Overcoming the Challenge: A Guide to Amplifying Trinucleotide Repeat Regions with PCR Additives
Amplifying trinucleotide repeat (TNR) regions presents significant challenges in PCR due to their propensity to form complex secondary structures, leading to poor yield, specificity, and fidelity.
Overcoming the Challenge: A Guide to Amplifying Trinucleotide Repeat Regions with PCR Additives
Abstract
Amplifying trinucleotide repeat (TNR) regions presents significant challenges in PCR due to their propensity to form complex secondary structures, leading to poor yield, specificity, and fidelity. This article provides a comprehensive guide for researchers and drug development professionals, covering the foundational biology of TNR instability, detailed methodological protocols incorporating specialized PCR additives, advanced troubleshooting strategies, and validation techniques. By synthesizing current research and practical optimization tips, this resource aims to equip scientists with the knowledge to reliably amplify these difficult sequences, thereby supporting advancements in the study and therapeutic targeting of neurodegenerative diseases and other TNR-associated disorders.
Understanding Trinucleotide Repeat Instability and Its Amplification Hurdles
The Biological Significance of Trinucleotide Repeats in Disease and Diagnostics
Trinucleotide repeats (TNRs) are simple sequences of three DNA bases repeated tens to hundreds of times throughout the human genome. While many such repeats are biologically stable, pathological expansion beyond a critical threshold is directly linked to numerous neurodegenerative diseases and other disorders [1]. The dynamic nature of these DNA repeat expansions stems from their capability to form alternative secondary structures that interfere with cellular mechanisms of replication, repair, recombination, and transcription [1]. Approximately 50 neurodegenerative diseases are associated with pathological expansion of these repeat tracks, with the illnesses believed to result directly from RNA and/or protein products of the affected genes [2].
Strikingly, different trinucleotide repeat sequences exhibit dramatically different expansion potentials. CAG repeat expansions cause Huntington's disease (HD), dentatorubral-pallidoluysian atrophy (DRPLA), spinal and bulbar muscular atrophy (SBMA), and several spinocerebellar ataxias (SCAs). Similarly, CTG repeat expansions are associated with myotonic dystrophy and spinocerebellar ataxia type 8 (SCA8) [2]. In stark contrast, the reversed sequences, GAC and GTC, do not undergo large-scale expansions, with only one disorder involving GAC repeats (skeletal dysplasia) resulting from only small changes in a normal (GAC)5 sequence [2]. This sequence-specific expansion behavior provides crucial constraints for understanding the fundamental mechanisms behind TNR-related pathogenesis.
Table 1: Major Trinucleotide Repeat Disorders and Their Genetic Characteristics
| Disease Name | Repeat Type | Repeat Location | Normal Repeat Length | Disease Repeat Length |
|---|---|---|---|---|
| Huntington's Disease (HD) | CAG | Protein coding region (polyQ) | 6–34 | 36–121 |
| Spinal and Bulbar Muscular Atrophy (SBMA) | CAG | Protein coding region (polyQ) | 9–36 | 38–62 |
| Spinocerebellar Ataxia 3 (SCA3/MJD) | CAG | Protein coding region (polyQ) | 10–51 | 55–87 |
| Myotonic Dystrophy Type 1 (DM1) | CTG | 3'UTR | 5–37 | 90–6500 |
| Friedreich Ataxia (FRDA) | GAA | Intron | 6–32 | >200 |
| Skeletal Dysplasias (COMP) | GAC | Protein coding region (polyaspartate) | 5 | 4, 6, 7 |
Structural Dynamics and Pathological Mechanisms
Hairpin Formation and Slippage Dynamics
The pathological potential of trinucleotide repeats is intimately connected to their structural properties. When duplex DNA containing TNR sequences transiently unwinds during replication, transcription, or repair, the separated repeat-containing strands can form various non-canonical structures including hairpins, cruciforms, and G-quadruplexes [2]. The propensity of these hairpins to slip along their corresponding strands is widely considered fundamental to expansion mechanisms [2].
Single-molecule FRET (smFRET) experiments and molecular dynamics simulations have revealed significant structural differences between disease-associated and non-disease associated TNR sequences. Specifically, CAG, CTG, and GTC hairpins strongly favor tetraloop configurations (89%, 89%, and 69% respectively), while GAC prefers triloop configurations [2]. These preferences have profound implications for the stability of secondary structures formed during DNA metabolic processes.
The slipping dynamics of TNR hairpins occur in steps of multiples of the trinucleotide unit. In a "0 slip" configuration, the hairpin has fully base-paired triplets with either a tetraloop (in even-numbered repeats) or triloop (in odd-numbered repeats). Hairpins can slip in either direction ("+" or "-"), creating unpaired triplets at either end while maintaining the fundamental bonding pattern [2]. This slipping behavior is hypothesized to contribute directly to repeat expansion during DNA replication and repair.
Sequence-Specific Structural Frustration
A critical finding from recent structural studies is the concept of structural frustration in opposing hairpins. When duplex (CAG)·(CTG) unwinds, both opposing CAG and CTG hairpins (of identical length) naturally prefer tetraloop configurations, creating a stable, symmetrical cruciform structure. In contrast, when duplex (GAC)·(GTC) opens into opposing hairpins of identical lengths, the different loop tendencies (GTC preferring tetraloops and GAC preferring triloops) create frustration between the arms of such a cruciform structure [2].
This frustration effect would influence the lifetime and migration tendency of the cruciform structure, with direct implications for expansion potential. The matched stability in (CAG)·(CTG) opposing hairpins encourages their persistence, providing greater opportunity for expansion mechanisms to operate, whereas the unmatched stability in (GAC)·(GTC) structures encourages more rapid resolution back to duplex DNA [2]. This biophysical difference may explain why CAG and CTG repeats undergo large, disease-related expansions while GAC and GTC sequences do not.
Diagnostic Approaches and Technical Protocols
PCR Amplification of Trinucleotide Repeat Regions
The polymerase chain reaction (PCR) has become the most widely used technique in molecular biology for amplifying specific target DNA fragments from small amounts of DNA or RNA source material [3]. For trinucleotide repeat regions, which present particular challenges due to their repetitive nature and secondary structure formation, careful optimization of PCR components and conditions is essential.
A typical PCR protocol for TNR amplification includes three fundamental steps repeated over 25-40 cycles:
- Denaturation: 30-60 seconds at 95°C to separate DNA strands
- Annealing: 30-60 seconds at 45-60°C (optimized based on primer Tm) to allow primer binding
- Extension: 20-60 seconds per kb at 72°C for DNA synthesis [3]
For trinucleotide repeats, additional considerations include:
- Extended denaturation times to resolve stable secondary structures
- Optimized MgCl₂ concentrations (typically 1.5-5.5 mM, titrated in 0.5 mM increments)
- Specialized polymerase selection with high processivity for GC-rich regions
- Template-specific optimization (5-50 ng genomic DNA per 20-100 μL reaction) [3] [4]
Table 2: Optimal PCR Components for Amplifying Challenging Trinucleotide Repeat Regions
| Component | Standard Concentration | Considerations for TNR Amplification | Potential Modifications |
|---|---|---|---|
| Template DNA | 10 ng–1 μg (gDNA) | Secondary structures may require increased amount | 50-100 ng gDNA; pre-denaturation at 98°C |
| DNA Polymerase | 1-2 units/50 μL reaction | High secondary structure resistance needed | Polymerases with strong strand displacement |
| Primers | 0.1–1 μM | Tm 55–70°C; avoid 3' end complementarity | Increased length (25-30 nt); elevated Tm |
| dNTPs | 200 μM each | Balanced concentration critical for fidelity | Increase to 250-300 μM for long repeats |
| MgCl₂ | 1.5–5.5 mM | Critical for enzyme processivity in GC-rich regions | Titrate 2.0-6.0 mM in 0.5 mM steps |
Advanced Detection and Sizing Methods
Accurate size determination of expanded repeats is essential for diagnosis and prognosis of TNR disorders. While conventional gel electrophoresis can resolve normal and moderately expanded alleles, very large expansions often require specialized techniques:
- Southern Blotting: The historical gold standard for large repeat expansions, providing accurate sizing but requiring large DNA amounts
- Triplet-Primed PCR (TP-PCR): Allows amplification despite the presence of very large expansions by using repeat-specific primers
- Long-Range PCR: Utilizing specialized polymerases and optimized conditions to amplify through extensive repeat regions
- Next-Generation Sequencing: Emerging approaches providing single-molecule resolution of repeat length [5]
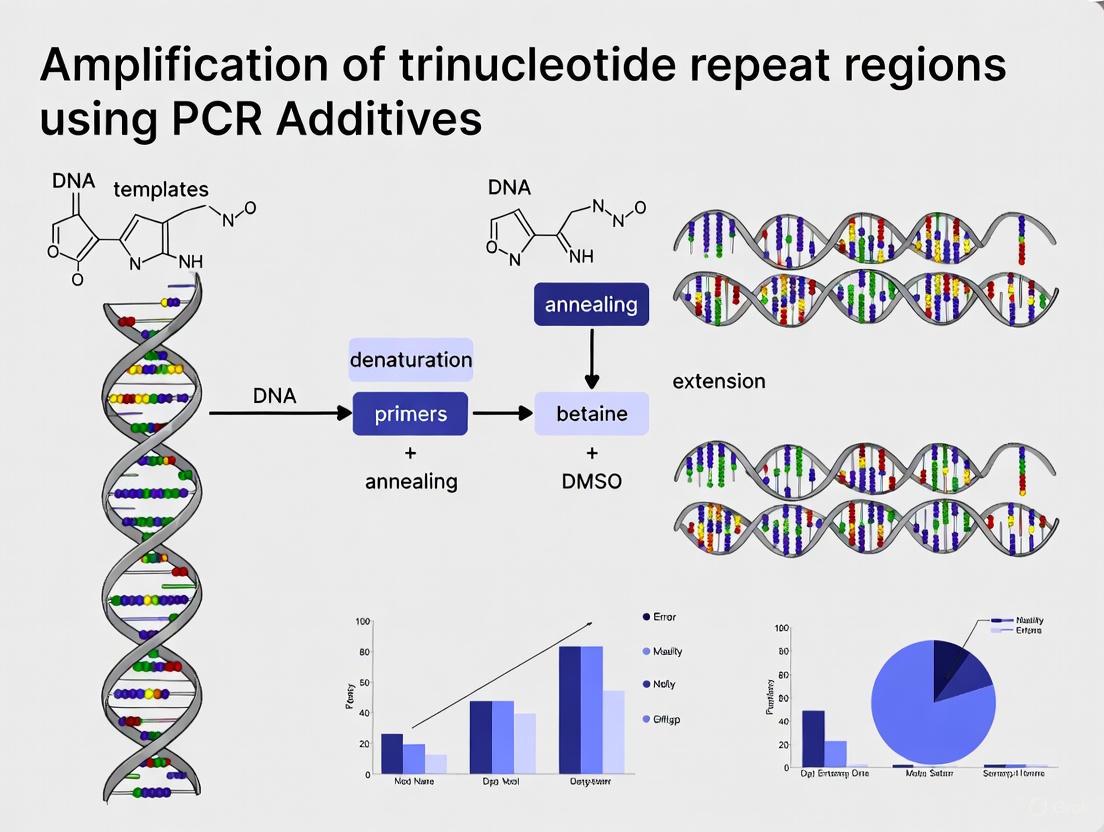
Recent advances incorporate slippage-suppressing additives such as betaine (5-Aza-2'-deoxycytidine) and DMSO (2-10%) to improve amplification efficiency through GC-rich repetitive regions. These additives help denature stable secondary structures that would otherwise cause polymerase stalling or premature termination [3] [4].
Research Reagent Solutions for TNR Studies
The following toolkit represents essential materials and reagents for investigating trinucleotide repeat biology and developing diagnostic applications:
Table 3: Essential Research Reagent Solutions for Trinucleotide Repeat Studies
| Reagent Category | Specific Examples | Application in TNR Research |
|---|---|---|
| Specialized DNA Polymerases | High-fidelity PCR systems, Long-range amplification kits | Faithful amplification of repetitive sequences without slippage artifacts |
| PCR Additives | Betaine, DMSO, 7-deaza-dGTP, Q-Solution | Disruption of secondary structures; enhancement of amplification efficiency |
| smFRET Components | Cy3/Cy5 or Atto647N fluorophores, biotin anchoring system | Real-time observation of hairpin dynamics and slipping events [2] |
| Molecular Cloning Systems | TA/Blunt-end cloning vectors, recombination-based kits | Propagation of repetitive sequences for mechanistic studies |
| CRISPR-Cas Systems | Cas9 nucleases, guide RNA design tools | Investigation of repeat instability mechanisms; potential therapeutic applications [6] |
| Next-Generation Sequencing | Long-read platforms (Oxford Nanopore, PacBio) | Complete resolution of expanded repeat regions at single-molecule level |
Visualization of Structural Dynamics and Experimental Workflows
Diagram 1: Structural Basis of Differential Expansion Potential Between CAG/CTG and GAC/GTC Repeats
Diagram 2: Experimental Workflow for Reliable Amplification of Trinucleotide Repeat Regions
The study of trinucleotide repeats represents a critical intersection of structural biology, genetics, and molecular diagnostics. The sequence-specific biophysical properties of these repeats, particularly their propensity to form stable non-canonical structures, directly influence their expansion potential and consequently their pathological impact. The differential behavior between disease-associated repeats (CAG, CTG) and non-disease associated repeats (GAC, GTC) provides both insight into disease mechanisms and opportunities for therapeutic intervention.
