Precision Genome Editing with CRISPR-Cas9: Mechanisms, Methodologies, and Clinical Translation
This article provides a comprehensive analysis of CRISPR-Cas9 technology for precise genome editing, tailored for researchers and drug development professionals.
Precision Genome Editing with CRISPR-Cas9: Mechanisms, Methodologies, and Clinical Translation
Abstract
This article provides a comprehensive analysis of CRISPR-Cas9 technology for precise genome editing, tailored for researchers and drug development professionals. It explores the foundational mechanisms of CRISPR-Cas9, from its bacterial origins to its function as programmable molecular scissors. The content details advanced delivery methodologies including viral vectors and lipid nanoparticles, alongside therapeutic applications across genetic disorders and oncology. Critical examination of off-target effects covers both predictive computational tools and empirical detection methods, while validation strategies from T7E1 to NGS are comparatively evaluated. The synthesis offers a roadmap for implementing precise CRISPR editing in biochemical research and clinical development.
The CRISPR-Cas9 Revolution: From Bacterial Immunity to Programmable Genome Editing
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and associated Cas proteins constitute an adaptive immune system that protects bacteria and archaea from invasive genetic elements such as viruses and plasmids [1]. This system provides sequence-specific, acquired immunity against foreign invaders, fundamentally shaping our understanding of virus-host interactions in prokaryotes [2]. The discovery that this bacterial defense mechanism could be repurposed for precise genome editing has revolutionized biomedical research and therapeutic development [3].
Conceptually, the CRISPR-Cas system shares remarkable functional features with the mammalian adaptive immune system, while also exhibiting characteristics of Lamarckian evolution [1]. The system functions by integrating short sequences from invading genetic elements into the host's CRISPR locus, creating a genetic record of immunization events that can be inherited by progeny [1]. This molecular memory allows prokaryotes to maintain immunity against previously encountered pathogens across generations.
The repurposing of this system, particularly the Type II CRISPR-Cas9 system, for genome engineering has transformed biochemical research, enabling unprecedented precision in manipulating genetic sequences in eukaryotic cells [3]. This application note explores the biological foundations of the CRISPR-Cas system, its molecular mechanisms, and its translation into powerful tools for biochemical research and drug development.
Biological Mechanisms of Native CRISPR-Cas Systems
Architecture and Distribution in Prokaryotes
CRISPR loci contain short, partially palindromic DNA repeats that occur at regular intervals, forming arrays that alternate repeated elements (CRISPR repeats) and variable sequences (CRISPR spacers) [1]. These peculiar loci are typically flanked by accompanying cas genes that encode the protein machinery responsible for CRISPR-mediated immunity [1]. The biological role of these sequences remained elusive until 2005, when computational analyses revealed that the spacers were homologous to foreign genetic elements, including viruses and plasmids, leading to the hypothesis that CRISPRs might function as an immune system [1].
Quantitatively, CRISPR arrays can vary significantly across organisms. While arrays containing up to 588 repeats have been reported in Haliangium ochraceum, most contain fewer than 50 units [1]. According to the CRISPRdb database, CRISPRs occur in nearly half (approximately 45%) of bacterial genomes and the large majority (approximately 83%) of archaea [1].
Table 1: Classification of Major CRISPR-Cas System Types
| Type | Signature Gene | Effector Complex | Target | PAM Location |
|---|---|---|---|---|
| Type I | cas3 | Multi-subunit (Cascade) | DNA | 5' end of protospacer |
| Type II | cas9 | Single protein (Cas9) | DNA | 3' end of protospacer |
| Type III | cas10 | Multi-subunit | RNA/DNA | Not characterized |
| Type IV | csf1 | Multi-subunit | DNA | Variable |
| Type V | cas12 | Single protein | DNA | 5' end of protospacer |
| Type VI | cas13 | Single protein | RNA | None required |
| Type VII | cas14 | Multi-subunit | RNA | Not characterized |
The known diversity of CRISPR-Cas systems continues to expand, with the current classification encompassing 2 classes, 7 types, and 46 subtypes [4]. Class 1 systems (Types I, III, IV, and VII) utilize multi-subunit effector complexes, while Class 2 systems (Types II, V, and VI) employ single protein effectors, making them particularly suitable for biotechnological applications [4].
Molecular Mechanism of Adaptive Immunity
The CRISPR-Cas immune system functions through three distinct stages that provide progressive immunization against invasive genetic elements.
Adaptation: Spacer Acquisition
The first step, adaptation, involves the acquisition of new spacers from exogenous nucleic acids into the CRISPR locus [1]. When a virus or plasmid invades a bacterial cell, fragments of the foreign DNA (approximately 30-40 base pairs in length) are selected as "spacers" and integrated into the CRISPR array in a polarized fashion at the leader end [1]. This process requires the universal Cas1-Cas2 protein complex, which catalyzes the integration of new spacers between two repeats, effectively creating a genetic memory of the infection [1].
The selection of protospacers from the invader's genome is not random; recent studies indicate sampling is biased, potentially due to DNA structural or composition features [1]. A critical feature in this process is the protospacer adjacent motif (PAM), typically a 2-5 nucleotide highly conserved sequence motif immediately flanking one side of the protospacer [1]. PAMs are essential for distinguishing self from non-self, preventing the CRISPR system from targeting the bacterial genome itself.
crRNA Biogenesis: Expression and Processing
The second stage, crRNA biogenesis, occurs when the CRISPR locus is transcribed as a long precursor CRISPR RNA (pre-crRNA) that is subsequently processed into small, mature CRISPR RNAs (crRNAs) [1]. Each crRNA contains a spacer sequence derived from the previously encountered foreign DNA flanked by partial repeat sequences. The processing mechanism varies between CRISPR types: in Type II systems, a trans-activating crRNA (tracrRNA) hybridizes with the repeat regions of the pre-crRNA, facilitating RNase III-mediated processing, while in other systems, Cas6 or Cas5d enzymes perform this function [4].
Interference: Target Degradation
The final stage, interference, involves the crRNA-guided destruction of invading nucleic acids. The mature crRNAs assemble with Cas proteins to form effector complexes that surveil the cell for sequences complementary to the crRNA spacer [1]. When a match is identified, the complex binds to the target sequence and Cas nucleases cleave the invading DNA or RNA, neutralizing the threat [2]. The specificity of this system is remarkable, with single-nucleotide mismatches often sufficient to abolish cleavage, providing exquisite discrimination between self and non-self targets.
The following diagram illustrates the complete three-stage mechanism of the native CRISPR-Cas immune system in prokaryotes:
CRISPR-Based Diagnostic Applications
The programmability and specificity of CRISPR-Cas systems have been harnessed for developing novel diagnostic platforms that outperform traditional methods in speed, sensitivity, and cost-effectiveness [5]. These applications leverage the collateral cleavage activities of certain Cas proteins, such as Cas12 and Cas13, which upon recognition of their target sequences, become activated to non-specifically cleave surrounding reporter molecules, enabling highly sensitive detection [5].
Table 2: Performance Comparison of CRISPR-Based Diagnostic Platforms
| Platform | Cas Protein | Target | Sensitivity | Time to Result | Key Features |
|---|---|---|---|---|---|
| SHERLOCK | Cas13 | RNA | aM level | 60-90 minutes | RNA detection, portable |
| DETECTR | Cas12 | DNA | aM level | 30-60 minutes | DNA detection, clinical validation |
| HOLMESv2 | Cas12b | DNA/RNA | aM level | 60 minutes | Dual detection, one-pot reaction |
The SHERLOCK (Specific High Sensitivity Enzyme Reporter Unlocking) platform utilizes Cas13 for RNA detection, while DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter) employs Cas12 for DNA detection [5]. These platforms have demonstrated the ability to detect trace amounts of pathogen nucleic acids in clinical samples, with sensitivities comparable to or exceeding those of PCR-based methods, but without requiring sophisticated equipment or trained technicians [5].
The following diagram illustrates the molecular mechanism of CRISPR-based diagnostics utilizing the trans-cleavage activity of Cas proteins:
Research Protocols: Implementing CRISPR-Cas9 in Neuronal Models
The following protocol details the application of CRISPR-Cas9 for gene knockdown and endogenous tagging in cultured mouse hippocampal neurons, an established model system for synaptic biology [6]. This approach enables the investigation of native protein function without the confounding effects of overexpression.
Protocol: CRISPR-Cas9-Mediated Gene Knockdown in Hippocampal Neurons
Preparation of Reagents and Culture Medium
Neuronal Culture Media (NB/B27 medium)
- Combine 500 mL Neurobasal (NB) medium with 10 mL B27 supplement, 5 mL penicillin-streptomycin, and 5 mL GlutaMAX
- Filter-sterilize using a 0.22 μm filter
- Store at 4°C for up to one month
- Warm to 37°C before use [6]
Neuronal Lysis Buffer
- Combine N-PER Neuronal Protein Extraction Reagent with 100X Protease and Phosphatase Inhibitor Cocktail to achieve final 1X concentration
- Vortex thoroughly and keep on ice
- Prepare fresh for each experiment [6]
Western Blot Buffers
- Running Buffer: Dilute 20X NuPAGE MES SDS Running Buffer to 1X with Milli-Q H₂O
- Transfer Buffer: Dilute 20X NuPAGE Transfer Buffer with 1500 mL Milli-Q H₂O and 400 mL methanol
- Store at room temperature or 4°C until use [6]
gRNA Design and CRISPR Vector Construction
- Design gRNAs targeting early exons of the gene of interest to maximize probability of frameshift mutations
- Select targets with high on-target efficiency scores and minimal off-target potential using validated algorithms
- Clone gRNA sequences into appropriate CRISPR vectors containing Cas9 and selection markers
- For endogenous tagging, include homology-directed repair (HDR) templates with desired tag sequences [6]
Viral Packaging and Transduction
- Package CRISPR constructs into adeno-associated virus (AAV) or lentivirus vectors depending on payload size and tropism requirements
- Purify and concentrate viral particles using ultracentrifugation or column-based methods
- Transduce cultured hippocampal neurons at appropriate multiplicity of infection (MOI) during early maturation stages (DIV 3-7)
- Include untransduced controls and fluorescence-only reporters to assess transduction efficiency and specificity [6]
Validation of Genome Editing
Assessment of Editing Efficiency
- Harvest cells 7-14 days post-transduction
- Extract genomic DNA using standard protocols
- Amplify target regions by PCR and analyze using T7 endonuclease I assay or tracking of indels by decomposition (TIDE)
- Confirm editing efficiency exceeds 70% for robust phenotypic analysis [6]
Protein Level Validation
- Lyse neurons in freshly prepared lysis buffer
- Perform Western blotting with target-specific antibodies
- Process samples using prepared running and transfer buffers
- Quantify protein reduction compared to controls using densitometry [6]
Off-Target Analysis
- Use computational prediction tools to identify potential off-target sites
- Amplify and sequence top candidate sites to confirm specificity
- Employ CRISPR-detector or similar bioinformatic tools for comprehensive analysis of editing outcomes [7]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for CRISPR-Cas9 Experiments
| Reagent/Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Cas Proteins | Cas9, Cas12, Cas13 | Target DNA/RNA cleavage | Choose based on PAM requirements and editing application |
| Guide RNA | crRNA, tracrRNA, sgRNA | Target recognition and Cas protein guidance | Design with minimal off-target potential |
| Delivery Vectors | AAV, Lentivirus, Electroporation | Introduction of CRISPR components into cells | Select based on payload size and cell type |
| Cell Culture Media | Neurobasal + B27 | Neuronal survival and maturation | Essential for primary neuronal cultures |
| Editing Validation | T7E1 assay, Western blot, Sequencing | Confirmation of genome editing efficiency | Critical for experimental quality control |
| Anti-CRISPR Proteins | AcrIIA4, AcrIIC1 | Inhibition of Cas9 activity | Enhance specificity; reduce off-target effects [8] |
Advanced Applications and Technological Innovations
Precision Control of CRISPR-Cas9 Activity
Recent advances have addressed a critical safety risk in therapeutic applications: the prolonged activity of Cas9 in cells, which can cause unintended DNA breaks at off-target sites [8]. Researchers have developed LFN-Acr/PA, the first cell-permeable anti-CRISPR protein system that rapidly shuts down Cas9 activity after genome editing is complete [8].
This system uses a component derived from anthrax toxin to introduce anti-CRISPR proteins (Acrs) into human cells within minutes, significantly reducing off-target effects and improving genome-editing specificity by up to 40% [8]. Even at picomolar concentrations, LFN-Acr/PA effectively inhibits Cas9 activity, representing a safer, more controllable means of harnessing CRISPR-Cas9 for therapeutic applications.
Computational Tools for Editing Analysis
The advancement of CRISPR technologies has necessitated the development of specialized bioinformatic tools for analyzing editing outcomes. CRISPR-detector is a comprehensive tool that enables fast and accurate detection, visualization, and annotation of genome-wide mutations induced by genome editing events [7]. This pipeline performs co-analysis of treated and control samples to remove existing background variants prior to genome editing, providing integrated structural variation calling and functional annotations of editing-induced mutations [7].
The CRISPR-Cas system represents a remarkable example of how fundamental biological research into prokaryotic adaptive immunity can yield transformative tools for biochemical research and therapeutic development. From its origins as a bacterial defense mechanism against viral infection, CRISPR technology has evolved into a precise genome-editing platform with broad applications across biomedicine.
The ongoing elucidation of new CRISPR types and subtypes, coupled with engineering advances that enhance specificity and control, continues to expand the utility of these systems. As research progresses, the integration of CRISPR tools with other technologies, including single-cell analysis, structural biology, and computational prediction, will further advance our ability to precisely manipulate genetic sequences for both basic research and clinical applications.
The protocols and applications detailed in this document provide a foundation for implementing CRISPR-based approaches in biochemical research, with particular emphasis on neuronal model systems. By understanding the natural origins and mechanisms of CRISPR-Cas systems, researchers can better harness their capabilities while innovating new applications that push the boundaries of genetic engineering.
The CRISPR-Cas9 system has revolutionized genome editing by providing an unprecedented tool for precise genetic modifications. This bacterial adaptive immune system has been repurposed to enable targeted DNA cleavage in various organisms, making it indispensable for biochemical research and therapeutic development [9]. The system's core components consist of the Cas9 nuclease and a guide RNA that directs the nuclease to specific genomic loci [10]. The simplicity and programmability of CRISPR-Cas9 have accelerated research across diverse fields, from functional genomics to gene therapy development, by allowing researchers to manipulate genes with exceptional precision and efficiency [11].
The molecular mechanism of CRISPR-Cas9 involves a sophisticated interplay between the guide RNA, Cas9 nuclease, and target DNA, culminating in a double-strand break at the predetermined site. Understanding this mechanism is crucial for optimizing editing efficiency and specificity, particularly for therapeutic applications where off-target effects present significant safety concerns [10] [12]. This application note details the molecular mechanisms underlying sgRNA guidance, PAM recognition, and DNA cleavage, providing researchers with comprehensive protocols and analytical frameworks for implementing CRISPR-Cas9 in their experimental workflows.
Molecular Components and Structural Foundations
Guide RNA: sgRNA Structure and Function
The single-guide RNA (sgRNA) is a chimeric RNA molecule that combines two natural RNA components: the CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) [9]. The crRNA segment contains a customizable 17-20 nucleotide sequence that is complementary to the target DNA, providing the specificity for DNA recognition. The tracrRNA serves as a structural scaffold that facilitates binding to the Cas9 nuclease. These two components are linked by a tetraloop structure to form the functional sgRNA, which simplifies the system for experimental applications [13].
The sgRNA's secondary and tertiary structures play critical roles in the stability and efficiency of the CRISPR-Cas9 complex. Recent structural studies of Cas9d, a compact Cas9 ortholog, have revealed that specific segments of the sgRNA scaffold interact with the REC domain of Cas9 to form a hybrid functional module termed the "RNA-coordinated target Engagement Module" (REM) [13]. This REM module is essential for target recognition and undergoes coordinated conformational rearrangement upon target binding, enabling heteroduplex propagation and nuclease activation.
Figure 1: sgRNA Structural Components and Functional Modules. The sgRNA consists of crRNA and tracrRNA elements connected by a tetraloop linker. A segment of the sgRNA scaffold interacts with the Cas9 REC domain to form the RNA-coordinated target Engagement Module (REM), essential for target recognition [9] [13].
Cas9 Nuclease: Domain Architecture
The Cas9 nuclease exhibits a bilobed architecture consisting of recognition (REC) and nuclease (NUC) lobes [13]. The REC lobe contains multiple domains (REC1, REC2, REC3) that facilitate binding to the sgRNA and target DNA. The NUC lobe encompasses the RuvC and HNH nuclease domains responsible for DNA cleavage, along with the PAM-interacting (PI) domain and wedge (WED) domain that participate in PAM recognition [14].
Structural analyses using cryo-electron microscopy have revealed that the REC3 domain serves as an allosteric hub that relays signals between the PAM-binding site and the HNH nuclease domain [14]. This allosteric network ensures that DNA cleavage occurs only when the correct PAM is present and sufficient complementarity exists between the sgRNA and target DNA. The dynamic nature of Cas9's domain arrangement, particularly the flexible HNH domain, is crucial for its catalytic activity and specificity.
Protospacer Adjacent Motif (PAM) Recognition
The protospacer adjacent motif (PAM) is a short, specific DNA sequence adjacent to the target site that is essential for Cas9 activation [15]. The PAM requirement is a fundamental constraint that defines the targetable genomic space for each Cas nuclease variant. Different Cas9 orthologs and engineered variants recognize distinct PAM sequences, which directly influences their targeting scope and applications [14].
For the commonly used Streptococcus pyogenes Cas9 (SpCas9), the canonical PAM sequence is 5'-NGG-3', where "N" can be any nucleotide [9]. Structural studies have elucidated that PAM recognition involves both the WED and PI domains of Cas9, which form specific hydrogen bonds and electrostatic interactions with the PAM nucleotides [13]. Key residues such as Asn651, Lys649, and Lys715 in Cas9d (equivalent to residues in SpCas9) directly contact the PAM sequence, with mutations at these positions altering PAM specificity or abolishing cleavage activity [13].
Table 1: PAM Specificities of Commonly Used Cas Nucleases
| Cas Nuclease | Source Organism | Canonical PAM Sequence | Additional PAM Variants | Reference |
|---|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | 5'-NGG-3' | 5'-NAG-3' (weaker) | [9] |
| SaCas9 | Staphylococcus aureus | 5'-NNGRR(N)-3' | 5'-NNAGT-3', 5'-NNAGGT-3' | [9] [15] |
| Cas9d | Deltaproteobacteria | 5'-NGG-3' | 5'-GAG-3', 5'-GGA-3' | [13] |
| AsCas12a | Acidaminococcus sp. | 5'-TTTV-3' | Various T-rich sequences | [15] |
| VQR variant | Engineered SpCas9 | 5'-NGA-3' | - | [14] |
| VRER variant | Engineered SpCas9 | 5'-NGCG-3' | - | [14] |
| EQR variant | Engineered SpCas9 | 5'-NGAG-3' | - | [14] |
Detailed Molecular Mechanism
Target Recognition and sgRNA-DNA Hybridization
The process of target recognition begins with PAM binding, which triggers local DNA melting and enables the seed region of the sgRNA (approximately 10-12 nucleotides adjacent to the PAM) to initiate hybridization with the target DNA strand [13]. This initial binding is followed by progressive zippering of the sgRNA along the target DNA, forming an RNA-DNA heteroduplex. Structural studies indicate that at least 17 base pairs in the guide-target heteroduplex are required for nuclease activation in Cas9d systems [13].
The REM module, composed of the sgRNA scaffold and REC domain, plays a critical role in monitoring heteroduplex complementarity. This hybrid module undergoes coordinated conformational rearrangement upon target binding, enabling heteroduplex propagation and facilitating nuclease activity [13]. The allosteric coupling between the REM module and the HNH nuclease domain ensures that DNA cleavage occurs only when sufficient complementarity exists between the sgRNA and target DNA.
DNA Cleavage Mechanism
Once successful target recognition and heteroduplex formation occur, the Cas9 nuclease introduces a double-strand break in the target DNA. The cleavage is catalyzed by two distinct nuclease domains: the HNH domain cleaves the target strand (complementary to the sgRNA), while the RuvC domain cleaves the non-target strand [13].
The HNH domain cleaves the target strand three nucleotides upstream of the PAM sequence, while the RuvC domain cleaves the non-target strand six nucleotides upstream of the PAM, generating blunt ends or slight overhangs depending on the specific Cas9 variant [13]. For Cas9d, this cleavage pattern produces sticky ends with three-nucleotide 5' overhangs, distinguishing it from SpCas9 which typically generates blunt ends [13].
The DNA cleavage activity is magnesium-dependent, with Mg²⁺ ions playing a crucial role in the catalytic mechanism of both nuclease domains [13]. Alanine substitutions at conserved catalytic residues in either nuclease domain convert Cas9 into a nickase that cleaves only one DNA strand, which can be utilized to enhance editing specificity [13].
Figure 2: CRISPR-Cas9 DNA Recognition and Cleavage Mechanism. The process initiates with PAM recognition, followed by local DNA melting, seed region hybridization, heteroduplex zippering, conformational rearrangement of the REM module, and culminates in coordinated DNA cleavage by the HNH and RuvC nuclease domains [13].
Experimental Protocols and Methodologies
PAM Recognition Profiling Using PAM-readID
The PAM-readID method provides a rapid, simple, and accurate approach for determining the PAM recognition profiles of CRISPR-Cas nucleases in mammalian cells [15]. This protocol eliminates the need for fluorescent reporters and fluorescence-activated cell sorting (FACS), making it more accessible for broad adoption.
Protocol Steps:
- Plasmid Construction: Generate two plasmids - (I) a target plasmid bearing the target sequence flanked by randomized PAMs (typically 6-8N), and (II) an expression plasmid for the Cas nuclease and sgRNA.
- Cell Transfection: Co-transfect mammalian cells (e.g., HEK293T) with the two plasmids and double-stranded oligodeoxynucleotides (dsODN) using standard transfection methods.
- Genomic DNA Extraction: Harvest cells after 72 hours to allow sufficient time for Cas nuclease cleavage and non-homologous end joining (NHEJ)-mediated dsODN integration.
- PCR Amplification: Amplify the integrated fragments using a forward primer specific to the dsODN tag and a reverse primer specific to the target plasmid.
- Sequencing and Analysis: Perform high-throughput sequencing of the amplicons and analyze the sequences to determine the PAM recognition profile. For a cost-effective alternative, Sanger sequencing can be used with analysis of signal peak ratios in the chromatograph.
Key Considerations:
- The dsODN integration efficiently tags cleaved DNA fragments bearing recognized PAMs.
- For SpCas9, analysis with extremely low sequence depth (as few as 500 reads) can accurately identify PAM preferences.
- The method has been successfully validated for SaCas9, SaHyCas9, Nme1Cas9, SpCas9, SpG, SpRY, and AsCas12a [15].
Off-Target Assessment Using CHANGE-seq and GUIDE-seq
Accurate assessment of off-target activity is crucial for therapeutic applications of CRISPR-Cas9. Both CHANGE-seq (in vitro) and GUIDE-seq (in cellula) provide comprehensive methods for identifying potential off-target sites [16].
CHANGE-seq Protocol (in vitro):
- Library Preparation: Create a DNA library containing the target sequence with flanking randomized regions.
- Cas9 RNP Cleavage: Incubate the library with Cas9 ribonucleoprotein (RNP) complexes.
- Adapter Ligation: Capture cleaved fragments by ligating adapters to the cleavage sites.
- High-Throughput Sequencing: Sequence the captured fragments using Illumina platforms.
- Bioinformatic Analysis: Map sequences to the reference genome and identify off-target sites with computational tools.
GUIDE-seq Protocol (in cellula):
- dsODN Transfection: Co-deliver Cas9-sgRNA RNP complexes with tagged dsODN into cells.
- Genomic Integration: Allow NHEJ-mediated integration of dsODN into Cas9-induced double-strand breaks.
- Genomic DNA Extraction: Harvest cells and extract genomic DNA after 72 hours.
- PCR Amplification: Amplify integration sites using dsODN-specific and genome-specific primers.
- Sequencing and Analysis: Perform high-throughput sequencing and map integration sites to the reference genome to identify on-target and off-target activities.
Table 2: Comparison of Off-Target Assessment Methods
| Method | Environment | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| CHANGE-seq | in vitro | High | Comprehensive profiling; No cellular constraints | May not reflect cellular context |
| GUIDE-seq | in cellula | High | Captures cellular context; Identifies genomic off-targets | Requires efficient dsODN delivery |
| TTISS | in cellula | Moderate | High-throughput; Multiple sgRNAs simultaneously | Complex data analysis |
| Computational Prediction | in silico | Variable | Fast and inexpensive; Guide design stage | Dependent on algorithm accuracy |
Computational Tools for sgRNA Design and Off-Target Prediction
Advanced computational tools are essential for designing specific sgRNAs and predicting potential off-target effects. Several deep learning approaches have recently been developed to enhance prediction accuracy by incorporating genomic context and epigenetic features.
DNABERT-Epi: Integrating Epigenetic Features
DNABERT-Epi represents a novel approach that integrates a pre-trained DNA foundation model (DNABERT) with epigenetic features including H3K4me3, H3K27ac, and ATAC-seq data [16]. This multi-modal model significantly enhances off-target prediction accuracy by capturing both sequence determinants and chromatin accessibility influences on Cas9 activity.
Implementation Protocol:
- Data Collection: Obtain sgRNA sequences and corresponding off-target data from curated databases.
- Epigenetic Feature Processing: For each potential off-target site, extract epigenetic signals within a 1000 bp window centered on the cleavage site. Process signals by capping outliers, applying Z-score normalization, and binning into 100 bp intervals.
- Model Training: Fine-tune the pre-trained DNABERT model on CRISPR off-target data while incorporating the 300-dimensional epigenetic feature vector (100 bins × 3 marks).
- Model Validation: Perform rigorous cross-validation using multiple off-target datasets to assess prediction performance.
- Interpretation Analysis: Apply SHAP and Integrated Gradients to identify influential sequence motifs and epigenetic features driving predictions.
CRISPR-Embedding: k-mer Based Prediction
CRISPR-Embedding utilizes a 9-layer convolutional neural network (CNN) with DNA k-mer embeddings for off-target activity prediction [17]. This approach addresses data imbalance issues through data augmentation and under-sampling strategies, achieving 94.07% accuracy in cross-validation studies.
Application Workflow:
- Sequence Representation: Convert DNA sequences into k-mer embeddings using pre-trained DNA2Vec models.
- Data Balancing: Apply random under-sampling to address class imbalance between active and inactive off-target sites.
- Model Training: Train the CNN architecture using the balanced dataset with standard deep learning frameworks.
- Performance Evaluation: Assess model performance using metrics including AUC-ROC, precision, recall, and F1-score across multiple test datasets.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for CRISPR-Cas9 Experiments
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Cas9 Nucleases | SpCas9, SaCas9, Cas9d, AsCas12a | DNA cleavage effector | Size, PAM specificity, editing efficiency |
| sgRNA Format | Synthetic sgRNA, IVT sgRNA, plasmid-expressed sgRNA | Target recognition and Cas9 guidance | Editing efficiency, off-target effects, delivery method |
| Delivery Tools | Lipid nanoparticles (LNPs), Viral vectors (AAV), Electroporation | Introducing components into cells | Cell type specificity, efficiency, toxicity |
| Detection Assays | GUIDE-seq, CHANGE-seq, T7E1 assay, NGS | Assessing on-target and off-target activity | Sensitivity, throughput, cost |
| Validation Tools | Sanger sequencing, NGS, Digital PCR | Confirming edits | Accuracy, quantitative capability |
| Control Elements | Non-targeting sgRNAs, Mock transfections | Experimental controls | Specificity assessment, background measurement |
| Software Tools | CHOPCHOP, Synthego Design Tool, DNABERT-Epi | sgRNA design and off-target prediction | Algorithm accuracy, user interface |
Troubleshooting and Optimization Guidelines
Enhancing Editing Specificity
Off-target effects remain a significant challenge for CRISPR-Cas9 applications, particularly in therapeutic contexts. Several strategies can mitigate off-target activity:
- High-Fidelity Cas9 Variants: Utilize engineered Cas9 variants with enhanced specificity, such as eSpCas9 or SpCas9-HF1, which reduce off-target effects by weakening non-specific interactions with DNA [10].
- Modified sgRNA Formats: Implement chemically modified synthetic sgRNAs with improved stability and specificity profiles compared to in vitro transcribed or plasmid-expressed guides [9].
- RNP Delivery: Use ribonucleoprotein (RNP) complexes rather than plasmid-based expression to limit the temporal window of Cas9 activity, reducing off-target effects [9].
- Dual Nickase Strategy: Employ paired Cas9 nickases with offset sgRNAs to create staggered cuts only when both guides bind in close proximity, dramatically increasing specificity [12].
Expanding Targeting Scope
The PAM requirement constrains the targetable genomic space. Multiple approaches can overcome this limitation:
- PAM-Relaxed Variants: Utilize engineered Cas9 variants like SpRY (recognizes 5'-NRN-3' and 5'-NYN-3') or xCas9 with broadened PAM compatibility [15] [14].
- Alternative Cas Nucleases: Implement naturally occurring Cas variants with diverse PAM specificities, such as Cas9d (NGG), Cas12a (TTTV), or ScCas9 (NNNVRYM) [13].
- PAM Engineering: Develop custom Cas9 variants with altered PAM specificities through structure-guided engineering or directed evolution [14].
Molecular dynamics simulations combined with graph-theory analyses have revealed that efficient PAM recognition involves not only direct contacts between PAM-interacting residues and DNA but also a distal network that stabilizes the PAM-binding domain and preserves long-range communication with the REC3 domain [14]. This understanding enables more rational engineering of Cas9 variants with expanded PAM compatibility.
The molecular mechanism of sgRNA guidance, PAM recognition, and DNA cleavage in CRISPR-Cas9 systems represents a sophisticated biological process that has been harnessed for precise genome engineering. Understanding these mechanisms at structural and biochemical levels enables researchers to optimize editing efficiency, specificity, and applicability across diverse biological contexts.
Recent advances in structural biology, particularly cryo-EM analyses of compact Cas9 systems like Cas9d, have revealed novel functional modules such as the REM that coordinates target recognition and cleavage activation [13]. Concurrent developments in computational prediction, exemplified by DNABERT-Epi, provide powerful tools for anticipating off-target effects by integrating sequence context and epigenetic features [16]. These advancements, coupled with improved experimental methods for PAM determination and off-target profiling, continue to expand the capabilities and safety of CRISPR-based genome editing.
As CRISPR therapeutics progress through clinical trials, with recent successes in treating sickle cell disease, beta thalassemia, and hereditary transthyretin amyloidosis [11], the fundamental mechanisms detailed in this application note become increasingly important for translating basic research into clinical applications. The ongoing development of more precise editing tools, enhanced delivery methods, and comprehensive safety assessments will further establish CRISPR-Cas9 as an indispensable technology for biochemical research and therapeutic development.
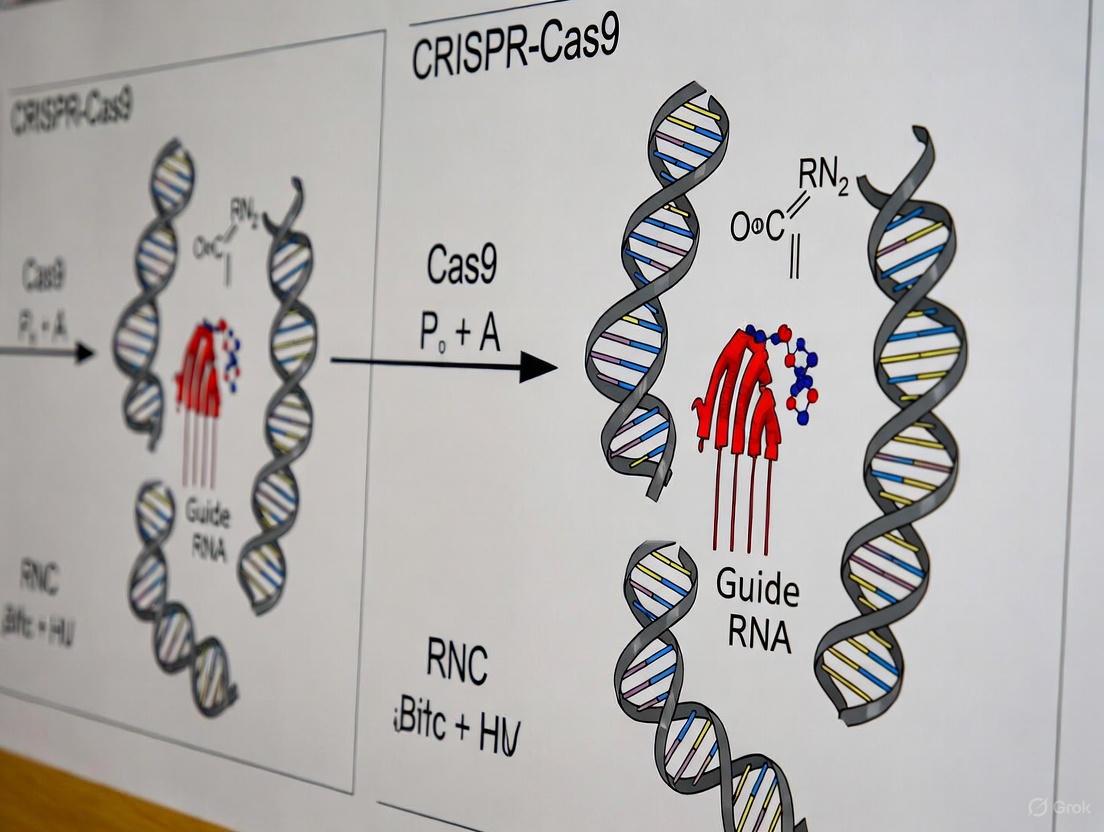
The CRISPR-Cas9 system has revolutionized genome editing, providing researchers with an unprecedented tool for precise genetic modifications. This technology, adapted from an adaptive immune system in bacteria, allows for targeted double-strand breaks in DNA, enabling gene knockout, knock-in, and transcriptional regulation. The system's core consists of two fundamental components: the Cas9 nuclease, which acts as the molecular scissor, and a guide RNA (gRNA), which functions as a homing device to direct Cas9 to specific genomic locations. Understanding the structural basis of Cas9 function and the principles governing effective gRNA design is paramount for successful genome editing experiments in biochemical research and drug development. This application note details the structural mechanisms of the Cas9 nuclease and provides comprehensive protocols for designing and validating guide RNAs to achieve precise genomic edits.
Structural Basis of Cas9 Nuclease Function
The Cas9 nuclease from Streptococcus pyogenes (SpCas9) exhibits a bilobed architecture consisting of two primary lobes: the recognition lobe (REC) and the nuclease lobe (NUC) [18]. These lobes form a central groove that accommodates the gRNA:DNA heteroduplex during target recognition and cleavage. The recognition lobe is predominantly α-helical and is crucial for binding both the sgRNA and the target DNA. The nuclease lobe contains the catalytic domains responsible for DNA cleavage and includes the PAM-interacting domain at its C-terminal end, which is essential for recognizing the protospacer adjacent motif in the target DNA [18].
Key Functional Domains
- HNH Nuclease Domain: Cleaves the DNA strand complementary to the gRNA (target strand) [19].
