western ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5138151/]
- 87. Western Blot Troubleshooting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-gel-electrophoresis-information/western-blot-troubleshooting.html]
- 88. Western Blot SDS-PAGE [https://www.novusbio.com/support/support-by-application/western-blot-sds-page?srsltid=AfmBOooPFh6B8KH_EuU-g50tJkzH6BHYf1tzPI4YI-x64yyC3tIfcog1]
- 89. How to reduce errors when running Western Blots [https://precisionbiosystems.com/how-to-reduce-errors-when-running-western-blots/]
- 90. Western blot stripping: Techniques and advantages [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-stripping]
- 91. The use of “lab-on-a-chip” microfluidic SDS electrophoresis ... [https://www.sciencedirect.com/science/article/abs/pii/S0958694608002045]
- 92. SDS-PAGE Electrophoresis Market Size & Share 2025-2032 [https://www.360iresearch.com/library/intelligence/sds-page-electrophoresis]
- 93. Microfluidic 2-D PAGE using multifunctional in situ ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/19190795/]
- 94. SDS-PAGE Electrophoresis Buffer 3.2 CAGR Growth ... [https://www.archivemarketresearch.com/reports/sds-page-electrophoresis-buffer-76652]