Future directions in TNR research will likely focus on several key areas:
- Novel therapeutic approaches leveraging CRISPR-Cas systems and other gene editing technologies to target expanded repeats [6]
- Advanced biomarker development incorporating TNR sizing into personalized treatment strategies [5] [7]
- High-resolution structural studies further elucidating the molecular basis of repeat instability
- Small molecule interventions designed to disrupt the pathogenic secondary structures that drive expansion
As our understanding of trinucleotide repeat biology continues to deepen, so too will our ability to diagnose, monitor, and ultimately treat the devastating disorders they cause. The integration of basic structural insights with advanced diagnostic methodologies represents the most promising path forward in addressing these challenging genetic conditions.
The polymerase chain reaction (PCR) stands as one of the most pivotal techniques in molecular biology, enabling the exponential amplification of specific DNA sequences. However, standard PCR protocols frequently encounter limitations when confronted with structurally complex templates. Secondary structures—such as hairpins, stem-loops, and G-quadruplexes—that form within nucleic acid templates present a significant barrier to efficient amplification by hindering polymerase progression and primer annealing. These challenges are particularly pronounced in trinucleotide repeat regions, which are not only associated with numerous human neurological disorders but also exhibit strong tendencies to form stable secondary structures due to their high GC content and repetitive nature.
The presence of these structures causes polymerase pausing, premature dissociation, and truncated amplification products, ultimately leading to PCR failure or biased results. For researchers investigating myotonic dystrophies, Huntington's disease, fragile X syndrome, and other repeat expansion disorders, these limitations directly impact diagnostic accuracy and research validity. This application note examines the mechanistic basis of how secondary structures interfere with standard PCR and provides optimized protocols incorporating specialized additives to overcome these challenges, with particular emphasis on amplifying CTG/CCTG-repeat regions relevant to myotonic dystrophies.
The Impact of Secondary Structures on PCR Efficiency
Mechanisms of PCR Inhibition
Secondary structures in DNA templates interfere with PCR amplification through multiple mechanisms that impact various stages of the reaction process:
Impaired polymerase progression: DNA polymerase enzymes exhibit reduced processivity when encountering secondary structures, leading to incomplete elongation and truncated products. This effect is particularly pronounced with proofreading enzymes that possess strong 3'→5' exonuclease activity [8].
Competitive primer binding: Stable secondary structures within the template create physical barriers that prevent primers from accessing their complementary binding sites, thereby reducing annealing efficiency and specificity [9].
Adapter-mediated self-priming: Recent deep learning analyses of multi-template PCR efficiency have identified that specific motifs adjacent to adapter priming sites facilitate self-priming events, which significantly reduce target amplification efficiency by creating competitive amplification pathways [10].
The effect of these inhibitory mechanisms becomes exponentially problematic with increasing cycle numbers, as even minor inefficiencies in early cycles result in substantial representation biases in later cycles. Research demonstrates that sequences with amplification efficiencies just 5% below the average can be underrepresented by a factor of approximately two after only 12 PCR cycles [10].
Structural Challenges in Trinucleotide Repeat Regions
Trinucleotide repeat regions present unique challenges for PCR amplification due to their sequence composition and structural properties:
High GC content: Many trinucleotide repeats, particularly those associated with disease, are GC-rich, leading to stronger hydrogen bonding between guanine and cytosine bases. This results in elevated melting temperatures that exceed standard PCR conditions [8].
Stable hairpin formation: Repeat sequences such as CAG/CTG and CGG/CCG readily form stable hairpin structures through intramolecular base pairing, creating significant obstacles for polymerase processivity [11].
Length-dependent instability: Longer repeat tracts form increasingly stable secondary structures, creating a technical barrier to amplifying the expanded alleles most relevant to disease pathology [11].
Table 1: Common Problematic Secondary Structures in PCR
| Structure Type | Formation Mechanism | Impact on PCR |
|---|---|---|
| Hairpin loops | Intra-strand base pairing in repeat regions | Polymerase pausing, primer binding competition |
| G-quadruplexes | Hoogsteen bonding between guanine tetrads | Complete polymerase arrest, particularly in GC-rich regions |
| Cruciform structures | Inverted repeat sequences | Template deformation, primer misannealing |
| Slipped-strand DNA | Misalignment of complementary repeats | Product heterogeneity, amplification bias |
Quantitative Analysis of Structural Interference
Efficiency Metrics and Amplification Bias
The impact of secondary structures on PCR performance can be quantified through specific efficiency metrics. In multi-template PCR applications—such as those used in DNA data storage systems and metagenomic studies—sequence-specific amplification efficiencies create substantial representation biases. Research utilizing synthetic DNA pools has demonstrated that approximately 2% of sequences exhibit severely compromised amplification efficiencies as low as 80% relative to the population mean. This efficiency reduction translates to a halving of relative abundance every 3 cycles, effectively eliminating these sequences from the amplification pool after 60 cycles [10].
Deep learning models trained to predict sequence-specific amplification efficiency have achieved notable performance (AUROC: 0.88, AUPRC: 0.44) based solely on sequence information, confirming the deterministic relationship between sequence features and amplification success [10]. The CluMo (Motif Discovery via Attribution and Clustering) interpretation framework has further identified specific motifs adjacent to adapter priming sites as strongly associated with poor amplification efficiency, challenging conventional PCR design assumptions [10].
Experimental Validation of Structural Inhibition
Orthogonal validation experiments comparing sequences with different predicted amplification efficiencies confirm the critical role of secondary structures:
- qPCR efficiency correlation: Sequences identified as having low amplification efficiency in multi-template PCR also demonstrated significantly lower efficiency in single-template qPCR validation experiments [10].
- GC-content independence: The progressive skewing of coverage distribution with increased PCR cycles persisted even in GC-controlled pools (constrained to 50% GC content), indicating that factors beyond overall GC content—likely specific structural motifs—drive amplification inefficiency [10].
- Reproducibility across pools: When sequences with low attributed amplification efficiencies were synthesized in new oligonucleotide pools, they consistently showed drastic under-representation after just 30 PCR cycles and were effectively eliminated by cycle 60, confirming that the amplification defects are sequence-specific rather than pool-dependent [10].
Table 2: Impact of PCR Cycle Number on Sequence Representation Based on Amplification Efficiency
| Amplification Efficiency Category | Relative Representation After 30 Cycles | Relative Representation After 60 Cycles |
|---|---|---|
| High (>95% of mean) | 100% | 100% |
| Average (90-95% of mean) | ~80% | ~65% |
| Low (<90% of mean) | ~35% | <5% |
Mechanistic Workflow of Structural Interference
The following diagram illustrates the sequential mechanisms through which secondary structures interfere with standard PCR amplification, culminating in reaction failure:
Secondary Structure Interference in PCR - This workflow illustrates how template structures lead to amplification failure through multiple inhibitory pathways.
Research Reagent Solutions for Structural Challenges
Successful amplification of structured templates requires specialized reagents that address the specific mechanisms of PCR inhibition. The following table details key research reagent solutions and their functional mechanisms:
Table 3: Essential Research Reagents for Amplifying Structured Templates
| Reagent Category | Specific Examples | Functional Mechanism | Optimal Concentration Range |
|---|---|---|---|
| Structure-Disrupting Additives | Betaine, DMSO, Formamide | Reduce secondary structure stability by lowering DNA melting temperature; betaine eliminates base composition dependence during denaturation [8] [9] | Betaine: 1-1.7 M; DMSO: 2-10%; Formamide: 1-5% |
| Polymerase Selection | DyNAzyme EXT DNA polymerase, Proofreading enzymes | Enhanced processivity through structured regions; proofreading activity maintains fidelity in challenging templates [11] | 1.6-2.0 units per 25 μL reaction |
| Cofactor Optimization | Magnesium ions (Mg²⁺) | Essential DNA polymerase cofactor that affects enzyme activity, primer annealing, and product specificity; concentration critically influences structured template amplification [9] | 1.0-4.0 mM (optimization required) |
| Specificity Enhancers | Tetramethylammonium chloride (TMAC) | Increases hybridization specificity through charge shielding, reducing non-specific amplification and primer-dimer formation [9] | 15-100 mM |
| Inhibitor Neutralizers | Bovine Serum Albumin (BSA) | Binds and neutralizes PCR inhibitors commonly present in nucleic acid preparations, particularly beneficial for clinical samples [9] | ~0.8 mg/mL |
Optimized Experimental Protocols
Protocol for Amplifying GC-Rich Trinucleotide Repeat Regions
This optimized protocol has been specifically validated for the amplification of CTG/CCTG-repeat expansions in myotonic dystrophy research and can be adapted for other structured templates [8] [11]:
Reaction Setup:
- Prepare a 25 μL reaction mixture containing:
- 1× PCR buffer (supplied with polymerase)
- 200 μmol/L of each dNTP
- 1.6 units of DyNAzyme EXT DNA polymerase or similar high-processivity enzyme
- 10 pmol of each forward and reverse primer
- 50 ng of template genomic DNA
- 1 M betaine (or 5% DMSO)
- 1.5 mM MgCl₂ (optimize between 1.0-4.0 mM)
Primer Design Considerations:
- Design primers with length of 18-24 nucleotides
- Maintain GC content between 40-60%
- Ensure melting temperature (Tm) of 65-75°C
- Avoid runs of 4+ identical bases or dinucleotide repeats
- Prevent intra-primer homology (self-complementarity) and inter-primer homology (primer-dimer formation) [12] [13]
Thermal Cycling Conditions:
- Initial denaturation: 95°C for 5 minutes
- 30 cycles of:
- Denaturation: 95°C for 45 seconds
- Annealing: 65-66°C for 8 seconds
- Extension: 75-78°C for 3 minutes
- Final extension: 72°C for 10 minutes
- Hold at 4°C
Critical Notes:
- The abbreviated annealing time reduces off-target binding while maintaining specificity
- The extended elongation time accommodates polymerase pausing at structured regions
- Betaine concentration may require optimization between 0.5-1.7 M depending on template structure
- Template quality is essential; use DNA with 260/280 ratio of ~1.8 [14]
Additive Optimization Strategy
When adapting this protocol for new target sequences, implement the following optimization workflow:
- Initial screening: Test DMSO (2-10%), formamide (1-5%), and betaine (1-1.7 M) in separate reactions
- Combination approach: Evaluate synergistic effects of DMSO + betaine or formamide + betaine
- Magnesium titration: Optimize MgCl₂ concentration (1.0-4.0 mM in 0.5 mM increments) after selecting additives
- Thermal profiling: Fine-tune annealing temperature (±5°C from calculated Tm) and extension time
This systematic optimization approach has been shown to improve amplification efficiency of challenging templates by 3- to 10-fold compared to standard protocols [8] [14].
The amplification of structured DNA templates, particularly trinucleotide repeat regions associated with neurological disorders, requires specialized approaches that address the unique challenges posed by secondary structures. Through strategic implementation of structure-disrupting additives, polymerase selection, and optimized thermal cycling parameters, researchers can successfully overcome these limitations.
The emerging integration of deep learning frameworks for predicting sequence-specific amplification efficiency represents a promising direction for future assay development. These computational approaches, combined with mechanistic insights into adapter-mediated self-priming and other inhibition pathways, will enable more rational design of amplification strategies for the most challenging templates. As research continues to elucidate the complex relationship between sequence features and amplification success, the protocols and principles outlined in this application note provide a foundation for reliable amplification of structured targets in both research and diagnostic contexts.
The Role of DNA Polymerase Processivity and Fidelity in TNR Amplification
Trinucleotide repeat (TNR) disorders represent a class of over a dozen hereditary neurological diseases, including fragile X syndrome, Huntington's disease, and myotonic dystrophy, whose molecular basis lies in the unstable expansion of repetitive DNA sequences beyond a critical threshold of approximately 25 repeats [15]. The inheritance of these conditions is characterized by genetic anticipation, a phenomenon where the probability, onset, and severity of the disease increase through successive generations as the repetitive tracts expand [15]. Amplifying these regions using polymerase chain reaction (PCR) presents substantial technical challenges due to the unique structural properties of TNRs. These repetitive sequences exhibit a strong propensity to form stable secondary structures, such as hairpin loops, which impede the progressive movement of DNA polymerases during amplification—a fundamental challenge that this application note addresses through detailed protocol optimization and polymerase selection guidance [16].