- RuvC Nuclease Domain: Cleaves the non-complementary DNA strand (non-target strand) [19].
- PAM-Interacting Domain: Recognizes the 5'-NGG-3' protospacer adjacent motif adjacent to the target sequence, a critical step for initiating DNA binding [18].
The coordination between these domains ensures that DNA cleavage occurs approximately 3-4 nucleotides upstream of the PAM sequence on both DNA strands, generating a blunt-ended double-strand break [19].
Table 1: Core Functional Domains of SpCas9 Nuclease
| Domain | Structural Location | Primary Function |
|---|---|---|
| REC Lobe | Recognition Lobe | sgRNA and target DNA binding, conformational activation |
| HNH Domain | Nuclease Lobe | Cleavage of target DNA strand (complementary to gRNA) |
| RuvC Domain | Nuclease Lobe | Cleavage of non-target DNA strand (non-complementary to gRNA) |
| PAM-Interacting Domain | C-terminal of Nuclease Lobe | Recognition of NGG protospacer adjacent motif |
Mechanism of DNA Recognition and Cleavage
Target recognition begins when Cas9 scans the genome for PAM sequences (5'-NGG-3' for SpCas9) [20]. Upon PAM recognition, Cas9 unwinds the DNA duplex, allowing the seed sequence (8-10 bases at the 3' end of the gRNA) to initiate annealing to the target DNA [19]. If sufficient complementarity exists, complete zippering of the gRNA to the target DNA occurs, triggering conformational changes that position the HNH and RuvC domains to cleave their respective DNA strands [18]. This mechanism ensures that DNA cleavage only occurs when the gRNA exhibits sufficient complementarity to the target sequence, particularly in the seed region proximal to the PAM.
Figure 1: Cas9 DNA Recognition and Cleavage Mechanism. The process initiates with PAM recognition, followed by DNA unwinding, seed sequence annealing, conformational activation of nuclease domains, and double-strand break formation.
Guide RNA Design Principles
Fundamental Requirements for gRNA Design
The single guide RNA (sgRNA) is a chimeric RNA molecule comprising a CRISPR RNA (crRNA) component, which contains the 20-nucleotide target-specific sequence, and a trans-activating crRNA (tracrRNA) scaffold that facilitates Cas9 binding [21]. Effective gRNA design must adhere to several fundamental requirements:
- PAM Proximity: The target sequence must be immediately adjacent to a 5'-NGG-3' PAM sequence, with the 20 nucleotides upstream of the PAM comprising the gRNA targeting region [21].
- Sequence Uniqueness: The 20-nucleotide spacer sequence should be unique within the genome to minimize off-target effects [19].
- Strand Flexibility: The target sequence can be located on either DNA strand relative to the PAM [21].
Extensive research on gRNA efficiency has revealed significant position-dependent nucleotide preferences that influence Cas9 cleavage activity:
Table 2: Position-Specific Nucleotide Preferences for gRNA Efficiency [22]
| Position | Preferred | Avoid | Impact on Efficiency |
|---|---|---|---|
| 1 (5' end) | G | C, U | G at position 1 enhances stability and transcription |
| 16 | C | G | C at position 16 correlates with higher efficiency |
| 17 | A, T | G, C | A or T at position 17 increases efficiency |
| 18 | C | U | C at position 18 improves binding stability |
| 19 | G, A | - | G or A at position 19 enhances activity |
| 20 (3' end) | G | C, U | G adjacent to PAM improves recognition |
| PAM | CGG | TGG | CGG PAM more efficient than TGG |
Additionally, certain sequence motifs significantly impact gRNA performance. Efficient gRNAs often contain AG, CA, AC, or UA dinucleotides, while inefficient gRNAs tend to have high counts of U or G nucleotides, particularly GGG or GGGG motifs that may form stable secondary structures [22]. The GC content should ideally be between 40-60%, as extremes outside this range (particularly >80%) can dramatically reduce efficiency [22].
Application-Specific gRNA Design Strategies
Gene Knockout (NHEJ-Mediated)
For gene knockout experiments utilizing non-homologous end joining (NHEJ), gRNAs should target early exonic regions (between 5-65% of the protein-coding region) to maximize the probability of generating frameshift mutations that disrupt protein function [23]. Targeting sequences near the N-terminus should be avoided to prevent the use of alternative start codons, while targeting near the C-terminus may produce partially functional protein fragments [24].
Gene Editing (HDR-Mediated)
For precision editing via homology-directed repair (HDR), the gRNA must be positioned within ~30 nucleotides of the intended edit due to the dramatically decreased efficiency when the cut site is farther from the repair template [23]. This locational constraint often limits gRNA options, requiring prioritization of proximity over optimal sequence features.
CRISPRa and CRISPRi
For transcriptional activation (CRISPRa) or interference (CRISPRi) using catalytically dead Cas9 (dCas9), gRNAs should target ~100 nucleotides upstream of the transcription start site (TSS) for activation and ~100 nucleotides downstream of the TSS for repression [23]. Accurate TSS annotation using databases like FANTOM, which employs CAGE-seq data, is crucial for success [23].
Experimental Protocols
Protocol: gRNA Design and Validation Workflow
gRNA Design Phase
- Target Identification: Identify the genomic region to be targeted based on experimental goal (knockout, HDR, transcriptional modulation).
- PAM Location: Scan for all 5'-NGG-3' PAM sequences in the target region using bioinformatic tools.
- gRNA Selection: For each PAM, extract the 20 nucleotides immediately upstream as potential gRNA targets.
- Efficiency Scoring: Input candidate gRNA sequences into prediction algorithms (e.g., CRISPRscan, Doench2016 algorithm) to score predicted efficiency [22].
- Specificity Analysis: Evaluate potential off-target sites using mismatch tolerance algorithms. Prioritize gRNAs with minimal off-target potential, particularly in the seed region [19].
gRNA Validation Phase
- In Vitro Transcription: Synthesize sgRNAs using systems like the Guide-it sgRNA In Vitro Transcription Kit, which utilizes PCR to generate template DNA with T7 promoter followed by in vitro transcription [21].
- In Vitro Cleavage Assay: Incubate synthesized gRNA with purified Cas9 protein and target DNA plasmid. Assess cleavage efficiency via gel electrophoresis [25].
- Cell-Based Validation: Transfert candidate gRNAs with Cas9 expression vector into relevant cell lines (e.g., HCT116, HEK293T) using appropriate methods (lipofection, electroporation) [26].
- Efficiency Quantification: Extract genomic DNA 72-96 hours post-transfection. Analyze editing efficiency using T7E1 assay, TIDE analysis, or qEva-CRISPR for more quantitative, multiplexable assessment [26].
Protocol: Quantitative Evaluation of Editing Efficiency Using qEva-CRISPR
The qEva-CRISPR method provides a quantitative, sensitive approach for evaluating CRISPR/Cas9-induced modifications, capable of detecting all mutation types including point mutations and large deletions [26].
- Probe Design: Design specific oligonucleotide probes for each target site, consisting of two oligonucleotides that hybridize to adjacent sequences of the target DNA.
- Probe Hybridization: Mix 100-200ng of genomic DNA with probe sets for both target and control regions. Denature at 98°C for 1 minute and hybridize at 60°C for 16 hours.
- Ligation Reaction: Add ligation mixture containing ligase to connected hybridized probes. Incubate at 54°C for 15 minutes.
- PCR Amplification: Amplify ligated products using fluorescently labeled primers with the following cycling conditions: 35 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 60 seconds.
- Fragment Analysis: Separate amplified products by capillary electrophoresis and quantify peak areas. Calculate editing efficiency as the ratio of mutant to wild-type signal.
- Multiplex Analysis: For simultaneous evaluation of multiple targets or off-target sites, design probes with different lengths to enable separation and individual quantification [26].
Figure 2: qEva-CRISPR Workflow for Quantitative Assessment of Genome Editing Efficiency. This ligation-based method enables sensitive, multiplex evaluation of editing efficiency at multiple genomic targets.
Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas9 Genome Editing Experiments
| Reagent Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Cas9 Expression Systems | pSpCas9(BB)-2A-GFP (PX458), Cas9 mRNA, Cas9 protein | Provides nuclease function | Plasmid delivery is simplest; mRNA offers transient expression; protein enables immediate activity |
| gRNA Expression Systems | U6-promoter vectors, in vitro transcription kits | Delivers targeting component | U6 vectors for stable expression; synthetic gRNAs for rapid screening |
| gRNA Design Tools | CHOPCHOP, CRISPR Design Tool, Synthego Design Tool | Bioinformatics design assistance | Vary in scoring algorithms, species coverage, and user interface |
| Delivery Methods | Lipofectamine LTX, Electroporation (Neon), Lentiviral transduction | Introduces CRISPR components into cells | Efficiency and cytotoxicity vary by cell type; viral methods enable difficult-to-transfect cells |
| Validation Kits | Guide-it sgRNA Screening Kit, T7E1 kits, qEva-CRISPR reagents | Assesses gRNA efficiency and editing success | Vary in sensitivity, quantification capability, and multiplexing capacity |
| High-Fidelity Cas9 Variants | eSpCas9(1.1), SpCas9-HF1, HypaCas9 | Reduces off-target effects | Trade-off between specificity and on-target efficiency should be evaluated |
| PAM-Flexible Cas9 Variants | xCas9, SpCas9-NG, SpRY | Expands targeting range | Recognize non-NGG PAMs (NG, GAA, NRN) but may have reduced efficiency |
The structural elegance of the Cas9 nuclease and the strategic design of guide RNAs form the foundation of successful CRISPR-Cas9 genome editing. The bilobed architecture of Cas9, with its specialized domains for PAM recognition, DNA unwinding, and strand-specific cleavage, enables precise DNA targeting when coupled with appropriately designed gRNAs. Critical to this process is the selection of gRNA sequences with favorable nucleotide compositions, optimal GC content, and minimal off-target potential, tailored to the specific experimental application—whether gene knockout, precise editing, or transcriptional modulation. The protocols and reagents outlined in this application note provide researchers with a comprehensive framework for designing, implementing, and validating CRISPR-Cas9 experiments, advancing the potential for precise genome editing in biochemical research and therapeutic development. As CRISPR technology continues to evolve, the fundamental principles of Cas9 structure and gRNA design remain essential knowledge for researchers harnessing this powerful technology.
The CRISPR-Cas9 system has revolutionized biological research by providing an unprecedented platform for precise genome engineering. This technology operates by creating targeted double-strand breaks (DSBs) in the DNA, which subsequently activate the cell's innate DNA repair machinery [27]. The outcome of a gene editing experiment is fundamentally determined by which of the two primary DNA repair pathways—non-homologous end joining (NHEJ) or homology-directed repair (HDR)—is employed to resolve these breaks [27] [28]. Understanding and strategically directing this cellular choice is therefore paramount for success in biochemical research and therapeutic development.
NHEJ is an error-prone repair mechanism that functions throughout the cell cycle by directly ligating broken DNA ends, often resulting in small insertions or deletions (indels) that disrupt the gene's open reading frame [29] [28]. This makes it the ideal pathway for generating gene knockouts. In contrast, HDR is a high-fidelity, template-dependent repair pathway that is primarily active in the S and G2 phases of the cell cycle. It utilizes a donor DNA template with homology to the regions flanking the break to facilitate precise edits, including specific insertions, point mutations, and gene corrections [27] [30]. The natural competition between these two pathways, with NHEJ typically dominating, presents a significant challenge for applications requiring precision [27]. This application note delineates robust protocols and strategies to harness the NHEJ pathway for efficient gene knockout and to enhance the efficiency of the HDR pathway for precise genome editing, specifically framed for biochemical research and drug discovery.
Pathway Mechanisms and Experimental Workflows
Visualizing the CRISPR-Cas9 DNA Repair Pathway Decision
The following diagram illustrates the critical decision point cellular machinery faces after a CRISPR-Cas9-induced double-strand break, guiding researchers in strategically directing the outcome toward their experimental goals.
Non-Homologous End Joining (NHEJ) for Gene Knockout
The NHEJ pathway is the cell's primary, rapid-response mechanism for repairing DSBs. Its key advantage for researchers is that it is active throughout all phases of the cell cycle and does not require a homologous repair template [28]. The process begins with the Ku70/Ku80 heterodimer recognizing and binding to the broken DNA ends. This is often followed by processing of the ends by nucleases like Artemis, and finally, ligation by the DNA ligase IV complex [27]. This end-processing and ligation is inherently error-prone, frequently resulting in small insertions or deletions (indels). When these indels occur within the coding sequence of a gene, they can cause frameshift mutations that lead to premature stop codons and effectively knockout the gene's function [28].
Experimental Workflow for NHEJ-Mediated Knockout:
- Design and Synthesis: Design sgRNAs targeting an early exon of the gene of interest to maximize the likelihood of a disruptive indel. Synthesize the sgRNA and procure high-fidelity Cas9 nuclease.
- Complex Formation: Form the Ribonucleoprotein (RNP) complex by pre-incubating synthetic sgRNA with Cas9 protein (typically Alt-R S.p. HiFi Cas9 V3) at room temperature for 20-30 minutes. The RNP delivery format offers high efficiency and reduced off-target effects [31] [32].
- Delivery: Deliver the RNP complex into the target cells. Electroporation is a common method, though emerging microfluidic technologies like the Droplet Cell Pincher (DCP) have demonstrated a ~6.5-fold increase in knockout efficiency compared to electroporation [33].
- Culture and Validate: Culture the transfected cells for several days to allow for expression of the knockout. Validate editing efficiency using T7E1 or TIDE assays, and confirm knockout via Sanger sequencing and functional protein assays (e.g., western blot) [32].
Homology-Directed Repair (HDR) for Precise Editing
HDR facilitates precise genome editing by using an exogenous donor DNA template to repair the DSB. This template contains the desired edit (e.g., a point mutation, GFP sequence, or corrected exon) flanked by homology arms complementary to the sequence around the cut site. This pathway is inherently less efficient than NHEJ because it is restricted primarily to the S and G2 phases of the cell cycle and is in direct competition with the NHEJ machinery [27] [30].
Experimental Workflow for HDR-Mediated Precise Editing:
- Design Donor Template: Design a single-stranded oligodeoxynucleotide (ssODN) or double-stranded donor template. The homology arms (typically 60-90 nt each for ssODNs) should be complementary to the region immediately surrounding the cut site. To prevent re-cleavage of the edited locus, incorporate silent mutations in the PAM sequence or the sgRNA seed region within the donor template [31].
- Synchronize Cell Cycle: To increase the proportion of cells in S/G2 phase, synchronize the cell population using chemicals like thymidine or nocodazole. This simple step can significantly boost HDR efficiency [27].
- Co-deliver RNP and Donor: Co-transfect the pre-formed RNP complex (Cas9 + sgRNA) along with the HDR donor template. The use of RNP complexes is crucial as it leads to rapid degradation of the editing machinery, minimizing extended exposure and potential off-target effects [31].
- Modulate Cellular Environment: To further enhance HDR rates, supplement the culture media with small molecule inhibitors that favor HDR. A highly effective strategy involves the transient knockdown of p53 using shRNA, combined with pro-survival factors like CloneR. This approach has been shown to achieve HDR efficiencies exceeding 90% in induced pluripotent stem cells (iPSCs) [31].
- Validate Precise Editing: Screen for precise edits using techniques such as droplet digital PCR (ddPCR) or next-generation sequencing (NGS) of the target locus to confirm the intended modification without collateral indels [30].
Quantitative Data and Reagent Solutions
The table below summarizes key strategies and their demonstrated effectiveness in enhancing either NHEJ or HDR efficiency, based on recent research.
Table 1: Quantified Efficiency Enhancements for NHEJ and HDR Pathways
| Strategy | Pathway | Reported Effect | Experimental Context |
|---|---|---|---|
| Repsox (TGF-β inhibitor) [29] | NHEJ | 3.16-fold increase | Porcine PK15 cells, RNP delivery |
| p53 inhibition + Pro-survival molecules [31] | HDR | >90% HDR rate | Human iPSCs, multiple genetic loci |
| Microfluidic DCP Delivery [33] | Knockout (NHEJ) | 6.5-fold higher vs. electroporation | K562 cells, RNP delivery |
| Microfluidic DCP Delivery [33] | Knock-in (HDR) | 3.8-fold higher vs. electroporation | K562 cells, RNP + donor delivery |
| Zidovudine (AZT) [29] | NHEJ | 1.17-fold increase | Porcine PK15 cells, RNP delivery |
The Scientist's Toolkit: Essential Research Reagents
A successful genome editing experiment relies on carefully selected, high-quality reagents. The following table outlines essential materials and their functions.
Table 2: Key Reagent Solutions for CRISPR Genome Editing
| Research Reagent | Function/Description | Key Considerations |
|---|---|---|
| Alt-R S.p. HiFi Cas9 Nuclease V3 [31] | High-fidelity Cas9 protein for RNP formation. | Reduces off-target effects while maintaining robust on-target activity. |
| Synthetic sgRNA [32] | Chemically synthesized guide RNA for RNP formation. | Higher purity and efficiency, lower immune response compared to IVT RNA. |
| ssODN (Ultramer) | Single-stranded DNA donor template for HDR. | Homology arms should be 60-90 nt; include silent PAM-disrupting mutations. |
| HDR Enhancer (e.g., IDT) [31] | Small molecule cocktail to boost HDR efficiency. | Used during cell recovery post-transfection. |
| CloneR [31] | Supplement that improves survival of single-cell cloned iPSCs. | Critical for outgrowth of edited cells after HDR transfection. |
| pCXLE-hOCT3/4-shp53-F Plasmid [31] | Plasmid for transient p53 knockdown. | Co-transfection dramatically increases HDR efficiency in iPSCs. |
The strategic harnessing of NHEJ and HDR pathways empowers researchers to tailor CRISPR-Cas9 editing for a wide spectrum of applications, from complete gene knockout to precise nucleotide-level changes. The protocols and data outlined herein provide a framework for optimizing these experiments in a biochemical research context. While NHEJ offers a straightforward and efficient route to gene disruption, achieving high-efficiency HDR requires a multi-faceted approach involving cell cycle synchronization, optimized delivery systems like RNPs, and chemical modulation of the DNA repair machinery. As the field advances, the integration of novel technologies such as microfluidic delivery and the development of next-generation editors like base and prime editors [30] will further enhance the precision and efficacy of genome editing, solidifying its role as a cornerstone of modern biochemistry and therapeutic development.
The development of CRISPR-Cas9 from a fundamental observation in bacterial immunology into a programmable genome-editing technology represents one of the most significant breakthroughs in modern biochemistry. This technology provides researchers, scientists, and drug development professionals with an unprecedented ability to perform precise modifications to the genetic code of virtually any organism. Its simplicity, cost-effectiveness, and high efficiency have revolutionized basic research, functional genomics, and therapeutic development [34] [35]. Framing this journey from discovery to recognition is essential for understanding how a bacterial defense mechanism was harnessed to create a versatile toolkit for precise genome editing, enabling the rewriting of the code of life itself [36].
Historical Timeline of Key Discoveries
The path to the CRISPR-Cas9 genome-editing tool was paved by the work of many scientists across the globe, whose cumulative discoveries elucidated the mechanism of a unique microbial adaptive immune system. The table below summarizes the critical milestones that transformed a curious genetic sequence into a programmable genetic scissor.
Table 1: Key Historical Milestones in the Development of CRISPR-Cas9
| Year | Key Discovery | Lead Scientist(s) | Significance |
|---|---|---|---|
| 1987/1993 | Initial observation of CRISPR sequences | Ishino et al.; Francisco Mojica [34] [37] | First characterization of unusual repetitive DNA structures in prokaryotes. |
| 2005 | CRISPR as an adaptive immune system; Cas9 & PAM identification | Mojica et al.; Alexander Bolotin [34] | Hypothesis that CRISPR fights viruses using stored phage DNA snippets; discovery of the core Cas9 protein and PAM sequence. |
| 2007 | Experimental proof of adaptive immunity | Philippe Horvath [34] | Demonstrated that CRISPR integrates new phage DNA to confer immunity in S. thermophilus. |
| 2011 | Discovery of tracrRNA | Emmanuelle Charpentier [34] [36] | Identified a second, trans-activating RNA (tracrRNA) essential for the CRISPR-Cas9 system. |
| 2012 | Reprogrammable Genetic Scissors | Charpentier & Doudna [34] [36] | Seminal Science paper: recreated system in vitro, fused crRNA and tracrRNA into a single-guide RNA (sgRNA), and proved it could be programmed to cut any DNA target. |
| 2013 | Genome editing in eukaryotic cells | Feng Zhang, George Church [34] | First adaptation of CRISPR-Cas9 for precise genome engineering in human and mouse cells. |
| 2020 | Nobel Prize in Chemistry | Emmanuelle Charpentier & Jennifer A. Doudna [38] [36] | Awarded for the development of the CRISPR-Cas9 "genetic scissors." |
Detailed Experimental Protocols
The transition of CRISPR-Cas9 from a natural system to a lab tool required key experiments that deciphered and reconfigured its molecular components. The following protocols detail the critical methodologies that enabled this breakthrough.
Protocol 1: In Vitro Reconstitution and Reprogramming of CRISPR-Cas9
This protocol is based on the seminal 2012 experiment by Charpentier and Doudna, which demonstrated that the CRISPR-Cas9 system could be simplified and reprogrammed in vitro [34] [36] [37].
- Objective: To recreate the bacterial CRISPR-Cas9 system in a test tube and demonstrate its programmability to cleave specific DNA sequences.
- Key Materials:
- Purified Cas9 nuclease from Streptococcus pyogenes.
- DNA templates for in vitro transcription of guide RNAs.
- Target DNA plasmids containing the desired protospacer adjacent motif (PAM) sequence (5'-NGG-3').
- In vitro transcription and RNA purification kits.
- Standard reagents for PCR and gel electrophoresis.
- Methodology:
- Component Preparation: Express and purify the Cas9 protein. Synthesize the two-RNA guide system: the CRISPR RNA (crRNA) containing the 20-nucleotide spacer sequence complementary to the target DNA, and the trans-activating crRNA (tracrRNA).
- System Assembly: Combine the purified Cas9 protein with the crRNA and tracrRNA to form a ribonucleoprotein (RNP) complex in a suitable reaction buffer. Incubate to allow complex formation.
- Target Cleavage: Add the target DNA plasmid to the RNP complex. The guide RNA directs Cas9 to the complementary DNA sequence adjacent to a PAM site.
- Analysis: Resolve the reaction products using agarose gel electrophoresis. Successful cleavage is indicated by the conversion of supercoiled plasmid DNA into linearized DNA.
- Key Innovation: The researchers fused the crRNA and tracrRNA into a single-guide RNA (sgRNA), drastically simplifying the system. They proved that by simply changing the 20-nucleotide sequence within the sgRNA, they could direct Cas9 to cleave any DNA target of their choosing [34] [36].
Protocol 2: Application of CRISPR-Cas9 for Genome Editing in Eukaryotic Cells
This protocol outlines the foundational 2013 experiment that adapted CRISPR-Cas9 for use in mammalian cells, opening the door to therapeutic applications [34].
- Objective: To demonstrate targeted genome modification in human and mouse cells using the CRISPR-Cas9 system.
- Key Materials:
- Mammalian cell lines (e.g., HEK293T).
- Plasmids for expression of codon-optimized Cas9 for eukaryotes and the sgRNA.
- Transfection reagent.
- PCR reagents and genotyping primers flanking the target locus.
- Surveyor or T7 Endonuclease I for detection of induced mutations.
- Methodology:
- Vector Construction: Clone the gene for a mammalian-codon-optimized Cas9 nuclease into an expression plasmid with a strong eukaryotic promoter (e.g., CMV). Clone the target-specific sgRNA sequence into a separate expression plasmid under a U6 promoter.
- Cell Transfection: Co-transfect the Cas9 and sgRNA plasmids into the mammalian cell line using a standard transfection method (e.g., lipofection).
- Genome Editing: The expressed Cas9 and sgRNA form a complex in the cell nucleus and create a double-strand break (DSB) at the targeted genomic locus.
- Analysis of Editing:
- Disruption (Knock-out): Harvest genomic DNA 48-72 hours post-transfection. Amplify the target region by PCR. Use the T7 Endonuclease I assay, which cleaves heteroduplex DNA formed by wild-type and indel-containing strands, to detect non-homologous end joining (NHEJ)-mediated mutagenesis.
- Precise Editing (Knock-in): Co-transfect a donor DNA template containing the desired edit along with the Cas9 and sgRNA plasmids. Use PCR and sequencing to confirm homology-directed repair (HDR)-mediated precise gene modification.
- Key Finding: This experiment confirmed that CRISPR-Cas9 functions as a highly versatile and programmable genome-editing tool in eukaryotic cells, capable of multiplexed editing and driving both NHEJ and HDR repair pathways [34].
Visualizing the CRISPR-Cas9 Mechanism
The following diagrams illustrate the core components and workflow of the engineered CRISPR-Cas9 system for genome editing.
Diagram 1: CRISPR-Cas9 Core System Components
Diagram Title: Core Components of the Engineered CRISPR-Cas9 System
Diagram 2: Genome Editing Workflow via Double-Strand Break Repair
Diagram Title: Cellular DNA Repair Pathways Following CRISPR Cleavage
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of CRISPR-Cas9 experiments relies on a core set of reagents. The table below details these essential materials and their functions.
Table 2: Key Research Reagent Solutions for CRISPR-Cas9 Experiments
| Reagent / Material | Function & Description | Key Considerations |
|---|---|---|
| Cas9 Nuclease | The effector protein that creates double-strand breaks in target DNA. | Can be delivered as a protein, mRNA, or encoded in a plasmid. High-fidelity variants are available to reduce off-target effects [39]. |
| Guide RNA (sgRNA) | A synthetic RNA chimera that directs Cas9 to a specific genomic locus via Watson-Crick base pairing. | The 20-nucleotide spacer sequence is user-defined. Specificity and efficiency are critical and must be validated [35]. |
| Delivery Vector | A plasmid or viral vector (e.g., lentivirus, AAV) used to express Cas9 and sgRNA in cells. | Choice depends on the target cell type (e.g., dividing vs. non-dividing) and application (e.g., in vitro vs. in vivo) [40]. |
| Donor DNA Template | A single-stranded or double-stranded DNA molecule containing the desired edit, flanked by homology arms. | Used for precise HDR-mediated editing (knock-ins). Not required for NHEJ-mediated gene knockout [40]. |
| Lipid Nanoparticles (LNPs) | A delivery method for in vivo applications, encapsulating CRISPR components for systemic administration. | Particularly effective for targeting liver cells and have been successfully used in clinical trials [11]. |
Delivery Systems and Therapeutic Applications: From Bench to Bedside
The advent of CRISPR-Cas9 technology has revolutionized precise genome editing in biochemistry research, enabling targeted modifications across diverse biological systems. The effectiveness of these editing tools is fundamentally dependent on the delivery system used to introduce them into target cells. Among the most powerful and widely adopted delivery vehicles are viral vectors, with adeno-associated virus (AAV) and lentivirus (LV) standing as the two predominant platforms for achieving stable gene expression in both dividing and non-dividing cells. These engineered viruses facilitate the efficient transduction of CRISPR machinery, but they possess distinct structural and functional characteristics that dictate their specific applications in research and therapy.
Viral vectors account for over 80% of approved gene therapy products, underscoring their critical role in modern genetic medicine [41]. Their powerful innate ability to infect cells and deliver genetic material makes them indispensable tools. In the context of CRISPR-Cas9 workflows, the choice between AAV and lentiviral vectors is pivotal and hinges on experimental goals, such as the need for long-term stable expression versus transient editing activity, the size of the genetic cargo, and the target cell type. AAV vectors are celebrated for their low immunogenicity and predominantly episomal persistence, whereas lentiviral vectors are distinguished by their capacity for stable genomic integration and larger cargo space. This application note delineates the fundamental properties, protocols, and practical applications of both vector systems to guide researchers in selecting and implementing the optimal strategy for their genome editing projects.
Comparative Analysis of AAV and Lentiviral Vector Systems
Structural and Functional Distinctions
AAV and lentiviral vectors are engineered from their wild-type viruses through the removal of pathogenic and replication genes, repurposing them as safe, replication-deficient gene delivery vehicles. However, their underlying virology confers distinct attributes.
Adeno-Associated Virus (AAV) is a small (approx. 20 nm), non-enveloped virus with an icosahedral capsid composed of VP1, VP2, and VP3 proteins [42]. Its single-stranded DNA (ssDNA) genome is flanked by inverted Terminal Repeats (ITRs), which are the only viral sequences retained in recombinant AAV (rAAV) vectors. The ITRs are essential for genome replication, packaging, and facilitating the formation of double-stranded DNA after transduction [43]. A key safety feature of rAAV is that it does not integrate into the host genome but persists in the nucleus as an episomal circular molecule (monomer or concatemer), leading to long-term expression in non-dividing cells but eventual dilution in rapidly dividing cells [43] [44]. A significant limitation is its constrained cargo capacity of approximately 4.7 kb [45] [44].
Lentivirus (LV), a subset of retroviruses, is an enveloped virus. Its lipid bilayer, derived from the host cell membrane, is studded with envelope glycoproteins such as the Vesicular Stomatitis Virus G-glycoprotein (VSV-G) commonly used for pseudotyping to broaden cellular tropism [44] [42]. Its core contains two copies of the single-stranded RNA genome. Unlike AAV, lentiviral vectors are designed for stable integration into the host genome. This is mediated by the viral enzyme integrase, which facilitates the insertion of the reverse-transcribed DNA copy into the host chromatin, enabling permanent genetic modification and long-term expression in both dividing and non-dividing cells [44] [42]. Lentivectors offer a more generous cargo capacity, typically accommodating 8-12 kb of foreign genetic material [42].
Table 1: Fundamental Characteristics of AAV and Lentiviral Vectors
| Characteristic | Adeno-Associated Virus (AAV) | Lentivirus (LV) |
|---|---|---|
| Virus Family | Parvoviridae | Retroviridae |
| Capsid Structure | Non-enveloped, Icosahedral | Enveloped |
| Genetic Material | Single-stranded DNA (ssDNA) | Single-stranded RNA (ssRNA) |
| Genomic Integration | Non-integrating (episomal) | Integrating |
| Key Genomic Elements | Inverted Terminal Repeats (ITRs) | Long Terminal Repeats (LTRs) |
| Typical Cargo Capacity | ~4.7 kb | ~8-12 kb |
Selection Criteria for Research Applications
Choosing between AAV and LV systems requires a careful assessment of the experimental parameters. The decision matrix below outlines the primary considerations for selection based on common research scenarios.
The cargo capacity is often the first and most critical deciding factor. The ~4.7 kb limit of AAV is a major constraint for delivering large genetic elements. While the canonical Streptococcus pyogenes Cas9 (SpCas9) gene can be packaged into AAV, it leaves little room for additional regulatory elements or multiple gRNAs. This has spurred the development of smaller Cas orthologs (e.g., SaCas9) for AAV delivery [45]. In contrast, the larger capacity of LV makes it suitable for delivering SpCas9 alongside gRNA expression cassettes and even donor DNA templates in a single vector.
The need for persistent gene expression is another crucial factor. LV's integrating nature ensures that the transgene is passed on to daughter cells, making it the preferred choice for creating stable cell lines or for long-term studies in dividing cell populations. Conversely, AAV's episomal DNA provides sustained expression in non-dividing cells (e.g., neurons) but is diluted and lost in rapidly proliferating cell cultures. For short-term expression or in vivo applications where the risk of insertional mutagenesis must be minimized, AAV's non-integrating profile is a significant safety advantage [44] [42].
Finally, the target cell type dictates the choice of serotype (for AAV) or envelope (for LV). Different AAV serotypes (e.g., AAV1, AAV2, AAV6, AAV8, AAV9) exhibit distinct tissue tropisms based on their capsid's interaction with specific cell surface receptors [43]. Similarly, LV can be pseudotyped with various envelope proteins (e.g., VSV-G, Rabies-G) to alter and direct its tropism toward specific cell types [44].
Table 2: Application-Based Selection Guide for Viral Vectors
| Research Goal | Recommended Vector | Rationale |
|---|---|---|
| Stable Cell Line Generation | Lentivirus | Genomic integration ensures heritability in dividing cells. |
| In vivo Gene Therapy (Non-dividing cells) | AAV | Low immunogenicity and long-term episomal expression in quiescent cells. |
| Delivery of Large Genetic Constructs (>5 kb) | Lentivirus | Larger cargo capacity accommodates big genes or complex cassettes. |
| CRISPR Knock-in with Large Donor DNA | Lentivirus | Can deliver Cas9, gRNA, and a sizable donor template simultaneously. |
| Transient CRISPR Editing (e.g., Gene Knockout) | AAV | High transduction efficiency with transient activity reduces off-target risk. |
| Studies with High Safety Priority | AAV | Non-integrating nature minimizes risk of insertional mutagenesis. |
Experimental Protocols
AAV Vector Production and Titration Protocol
The production of high-titer, high-purity recombinant AAV is crucial for successful experimentation. The following protocol outlines a standard method using the triple-plasmid transfection system in HEK293T cells.
Principle: Recombinant AAV is produced by co-transfecting three plasmids into a producer cell line (typically HEK293T): the Transfer Plasmid (containing the gene of interest flanked by ITRs), the Packaging Plasmid (providing AAV rep and cap genes), and the Helper Plasmid (providing essential adenoviral genes E2A, E4, and VA required for AAV replication) [42]. The AAV capsids assemble in the nucleus, and viral particles are released via cell lysis.
Materials:
- Plasmids: pAAV-[GOI] (Transfer Plasmid), pAAV-RC (Packaging Plasmid), pHelper (Helper Plasmid)
- Cell Line: HEK293T cells (adherent or suspension)
- Transfection Reagent: Polyethylenimine (PEI) or commercial equivalent
- Culture Medium: DMEM or Freestyle 293 Expression Medium, supplemented with FBS if needed
- Lysis Buffer: 150 mM NaCl, 50 mM Tris-HCl, pH 8.5
- Benzonase Nuclease
- Purification Reagents: Iodixanol for gradient ultracentrifugation or affinity chromatography resins
- Quantification Kit: qPCR kit with primers targeting the ITR region
Step-by-Step Workflow:
Detailed Procedure:
- Cell Preparation: Seed HEK293T cells in cell culture vessels. For adherent culture, aim for 60-70% confluence at the time of transfection. For large-scale production, use suspension cultures in shake flasks or bioreactors.
- Transfection Complex Formation: For a standard 15 cm plate, prepare a DNA mix containing 7.5 µg pAAV-[GOI], 7.5 µg pAAV-RC, and 10 µg pHelper in a suitable volume of serum-free medium. In a separate tube, dilute PEI transfection reagent (at a 3:1 PEI:Total DNA ratio) in the same medium. Combine the two mixtures, vortex, and incubate for 15-20 minutes at room temperature to allow complex formation. Add the complexes dropwise to the cells.
- Harvest and Lysis: Incubate cells for 48-72 hours. Harvest cells and media by scraping and centrifugation. Resuspend the cell pellet in lysis buffer and subject to three cycles of freeze-thaw (alternating between liquid nitrogen/ethanol and a 37°C water bath) to release the viral particles.
- Benzonase Treatment: Add Benzonase nuclease (e.g., 50 U/mL) to the lysate and incubate at 37°C for 30-60 minutes. This step degrades unpackaged cellular and plasmid DNA, a critical step for reducing impurities.