The amplification of TNR regions is crucial for both diagnostic applications and basic research into the mechanisms of repeat expansion. However, the complex structural nature of these sequences often leads to polymerase dissociation from the template, resulting in incomplete fragments that act as megaprimers. These megaprimers subsequently anneal nonspecifically, generating a diverse library of undesired artefacts and manifesting as a characteristic laddering effect on electrophoretic gels [16]. Moreover, templates with high GC content (frequently exceeding 65%), which are common in TNR regions, further exacerbate these issues, promoting nonspecific amplification and multiple band formation [16]. Understanding and overcoming these challenges requires a comprehensive approach that addresses both polymerase characteristics and reaction conditions.
DNA Polymerase Characteristics Critical for TNR Amplification
Fidelity and Proofreading Mechanisms
The fidelity of DNA polymerases refers to the accuracy with which these enzymes copy DNA sequences, a parameter critically important for maintaining sequence integrity during TNR amplification. Fidelity is commonly expressed as error rates per base incorporated, with different polymerases exhibiting substantially different fidelity profiles [17]. For instance, while Taq DNA polymerase demonstrates an error rate of approximately 1.5 × 10⁻⁴ substitutions per base per doubling, high-fidelity enzymes like Q5 DNA Polymerase show dramatically lower error rates around 5.3 × 10⁻⁷—approximately 280-fold higher fidelity than Taq [17]. This exceptional accuracy is particularly crucial when amplifying TNR regions, where even minor replication errors can compound through successive amplification cycles.
The mechanisms underlying polymerase fidelity operate at multiple levels. Initial nucleotide selection depends heavily on the geometry of the polymerase active site, which optimally aligns correct nucleotides for efficient incorporation while slowing the incorporation of incorrect nucleotides due to suboptimal architecture [17]. Additionally, many high-fidelity polymerases possess a 3´→5´ exonuclease domain that confers proofreading capability. This domain detects and excises misincorporated nucleotides from the growing DNA strand before permanent incorporation occurs, providing an essential corrective mechanism that enhances replication accuracy [17]. The presence of proofreading activity can dramatically impact error rates, as demonstrated by the 125-fold decrease in error rate observed when comparing exonuclease-proficient Deep Vent DNA Polymerase (4.0 × 10⁻⁶) to its exonuclease-deficient counterpart (5.0 × 10⁻⁴) [17].
Processivity and Structured Template Amplification
Processivity refers to the number of nucleotides a DNA polymerase can incorporate during a single template binding event before dissociating. This characteristic becomes particularly critical when amplifying through repetitive sequences prone to forming stable secondary structures. TNR regions, especially those with high GC content, readily form hairpin loop structures that present significant physical barriers to polymerase progression [16]. Low-processivity enzymes frequently dissociate from these structured templates, leading to incomplete amplification products that can then act as megaprimers and generate artefactual amplification patterns [16].
The proofreading activity of high-fidelity polymerases contributes not only to fidelity but also to processivity on structured templates. When a polymerase encounters a structural impediment, the resulting perturbation and delayed progression increases the likelihood that the enzyme will engage its proofreading domain [17]. This process involves transferring the 3' end of the growing DNA strand into the exonuclease domain for corrective processing before returning it to the polymerase active site for continued extension [17]. While this proofreading cycle introduces a temporary pause in synthesis, it ultimately facilitates more successful navigation through challenging template structures by correcting misalignments that might otherwise lead to dissociation or replication errors.
Table 1: DNA Polymerase Fidelity Comparisons Based on PacBio SMRT Sequencing Data
| DNA Polymerase | Substitution Rate (per base per doubling) | Accuracy (bases per one error) | Fidelity Relative to Taq |
|---|---|---|---|
| Taq | 1.5 × 10⁻⁴ | 6,456 | 1x |
| Q5 | 5.3 × 10⁻⁷ | 1,870,763 | 280x |
| Phusion | 3.9 × 10⁻⁶ | 255,118 | 39x |
| Deep Vent | 4.0 × 10⁻⁶ | 251,129 | 44x |
| Pfu | 5.1 × 10⁻⁶ | 195,275 | 30x |
| PrimeSTAR GXL | 8.4 × 10⁻⁶ | 118,467 | 18x |
| KOD | 1.2 × 10⁻⁵ | 82,303 | 12x |
| Deep Vent (exo-) | 5.0 × 10⁻⁴ | 2,020 | 0.3x |
Data sourced from PacBio SMRT sequencing analysis [17]
Experimental Approaches for TNR Analysis
Current Methodological Limitations
Traditional methods for analyzing TNR expansions, including Southern blotting and conventional PCR, face significant limitations in accurately characterizing these challenging genomic regions. Southern blot analysis, while useful for determining approximate expansion sizes, provides limited sequence information and cannot detect sequence interruptions within the repetitive tract [18]. Similarly, standard PCR protocols frequently fail to amplify through long TNR regions due to the structural challenges described previously, often resulting in preferential amplification of smaller alleles and incomplete representation of the true genetic heterogeneity [16]. These technical limitations have profound implications for genetic counseling and prognostic accuracy in TNR disorders, as both repeat length and the presence of interruption sequences significantly influence disease presentation and progression [18].
Triplet-primed PCR methodologies represent an improvement over conventional approaches, offering enhanced capability to detect the presence of interruptions at the 5' and 3' ends of TNR expansions [18]. However, this technique still fails to provide comprehensive information about the internal structure of long repetitive sequences, particularly for larger expansions that exceed several hundred repeats. These methodological gaps underscore the necessity for both improved amplification strategies and advanced sequencing technologies to fully characterize TNR regions and understand their role in disease pathogenesis and inheritance patterns.
Advanced Sequencing Technologies
The emergence of single-molecule, real-time (SMRT) sequencing technologies (PacBio) has revolutionized TNR analysis by enabling comprehensive characterization of repeat expansions, including their length, sequence composition, and interruption patterns. This platform has demonstrated remarkable capability in sequencing extremely long TNR tracts, with successful analysis of repeats exceeding 1,000 triplets in length [18]. Unlike short-read sequencing technologies that struggle with repetitive elements, SMRT sequencing provides the long read lengths necessary to span entire expanded regions, thereby preserving the contextual information needed for accurate genotyping and interruption mapping.
A key advantage of SMRT sequencing for TNR analysis lies in its ability to detect interruption patterns within expanded repeats, which has important implications for disease variability and prognostic assessment. For instance, in myotonic dystrophy type 1 (DM1) caused by CTG repeat expansions, researchers have utilized SMRT sequencing to identify de novo CCG interruptions associated with CTG stabilization/contraction across generations within affected families [18]. This technology has also revealed substantial heterogeneity in the number and type of interruptions within expanded alleles, suggesting novel mechanisms involving DNA damage response, repair processes, and polymerase errors occurring throughout development and aging [18]. These insights would be difficult or impossible to obtain using conventional methodologies, highlighting the transformative potential of long-read sequencing for TNR disorder research.
Optimized Protocols for TNR Amplification
PCR Component Optimization for Repetitive Sequences
Successful amplification of TNR regions requires careful optimization of all PCR components, with particular attention to DNA polymerase selection, template quality, and reaction additives. The following protocol has been specifically developed for challenging TNR templates, incorporating empirical observations from successful amplification of the MaSp1 gene—a model repetitive sequence characterized by 68.8% GC content and poly-alanine-glycine motifs that exemplify the difficulties encountered with structured templates [16].
Table 2: Optimized PCR Reaction Components for TNR Amplification
| Component | Standard Concentration | TNR-Optimized Concentration | Notes |
|---|---|---|---|
| DNA Polymerase | 1–2 units/50 µL | 2–2.5 units/50 µL | Use high-fidelity, proofreading enzymes |
| Template DNA | 5–50 ng (gDNA) | 10–100 ng (gDNA) | Higher amounts counterbalance polymerase dissociation |
| Primers | 0.1–1 µM | 0.3–0.5 µM | Avoid excess to minimize mispriming |
| dNTPs | 0.2 mM each | 0.2–0.25 mM each | Balanced concentration critical for fidelity |
| MgCl₂ | 1.5–2.5 mM | 2–3 mM | Optimize based on polymerase and template |
Adapted from general PCR optimization guidelines [19] and TNR-specific modifications [16]
DNA Template Preparation: For TNR amplification, use high-quality, intact genomic DNA or plasmid template. When using plasmid templates containing TNR inserts, optimal DNA concentrations typically range from 250- to 1000-fold dilutions of stock solutions (approximately 100 μg/mL), with the specific optimal concentration depending on the length and GC content of the target repeat region [16]. For genomic DNA templates, increase input amounts to 10–100 ng per 50 μL reaction to counterbalance potential polymerase dissociation events and ensure representative amplification of all target alleles.
Polymerase Selection: Employ high-fidelity DNA polymerases with strong processivity and proofreading capabilities, such as Q5, Phusion, or Pfu variants [17] [20]. These enzymes demonstrate significantly lower error rates (10⁻⁶ to 10⁻⁷ range) compared to standard Taq polymerase (10⁻⁴ range), which is critical for accurate TNR amplification [17]. Slightly increase polymerase concentrations to 2–2.5 units per 50 μL reaction to enhance processivity through structured regions, but avoid excessive concentrations that may promote nonspecific amplification [19].
Primer Design Considerations: Design primers with melting temperatures (Tm) of 55–70°C, ensuring that both forward and reverse primers have Tms within 5°C of each other [19]. Maintain GC content between 40–60% with uniform nucleotide distribution to prevent secondary structure formation [19]. Position primers to avoid complementarity at 3' ends, which promotes primer-dimer formation, and include one G or C nucleotide at the 3' terminus to enhance priming specificity without increasing mispriming risk [19]. When possible, design primers to anneal outside the repetitive region itself, as this improves amplification specificity and reduces artefact generation.
Thermal Cycling Parameters for Structured Templates
Thermal cycling parameters represent a critical determinant of success in TNR amplification, with denaturation temperature proving particularly important for overcoming the structural challenges posed by repetitive sequences. The following optimized protocol is adapted from successful amplification of the highly repetitive and GC-rich MaSp1 gene, with specific modifications to address TNR-specific amplification hurdles [16].
Initial Denaturation: 98°C for 30 seconds. This higher initial denaturation temperature helps disrupt stable secondary structures in GC-rich TNR regions that might resist complete separation at lower temperatures.
Amplification Cycling (30–35 cycles):
- Denaturation: 98°C for 10–20 seconds. The elevated denaturation temperature is crucial for complete separation of template strands in GC-rich repetitive regions [16]. While standard protocols often use 94–95°C, increasing to 98°C significantly improves amplification efficiency for structured templates.
- Annealing: 55–65°C for 10–30 seconds. Determine the optimal temperature based on primer Tm, with higher temperatures favoring increased specificity. For TNR amplification, slightly higher annealing temperatures (60–65°C) often improve specificity without compromising yield [16].
- Extension: 68°C for 40–60 seconds per kilobase. For longer TNR regions (>1 kb), extend the extension time to 2–3 minutes per kilobase to accommodate potentially slowed polymerization through structured regions.
Final Extension: 72°C for 5–10 minutes to ensure complete extension of all products, particularly important for structured templates where polymerase progression may be impeded.
Reaction Additives: Incorporate betaine (1–1.5 M final concentration) or DMSO (3–10%) to reduce secondary structure formation in GC-rich templates [16]. These additives enhance amplification efficiency by destabilizing hairpin structures that would otherwise impede polymerase progression through TNR regions.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for TNR Amplification and Analysis
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| High-Fidelity DNA Polymerases | Q5, Phusion, Pfu, Deep Vent | Accurate amplification with proofreading capability for TNR regions |
| PCR Additives | Betaine, DMSO, GC enhancers | Disruption of secondary structures in GC-rich repetitive sequences |
| Long-Range Amplification Kits | TaKaRa LA Taq, KAPA LongRange | Enhanced processivity for amplifying longer TNR expansions |
| SMRT Sequencing | PacBio Sequel II System | Comprehensive characterization of repeat length and interruptions |
| Specialty dNTPs | dUTP for carryover prevention | Contamination control in diagnostic applications |
| Cloning Systems | Gateway, restriction-based | Downstream manipulation and analysis of amplified TNR products |
Troubleshooting and Quality Assessment
Common Amplification Challenges
Amplification of TNR regions frequently encounters specific technical challenges that require systematic troubleshooting approaches. The most common issue observed during TNR PCR is the laddering effect on electrophoretic gels, characterized by multiple bands of varying sizes rather than a single discrete amplicon [16]. This artefact results from polymerase dissociation from the template due to structural impediments, generating incomplete fragments that function as megaprimers in subsequent cycles [16]. To address this challenge, first verify that denaturation temperatures reach 98°C, as this parameter critically influences the successful amplification of structured templates [16]. Additionally, evaluate magnesium concentrations (typically 2–3 mM for TNR targets) and consider incorporating betaine (1–1.5 M) to destabilize secondary structures without compromising polymerase activity.
Another frequent challenge in TNR work is preferential amplification of shorter alleles, which can lead to inaccurate genotyping and failure to detect expanded repeats. This bias stems from the increased difficulty polymerases experience when traversing longer repetitive sequences, resulting in more efficient amplification of shorter alleles. To minimize this bias, ensure template DNA quality and integrity, optimize polymerase concentrations to enhance processivity, and carefully control cycle numbers to prevent plateau phase amplification where size-based biases become exaggerated. For critical applications, validate amplification efficiency across different allele sizes using control templates with known repeat lengths.
Analytical Validation Methods
Given the technical challenges associated with TNR amplification, rigorous validation of amplification products is essential before proceeding to downstream applications. Electrophoretic analysis provides initial quality assessment, with successful amplification typically yielding a single discrete band of expected size, while failed reactions show smearing, multiple bands, or a characteristic laddering pattern [16]. For precise size determination, especially with larger expansions, pulsed-field gel electrophoresis may be necessary to resolve longer amplicons that conventional agarose gels cannot adequately separate.
Sequencing validation remains the gold standard for confirming TNR amplification accuracy. While Sanger sequencing suffices for shorter repeats, long-read sequencing technologies like PacBio SMRT sequencing are indispensable for characterizing larger expansions and detecting interruption patterns within the repetitive tract [18]. This platform has demonstrated exceptional capability in sequencing extremely long TNR regions (exceeding 1,000 triplets) while simultaneously identifying sequence interruptions that significantly influence disease presentation and inheritance patterns [18]. When employing SMRT sequencing for TNR analysis, prepare PCR products using standard protocols, then generate SMRTbell libraries according to manufacturer recommendations, ensuring that library insert sizes adequately encompass the entire repetitive region of interest.
Visualizing the Relationship Between Polymerase Properties and TNR Amplification
The following diagram illustrates the critical relationship between DNA polymerase characteristics and successful trinucleotide repeat amplification, highlighting how enzyme properties influence experimental outcomes:
Figure 1: Relationship between DNA polymerase properties and TNR amplification success. High-fidelity, high-processivity polymerases with proofreading capability (left pathway) enable accurate amplification of structured TNR templates, yielding specific products with correct sequences. In contrast, standard polymerases (right pathway) struggle with structured templates, resulting in characteristic amplification artefacts including laddering patterns, incomplete products, and sequence errors. The diagram highlights how specific polymerase characteristics directly influence experimental outcomes when working with challenging TNR templates.
The successful amplification of trinucleotide repeat regions demands careful consideration of DNA polymerase characteristics, with particular emphasis on both fidelity and processivity. High-fidelity enzymes possessing robust proofreading capabilities, such as Q5 and Phusion DNA polymerases, provide the necessary accuracy to maintain sequence integrity during TNR amplification, while simultaneously offering the processivity required to navigate through stable secondary structures that characterize these repetitive sequences [17]. The optimized protocols presented in this application note, incorporating elevated denaturation temperatures, strategic reaction additives, and validated thermal cycling parameters, provide researchers with a foundational framework for overcoming the technical challenges associated with TNR amplification [16].
As research into triplet expansion disorders continues to advance, the integration of improved amplification methodologies with long-read sequencing technologies promises to unlock new insights into the molecular mechanisms underlying repeat instability and its relationship to disease pathogenesis [18]. The strategic approach outlined in this document—combining polymerase biochemistry, reaction optimization, and appropriate analytical validation—empowers researchers to reliably amplify and characterize these challenging genomic regions, thereby accelerating both basic research and diagnostic applications for TNR disorders. Through continued refinement of these methodologies and thoughtful application of emerging technologies, the scientific community moves closer to comprehensive understanding and effective intervention for this complex class of genetic conditions.
Current Models of TNR Mutagenesis and Implications for PCR
Trinucleotide repeat (TNR) instability represents a major mutational mechanism underlying numerous severe neurological disorders, including Huntington's disease, myotonic dystrophy type 1, Friedreich's ataxia, and various spinocerebellar ataxias. These disorders collectively affect approximately 1 in 283 individuals based on recent population-scale genomic studies, indicating they are significantly more common than previously recognized [21]. The term "TNR mutagenesis" refers to the process where these repetitive DNA sequences become unstable and undergo expansion or contraction, ultimately leading to gene dysfunction and disease pathogenesis.
Understanding TNR mutagenesis is particularly crucial for PCR-based research, as the technical challenges of amplifying these repetitive, structure-forming regions can lead to significant artifacts and inaccuracies. These GC-rich repetitive sequences tend to form complex secondary structures—including hairpins, cruciforms, and quadruplexes—that impede polymerase processivity and promote slippage during amplification. This review integrates current molecular models of TNR instability with practical experimental strategies to overcome these challenges in PCR-based assays, providing a framework for more accurate genotyping and analysis of these difficult genomic regions.
Molecular Mechanisms of TNR Instability
DNA Repair Pathways in Repeat Expansion
Recent research has elucidated sophisticated molecular mechanisms governing TNR expansion and contraction, primarily involving specialized DNA repair pathways. The current model reveals that MutSβ (MSH2-MSH3) and MutLγ (MLH1-MLH3) complexes drive repeat expansion through a defined biochemical pathway [22].
The expansion mechanism begins when MutSβ recognizes and binds to extrahelical loops formed by expanded repeats. This recognition event triggers the recruitment of MutLγ, which incises the DNA strand opposite the extrahelical loop. RFC-loaded PCNA then plays a critical role in confining these MutLγ incisions to regions near the loop. Finally, Polδ utilizes the loop as a template for displacement synthesis, resulting in net repeat expansion [22].
In a protective counterbalance, the FAN1 nuclease promotes repeat contraction through a distinct pathway. FAN1 preferentially targets the looped strand itself, with its activity stimulated and directed by RFC-PCNA to the 3' boundary of the loop. Following FAN1-RFC-PCNA action, Polδ removes the loop and resynthesizes the DNA, resulting in contraction. FAN1 also directly inhibits MutLγ, providing dual protection against pathological expansions [22].
Table 1: Key Protein Complexes in TNR Instability
| Protein Complex | Components | Primary Function in TNR Instability |
|---|---|---|
| MutSβ | MSH2-MSH3 | Recognizes and binds extrahelical loops |
| MutLγ | MLH1-MLH3 | Incises DNA strand opposite extrahelical loops |
| FAN1 | Structure-specific nuclease | Promotes repeat contraction via loop excision |
| RFC-PCNA | RFC + PCNA trimer | Regulates incision positioning and FAN1 activity |
| Polδ | DNA polymerase delta | Conducts displacement synthesis (expansion) or resynthesis (contraction) |
Translesion Synthesis and TNR Protection
Beyond the core repair pathways, translesion synthesis (TLS) polymerases have emerged as important modulators of TNR stability. Recent evidence indicates that the TLS polymerase REV1 plays a protective role against TNR mutagenesis in human cells. Experiments using a quantitative GFP reporter system with expanded CAG repeats demonstrated that REV1 inhibition through either the chemical inhibitor JH-RE-06 or siRNA knockdown consistently increased TNR instability and underlying mutability [23].
This finding suggests that REV1 facilitates continuous DNA synthesis when replicative polymerases stall ahead of repeat secondary structures, providing a stabilizing influence. The protective function appears to involve bypassing difficult-to-replicate DNA regions caused by the complex structures formed by trinucleotide repeats, highlighting another layer of complexity in TNR mutagenesis that has implications for PCR amplification strategies.
Figure 1: Molecular Pathways in TNR Instability. MutSβ-MutLγ drive expansion, FAN1 promotes contraction and inhibits expansion, and REV1 provides stabilization.
Experimental Models and Methodologies
Quantitative TNR Instability Assays
The study of TNR mutagenesis requires specialized experimental approaches capable of quantitatively measuring repeat length changes. A leading method employs a quantitative GFP reporter system with expanded CAG repeats in human cell cultures [23]. This sophisticated approach enables researchers to:
- Monitor repeat instability in real-time through fluorescent readouts
- Quantify expansion and contraction rates simultaneously
- Test pharmacological interventions like REV1 inhibitors
- Combine with gene knockdown techniques (e.g., siRNA) for mechanistic studies
The protocol for this assay involves transfecting cells with the GFP-TNR reporter construct, treating with experimental conditions (e.g., JH-RE-06 for REV1 inhibition), and analyzing changes in fluorescence patterns that correspond to repeat length alterations. Flow cytometry then facilitates high-throughput quantification of these changes across large cell populations.
Base Editing for TNR Stabilization
Innovative genome editing approaches have recently been applied to modify TNR tracts and study their stability. Cytosine base editing (CBE) and adenine base editing (ABE) technologies can introduce nucleotide interruptions within pure TNR tracts, mimicking naturally occurring stable alleles found in unaffected individuals [24].
The experimental workflow for TNR base editing includes:
- Design of guide RNAs targeting CTG repeats on the opposite strand (for CAG repeats)
- Delivery of base editor components to patient-derived cells or animal models
- Validation of editing efficiency using high-throughput sequencing and specialized analysis tools like powTNRka
- Assessment of repeat stability through long-term culture and molecular analyses
This approach has demonstrated significant success in mouse models of Huntington's disease (Htt.Q111) and Friedreich's ataxia (YG8s), where AAV9 delivery of optimized base editors resulted in efficient editing in transduced tissues and significantly reduced repeat expansion in the central nervous system [24].
Table 2: Experimental Models for Studying TNR Mutagenesis
| Experimental System | Key Applications | Readout Methods | Advantages |
|---|---|---|---|
| GFP Reporter with CAG Repeats | Quantifying instability, drug screening | Flow cytometry, sequencing | High-throughput, quantitative |
| Base Editing in Patient Cells | Introducing stabilizing interruptions | HTS, powTNRka analysis | Therapeutic relevance, precise editing |
| Mouse Models (Htt.Q111, YG8s) | In vivo stability studies, therapeutic testing | AAV delivery, tissue analysis | Physiological context, translational value |
| Purified Protein Systems | Mechanistic biochemical studies | Southern blotting, nuclease assays | Defined molecular pathways, no cellular complexity |
Advanced Detection Methodologies
Sequencing-Based Detection Approaches
Accurate detection of TNR expansions has been transformed by advanced sequencing technologies and bioinformatic tools. Multiple approaches now enable comprehensive analysis:
Short-read whole-genome sequencing (WGS) coupled with specialized bioinformatic tools like ExpansionHunter can identify known repeat expansions with high sensitivity and specificity [21] [25]. This approach has enabled large-scale population studies revealing the true frequency of REDs across diverse populations.
Long-read technologies including Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) HiFi sequencing overcome the limitations of short reads for spanning long repetitive regions [25]. These methods are particularly valuable for characterizing complex expansion structures and detecting interruptions within repeat tracts.
Error-corrected sequencing methods like nanorate sequencing (NanoSeq) achieve ultra-low error rates (below 5 errors per billion base pairs) and enable detection of somatic mutations in small clones [26]. This exquisite sensitivity permits the identification of early mutational events in TNR instability.
Traditional Molecular Detection Methods
Despite advances in sequencing, traditional methods remain relevant in TNR analysis:
- Southern blotting provides quantitative information about large expansions but offers limited sequence context
- Repeat-primed PCR specifically detects expanded alleles but doesn't provide precise sizing
- Long-range PCR followed by sequencing enables targeted analysis of specific loci
- CRISPR-Cas9 enrichment combined with long-read sequencing offers a balanced approach for focused studies [25]
Each method presents distinct advantages for particular applications, and often a combination approach provides the most comprehensive understanding of TNR status in research or diagnostic contexts.
Figure 2: TNR Expansion Detection Methodologies. Multiple complementary approaches enable comprehensive analysis of repeat expansions.
Implications for PCR-Based TNR Research
Technical Challenges in TNR Amplification
Amplifying trinucleotide repeat regions presents substantial technical challenges that can compromise experimental results. These difficulties arise from the fundamental biochemical properties of repetitive DNA sequences:
- Secondary structure formation: GC-rich TNRs form stable hairpins and other structures that block polymerase progression
- Replication slippage: Repeated motifs promote misalignment between template and nascent strands during synthesis
- Premature termination: Polymerases frequently stall within long repetitive tracts, yielding incomplete products
- Amplification bias: Shorter alleles amplify more efficiently than expanded ones, skewing quantification
- Artifact generation: Slippage events create artificial expansion/contraction products that don't reflect biological reality
These challenges are particularly problematic for accurate genotyping of pathological expansions, where precise repeat length determination directly impacts clinical interpretation and molecular diagnosis.
Strategic Approaches for Improved TNR PCR
Based on current understanding of TNR mutagenesis mechanisms, several strategic approaches can significantly enhance PCR performance for these difficult templates:
PCR Additive Optimization: Incorporating specific additives that disrupt secondary structures is essential. Betaine, DMSO, formamide, and 7-deaza-dGTP can destabilize hairpin formations and improve amplification efficiency. The concentration of these additives requires empirical optimization for each specific TNR locus.
Polymerase Selection: Polymerases with high processivity and strong strand displacement activity are critical for traversing repetitive regions. Specialty polymerases or custom blends often outperform standard Taq polymerase for these applications.
Thermal Cycling Modifications: Implementing slow ramping rates between denaturing and annealing steps, incorporating prolonged extension times, and using touchdown protocols can dramatically improve specificity and yield for TNR amplicons.