- Purification and Concentration: Clarify the lysate by centrifugation. Purify the virus from the supernatant using iodixanol density gradient ultracentrifugation. Alternatively, use affinity chromatography columns for higher purity and scalability. Concentrate the purified virus using centrifugal filter units (e.g., 100 kDa MWCO).
- Titration and QC: Determine the genomic titer (vector genomes/mL, vg/mL) by quantitative PCR (qPCR) using primers and a probe specific to the ITR region or a ubiquitous sequence within the transgene. Analyze the purity by running an SDS-PAGE gel and staining for the viral capsid proteins VP1, VP2, and VP3 (approximate weights: 87 kDa, 73 kDa, 62 kDa). Analytical ultracentrifugation or ELISA can be used to assess the proportion of empty vs. full capsids, a key quality metric.
Lentiviral Vector Production and Transduction Protocol
This protocol describes the production of third-generation, replication-incompetent lentiviral vectors using a multi-plasmid transfection system, which offers an enhanced safety profile.
Principle: Lentiviral production involves co-transfection of four plasmids: the Transfer Plasmid (containing the gene of interest flanked by LTRs and with a self-inactivating (SIN) design), the Packaging Plasmid (pMDLg/pRRE) providing Gag and Pol proteins, the Rev-Encoding Plasmid (pRSV-Rev), and the Envelope Plasmid (pMD2.G) which provides the VSV-G glycoprotein for pseudotyping [42]. The virus assembles in the cytoplasm and buds from the cell membrane, acquiring its lipid envelope.
Materials:
- Plasmids: Transfer Plasmid (e.g., pLenti-[GOI]), pMDLg/pRRE, pRSV-Rev, pMD2.G
- Cell Line: HEK293T cells
- Transfection Reagent: PEI
- Culture Medium: High-glucose DMEM with 10% FBS
- Concentration Reagent: Lenti-X Concentrator or equivalent PEG solution
- Transduction Reagent: Polybrene (hexadimethrine bromide, 4-8 µg/mL)
- Titration Reagents: Puromycin for selection or qPCR kit for genomic DNA integration analysis
Step-by-Step Workflow:
Detailed Procedure:
- Production: Seed HEK293T cells to reach 70-80% confluence at transfection. For a 10 cm plate, prepare a DNA mix with 10 µg Transfer Plasmid, 7.5 µg pMDLg/pRRE, 3 µg pRSV-Rev, and 5 µg pMD2.G. Complex the DNA with PEI (as described in the AAV protocol) and add to the cells. Replace the medium 6-8 hours post-transfection.
- Harvest and Concentration: Collect the virus-containing supernatant at 48 and 72 hours post-transfection. Pool the harvests, clarify by filtration through a 0.45 µm PES filter, and concentrate. A common method is to mix the supernatant with Lenti-X Concentrator (1:3 ratio), incubate at 4°C overnight, and centrifuge at 1500 x g for 45 minutes. Resuspend the viral pellet in a small volume of cold PBS or medium.
- Functional Titer Determination (CFU/mL): Seed HEK293T cells in a 24-well plate. The next day, infect the cells with serial dilutions of the concentrated virus in the presence of 8 µg/mL Polybrene. If the transfer plasmid contains a fluorescent reporter (e.g., GFP), analyze the cells by flow cytometry 72-96 hours later. Calculate the titer using the formula: Titer (CFU/mL) = (F × C / V) × D, where F is the frequency of GFP+ cells, C is the total number of cells at the time of infection, V is the volume of inoculum (mL), and D is the dilution factor. For non-fluorescent constructs, genomic DNA can be extracted post-transduction and the number of integrated vector copies per cell can be quantified by qPCR.
- Transduction of Target Cells: Plate the target cells at a density that will be 30-50% confluent at the time of transduction. Add the appropriate volume of viral supernatant (based on the determined titer and desired Multiplicity of Infection, MOI) and Polybrene (4-8 µg/mL) to enhance transduction efficiency. Spinoculation—centrifuging the plate at 800-1000 x g for 30-60 minutes at 32°C—can significantly improve infection rates for many cell types. Replace the medium with fresh culture medium 6-24 hours post-transduction.
- Selection and Analysis: If the vector contains a selectable marker (e.g., puromycin resistance), begin antibiotic selection 48 hours post-transduction. Maintain selection pressure for 3-7 days, until all cells in the negative control (untransduced) have died. Validate editing efficiency via genomic DNA sequencing, Western blot, or functional assays.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Viral Vector Production and Application
| Reagent / Material | Function | Example/Catalog Consideration |
|---|---|---|
| HEK293T Cell Line | Producer cell line for virus packaging due to high transfection efficiency and provision of SV40 T-antigen. | ATCC CRL-3216, Thermo Fisher R70007 |
| Polyethylenimine (PEI) | Cationic polymer transfection reagent for efficient plasmid DNA delivery into producer cells. | Linear PEI, MW 25,000; Polysciences 23966 |
| Iodixanol | Inert density gradient medium for the purification of AAV vectors via ultracentrifugation. | OptiPrep Density Gradient Medium, Sigma D1556 |
| Lenti-X Concentrator | A PEG-based solution that precipitates lentiviral particles for easy concentration and buffer exchange. | Takara Bio 631231 |
| Polybrene | Cationic polymer that reduces electrostatic repulsion between viral particles and the cell membrane, enhancing transduction efficiency. | Hexadimethrine bromide, Sigma H9268 |
| Benzonase Nuclease | Endonuclease that degrades all forms of DNA and RNA; used to clean up AAV lysates by digesting unpackaged nucleic acids. | Millipore Sigma E1014 |
| Puromycin Dihydrochloride | Antibiotic for the selection of successfully transduced cells following infection with a puromycin-resistant lentiviral vector. | Thermo Fisher A1113803 |
| ITR-specific qPCR Assay | For accurate quantification of AAV genomic titer (vg/mL) by targeting the conserved inverted terminal repeat sequence. | Commercial kits or custom-designed assays |
AAV and lentiviral vectors are cornerstone technologies for achieving stable gene expression in CRISPR-Cas9-mediated genome editing. The choice between these systems is not one of superiority but of strategic application. Lentiviral vectors excel in scenarios demanding permanent genomic integration, such as the generation of stable cell lines or long-term functional studies in dividing cells, and they offer the advantage of a larger cargo capacity. In contrast, AAV vectors provide a superior option for in vivo applications and experiments in non-dividing cells where high transduction efficiency and a favorable safety profile, due to their non-integrating nature, are paramount, albeit with a strict cargo limit.
A thorough understanding of their distinct structural properties, production protocols, and application-specific strengths, as detailed in this application note, is indispensable for researchers. By aligning the unique advantages of each vector system with their experimental objectives—whether it involves creating a novel disease model, performing high-throughput genetic screens, or developing a therapeutic candidate—scientists can fully leverage the power of CRISPR-Cas9 to drive innovation in biochemistry research and drug development.
The efficacy of CRISPR-Cas9 genome editing is fundamentally governed by the choice of delivery cargo and vehicle. The three primary forms of CRISPR cargo include plasmid DNA (pDNA), messenger RNA (mRNA) combined with guide RNA (gRNA), and pre-assembled Ribonucleoprotein (RNP) complexes [45] [46]. The RNP complex, consisting of the Cas9 protein complexed with a gRNA, has emerged as a superior cargo for non-viral delivery strategies due to its rapid activity, high editing specificity, and reduced off-target effects [45]. Unlike DNA-based cargos, which require transcription and translation, RNPs are immediately active upon nuclear entry and are rapidly degraded, minimizing prolonged Cas9 exposure and improving safety profiles [46]. This application note details protocols and quantitative comparisons for the two leading non-viral RNP delivery methods: lipid nanoparticle (LNP) encapsulation and electroporation.
Comparative Analysis of Delivery Methods
The selection of a delivery method is critical for experimental success. The table below provides a structured quantitative comparison of LNP and electroporation methods based on recent research, highlighting their performance across key metrics.
Table 1: Performance Comparison of Non-Viral RNP Delivery Methods
| Delivery Method | Reported Editing Efficiency | Key Applications & Cell Types | Notable Advantages | Key Limitations |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | ~25% in DLB-1 marine teleost cells [47]. Efficient editing in mouse liver and lung with tissue-specific LNPs [46]. | In vivo systemic delivery; liver-targeting; difficult-to-transfect cell lines [46] [48]. | Minimal immunogenicity; organ-targeting potential (e.g., SORT-LNPs); suitable for in vivo use [45] [11]. | Must escape endosomes to avoid degradation; editing efficiency can be cell-line dependent [45] [47]. |
| Electroporation | Up to 95% in SaB-1 marine teleost cells [47]. >90% knock-out and ~40% knock-in in human T cells [49]. Up to 90% indels in hematopoietic stem cells (HSCs) for CASGEVY [46]. | Ex vivo editing of immune cells (T cells, HSCs); zygotes; cell lines amenable to physical manipulation [46] [49]. | High efficiency for ex vivo applications; scalable to 1 billion cells; proven clinical success (CASGEVY) [46] [49]. | High cytotoxicity if not optimized; requires specialized equipment; not suitable for in vivo delivery [46]. |
Experimental Protocols
Protocol 1: LNP-Mediated RNP Delivery forIn Vivo/In VitroEditing
This protocol outlines the procedure for formulating LNPs encapsulating Cas9 RNPs and their subsequent application, adapted for both in vivo and in vitro use [46] [48].
Workflow Overview:
Detailed Steps:
- RNP Complex Assembly: Pre-complex the recombinant Cas9 protein with synthetic gRNA at a molar ratio of 1:1.2 to 1:1.5 (Cas9:gRNA). Incubate at room temperature for 10-20 minutes to form the active RNP complex [46].
- Lipid Formulation Preparation: Prepare an ethanol phase containing ionizable cationic lipid, phospholipid, cholesterol, and PEG-lipid at a defined molar ratio (e.g., 50:10:38.5:1.5). The ionizable lipid is critical for endosomal escape [45] [50].
- Nanoassembly: Combine the aqueous RNP solution with the ethanol lipid phase using a microfluidic mixer or rapid pipetting. This induces spontaneous nanoparticle formation as the ethanol diffuses, encapsulating the RNP cargo [50].
- Buffer Exchange and Purification: Dialyze or use tangential flow filtration against a phosphate-buffered saline (PBS) solution at pH 7.4 to remove ethanol and any unencapsulated components. Concentrate the LNP formulation to the desired concentration [48].
- Quality Control: Characterize the final LNP product by measuring:
- Administration:
- For in vitro delivery: Add LNPs to cells in culture, typically at a final concentration of 0.5-2.0 mg/mL total lipid. Transfection efficiency can be enhanced using devices that induce convective flow [48].
- For in vivo delivery: Administer systemically via intravenous (IV) injection. LNPs naturally accumulate in the liver; for other organs, use targeted (SORT) LNPs [45] [11].
- Efficiency Analysis: Allow 48-72 hours for editing, then harvest genomic DNA. Assess editing efficiency via T7E1 assay, TIDE analysis, or next-generation sequencing (NGS) [47].
Protocol 2: Scalable Electroporation of RNPs for T-Cell Engineering
This protocol details the efficient, large-scale electroporation of Cas9 RNPs for ex vivo cell engineering, such as CAR-T cell manufacturing [49].
Workflow Overview:
Detailed Steps:
- Cell Preparation: Expand human primary T-cells in culture using IL-2 and CD3/CD28 activation. Harvest cells during the log growth phase and wash with PBS to remove serum and antibiotics [49].
- RNP Complex Assembly: As described in Protocol 1, section 1.
- Cell Resuspension: Resuspend the washed T-cell pellet in a specialized electroporation buffer. The cell density is critical; for large-scale transfection of up to 1 billion cells, optimize density to ~1x10^7 cells/mL [49].
- Cargo Mixing: Combine the cell suspension with the pre-assembled RNP complex. For knock-in experiments, include a donor DNA template (e.g., single-stranded or double-stranded DNA) at this stage [49].
- Electroporation: Transfer the cell-cargo mixture to an electroporation cuvette or a large-scale cartridge. Apply an optimized electrical pulse sequence. For primary T cells, a specific program on a 4D-Nucleofector system is often used. The pulse creates transient pores in the cell membrane, allowing RNP entry [46] [49].
- Post-Transfection Recovery: Immediately after pulsing, add pre-warmed culture medium to the cells. Transfer the cells to a culture plate and incubate at 37°C for 15-30 minutes to allow membrane resealing and recovery [49].
- Cell Culture and Expansion: Culture the edited cells in growth medium supplemented with cytokines (e.g., IL-2). Monitor cell viability and expand as needed for the downstream application.
- Efficiency Validation: After 3-5 days, analyze a sample of the cell population. Use flow cytometry to assess knock-in efficiency (if applicable) and genomic DNA extraction followed by NGS or TIDE analysis to quantify indel percentages at the target locus [49].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of the above protocols requires high-quality, functional reagents. The table below lists key materials and their critical functions.
Table 2: Essential Reagents for Non-Viral RNP Delivery
| Reagent / Material | Function & Importance | Notes for Selection |
|---|---|---|
| Recombinant Cas9 Protein | The core nuclease enzyme. High-purity, endotoxin-free protein with high specific activity is essential for forming functional RNPs and minimizing cellular toxicity [46]. | Avoid aggregated protein, which compromises delivery efficiency and editing [46]. |
| Synthetic gRNA | Guides the Cas9 protein to the specific genomic target sequence. Chemical modifications can enhance stability and reduce off-target effects [46]. | Ensure high-performance liquid chromatography (HPLC) purification for consistency. |
| Ionizable Cationic Lipid | The key component of LNPs; promotes self-assembly, encapsulation, and endosomal escape via the proton sponge effect [45] [50]. | Examples include DLin-MC3-DMA. The structure determines efficacy and toxicity. |
| Electroporation Buffer | A low-conductivity solution that maintains cell viability while enabling efficient electroporation by minimizing arcing and heat generation [49]. | Use cell-type-specific, commercially available buffers for best results. |
| Donor DNA Template | Provides the homologous repair template for HDR-mediated precise gene knock-in or correction during co-delivery with RNP [49]. | Can be single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA (dsDNA). |
Lipid nanoparticles and electroporation represent two powerful and complementary non-viral strategies for delivering CRISPR-Cas9 RNP complexes. LNPs offer a versatile platform for in vivo applications and targeting specific organs, while electroporation remains the gold standard for achieving high editing efficiencies in ex vivo settings, such as the engineering of therapeutic cell products. The choice between them should be guided by the specific research goal, target cell type, and required scalability. By following the detailed protocols and utilizing the essential reagents outlined in this application note, researchers can robustly integrate these non-viral delivery methods into their genome editing workflows.
{# The Application Notes}
Ex Vivo vs. In Vivo Editing Strategies: Technical Considerations and Workflows
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas system represents a groundbreaking technology for precise genome editing, revolutionizing biochemical research and therapeutic development [51]. The application of this technology primarily proceeds along two distinct methodological pathways: ex vivo and in vivo gene editing. The ex vivo strategy involves harvesting cells from a patient, genetically modifying them outside the body using CRISPR-Cas9, and then reinfusing the edited cells back into the patient [52]. In contrast, the in vivo strategy involves the direct administration of the CRISPR-Cas9 editing machinery into the patient's body to modify cells within their native physiological context [51].
The choice between these strategies is fundamental to experimental and therapeutic design, with profound implications for technical workflow, editing efficiency, safety, and clinical translatability. This article provides a detailed comparison of ex vivo and in vivo editing strategies, framed within the context of using CRISPR-Cas9 for precise genome editing in biochemistry research. It is structured to serve researchers, scientists, and drug development professionals by summarizing key quantitative data in structured tables, providing detailed experimental protocols, and visualizing critical workflows and signaling pathways.
Technical Comparison of Editing Strategies
The ex vivo and in vivo approaches present a series of contrasting technical considerations, from delivery methods to inherent advantages and challenges. A head-to-head comparison of their core characteristics is essential for strategic planning.
Table 1: Core Characteristics of Ex Vivo and In Vivo Editing Strategies
| Characteristic | Ex Vivo Editing | In Vivo Editing |
|---|---|---|
| Definition | Cells are edited outside the organism and then reintroduced [52]. | Editing machinery is delivered directly to target cells inside the organism [51]. |
| Primary Delivery Vehicles | Electroporation, viral vectors (e.g., Lentiviral Vectors) [45]. | Viral vectors (e.g., rAAV), Lipid Nanoparticles (LNPs) [51] [45]. |
| Key Advantage | High precision and control over editing conditions; easier to validate editing outcomes [52] [53]. | Avoids complex cell harvesting and reinfusion; potential to target inaccessible tissues [11] [51]. |
| Key Challenge | Requires robust cell culture and transplantation protocols; potential for cell damage or loss during process [53] [54]. | Limited by delivery vehicle efficiency, tissue tropism, and potential immune responses [51] [45]. |
| Ideal Cargo Format | Ribonucleoprotein (RNP) complexes for transient activity and reduced off-target effects [45]. | DNA (for rAAV) or mRNA (for LNPs) for sustained or transient expression of editors [45]. |
| Therapeutic Example | CASGEVY (exagamglogene autotemcel) for SCD/TDT [52] [55]. | EDIT-101 for Leber Congenital Amaurosis; LNP-editors for hATTR [11] [51]. |
A critical differentiator is the choice of delivery vehicles, each with distinct properties that influence their suitability for either strategy.
Table 2: Common Delivery Vehicles in CRISPR-Cas9 Editing
| Delivery Vehicle | Type | Packaging Capacity | Key Advantages | Key Disadvantages | Best Suited For |
|---|---|---|---|---|---|
| Adeno-associated Virus (rAAV) | Viral | <4.7 kb [51] [45] | Favorable safety profile, high tissue specificity, sustained expression [51]. | Very limited packaging capacity, potential pre-existing immunity [51] [45]. | In vivo delivery (requires compact editors) [51]. |
| Lentiviral Vector (LV) | Viral | ~8 kb [45] | Large cargo capacity, infects dividing and non-dividing cells [45]. | Integrates into host genome, raising safety concerns for therapeutics [45]. | Ex vivo delivery (e.g., for CAR-T cells) [45]. |
| Lipid Nanoparticle (LNP) | Non-viral | Varies (mRNA/RNP) | Low immunogenicity, tunable for organ targeting, suitable for re-dosing [11] [45]. | Endosomal escape challenge, primarily targets liver without modification [45]. | In vivo mRNA/RNP delivery [11] [55]. |
| Electroporation | Physical | N/A (direct delivery) | Highly efficient for RNP delivery into cells in vitro; immediate activity [45]. | Not suitable for in vivo delivery; can cause significant cell death [45]. | Ex vivo delivery of RNPs to primary cells (e.g., HSCs) [45]. |
Experimental Protocols
Detailed Protocol: Ex Vivo Editing of Hematopoietic Stem and Progenitor Cells (HSPCs)
This protocol details the optimization of culture conditions for CRISPR-Cas9-mediated gene editing in human HSPCs, a key process for therapies like CASGEVY. A critical challenge is that long ex vivo culture to engage homology-directed repair (HDR) can induce detrimental cellular responses, compromising the long-term functionality of edited HSPCs [53] [54]. The integration of a p38 mitogen-activated protein kinase (MAPK) inhibitor in the culture medium has been shown to reduce this proliferation stress and DNA damage, thereby preserving stem cell fitness [54].
Diagram 1: Ex Vivo HSPC Editing Workflow
3.1.1 Materials and Reagents
- Human CD34+ HSPCs: Sourced from mobilized peripheral blood or cord blood.
- Stimulation Media: StemSpan SFEM II, supplemented with human cytokines (e.g., SCF, TPO, FLT3-L) [53].
- p38 MAPK Inhibitor: e.g., SB203580, dissolved in DMSO.
- CRISPR Components: Cas9 nuclease (as protein or mRNA), synthetic sgRNA, and optional single-stranded DNA (ssDNA) HDR donor template.
- Electroporation Buffer: Proprietary buffers like P3 Primary Cell Solution.
3.1.2 Step-by-Step Methodology
- Thawing and Pre-stimulation: Thaw frozen HSPCs rapidly and plate in pre-warmed Stimulation Media. Culture for 24-48 hours to activate the cells and bring them into the cell cycle, which is crucial for subsequent gene editing efficiency [53].
- RNP Complex Formation: Formulate the ribonucleoprotein (RNP) complex by pre-incubating purified Cas9 protein with synthetic sgRNA at room temperature for 10-20 minutes. Using RNP complexes ensures immediate editing activity upon delivery and reduces off-target effects compared to plasmid DNA [45].
- Electroporation: Harvest pre-stimulated HSPCs and resuspend them in the appropriate electroporation buffer. Mix the cell suspension with the prepared RNP complex (and ssDNA HDR donor if performing knock-in). Electroporate using a certified device (e.g., 4D-Nucleofector) with an optimized program for human HSPCs.
- Post-Editing Recovery with p38 Inhibition: Immediately after electroporation, transfer the cells into recovery media (Stimulation Media) supplemented with a p38 MAPK inhibitor (e.g., 5-10 µM SB203580). This step is critical to mitigate the p38 MAPK-driven proliferation stress and DNA damage response triggered by the ex vivo culture and editing process, thereby preserving the long-term repopulating capacity of the HSPCs [54]. Culture the cells for an additional 48-72 hours before analysis or transplantation.
- Downstream Analysis:
- In Vitro: Assess editing efficiency (e.g., via T7E1 assay or NGS), cell viability, and immunophenotype to confirm stem/progenitor markers.
- In Vivo: Transplant the edited HSPCs into immunodeficient mouse models (e.g., NSG mice) to validate their in vivo repopulating capacity and long-term multilineage potential, the gold-standard functional assay [53] [54].
Detailed Protocol: In Vivo Genome Editing via rAAV Delivery
This protocol outlines the strategy for in vivo therapeutic genome editing using recombinant adeno-associated virus (rAAV) vectors, which are favored for their tissue specificity and sustained expression [51]. The primary challenge is the limited packaging capacity (<4.7 kb) of rAAV, which is insufficient for the standard SpCas9 (~4.2 kb) plus sgRNA and regulatory elements. The strategy employs dual rAAV vectors or compact Cas orthologs to overcome this hurdle [51].
Diagram 2: In Vivo rAAV Editing Strategy
3.2.1 Materials and Reagents
- CRISPR Components:
- Path A (All-in-one): Plasmid encoding a compact Cas ortholog (e.g., SaCas9, ~3.2 kb) and its sgRNA.
- Path B (Dual-Vector): Separate plasmids for full-length SpCas9 (or other nuclease) and the sgRNA expression cassette.
- rAAV Production System: HEK293T cells, packaging plasmid (pDGM), transfection reagent.
- Purification and Quantification Kits: For purifying viral vectors and quantifying genomic titer (e.g., via qPCR).
- Animal Model: Suitable mouse or other model for the disease of interest.
3.2.2 Step-by-Step Methodology
- Vector Design and Production:
- For Path A (Compact Cas): Clone the expression cassette for the compact Cas nuclease (e.g., SaCas9) and its target-specific sgRNA into a single AAV transfer plasmid. Ensure the total size remains within the ~4.7 kb packaging limit [51].
- For Path B (Dual-Vector): Clone the Cas9 nuclease into one AAV transfer plasmid and the sgRNA expression cassette into a separate AAV transfer plasmid. Co-transfect these plasmids with the packaging and helper plasmids into HEK293T cells to produce the two separate rAAV stocks [51].
- Vector Purification and QC: Harvest the viral particles from the cell culture supernatant and lysate. Purify the rAAV using iodixanol gradient centrifugation or affinity chromatography. Quantify the genomic titer of each preparation using digital PCR or qPCR.
- Administration In Vivo: Administer the rAAV vector(s) to the animal model. The route of administration (e.g., systemic intravenous injection for liver targeting, subretinal injection for retinal targeting) depends on the rAAV serotype selected (e.g., AAV8/9 for liver, AAV5 for retina) [51]. For the dual-vector approach, ensure the two rAAVs are co-administered at equivalent titers.
- Analysis of Editing Outcomes:
- Molecular Analysis: After a sufficient period (e.g., 2-4 weeks), harvest the target tissue. Isolate genomic DNA and analyze editing efficiency at the target locus using next-generation sequencing (NGS) to quantify indels and/or HDR.
- Functional Analysis: Assess the phenotypic outcome, which may include quantification of target protein reduction in serum (e.g., TTR for hATTR), histological examination of tissue sections, or measurement of physiological improvements [11] [51].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of CRISPR-Cas9 experiments requires a suite of specialized reagents. The following table lists key solutions and their critical functions in the editing workflow.
Table 3: Essential Reagents for CRISPR-Cas9 Genome Editing
| Research Reagent | Function/Application | Key Considerations |
|---|---|---|
| Purified Cas9 Nuclease | Core enzyme for inducing double-strand breaks in DNA. | Available as wild-type or high-fidelity variants. Delivery format (DNA, mRNA, protein) dictates kinetics and off-target profile [45]. |
| Synthetic sgRNA | Guides Cas9 nuclease to the specific genomic target sequence. | Chemically modified sgRNAs can enhance stability and editing efficiency in primary cells [54]. |
| HDR Donor Template | Provides a template for precise gene correction or insertion. | Can be single-stranded oligodeoxynucleotide (ssODN) for small edits or double-stranded DNA (dsDNA) for larger inserts. Must contain homology arms [52]. |
| p38 MAPK Inhibitor | Improves fitness and long-term functionality of ex vivo edited HSPCs. | Added to culture media post-electroporation to reduce detrimental cellular responses to editing and culture [54]. |
| Lipid Nanoparticles (LNPs) | A leading non-viral vehicle for in vivo delivery of CRISPR cargo (especially mRNA/RNP). | Enable redosing; can be engineered for selective organ targeting (SORT) beyond the liver [11] [45]. |
| rAAV Vectors | A leading viral vehicle for in vivo delivery of CRISPR cargo. | Serotype determines tissue tropism. Limited packaging capacity necessitates use of compact Cas proteins or dual-vector systems [51]. |
Emerging Technologies and Future Directions
The field of CRISPR-based genome editing is evolving rapidly, with new technologies addressing the limitations of early systems.
- Prime Editing: This system uses a Cas9 nickase fused to a reverse transcriptase to directly write new genetic information into a target DNA site without requiring double-strand breaks or donor templates, offering greater precision and versatility [56]. Recent advances in 2025 have dramatically lowered its error rate by using modified Cas9 proteins that destabilize the old DNA strand, favoring incorporation of the edited strand and reducing errors from approximately 1 in 7 edits to 1 in 101 for the most-used mode [56].
- Base Editing: This technology enables the direct, irreversible chemical conversion of one DNA base pair into another (e.g., C•G to T•A) without making a double-strand break, thereby minimizing indel formation [51]. It has shown success in clinical settings for treating genetic disorders like severe carbamoyl-phosphate synthetase 1 deficiency [11].
- Advanced Delivery Platforms: Innovations in delivery are crucial for both strategies. For in vivo editing, the development of novel lipid nanoparticles (LNPs) that can be re-dosed and targeted to organs beyond the liver is a major focus [11] [55]. Virus-like particles (VLPs) are also being explored as a potentially safer alternative to traditional viral vectors, as they are non-replicative and non-integrating [45].
- Ultra-Compact CRISPR Systems: The discovery and engineering of putative ancestors of Cas proteins, such as IscB and TnpB, offer ultra-compact editing tools that are highly compatible with rAAV delivery and may have a reduced immunogenicity profile [51].
The strategic decision between ex vivo and in vivo CRISPR-Cas9 editing is pivotal, with each path offering a distinct set of technical workflows, advantages, and challenges. Ex vivo editing provides unparalleled control and is the established paradigm for cell-based therapies like CASGEVY, but it demands complex and costly cell processing. In vivo editing promises a more straightforward interventional strategy but is critically constrained by the efficiency and safety of delivery vehicles. For the biochemical researcher, the choice hinges on the specific research question, the target cell type, and the required precision. As the field advances, emerging technologies like enhanced prime editing, base editing, and next-generation delivery vectors are poised to expand the capabilities of both strategies, enabling more precise, safe, and effective genome editing applications across basic research and therapeutic development.
Application Note: CRISPR-Cas9 in Clinical Trials
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system represents a transformative technology for precise genome editing in biochemistry and therapeutic development [25]. This two-component system utilizes a Cas9 nuclease and a synthetic guide RNA (sgRNA) that can be readily targeted to specific DNA sequences, creating double-strand breaks that trigger the cell's endogenous DNA repair machinery [25]. The advent of CRISPR-Cas9 has revolutionized the development of potential cures for genetic disorders, with notable clinical successes emerging for sickle cell disease (SCD) and transthyretin (ATTR) amyloidosis.
Sickle Cell Disease and CASGEVY
Sickle cell disease is an inherited blood disorder caused by a single mutation in the hemoglobin gene, leading to the production of abnormal hemoglobin that distorts red blood cells into a sickle shape [57]. These misshapen cells cause vaso-occlusive crises (VOCs), severe pain, organ damage, and reduced life expectancy, with patients rarely surviving beyond their 40s [57].
CASGEVY (exagamglogene autotemcel), developed by Vertex and CRISPR Therapeutics, is the first FDA-approved therapy based on CRISPR-Cas9 technology [57]. This one-time autologous treatment edits a patient's own blood stem cells to produce high levels of fetal hemoglobin (HbF), which does not sickle, thereby addressing the root cause of the disease [58] [59].
Table: Key Efficacy Outcomes from CASGEVY Clinical Trial
| Parameter | Result | Follow-up Period |
|---|---|---|
| Patients free of severe VOCs | 29 out of 31 (93.5%) | 12 consecutive months [58] |
| Patients free of VOC-related hospitalizations | 30 out of 30 (100%) | 12 consecutive months [58] |
| Average time free of severe VOCs (median) | 22.2 months | Ongoing assessment [58] |
Clinical outcomes have been transformative. Over 90% of treated individuals remain free of painful crises following therapy [57]. Patients like Marie-Chantal and 17-year-old Carlos have reported being freed from the debilitating pain crises that previously dominated their lives, allowing them to envision normal futures [57] [59].
Transthyretin Amyloidosis and Nexiguran Ziclumeran
Transthyretin (ATTR) amyloidosis is a rare, progressive, and fatal disease characterized by the buildup of misfolded transthyretin protein as amyloid deposits in tissues, including the heart and nerves [60]. Nexiguran ziclumeran (nex-z, formerly NTLA-2001) is an investigational CRISPR-Cas9-based therapy designed as a one-time treatment to inactivate the TTR gene in the liver, reducing the production of the disease-causing protein [60].
Table: Key Efficacy Outcomes from Phase 1 Study of Nex-z in ATTR Amyloidosis with Cardiomyopathy
| Parameter | Result | Follow-up Period |
|---|---|---|
| Mean reduction in serum TTR | 87% | 36 months (n=9) [60] |
| Patients stable/improved on NT-proBNP (cardiac biomarker) | 70% | 24 months [60] |
| Patients stable/improved on 6-minute walk test | 69% | 24 months [60] |
| Patients stable/improved in NYHA Class (cardiac function) | 81% | 24 months [60] |
Longer-term data show that a single dose of nex-z leads to a rapid, deep, and durable reduction in serum TTR, with evidence of disease stabilization or improvement across multiple measures of cardiomyopathy through 24-36 months of follow-up [60]. In October 2025, the MAGNITUDE Phase 3 trials for nex-z were temporarily paused after a participant experienced Grade 4 elevations in liver enzymes, highlighting the critical importance of ongoing safety monitoring in gene editing therapies [61].
Experimental Protocols
Protocol for Clinical-Scale CRISPR-Cas9 Gene Therapy
The following detailed protocol outlines the key steps for the administration of autologous CRISPR-Cas9-edited cell therapies, as used in CASGEVY treatment [57] [58] [59].
Patient Eligibility and Pre-treatment Screening
- Inclusion Criteria: Patients aged 12 years and older with severe SCD (e.g., ≥2 severe VOCs annually) or a diagnosis of ATTR amyloidosis, adequate organ function, and eligibility for stem cell transplant [58].
- Exclusion Criteria: Active uncontrolled infection, history of malignancy, contraindications to conditioning chemotherapy, or positive HIV, hepatitis B, or hepatitis C status.
- Pre-treatment Workup: Comprehensive medical history, physical examination, cardiac/pulmonary/renal/liver function testing, infectious disease screening, and fertility preservation counseling [58].
Stem Cell Mobilization and Collection (Apheresis)
- Mobilization: Administer a mobilization medicine (e.g., plerixafor, G-CSF) to move hematopoietic stem cells (HSCs) from the bone marrow into the peripheral blood bloodstream [58].
- Apheresis: Collect the mobilized blood stem cells via leukapheresis. This process may need to be repeated over several days to obtain a sufficient CD34+ HSC dose (target typically > 2.0 x 10^6 CD34+ cells/kg) [58].
- Rescue Cell Collection: A portion of the collected cells is set aside and cryopreserved as unmanipulated "rescue cells" to be reinfused in case of treatment failure [58].
CRISPR-Cas9 Genome Editing and Manufacturing
- sgRNA Design and Delivery: For SCD, design sgRNAs targeting the BCL11A gene, a key repressor of fetal hemoglobin production [57]. For ATTR amyloidosis, design sgRNAs targeting the TTR gene in the liver [60].
- Ex Vivo Editing: Electroporation of patient CD34+ cells with CRISPR-Cas9 components (e.g., Cas9 nuclease and sgRNA). The process involves:
- Cell Preparation: Isolate and activate CD34+ cells from the apheresis product.
- Electroporation: Introduce CRISPR-Cas9 ribonucleoprotein (RNP) complexes into the cells.
- Quality Control: Assess cell viability, editing efficiency (e.g., via T7 Endonuclease I assay or NGS), and sterility [25].
- Manufacturing and Release Testing: The final drug product (e.g., CASGEVY) is formulated, cryopreserved, and subjected to rigorous quality control testing, including viability, potency, identity, and freedom from contaminants. This manufacturing process can take up to 6 months [58].
Myeloablative Conditioning
- Patient Admission: Admit the patient to the hospital for the conditioning regimen.
- Chemotherapy Administration: Administer myeloablative conditioning (e.g., busulfan) for several days to clear the bone marrow of native HSCs and create "space" for the engraftment of the edited cells [58] [59].
- Supportive Care: Provide prophylactic anti-emetics, seizure prophylaxis (for busulfan), and hydration.
Infusion of Gene-Edited Cells and Post-Infusion Monitoring
- Product Thaw and Infusion: Thaw the cryopreserved CASGEVY product at the bedside and administer via intravenous infusion over a short period (e.g., less than one hour) [59].
- Engraftment Monitoring: Monitor blood counts daily for evidence of neutrophil and platelet engraftment. Neutrophil engraftment typically occurs within 20-30 days post-infusion [58].
- Supportive Care: Provide stringent supportive care, including:
- Infection Prophylaxis and Management: Antibacterial, antiviral, and antifungal prophylaxis. Monitor for and treat febrile neutropenia aggressively.
- Transfusion Support: Provide irradiated, leukoreduced packed red blood cells and platelet transfusions as needed.
- Pain and Symptom Management.
- Hospital Discharge: Discharge the patient once they have achieved neutrophil engraftment, are afebrile, and are stable on oral medications.
Long-Term Follow-Up
- Schedule regular follow-up visits for at least 15 years to monitor:
- Disease-specific Efficacy: Hemoglobin electrophoresis, HbF levels, VOC frequency (for SCD); serum TTR levels, cardiac and neurological assessments (for ATTR amyloidosis) [58] [60].