Template Modification: Prior to PCR, template DNA can be treated with single-strand binding proteins or denaturing agents to minimize secondary structure formation during the initial amplification cycles.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for TNR Mutagenesis Studies
| Reagent / Tool | Specific Examples | Primary Application | Mechanistic Basis |
|---|---|---|---|
| REV1 Inhibitor | JH-RE-06 | Studying TLS in TNR stability | Blocks REV1 polymerase activity, increasing instability |
| Base Editors | CBE (C>T), ABE (A>G) | Introducing TNR interruptions | Mimics natural stabilizing variants in repeat tracts |
| TNR Reporter | GFP-CAG construct | Quantitative instability measurement | Fluorescent readout correlates with repeat length changes |
| Specialized Polymerases | Long-range PCR enzymes | Amplifying expanded repeats | Enhanced processivity through structured regions |
| PCR Additives | Betaine, DMSO, 7-deaza-dGTP | Improving TNR amplification | Destabilizes secondary structures, reduces slippage |
| Mismatch Repair Proteins | Recombinant MutSβ, MutLγ | In vitro expansion assays | Reconstitutes core expansion machinery |
| Nuclease Proteins | Recombinant FAN1 | In vitro contraction assays | Reconstitutes loop excision activity |
Current models of TNR mutagenesis reveal a complex interplay between DNA repair pathways, polymerase fidelity, and DNA secondary structure. The elaborate molecular machinery involving MutSβ-MutLγ-driven expansion counterbalanced by FAN1-mediated contraction provides a sophisticated framework for understanding repeat instability [22]. The emerging protective role of REV1 highlights additional complexity in how translesion synthesis polymerases modulate TNR stability [23].
For PCR-based research on trinucleotide repeat regions, these mechanistic insights directly inform experimental strategies. Understanding that repetitive sequences form structural impediments to polymerases justifies the use of structure-disrupting additives and specialized polymerases. Recognizing the propensity for replication slippage emphasizes the need for optimized thermal cycling conditions and careful validation of amplification products.
Future directions in TNR research will likely focus on translating these mechanistic insights into therapeutic strategies, such as the promising base editing approaches that can introduce stabilizing interruptions [24]. Additionally, continued refinement of detection methodologies will enable more comprehensive population screening and earlier diagnosis of repeat expansion disorders. For PCR-based applications, development of novel polymerase variants with enhanced ability to traverse repetitive sequences and novel additive combinations that more effectively destabilize secondary structures will further improve the accuracy and reliability of TNR genotyping.
The integration of mechanistic understanding with practical methodological optimization represents the most productive path forward for both basic research and clinical applications related to trinucleotide repeat disorders.
Building a Robust PCR Protocol: Additives, Enzymes, and Cycling Conditions
The selection of an appropriate DNA polymerase is a critical determinant for the success of the polymerase chain reaction (PCR), particularly when amplifying challenging genetic targets such as trinucleotide repeat regions. These sequences, often characterized by high Guanine-Cytosine (GC) content, are prevalent in promoters of housekeeping and tumor suppressor genes and are implicated in numerous genetic disorders including Fragile X syndrome (FMR1 gene), Huntington's disease, and myotonic dystrophy [27] [28]. Amplification of these regions is notoriously difficult due to the formation of stable secondary structures—hairpins and loops—that resist denaturation and cause polymerases to stall [27] [28]. This application note provides a structured framework for selecting from three key polymerase categories—hot-start, high-fidelity, and high-processivity enzymes—with a specific focus on protocols and reagent solutions optimized for trinucleotide repeat research and drug development.
DNA Polymerase Categories and Characteristics
Core Enzyme Types and Their Applications
Hot-Start Polymerases: These enzymes are engineered to remain inactive at room temperature, preventing non-specific amplification and primer-dimer formation during reaction setup. Activity is restored only after an initial high-temperature activation step (often >90°C) [29]. This mechanism is invaluable for multiplex PCR and for reactions set up at ambient temperature, ensuring high specificity and yield [30] [29].
High-Fidelity Polymerases: Enzymes like Q5 High-Fidelity DNA Polymerase possess 3′→5′ exonuclease (proofreading) activity, enabling them to correct misincorporated nucleotides during DNA synthesis [31] [27]. With error rates up to 280 times lower than Taq polymerase, they are essential for applications demanding high accuracy, such as cloning, sequencing, and functional studies of genetic targets [27].
High-Processivity Polymerases: Processivity refers to the number of nucleotides a polymerase can incorporate per binding event. Enzymes with high processivity remain bound to the template for longer, which allows them to efficiently amplify long targets and navigate through complex secondary structures and inhibitor-laden samples, such as those encountered in direct PCR from whole blood [32] [29].
Comparative Analysis of DNA Polymerases
Table 1: Key Characteristics and Applications of Different DNA Polymerase Types
| Polymerase Type | Key Feature | Primary Application | Example Enzymes | Best for Trinucleotide Repeats? |
|---|---|---|---|---|
| Standard Taq | No proofreading; low processivity | Routine amplification of simple templates | Taq DNA Polymerase | No - Often fails due to stalling. |
| Hot-Start | Inactive at room temperature | Multiplex PCR; high-specificity assays | Platinum II Taq Hot-Start [29] | Yes - Reduces non-specific products from complex repeats. |
| High-Fidelity | Proofreading; low error rate | Cloning, sequencing, mutation detection | Q5 High-Fidelity, Pfu [31] [27] | Yes - Critical for accurate genotyping. |
| High-Processivity | High nucleotides/binding event | Long amplicons; direct PCR from crude samples | OneTaq DNA Polymerase [29] | Yes - Navigates through secondary structures. |
Experimental Protocols for Amplifying Challenging Targets
Protocol 1: Direct PCR from Whole Blood for Genetic Screening
This protocol enables the rapid detection of short tandem repeats, such as those in the myotonic dystrophy type 1 (DM1) gene, directly from whole blood, bypassing DNA purification [32].
Reagent Setup:
- Polymerase Master Mix: 1X NEBNext High-Fidelity 2X PCR Master Mix (or other inhibitor-resistant master mix) [32].
- Primers: 0.5 µM each forward and reverse.
- Template: 10% whole blood (K₂EDTA anticoagulated) in a 25 µL total reaction volume.
- MgCl₂: Optional adjustment to 3 mM final concentration for higher blood volumes [32].
Thermal Cycling (Rapid Two-Step Protocol):
- Instrument: Use a thermal cycler with fast ramp rates (e.g., Streck Philisa) [32].
- Hot-Start/Cell Lysis: 98°C for 3 minutes.
- Amplification (30 cycles):
- Denaturation: 98°C for 6 seconds.
- Annealing/Extension: 68°C for 12 seconds.
Analysis: Analyze PCR products via agarose gel electrophoresis or an Agilent 2100 Bioanalyzer [32]. A successful reaction will show clear bands of the expected size (e.g., 114 + 3N bp for DM1), allowing for the exclusion of DM1-negative genotypes without further testing.
Diagram 1: Direct blood PCR workflow.
Protocol 2: Optimized PCR for GC-Rich Trinucleotide Repeats
This protocol is optimized for amplifying high-GC content targets like the FMR1 gene, using a combination of a robust polymerase and PCR additives [28].
Reagent Setup:
Thermal Cycling:
- Initial Denaturation: 98°C for 30-60 seconds.
- Amplification (35 cycles):
- Denaturation: 98°C for 10-20 seconds.
- Annealing: Temperature gradient from 65°C to 58°C (Touchdown), then maintained at optimal Ta [29].
- Extension: 72°C for 30-60 seconds/kb.
- Final Extension: 72°C for 2 minutes.
Analysis: Verify amplification and specificity on an agarose gel. The combination of betaine (destabilizes secondary structures) and DMSO (lowers melting temperature) is highly effective for reproducible amplification of GC-rich templates [28].
The Scientist's Toolkit: Essential Reagents for Success
Table 2: Key Research Reagent Solutions for Trinucleotide Repeat PCR
| Reagent Category | Specific Example | Function / Rationale |
|---|---|---|
| Specialized Polymerases | OneTaq DNA Polymerase with GC Buffer [27] | Optimized buffer system for high yields of difficult amplicons. |
| Q5 High-Fidelity DNA Polymerase with GC Enhancer [27] | High accuracy and enhanced amplification of GC-rich targets up to 80% GC. | |
| Q5 Blood Direct 2X Master Mix [27] | Enables direct PCR from up to 30% whole blood, resistant to inhibitors. | |
| PCR Additives | Betaine (1 M) [28] | Equalizes base-stacking energy, destabilizes secondary structures, reduces melting temperature. |
| DMSO (5-10%) [27] [28] | Disrupts base pairing, aiding in denaturation of stable GC-rich duplexes. | |
| 7-deaza-dGTP [28] | dGTP analog that reduces hydrogen bonding, preventing stable Hoogsteen base pairing. | |
| Sample Preparation | Tricine Buffer (pH 8.6) with Tween 20 & Trehalose [30] | High-pH buffer with detergent and stabilizer for direct PCR from anticoagulated blood. |
Optimization Strategies and Data Analysis
Systematic Optimization of Reaction Components
Amplification of trinucleotide repeats often requires fine-tuning beyond standard protocols. A systematic approach is key to success.
- Magnesium Concentration: Mg²⁺ is a critical cofactor. While 1.5-2.0 mM is standard, GC-rich templates may require optimization. Test a gradient from 1.0 mM to 4.0 mM in 0.5 mM increments to find the concentration that maximizes yield without compromising specificity [27] [28].
- Annealing Temperature: Employ a touchdown PCR strategy. Start with an annealing temperature 5-10°C above the calculated Tm and decrease it by 1°C per cycle for the first 10-15 cycles. This enriches the desired specific product early in the reaction [29].
- Polymerase Blends: For very long or complex trinucleotide repeats, consider using a blend of a high-processivity enzyme (e.g., Taq) and a high-fidelity enzyme (e.g., Pfu). This can combine the benefits of robust amplification with improved accuracy [29].
Diagram 2: GC-rich PCR optimization.
Quantitative Data from Experimental Studies
Table 3: Summary of Experimental Results from Literature
| Experimental Goal | Polymerase & Key Condition | Result / Detection Limit | Source |
|---|---|---|---|
| Direct pathogen detection in blood | EcoliTaq with high-pH Tricine buffer | 200 CFU/mL Salmonella typhimurium; 640 CFU/mL Shigella flexneri | [30] |
| Rapid DM1 screening from blood | NEBNext High-Fidelity Master Mix (15-min PCR) | 100% concordance with commercial kit; identified 23/40 (57.5%) as DM1 negative | [32] |
| Amplification of FMR1 (GC-rich) | Standard Taq with 1M Betaine + 5% DMSO | Reproducible amplification of >80% GC-rich 5' UTR of FMR1 gene | [28] |
| HLA-B27 genotyping | EcoliTaq (unpurified) | 100% concordance (55/55 positive & 55/55 negative) with commercial kit | [30] |
The strategic selection and application of DNA polymerases—harnessing the specificity of hot-start, the accuracy of high-fidelity, and the resilience of high-processivity enzymes—are fundamental to overcoming the challenges inherent in amplifying trinucleotide repeat regions. By integrating the detailed protocols, reagent solutions, and optimization strategies outlined in this application note, researchers and drug development professionals can significantly enhance the reliability and efficiency of their genetic analyses. This approach not only facilitates the screening and diagnosis of GC-rich repeat expansion disorders but also paves the way for advanced research into the mechanisms and potential therapeutics for these conditions.
Within the context of amplifying trinucleotide repeat (TNR) regions—a critical step in researching numerous neurodegenerative diseases and cancer—standard Polymerase Chain Reaction (PCR) conditions often prove inadequate [33] [34]. These GC-rich sequences are prone to forming stable secondary structures, such as hairpins, which hinder polymerase progression and lead to amplification failure, nonspecific products, or biased amplification of shorter alleles [35] [36]. This application note details the use of four essential additives—Dimethyl Sulfoxide (DMSO), formamide, betaine, and Bovine Serum Albumin (BSA)—to overcome these challenges, providing structured data, detailed protocols, and visual workflows to support researchers and drug development professionals in this specialized field.
Additive Mechanisms and Quantitative Data
The following table summarizes the core characteristics and mechanisms of action for each key additive.
Table 1: Essential PCR Additives for Amplifying GC-Rich and Trinucleotide Repeat Regions
| Additive | Recommended Concentration | Primary Mechanism of Action | Key Applications & Benefits |
|---|---|---|---|
| DMSO | 3-10% [36]; 5% used effectively in combinations [35] | Disrupts secondary structures, decreases DNA melting temperature (Tm), and reduces DNA supercoiling [36]. | Amplification of high GC-content DNA [36] [37]; Essential in combinations for specific TNR disease gene analysis (e.g., RET, LMX1B) [35]. |
| Betaine | 1.3 M [35] | Equalizes the melting temperature between GC- and AT-rich regions by acting as an isostabilizer [38] [37]. | Ameliorates amplification of GC-rich DNA sequences [38]; Critical for clean amplification in multi-additive cocktails [35]. |
| Formamide | 0-10% (effective range broadened with BSA) [39] | Destabilizes DNA double helix, likely by binding to DNA grooves, improving initial melting [39]. | Increases specificity in GC-rich amplification; More effective for fragments up to ~2.5 kb [39]. |
| BSA | 0.1-0.8 mg/mL [40]; 1-10 µg/µL as co-additive [39] | Binds to reaction inhibitors, stabilizes DNA polymerase against thermal denaturation, and protects DNA template [39] [40]. | Powerful co-enhancer with organic solvents (DMSO/formamide) for GC-rich targets over a broad size range [39]. |
Experimental Protocols
Protocol 1: Combined Additive Cocktail for GC-Rich Disease Genes
This protocol is adapted from research that successfully amplified GC-rich sequences (67-79% GC) of genes like RET, LMX1B, and PHOX2B, the latter being relevant to congenital central hypoventilation syndrome (CCHS) and involving triplet GCN expansion [35].