- Safety: Comprehensive metabolic panel, complete blood count, assessment for off-target editing, clonal dominance, or secondary malignancies.
- Product Persistence: Peripheral blood vector copy number and continued presence of edited cells.
Protocol for Rapid Screening of CRISPR-Cas9 Editing Outcomes
This protocol provides a method for rapidly quantifying the efficiency of CRISPR-Cas9 gene editing in a cell population using a fluorescent reporter system [62].
Generation of eGFP-Positive Reporter Cell Line
- Lentiviral Transduction: Generate a stable cell line expressing enhanced Green Fluorescent Protein (eGFP) via lentiviral transduction.
- Culture the target cells (e.g., HEK293T, iPSCs) under standard conditions.
- Produce lentiviral particles encoding the eGFP gene.
- Transduce the target cells with the eGFP lentivirus in the presence of a transduction enhancer (e.g., polybrene).
- Select and expand eGFP-positive cells using fluorescence-activated cell sorting (FACS) or antibiotic selection.
Design and Transfection of Gene-Editing Reagents
- sgRNA Design: Design sgRNAs targeting critical regions of the eGFP gene to disrupt its function. A positive control sgRNA can be designed to convert eGFP to Blue Fluorescent Protein (BFP) via homology-directed repair (HDR) [62].
- Preparation of Editing Reagents: Complex CRISPR-Cas9 ribonucleoprotein (RNP) by combining purified Cas9 protein with the synthesized sgRNA. If performing HDR, also include a single-stranded DNA (ssODN) repair template.
- Cell Transfection: Transfect the eGFP-positive cells with the RNP complexes (and repair template if applicable) using a high-efficiency method like electroporation.
Analysis of Editing Outcomes via Flow Cytometry
- Cell Handling Post-Transfection: Culture the transfected cells for 3-7 days to allow for expression of editing outcomes.
- Sample Preparation: Harvest cells, wash with PBS, and resuspend in flow cytometry buffer containing a viability dye.
- Flow Cytometry Analysis: Analyze the cells using a flow cytometer equipped with lasers and filters for detecting eGFP (e.g., 488 nm laser, 530/30 nm filter) and BFP (e.g., 405 nm laser, 450/50 nm filter).
- Gate on viable, single cells.
- Quantify the percentage of non-fluorescent cells: Loss of eGFP signal indicates successful gene knockout via NHEJ.
- Quantify the percentage of BFP-positive cells: Appearance of BFP signal indicates successful knock-in via HDR [62].
Data Interpretation
- Editing Efficiency: Calculate NHEJ efficiency as (% of non-fluorescent cells in test sample) - (% of non-fluorescent cells in untreated control).
- HDR Efficiency: Calculate HDR efficiency as (% of BFP-positive cells in test sample).
- Statistical Analysis: Perform experiments in triplicate and report data as mean ± standard deviation.
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents and Materials for CRISPR-Cas9 Gene Editing Research
| Reagent/Material | Function | Examples and Notes |
|---|---|---|
| Cas9 Nuclease | Creates double-strand breaks at target DNA sites. | Recombinant S. pyogenes Cas9 protein; used as purified protein for RNP formation or encoded in plasmid/mRNA [25]. |
| Synthetic Guide RNA (sgRNA) | Directs Cas9 to a specific genomic locus via Watson-Crick base pairing. | Chemically synthesized crRNA and tracrRNA or a single-guide RNA (sgRNA); requires careful design to maximize on-target and minimize off-target activity [25]. |
| Repair Templates | Serves as a donor DNA for introducing specific mutations via HDR. | Single-stranded oligodeoxynucleotides (ssODNs) for small edits; double-stranded DNA plasmids for larger insertions [25]. |
| Delivery Vectors | Vehicles for introducing CRISPR components into cells. | Electroporation for RNPs; lentiviral/AAV vectors for hard-to-transfect cells; plasmids for simple transfection [25]. |
| Stem Cell Culture Reagents | Supports the growth and maintenance of pluripotent stem cells. | Defined culture media (e.g., mTeSR), recombinant growth factors, Geltrex/Matrigel for feeder-free culture [25]. |
| Editing Efficiency Assays | Measures the frequency and accuracy of genomic modifications. | T7 Endonuclease I or Surveyor assay for initial screening; barcoded deep sequencing for comprehensive quantification of indels and HDR [25]. |
| Fluorescent Reporters | Enables rapid, visual screening of editing outcomes. | Cell lines with integrated eGFP; conversion to BFP or loss of fluorescence serves as a readout for HDR or NHEJ efficiency, respectively [62]. |
| Selection Markers | Enriches for successfully transfected or edited cells. | Antibiotic resistance genes (puromycin, neomycin) or fluorescent proteins co-expressed with CRISPR machinery [25]. |
Workflow and Mechanism Diagrams
CASGEVY Mechanism of Action
CRISPR-Cas9 Gene Editing Workflow
Application Note: CRISPR-Cas9 in Cancer Immunotherapy
CRISPR-Cas9 technology is revolutionizing cancer immunotherapy by enabling precise genomic modifications that enhance the body's immune response against tumors. This approach primarily focuses on engineering immune cells, such as T cells, to improve their ability to recognize, attack, and persist within the immunosuppressive tumor microenvironment [63] [64]. The technology allows for the disruption of immune checkpoint molecules that tumors exploit to evade detection, and the creation of more potent "off-the-shelf" cellular therapies [65]. By overcoming key limitations of conventional immunotherapies, CRISPR-Cas9 is pushing the boundaries of treatable cancers, particularly for solid tumors that have previously been resistant to adoptive cell transfer approaches [63] [64].
Key Quantitative Data and Clinical Progress
Table 1: Clinical Trial Data for CRISPR-Enhanced Cancer Immunotherapies
| Application Area | Target Gene(s) | Phase | Key Outcome Measures | Reported Efficacy/Results |
|---|---|---|---|---|
| Universal CAR-T Cells | TRAC, β2M, PD-1 [65] | Early-phase trials | GVHD incidence, tumor response | Successful multiplex editing; reduced alloreactivity in preclinical models [65] |
| Enhanced CAR-T Function | PD-1, CTLA-4, LAG-3 [63] | Preclinical/early clinical | Tumor clearance, T-cell persistence | Augmented tumor killing in PD-L1+ xenograft models [65] |
| TCR-T Cell Therapy | Endogenous TCR, HLA [65] | Early-phase trials | Target antigen response, off-tumor toxicity | Improved tumor-specific targeting in preclinical studies [65] |
Table 2: CRISPR Screen-Discovered Immunotherapy Targets
| Target Gene | Cancer Type | Therapeutic Approach | Effect of Modulation |
|---|---|---|---|
| DGK [65] | Multiple | CAR-T with DGK ablation | Improved anti-tumor immunity [65] |
| GM-CSF [65] | Multiple | CAR-T with GM-CSF knockout | Enhanced CAR-T function, reduced CRS risk [65] |
| TGF-β Receptor [65] | Solid tumors | TGFBR2 knockout in CAR-T | Resistance to immunosuppressive TGF-β signaling [65] |
Experimental Protocol: Generation of PD-1 Knockout CAR-T Cells
Objective: To enhance antitumor activity of CAR-T cells by disrupting the PD-1 immune checkpoint gene.
Materials:
- Cas9 Source: Streptococcus pyogenes Cas9 nuclease (mRNA or protein) [63]
- Guide RNA: Synthetic sgRNA targeting PDCD1 exon 1 [65]
- Delivery System: Electroporation for RNPs or lentiviral vectors for Cas9/gRNA constructs [65]
- T Cells: Human primary T cells isolated via leukapheresis
- Culture Media: X-VIVO 15 serum-free media with IL-2 and IL-15 [65]
- Activation Beads: Anti-CD3/CD28 magnetic beads
- CAR Transduction: Lentiviral vectors encoding CAR construct
Procedure:
- T Cell Activation: Isolate PBMCs from leukapheresis product using Ficoll density gradient centrifugation. Activate T cells with anti-CD3/CD28 beads in complete media supplemented with 100 U/mL IL-2 and 10 ng/mL IL-15 for 24-48 hours [65].
- CRISPR Component Preparation: Complex Cas9 protein with PD-1-targeting sgRNA at 3:1 molar ratio in PBS buffer to form ribonucleoproteins (RNPs). Incubate at room temperature for 15 minutes [65].
- Electroporation: Wash activated T cells and resuspend in electroporation buffer. Electroporate 1-2 million cells with 5-10 µg RNP complex using manufacturer-recommended settings for primary T cells [65].
- CAR Transduction: Transduce cells with CAR-encoding lentivirus 24 hours post-electroporation at MOI of 5-10 in the presence of 8 µg/mL polybrene [65].
- Expansion and Validation: Expand edited T cells for 10-14 days with cytokine support. Validate PD-1 knockout efficiency via flow cytometry and DNA sequencing. Assess CAR expression via flow cytometry [65].
Timeline: Complete protocol requires 3-4 weeks from T cell isolation to validated product.
Troubleshooting:
- Low editing efficiency: Optimize RNP concentration and electroporation parameters
- Reduced cell viability: Decrease RNP concentration, increase cytokine support
- Variable CAR expression: Titrate viral titer and optimize transduction protocol
Signaling Pathways in CRISPR-Enhanced Cancer Immunotherapy
Diagram 1: CRISPR engineering creates universal, enhanced CAR-T cells.
Application Note: CRISPR-Cas9 for Rare Genetic Disorders
CRISPR-Cas9 has emerged as a transformative therapeutic platform for rare genetic disorders, enabling precise correction of disease-causing mutations at the DNA level [66]. The technology offers particular promise for monogenic diseases that have historically lacked effective treatments, with recent clinical successes demonstrating both ex vivo and in vivo editing approaches [11] [67]. The 2023 approval of Casgevy for sickle cell disease and beta thalassemia marked a watershed moment, validating CRISPR's therapeutic potential [68]. More recently, the development of a personalized CRISPR therapy for CPS1 deficiency has demonstrated the feasibility of creating bespoke gene editing treatments for ultra-rare genetic conditions, opening new possibilities for addressing the vast landscape of orphan diseases [67].
Key Quantitative Data and Clinical Progress
Table 3: Clinical Outcomes for CRISPR Therapies in Genetic Disorders
| Disease | Therapeutic Approach | Phase | Key Efficacy Endpoints | Reported Outcomes |
|---|---|---|---|---|
| Sickle Cell Disease | BCL11A enhancer editing (ex vivo) [68] | Approved (2023) | Freedom from vaso-occlusive crises | 94.1% remained crisis-free at 12 months [11] |
| β-Thalassemia | BCL11A enhancer editing (ex vivo) [68] | Approved (2023) | Transfusion independence | 89.5% achieved transfusion independence [11] |
| hATTR Amyloidosis | TTR knockout (in vivo LNP) [11] | Phase III | Serum TTR reduction | ~90% sustained TTR reduction at 2 years [11] |
| Hereditary Angioedema | Kallikrein knockout (in vivo LNP) [11] | Phase I/II | Kallikrein reduction, attack rate | 86% kallikrein reduction; 8/11 attack-free [11] |
| CPS1 Deficiency | Base editing (in vivo LNP) [67] | Case study | Protein tolerance, ammonia levels | Improved dietary protein tolerance, reduced medications [67] |
Table 4: Delivery Systems for Genetic Disorder Therapies
| Delivery Method | Advantages | Limitations | Clinical Examples |
|---|---|---|---|
| Lipid Nanoparticles (LNPs) [11] | Target hepatocytes efficiently, redosing possible [11] | Primarily hepatic tropism, transient expression | hATTR, HAE, CPS1 therapies [11] [67] |
| AAV Vectors [69] | Long-term expression, broad tropism | Immune concerns, limited payload capacity | Preclinical studies [69] |
| Ex Vivo Electroporation [68] | High editing efficiency, controlled conditions | Complex manufacturing, autologous only | Casgevy for SCD and β-thalassemia [68] |
Experimental Protocol: In Vivo Base Editing via LNP Delivery
Objective: To correct point mutations in hepatocytes using LNP-delivered base editors.
Materials:
- Base Editor: ABE8e or BE4max mRNA with optimized codons [67]
- Guide RNA: Chemically modified sgRNA with 3' polyA tail [67]
- LNP Formulation: Ionizable lipid (e.g., DLin-MC3-DMA), DSPC, cholesterol, PEG-lipid [11]
- Animal Model: Mouse model with humanized disease mutation
- Analytical Methods: NGS for editing efficiency, ELISA for protein correction, metabolic assays
Procedure:
- CRISPR Formulation: Complex base editor mRNA and sgRNA at 1:2 mass ratio in citrate buffer (pH 4.0). encapsulate in LNPs using microfluidic mixing with lipid mixture at 3:1 aqueous:organic flow rate ratio [11].
- LNP Characterization: Determine particle size (target 70-100 nm) via dynamic light scattering. Measure encapsulation efficiency using RiboGreen assay. Confirm sterility through endotoxin testing [11].
- In Vivo Administration: Adminylate LNPs via tail vein injection at 1-3 mg/kg mRNA dose in adult mice. For large animal models, use peripheral venous catheter with physiological monitoring [67].
- Efficacy Assessment: Collect tissue biopsies and plasma samples at 48 hours (peak editing) and 2-4 weeks (stable correction). Quantify editing efficiency via amplicon sequencing. Assess functional correction through metabolite analysis or protein quantification [67].
- Safety Monitoring: Perform serial CBC, chemistry panels. Assess liver enzymes (ALT, AST) weekly. Conduct histopathological examination of liver tissue at endpoint [67].
Timeline: From LNP formulation to initial efficacy readout requires 4-6 weeks.
Troubleshooting:
- Low editing efficiency: Optimize LNP composition, increase guide RNA stability modifications
- Elevated liver enzymes: Reduce LNP dose, extend dosing interval
- Inconsistent delivery: Characterize LNP biodistribution, optimize injection protocol
Therapeutic Mechanism of In Vivo Gene Editing
Diagram 2: In vivo base editing corrects mutations to restore function.
Application Note: CRISPR-Cas9 in Infectious Diseases
CRISPR-Cas9 technology provides powerful new approaches for combating infectious diseases through direct targeting of pathogen genomes, enhanced diagnostic capabilities, and engineering of infection-resistant cells [70] [71]. The discovery of RNA-targeting Cas enzymes (particularly Cas13) has been particularly valuable for detecting and treating RNA viruses like SARS-CoV-2 [70] [71]. CRISPR-based diagnostic platforms such as SHERLOCK and DETECTR offer rapid, sensitive pathogen detection that is deployable in resource-limited settings [70]. Meanwhile, therapeutic applications range from excising integrated HIV provirus from host genomes to engineering bacteriophages that selectively destroy antibiotic-resistant bacteria, representing a paradigm shift in our approach to infectious disease management [70] [71].
Key Quantitative Data and Technical Performance
Table 5: CRISPR-Based Applications in Infectious Diseases
| Application | Pathogen Target | Cas System | Key Performance Metrics | Reported Efficacy |
|---|---|---|---|---|
| Diagnostic Platform | SARS-CoV-2 [70] | Cas12, Cas13 | Sensitivity, specificity, time | 95% positive predictive agreement with PCR, 45 min turnaround [70] |
| Viral Excision Therapy | HIV-1 provirus [71] | Cas9 | Excision efficiency, viral rebound | Successful proviral excision in preclinical models; EBT-101 in clinical trials [71] |
| Antiviral Host Targeting | SARS-CoV-2 [71] | Cas9 | Viral replication inhibition | Identified host factors (FASN, DAXX) via CRISPR screens [71] |
| CRISPR-Phage Therapy | E. coli [71] | Cas3 | Bacterial killing specificity | Phase1b trial completion for UTIs; selective pathogen targeting [71] |
| HPV Oncogene Targeting | HPV E6/E7 [71] | Cas9 | Tumor growth inhibition | Cleared HPV16 tumors in mice without toxicity [71] |
Table 6: CRISPR Diagnostic Platforms for Infectious Diseases
| Platform | Cas Enzyme | Target Type | Detection Method | Applications |
|---|---|---|---|---|
| SHERLOCK [70] | Cas13 [70] | RNA | Fluorescent reporter | SARS-CoV-2, Ebola, Zika, Dengue [70] |
| DETECTR [70] | Cas12a [70] | DNA | Fluorescent reporter | HPV, SARS-CoV-2 [70] |
| DASH [70] | Cas9 | DNA | NGS enrichment | Fungal, parasitic sequences in CSF [70] |
| FLASH [70] | Cas9 | DNA | NGS enrichment | Antibiotic resistance genes [70] |
Experimental Protocol: CRISPR Diagnostic Test (DETECTR)
Objective: To detect pathogen DNA in clinical samples using Cas12a-based DETECTR platform.
Materials:
- Cas Enzyme: LbaCas12a (Cpf1) nuclease [70]
- Guide RNA: crRNA designed against pathogen-specific sequence [70]
- Sample Preparation: DNA extraction kit, isothermal amplification reagents
- Reporter System: Fluorescently quenched ssDNA reporter (e.g., FAM-TTATT-BHQ1) [70]
- Equipment: Tube scanner or lateral flow strip reader, heating block or water bath
Procedure:
- Sample Preparation: Extract nucleic acids from clinical samples (swabs, blood, tissue) using silica column-based method. For RNA viruses, include reverse transcription step with specific primers [70].
- Isothermal Amplification: Amplify target sequence using recombinase polymerase amplification (RPA). Prepare 50 μL reaction with TwistAmp Basic kit according to manufacturer instructions. Incubate at 37-42°C for 15-20 minutes [70].
- CRISPR Detection: Prepare Cas12a detection mix containing 50 nM LbaCas12a, 50 nM crRNA, and 100 nM ssDNA reporter in NEBuffer 2.1. Combine 10 μL detection mix with 5 μL amplified product in 96-well plate [70].
- Signal Generation and Readout: Incubate reaction at 37°C for 10-15 minutes. Measure fluorescence using plate reader (Ex/Em: 485/535 nm) or visualize via lateral flow dipstick. Include positive and negative controls in each run [70].
- Data Interpretation: Calculate cutoff threshold based on negative control mean + 3 standard deviations. Samples exceeding threshold are considered positive [70].
Timeline: Complete protocol from sample to result takes approximately 45-60 minutes.
Troubleshooting:
- High background: Titrate crRNA concentration, optimize reporter design
- Low sensitivity: Check RPA primer design, increase amplification time
- Inconsistent results: Standardize sample input, include extraction controls
CRISPR Antimicrobial Mechanism of Action
Diagram 3: CRISPR-phage therapy selectively eliminates bacterial pathogens.
The Scientist's Toolkit: Essential Research Reagents
Table 7: Essential Research Reagents for CRISPR-Cas9 Applications
| Reagent Category | Specific Examples | Key Function | Application Notes |
|---|---|---|---|
| Cas Nucleases | SpCas9, LbCas12a, Cas13a [63] [70] | Target DNA/RNA cleavage | SpCas9 requires NGG PAM; Cas12a targets T-rich PAM; Cas13 cleaves RNA [63] [70] |
| Delivery Systems | LNPs, AAVs, Electroporation [69] [11] | CRISPR component delivery | LNPs optimal for in vivo; AAVs for long-term expression; electroporation for ex vivo [69] [11] |
| Editing Enhancers | HDR enhancers, NHEJ inhibitors [63] | Modulate repair outcomes | Increase HDR efficiency for precise edits; enhance NHEJ for gene knockouts [63] |
| Detection Assays | T7E1, TIDE, NGS [65] | Editing efficiency quantification | NGS most accurate; T7E1 for quick validation; TIDE for smaller indels [65] |
| Cell Culture | IL-2, IL-15, CD3/CD28 beads [65] | T cell activation/expansion | Critical for primary immune cell editing and CAR-T generation [65] |
| gRNA Design Tools | CRISPOR, CHOPCHOP [65] | Guide RNA selection | Optimize for on-target efficiency, minimize off-target effects [65] |
The field of genome editing has been revolutionized by the CRISPR-Cas system, but traditional CRISPR-Cas9 approaches face significant limitations for therapeutic applications requiring precise nucleotide changes. These methods rely on creating double-strand breaks (DSBs) in DNA, which are primarily repaired by error-prone non-homologous end joining (NHEJ), often resulting in a high frequency of insertions and deletions (indels) [72]. While homology-directed repair (HDR) can achieve precise edits, this pathway is inefficient, restricted to dividing cells, and often outperformed by NHEJ, making precise editing challenging in therapeutically relevant cell types [72] [73].
To overcome these limitations, two groundbreaking technologies have emerged: base editing and prime editing. These next-generation editors enable precise genome modifications without requiring DSBs or donor DNA templates, significantly expanding the toolbox for precise genetic manipulation [72] [73]. Base editing, first introduced in 2016, allows direct conversion of one DNA base to another through chemical deamination [74]. Prime editing, developed in 2019, provides even greater versatility by performing all types of point mutations, small insertions, and deletions through a "search-and-replace" mechanism [75] [76]. These technologies are particularly valuable for correcting pathogenic mutations, with base editors potentially addressing over 25% of known pathogenic single-nucleotide polymorphisms (SNPs), and prime editors theoretically capable of correcting up to 89% of disease-associated genetic variants [72].
Base Editing Technology
Molecular Mechanism and Architecture
Base editors are sophisticated protein complexes that combine a catalytically impaired Cas protein with a nucleotide-modifying enzyme. The core architecture consists of a Cas9 nickase (nCas9) that contains a single active cutting domain, fused to a deaminase enzyme that catalyzes chemical conversion of nucleobases [73]. Unlike traditional CRISPR-Cas9 that creates double-strand breaks, base editors leverage the Cas component primarily for programmable DNA binding rather than cleavage, positioning the deaminase domain precisely at the target nucleotide [74].
Two primary classes of base editors have been developed: Cytosine Base Editors (CBEs) and Adenine Base Editors (ABEs). CBEs incorporate a cytidine deaminase enzyme (such as APOBEC1) that converts cytosine (C) to uracil (U), which is subsequently processed by cellular repair machinery to thymine (T), effectively achieving C•G to T•A conversions [72] [73]. To prevent uracil excision and improve editing efficiency, CBEs typically include a uracil glycosylase inhibitor (UGI) [73]. ABEs utilize an engineered tRNA adenosine deaminase (TadA) that catalyzes the deamination of adenine (A) to inosine (I), which is read as guanine (G) by DNA polymerases, resulting in A•T to G•C conversions [72] [73]. The editing activity occurs within a defined "editing window" of approximately 4-5 nucleotides in the spacer region where the single-stranded DNA is accessible to the deaminase enzyme [77].
Advanced Base Editor Engineering
Recent engineering efforts have focused on improving the precision and reducing unwanted editing byproducts of base editors. A significant challenge has been the phenomenon of bystander editing, where multiple editable nucleotides within the activity window undergo unintended conversion [77]. For ABE8e, the most efficient ABE variant to date, the editing window spans approximately 10 base pairs, increasing the likelihood of bystander edits [77]. Approximately 82.3% of human disease-associated mutations correctable by ABEs are located in regions containing multiple adenines, highlighting the clinical significance of this limitation [77].
Recent breakthroughs in 2025 have addressed this challenge through innovative protein engineering. Researchers developed TadA-NW1, an engineered deoxyadenosine deaminase that incorporates a naturally occurring oligonucleotide binding module into the active center of TadA-8e [77]. This modification enhances binding specificity with the DNA nontarget strand, resulting in a substantially narrowed editing window of just four nucleotides (protospacer positions 4-7) compared to the 10-bp window of ABE8e [77]. The resulting editor, ABE-NW1, maintains robust on-target editing efficiency while reducing bystander editing by up to 97.1-fold at certain genomic sites and demonstrating significantly decreased Cas9-dependent and independent off-target activity [77].
Table 1: Comparison of Base Editing Platforms
| Editor Type | Base Conversion | Key Components | Editing Window | Therapeutic Potential |
|---|---|---|---|---|
| Cytosine Base Editor (CBE) | C•G to T•A | nCas9 + cytidine deaminase + UGI | 4-5 nucleotides | Corrects ~25% of pathogenic SNPs [72] |
| Adenine Base Editor (ABE) | A•T to G•C | nCas9 + engineered TadA | 4-5 nucleotides | Corrects ~25% of pathogenic SNPs [72] |
| ABE8e | A•T to G•C | nCas9 + TadA-8e | ~10 nucleotides | High efficiency but increased bystander editing [77] |
| ABE-NW1 | A•T to G•C | nCas9 + TadA-NW1 | 4 nucleotides | Reduced bystander editing; improved specificity [77] |
| CGBE | C•G to G•C | nCas9 + cytidine deaminase + UGI + additional enzymes | Varies | Expands transversion capabilities [77] |
| ACBE | A•T to C•G | nCas9 + engineered deaminase | Varies | Expands transversion capabilities [77] |
Diagram 1: Base Editing Mechanism. The base editor complex binds DNA via sgRNA guidance, recognizing a PAM sequence. Deamination occurs within a defined editing window, with potential bystander edits at non-target nucleotides within this window.
Prime Editing Technology
Molecular Mechanism and Architecture
Prime editing represents a revolutionary advance in precision genome editing by enabling virtually all possible types of DNA substitutions, small insertions, and deletions without requiring double-strand breaks or donor DNA templates [75] [78]. The technology functions as a "search-and-replace" system that directly writes new genetic information into a target DNA site [76].
A prime editor consists of two key components: (1) the prime editor protein, a fusion of a Cas9 nickase (H840A) with an engineered reverse transcriptase (RT), and (2) a specialized prime editing guide RNA (pegRNA) [75] [78]. The pegRNA serves dual functions: it contains a spacer sequence that directs the complex to the target genomic locus, and an extension that encodes the desired edit through a reverse transcription template (RTT) and primer binding site (PBS) [75] [76].
The prime editing process occurs through several coordinated steps. First, the prime editor complex binds to the target DNA sequence specified by the pegRNA spacer. The Cas9 nickase then creates a single-strand nick in the DNA, exposing a 3'-hydroxyl group that serves as a primer for reverse transcription [75] [78]. The reverse transcriptase uses the RTT region of the pegRNA as a template to synthesize a new DNA strand containing the desired edit. This creates a branched DNA intermediate with both the original unedited strand and the newly synthesized edited strand [78]. Cellular repair mechanisms then resolve this intermediate by removing the unedited 5' flap and ligating the edited 3' flap into the genome, resulting in permanent incorporation of the genetic modification [75] [78].
Prime Editor Evolution and Optimization
Since the initial development of PE1, prime editors have undergone substantial optimization to improve editing efficiency and specificity. PE2 incorporated an engineered reverse transcriptase with enhanced processivity and DNA-RNA hybrid affinity, significantly improving editing efficiency [75] [78]. PE3 further increased efficiency by incorporating an additional sgRNA that nicks the non-edited DNA strand, encouraging the cellular repair machinery to use the edited strand as a template [75] [78].
More recent versions have addressed additional limitations. PE4 and PE5 incorporate dominant-negative MLH1 (MLH1dn) to inhibit mismatch repair pathways that often reverse prime edits, increasing efficiency from 50-70% (PE4) to 60-80% (PE5) in HEK293T cells [78]. PE6 introduced compact RT variants and enhanced Cas9 variants combined with engineered pegRNAs (epegRNAs), achieving 70-90% editing efficiency [78]. PE7 further improved performance by fusing the prime editor complex with La protein, enhancing pegRNA stability and editing outcomes in challenging cell types [78].
pegRNA engineering has been particularly important for improving prime editing efficiency. Initial pegRNAs suffered from degradation due to their extended length, leading to the development of engineered pegRNAs (epegRNAs) with stabilizing secondary structures at their 3' end, such as evopreQ and mpknot motifs [75]. These modifications improve editing efficiency by 3-4-fold across multiple human cell lines and primary human fibroblasts without increasing off-target effects [75]. Additional innovations include the development of split prime editors (sPE) that separate nCas9 and RT components to facilitate delivery, and zip-editing approaches that enhance editing rates through optimized designs [75].
Table 2: Evolution of Prime Editing Systems
| Editor Version | Key Improvements | Typical Efficiency in HEK293T | Applications and Advantages |
|---|---|---|---|
| PE1 | Initial proof-of-concept | ~10-20% | Demonstrated search-and-replace editing [78] |
| PE2 | Engineered reverse transcriptase | ~20-40% | Improved processivity and stability [78] |
| PE3/PE3b | Additional nicking sgRNA | ~30-50% | Enhanced editing efficiency via strand bias [78] |
| PE4 | MLH1dn to inhibit MMR | ~50-70% | Reduced repair-mediated reversal [78] |
| PE5 | MLH1dn + additional nicking sgRNA | ~60-80% | Further improved efficiency and precision [78] |
| PE6 | Compact RT variants, enhanced Cas9, epegRNAs | ~70-90% | Better delivery, reduced degradation [78] |
| PE7 | La protein fusion for stability | ~80-95% | Enhanced outcomes in challenging cells [78] |
| Cas12a PE | Cas12a nickase, circular pegRNA | Up to 40.75% | Smaller size, T-rich PAM targeting [78] |
Diagram 2: Prime Editing Mechanism. The prime editor complex binds DNA via pegRNA guidance, creates a single-strand nick, and uses reverse transcription to synthesize DNA containing desired edits, which are then incorporated via cellular repair mechanisms.
Comparative Analysis of Editing Technologies
Technical Performance Metrics
When selecting genome editing technologies for specific applications, researchers must consider multiple performance characteristics. Traditional CRISPR-Cas9 nucleases excel at gene disruption but produce unpredictable indels and have higher rates of off-target effects due to double-strand break formation [72] [73]. Base editors offer higher precision for transition mutations with significantly reduced indel formation, but are limited to specific base conversions and suffer from bystander editing within their activity window [72] [77]. Prime editors provide the broadest editing capabilities, including all transition and transversion mutations, small insertions, and deletions, with exceptionally high precision and minimal off-target effects, though often with lower efficiency than base editors [75] [78].
Editing efficiency varies substantially across cell types and genomic contexts. Base editors typically achieve higher editing rates (often 30-70% in amenable cell types) but are constrained by sequence context and editing window limitations [77]. Prime editing efficiencies range widely from 10-90% depending on the specific editor version, pegRNA design, and target locus, with newer generations (PE6, PE7) consistently achieving higher efficiencies [78]. Both technologies are continually being optimized through protein engineering and improved guide RNA designs to expand their targeting scope and efficiency.
Table 3: Performance Comparison of Genome Editing Technologies
| Characteristic | CRISPR-Cas9 Nuclease | Base Editing | Prime Editing |
|---|---|---|---|
| Editing Types | Indels (NHEJ), precise edits with HDR template | C•G to T•A (CBE), A•T to G•C (ABE), some transversions | All 12 base-to-base conversions, insertions, deletions |
| DSB Formation | Yes, required for activity | No | No |
| Donor DNA Required | For HDR-mediated editing | No | No |
| Typical Efficiency | High for indels (often >70%), low for HDR (<10%) | Moderate to high (30-70%) | Variable (10-90%), version-dependent [78] |
| Off-Target Effects | Higher (DSB-dependent) | Reduced, but Cas-independent RNA/DNA off-targets possible | Lowest (requires 3 hybridization events) [75] |
| Bystander Editing | Not applicable | Yes, within editing window | Minimal [75] |
| Theoretical Coverage of Pathogenic SNPs | All mutations | ~25% [72] | Up to 89% [72] |
| Key Limitations | Unpredictable indels, low HDR efficiency, restricted to dividing cells for HDR | Restricted to specific base changes, bystander editing, sequence context dependence | Efficiency varies by locus, large size challenges delivery |
Application-Specific Recommendations
The choice of genome editing technology should be guided by the specific research or therapeutic objective. For gene knockout studies, traditional CRISPR-Cas9 remains the most efficient and straightforward approach. For disease modeling or therapeutic correction of point mutations that match base editing capabilities (C•G to T•A or A•T to G•C), base editors offer higher efficiency and simpler implementation. However, for mutations involving transversions (C•G to G•C, A•T to C•G, etc.), insertions, deletions, or when maximal precision is required without bystander editing, prime editors are the superior choice despite potentially lower efficiency.
Recent advances have expanded the capabilities of both platforms. For base editing, the development of ABE-NW1 with its narrowed editing window makes it particularly valuable for correcting disease mutations in sequence contexts with multiple editable nucleotides [77]. For prime editing, the evolution through PE6 and PE7 systems has substantially improved efficiency while maintaining high precision [78]. The emergence of Cas12a-based prime editors offers alternative PAM preferences and smaller size for improved delivery [78].
Experimental Protocols
Base Editing Workflow for Precision Correction
Protocol: ABE-NW1 Mediated Correction of CFTR W1282X Mutation
This protocol describes the application of the novel ABE-NW1 base editor for precise correction of the CFTR W1282X mutation, one of the most common cystic fibrosis-causing mutations, in a lung epithelial cell model [77].
Materials and Reagents:
- ABE-NW1 expression plasmid (Addgene #XXXXX)
- sgRNA expression vector targeting CFTR W128X locus
- Human lung epithelial cells (e.g., CFBE41o-)
- Transfection reagent (Lipofectamine 3000 or similar)
- Genomic DNA extraction kit
- PCR reagents for amplification of target locus
- Next-generation sequencing library preparation kit
- T7 Endonuclease I (for quick efficiency assessment)
Procedure:
- sgRNA Design and Cloning: Design sgRNA with target adenine positioned within protospacer positions 4-7, counting PAM as positions 21-23 [77]. Clone sgRNA expression cassette into ABE-NW1 plasmid or co-expression vector.
- Cell Culture and Transfection: Culture lung epithelial cells in appropriate medium. At 70-80% confluence, transfect with ABE-NW1 and sgRNA plasmids using lipid-based transfection. Include untransfected and sgRNA-only controls.
- Harvest and Analysis: Harvest cells 72 hours post-transfection. Extract genomic DNA using commercial kits.
- Editing Efficiency Assessment: Amplify target region by PCR and analyze by:
- Next-generation sequencing (most accurate): Prepare sequencing libraries and sequence on Illumina platform to quantify A•T to G•C conversion rates and bystander editing.
- T7E1 assay (quick assessment): Hybridize PCR products, digest with T7 Endonuclease I, and analyze by gel electrophoresis.
- Functional Validation: For corrected clones, assess CFTR function by membrane localization and chloride channel activity assays.
Troubleshooting:
- Low editing efficiency: Optimize sgRNA positioning to place target adenine within optimal window (positions 4-7).
- High bystander editing: Verify ABE-NW1 sequence and consider further narrowing editing window with additional protein engineering.
- Cell toxicity: Reduce plasmid amounts and include viability controls.
Protocol: PE6-Mediated Installation of Oncogenic Mutations
This protocol describes using PE6 systems for precise installation of oncogenic mutations in cell lines for functional studies, based on the competitive precision genome editing approach [79].
Materials and Reagents:
- PE6 expression plasmid (Addgene #XXXXX)
- pegRNA design and expression system
- Target cell line (e.g., HAP1, KBM-7, or other relevant models)
- Transfection reagents
- Genomic DNA extraction kit
- High-fidelity PCR enzymes
- Next-generation sequencing platform
- Flow cytometry equipment (if including selection markers)
Procedure:
- pegRNA Design: Design pegRNA with:
- Spacer sequence (20 nt) complementary to target site
- Reverse transcription template (RTT, 25-40 nt) encoding desired edit plus homology arm
- Primer binding site (PBS, 10-15 nt) complementary to nicked genomic DNA
- Consider engineered pegRNA (epegRNA) designs with 3' stability motifs [75]
- Vector Assembly: Clone pegRNA into appropriate expression vector compatible with PE6 protein expression.