Research Reagent Solutions:
- Taq Polymerase: Use standard Taq (e.g., Eppendorf-5 Prime) or Hot-Start Taq (e.g., Applied Biosystems Gold Taq) for difficult templates [35].
- PCR Buffer: Use the manufacturer's supplied buffer, typically supplemented with MgCl₂ to a final concentration of 2-2.5 mM [35].
- dNTPs: 200 µM of each dNTP [35].
- Primers: 10 nmol of each forward and reverse primer [35].
- Template DNA: 100 ng of genomic DNA [35].
- Additive Cocktail:
Methodology:
- Prepare a master mix on ice containing, per 25 µL reaction:
- 1× PCR Buffer
- 2.5 mM MgCl₂
- 200 µM of each dNTP
- 50 µM 7-deaza-dGTP
- 10 nmol of each primer
- 1.3 M Betaine
- 5% DMSO
- 1.25 units of Taq Polymerase
- Nuclease-free water
- Aliquot the master mix and add 100 ng of template DNA.
- Perform PCR amplification using the following cycling conditions, optimized for the RET promoter region [35]:
- Initial Denaturation: 94°C for 5 minutes
- Amplification (40 cycles):
- Denaturation: 94°C for 30 seconds
- Annealing: 60°C for 30 seconds
- Extension: 72°C for 45 seconds
- Final Extension: 72°C for 5 minutes
- Analyze 5 µL of the PCR product by agarose gel electrophoresis.
Protocol 2: BSA as a Co-Enhancer with Organic Solvents
This protocol leverages BSA to boost the performance of DMSO or formamide, especially for long GC-rich amplicons (0.4 kb to 7.1 kb) from bacterial genomes [39].
Research Reagent Solutions:
- High-Fidelity DNA Polymerase: Essential for accurate amplification of long fragments.
- PCR Buffer: As supplied with the polymerase.
- Template DNA: e.g., Genomic DNA from Azospirillum brasilense (GC >65%) or other high-GC DNA [39].
- Organic Solvent: Either DMSO (2.5-5%) or formamide (≤10%) [39].
- BSA Solution: Molecular biology grade.
Methodology:
- Prepare a master mix on ice containing, per reaction:
- 1× PCR Buffer
- dNTPs, Mg²⁺, and primers per standard protocol
- Organic solvent (DMSO or formamide) at optimized concentration
- BSA at a concentration of 1-10 µg/µL
- High-Fidelity DNA Polymerase
- Nuclease-free water
- Add template DNA.
- Run PCR using cycling parameters appropriate for the amplicon length and primer Tₘ.
- Optional Pause-and-Add Enhancement: For very challenging amplicons, the protocol can be paused after the first 10 cycles to add a fresh aliquot of BSA, which counteracts its thermal denaturation and further enhances yield [39].
Diagram 1: Problem-Solution Framework for PCR Additives
Advanced Research Applications
Analyzing Trinucleotide Repeat Instability
Research into TNR instability, which drives disease progression in conditions like Huntington's disease, employs specialized in vitro methods to elucidate the role of DNA repair pathways [33].
Key Workflow: Oligonucleotide-Based Method for Base Excision Repair (BER) Studies This method assesses how site-specific DNA base lesions within a TNR tract influence repeat instability during repair [33].
Research Reagent Solutions:
- Synthesized Oligonucleotides: 100 nt long, containing a TNR tract (e.g., (CAG)₂₀) with a specific base lesion (e.g., 8-oxoguanine or an abasic site analog) inserted at the 5'-side, middle, or 3'-side of the repeats [33].
- Template Strand: Complementary oligonucleotide with a 5'-biotin tag [33].
- Purified BER Enzymes: Recombinant human APE1, Pol β, FEN1, DNA Ligase I [33].
- Binding Buffer: 0.1 M phosphate, 0.15 M NaCl, 1% Nonidet P-40, pH 7.2 [33].
- Avidin-Agarose: For capturing biotinylated templates [33].
- Fluorescence-labeled (6-FAM) PCR Primers: For subsequent fragment analysis [33].
Methodology:
- Substrate Preparation: Anneal the lesion-containing oligonucleotide to its biotinylated template strand [33].
- Reconstituted BER: Incubate the substrate with purified BER core enzymes and cofactors (e.g., in 50 mM Tris-HCl, 50 mM KCl, 0.1 mg/mL BSA, 1 mM DTT, 0.1 mM EDTA) to initiate repair [33].
- Product Isolation: Bind the reaction mixture to avidin-agarose. Denature with a low concentration of NaOH (e.g., 0.15 M) to elute the repaired strand, separating it from the biotin-bound template [33].
- Analysis: PCR-amplify the eluted repaired products using 6-FAM-labeled primers. Analyze the PCR products by capillary electrophoresis to determine changes in the TNR length (expansion or deletion) resulting from BER [33].
Diagram 2: Workflow for TNR Instability Analysis
The Scientist's Toolkit
Table 2: Essential Research Reagents for TNR Amplification and Analysis
| Reagent / Solution | Function / Application |
|---|---|
| Betaine (1.3 M Stock) | Isostabilizing agent for standard GC-rich and TNR PCR [35]. |
| DMSO (Molecular Grade) | Standard additive for disrupting secondary structures [35] [36]. |
| Molecular Grade BSA | Co-enhancer for use with organic solvents, especially for long amplicons [39]. |
| 7-deaza-dGTP | Modified nucleotide used to reduce secondary structure stability in extreme cases [35]. |
| Avidin-Agarose Beads | Critical for isolating biotin-labeled DNA strands in BER assays [33]. |
| Fluorescently Labeled Primers (e.g., 6-FAM) | Enable high-resolution fragment analysis for sizing TNR repeats [33] [34]. |
| Purified BER Enzymes | Required for reconstituting DNA repair pathways in mechanistic studies [33]. |
The amplification of trinucleotide repeat (TNR) regions presents a significant challenge in molecular diagnostics and research for hereditary neurodegenerative disorders. These regions, associated with over 50 human diseases, including Huntington's disease, Friedreich's ataxia, and various ataxias, exhibit unique biochemical properties that complicate standard polymerase chain reaction (PCR) protocols [41]. A core characteristic of expandable repeats is their propensity to form stable non-B DNA secondary structures—such as hairpins, slipped-strand DNA, and quadruplexes—which can block polymerase progression and promote replication slippage [41]. This application note provides a detailed framework for optimizing critical reaction components—Mg2+ concentration, dNTP balance, and primer design—to reliably amplify these problematic sequences within the context of ongoing research into PCR additives.
The Challenge of Trinucleotide Repeat Amplification
The instability of TNR regions is not merely a genetic phenomenon but a direct source of technical difficulty in PCR. DNA polymerases struggle to replicate through the stable secondary structures formed by repetitive sequences, often resulting in polymerase stalling, incomplete synthesis, and preferential amplification of smaller, non-pathogenic alleles over larger, expanded ones [41]. Furthermore, these regions are prone to expansion even during in vitro replication, a process influenced by reaction conditions such as temperature and cation concentration [42]. Slippage events and structure formation can lead to PCR artifacts, smearing on gels, and complete amplification failure, underscoring the need for finely tuned reaction conditions.
Optimizing Critical Reaction Components
Magnesium Ion (Mg2+) Concentration
Mg2+ serves as an essential cofactor for DNA polymerase, catalyzing the phosphodiester bond formation between the 3'-OH of a primer and the phosphate group of an incoming dNTP [19]. It also stabilizes the double-stranded DNA structure by neutralizing negative charges on the phosphate backbone. However, its concentration requires precise optimization, as it profoundly influences enzyme activity and reaction specificity [43].
Table 1: Effects and Optimization of Mg2+ Concentration in TNR PCR
| Parameter | Effect of Low [Mg2+] | Effect of High [Mg2+] | Recommended Optimization Strategy for TNRs |
|---|---|---|---|
| DNA Polymerase Activity | Drastically reduced enzymatic activity; low or failed amplification. | Increased activity but also higher rates of misincorporation. | Titrate in 0.5 mM increments from a starting point of 1.5 mM. |
| Reaction Fidelity | High fidelity but insufficient product yield. | Reduced fidelity; increased error rate due to stabilized mismatches. | Balance yield with specificity; aim for the lowest concentration that gives robust yield. |
| Secondary Structure | May not effectively destabilize stable hairpins in GC-rich repeats. | Can over-stabilize non-B DNA structures (e.g., hairpins), hindering replication. | Use additives like DMSO in conjunction with optimized Mg2+. |
| Specificity | High specificity but potentially low yield. | Stabilizes nonspecific primer-template binding; increases smearing and primer-dimers. | Use Mg2+ concentrations 1-2 mM higher than the total dNTP concentration [19]. |
A foundational study on TNR expansion in vitro demonstrated that varying Mg2+ concentration could trigger "dramatic expansions of repeat size during DNA replication," with expansions of up to 1000-fold observed [42]. This highlights the direct and powerful influence this cation has on repeat tract stability.
dNTP Balance and Concentration
Deoxynucleoside triphosphates (dNTPs) are the building blocks of DNA synthesis. Their concentration and balance are critical for maintaining replication fidelity and preventing mutations. A crucial factor in maintaining genome stability is establishing dNTP levels within a range that is optimal for chromosomal replication [44].
Table 2: Guidelines for dNTP Usage in TNR PCR
| Aspect | Standard Recommendation | Consideration for TNR PCR | Rationale |
|---|---|---|---|
| Final Concentration | 0.2 mM of each dNTP is common [19]. | May require slight optimization. Higher concentrations can be inhibitory. | High dNTP levels reduce replication fidelity by decreasing proofreading efficiency [44]. |
| Balance | Four dNTPs should be in equimolar amounts. | Strictly maintain equimolar ratios. | Imbalances can increase misincorporation by DNA polymerase, exacerbating slippage in repeats. |
| Km Consideration | Free dNTPs should be >0.010–0.015 mM [19]. | Ensure Mg2+ concentration is higher than [dNTP] to account for binding. | Mg2+ binds dNTPs; insufficient free Mg2+ can inhibit polymerase. |
| Fidelity | Lower dNTP (0.01–0.05 mM) can improve fidelity with non-proofreading enzymes [19]. | Consider for high-fidelity amplification of normal-length repeats. | Reduces the rate of mis-extension of mismatched primers. |
Abnormal dNTP levels have profound consequences. Elevated dNTP pools can stimulate error-prone translesion synthesis (TLS) polymerases, which have a higher Km for dNTPs and are more prone to slippage on repetitive tracts [44]. Conversely, low dNTP levels cause replication fork stalling, which is particularly problematic when the fork is already impeded by secondary structures, potentially leading to double-strand breaks and large-scale instability [44] [41].
Primer Design for Repeat Regions
Primers must be meticulously designed to ensure specific and efficient amplification of the target TNR locus while avoiding artifacts. The guiding principles are specificity, stability, and the avoidance of structures that promote slippage.
Table 3: Primer Design Specifications for TNR Amplification
| Design Feature | Optimal Specification | Rationale in TNR Context | Tool/Calculation |
|---|---|---|---|
| Length | 18–30 nucleotides [19] [13]. | Provides sufficient sequence for unique binding outside the repetitive region. | - |
| Melting Temperature (Tm) | 55–70°C; forward and reverse primers within 5°C of each other [19] [13]. | Ensures both primers anneal efficiently at the same temperature. | SantaLucia 1998 parameters are the default in NCBI Primer-BLAST [45]. |
| GC Content | 40–60% with uniform distribution [19]. | Avoids AT-rich regions with low binding stability and GC-rich regions with very high Tm. | - |
| GC Clamp | One G or C base at the 3'-end [13]. | Strengthens the terminal binding due to stronger hydrogen bonding. | - |
| Specificity Check | BLAST against the appropriate genome database [45]. | Confirms primers bind uniquely to the flanking sequence of the target gene (e.g., FXN for Friedreich's ataxia). | NCBI Primer-BLAST [45]. |
| Repeat Sequences | Avoid runs of 4+ identical bases or dinucleotide repeats (e.g., ACCCC, ATATAT) [13]. | Prevents mispriming within the repetitive tract itself or other satellite sequences. | - |
| Self-Complementarity | Avoid intra-primer homology (>3 bases) or inter-primer homology [13]. | Prevents formation of primer-dimers and self-hairpins that compete with target amplification. | - |
A critical rule is to design primers to unique, non-repetitive sequences that flank the TNR region. The primer should never be complementary to the repeat tract itself, as research has shown that "expansions were only detected when the primer was complementary to the repeat tract rather than the flanking sequence" [42].
Integrated Experimental Protocols
Protocol 1: Optimization of Mg2+ Concentration
Objective: To empirically determine the optimal MgCl2 concentration for the specific amplification of a TNR region. Materials:
- Template DNA (e.g., 50 ng human gDNA)
- Forward and Reverse Primers (10 µM stock each)
- 10x PCR Buffer (without MgCl2)
- 25 mM MgCl2 stock solution
- 10 mM dNTP mix
- DNA Polymerase (e.g., high-fidelity enzyme)
- Nuclease-free water
Method:
- Prepare a master mix for n+1 reactions, containing: 1x PCR Buffer, 0.2 mM dNTPs, 0.3 µM of each primer, 1 unit of DNA polymerase, and template DNA.
- Aliquot equal volumes of the master mix into 5 separate PCR tubes.
- Add the 25 mM MgCl2 stock to each tube to achieve final concentrations of: 1.0 mM, 1.5 mM, 2.0 mM, 2.5 mM, and 3.0 mM.
- Run the following touchdown PCR program:
- Initial Denaturation: 98°C for 2 min.
- 10x Cycles: 98°C for 20 sec, 65°C for 20 sec (decreasing by 1°C per cycle), 72°C for 1 min/kb.
- 25x Cycles: 98°C for 20 sec, 55°C for 20 sec, 72°C for 1 min/kb.
- Final Extension: 72°C for 5 min.
- Analyze 5 µL of each reaction on an agarose gel. The optimal condition produces a single, intense band of the expected size with minimal background smearing.
Protocol 2: Long-Range PCR for Large Expanded Repeats
Objective: To amplify long, pathogenic TNR alleles (e.g., >200 GAA repeats in Friedreich's ataxia) that are difficult to amplify with standard PCR [46]. Materials:
- Specialized reagents for long-range PCR
- Template DNA: 100-200 ng of high-quality gDNA.
- Primers: Designed per Table 3, with Tms around 70°C.
- DNA Polymerase: Use a high-fidelity, processive enzyme blend designed for long amplicons.
- PCR Additives: Include 5% (v/v) DMSO or 1 M Betaine to destabilize secondary structures.
Method:
- Set up a 50 µL reaction containing: 1x proprietary long-range PCR buffer, 2.0 mM MgSO4 (or as optimized), 0.3 mM of each dNTP, 0.5 µM of each primer, 5% DMSO, 2.5 units of DNA polymerase blend, and template DNA.
- Run the following PCR protocol:
- Initial Denaturation: 94°C for 2 min.
- 35x Cycles: 94°C for 20 sec, 68°C for 20 sec, 68°C for 10-15 min (extend time based on product length).
- Final Extension: 72°C for 10 min.
- For very large expansions (>1000 repeats), the extension time may need to be increased significantly.
- Analyze the product by 0.8-1.0% agarose gel electrophoresis. Expect a diffuse or discrete high-molecular-weight band.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for TNR PCR Research
| Reagent / Material | Function & Importance | Example Application |
|---|---|---|
| High-Fidelity DNA Polymerase Blends | Engineered for processivity and high tolerance to inhibitors; often includes a proofreading subunit for superior accuracy. | Essential for long-range amplification of expanded repeats and minimizing PCR-induced errors [19]. |
| PCR Additives (DMSO, Betaine, BSA) | DMSO and betaine destabilize GC-rich secondary structures. BSA stabilizes enzymes and blocks nonspecific surface binding. | Critical for denaturing stable hairpins in CAG or GAA repeat tracts to allow polymerase passage [43]. |
| TR-PCR & LR-PCR Assays | Triple-repeat primed PCR (TR-PCR) detects the presence of expansions. Long-range PCR (LR-PCR) sizes large alleles [46]. | Two-tier diagnostic genotyping for diseases like Friedreich's ataxia [46]. |
| dNTP Solutions (High-Purity) | Provides balanced, high-quality nucleotides for efficient and accurate DNA synthesis. | Prevents amplification bias and errors that can be exacerbated in repetitive sequences. |
| Mg2+ Optimization Kits | Provides a range of MgCl2 or MgSO4 concentrations in a pre-mixed format for systematic titration. | Enables rapid optimization of reaction specificity and yield for a new TNR target. |
Signaling Pathways and Workflow Diagrams
The reliable amplification of trinucleotide repeat regions demands a methodical approach to PCR optimization that accounts for their unique biochemical properties. By systematically tuning Mg2+ concentration, maintaining precise dNTP balance, and adhering to strict primer design rules, researchers can overcome the challenges of secondary structure formation and replication slippage. The protocols and frameworks provided here serve as a foundation for generating robust, reproducible data in both research and diagnostic settings. As the field advances, integrating these optimized conditions with emerging PCR technologies and a deeper understanding of repeat instability mechanisms will be crucial for developing next-generation assays for repeat expansion diseases.
Tailoring Thermal Cycler Parameters for TNR Templates
Amplifying trinucleotide repeat (TNR) regions represents a significant challenge in molecular biology due to their propensity to form stable secondary structures and their characteristically high GC content. These regions are clinically relevant for numerous neurological disorders, making their reliable amplification essential for both basic research and diagnostic applications within pharmaceutical development [8]. This application note details a optimized thermal cycler parameters and specialized reagent formulations to overcome the inherent difficulties of TNR amplification, providing robust protocols validated for challenging templates.
Experimental Protocols
Optimized Thermal Cycling Protocol for GC-Rich TNR Templates
The following procedure is adapted from established methods for GC-rich nicotinic acetylcholine receptor subunits and incorporates critical modifications for successful TNR amplification [8].
Reagents and Materials
- Template DNA: 100 ng/μL of genomic DNA or plasmid containing the TNR region.
- Primers: Forward and reverse primers (10 μM each), designed with a Tm of 55–65°C.
- PCR Additives: 5 M Betaine, 100% DMSO, 10x GC Enhancer (if provided with polymerase).
- Polymerase: High-fidelity, proofreading DNA polymerase (e.g., Platinum SuperFi II, Phusion High-Fidelity).
- Nucleotides: dNTP mix (10 mM each).
- Buffer: Appropriate 10x reaction buffer supplied with the polymerase.
- Equipment: Thermal cycler with gradient block capability and precise temperature control.
Procedure
- Prepare Reaction Master Mix (50 μL total volume):
- Sterile water: 38 μL
- 10x Reaction Buffer: 5 μL
- dNTP Mix (10 mM): 1 μL
- Forward Primer (10 μM): 2 μL
- Reverse Primer (10 μM): 2 μL
- Betaine (5 M): 5 μL (Final concentration: 0.5-1 M)
- DMSO: 1.25 μL (Final concentration: 2.5%)
- DNA Template (100 ng/μL): 1 μL
- DNA Polymerase (0.5 U/μL): 1 μL
Thermal Cycling Conditions:
- Initial Denaturation: 98°C for 3–5 minutes [47]
- Amplification Cycles (35 cycles):
- Denaturation: 98°C for 30 seconds
- Annealing: Gradient from 55–72°C for 30 seconds (optimize using gradient function)
- Extension: 72°C for 1–2 minutes per kb of amplicon
- Final Extension: 72°C for 10–15 minutes [47]
- Hold: 4°C indefinitely
Product Analysis:
- Analyze 5 μL of PCR product by 1% agarose gel electrophoresis with appropriate DNA size markers.
- Visualize using ethidium bromide or safer alternatives like Red Safe.
Troubleshooting Notes
- If nonspecific amplification occurs: Increase annealing temperature in 2–3°C increments.
- If no product is observed: Reduce annealing temperature, increase betaine concentration to 1 M, or extend extension time.
- For very long TNR regions (>1 kb): Consider using a polymerase mix specifically designed for long-range amplification.
Parameter Optimization Experiment
To systematically determine optimal conditions for TNR amplification, a multi-factorial experiment is recommended.
Experimental Design
- Tested Variables:
- Annealing Temperature: 55°C, 58°C, 60°C, 63°C, 65°C, 68°C
- Betaine Concentration: 0 M, 0.5 M, 1 M, 1.5 M
- DMSO Concentration: 0%, 2.5%, 5%, 7.5%
- Polymerase Type: Standard Taq, High-Fidelity, GC-Rich Specialized
- Evaluation Metrics:
- Amplification Specificity (gel band sharpness)
- Product Yield (band intensity)
- Presence of Secondary Products
- Reproducibility across replicates
Results and Data Presentation
Quantitative Parameter Comparison
Table 1: Optimization of Thermal Cycler Parameters for TNR Amplification
| Parameter | Standard Value | GC-Rich Optimized Value | TNR-Specific Recommendation | Effect on Amplification |
|---|---|---|---|---|
| Initial Denaturation | 94°C, 2 min [48] | 98°C, 3-5 min [47] | 98°C, 5 min | Ensures complete separation of high-GC templates |
| Denaturation Cycles | 94°C, 30 sec [48] | 98°C, 30 sec [47] | 98°C, 30 sec | Prevents reformation of secondary structures |
| Annealing Temperature | Calculated Tm-5°C [47] | Gradient optimization | Tm+2°C with additives | Balances specificity and efficiency |
| Annealing Time | 30 sec [48] | 30 sec | 45 sec | Accommodates slower primer binding with additives |
| Extension Time | 1 min/kb [47] | 1-2 min/kb | 2 min/kb | Compensates for polymerase slowing with additives |
| Cycle Number | 25-30 [47] | 35 | 35-40 | Increases yield for difficult templates |
| Final Extension | 72°C, 5 min [48] | 72°C, 10-15 min [47] | 72°C, 15 min | Ensures complete full-length products |
Table 2: Additive Effects on TNR Amplification Efficiency
| Additive | Recommended Concentration | Mechanism of Action | Effect on Tm | Compatibility with Polymerases |
|---|---|---|---|---|
| Betaine | 0.5-1.5 M [8] | Equalizes base-pair stability, disrupts secondary structures | Decreases | Compatible with most polymerases |
| DMSO | 2.5-10% [8] | Disrupts base pairing, prevents secondary structures | Decreases 5.5-6.0°C per 10% [47] | May inhibit some polymerases at >10% |
| Formamide | 1-5% | Denaturant, prevents secondary structures | Decreases | Concentration-dependent inhibition |
| 7-deaza-dGTP | Substitute for dGTP | Replaces dGTP, reduces Hoogsteen bonding | Decreases | Compatible with most polymerases |
| GC Enhancer | As per manufacturer | Proprietary formulations | Varies | Polymerase-specific |
Polymerase Performance Comparison
Table 3: DNA Polymerase Selection for TNR Amplification
| Polymerase Type | Processivity | Proofreading | Recommended Additives | Optimal Extension Time |
|---|---|---|---|---|
| Standard Taq | Low | No | Betaine, DMSO | 1 min/kb [47] |
| High-Fidelity | Medium | Yes | Betaine, DMSO | 2 min/kb [47] |
| GC-Rich Specialized | High | Variable | Included in buffer | As recommended |
| Fast Polymerases | High | Variable | Betaine | 15 sec/kb |
Visualization of Workflows and Relationships
Experimental Workflow for TNR Amplification
Additive Mechanisms in TNR Amplification
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for TNR Amplification
| Reagent | Function | Optimized Concentration | Key Considerations |
|---|---|---|---|
| Betaine | Destabilizes secondary structures, equalizes GC/AT bonding stability [8] | 0.5-1.5 M | Stock solution: 5M; non-inhibitory to polymerases |
| DMSO | Disrupts hydrogen bonding, prevents secondary structure formation [8] | 2.5-5% | Higher concentrations may inhibit polymerase activity |
| 7-deaza-dGTP | Replaces dGTP, reduces Hoogsteen bonding in GC-rich regions | 1:1 substitution for dGTP | Requires adjustment of dNTP mix composition |
| High-Fidelity Polymerase | Accurate amplification with proofreading activity | As manufacturer recommends | Essential for sequencing applications |
| GC Enhancer Buffer | Proprietary formulations to improve GC-rich amplification | As manufacturer recommends | Polymerase-specific formulations |
| Single-Strand Binding Protein | Stabilizes single-stranded DNA, prevents reannealing [49] | 50-200 ng/μL | Particularly useful for long TNR regions |
Discussion
The optimized thermal cycler parameters and reagent formulations presented here address the fundamental challenges of TNR amplification through a multi-faceted approach. The combination of elevated denaturation temperatures, extended cycling times, and strategic additive use creates conditions that counteract the strong secondary structures and high melting temperatures characteristic of TNR regions [8] [47].
The critical importance of betaine and DMSO in these protocols cannot be overstated. These additives work through complementary mechanisms—betaine by equalizing the stability of GC and AT base pairs, and DMSO by directly interfering with hydrogen bonding networks that stabilize secondary structures [8]. This dual approach allows standard polymerases to successfully amplify templates that would otherwise be refractory to amplification.
Recent advancements in enzymatic amplification technologies, particularly engineered helicase systems like SHARP, offer promising alternatives to conventional thermal cycling for challenging templates [49]. While not yet widely adopted, these isothermal methods demonstrate the potential for amplifying long GC-rich targets (up to 6000 bp) without the need for extreme denaturation temperatures, potentially preserving polymerase activity and improving yields for particularly difficult TNR regions.