- Cell Transfection: Transfect target cells with PE6 and pegRNA plasmids. For competitive editing approaches, include multiple repair templates with sequence tags for lineage tracing [79].
- Editing Validation: Harvest cells 96 hours post-transfection. Extract genomic DNA and amplify target locus with barcoded primers for NGS.
- Efficiency Quantification: Analyze sequencing data for precise edit incorporation, calculating efficiency as percentage of reads containing desired edit without byproducts.
- Functional Assessment: For successful edits, isolate single-cell clones and validate functional consequences of mutations through phenotype-specific assays.
Optimization Tips:
- Test multiple pegRNA designs for each target, varying PBS length (10-15 nt) and RTT length.
- Co-express MLH1dn to inhibit mismatch repair and improve efficiency [78].
- For difficult targets, consider dual-pegRNA strategies or the PE7 system with La fusion protein [78].
Research Reagent Solutions
Table 4: Essential Reagents for Base and Prime Editing Research
| Reagent Category | Specific Examples | Function and Applications | Key Considerations |
|---|---|---|---|
| Base Editor Plasmids | ABE8e, ABE-NW1, BE4max, Target-AID | Express base editor proteins in target cells | Choose based on editing type (CBE vs ABE) and specificity requirements [77] |
| Prime Editor Systems | PE2, PE3, PE6, PE7, Cas12a-PE | Express prime editor proteins with reverse transcriptase activity | Later versions (PE6/PE7) offer higher efficiency but larger size [78] |
| Guide RNA Systems | sgRNA for BEs, pegRNA for PEs | Target editors to specific genomic loci | pegRNAs require specialized design with PBS and RTT elements [76] |
| Delivery Vehicles | AAV vectors, lipid nanoparticles (LNPs), electroporation | Deliver editing components to target cells | Consider size constraints (AAV ~4.7kb) and cell type compatibility [72] [11] |
| Validation Tools | T7E1 assay, next-generation sequencing, digital PCR | Assess editing efficiency and specificity | NGS provides most comprehensive analysis of on-target and bystander editing [80] |
| Cell Lines | HEK293T, HAP1, KBM-7, iPSCs, primary cells | Model systems for editing optimization | Choosing therapeutically relevant cells is crucial for translational research |
| Efficiency Enhancers | MLH1dn, Rad51 inhibitors, La protein | Improve editing rates by modulating DNA repair | Particularly valuable for prime editing systems [78] |
Clinical Applications and Future Perspectives
The therapeutic potential of base editing and prime editing is rapidly being realized in clinical development. Base editors have shown remarkable success in clinical trials for hereditary transthyretin amyloidosis (hATTR), where an LNP-delivered ABE reduced disease-causing TTR protein levels by approximately 90% in study participants, with sustained response over two years [11]. Similarly, ABE-mediated knockdown of kallikrein for hereditary angioedema (HAE) reduced attack frequency, with 8 of 11 participants in the high-dose group remaining attack-free during the 16-week study period [11].
Prime editing, though earlier in development, has demonstrated compelling preclinical results. Correction of the sickle cell disease mutation in patient-derived stem cells achieved 40% correction rates, with potential for long-term clinical efficacy in mouse models [74]. The technology's versatility was further highlighted by the first personalized in vivo CRISPR therapy for an infant with CPS1 deficiency, developed and delivered in just six months, establishing a regulatory precedent for rapid approval of bespoke genomic medicines [11].
Future development will focus on addressing remaining challenges, particularly in delivery efficiency and editing precision. For base editing, further narrowing of editing windows and reducing Cas-independent off-target activity remain priorities [77]. For prime editing, improving efficiency across diverse genomic contexts and cell types, while minimizing the large size of editing constructs for delivery, are key focus areas [75] [78]. The emergence of novel delivery platforms, including engineered lipid nanoparticles and viral vectors, will be crucial for expanding the therapeutic reach of these technologies beyond currently accessible tissues [11].
As the field progresses, base editing and prime editing are poised to transform therapeutic development for genetic diseases, enabling precise correction of pathogenic mutations with minimized risk of genotoxic effects. These technologies represent a paradigm shift in precision medicine, offering unprecedented capabilities to rewrite the human genome with growing sophistication and safety.
Mitigating Off-Target Effects and Optimizing Editing Efficiency
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 system has revolutionized biochemical research by enabling precise, programmable genome editing. This ribonucleoprotein complex, consisting of a Cas9 nuclease and a single guide RNA (sgRNA), creates site-specific double-strand breaks (DSBs) in DNA by recognizing sequences adjacent to a protospacer adjacent motif (PAM) [81] [19]. Despite its transformative potential, a significant challenge persists: off-target effects, where unintended genomic locations are cleaved, leading to potentially adverse outcomes [81] [12]. For researchers and drug development professionals, understanding and mitigating these effects is paramount to ensuring experimental validity and therapeutic safety.
The clinical relevance of off-target effects was highlighted during the development and regulatory review of Casgevy (exa-cel), the first CRISPR-based therapy approved to treat sickle cell disease. Regulatory agencies like the FDA emphasized thorough characterization of off-target editing, noting that patients with rare genetic variants might face higher risks [82]. This underscores the critical need for comprehensive off-target assessment in preclinical and clinical development pipelines. The consequences of off-target editing range from confounding experimental results in functional genomics studies to potentially initiating oncogenic transformations if tumor suppressor genes are disrupted [82] [83].
Mechanisms and Causes of Off-Target Editing
Fundamental Mechanisms
Off-target effects primarily occur due to the biochemical promiscuity of the Cas9-sgRNA complex. While designed for perfect complementarity between the sgRNA spacer and target DNA, the system can tolerate mismatches, bulges, and base-pairing imperfections under specific conditions [12] [19]. The wild-type Streptococcus pyogenes Cas9 (SpCas9) can tolerate between three and five base pair mismatches while still catalyzing cleavage, creating potential off-target sites across the genome [82].
The position of mismatches significantly influences their tolerance. The seed sequence (8-10 bases at the 3' end of the gRNA targeting sequence) is particularly critical for target recognition [19]. Mismatches in this PAM-proximal region typically inhibit target cleavage, while those toward the 5' end (PAM-distal) are more readily tolerated [81] [19]. This positional tolerance varies among different Cas enzymes and engineered variants, influencing their overall specificity profiles.
Contributing Factors
Multiple factors influence off-target activity beyond simple sequence homology:
- Guide RNA secondary structure: The structural conformation of sgRNA can affect its binding specificity and efficiency [12].
- Chromatin accessibility and epigenetic context: Nuclear microenvironment factors such as histone modifications, DNA methylation, and chromatin organization influence Cas9 binding and cleavage efficiency [81] [84].
- Cellular delivery method and persistence: The format of CRISPR cargo (plasmid DNA, mRNA, or ribonucleoprotein) and delivery vehicle affect how long editing components remain active, with prolonged exposure increasing off-target risks [82].
- Enzyme concentration: Higher cellular concentrations of Cas9 can increase the likelihood of off-target binding and cleavage [12].
- Cellular repair pathway dynamics: The balance between non-homologous end joining (NHEJ) and homology-directed repair (HDR) pathways influences the mutational outcomes at both on-target and off-target sites [81].
Figure 1: Mechanisms and contributing factors to CRISPR off-target effects. The seed region near the PAM site is particularly critical for specificity.
Detection and Analysis Methods
In Silico Prediction Tools
Computational prediction represents the first line of defense against off-target effects. These tools identify potential off-target sites during experimental design, enabling selection of optimal sgRNAs. The field has evolved from simple alignment-based algorithms to sophisticated deep learning approaches.
Table 1: Classification of CRISPR Off-Target Prediction Tools
| Category | Representative Tools | Key Features | Limitations |
|---|---|---|---|
| Alignment-Based | Cas-OFFinder, CHOPCHOP, CasOT [81] [85] | Genome-wide scanning with adjustable PAM and mismatch parameters; fast processing | Bias toward sgRNA-dependent effects; insufficient epigenetic consideration |
| Scoring-Based | MIT, CCTop, CROP-IT, CFD [81] [84] | Position-specific mismatch weighting; experimentally validated datasets | Limited generalization across diverse sequences |
| Energy-Based | CRISPRoff [84] | Models binding energy of Cas9-gRNA-DNA complex | Computational intensity; approximation requirements |
| Learning-Based | DeepCRISPR, CCLMoff, CRISPR-Net [81] [84] | Automatic feature extraction from comprehensive datasets; superior performance | Training data requirements; computational complexity |
Recent advances in deep learning have significantly improved prediction accuracy. The CCLMoff framework, introduced in 2025, incorporates a pretrained RNA language model and demonstrates strong generalization across diverse next-generation sequencing (NGS)-based detection datasets [84]. This approach captures mutual sequence information between sgRNAs and target sites, with model interpretation confirming the biological importance of the seed region.
Experimental Detection Methods
Experimental validation remains essential for comprehensive off-target assessment. These methods can be categorized based on their detection focus and cellular context.
Table 2: Experimental Methods for Detecting CRISPR Off-Target Effects
| Method | Detection Principle | Cellular Context | Advantages | Limitations |
|---|---|---|---|---|
| GUIDE-seq [81] [82] | DSB capture via dsODN integration | In vivo | Highly sensitive; low false positive rate | Limited by transfection efficiency |
| CIRCLE-seq [81] [84] | Circularized DNA cleavage detection | In vitro (cell-free) | Highly sensitive; minimal background | May miss in vivo contextual factors |
| DISCOVER-seq [81] [84] | MRE11 DNA repair factor recruitment | In vivo | Highly sensitive; captures cellular repair context | Potential false positives |
| Digenome-seq [81] | Cas9-digested genomic DNA sequencing | In vitro (cell-free) | Highly sensitive; comprehensive | Expensive; requires high sequencing coverage |
| Whole Genome Sequencing [81] [83] | Comprehensive genome analysis | In vivo | Unbiased; detects chromosomal abnormalities | Costly; computationally intensive |
| ChIP-seq [81] | dCas9 binding site mapping | In vivo | Identifies binding sites genome-wide | Does not distinguish cleavage from binding |
Figure 2: Experimental workflow for CRISPR off-target detection, spanning computational prediction to comprehensive genomic analysis.
Detailed Protocol: GUIDE-seq for Off-Target Detection
Principle: GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) utilizes double-stranded oligodeoxynucleotides (dsODNs) that integrate into DSBs via the NHEJ pathway, enabling genome-wide identification of off-target sites [81] [82].
Materials:
- dsODN tag (24-bp with phosphorothioate modifications on 5' and 3' ends)
- Cas9 protein and in vitro transcribed sgRNA or plasmid expression systems
- Cells amenable to transfection (HEK293T, U2OS, or other relevant models)
- PCR reagents and NGS library preparation kit
- Bioinformatics pipeline for GUIDE-seq data analysis
Procedure:
- Transfection: Co-transfect cells with Cas9-sgRNA complex and dsODN tag using an appropriate method (lipofection, electroporation). Include untransfected controls.
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract genomic DNA using a method that yields high-molecular-weight DNA.
- Library Preparation:
- Fragment DNA to ~500 bp using a focused-ultrasonicator.
- Repair ends and ligate sequencing adaptors.
- Perform PCR amplification with primers specific to the dsODN tag and sequencing adaptors.
- Sequencing and Analysis:
- Sequence libraries on an appropriate NGS platform (Illumina recommended).
- Align sequences to the reference genome.
- Identify dsODN integration sites using dedicated GUIDE-seq analysis software.
- Compare integration sites with predicted off-target loci from in silico tools.
Troubleshooting Notes:
- Low dsODN integration efficiency can be improved by optimizing transfection conditions and dsODN concentration.
- High background may indicate inadequate controls or PCR amplification bias.
- Validation of top candidate off-target sites via targeted sequencing is recommended.
Strategies for Minimizing Off-Target Effects
CRISPR System Engineering
High-Fidelity Cas Variants: Several engineered Cas9 variants with enhanced specificity have been developed. These include:
- eSpCas9(1.1): Weakened interactions between Cas9 and the non-target DNA strand reduce off-target editing [19].
- SpCas9-HF1: Disrupted interactions with the DNA phosphate backbone enhance discrimination against mismatched targets [19] [83].
- HypaCas9: Improved proofreading capability increases discrimination between perfectly matched and mismatched targets [19] [83].
- evoCas9: Engineered through directed evolution for reduced off-target effects while maintaining on-target activity [19] [83].
A 2022 study comparing wildtype Cas9 and a high-fidelity variant in chicken primordial germ cells demonstrated significantly improved performance with the high-fidelity version, which achieved 69% deletion efficiency compared to 29% with wildtype Cas9 [86].
Alternative Cas Enzymes: Beyond SpCas9, other naturally occurring or engineered nucleases such as Cas12a (Cpf1) offer different PAM requirements and potentially reduced off-target profiles [81] [82]. Additionally, Cas9 nickases (Cas9n) that create single-strand breaks instead of DSBs can be used in pairs to enhance specificity, as this requires two proximal binding events for DSB formation [19].
Experimental Design Optimization
Guide RNA Design Considerations:
- Select sgRNAs with minimal off-target potential using multiple prediction algorithms
- Prioritize sgRNAs with higher GC content in the seed region, which stabilizes the DNA:RNA duplex [82]
- Consider truncated sgRNAs (17-18 nt instead of 20 nt) to reduce off-target activity while potentially maintaining on-target efficiency [82]
- Incorporate chemical modifications such as 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bonds (PS) to reduce off-target edits and increase on-target efficiency [82]
Delivery Method Optimization:
- Utilize ribonucleoprotein (RNP) complexes rather than plasmid-based expression to reduce the duration of nuclease activity [82]
- Optimize delivery conditions to minimize cargo amount while maintaining editing efficiency
- Consider the timing of analysis to capture both immediate and delayed off-target effects
Table 3: Research Reagent Solutions for Off-Target Assessment
| Reagent Type | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| High-Fidelity Cas9 Variants | SpCas9-HF1, eSpCas9(1.1), HypaCas9, evoCas9 [19] [83] [86] | Reduced off-target cleavage while maintaining on-target activity | Engineered protein structures with enhanced specificity |
| Alternative Cas Enzymes | Cas12a (Cpf1), Cas9 nickase [81] [19] | Different PAM requirements; reduced off-target profiles | Cas12a utilizes T-rich PAM; nickase requires dual guides for DSBs |
| Chemically Modified gRNAs | 2'-O-methyl analogs, 3' phosphorothioate bonds [82] | Enhanced stability and reduced off-target effects | Improved nuclease resistance and binding specificity |
| Detection Kits | GUIDE-seq, CIRCLE-seq, DISCOVER-seq reagents [81] [84] | Experimental identification of off-target sites | Specialized reagents for genome-wide off-target mapping |
| Prediction Software | CCLMoff, Cas-OFFinder, CRISPOR [81] [85] [84] | In silico off-target nomination during guide design | Algorithm-based risk assessment before experiments |
| Analysis Tools | CRISPResso, ICE (Inference of CRISPR Edits) [85] [82] | Computational analysis of editing efficiency and specificity | Processing of NGS data for quantitative assessment |
The advancing clinical application of CRISPR-based therapies necessitates increasingly sophisticated approaches to off-target assessment. While current methods provide robust detection capabilities, emerging technologies promise enhanced sensitivity and comprehensiveness. The integration of deep learning approaches like CCLMoff represents a significant step toward predictive accuracy, potentially enabling preemptive off-target minimization during guide design [84].
For biochemical researchers and drug development professionals, a multipronged strategy combining computational prediction, careful experimental design, and empirical validation remains essential. The selection of appropriate Cas variants, guide designs, and detection methods should be guided by specific application requirements, with therapeutic applications demanding the most stringent approaches. As the field progresses toward "PAM-less" Cas variants with expanded targeting ranges and enhanced specificities, the fundamental goal remains unchanged: achieving precise genetic modifications without compromising genomic integrity [19].
The continued evolution of CRISPR technology will undoubtedly yield increasingly sophisticated solutions to the challenge of off-target effects, further establishing genome editing as a cornerstone of biochemical research and therapeutic development.
CRISPR-Cas9 genome editing has revolutionized biochemical research, yet its therapeutic application has been constrained by off-target effects—unintended cleavages at genomic sites with sequence similarity to the intended target. These off-target mutations can confound experimental results and pose significant safety risks in therapeutic contexts [81]. The fundamental mechanism behind these effects lies in the Cas9-sgRNA complex's ability to tolerate mismatches between the guide RNA and target DNA, particularly in the PAM-distal region [87]. To address this critical limitation, researchers have developed high-fidelity Cas9 variants through rational protein engineering. These engineered nucleases maintain robust on-target activity while dramatically reducing off-target effects by minimizing non-specific DNA contacts during the recognition process [88] [89]. This application note details the mechanisms, performance characteristics, and implementation protocols for these high-fidelity variants, providing biochemical researchers with essential tools for achieving precise genome editing.
Mechanisms of Off-Target Reduction
Structural Basis for Improved Specificity
High-fidelity Cas9 variants operate on the principle of reducing non-specific DNA contacts while preserving essential interactions for on-target recognition. Structural studies reveal that wild-type SpCas9 forms multiple hydrogen bonds with the target DNA phosphate backbone through residues including N497, R661, Q695, and Q926 [89]. These extensive contacts provide excess binding energy that enables tolerance of mismatched off-target sites. The strategic approach to engineering high-fidelity variants involves disrupting a subset of these non-specific contacts while maintaining those crucial for recognizing perfectly matched target sequences [88] [87].
Cryo-EM structural analyses of Cas9-sgRNA-DNA ternary complexes have revealed that Cas9 activation involves a conformational transition from a linear to a kinked duplex conformation in the target DNA-sgRNA hybrid. This transition is essential for proper positioning of the HNH nuclease domain and subsequent DNA cleavage [87]. Mismatches in the PAM-distal region, particularly between positions 12-17, inhibit this conformational transition and HNH docking, thereby preventing cleavage. High-fidelity variants exploit this mechanism by requiring more precise complementarity to achieve the fully active conformation [87].
Engineering Strategies for Enhanced Fidelity
The primary engineering strategy for developing SpCas9-HF1 ("High-Fidelity 1") involved systematic alanine substitution of key DNA-binding residues. Researchers created 15 variants with single, double, triple, and quadruple alanine substitutions at positions N497, R661, Q695, and Q926 [89]. Initial screening against mismatched target sites revealed that the quadruple mutant (N497A/R661A/Q695A/Q926A) and one triple mutant (R661A/Q695A/Q926A) exhibited the greatest discrimination against off-target sites while maintaining on-target activity, leading to the selection of the quadruple mutant as SpCas9-HF1 [89].
Additional high-fidelity variants employ complementary strategies. For instance, eSpCas9(1.1) was engineered to neutralize positive charges in the non-target DNA strand groove, reducing non-specific interactions with the DNA backbone [90]. These variants collectively demonstrate that strategic weakening of non-essential DNA contacts can raise the energy threshold for cleavage, thereby enforcing stricter complementarity requirements while preserving on-target efficiency [88] [89] [90].
Diagram Title: Mechanism of High-Fidelity Cas9 Variants
Quantitative Comparison of High-Fidelity Cas9 Variants
Performance Metrics of SpCas9-HF1
SpCas9-HF1 represents a breakthrough in specificity optimization. Comprehensive testing across 37 different sgRNAs targeting both reporter genes and endogenous human loci demonstrated that SpCas9-HF1 retains ≥70% of wild-type on-target activity for 86% (32/37) of sgRNAs tested [89]. In many cases, the variant exhibited activities between 90-140% of wild-type SpCas9, indicating not just preservation but occasional enhancement of editing efficiency at certain targets [89].
The most striking improvement lies in off-target reduction. Genome-wide assessment using GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing) revealed that for six of eight sgRNAs tested, SpCas9-HF1 rendered all off-target events undetectable [88] [89]. In these experiments, wild-type SpCas9 induced 2-25 off-target sites per sgRNA, while SpCas9-HF1 eliminated nearly all such events. For the remaining two sgRNAs, only a single off-target site was detected with SpCas9-HF1—one that contained just a single mismatch in the protospacer seed sequence [89]. Deep sequencing validation confirmed that indel frequencies at 34 of 36 previously identified off-target sites were reduced to background levels with SpCas9-HF1 [89].
Table 1: Performance Comparison of High-Fidelity Cas9 Variants
| Variant | On-Target Efficiency (% of WT) | Off-Target Reduction | Key Mutations | Applications Demonstrated |
|---|---|---|---|---|
| SpCas9-HF1 | >85% of sgRNAs show ≥70% of WT activity [89] | All or nearly all off-target events eliminated for standard non-repetitive targets [88] | N497A, R661A, Q695A, Q926A [89] | Gene knockout, therapeutic targeting of KRAS mutations [91] |
| eSpCas9(1.1) | Data not fully available in sources | Improved specificity | Charge-neutralizing mutations in NT-groove [90] | General genome editing |
| SuperFi-Cas9 | Near wild-type cleavage efficiency [87] | Extreme-low mismatch rates [87] | Engineered based on structural mismatch recognition [87] | Preclinical development |
| HiFiCas9 | Maintained high efficiency in KRAS targeting [91] | Single-nucleotide discrimination between KRAS mutant and wild-type alleles [91] | Proprietary high-fidelity mutations | Specific oncogene targeting |
Therapeutic Application: KRAS Mutation Targeting
Recent therapeutic applications demonstrate the critical importance of high-fidelity variants in discriminating between single-nucleotide differences. Researchers developed a CRISPR-HiFiCas9 strategy to specifically target oncogenic KRASG12C and KRASG12D mutations while preserving the wild-type KRAS allele [91]. These mutations differ by only a single nucleotide from the wild-type sequence, presenting an extreme challenge for specificity.
The HiFiCas9 system successfully distinguished mutant from wild-type KRAS alleles across multiple cell lines and in genetically engineered mouse embryonic fibroblast models [91]. Deep sequencing analysis confirmed specific disruption of mutant KRAS open reading frames without affecting the wild-type version, demonstrating single-nucleotide discrimination capability [91]. This precision enabled significant tumor growth suppression in preclinical non-small cell lung cancer models, highlighting the therapeutic potential of high-fidelity editing systems [91].
Table 2: Detection Methods for CRISPR-Cas9 Off-Target Effects
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| GUIDE-seq [81] [90] | Captures DSBs with double-stranded oligonucleotides followed by sequencing | Highly sensitive, low false positive rate, genome-wide | Requires efficient dsODN delivery, potential cytotoxicity |
| Digenome-seq [81] | Cell-free digestion of purified genomic DNA with Cas9 RNP followed by whole genome sequencing | Highly sensitive, in vitro | Expensive, requires high sequencing coverage |
| BLESS/BLISS [81] [90] | Direct in situ capture of DSBs with biotinylated adaptors | Captures breaks in native context, applicable to tissues | Only identifies breaks at time of detection |
| CIRCLE-seq [81] | Circularization of sheared genomic DNA followed by Cas9 digestion and sequencing | Sensitive, in vitro, low background | Does not account for cellular context |
| Computational Prediction (Cas-OFFinder, Off-Spotter) [81] [91] | In silico prediction based on sequence homology and scoring models | Convenient, rapid, inexpensive | Biased toward sgRNA-dependent effects, variable accuracy |
Experimental Protocols for High-Fidelity Cas9 Implementation
Ribonucleoprotein (RNP) Delivery for Optimal Specificity
RNP delivery represents the gold standard for achieving maximal specificity with high-fidelity Cas9 variants. This approach involves pre-complexing the purified Cas9 protein with in vitro-transcribed sgRNA before delivery into cells, leading to rapid editing and reduced off-target effects due to limited persistence time [91] [92].
Protocol: RNP Complex Formation and Delivery
sgRNA Preparation: Design sgRNAs with specificity-checking using Cas-OFFinder or Off-Spotter algorithms [91]. For high-fidelity variants, ensure optimal 5' nucleotide (G or A) to maximize activity [93]. Synthesize sgRNA via in vitro transcription or commercial synthesis.
RNP Complex Assembly:
- Combine 10μL of 60μM purified HiFi Cas9 protein with 10μL of 60μM sgRNA in optimized buffer (e.g., 150mM KCl, 20mM HEPES pH 7.5, 1mM DTT, 1mM MgCl₂).
- Incubate at 37°C for 15 minutes to allow proper RNP formation.
Cell Delivery via Nucleofection (for immortalized cell lines):
- Harvest and count cells, resuspend in appropriate nucleofection solution.
- Mix 100μL cell suspension (1-5×10⁶ cells) with 20μL RNP complex.
- Transfer to nucleofection cuvette and apply recommended program (e.g., CM-130 for HEK293).
- Immediately add pre-warmed culture media and transfer to plates.
Efficiency Validation:
- Assess editing efficiency 48-72 hours post-delivery via T7 Endonuclease I assay or targeted amplicon sequencing.
- Validate specificity through GUIDE-seq or targeted sequencing of predicted off-top sites [91].
Diagram Title: RNP Delivery Workflow
Activity Enhancement for Challenging Targets
Some high-fidelity variants exhibit reduced activity with particular sgRNAs or at challenging genomic loci. To address this limitation, researchers have developed enhancement strategies that boost editing efficiency without compromising specificity.
Protocol: tRNA-sgRNA Fusion System for Enhanced Activity
tRNA-sgRNA Fusion Design:
- Fuse the sgRNA sequence to the 3' end of a tRNAGln sequence using overlap PCR or synthetic gene synthesis.
- Ensure the sgRNA maintains the optimal GN20 format following the tRNA sequence.
Vector Construction:
- Clone the tRNA-sgRNA fusion construct into an appropriate expression vector under a U6 or H1 promoter.
- For maximal efficiency, incorporate nuclear localization signals in the Cas9 expression construct.
Delivery and Processing:
- Co-transfect the tRNA-sgRNA fusion vector with HiFi Cas9 expression plasmid into target cells.
- The endogenous tRNA processing machinery cleaves the fusion transcript, generating mature sgRNA with precise ends.
- This processing enhances Cas9 activity 6- to 8-fold for certain high-fidelity variants while maintaining specificity [93].
Therapeutic Application:
- This system has been successfully applied to correct pathogenic mutations in the retinoschisin 1 (RS1) gene, demonstrating its utility for therapeutic genome editing [93].
Table 3: Research Reagent Solutions for High-Fidelity Genome Editing
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| SpCas9-HF1 Plasmid | High-fidelity nuclease expression | Available from Addgene (www.addgene.org/crispr-cas) [88] |
| HiFi Cas9 Protein | Purified protein for RNP formation | Commercial sources available; essential for RNP delivery protocols |
| tRNA-sgRNA Fusion System | Enhances activity of high-fidelity variants | Boosts efficiency 6- to 8-fold for challenging targets [93] |
| GUIDE-seq Kit | Genome-wide off-target detection | Critical for comprehensive specificity validation [81] |
| Cas-OFFinder Software | Computational off-target prediction | Web-based tool for identifying potential off-target sites [81] [91] |
| Nucleofection System | Physical delivery method for RNPs | Higher efficiency for difficult-to-transfect cells [92] |
High-fidelity Cas9 variants represent a significant advancement in precision genome editing, effectively addressing the critical challenge of off-target effects that has limited therapeutic applications. Through rational engineering of DNA recognition interfaces, these variants maintain robust on-target activity while dramatically reducing off-target cleavages. The implementation of RNP delivery and enhancement strategies like tRNA-sgRNA fusions further optimizes the efficiency and specificity of these systems.
As structural insights into mismatch recognition mechanisms continue to emerge [87], next-generation variants with even greater precision are anticipated. Combined with advanced delivery methods and comprehensive off-target assessment protocols, high-fidelity CRISPR systems are poised to enable new generations of precise genome editing applications in both basic research and clinical therapeutics.
The CRISPR-Cas9 system has revolutionized precise genome editing in biochemistry research; however, its therapeutic application is significantly limited by the inherent mismatch tolerance of Cas9-gRNA complexes, which can lead to unintended off-target effects [94]. Achieving high specificity is particularly crucial for discriminating between wild-type and mutant alleles that differ by only a single nucleotide, a common scenario in modeling genetic diseases and developing targeted therapies [94]. This application note provides a comprehensive framework of strategies and detailed protocols for optimizing gRNA design to enhance editing specificity, reduce mismatch tolerance, and validate editing outcomes for robust biochemical research.
Core Optimization Strategies
ARROW: Strategic Mismatch Incorporation
The ARROW (Allele-specific Recombined gRNA design for Reduced Off-target With enhanced specificity) strategy systematically enhances single-nucleotide discrimination by deliberately introducing intentional mismatches into gRNAs targeting mutant alleles [94]. This approach reduces Cas9 sequence tolerance and enables precise mutant allele editing while avoiding cleavage of the corresponding wild-type allele.
Key Design Principles:
- Introduce mismatches at specific positions within the gRNA seed region (-1, -3, -6 bp from PAM) or non-seed regions (-9, -12, -15 bp from PAM) [94]
- Systematically evaluate the impact of different mismatch types (A, T, C, G) on editing outcomes
- Balance specificity enhancement with maintained on-target efficiency
Table 1: Performance of ARROW gRNAs on Cancer-Associated Mutations
| Target Mutation | gRNA Type | Mutant Allele Indel Rate (%) | Wild-Type Allele Indel Rate (%) | Specificity Enhancement |
|---|---|---|---|---|
| EGFR L858R | Perfect Match | >40 | 11 | Baseline |
| EGFR L858R | ARROW | 1-10 | <1 | >10-fold |
| KRAS G12V | Perfect Match | 77 | 66 | Baseline |
| KRAS G12V | ARROW | 40-60 | <5 | >13-fold |
Data adapted from [94] demonstrating allele discrimination using the dual fluorescence reporter vector system in HEK293T cells.
gRNA Design and Nuclease Selection
Optimal gRNA design begins with bioinformatic selection and extends to choosing appropriate nuclease variants with enhanced fidelity properties.
gRNA Design Considerations:
- Target Specificity: Ensure minimal similarity to other genomic sites using tools like CRISPOR, CHOPCHOP, or CRISPR Design Tool [95] [25]
- GC Content: Higher GC content (40-60%) stabilizes the DNA:RNA duplex, increasing on-target efficiency [82]
- Guide Length: Standard 20-nucleotide guides offer optimal balance between specificity and efficiency [95] [82]
- Chemical Modifications: Incorporate 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bonds (PS) to reduce off-target edits and enhance stability [82]
Nuclease Selection Strategy:
- High-Fidelity Cas9 Variants: eSpCas9, SpCas9-HF1 exhibit reduced off-target cleavage while maintaining on-target efficiency [82]
- Alternative Cas Proteins: Cas12a (Cpf1) offers different PAM requirements and potentially higher specificity [82]
- Nickase Systems: Paired nCas9 nickases with offset gRNAs create staggered cuts, significantly reducing off-target effects [82]
- Base and Prime Editors: Catalytically impaired dCas9 or nCas9 fused to deaminases enable precise editing without double-strand breaks [96]
Delivery and Expression Optimization
The method of CRISPR component delivery significantly impacts editing specificity by controlling the duration and concentration of nuclease exposure.
Optimal Delivery Approaches:
- RNP Complexes: Pre-formed ribonucleoprotein complexes with synthetic gRNAs enable rapid activity and degradation, minimizing off-target exposure [82]
- mRNA Delivery: Cas9 mRNA with modified gRNAs provides transient expression compared to plasmid-based systems [82]
- Chemical Modifications: Modified gRNAs (2'-O-Me, PS bonds) enhance stability and reduce immune responses [82]
- Dose Optimization: Titrate components to the minimum effective concentration to reduce off-target editing [82]
gRNA Optimization and Delivery Workflow: Strategic path from initial design to validated editors, highlighting the impact of delivery format on off-target rates.
Experimental Protocols
Protocol: ARROW gRNA Design and Testing
This protocol details the systematic introduction of mismatches into gRNAs and evaluation of their allele discrimination capability [94].
Materials:
- Dual fluorescence reporter vector system (mRFP and eGFP)
- HEK293T or other relevant cell lines
- Lipofectamine 2000 transfection reagent
- SpCas9 plasmid (Addgene #104171)
- gRNA cloning plasmid (Addgene #104174)
- Flow cytometer for FACS analysis
- Next-generation sequencing platform
Procedure:
- Design Mismatched gRNA Variants
- Identify the single-nucleotide difference between wild-type and mutant alleles
- Generate gRNA sequences perfectly matched to the mutant allele sequence
- Systematically introduce single-base mismatches at seed region positions (-1, -3, -6 from PAM) and non-seed positions (-9, -12, -15 from PAM)
- For each position, design all four possible nucleotide variants (A, T, C, G)
Clone gRNA Expression Constructs
- Synthesize sense and antisense DNA strands for each gRNA variant
- Anneal oligonucleotides and ligate into BsmBI-digested pSpCas9(BB)-2A-GFP (PX458) plasmid [26]
- Transform chemically competent E. coli GT116 cells and plate on ampicillin selection plates
- Isolate plasmids using standard miniprep kits and verify by Sanger sequencing with U6-Fwd primer
Evaluate Editing Specificity Using Dual Fluorescence Reporter System
- Seed 1.5 × 10^5 HEK293T cells in 24-well plates and culture for 24 hours
- Transfect with 200 ng SpCas9 plasmid, 200 ng gRNA plasmid, and 100 ng reporter vector using Lipofectamine 2000
- Culture cells for 48 hours post-transfection
- Analyze mRFP and eGFP expression by FACS to quantify editing efficiency
- Calculate the ratio of mutant to wild-type allele editing for each gRNA variant
Validate Editing by Targeted Deep Sequencing
- Amplify on-target and potential off-target sites using Phusion Hot Start II polymerase
- Perform secondary PCR with NGS adapter primers, followed by a third PCR with indexing primers
- Purify PCR amplicons and sequence by Illumina paired-end sequencing
- Analyze sequencing data using Cas-Analyzer to quantify indel frequencies
- Compare editing efficiency between wild-type and mutant alleles for each gRNA variant
Troubleshooting:
- If specificity is insufficient, test dual mismatch combinations or adjust mismatch positions
- If on-target efficiency is compromised, select alternative mismatch types or positions
- Include perfectly matched gRNA controls for both wild-type and mutant alleles in all experiments
Protocol: Comprehensive Off-Target Assessment
Rigorous off-target profiling is essential for validating gRNA specificity in biochemical applications [82] [26].
Materials:
- Cas-OFFinder software or similar prediction tool
- T7 Endonuclease I (T7E1) or Surveyor nuclease
- PCR amplification reagents
- qEva-CRISPR assay components (synthesized probes, ligation reagents)
- Next-generation sequencing platform
Procedure:
- Bioinformatic Off-Target Prediction
- Use Cas-OFFinder (http://www.rgenome.net) to identify potential off-target sites with up to 8 nucleotide mismatches
- Include sites with DNA or RNA bulges (up to 2 nucleotides) in the analysis
- Rank potential off-target sites by similarity score and genomic context
T7E1 Assay for Candidate Off-Target Sites [94]
- Design PCR primers flanking each predicted off-target site (amplicon size 300-600 bp)
- Amplify genomic DNA from edited cells using Phusion Hot Start II polymerase
- Denature and re-anneal PCR products in a thermal cycler (95°C for 5 min, ramp to 85°C at -2°C/sec, then to 25°C at -0.1°C/sec)
- Digest heteroduplex DNA with T7E1 enzyme at 37°C for 25 minutes
- Terminate reaction by adding stop buffer (100 mM EDTA, 1.2% SDS)
- Separate cleaved DNA fragments by 2% agarose gel electrophoresis
- Quantify cleavage efficiency using gel imaging software
qEva-CRISPR for Quantitative Off-Target Analysis [26]
- Design specific oligonucleotide probes for each target and off-target site
- Perform multiplex ligation-based probe amplification (MLPA) according to established protocols
- Use capillary electrophoresis to separate and quantify amplification products
- Calculate editing efficiencies by comparing peak areas between edited and control samples
- This method detects all mutation types, including point mutations and large deletions, with sensitivity to 0.1% variant frequency
Whole Genome Sequencing for Comprehensive Analysis
- Extract high-quality genomic DNA from edited cells
- Prepare sequencing libraries using standard WGS protocols
- Sequence to appropriate coverage (typically 30-50x) for confident variant calling
- Analyze data for unexpected editing events, including large structural variations
Table 2: Off-Target Analysis Methods Comparison
| Method | Sensitivity | Multiplexing Capacity | Detection Capabilities | Cost and Complexity |
|---|---|---|---|---|
| T7E1 Assay | ~5% | Low | Indels >5bp | Low |
| Targeted Sequencing | 0.1-1% | Medium | All mutation types | Medium |
| qEva-CRISPR | 0.1% | High | All mutations, works in difficult regions | Medium |
| GUIDE-seq | <0.1% | Low | Genome-wide integration sites | High |
| Whole Genome Sequencing | <0.1% | N/A | All variants, chromosomal rearrangements | Very High |
The Scientist's Toolkit
Table 3: Essential Reagents for gRNA Optimization Studies
| Reagent / Tool | Function | Example Sources |
|---|---|---|
| Dual Fluorescence Reporter System | Quantifies allele-specific editing efficiency in live cells | Custom construction [94] |
| High-Fidelity Cas9 Variants | Reduces off-target editing while maintaining on-target activity | eSpCas9, SpCas9-HF1 [82] |
| Synthetic gRNA with Modifications | Chemically modified guides with enhanced stability and reduced off-target effects | Synthego [82] |
| Cas-Analyzer | Web tool for analyzing deep sequencing data from CRISPR experiments | http://www.rgenome.net [94] |
| ICE (Inference of CRISPR Edits) | Software for analyzing CRISPR editing efficiency from Sanger sequencing | Synthego [97] |
| CRISPOR / CHOPCHOP | Bioinformatics tools for gRNA design and off-target prediction | Open source web tools [95] [25] |
| T7 Endonuclease I | Enzyme for mismatch cleavage assays to detect editing events | Commercially available [94] |
| qEva-CRISPR Assay Components | Reagents for quantitative, multiplex evaluation of editing at multiple sites | Custom synthesis [26] |
Data Analysis and Validation
ICE Analysis for CRISPR Editing Assessment
The Inference of CRISPR Edits (ICE) tool enables rapid, cost-effective analysis of editing efficiency from Sanger sequencing data [97].
Procedure:
- Prepare Sequencing Samples
- Extract genomic DNA from edited cells using standard protocols
- Design PCR primers flanking the target site (amplicon size 300-800 bp)
- Purify PCR products and submit for Sanger sequencing
ICE Analysis Workflow
- Upload Sanger sequencing files (.ab1 format) to the ICE web tool (synthego.com)
- Input the gRNA target sequence (20 nt, excluding PAM)
- Select the appropriate nuclease (SpCas9, Cas12a, etc.) from dropdown menu
- For knockout analysis, review the Knockout Score (proportion of frameshift or 21+ bp indels)
- For knock-in analysis, provide the donor sequence and review the Knock-in Score
- Evaluate the Model Fit (R²) score - values >0.9 indicate high-confidence results
- Download detailed reports including indel spectra and alignment data
Interpretation of Results
- Indel Percentage: Overall editing efficiency in the population
- Knockout Score: Proportion of edits likely to cause functional gene disruption
- R² Value: Quality metric for the sequencing data and model fit
- Use the "contributions" tab to view specific indel sequences and their frequencies
Quantitative Analysis of Editing Specificity
Editing Specificity Analysis Workflow: Comprehensive process from edited cells to optimized gRNA selection, highlighting key analysis steps.
Calculate specificity metrics using quantitative data from deep sequencing:
- Specificity Index = (Mutant Allele Editing %) / (Wild-Type Allele Editing %)
- Discrimination Factor = (Mutant indel rate) / (Wild-type indel rate)
- Therapeutic Window = (On-target efficiency) × (Specificity Index)
For clinical applications, aim for a Specificity Index >10 and absolute wild-type editing <0.1% to minimize potential adverse effects [94] [82].
Optimizing gRNA specificity through strategic design, including the innovative ARROW approach of intentional mismatch incorporation, represents a crucial advancement for precise genome editing in biochemical research and therapeutic development [94]. The comprehensive strategies and detailed protocols presented here provide researchers with a systematic framework to enhance allele discrimination while minimizing off-target effects. As CRISPR-based therapies continue to advance toward clinical application, rigorous specificity validation using the described methods will be essential for ensuring both experimental accuracy and therapeutic safety [82] [98]. Implementation of these gRNA optimization principles will enable researchers to achieve the high-precision editing required for modeling genetic diseases, functional genomics studies, and developing targeted genetic therapies.
The CRISPR-Cas9 system has revolutionized genetic engineering by providing unprecedented capability for targeted DNA manipulation. However, precise spatial and temporal control over CRISPR-Cas9 activity remains crucial for therapeutic applications, particularly in reducing off-target effects and improving safety profiles [99]. Cell-permeable anti-CRISPR (Acr) proteins represent a cutting-edge technology for controlled deactivation of CRISPR systems, offering researchers the ability to precisely terminate gene editing processes once desired modifications are complete.
These inhibition systems are particularly valuable in the context of a broader thesis on precise genome editing for biochemistry research, as they enable unprecedented control over editing windows. This document provides detailed application notes and experimental protocols for implementing cell-permeable Acr proteins, with specific focus on quantitative performance characteristics and standardized methodologies for the research community.
Quantitative Analysis of Anti-CRISPR Inhibition Systems
The tables below summarize key quantitative data for selected anti-CRISPR proteins and their functional characteristics in controlled deactivation of CRISPR-Cas systems.
Table 1: Anti-CRISPR Proteins and Their Characteristics
| Anti-CRISPR Protein | Target CRISPR System | Inhibition Mechanism | Key Structural Features | Reported Inhibition Efficiency |
|---|---|---|---|---|
| AcrIF24 | Type I-F | Csy complex dimerization; blocks DNA hybridization; induces non-specific dsDNA binding [100] | HTH motif (residues 181-198); N-terminal domain (residues 1-72); C-terminal domain (residues 136-228) [100] | >90% in vitro cleavage inhibition [100] |
| AcrIF9 | Type I-F | Sterically inhibits base-pairing; tethers non-specific DNA to Csy complex [100] | Not specified in results | High (specific quantitative data not provided) |
| AcrIIA1 | Type II-A | Dual Acr-Aca function [100] | HTH motif [100] | Not specified |
Table 2: Comparison of CRISPR Inhibition Platforms
| Inhibition Platform | Control Mechanism | Dynamic Range | Activation/Deactivation Time | Special Requirements |
|---|---|---|---|---|
| Ligand-activated sgRNA [101] | Small-molecule control of sgRNA function | >10-fold [101] | Not specified | RNA aptamers inserted into sgRNA |
| Optically controlled gRNA [99] | Light-activated CRISPR-OFF switch | Not specified | Not specified | Vinyl ether RNA modification; phenanthrenequinone derivative; visible light |
| AcrIF24 [100] | Direct protein interaction with Csy complex | Not applicable | Not specified | Recombinant protein expression and delivery |
Experimental Protocols
Protocol 1: In Vitro Cleavage Inhibition Assay for AcrIF24
Purpose: To quantitatively assess the inhibition efficiency of AcrIF24 against type I-F CRISPR-Cas system.
Materials:
- Purified type I-F CRISPR-Cas components (Cas1, Cas2/3 fusion, Cas8f, Cas5f, Cas7f, Cas6f)
- Target DNA substrate with appropriate PAM sequence
- crRNA complementary to target DNA
- Recombinant AcrIF24 protein (full-length, 1-228 amino acids)
- Reaction buffer (20 mM HEPES pH 7.5, 150 mM KCl, 10 mM MgCl₂, 1 mM DTT)
- Stop solution (10% SDS, 50 mM EDTA)
Procedure:
- Assemble the Csy complex by incubating Cas8f, Cas5f, six Cas7f subunits, and Cas6f-bound crRNA in reaction buffer at 25°C for 30 minutes.
- Prepare serial dilutions of AcrIF24 protein (0-500 nM) in reaction buffer.
- In separate tubes, pre-incubate 50 nM Csy complex with varying concentrations of AcrIF24 for 15 minutes at 37°C.
- Initiate cleavage reactions by adding 10 nM target DNA substrate and 20 nM Cas2/3 fusion protein.
- Incubate at 37°C for 45 minutes.
- Stop reactions with 2× stop solution.
- Analyze DNA cleavage by agarose gel electrophoresis or capillary electrophoresis.
- Quantify inhibition efficiency by comparing intact substrate in AcrIF24-treated samples versus untreated controls.
Validation Notes: AcrIF24ΔMD (deletion of residues 73-135) shows complete loss of inhibitory function, confirming the essential role of the middle domain [100].
Protocol 2: Electrophoretic Mobility Shift Assay (EMSA) for Acr-DNA Interaction
Purpose: To evaluate DNA binding capability of AcrIF24 to its promoter region.
Materials:
- Recombinant AcrIF24 (full-length or ΔMD variant)
- IR23-24 dsDNA fragment (23-bp containing inverted repeat from acrIF23-24 promoter region)
- Binding buffer (10 mM Tris pH 7.5, 50 mM NaCl, 1 mM DTT, 5% glycerol)
- 6% non-denaturing polyacrylamide gel
- SYBR Gold nucleic acid stain
Procedure:
- Prepare 100 nM IR23-24 dsDNA in binding buffer.
- Mix with increasing concentrations of AcrIF24 (0-1 µM) in 20 µL reaction volume.
- Incubate at 25°C for 30 minutes.
- Load samples onto 6% non-denaturing polyacrylamide gel.
- Run electrophoresis at 100V for 60 minutes in 0.5× TBE buffer at 4°C.
- Stain gel with SYBR Gold and visualize under UV transillumination.
- Confirm HTH-dependent binding by comparing full-length AcrIF24 with CTD-truncated variants.
Protocol 3: Cell-Penetrating Peptide-Mediated Acr Delivery
Purpose: To facilitate intracellular delivery of Acr proteins using cell-penetrating peptides (CPPs).
Materials:
- Purified Acr protein
- Cell-penetrating peptides (CPPs)
- Complexation buffer (PBS pH 7.4)
- Target cells (adherent or suspension)
- Serum-free cell culture medium
Procedure:
- Formulate CPP-Acr complexes at optimal mass ratio (typically 10:1 to 20:1 CPP:protein).
- Incubate CPP and Acr protein in complexation buffer for 30 minutes at room temperature.
- Wash target cells with PBS and replace medium with serum-free medium.
- Add CPP-Acr complexes to cells and incubate at 37°C for 2-4 hours.
- Replace with complete growth medium and continue incubation.
- Assess intracellular delivery efficiency by fluorescence microscopy (for labeled proteins) or functional inhibition assays.
- Evaluate CRISPR inhibition by measuring reduction in editing efficiency at target loci.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Anti-CRISPR Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Anti-CRISPR Proteins | AcrIF24, AcrIF9, AcrIIA1 [100] | Direct inhibition of CRISPR-Cas complexes; controlled deactivation of gene editing |
| CRISPR System Components | Cas1, Cas2/3, Cas8f, Cas5f, Cas7f, Cas6f (Type I-F) [100] | Reconstitution of functional CRISPR systems for inhibition studies |
| Cell-Penetrating Peptides | Not specified (various CPPs) [102] | Intracellular delivery of ribonucleoprotein complexes including anti-CRISPR proteins |
| Control Systems | Ligand-activated sgRNA [101]; Optically controlled gRNA [99] | Alternative methods for regulating CRISPR activity; comparison platforms |
| Analytical Tools | Electrophoretic mobility shift assay; In vitro cleavage assays; Gel filtration with SLS [100] | Characterization of protein-DNA interactions; inhibition efficiency quantification; oligomeric state determination |
Visualizing Anti-CRISPR Mechanisms and Workflows
The following diagrams illustrate the molecular mechanism of AcrIF24 action and the experimental workflow for evaluating anti-CRISPR proteins.
Diagram 1: Anti-CRISPR Molecular Mechanism
Diagram 2: Experimental Workflow
The therapeutic application of CRISPR-Cas9 genome editing is fundamentally constrained by off-target effects, which pose substantial genotoxicity risks and delay clinical translation [103]. While bioinformatics tools for guide RNA (gRNA) design and engineered high-fidelity Cas variants have improved specificity, the kinetic profile of CRISPR component delivery and activity represents an underutilized layer of control [104] [105]. Kinetic control strategies aim to minimize the time window during which Cas nuclease concentrations are sufficiently high to bind and cleave at off-target sites with imperfect complementarity, while maintaining efficient on-target editing [104].
The principle is grounded in the biophysical mechanism of Cas9-DNA interaction: the Cas9-sgRNA complex first binds to a protospacer adjacent motif (PAM) sequence, then unwinds the adjacent DNA to form an RNA-DNA heteroduplex. Mismatches, particularly in the PAM-distal region, are better tolerated when nuclease concentrations are high and exposure is prolonged [104]. Transient, high-efficiency delivery of CRISPR components creates a kinetic profile that favors specific on-target editing over non-specific off-target activity. This application note details practical methodologies for implementing kinetic control through advanced delivery timing strategies.
Kinetic Control Strategies and Mechanisms
Strategic Approaches to Temporal Control
Table 1: Comparison of Kinetic Control Strategies for Minimizing Off-Target Effects
| Strategy | Mechanism | Key Parameters | Editing Efficiency | Specificity Improvement | Primary Applications |
|---|---|---|---|---|---|
| RNP Electroporation | Direct delivery of pre-formed Cas9-gRNA complexes; immediate activity and rapid degradation [106] [45] | Cas9 concentration, gRNA:Cas9 ratio, pulse parameters | High (e.g., 73% HDR in BEL-A cells [106]) | High (reduced off-targets vs. plasmid DNA) | Primary immune cells, stem cells, clinical applications [105] |
| Drug-Inducible Degradation | Fusion of Cas9 to degron domains; small molecule-triggered protein degradation [107] | Degradation kinetics (e.g., 4 hrs for Cas9-d), inducer concentration | Modulatable (3–5-fold reduction with pomalidomide [107]) | High (precise temporal termination) | Research models, future therapeutics requiring precise timing |
| mRNA/LNP Delivery | Encapsulated mRNA translated into transient Cas9 protein [105] | LNP formulation, mRNA stability, translation kinetics | High (up to 70% knock-in in iPSCs with cssDNA [105]) | Moderate to High (transient expression window) | In vivo therapeutic delivery, hard-to-transfect cells [105] |
| Cold Shock | Temporary reduction of cellular metabolism to influence repair pathways [106] | Temperature, duration of shock | Variable (no significant PGE increase in BEL-A cells [106]) | Inconclusive | In vitro studies, combination with other methods |
| Cell Cycle Synchronization | Enrichment of cells in G2/M phase where HDR is preferred [106] | Synchronization agent (e.g., nocodazole), release timing | High HDR but reduced viability (e.g., with nocodazole [106]) | Indirect (via enhanced HDR) | Experiments requiring high precision editing |
The underlying logic for selecting a kinetic control strategy depends on the experimental goals and cellular context, as visualized in the decision workflow below.
Molecular Mechanisms of Off-Target Effects
Understanding the molecular basis of off-target activity is crucial for designing effective kinetic control strategies. The Cas9-sgRNA complex recognizes target DNA through a multi-step process initiated by PAM interaction, followed by DNA unwinding and RNA-DNA heteroduplex formation [104]. Off-target binding occurs when the complex tolerates mismatches, bulges, or DNA/RNA imperfections, particularly in the PAM-distal region [104].
The relationship between nuclease concentration, exposure time, and off-target activity is governed by the allosteric regulation of Cas9. Structural studies reveal that Cas9 undergoes conformational changes that control its nuclease activity. Prolonged presence of high Cas9 concentrations increases the probability of these conformational shifts occurring at off-target sites, facilitating non-specific cleavage [104]. Transient delivery methods, such as RNP electroporation, exploit this mechanism by limiting the time window for these non-productive interactions, thereby enhancing specificity without compromising on-target efficiency [106] [45].
Experimental Protocols for Kinetic Control
Protocol 1: RNP Electroporation with Small Molecule Enhancers
This protocol details the methodology for introducing specific mutations via ribonucleoprotein (RNP) electroporation in human erythroid cell line BEL-A, achieving high-precision editing efficiency (73%) with minimal off-target effects [106].
Research Reagent Solutions:
- CRISPR-Cas9 RNP Complex: 3 µg Cas9 protein combined with sgRNA at a 1:2.5 ratio (gRNA:Cas9) [106]
- HDR Enhancer: 0.25 µM Nedisertib (DNA-PK inhibitor) in DMSO [106]
- Electroporation Buffer: Cell-type specific nucleofection solution
- ssODN Donor Template: 100 pmol single-stranded oligodeoxynucleotide with homology arms (36-nt PAM-distal, 91-nt PAM-proximal) [106]
- Cell Culture Medium: Complete growth medium with appropriate cytokines and supplements
Procedure:
- RNP Complex Formation: Incubate 3 µg of high-fidelity Cas9 protein with sgRNA at a 1:2.5 mass ratio (gRNA:Cas9) in nuclease-free buffer for 15-20 minutes at room temperature to form RNP complexes [106].
- Cell Preparation: Harvest and count 5 × 10^4 BEL-A cells per condition. Centrifuge at 300 × g for 5 minutes and resuspend in electroporation buffer [106].
- Nucleofection Mixture: Combine RNP complexes with 100 pmol ssODN donor template and resuspended cells. Mix gently by pipetting.
- Electroporation: Transfer mixture to a 16-well nucleocuvette strip. Electroporate using the Amaxa 4D-Nucleofector with program DZ-100 [106].
- Post-Transfection Processing: Immediately add pre-warmed culture medium containing 0.25 µM Nedisertib. Transfer cells to a culture plate.
- Incubation and Analysis: Culture at 37°C, 5% CO₂ for 48-72 hours before assessing editing efficiency via flow cytometry or sequencing.
Protocol 2: Drug-Inducible Cas9 Degradation System
This protocol implements a degradable Cas9 system (Cas9-d) for precise temporal control over editing activity, reducing on-target edits 3-5-fold within hours of inducer addition [107].
Research Reagent Solutions:
- Degradable Cas9 Construct: Cas9-d fusion (Cas9 coupled to degron domain)
- Degradation Inducer: 1-5 µM pomalidomide or analogous small molecule
- Delivery Vector: AAV or lentiviral vector encoding Cas9-d system
- Control Vector: Non-degradable Cas9 construct
Procedure:
- System Delivery: Transduce target cells with viral vector encoding the Cas9-d construct at appropriate MOI. Include control cells with non-degradable Cas9.
- Expression Validation: Confirm Cas9 expression via Western blot or fluorescence 48 hours post-transduction.
- Editing Initiation: Introduce sgRNA complexed with Cas9-d via preferred delivery method (e.g., RNP electroporation).
- Kinetic Control Point: Add 1-5 µM pomalidomide to culture medium at predetermined optimal timepoint (typically 4-24 hours post-editing initiation) [107].
- Degradation Monitoring: Monitor Cas9-d degradation over 4-24 hours post-induction via Western blot.
- Efficiency Assessment: Harvest cells at 72 hours post-editing for sequencing analysis of on-target and predicted off-target sites.
Quantitative Analysis of Kinetic Control Efficacy
Table 2: Quantitative Assessment of Kinetic Control Methods in Model Systems
| Cell Type | Kinetic Control Method | On-Target Efficiency | Off-Target Reduction | Key Experimental Parameters | Viability Impact |
|---|---|---|---|---|---|
| BEL-A (Erythroid) [106] | RNP + Nedisertib (0.25 µM) | 73% precise gene editing | ~21% increase in PGE vs. control | 3 µg Cas9, 1:2.5 gRNA:Cas9, 100 pmol ssODN | 74% viability |
| BEL-A (Erythroid) [106] | RNP + NU7441 (1 µM) | ~11% increase in PGE | ~11% increase in PGE vs. control | Same as above | Moderate reduction |
| BEL-A (Erythroid) [106] | Cell Sync. (Nocodazole) | No significant PGE increase | No significant change | 18 hr pre-treatment, release at transfection | Marked reduction |
| HEK293T [107] | Cas9-d + Pomalidomide | 3-5-fold reduction (inducible) | Not quantified | 4 hr to degradation, 24 hr to restoration | Low toxicity |
| Primary T Cells [105] | mRNA/LNP + cssDNA | Up to 70% knock-in | Not quantified | LNP formulation, cssDNA donor | Favorable profile |
| iPSCs [105] | mRNA/LNP + cssDNA | Up to 70% knock-in | Not quantified | LNP formulation, cssDNA donor | Maintained pluripotency |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Kinetic Control Experiments
| Reagent / Tool | Function in Kinetic Control | Example Specifications | Experimental Considerations |
|---|---|---|---|
| High-Fidelity Cas9 Protein | RNP complex formation for transient expression | >90% purity, endotoxin-free | Aliquot to prevent freeze-thaw cycles; titrate for cell-type specificity [106] |
| Nedisertib (M3812) | DNA-PK inhibitor enhancing HDR efficiency | 0.25 µM working concentration [106] | Cytotoxicity observed at higher concentrations (≥2 µM); optimal window narrow [106] |
| Nocodazole | Microtubule polymerization inhibitor for cell cycle synchronization | 100 ng/mL, 18 hr treatment [106] | Causes significant viability reduction; requires careful timing of release [106] |
| Pomalidomide | Triggers degradation of Cas9-d constructs | 1-5 µM working concentration [107] | Rapid effect (4 hr); reversible upon removal; cell permeability important [107] |
| Selective Organ Targeting (SORT) LNPs | Tissue-targeted mRNA delivery for in vivo applications | Tunable formulations for lung, spleen, liver [45] | Requires optimization for each tissue type; encapsulation efficiency critical [105] |
| ssODN Donor Templates | Homology-directed repair template for precise edits | 127-nt, 36-nt/91-nt homology arms [106] | HPLC purification recommended; position relative to PAM critical for efficiency [106] |
| Cas9-d Degron System | Drug-inducible Cas9 degradation for precise timing | Based on FKBP12F36V degradation domain [107] | Requires viral delivery; baseline expression levels affect dynamic range [107] |
The relationship between these reagents and their points of application in the kinetic control workflow is illustrated below.
Kinetic control through advanced delivery timing represents a paradigm shift in precision genome editing. By moving beyond static sequence design to incorporate temporal dimensions of nuclease activity, researchers can achieve unprecedented specificity in CRISPR-Cas9 applications. The strategies outlined here—from RNP delivery with small molecule enhancers to degradable Cas9 systems—provide a toolkit for minimizing off-target exposure while maintaining high on-target efficiency.
Future developments will likely focus on orthogonal control systems that combine multiple kinetic modalities, such as LNPs with built-in degradation triggers or cell-cycle specific editors that automatically limit their activity window. As the field progresses toward clinical applications, integrating these kinetic control strategies with improved bioinformatics prediction tools [85] [17] and enhanced fidelity Cas variants will establish a comprehensive safety framework for therapeutic genome editing. The quantitative frameworks and standardized protocols provided here offer researchers a foundation for implementing these advanced timing strategies across diverse experimental and therapeutic contexts.
The success of CRISPR-Cas9 genome editing experiments is fundamentally dependent on the careful selection of highly specific guide RNAs (gRNAs). Computational prediction tools have become indispensable for identifying potential off-target sites, thereby minimizing unintended genomic alterations. This application note focuses on three prominent bioinformatics tools—COSMID, CCTop, and Cas-OFFinder—detailing their algorithms, appropriate use cases, and implementation protocols for effective gRNA selection within biochemical research and drug development pipelines. These tools leverage homology-based searches to scan reference genomes for DNA sequences similar to the intended gRNA target, flagging sites with partial complementarity that might be cleaved by the Cas9 nuclease. Their integration into the experimental design workflow is crucial for enhancing the specificity and safety of CRISPR applications, particularly in translational research where off-target effects could have significant consequences [108] [109] [110].
Table 1: Overview of Featured CRISPR Off-target Prediction Tools
| Tool Name | Primary Search Features | Key Algorithm/Interface | PAM Flexibility |
|---|---|---|---|
| COSMID | Mismatches, insertions, and deletions (indels) | Web interface; FetchGWI search algorithm | User-defined |
| CCTop | Mismatches, intuitive ranking | Web interface; Bowtie alignment; Off-target score | Standard SpCas9 (NGG) |
| Cas-OFFinder | Mismatches, flexible PAM | Command-line or web; OpenCL for parallel processing | Highly flexible, user-definable |
Tool Capabilities and Comparative Analysis
Core Functionalities and Distinguishing Features
A head-to-head comparative analysis of these tools in primary human hematopoietic stem and progenitor cells (HSPCs) revealed that all successfully identified the rare off-target sites generated by high-fidelity Cas9, demonstrating high sensitivity [108]. However, their underlying approaches and capabilities differ.
COSMID (CRISPR Off-target Sites with Mismatches, Insertions, and Deletions): A key differentiator for COSMID is its ability to account for potential off-target sites caused not only by base mismatches but also by insertions or deletions (indels) between the gRNA and the genomic DNA sequence. Its search algorithm, FetchGWI, uses indexed genome sequences for exhaustive and rapid searching. The tool provides a ranked list of potential off-target sites and can output optimized primers for experimental validation [109].
CCTop (CRISPR/Cas9 target online predictor): CCTop is designed for intuitive use, offering reasonable default parameters that can be customized by experts. It employs the Bowtie alignment algorithm to search for off-target sites and ranks candidates using a proprietary off-target mismatch score. This score weights mismatches more heavily if they occur closer to the Protospacer Adjacent Motif (PAM), reflecting the established understanding that mismatches in this region are more disruptive to Cas9 binding. The tool provides a comprehensive output, including the location of off-target sites and their proximity to annotated exons [111].
Cas-OFFinder: This tool stands out for its high speed and versatility. Written in OpenCL, it can leverage parallel processing on CPUs, GPUs, and DSPs, making genome-wide searches very fast. A significant advantage is its flexibility with PAM sequences; it is not limited to SpCas9's NGG PAM but can accommodate the PAM requirements of other Cas9 orthologs (e.g., NmCas9, StCas9) and even other engineered nucleases like ZFNs and TALENs. It allows users to specify the number of mismatches without a hard上限 (upper limit) [110].
Performance and Practical Considerations
The performance of these tools is critical for practical application. The 2023 study by Cromer et al. found that COSMID, alongside empirical methods like DISCOVER-Seq and GUIDE-Seq, attained the highest positive predictive value (PPV), meaning it reported fewer false positives [108]. Cas-OFFinder demonstrates a clear performance benefit when utilizing a GPU, completing searches for 1000 target sites in approximately 3 seconds—20 times faster than a CPU-based calculation [110].
Table 2: Practical Application and Experimental Validation
| Tool Name | Reported Performance/Speed | Experimental Validation Cited | Key Practical Strength |
|---|---|---|---|
| COSMID | High Positive Predictive Value [108] | K-562 cells; T7EI assay [109] | Identifies bulge-type off-target sites |
| CCTop | Intuitive interface with detailed documentation [111] | Gene inactivation, NHEJ, and HDR in medaka fish [111] | User-friendly with excellent off-target ranking |
| Cas-OFFinder | GPU: ~3 sec for 1000 targets; CPU: ~60 sec [110] | Cited in analysis of RGEN off-target effects [110] | Extreme speed and PAM flexibility |
Experimental Protocol for Off-Target Assessment
This protocol outlines a standard workflow for using computational tools to predict and validate off-target sites for a candidate gRNA.
Stage 1: In Silico gRNA Design and Off-Target Nomination
Step 1: Define gRNA and Parameters
- Input your 20-nucleotide gRNA spacer sequence into your chosen tool(s).
- Select the relevant reference genome (e.g., GRCh38 for human).
- Set the search parameters. For an initial screen, a common setting is to allow up to 3-5 nucleotide mismatches. For COSMID, you can choose to include searches for sites with 1-base indels and 2 mismatches [109]. CCTop allows setting the core sequence length (e.g., 12 bases adjacent to the PAM) and the maximum number of mismatches within it and the full gRNA [111].
Step 2: Execute Search and Analyze Results
- Run the search algorithm. The output will be a ranked list of potential off-target sites.
- Prioritize sites based on the tool's scoring system. High-priority sites typically have fewer mismatches, especially those with mismatches far from the PAM sequence. Also, prioritize sites located within protein-coding exons or regulatory regions over those in intergenic or intronic regions [111].
- It is considered best practice to run the candidate gRNA through multiple tools (e.g., both CCTop and Cas-OFFinder) to create a comprehensive list of potential off-target sites for validation [108].
Stage 2: Experimental Validation of Predicted Off-Target Sites
Step 3: Amplify Genomic Loci
- Design PCR primers flanking each nominated off-target site (and the on-target site as a positive control). Many tools, including COSMID, can design these primers automatically [109].
- Perform PCR on genomic DNA extracted from edited cells and control (un-edited) cells. Use high-fidelity polymerase to minimize PCR-derived errors.
Step 4: Detect Mutational Indels
- Utilize a mutation detection assay to assess cleavage efficiency at each locus. Common methods include:
- T7 Endonuclease I (T7EI) Assay: This enzyme cleaves heteroduplex DNA formed by annealing wild-type and indel-containing PCR products. The cleavage fragments, visualized by gel electrophoresis, indicate the presence of mutations [109].
- Targeted Next-Generation Sequencing (NGS): This is the gold standard. Amplify the loci of interest and submit for deep sequencing (>10,000x coverage). Analyze the resulting data with a variant-calling algorithm (e.g., CRISPResso2) to quantify the precise frequency of indels at each site [108].
The following workflow diagram illustrates the complete process from gRNA design to final validation:
The Scientist's Toolkit: Essential Research Reagents
The following table lists key materials and reagents required for the computational prediction and subsequent experimental validation of CRISPR gRNA specificity.
Table 3: Essential Reagents and Resources for gRNA Off-Target Analysis
| Item Name | Specification / Example | Primary Function in Workflow |
|---|---|---|
| gRNA Design Tool | Synthego CRISPR Design Tool, Benchling | Initial on-target efficiency prediction and gRNA sequence generation [24]. |
| Off-Target Prediction Software | COSMID, CCTop, Cas-OFFinder | Nominates genomic loci with sequence homology for experimental screening [109] [110] [111]. |
| Reference Genome | GRCh38 (human), GRCm39 (mouse) | The sequence database against which computational tools search for off-target sites. |
| High-Fidelity DNA Polymerase | Q5 Hot-Start (NEB), KAPA HiFi | Accurate amplification of on-target and off-target genomic loci for sequencing validation. |
| Next-Generation Sequencer | Illumina MiSeq, NovaSeq | High-depth sequencing of amplified target sites to precisely quantify indel mutation frequencies [108]. |
| Analysis Software | CRISPResso2, BWA, GATK | Processes NGS data to align sequences and call insertions/deletions (indels) relative to the reference genome. |
| Cas9 Nuclease | Wild-type SpCas9, High-Fidelity Cas9 (HiFi Cas9) | The effector protein that creates double-strand breaks. HiFi variants can dramatically reduce off-target editing [108]. |
Validation Strategies and Method Comparison for Editing Assessment
The transformative potential of CRISPR-Cas9 in therapeutic applications is tempered by the risk of off-target effects—unintended modifications at genomic sites with sequence similarity to the target locus. These off-target events can disrupt vital genes, create cryptic splice sites, or even confer oncogenic potential, presenting significant safety concerns for clinical translation [112]. Consequently, comprehensive off-target screening has become an essential component of therapeutic development, with the U.S. FDA recently recommending multiple methods for off-target assessment, including genome-wide analysis [113]. This application note details three powerful genome-wide methods—GUIDE-seq, CIRCLE-seq, and DISCOVER-seq—that enable researchers to empirically define the off-target landscape of CRISPR-Cas9 nucleases with high sensitivity and precision.
Technology Comparison
The following table summarizes the key characteristics, advantages, and limitations of each genome-wide off-target detection method:
Table 1: Comparative Analysis of Genome-Wide Off-Target Detection Methods
| Parameter | GUIDE-Seq | CIRCLE-Seq | DISCOVER-Seq |
|---|---|---|---|
| Fundamental Principle | Tagging DSBs via NHEJ-mediated integration of dsODN in living cells [114] | In vitro cleavage of circularized genomic DNA followed by sequencing [115] | ChIP-seq of MRE11 repair protein recruited to DSBs in cells [116] |
| Detection Context | Native chromatin + cellular repair machinery | Purified genomic DNA (no chromatin influence) | Native chromatin + cellular repair machinery |
| Sensitivity | ~0.1% indel frequency [114] | Higher than GUIDE-seq and Digenome-seq [115] | ~0.3% indel frequency [116] |
| Sequencing Depth | 2-5 million reads (runs on benchtop sequencers) [114] | ~100-fold fewer reads than Digenome-seq [115] | ≥30 million reads [116] |
| Input Material | Living cells (transfected with dsODN) [114] | Purified genomic DNA (nanogram amounts) [113] | ≥5×10⁶ cells [116] |
| Key Advantages | Direct cell-based measurement; high validation rate; quantitative; captures biological relevance [114] | Ultra-sensitive; does not require reference genome; identifies SNP-influenced sites; no cell culture needed [115] | Works in primary cells and animal models; low false positive rate; captures in situ activity [116] |
| Primary Limitations | Requires dsODN transfection; some cell types sensitive to DNA ends [114] | Higher false positives due to lack of epigenetic context [112] | Temporally restricted detection window; requires more material [116] |
| Therapeutic Application | Ideal for cell types tolerant to dsODN transfection [114] | Excellent for initial comprehensive screening [115] | Suitable for patient-derived cells and in vivo models [116] |
Methodology and Workflows
GUIDE-Seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing)
GUIDE-seq employs a straightforward approach based on the non-homologous end joining (NHEJ) DNA repair pathway to tag and identify nuclease-induced double-strand breaks [114]. The method utilizes a short, end-protected double-stranded oligodeoxynucleotide (dsODN) tag that is co-delivered with CRISPR-Cas9 components into cells.
Table 2: Key Reagents for GUIDE-Seq
| Reagent | Specifications | Function |
|---|---|---|
| dsODN Tag | Blunt-ended, 34 bp, phosphorothioate modifications on 3' ends [114] | Integration into DSBs via NHEJ pathway |
| Cas9 Delivery | Plasmid DNA or ribonucleoprotein (RNP) complexes [114] | Genome editing machinery |
| PCR Primers | Contains portions complementary to dsODN tag and Illumina adapters [114] | Specific amplification of tag-integrated regions |
| UMI Adapters | 8-bp unique molecular identifiers [114] | Correction of PCR amplification bias |
Experimental Workflow:
- Cell Transfection: Co-deliver Cas9 (as plasmid or RNP) and dsODN tag into cultured cells using appropriate transfection methods [114].
- Incubation: Culture cells for 3 days to allow for genome editing and tag integration.
- Genomic DNA Isolation: Extract high-quality genomic DNA using standard purification methods.
- Library Preparation:
- Fragment genomic DNA (200-500 bp) via sonication or enzymatic digestion
- End-repair, A-tailing, and ligation of UMI-containing adapters
- Two rounds of PCR with primers complementary to ligated adapter and dsODN tag
- Sequencing and Analysis: Sequence on Illumina platforms (2-5 million reads sufficient) and analyze using GUIDEseq Bioconductor package [117].
The critical optimization parameter for GUIDE-seq is ensuring sufficient dsODN integration frequency (>5% of nuclease-induced mutations), which can be validated via NdeI restriction digestion of PCR amplicons containing the tag-specific restriction site [114].
CIRCLE-Seq (Circularization for In Vitro Reporting of Cleavage Effects by Sequencing)
CIRCLE-seq is an ultra-sensitive in vitro method that detects Cas9 cleavage sites using circularized genomic DNA, virtually eliminating the background noise that plagues other biochemical methods [115].
Experimental Workflow:
- Genomic DNA Preparation: Isolate genomic DNA from cells of interest (e.g., iPSCs). The protocol requires approximately 25 μg of gDNA (~2.0×10⁷ cells per sample) [112].
- DNA Shearing and Circularization:
- Randomly shear genomic DNA via focused ultrasonication
- Treat with exonuclease to remove linear DNA fragments
- Circularize remaining DNA fragments using ligase
- Purify circular DNA molecules with plasmid-safe DNase treatment [112]
- In Vitro Cleavage: Incubate circularized DNA with preassembled Cas9-gRNA ribonucleoprotein (RNP) complexes under optimized reaction conditions.
- Library Preparation and Sequencing:
- Ligate Illumina adapters to Cas9-cleaved ends
- Amplify libraries via PCR
- Sequence using paired-end chemistry (enables capture of both sides of cleavage event) [115]
A key advantage of CIRCLE-seq is its ability to identify off-target sites without requiring a reference genome sequence, enabling off-target profiling in genetically diverse populations or organisms with incomplete genomic sequences [115]. The method demonstrates approximately 180,000-fold better enrichment for nuclease-cleaved sequences compared to Digenome-seq, allowing for sensitive detection with 100-fold fewer sequencing reads [115].
DISCOVER-Seq (Discovery of In Situ Cas Off-Targets and Verification by Sequencing)
DISCOVER-seq leverages the endogenous DNA repair machinery to identify off-target sites by monitoring the recruitment of MRE11, a key protein in the MRN complex that responds to double-strand breaks [116].
Experimental Workflow:
- Genome Editing: Introduce CRISPR-Cas9 components into cells or living organisms via appropriate delivery methods. Timing is critical—for RNP delivery, DNA breaks happen almost immediately, while viral delivery requires time for expression [116].
- Crosslinking and Chromatin Preparation: At appropriate timepoints post-editing (typically 24-48 hours):
- Crosslink cells with formaldehyde to fix protein-DNA interactions
- Isolate and shear chromatin to ~200-500 bp fragments via sonication [116]
- Immunoprecipitation:
- Incubate with anti-MRE11 antibody (commercially available human/mouse cross-reactive antibodies suitable)
- Capture antibody-chromatin complexes with protein A/G beads
- Wash and elute bound DNA fragments
- Reverse crosslinks and purify DNA [116]
- Library Preparation and Sequencing:
- Prepare sequencing libraries using standard ChIP-seq protocols
- Sequence with sufficient depth (≥30 million reads recommended)
- Analyze using BLENDER bioinformatics pipeline [116]
DISCOVER-seq successfully identifies off-target sites in various contexts, including induced pluripotent stem cells (iPSCs) and during adenoviral in situ editing of mouse liver, making it particularly valuable for pre-clinical safety assessment [116].
Workflow Visualization
Application Guidelines
Selection Criteria
Choosing the appropriate off-target detection method depends on research goals, cell type, and resources:
- GUIDE-seq is ideal for cell lines tolerant to dsODN transfection when seeking biologically relevant off-target profiles with high validation rates [114].
- CIRCLE-seq provides the highest sensitivity for comprehensive screening and can identify potential off-target sites that might be missed by cell-based methods, though with potentially higher false positive rates [115] [112].
- DISCOVER-seq enables off-target detection in challenging systems including primary cells, patient-derived samples, and in vivo models where introducing exogenous tags is problematic [116].
Complementary Approaches
For comprehensive off-target assessment, consider a tiered approach:
- Initial comprehensive screening with CIRCLE-seq to identify potential off-target candidates
- Validation of biologically relevant sites in target cells using GUIDE-seq or DISCOVER-seq
- Final confirmation of top off-target sites via targeted amplicon sequencing in therapeutically relevant models
The empirical methods detailed herein—GUIDE-seq, CIRCLE-seq, and DISCOVER-seq—provide researchers with powerful tools for comprehensive off-target profiling of CRISPR-Cas9 genome editors. GUIDE-seq offers sensitive detection in living cells with high validation rates, CIRCLE-seq delivers ultra-sensitive in vitro screening, and DISCOVER-seq enables off-target identification in clinically relevant primary cells and in vivo models. As CRISPR-based therapies advance toward clinical application, employing these complementary genome-wide methods will be essential for ensuring the safety and efficacy of genome editing therapeutics. Researchers should select methods based on their specific experimental systems and development stage, with strategic implementation of multiple orthogonal approaches providing the most rigorous safety assessment.
The advent of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 technology has revolutionized precise genome editing in biochemistry research. However, the full potential of this technology can only be realized with rigorous, quantitative assessment of editing outcomes. Targeted deep sequencing has emerged as the gold standard methodology for quantifying two critical parameters: editing efficiency and insertion/deletion (indel) profiles. This application note details protocols and considerations for implementing targeted deep sequencing to characterize CRISPR-Cas9 genome editing outcomes, enabling researchers and drug development professionals to accurately measure on-target efficiency, identify off-target effects, and advance therapeutic applications.
The fundamental strength of targeted deep sequencing lies in its capacity to detect low-frequency editing events with high precision. Unlike traditional validation methods, it provides single-nucleotide resolution across thousands to millions of parallel sequencing reads, enabling comprehensive characterization of heterogeneous editing outcomes within cell populations. As CRISPR therapeutics advance toward clinical applications—with recent FDA approvals for sickle cell disease and ongoing trials for conditions like hereditary transthyretin amyloidosis (hATTR) and hereditary angioedema—robust quantification of editing outcomes becomes increasingly critical for both basic research and therapeutic development [11].
Quantitative Analysis of CRISPR Editing Efficiency
Establishing Baseline Editing Metrics
Targeted deep sequencing provides quantitative data essential for evaluating the performance of different CRISPR systems. Editing efficiency is typically calculated as the percentage of sequencing reads containing indels or specific sequence modifications at the target locus. Research comparing next-generation CRISPR systems has demonstrated the critical importance of these measurements. For instance, in the characterization of AI-designed editors such as OpenCRISPR-1, targeted deep sequencing revealed comparable or improved activity relative to the prototypical SpCas9, despite being 400 mutations away in sequence [118]. Such comparative analyses rely entirely on the precision of deep sequencing methodologies.
The context of editing also influences efficiency measurements. Studies investigating chromatin effects on CRISPR activity have revealed that Cas9 induces indels with higher frequencies at sites in open chromatin regions than at respective sites with identical DNA sequences in closed chromatin regions [119]. On average, indel frequencies at open chromatin sites were higher than those at closed chromatin sites by a factor of 1.6 ± 0.2 or 1.3 ± 0.2 in HEK 293T or HeLa cells, respectively [119]. These findings underscore the importance of genomic context when interpreting editing efficiency data.
Table 1: Factors Influencing CRISPR-Cas9 Editing Efficiency
| Factor | Impact on Editing Efficiency | Experimental Consideration |
|---|---|---|
| Chromatin State | 1.3-1.6x higher efficiency in open vs. closed chromatin | Account for genomic context in experimental design |
| sgRNA Design | Mismatched sgRNAs show 43-1100x reduced activity in closed chromatin | Optimize sgRNA specificity and binding affinity |
| Cas9 Variant | High-fidelity variants reduce off-targets but may affect on-target efficiency | Balance specificity with activity requirements |
| Delivery Method | Viral vs. non-viral delivery affects editing rates | Choose method based on cell type and application |
Advanced Applications: From Efficiency to Specificity
Beyond basic efficiency quantification, targeted deep sequencing enables sophisticated analyses of editing quality. The indel spectrum—the distribution and frequency of different insertion and deletion patterns—provides crucial insights into repair pathway preferences and potential functional consequences. Research utilizing deep sequencing has demonstrated that different CRISPR systems can produce distinct indel profiles, which may influence experimental outcomes and therapeutic safety [118].
Furthermore, targeted sequencing approaches have been adapted to quantify off-target effects, a critical safety parameter for therapeutic applications. Methods like DIG-seq (Digenome-seq using native chromatin DNA) leverage deep sequencing to identify genome-wide Cas9 off-target sites in a chromatin context, providing more physiologically relevant specificity profiles [119]. These approaches have revealed that chromatin accessibility significantly influences off-target activity, with mismatched sgRNAs showing dramatically reduced activity in closed chromatin regions compared to open chromatin regions [119].
Experimental Protocol for Targeted Deep Sequencing
Sample Preparation and Library Construction
The following protocol outlines a robust methodology for targeted deep sequencing of CRISPR-edited samples, adapted from established methods in the field [120]:
Step 1: DNA Extraction and Quality Control
- Extract genomic DNA from edited cells using standard methods (e.g., phenol-chloroform extraction or commercial kits)
- Quantify DNA concentration using fluorometric methods and assess quality via agarose gel electrophoresis or Bioanalyzer
- Ensure DNA integrity for optimal amplification of target regions
Step 2: Target Amplification
- Design primers flanking the on-target and validated off-target sites with overhangs compatible with Illumina sequencing platforms
- Amplify target regions using high-fidelity DNA polymerase (e.g., Phusion High-Fidelity DNA Polymerase, New England Biolabs) to minimize amplification errors
- Use approximately 100 ng of genomic DNA as template per PCR reaction
- Include control samples (untransfected cells and cells transfected with control sgRNA) to establish background mutation rates
Step 3: Library Preparation and Sequencing
- Purify PCR products with magnetic beads (e.g., Ampure XP, Agencourt)
- Normalize concentrations of amplified products and pool samples as needed
- Prepare Illumina-compatible sequencing libraries using a high-throughput library preparation kit (e.g., KAPA BioSystems)
- Sequence pooled libraries via 150-bp paired-end sequencing on an Illumina MiSeq or similar platform
Figure 1: Targeted Deep Sequencing Workflow. The process begins with genomic DNA extraction from CRISPR-edited cells, followed by target-specific amplification, library preparation, high-throughput sequencing, and bioinformatic analysis.
Bioinformatic Analysis Pipeline
Step 4: Data Processing and Variant Calling
- Align paired-end reads to the reference genome using alignment algorithms (e.g., bwa mem with default parameters)
- Filter high-quality reads (average quality score ≥ 30)
- Identify indel mutations overlapping the on- or off-target sites
- Include single-bp indels only if they occur directly adjacent to the predicted cleavage site
- Calculate editing efficiency as the percentage of reads containing indels at each target site
Step 5: Statistical Analysis
- Perform statistical comparisons between experimental conditions using appropriate tests (e.g., one-sided Fisher exact test)
- Adjust P-values for multiple comparisons using established methods (e.g., Benjamini and Hochberg correction)
- Generate comprehensive reports including editing efficiencies, indel spectra, and statistical significance
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of targeted deep sequencing for CRISPR analysis requires specific reagents and tools optimized for accuracy and reproducibility. The following table details essential components of the experimental pipeline:
Table 2: Research Reagent Solutions for Targeted Deep Sequencing
| Reagent/Material | Function | Example Product/Specification |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies target regions with minimal errors | Phusion High-Fidelity DNA Polymerase (NEB) |
| Magnetic Beads | Purifies PCR products and normalizes concentrations | Ampure XP Magnetic Beads (Agencourt) |
| Library Prep Kit | Prepares sequencing libraries from amplified products | KAPA HTP Library Preparation Kit (KAPA BioSystems) |
| Sequencing Platform | Generates high-quality sequence data | Illumina MiSeq (150-bp paired-end) |
| Alignment Software | Maps sequence reads to reference genome | bwa mem algorithm |
| Statistical Analysis Tool | Performs statistical tests on editing data | R software package (v3.2.2 or later) |
Integration with Broader CRISPR Workflows
Targeted deep sequencing does not operate in isolation but serves as a critical validation component within comprehensive CRISPR experimental designs. Recent advances in CRISPR technology highlight the growing importance of precise efficiency quantification. For instance, the development of anti-CRISPR protein systems such as LFN-Acr/PA, which can shut down Cas9 activity after editing is complete, requires precise measurement of editing efficiency and off-target reduction to validate performance [8]. Researchers using these systems rely on targeted deep sequencing to demonstrate the specificity improvements—reporting up to 40% reduction in off-target effects [8].
Similarly, the emergence of molecular glue degraders for controlling Cas9 activity (e.g., Cas9-degron systems that degrade Cas9 in the presence of FDA-approved pomalidomide) depends on accurate editing assessment to validate the reversible control of genome editing [121]. These systems demonstrate a 3- to 5-fold decrease in editing at on-target sites when degradation is initiated, measurements that require the sensitivity of targeted deep sequencing [121].
The growing application of artificial intelligence to CRISPR design further underscores the need for robust validation methods. AI-designed editors like those generated through large language models trained on CRISPR-Cas sequences represent a 4.8-fold expansion of diversity compared to natural proteins [118]. Validating the functionality of these novel editors necessitates precise quantification of editing efficiency and specificity, positioning targeted deep sequencing as an indispensable component of the AI-driven protein design pipeline.
Targeted deep sequencing represents an indispensable methodology for characterizing CRISPR-Cas9 genome editing outcomes in biochemical research and therapeutic development. By providing quantitative data on editing efficiency, indel profiles, and off-target effects, this approach enables researchers to make informed decisions about CRISPR system selection, optimization, and application. As CRISPR technology continues to evolve—with advances in AI-designed editors, controllable systems, and therapeutic applications—targeted deep sequencing will remain essential for validating new systems and ensuring their safe, effective implementation. The protocols and considerations outlined in this application note provide a foundation for researchers to implement this critical methodology in their genome editing workflows.
Within the framework of using CRISPR-Cas9 for precise genome editing in biochemistry research, validating editing outcomes is a critical step. The choice of detection method profoundly impacts the accuracy, reliability, and interpretation of experimental results. This application note provides a comparative analysis of two commonly used techniques: the T7 Endonuclease I (T7E1) assay and Next-Generation Sequencing (NGS). We evaluate their sensitivity, accuracy, and practical applicability to guide researchers and drug development professionals in selecting the optimal method for their specific needs. Evidence from systematic benchmarking reveals that while T7E1 offers a rapid initial assessment, NGS provides a comprehensive and quantitative readout, establishing it as the gold standard for sensitive and accurate editing detection [122] [123].
Methodological Comparison: T7E1 vs. NGS
Fundamental Principles and Workflows
The core principles of T7E1 and NGS are fundamentally different, which directly leads to their disparities in performance.
- T7 Endonuclease I (T7E1) Assay: This method is a PCR-based, non-sequencing approach. After PCR amplification of the target locus, the amplicons are denatured and re-annealed. This process creates heteroduplex DNA molecules when indel-containing strands pair with wild-type strands, resulting in mismatches. The T7E1 enzyme recognizes and cleaves these mismatched sites. The cleavage products are then separated by gel electrophoresis, and the editing efficiency is semi-quantitatively estimated based on the band intensities of the cleaved versus uncleaved products [124] [123].
- Next-Generation Sequencing (NGS): Also referred to as targeted amplicon sequencing (AmpSeq), NGS is a sequencing-based technique. The target site is PCR-amplified, and the resulting library is subjected to high-throughput sequencing. This provides the exact DNA sequence for thousands of individual DNA molecules, allowing for precise quantification of all insertion, deletion, and substitution events at the target site. It delivers absolute, nucleotide-level resolution of the editing outcomes [125] [122] [123].
The experimental workflow for these methods is outlined in the diagram below.
Performance Benchmarking: Quantitative Data
Systematic benchmarking studies, which use NGS as the reference standard, have quantitatively highlighted the limitations of the T7E1 assay. The following table summarizes key performance metrics.
Table 1: Performance Benchmarking of T7E1 vs. NGS
| Parameter | T7E1 Assay | NGS (AmpSeq) | Supporting Evidence |
|---|---|---|---|
| Accuracy & Dynamic Range | Low accuracy, particularly at high editing rates; compresses efficiency (>90% NGS appears as ~30-40% by T7E1) [122]. Underestimates low-frequency edits (<10%) [125]. | High accuracy across the entire dynamic range (0-100%) [125] [122]. | A study found T7E1 estimates averaged 22% across 19 targets, while NGS revealed the true average was 68% [122]. |
| Sensitivity | Low sensitivity; struggles to detect edits below ~0.1-1% allele frequency and is unreliable for low-frequency edits (<10%) [125]. | High sensitivity; can detect edits at frequencies as low as <0.1% [125]. | In plant studies, T7E1 failed to detect editing at less than 0.1% efficiency, whereas AmpSeq was consistently sensitive [125]. |
| Quantitative Output | Semi-quantitative. Provides an estimated mutation frequency based on gel band intensity [124] [123]. | Fully quantitative. Provides the exact percentage and type of every indel present in the population [125] [123]. | NGS provides a comprehensive profile of all CRISPR-mediated mutagenesis events [125]. |
| Information Richness | Low. Reveals only the presence of indels, not their specific sequences or sizes [123]. | High. Identifies the exact sequences, sizes, and spectrum of all indels and other sequence variations [125] [122]. | Targeted NGS provides a comprehensive view of the indels generated [123]. |
Detailed Experimental Protocols
Protocol: T7E1 Mismatch Cleavage Assay
Principle: Detect indels via enzyme cleavage of heteroduplex DNA formed between wild-type and mutant sequences [124].
Materials:
- T7 Endonuclease I (e.g., M0302, New England Biolabs)
- NEBuffer 2 (or compatible buffer supplied with enzyme)
- High-Fidelity PCR Master Mix
- Agarose Gel Electrophoresis System
- DNA Clean-up Kit
Procedure:
- PCR Amplification: Amplify the target genomic region (200-500 bp) from edited and control samples using high-fidelity PCR. Include a negative control (wild-type DNA).
- PCR Product Purification: Purify the PCR products to remove primers and enzymes.
- Heteroduplex Formation:
- Mix 8 µL of purified PCR product.
- Place in a thermocycler and run the following program:
- Denature: 95°C for 5 minutes.
- Re-anneal: Ramp down from 95°C to 25°C at a rate of -0.5°C per second.
- Hold at 4°C.
- T7E1 Digestion:
- To the re-annealed DNA, add:
- 1 µL of NEBuffer 2.
- 1 µL of T7 Endonuclease I enzyme.
- Mix gently and centrifuge briefly.
- Incubate at 37°C for 30 minutes.
- To the re-annealed DNA, add:
- Analysis:
- Terminate the reaction by adding a stop solution (e.g., EDTA) or loading dye.
- Separate the digestion products on a 1.5-2% agarose gel.
- Visualize bands under UV light. The editing efficiency is estimated using the formula:
- Editing Frequency (%) = [1 - (1 - (b + c)/(a + b + c))^{1/2}] × 100
- Where
ais the intensity of the undigested PCR product band, andbandcare the intensities of the cleavage products [124].
- Where
Protocol: Targeted Next-Generation Sequencing (Amplicon Sequencing)
Principle: Deep sequencing of PCR-amplified target loci for precise, quantitative analysis of all editing events [125] [122].
Materials:
- NGS Library Preparation Kit (e.g., Illumina, MGI)
- Indexed Adapters (for multiplexing)
- High-Fidelity DNA Polymerase
- Magnetic Beads (for clean-up and size selection)
- Bioinformatics Software (for read alignment and variant calling)
Procedure:
- First-Stage PCR (Target Amplification):
- Design primers with overhangs complementary to the NGS adapter sequences.
- Amplify the target region from genomic DNA. Use a high-fidelity polymerase to minimize PCR errors.
- Purify the amplicons.
- Second-Stage PCR (Indexing and Adapter Ligation/Addition):
- Attach unique dual indices (UDIs) and full sequencing adapters to the purified amplicons via a limited-cycle PCR.
- This step allows for the multiplexing of multiple samples in a single sequencing run.
- Library Quality Control and Pooling:
- Quantify the final libraries using a fluorometric method (e.g., Qubit).
- Assess library size distribution using a bioanalyzer or fragment analyzer.
- Normalize and pool libraries in equimolar ratios.
- Sequencing:
- Load the pooled library onto an NGS instrument (e.g., Illumina MiSeq, iSeq) following the manufacturer's instructions.
- Sequence with paired-end reads of sufficient length to cover the entire amplicon.
- Data Analysis:
- Demultiplexing: Assign sequences to individual samples based on their unique indices.
- Quality Control: Filter reads based on quality scores (e.g., using FastQC).
- Alignment: Map high-quality reads to the reference genome or target sequence (e.g., using BWA or Bowtie2).
- Variant Calling: Use specialized tools (e.g., CRISPResso2, AmpliCan) to identify and quantify indels relative to the expected cut site. The output provides the percentage of reads with indels (total editing efficiency) and a detailed spectrum of all mutation types [125] [122].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CRISPR Editing Detection
| Item | Function/Application |
|---|---|
| T7 Endonuclease I | The core enzyme for the T7E1 assay; cleaves heteroduplex DNA at mismatch sites [124]. |
| High-Fidelity PCR Master Mix | Essential for accurate amplification of the target locus prior to both T7E1 and NGS analysis, minimizing PCR-introduced errors. |
| NGS Library Prep Kit | Provides all necessary enzymes, buffers, and adapters for converting PCR amplicons into a sequencing-ready library. |
| Indexed Adapters | Enable sample multiplexing in NGS by assigning a unique barcode to each sample's amplicons. |
| Droplet Digital PCR (ddPCR) | An alternative quantitative method offering high sensitivity and precision for specific, known edits; useful for validation [125] [124]. |
The choice between T7E1 and NGS is a trade-off between speed/cost and accuracy/comprehensiveness. The following diagram summarizes the decision-making logic for method selection.
For biochemistry research and drug development applications where precision is paramount, NGS is the unequivocal gold standard. It is the recommended method for characterizing novel gRNAs, quantifying editing efficiencies in heterogeneous cell pools, and documenting the exact spectrum of edits for regulatory and publication purposes. The T7E1 assay serves best in early-stage, high-throughput gRNA screening where its speed and low cost are advantageous, provided researchers are aware of its significant quantitative limitations.
Within the framework of using CRISPR-Cas9 for precise genome editing in biochemistry research, validating editing outcomes is a critical step following the introduction of a double-strand break (DSB). The DSB is primarily repaired by the cell's endogenous non-homologous end joining (NHEJ) pathway, an error-prone mechanism that often results in small insertions or deletions (indels) at the target site [126] [127]. Characterizing the spectrum and frequency of these indels is essential for assessing the efficiency and specificity of the CRISPR-Cas9 reagents. While next-generation sequencing (NGS) is considered the gold standard for comprehensive analysis, its cost, time, and bioinformatic requirements can be prohibitive [123]. This application note details two rapid, cost-effective methods for the quantitative analysis of indel mutations: Tracking of Indels by DEcomposition (TIDE) and Indel Detection by Amplicon Analysis (IDAA).
TIDE (Tracking of Indels by Decomposition)
TIDE is a computational method that quantitatively decomposes complex Sanger sequencing traces from edited cell pools [128] [129]. It requires standard capillary sequencing data from both a control (non-edited) sample and the test (edited) sample. The TIDE algorithm aligns the sequence traces from the two samples and then analyzes the decomposition window downstream of the CRISPR-Cas9 cut site. Using non-negative linear regression modeling, it identifies the combination of indels that best explains the composite sequence trace of the edited sample, providing a detailed profile of the predominant indel types and their frequencies [128].
IDAA (Indel Detection by Amplicon Analysis)
IDAA is a fluorescence-based capillary electrophoresis method that detects length variations in PCR amplicons caused by indels [130]. Following PCR amplification of the target locus, one primer is labeled with a fluorescent dye. The amplicons are then analyzed by capillary electrophoresis, which separates DNA fragments by size. The resulting chromatogram displays a series of peaks, where the height and position of each peak correspond to the abundance and size of a specific indel, providing a direct quantification of the editing spectrum [130].
Table 1: Core Principle and Data Input of TIDE and IDAA
| Feature | TIDE Method | IDAA Method |
|---|---|---|
| Core Principle | Decomposition of Sanger sequencing traces via algorithm [129] | Capillary electrophoresis of fluorescently labeled PCR amplicons [130] |
| Primary Data Input | Sanger sequencing chromatograms (.ab1, .scf files) [128] | Fluorescently labeled PCR products |
| Typical Experiment Duration | 1-2 days (from DNA to result) | 1 day (from DNA to result) |
| Key Output | Indel spectrum, frequency, R² goodness-of-fit [128] | Electropherogram with peaks for each indel size [130] |
Experimental Workflow
The following diagram illustrates the core workflows for TIDE and IDAA, highlighting their parallel initial steps and divergent analytical paths.
Detailed Experimental Protocols
TIDE Protocol
Step 1: DNA Extraction and PCR Amplification
- Isolate genomic DNA from your control (e.g., non-transfected) and CRISPR-edited cell pools using a standard kit.
- Design PCR primers to amplify a region of ~500-700 bp enclosing the CRISPR target site. The projected break site should be located preferably ~200 bp downstream from the sequencing start site [128].
- Perform PCR amplification using a high-fidelity DNA polymerase to minimize PCR-induced errors.
Step 2: Sanger Sequencing
- Purify the PCR amplicons.
- Submit the purified PCR products for Sanger sequencing using one of the PCR primers. It is recommended to sequence the opposite strand as well to confirm results [128].
Step 3: TIDE Web Analysis
- Access the TIDE web tool at
https://tide.nki.nl[128]. - Input the sgRNA sequence: Enter the 20nt guide sequence (without the PAM) in the designated field.
- Upload chromatogram files: Upload the
.ab1or.scffiles for the control and test samples. - Set parameters: Use the default settings or adjust advanced parameters as needed:
- Alignment window: The sequence segment used to align the control and test samples (default left boundary: 100 bp).
- Decomposition window: The sequence segment used for decomposition (default is the maximum window possible).
- Indel size range: The maximum size of indels to be modeled (default: 10 bp).
- P-value threshold: Significance cutoff for decomposition (default: p < 0.001) [128].
- Click "Update View" to run the analysis.
Step 4: Interpret Results
- The main output is an Indel spectrum plot showing the types and frequencies of identified indels.
- Check the R² value as a measure of the goodness of fit; aim for R² > 0.9 [128].
- Review the Aberrant sequence signal plot for quality control. The control sample should show a low, evenly distributed signal, while the test sample should show a low signal before the break site and a higher signal after it [128].
IDAA Protocol
Step 1: DNA Extraction and Fluorescent PCR
- Isolate genomic DNA from control and edited cell pools.
- Design PCR primers to generate an amplicon of 150-500 bp covering the target site.
- Perform a multiplex PCR where one of the primers is labeled with a fluorescent dye (e.g., FAM, HEX, or TET) [130].
Step 2: Capillary Electrophoresis
- Pool a small aliquot of the fluorescently labeled PCR product with a DNA size standard and formamide.
- Denature the samples and run them on a capillary electrophoresis instrument (e.g., an ABI genetic analyzer).
- The instrument separates DNA fragments by single-base resolution, detecting the fluorescent signal of each fragment.
Step 3: Data Analysis
- The software generates an electropherogram for each sample.
- In the control sample, a single dominant peak represents the wild-type allele.
- In the edited sample, additional peaks appear, shifted relative to the wild-type peak. Peaks to the left indicate deletions, while peaks to the right indicate insertions.
- The relative area under each peak corresponds to the frequency of that specific indel in the cell population.
Performance Comparison and Data Output
Quantitative Performance and Limitations
When selecting an analysis method, understanding their comparative performance against the gold standard, Next-Generation Sequencing (NGS), is crucial for data interpretation.
Table 2: Comparison of Performance and Limitations of CRISPR Analysis Methods
| Aspect | TIDE | IDAA | NGS (Gold Standard) |
|---|---|---|---|
| Quantitative Accuracy | High correlation with NGS for pools [130]. Can miscall alleles in clones [130]. | Predictive for pools; may miscall in clones [130]. | Highest accuracy and sensitivity [123]. |
| Detection Limit | Accurately quantifies indels down to ~1-2% frequency. | Similar sensitivity to TIDE. | Can detect very rare indels (<0.1%). |
| Key Advantage | Provides explicit indel sequences and statistical significance [128]. | Rapid, high-throughput, and does not require sequencing [130]. | Comprehensive view of all sequence variations [123]. |
| Primary Limitation | Cannot detect large deletions or complex rearrangements [128]. | Does not provide actual DNA sequence, only indel size [130]. | High cost, time-consuming, requires bioinformatics [123]. |
| Best For | Rapid, sequence-specific quantification of small indels in heterogeneous pools. | High-throughput screening of sgRNA activity and quick quantification of common indels. | Definitive, comprehensive analysis of all editing outcomes, including complex mutations. |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for TIDE and IDAA
| Reagent / Material | Function / Description | Example / Note |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies the target genomic locus with minimal errors for downstream analysis. | Kits from suppliers like NEB, Thermo Fisher, or Takara. |
| Sanger Sequencing Service | Generates the chromatogram data files required for TIDE analysis. | In-house facility or commercial service (e.g., Genewiz). |
| Fluorescently Labeled Primer | One PCR primer is tagged with a fluorophore for detection in IDAA. | FAM, HEX, or TET dyes are commonly used. |
| Capillary Electrophoresis System | Separates fluorescently labeled amplicons by size with single-base resolution. | ABI 3500 or similar genetic analyzers. |
| TIDE Web Tool | The algorithm that decomposes sequencing traces and quantifies indels. | Freely available at https://tide.nki.nl [128]. |
| Genomic DNA Extraction Kit | Isolates high-quality, PCR-ready DNA from control and edited cell pools. | Kits from Qiagen, Macherey-Nagel, or similar. |
TIDE and IDAA represent powerful, accessible tools for the rapid characterization of indel profiles in CRISPR-Cas9 edited cell pools. Both methods effectively bridge the gap between rudimentary qualitative assays and comprehensive but resource-intensive NGS. TIDE is ideal for researchers who require explicit sequence information from standard Sanger data, while IDAA offers an exceptionally fast and simple workflow for size-based quantification. Integrating these validation strategies into a CRISPR genome editing workflow allows biochemistry researchers and drug development professionals to efficiently quantify and characterize editing outcomes, thereby accelerating the development of precise genetic tools and therapies. For the most critical applications or when complex mutations are suspected, confirmation of key results with targeted NGS is recommended.
In the realm of precise genome editing, biochemical assays serve as indispensable tools for the in vitro validation of CRISPR-Cas9 components prior to their use in complex cellular or in vivo environments. These assays translate biological interactions into quantifiable data, providing researchers with critical insights into the functionality, specificity, and efficiency of their gene-editing tools. Within CRISPR-Cas9 research, robust biochemical validation is paramount for de-risking experiments, optimizing reagents, and interpreting editing outcomes accurately. The fundamental components of the CRISPR-Cas9 system include the Cas nuclease, which acts as molecular scissors, and the guide RNA (gRNA), which confers sequence specificity [131]. Biochemical assays are uniquely positioned to probe the activity of these components in isolation, enabling the establishment of a foundational understanding of system performance that informs subsequent experimental stages.
Key Limitations of Biochemical Assays
Despite their utility, biochemical assays possess inherent limitations that researchers must acknowledge to ensure appropriate data interpretation. The following table summarizes the primary constraints encountered when employing these assays for CRISPR-Cas9 validation.
Table 1: Key Limitations of Biochemical Assays for CRISPR-Cas9 Validation
| Limitation Category | Specific Challenge | Impact on CRISPR-Cas9 Validation |
|---|---|---|
| Complexity of Biological Systems | Inability to fully recapitulate the intracellular environment (e.g., chromatin structure, DNA repair machinery) [132]. | An assay showing high cleavage efficiency in vitro may not predict on-target editing in cells, as nuclear access and local chromatin状态 are critical factors. |
| Genotoxic Risk Assessment | Difficulty in detecting rare, unintended editing outcomes like large insertions (LgIns) and structural variants without highly sensitive methods [133]. | Standard gel-based cleavage assays can miss LgIns of repetitive elements, regulatory sequences, or concatemeric donor DNA, which are a ubiquitous risk [133]. |
| Assay Variability and Validation | High variability due to biological materials and complex manufacturing processes [132]. | Complicates the validation of guide RNA (gRNA) potency and leads to inconsistent results across batches and laboratories. |
| Lack of Reference Materials | Absence of well-characterized, stable reference materials for newer gene-editing tools [132]. | Hinders standardization and cross-comparison of Cas nuclease activity or gRNA potency between different labs and studies. |
| Defining Potency and Activity | A single assay rarely captures the full functionality of a therapy with multiple mechanisms of action [132]. | For CRISPR-based therapeutics, an assay measuring DNA cleavage may not fully represent the final therapeutic potency, which depends on precise HDR. |
A significant and often overlooked genotoxic risk is the integration of unintended genetic material at the Cas9-induced break site. Recent sensitive analyses, such as Unique Molecular Identifier (UMI)-based long-read sequencing (IDMseq), have revealed that Cas9 editing—with or without donor templates—can consistently induce large insertions (LgIns). These insertions can originate from retrotransposable elements (LINEs, SINEs, LTRs), host genomic coding and regulatory sequences, and significant unintended full-length and concatemeric integrations of donor DNA [133]. The frequency of these LgIns can increase with the presence of donor templates, underscoring a critical limitation of standard biochemical assays that lack the sensitivity to detect these unintended outcomes.
Appropriate Use Cases and Validation Protocols
Biochemical assays excel in specific, well-defined use cases within the CRISPR-Cas9 workflow. They are particularly powerful for the initial, rapid pre-validation of system components.
Use Case 1: Pre-validation of Guide RNA (gRNA) Efficiency
A primary application is the in vitro cleavage assay to pre-validate the efficiency of designed gRNAs before committing to costly and time-consuming cellular experiments [134].
Protocol: In Vitro DNA Cleavage Assay using Cas9 Ribonucleoprotein (RNP) Complex
- Principle: The recombinant Cas9 protein is pre-complexed with gRNA (as a synthetic crRNA:tracrRNA duplex or sgRNA) to form an RNP complex. This complex is then incubated with a purified DNA substrate containing the target site. Successful cleavage is visualized by gel electrophoresis [134].
- Materials:
- Recombinant S. pyogenes Cas9 Nuclease
- Target-specific crRNA and tracrRNA (or synthetic sgRNA)
- Purified DNA substrate (e.g., PCR-amplified genomic region of interest)
- Nuclease reaction buffer (e.g., 10X Cas9 nuclease reaction buffer: 1 M NaCl, 0.1 M MgCl₂, 0.5 M Tris-HCl, 1 mg/ml BSA, pH 7.9)
- Thermocycler and electrophoresis equipment
- Step-by-Step Method:
- Annealing: Combine equimolar amounts of crRNA and tracrRNA (e.g., 5 µg of crRNA and 10 µg of tracrRNA). Heat the mixture to 95°C for 5 minutes and then cool slowly to 25°C in a thermocycler to form the duplex guide RNA [134].
- RNP Complex Formation: Mix the annealed RNA duplex with the recombinant Cas9 protein. This can be done by incubating before the assay or during the cleavage reaction itself. A typical pre-incubation might be 10-20 minutes at room temperature.
- Cleavage Reaction: Assemble a 30 µL reaction containing:
- 1x Cas9 nuclease reaction buffer (providing Mg²⁺ as a cofactor)
- The assembled RNP complex (e.g., 200-500 nM final concentration)
- The DNA substrate (e.g., 100-200 ng of PCR product)
- Nuclease-free water.
- Incubation: Incubate the reaction at 37°C for 1 hour.
- Analysis: Stop the reaction (e.g., with EDTA or by heating). Analyze the products by agarose gel electrophoresis. Successful cleavage is indicated by the disappearance of the full-length DNA substrate band and the appearance of two smaller, cleaved product bands.
- Advantages: This RNP-based format is rapid, highly efficient, and avoids the pitfalls of plasmid-based delivery, such as variable expression and prolonged Cas9 exposure, which can exacerbate off-target effects [134].
The following workflow diagram illustrates the key steps and decision points in this protocol:
Diagram 1: Workflow for in vitro gRNA pre-validation.
Use Case 2: Validation of Editing Outcomes in Cellular Systems
Following cellular editing, biochemical assays are crucial for quantifying on-target editing efficiency and characterizing the types of induced mutations.
Protocol: Enzymatic Mismatch Cleavage for Indel Detection
- Principle: After CRISPR editing, DNA is isolated from cells. The target locus is PCR-amplified and denatured then re-annealed. This process creates heteroduplexes—double-stranded DNA molecules with mismatches—if indels are present. Enzymes like T7 Endonuclease I or a specialized mixture like Authenticase cleave these heteroduplexes at the mismatch sites. The cleavage products are visualized on a gel, and the band intensities are used to estimate the indel frequency [135].
- Materials:
- Genomic DNA from edited cells
- PCR reagents and target-specific primers
- T7 Endonuclease I or Authenticase
- Appropriate reaction buffer
- Gel electrophoresis system
- Step-by-Step Method:
- PCR Amplification: Perform PCR on the purified genomic DNA to amplify the region surrounding the Cas9 target site.
- Heteroduplex Formation: Denature the PCR products by heating (e.g., 95°C for 5-10 minutes) and then re-anneal by slowly cooling to room temperature. This step allows strands from different alleles (wild-type and edited) to hybridize.
- Enzymatic Digestion: Treat the re-annealed PCR products with the mismatch-sensitive nuclease (e.g., T7 Endonuclease I) according to the manufacturer's protocol.
- Analysis: Separate the digested products by agarose or polyacrylamide gel electrophoresis. The cleavage products (additional bands of smaller size) indicate the presence of indels. The editing efficiency can be estimated by comparing the band intensities of the cleaved and uncleaved products.
Table 2: Comparison of Methods for Validating CRISPR Editing Efficiency
| Method | Principle | Key Advantage | Key Limitation | Best Use Case |
|---|---|---|---|---|
| In Vitro RNP Cleavage [134] | Direct DNA cleavage by pre-assembled RNP complex. | Rapid, cost-effective pre-validation of gRNA function. | Does not account for cellular context. | Initial gRNA screening and optimization. |
| Enzymatic Mismatch (T7E1/Authenticase) [135] | Cleavage of heteroduplex DNA at mismatch sites. | Simple, relatively inexpensive; does not require sequencing. | Low resolution; cannot identify specific sequence of indels. | Quick, semi-quantitative estimation of overall indel frequency. |
| Cas9 Digest Assay [135] | Re-digestion of PCR amplicons with Cas9 RNP. | Assesses locus modification at efficiencies >50%. | Requires a functional PAM site to remain post-editing. | Confirming highly efficient knock-outs. |
| Next-Generation Sequencing (NGS) | High-throughput sequencing of the target locus. | Gold standard for accuracy; provides exact indel sequences and frequency. | Higher cost and computational burden. | Definitive, quantitative analysis of all editing outcomes, including LgIns [133]. |
Use Case 3: Assessing and Mitigating Genotoxic Risks
As identified in Table 1, a major limitation of standard assays is the failure to detect Large Insertions (LgIns). Therefore, an appropriate use case for advanced biochemical and biophysical methods is the specific assessment of these genotoxic risks.
- Recommended Technique: Next-Generation Sequencing (NGS), particularly methods incorporating Unique Molecular Identifiers (UMIs) like IDMseq, is required for the sensitive and quantitative detection of LgIns and other complex structural variants [133].
- Mitigation Strategy: Research indicates that the choice of donor template and Cas9 variant influences the frequency of unwanted insertions. For example, using phosphorylated double-stranded DNA (dsDNA) donors has been shown to reduce large insertions and deletions by almost two-fold without compromising HDR efficiency. Similarly, high-fidelity Cas9 variants (e.g., HiFi Cas9) can reduce off-target effects and may lower the incidence of certain on-target artifacts [133].
The Scientist's Toolkit: Essential Reagents for Validation
Table 3: Key Research Reagent Solutions for CRISPR-Cas9 Biochemical Validation
| Reagent / Kit | Function | Specific Example & Use |
|---|---|---|
| Recombinant Cas9 Nuclease | The core enzyme for creating double-stranded breaks in DNA. | S. pyogenes Cas9 (NEB #M0386) used in in vitro cleavage and re-digestion assays [135]. |
| crRNA & tracrRNA | Short RNA components that form the functional guide RNA when complexed. | Alt-R CRISPR-Cas9 crRNA and tracrRNA (IDT) can be annealed to form a functional guide for RNP complexes, offering high efficiency and reduced immune response in cells [134]. |
| Mismatch Detection Enzymes | Detect insertions/deletions (indels) by cleaving heteroduplex DNA. | T7 Endonuclease I or the more robust Authenticase (NEB #M0689) used in enzymatic mismatch cleavage assays post-cellular editing [135]. |
| NGS Library Prep Kits | Prepare amplified target DNA for high-throughput sequencing. | NEBNext Ultra II DNA Library Prep Kits (e.g., NEB #E7645) used for preparing amplicons from edited genomic regions for deep sequencing analysis [135]. |
| High-Fidelity Cas9 Variants | Engineered nucleases with reduced off-target activity. | Alt-R S.p. HiFi Cas9 (IDT) can be used to minimize off-target cleavage while maintaining on-target efficiency, useful for both in vitro and cellular work [133]. |
| Phosphorylated dsDNA Donors | Double-stranded DNA templates for precise Homology-Directed Repair (HDR). | Using phosphorylated dsDNA donors instead of single-stranded or circular donors has been shown to reduce unintended LgIns and large deletions [133]. |
The assembly of the Cas9 Ribonucleoprotein (RNP) complex is a critical step for many biochemical assays and for efficient electroporation into cells. The following diagram depicts the structure of this core functional unit:
Diagram 2: Structure of the Cas9 RNP complex.
Biochemical assays are foundational for the rigorous validation of CRISPR-Cas9 genome editing tools, offering rapid, controlled insights into gRNA efficiency and nuclease activity. Their appropriate use cases are centered on the pre-validation of components and the initial characterization of editing outcomes. However, researchers must be acutely aware of their limitations, particularly the inability to fully model the cellular environment and the failure of standard methods to detect complex genotoxic outcomes like large insertions. A robust validation strategy must therefore leverage a tiered approach: initiating with in vitro biochemical assays (e.g., RNP cleavage) for screening, progressing to cellular assays (e.g., enzymatic mismatch detection) for initial confirmation, and culminating in sensitive, advanced molecular analyses (e.g., UMI-NGS) for a comprehensive safety profile. By understanding and respecting these boundaries, scientists can effectively harness the power of biochemical assays to accelerate the development of safer and more precise CRISPR-Cas9-based therapies.
The transition of CRISPR-Cas9 from a powerful research tool to a clinically approved therapeutic represents a paradigm shift in precision medicine. Clinical validation is the comprehensive process of generating robust evidence to demonstrate the safety, efficacy, and quality of a therapeutic product for human use. For CRISPR-based medicines, this process involves unique considerations specific to genome-editing technologies. The first CRISPR-based medicine, Casgevy (exagamglogene autotemcel), received approval for treating sickle cell disease (SCD) and transfusion-dependent beta thalassemia (TBT), establishing a foundational regulatory pathway for subsequent therapies [11] [136]. This application note outlines the key regulatory requirements and experimental protocols for the clinical validation of CRISPR-Cas9 therapeutics, providing a structured framework for researchers and drug development professionals.
The clinical development of CRISPR therapies operates within a rigorous regulatory framework designed to balance innovation with patient safety. Unlike conventional small-molecule drugs, CRISPR medicines involve permanent modification of the human genome, necessitating specialized assessment criteria. Regulatory agencies including the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) require comprehensive data packages demonstrating target engagement, editing precision, off-target profiling, and long-term safety monitoring [11] [137] [138]. The successful approval of Casgevy has created a precedent for the type and quality of evidence required, with ongoing clinical trials continuing to refine these standards across different therapeutic areas and delivery platforms.
Regulatory Framework and Clinical Trial Design
Clinical Trial Phases and Objectives
Clinical validation of CRISPR therapeutics follows a phased development approach similar to conventional pharmaceuticals but with genome-specific considerations at each stage. Each phase addresses distinct research questions and accumulates evidence for regulatory assessment.
Table 1: Clinical Trial Phases for CRISPR Therapeutics
| Phase | Primary Objectives | Key Endpoints | Patient Population | Duration |
|---|---|---|---|---|
| Phase I | Assess safety, tolerability, and preliminary dosing [11] | Frequency and severity of adverse events, pharmacokinetics, initial editing efficiency [11] [138] | Small cohort (20-100 patients) with advanced disease | 1-2 years |
| Phase II | Evaluate efficacy and optimal dosing [11] | Biomarker response, clinical outcome measures, further safety assessment [11] [137] | Larger cohort (100-300 patients) with defined disease characteristics | 2-3 years |
| Phase III | Confirm efficacy, monitor adverse reactions [11] | Clinically significant endpoints, risk-benefit assessment, comparison to standard of care [11] | Large population (300-1000+ patients) across multiple centers | 3-4 years |
| Phase IV | Post-marketing surveillance | Long-term safety, rare adverse events, additional population data [11] | All treated patients after approval | 5-15 years |
The phase I trial for Intellia Therapeutics' hereditary transthyretin amyloidosis (hATTR) treatment exemplifies this approach, establishing safety and demonstrating ~90% reduction in disease-related TTR protein levels [11]. This trial also illustrated the potential for long-lasting effects, with all 27 participants who reached two-year follow-up showing sustained response [11]. Recent trials have also explored redosing strategies, with Intellia reporting the first ever administration of multiple doses of an in vivo CRISPR therapy delivered by lipid nanoparticle (LNP) [11].
Regulatory Designations and Pathways
Several regulatory mechanisms can accelerate the development of promising CRISPR therapies. The FDA's Regenerative Medicine Advanced Therapy (RMAT) designation, granted to CRISPR Therapeutics' CTX112 for B-cell malignancies, facilitates efficient development and review of products addressing unmet medical needs [55]. The landmark case of a personalized CRISPR treatment for an infant with CPS1 deficiency demonstrated a rapid regulatory pathway, with development, FDA approval, and patient delivery achieved in just six months [11]. This case establishes a precedent for ultra-personalized CRISPR approaches for rare genetic disorders and highlights the importance of engaging regulatory agencies early in the development process.
Essential Analytical Methods and Validation Protocols
Editing Efficiency and Specificity Assessment
Comprehensive characterization of genome editing outcomes is fundamental to clinical validation. The following protocol outlines key experiments for assessing editing efficiency and specificity in clinical samples.
Table 2: Analytical Methods for Assessing CRISPR Editing
| Parameter | Method | Key Reagents | Validation Criteria | Regulatory Standard |
|---|---|---|---|---|
| On-target editing efficiency | NGS amplicon sequencing [137] | Target-specific primers, NGS library prep kits | >90% homology-directed repair efficiency for knock-ins; limit of detection: 0.1% | FDA guidance on human gene therapy products |
| Off-target editing | GUIDE-seq, CIRCLE-seq, DISCOVER-Seq [137] | Whole genome sequencing kits, Cas9-specific antibodies | Profile potential off-target sites predicted by in silico tools; experimental validation required | Demonstration of minimal off-target effects (<0.1% at any site) |
| Structural variants | Karyotyping, FISH, optical genome mapping | Metaphase chromosome spreads, fluorescent probes | Detection of large deletions (>100 bp) and translocations | Absence of clinically significant structural variations |
| Editing persistence | Long-term follow-up sequencing [11] | Patient-derived cells at multiple timepoints | Stable editing percentage over time (e.g., 2+ years) [11] | Continued expression of therapeutic effect without decline |
Next-generation sequencing (NGS) represents the gold standard for quantifying editing efficiency, with sensitivities down to 0.1% for minor indels. For the CPS1 deficiency case, researchers monitored editing percentage after each LNP-administered dose, observing incremental improvement with redosing [11]. Newer methods like AutoDISCO, a CRISPR-Cas-based tool for detecting off-target genome edits using minimal patient tissue, are emerging to meet regulatory demands for comprehensive off-target profiling in therapeutic workflows [137].
Functional Validation Assays
Beyond genomic analyses, functional assays demonstrate therapeutic activity at molecular and cellular levels. For Casgevy, successful editing of the BCL11A gene results in increased fetal hemoglobin (HbF) production, directly measured via HPLC of patient blood samples [55]. In Intellia's hereditary angioedema (HAE) trial, researchers used blood tests to quantify reduction in kallikrein protein (average 86% reduction in high-dose group) and tracked the frequency of inflammation attacks, with eight of eleven participants in the high-dose group being attack-free during the 16-week observation period [11].
Protocol: Functional Validation of CRISPR Editing in Hematopoietic Stem Cells
- Cell Collection and Processing: Collect CD34+ hematopoietic stem and progenitor cells (minimum 3×10^6 cells/kg patient weight) via apheresis after mobilization [55].
- CRISPR Delivery: Electroporate cells with CRISPR-Cas9 ribonucleoprotein complex targeting the therapeutic locus (e.g., BCL11A erythroid enhancer for Casgevy) using optimized parameters (1400V, 10ms pulse width, 3 pulses).
- Quality Control Assessment:
- Determine cell viability via trypan blue exclusion (>70% post-electroporation)
- Quantify editing efficiency via T7E1 assay or NGS (>25% indels required for progression)
- Verify sterility (bacterial/fungal culture, mycoplasma testing)
- Functional Potency Assay:
- Culture edited cells in erythroid differentiation medium (SCF, EPO, IL-3) for 14 days
- Measure HbF production via FACS (anti-HbF antibody) and HPLC
- Success criterion: >20% HbF-positive cells in differentiated erythroblasts
- Animal Model Validation:
- Transplant edited CD34+ cells into immunodeficient NSG mice (0.1×10^6 cells/mouse)
- Analyze human cell engraftment in bone marrow at 16 weeks via flow cytometry (anti-hCD45)
- Assess HbF expression in peripheral blood erythrocytes at 20 weeks
Safety Assessment and Risk Mitigation Strategies
Preclinical Safety Pharmacology
Comprehensive safety assessment begins with extensive preclinical studies evaluating potential risks unique to genome-editing therapies. The following areas require particular attention:
Immune Response Profiling: CRISPR-Cas systems derived from bacterial proteins pose immunogenicity risks. Assess anti-Cas9 antibody titers in patient serum pre- and post-treatment. Monitor for infusion-related reactions, particularly with LNP delivery where mild-to-moderate infusion-related events have been commonly observed [11]. Evaluate T-cell responses against Cas epitopes via ELISpot assays.
Genotoxicity Assessment: Conduct integrated genotoxicity testing including:
- In vitro transformation assays in edited primary cells
- Tumorigenicity studies in immunodeficient mice
- Comprehensive genomic analysis for large deletions, translocations, and chromothripsis
The recent pause in Intellia's Phase 3 trials for nexiguran ziclumeran after a patient experienced severe liver toxicity (Grade 4 elevated enzymes and bilirubin) highlights the importance of robust safety monitoring, even in late-stage trials [137]. Although delivery vectors were not immediately suspected, such events trigger comprehensive investigations and may require protocol modifications.
Long-Term Monitoring Requirements
CRISPR therapies necessitate extended follow-up periods due to the permanent nature of genomic modifications. The FDA requires a minimum 15-year long-term follow-up plan for all gene therapy products, with specific considerations for CRISPR-based treatments:
- Annual comprehensive physical examinations and laboratory assessments
- Periodic assessment of editing persistence in accessible tissues (e.g., blood)
- Monitoring for delayed adverse events including malignancy
- Reproductive impacts and potential for germline transmission assessment
- Offspring monitoring for patients receiving CRISPR therapies
The remarkable case of a pig kidney with 69 CRISPR edits functioning for 271 days in a human recipient (the longest duration for such a xenograft) demonstrates both the potential durability and eventual decline of edited tissues, highlighting the need for long-term monitoring [137].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Clinical Validation
| Reagent Category | Specific Examples | Function | Clinical Validation Application |
|---|---|---|---|
| Editing Controls | TRAC, RELA, CDC42BPB guide RNAs [138] | Positive control for editing efficiency | Verify transfection/electroporation efficiency; benchmark editing performance |
| Delivery Systems | Lipid nanoparticles (LNPs) [11] | In vivo delivery of CRISPR components | Liver-targeted editing (e.g., for hATTR, HAE, cardiovascular targets) |
| Viral vectors (AAV, lentivirus) | Ex vivo and in vivo delivery | Hematopoietic stem cell editing (Casgevy); CAR-T cell engineering (CTX112) | |
| Validation Tools | ICE (Inference of CRISPR Edits) [138] | Analysis of Sanger sequencing data | Quantify editing efficiency and indel patterns from clinical samples |
| GUIDE-seq reagents [137] | Genome-wide off-target detection | Comprehensive off-target profiling for regulatory submissions | |
| Cell Culture | Hematopoietic Stem Cell Media | Maintenance and expansion | Ex vivo editing for hematopoietic disorders (SCD, TBT) |
| T-cell Activation Cocktails | T-cell stimulation | CAR-T cell manufacturing (CTX112, CTX131) |
Experimental Workflows and Signaling Pathways
Clinical Development Pathway for CRISPR Therapeutics
The following diagram illustrates the complete clinical development pathway from preclinical research to post-marketing surveillance for CRISPR therapeutics:
CRISPR Therapeutic Validation Workflow
The following diagram outlines the key experimental workflow for validating CRISPR therapeutics from candidate selection through clinical lot release:
The clinical validation pathway for CRISPR-Cas9 therapeutics has evolved from theoretical concept to established regulatory framework with the approval of multiple therapies. Success requires meticulous attention to editing specificity, comprehensive safety assessment, and robust clinical trial design with appropriate controls. The field continues to advance rapidly, with next-generation approaches including epigenetic editing [136], base editing [137], and improved delivery systems expanding the therapeutic landscape. By adhering to these regulatory considerations and validation protocols, researchers can effectively translate CRISPR-based discoveries into transformative medicines for patients with genetic diseases, cancer, and other debilitating conditions.
Conclusion
CRISPR-Cas9 has revolutionized precise genome editing, transitioning from a fundamental bacterial immune mechanism to a powerful therapeutic tool with demonstrated clinical success. The foundational understanding of its molecular mechanisms enables sophisticated engineering of novel editors like base and prime editors. While delivery challenges persist, advances in LNP and viral vector technologies continue to expand therapeutic possibilities. Critical to clinical translation is the rigorous addressing of off-target effects through high-fidelity variants, optimized guide RNAs, and controlled inhibition systems. Comprehensive validation using NGS-based methods remains essential for accurately assessing editing outcomes. Future directions include developing tissue-specific delivery systems, refining single-base editing capabilities, expanding clinical applications for common diseases, and establishing standardized safety assessment protocols. As CRISPR technology continues to evolve, it promises to unlock unprecedented opportunities for treating genetic disorders, cancers, and other intractable diseases, fundamentally advancing biomedical research and therapeutic development.
References
- 1. CRISPR-Cas systems: prokaryotes upgrade to adaptive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4025954/]
- 2. The CRISPR-Cas immune system: Biology, mechanisms ... [https://www.sciencedirect.com/science/article/pii/S0300908415001042]
- 3. Recent applications, future perspectives, and limitations of ... [https://pubmed.ncbi.nlm.nih.gov/40777740/]
- 4. An updated evolutionary classification of CRISPR–Cas ... [https://www.nature.com/articles/s41564-025-02180-8]
- 5. CRISPR‐driven diagnostics: Molecular mechanisms, clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464570/]
- 6. Protocol for CRISPR-based manipulation and visualization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12305205/]
- 7. CRISPR-detector: fast and accurate detection, visualization ... [https://www.sciencedirect.com/science/article/pii/S1673852723000747]
- 8. A boost for the precision of genome editing [https://news.mit.edu/2025/boost-precision-genome-editing-0820]
- 9. The Complete Guide to Understanding CRISPR sgRNA [https://www.synthego.com/guide/how-to-use-crispr/sgrna/]
- 10. Off-target interactions in the CRISPR-Cas9 Machinery [https://pubmed.ncbi.nlm.nih.gov/40688512/]
- 11. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 12. Off-target interactions in the CRISPR-Cas9 Machinery [https://www.sciencedirect.com/science/article/pii/S2405580825002213]
- 13. Insights into the compact CRISPR–Cas9d system [https://www.nature.com/articles/s41467-025-57455-9]
- 14. Design Rules for Expanding PAM Compatibility in CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12424760/]
- 15. PAM-readID is a rapid, simple, and accurate ... [https://www.nature.com/articles/s42003-025-08984-y]
- 16. Improved CRISPR/Cas9 off-target prediction with DNABERT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12611124/]
- 17. CRISPR/Cas9 off-target activity prediction using DNA k- ... [https://www.sciencedirect.com/science/article/pii/S2950363925000146]
- 18. Crystal Structure of Cas9 in Complex with Guide RNA and ... [https://www.sciencedirect.com/science/article/pii/S0092867414001561]
- 19. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 20. Insights into the Mechanism of CRISPR/Cas9-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9850488/]
- 21. How to design sgRNA sequences [https://www.takarabio.com/learning-centers/gene-function/gene-editing/gene-editing-tools-and-information/how-to-design-sgrna-sequences?srsltid=AfmBOoqWzJNXiNDxFbIS32B1B-6KzGBTquWcEFL2oZMXq5Jp_jQOKeXx]
- 22. CRISPR–Cas9 gRNA efficiency prediction: an overview of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9023298/]
- 23. How to Design Your gRNA for CRISPR Genome Editing [https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing]
- 24. How To Design Guide RNA for CRISPR [https://www.synthego.com/guide/how-to-use-crispr/design-grna-crispr/]
- 25. Comprehensive protocols for CRISPR/Cas9-based gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4988528/]
- 26. qEva-CRISPR: a method for quantitative evaluation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6158505/]
- 27. CRISPR-Cas9-mediated homology-directed repair for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11531618/]
- 28. HDR vs NHEJ: DNA Repair Pathway Comparison [https://invivobiosystems.com/crispr/hdr-vs-nhej/]
- 29. Improving the Efficiency of CRISPR/Cas9-Mediated Non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384319/]
- 30. Precise genome-editing in human diseases [https://www.nature.com/articles/s41392-024-01750-2]
- 31. A high efficiency precision genome editing method with ... [https://www.nature.com/articles/s41598-024-60766-4]
- 32. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 33. Highly efficient CRISPR-mediated genome editing through ... [https://www.nature.com/articles/s41467-024-52493-1]
- 34. CRISPR Timeline [https://www.broadinstitute.org/what-broad/areas-focus/project-spotlight/crispr-timeline]
- 35. Application of CRISPR-Cas9 genome editing technology in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10916045/]
- 36. Press release: The Nobel Prize in Chemistry 2020 [https://www.nobelprize.org/prizes/chemistry/2020/press-release/]
- 37. CRISPR–Cas9: A History of Its Discovery and Ethical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9377665/]
- 38. Nobel Prize Awarded to Jennifer Doudna And Emmanuelle ... [https://www.synthego.com/blog/gene-editing-nobel-prize/]
- 39. Revolution of Biotechnology with CRISPR [https://www.nature.com/articles/s12276-025-01452-x]
- 40. ORANGE: A CRISPR/Cas9-based genome editing toolbox ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7176289/]
- 41. Viral and non-viral vectors in gene therapy: current state and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12271757/]
- 42. AAV vs Lentivirus Resources - BioInnovatise [https://bioinnovatise.com/articles/aav-vs-lentivirus-which-viral-vector-is-right-for-my-research/]
- 43. Genome engineering with Cas9 and AAV repair templates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12130156/]
- 44. AAV Vs. Lentiviral Vectors - Life in the Lab [https://www.thermofisher.com/blog/life-in-the-lab/aav-vs-lv/]
- 45. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 46. CRISPR/Cas9 Delivery Systems to Enhance Gene Editing ... [https://www.mdpi.com/1422-0067/26/9/4420]
- 47. Comparative Analysis of CRISPR/Cas9 Delivery Methods ... [https://pubmed.ncbi.nlm.nih.gov/41226739/]
- 48. Non-Invasive Delivery of CRISPR/Cas9 Ribonucleoproteins ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11859894/]
- 49. Non-viral, large-scale delivery of gene editing tools for T ... [https://www.insights.bio/cell-and-gene-therapy-insights/webinars/748/nonviral-largescale-delivery-of-gene-editing-tools-for-t-cell-manufacturing]
- 50. Advances in Nanoparticles as Non-Viral Vectors for ... [https://www.mdpi.com/1999-4923/16/9/1197]
- 51. innovations and challenges in rAAV vector-based CRISPR ... [https://www.nature.com/articles/s41434-025-00573-2]
- 52. CRISPR Technologies for In Vivo and Ex Vivo Gene Editing [https://www.ncbi.nlm.nih.gov/books/NBK609557/]
- 53. Protocol for optimizing culture conditions for ex vivo ... [https://www.sciencedirect.com/science/article/pii/S2666166725001285]
- 54. Protocol for optimizing culture conditions for ex vivo ... [https://pubmed.ncbi.nlm.nih.gov/40173037/]
- 55. CRISPR Therapeutics Highlights Strategic Priorities and ... [https://ir.crisprtx.com/news-releases/news-release-details/crispr-therapeutics-highlights-strategic-priorities-and]
- 56. A more precise way to edit the genome [https://news.mit.edu/2025/more-precise-way-edit-genome-0917]
- 57. Gene Therapy for Sickle Cell Disease: Marie-Chantal's Story [https://www.chop.edu/stories/gene-therapy-sickle-cell-disease-marie-chantal-s-story]
- 58. CASGEVY® Clinical Trial and Results [https://www.casgevy.com/sickle-cell-disease/study-information]
- 59. Nationwide Children's Hospital Delivers its First Infusion of ... [https://www.nationwidechildrens.org/newsroom/news-releases/2025/05/casgevy_scd]
- 60. Release Details [https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-presents-positive-longer-term-phase-1-data]
- 61. ATTR Amyloidosis MAGNITUDE Clinical Trials Temporarily ... [https://arci.org/magnitudehold/]
- 62. Protocol to rapidly screen CRISPR-Cas9 gene editing ... [https://www.sciencedirect.com/science/article/pii/S2666166725003569]
- 63. CRISPR/Cas9 technology for advancements in cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11490091/]
- 64. Recent advances and applications of CRISPR-Cas9 in ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01738-6]
- 65. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.674467/full]
- 66. CRISPR–Cas9 Gene Editing: Curing Genetic Diseases by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10980556/]
- 67. World's First Patient Treated with Personalized CRISPR ... [https://www.chop.edu/news/worlds-first-patient-treated-personalized-crispr-gene-editing-therapy-childrens-hospital]
- 68. CRISPR gene editing - Wikipedia [https://en.wikipedia.org/wiki/CRISPR_gene_editing]
- 69. CRISPR/Cas9 therapeutics: progress and prospects | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-023-01309-7]
- 70. CRISPR-based strategies in infectious disease diagnosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7779109/]
- 71. Fighting Infectious Disease with CRISPR [https://www.synthego.com/blog/crispr-infectious-disease/]
- 72. CRISPR-Cas9 DNA Base-Editing and Prime-Editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7503568/]
- 73. From bench to bedside: cutting-edge applications of base editing and prime editing in precision medicine | Journal of Translational Medicine | Full Text [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05957-3]
- 74. Back to Basics - Base & Prime Editing - Front Line Genomics [https://frontlinegenomics.com/back-to-basics-base-and-prime-editing/]
- 75. Emerging trends in prime editing for precision genome ... [https://www.nature.com/articles/s12276-025-01463-8]
- 76. Prime Editing as a Precision Gene Editing Tool [https://www.synthego.com/guide/crispr-methods/prime-editing/]
- 77. A streamlined base editor engineering strategy to reduce ... [https://www.nature.com/articles/s41467-025-63609-6]
- 78. Emerging trends in prime editing for precision genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322275/]
- 79. A competitive precision CRISPR method to identify the ... [https://www.nature.com/articles/s41587-022-01444-6]
- 80. Precise measurement of CRISPR genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11987616/]
- 81. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 82. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 83. CRISPR 101: Off-Target Effects [https://blog.addgene.org/crispr-101-off-target-effects]
- 84. A versatile CRISPR/Cas9 system off-target prediction tool ... [https://www.nature.com/articles/s42003-025-08275-6]
- 85. CRISPR-Cas9 and Its Bioinformatics Tools: A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109910/]
- 86. Quantitative analysis of CRISPR/Cas9-mediated provirus ... [https://www.nature.com/articles/s41598-022-19861-7]
- 87. promising for development of high-fidelity Cas9 variants [https://www.nature.com/articles/s41392-022-01139-z]
- 88. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects | Nature [https://www.nature.com/articles/nature16526]
- 89. High-fidelity CRISPR-Cas9 variants with undetectable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4851738/]
- 90. Methods for Optimizing CRISPR-Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4976696/]
- 91. High-fidelity Cas9-mediated targeting of KRAS driver ... [https://www.nature.com/articles/s41467-025-62350-4]
- 92. CRISPR Transfection Protocols Guide: How To Select The ... [https://www.synthego.com/guide/how-to-use-crispr/transfection-protocols/]
- 93. Boosting activity of high-fidelity CRISPR/Cas9 variants ... [https://www.sciencedirect.com/science/article/pii/S0021925820351747]
- 94. ARROW: Allele-Specific Recombined gRNA Design for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12649741/]
- 95. Typical Workflow of CRISPR-Cas9 Genome Editing [https://www.assaygenie.com/blog/typical-workflow-of-crispr-cas9-genome-editing-f680ce/?srsltid=AfmBOorZFlrDkOALZLZXUYGe5VznKi2Cg11FVzrDw_Glp2DYbGSckZBz]
- 96. CMN Weekly (17 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-17-october-2025-your-weekly-crispr-medicine-news/]
- 97. How To Use ICE: A Detailed Guide for Analyzing CRISPR ... [https://www.synthego.com/guide/how-to-use-crispr/ice-analysis-guide/]
- 98. Cleveland Clinic First-In-Human Trial of CRISPR Gene ... [https://newsroom.clevelandclinic.org/2025/11/08/cleveland-clinic-first-in-human-trial-of-crispr-gene-editing-therapy-shown-to-safely-lower-cholesterol-and-triglycerides]
- 99. Reducing CRISPR-Cas9 off-target effects by optically ... [https://www.sciencedirect.com/science/article/pii/S2451945624004021]
- 100. Insights into the inhibition of type I-F CRISPR-Cas system ... [https://www.nature.com/articles/s41467-022-29581-1]
- 101. Controlling CRISPR-Cas9 with ligand-activated and ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/31073154/]
- 102. Cell-Penetrating Peptides and CRISPR-Cas9: A Combined ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11511780/]
- 103. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 104. Off-target interactions in the CRISPR-Cas9 Machinery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272595/]
- 105. Optimizing CRISPR delivery for drug discovery [https://www.drugdiscoverynews.com/optimizing-crispr-delivery-for-drug-discovery-16487]
- 106. Optimizing CRISPR methodology for precise gene editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12580712/]
- 107. CMN Weekly (25 July 2025) - Your Weekly CRISPR ... [https://crisprmedicinenews.com/news/cmn-weekly-25-july-2025-your-weekly-crispr-medicine-news/]
- 108. Comparative analysis of CRISPR off-target discovery tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10124080/]
- 109. COSMID: A Web-based Tool for Identifying and Validating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4272406/]
- 110. Cas-OFFinder: a fast and versatile algorithm that searches for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4016707/]
- 111. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0124633]
- 112. CIRCLE-Seq for Interrogation of Off-Target Gene Editing [https://app.jove.com/t/67069/circle-seq-for-interrogation-of-off-target-gene-editing]
- 113. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 114. Defining genome-wide CRISPR-Cas genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9331158/]
- 115. CIRCLE-seq: a highly sensitive in vitro screen for genome- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5924695/]
- 116. CRISPR off-target detection with DISCOVER-Seq - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7305837/]
- 117. GUIDEseq: a bioconductor package to analyze GUIDE-Seq ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3746-y]
- 118. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 119. DIG-seq: a genome-wide CRISPR off-target profiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6280750/]
- 120. Targeted deep-sequencing [https://bio-protocol.org/bio101/r1198669]
- 121. Controlling CRISPR-Cas9 genome editing in human cells ... [https://www.sciencedirect.com/science/article/pii/S2162253125001945]
- 122. A Survey of Validation Strategies for CRISPR-Cas9 Editing [https://www.nature.com/articles/s41598-018-19441-8]
- 123. How to Pick the Best CRISPR Data Analysis Method for ... [https://www.synthego.com/guide/how-to-use-crispr/analysis-methods/]
- 124. Comparative Analysis of Methods for Assessing On-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932317/]
- 125. Comprehensive benchmarking of genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144422/]
- 126. Precision gene editing: The power of CRISPR-Cas in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590234/]
- 127. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 128. TIDE: Tracking of Indels by DEcomposition [https://apps.datacurators.nl/tide/]
- 129. Rapid Quantitative Evaluation of CRISPR Genome Editing ... [https://pubmed.ncbi.nlm.nih.gov/30912038/]
- 130. A Survey of Validation Strategies for CRISPR-Cas9 Editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC5772360/]
- 131. Gene Editing [https://crisprtx.com/gene-editing]
- 132. Understanding the Limitations of Potency Assays in ... [https://quatrelab.com/understanding-the-limitations-of-potency-assays-in-advanced-biological-products/]
- 133. Prevalent integration of genomic repetitive and regulatory ... [https://www.nature.com/articles/s42003-025-07539-5]
- 134. In Vitro Pre-validation of Gene Editing by CRISPR/Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6626505/]
- 135. Validation of CRISPR-based Gene Editing [https://www.neb.com/en-us/products/crispr-cas-gene-editing/crispr-validation?srsltid=AfmBOorG8MvSe4jq_rM6OTf1aTa688BXdAFI_Z7iV1zwY34Zak5bzSVG]
- 136. What is CRISPR? A bioengineer explains - Stanford Report [https://news.stanford.edu/stories/2024/06/stanford-explainer-crispr-gene-editing-and-beyond]
- 137. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 138. Ensure Proper Controls in Your CRISPR Experiments [https://www.synthego.com/blog/controls-crispr-experiments/]