For researchers in pharmaceutical development, these optimized protocols enable reliable genotyping of TNR expansions associated with neurological disorders, facilitating both basic research into disease mechanisms and the development of targeted therapeutics. The standardized parameters and comprehensive reagent toolkit provide a foundation for reproducible amplification of these challenging genetic targets across different laboratory settings.
Solving Common Problems: A Troubleshooting Guide for TNR PCR
Diagnosing No Amplification, Smears, or Multiple Bands
Amplifying trinucleotide repeat (TNR) regions presents unique challenges for molecular researchers. The propensity of these GC-rich, repetitive sequences to form complex secondary structures often leads to PCR artifacts such as failed amplification, smeared bands, or non-specific products. These issues are particularly prevalent in genetics research and drug development for TNR disorders like Huntington's disease and myotonic dystrophy. This application note provides a structured troubleshooting guide and optimized protocols to overcome these obstacles, enabling reliable analysis of these clinically significant genomic regions.
Troubleshooting Common PCR Amplification Issues
The table below summarizes the primary symptoms, their causes, and recommended solutions for PCR amplification of difficult trinucleotide repeat regions.
Table 1: Troubleshooting Guide for Trinucleotide Repeat PCR
| Symptom | Potential Cause | Recommended Solution |
|---|---|---|
| No Amplification [50] | PCR inhibitors present in template | Dilute template 100-fold or purify using a silica-membrane-based cleanup kit [50]. |
| Excessively stringent cycling conditions | Lower annealing temperature in 2°C increments; increase extension time [50]. | |
| Low abundance or inaccessible template | Increase number of PCR cycles (3-5 cycles at a time, up to 40 cycles) [50]. | |
| Nonspecific Bands (Multiple Bands) [50] | Insufficiently stringent PCR conditions | Increase annealing temperature in 2°C increments; use touchdown PCR; reduce cycle number [50]. |
| Excess template | Reduce the amount of template by 2- to 5-fold [50]. | |
| Non-optimal primers | Check primer specificity via BLAST; redesign primers if necessary [50]. | |
| Smear of Bands [50] | Contamination from previous PCR products | Use separate pre- and post-PCR areas; decontaminate pipettes and workstations with UV light or 10% bleach [50]. |
| Over-cycling or suboptimal conditions | Reduce number of cycles; reduce amount of template; increase annealing temperature [50]. |
Special Considerations for Polymerase Selection
The choice of DNA polymerase is critical for success. Standard polymerases often fail with complex TNR templates. Consider the following enzyme-specific adjustments [50]:
- For high-GC content templates (>65%): Use a polymerase specifically formulated for high GC content.
- For PrimeSTAR HS DNA Polymerase: Use ≤100 ng of human genomic DNA in a 50 µl reaction and an extension time of at least 1 min/kb.
- For SpeedSTAR HS DNA Polymerase: While the standard extension is 10 sec/kb, complex templates may require an increase to ~0.5 min/kb.
Advanced Methodologies for TNR Sizing and Quantification
Accurate sizing of expanded repeats is vital for molecular diagnosis and prognosis in TNR disorders. Traditional methods face challenges due to the inherent instability of repeats and preferential amplification of shorter alleles [51] [52].
Table 2: Advanced Methods for Analyzing Trinucleotide Repeat Expansions
| Method | Principle | Application & Advantage |
|---|---|---|
| Small-Pool PCR (SP-PCR) [51] | Amplification of diluted DNA samples to isolate and visualize individual expanded alleles. | Directly assesses somatic repeat instability from small amounts of DNA; captures heterogeneity in repeat length [51]. |
| Triplet Repeat Primed PCR (TP-PCR) [51] | Uses a repeat-specific primer enabling amplification regardless of repeat length. | Rapidly identifies the presence of expanded alleles; often used as a initial screening tool [51]. |
| µLAS (Micro-Laboratory for DNA Separation) [52] | Microfluidics-based system for high-efficiency DNA separation and sizing. | High-speed (minutes) and sensitive (femtomolar) sizing of expanded repeats across a broad size range [52]. |
| Long-Read Sequencing (e.g., Oxford Nanopore, PacBio) [51] | Sequences single DNA molecules, allowing for precise determination of long repeat tracts. | Provides comprehensive mapping of repeat length and sequence context; can be costly and require high DNA input [51]. |
Experimental Protocols
Protocol 1: PCR Amplification of Challenging Trinucleotide Repeat Regions
This protocol is optimized for amplifying GC-rich, structured TNR regions using a high-fidelity, GC-enhanced polymerase.
Materials:
- DNA Polymerase: A proofreading polymerase robust to high GC content (e.g., PrimeSTAR GXL).
- PCR Additives: Betaine (1-1.5 M final concentration) or DMSO (2-5% v/v).
- Primers: Designed with a Tm >55°C for better specificity [50].
- Template: 10-100 ng of high-quality, purified genomic DNA.
Procedure:
- Reaction Setup: Prepare a 50 µL master mix on ice.
- 1X Polymerase Buffer
- 200 µM of each dNTP
- 0.5 µM of each forward and reverse primer
- 1-1.5 M Betaine
- 1.25 U of DNA Polymerase
- Template DNA (10-100 ng)
- Thermal Cycling:
- Initial Denaturation: 98°C for 2 minutes.
- 35-40 Cycles of:
- Denaturation: 98°C for 10 seconds.
- Annealing: Use a gradient to determine the optimal temperature (start at 60-68°C for 5-15 seconds) [50].
- Extension: 68°C for 1-2 minutes per kb. For very long or complex repeats, extend this time.
- Final Extension: 68°C for 5-10 minutes.
- Analysis: Analyze 5-10 µL of the PCR product by agarose gel electrophoresis.
SP-PCR is used to detect the heterogeneity of expanded TNR alleles within a tissue or cell population.
Materials:
- DNA Sample: Genomic DNA from tissues of interest (e.g., leukocytes, corneal endothelium).
- Primers: Flanking the TNR region in the target gene (e.g., TCF4 for FECD).
- Specialized Kits: DNeasy Blood and Tissue Kit for DNA extraction.
Procedure:
- DNA Quantification: Precisely measure DNA concentration using a sensitive method like droplet digital PCR (ddPCR) to ensure accuracy for dilution [51].
- Sample Dilution: Serially dilute the DNA to a concentration that yields a statistical probability of amplifying a single molecule per reaction (e.g., picogram amounts).
- PCR Amplification: Perform a standard PCR protocol using the diluted DNA samples and primers flanking the repeat region.
- Product Analysis: Analyze the PCR products by Southern blotting or capillary electrophoresis to visualize the size distribution of the expanded alleles, revealing the degree of somatic instability [51].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Amplifying Trinucleotide Repeat Regions
| Reagent / Material | Function in TNR PCR |
|---|---|
| Betaine | PCR additive that destabilizes secondary structures, improving the efficiency of amplifying GC-rich repeats [50]. |
| High-Fidelity GC-Rich Polymerase | Engineered enzyme blends that remain stable and processive through difficult templates with high GC content or strong secondary structures [50]. |
| NucleoSpin Gel and PCR Clean-up Kit | For purifying template DNA to remove common PCR inhibitors (e.g., salts, organics) that can prevent amplification [50]. |
| Homotrimeric UMI Barcodes | Unique Molecular Identifiers synthesized in homotrimer blocks to enable error-correction in sequencing, countering PCR-introduced errors in quantitative assays [53]. |
| Terra PCR Direct Polymerase | A polymerase blend tolerant to inhibitors, useful for amplifying targets directly from crude samples without extensive DNA purification [50]. |
Experimental Workflow and Pathway Diagrams
The following diagram illustrates the logical decision-making process for diagnosing and resolving common PCR issues when working with trinucleotide repeats.
PCR Troubleshooting Pathway - This flowchart provides a systematic approach to diagnose and resolve the most common PCR failures when amplifying trinucleotide repeat regions.
Successfully amplifying trinucleotide repeat regions requires a meticulous approach to template quality, reagent selection, and cycling parameters. The protocols and troubleshooting guides provided here form a foundation for reliable analysis of these genetically and clinically significant sequences. By implementing specialized polymerases, PCR additives like betaine, and advanced sizing techniques such as SP-PCR, researchers and drug developers can overcome the inherent challenges of TNR amplification, thereby advancing our understanding and therapeutic targeting of repeat expansion disorders.
Optimizing Primer Design to Avoid Primer-Dimers and Mispriming
Primer-dimer formation and mispriming are prevalent challenges in polymerase chain reaction (PCR) that significantly reduce amplification efficiency, specificity, and yield. These artifacts arise when primers anneal to each other or to non-target sequences, consuming reaction resources and generating false-positive signals. Within research focused on amplifying challenging trinucleotide repeat regions—a critical area for studying neurological disorders like Huntington's disease and fragile X syndrome—optimized primer design becomes paramount. The structural peculiarities of these GC-rich, repetitive sequences exacerbate mispriming risks. This application note provides detailed protocols and data-driven strategies to overcome these obstacles, integrating advanced design principles with practical experimental optimization for research and diagnostic applications.
Understanding Primer-Dimers and Mispriming
Definitions and Formation Mechanisms
Primer-dimers are short, unintended DNA fragments amplified when primers anneal to each other instead of the target DNA template. They typically appear below 100 base pairs on an electrophoretic gel and have a smeary appearance rather than a sharp, defined band [54]. Two primary formation mechanisms exist:
- Self-dimerization: Occurs when a single primer molecule contains regions of self-complementarity, enabling it to fold and create a free 3' end that DNA polymerase can extend.
- Cross-dimerization: Happens when forward and reverse primers contain complementary regions, allowing them to anneal to each other and form extendable duplexes [54].
Mispriming refers to non-specific amplification resulting from primers binding to partially complementary, non-target sites in the template DNA. This leads to amplification of unintended products, compromising assay specificity and sensitivity, particularly in complex genomic samples.
Impact on PCR Performance
The consequences of primer-dimer formation and mispriming are particularly detrimental in sensitive applications:
- Resource Consumption: Primer-dimers compete for essential reaction components—DNA polymerase, dNTPs, and primers—reducing available resources for target amplification [55].
- Reduced Sensitivity: Non-specific products can overwhelm detection systems in quantitative PCR, leading to inaccurate quantification and false negatives, especially with low-abundance targets [56].
- Compromised Multiplexing: In multiplex PCR assays, primer-dimer formation between different primer pairs dramatically increases with primer complexity, potentially causing assay failure [55].
- Sequencing Interference: Primer-dimers consume sequencing resources in next-generation sequencing library preparations, reducing usable reads and increasing costs [10].
Primer Design Optimization Strategies
Computational Design Parameters
Strategic primer design represents the most effective approach to prevent primer-dimer formation. The following parameters should be rigorously evaluated during design:
Table 1: Critical Parameters for Optimal Primer Design
| Parameter | Optimal Range | Rationale | Design Tool Evaluation |
|---|---|---|---|
| Length | 18-25 nucleotides | Provides sufficient specificity while maintaining reasonable annealing kinetics | Check for uniqueness in target genome |
| Tm | 55-65°C | Enables specific annealing at standardized temperatures | Ensure Tm uniformity within ±1°C for primer pairs |
| GC Content | 40-60% | Balanced stability; avoids AT-rich instability or GC-rich secondary structures | Flag primers outside this range [57] |
| 3'-End Complementarity | ≤3 complementary bases | Minimizes primer-dimer initiation from extendable 3' ends | Critical parameter for dimer prevention [58] |
| Self-Complementarity | ΔG > -5 kcal/mol | Reduces intra-primer folding and self-dimerization | Evaluate hairpin formation potential |
| Cross-Complementarity | ΔG > -5 kcal/mol | Prevents inter-primer annealing and dimer formation | Check all possible pairings in multiplex assays |
Advanced design should incorporate nearest-neighbor thermodynamics rather than simple GC-content calculations. This method, based on SantaLucia's unified parameters (1998), accounts for sequence context and dinucleotide stacking interactions, providing Tm predictions with ±1-2°C accuracy compared to ±5-10°C for GC% methods [57]. Salt concentrations significantly impact melting temperature and must be considered in design parameters:
- Na⁺ concentration (50-200 mM): Increasing from 50 mM to 100 mM raises Tm by approximately 3-5°C [57]
- Mg²⁺ concentration (1.5-2.5 mM for standard PCR): Has a stronger stabilizing effect than Na⁺; increasing from 0 mM to 2 mM can raise Tm by 5-8°C [57]
- DMSO (0-10%): Lowers Tm by approximately 0.5-0.7°C per 1% added [57]
Advanced Sequence Engineering
For challenging applications like trinucleotide repeat amplification, innovative chemistries can overcome persistent dimerization:
Self-Avoiding Molecular Recognition Systems (SAMRS) incorporate modified nucleobases that pair strongly with natural DNA but weakly with other SAMRS nucleotides. This technology strategically replaces standard bases (G, A, C, T) with SAMRS analogs (g, a, c, t) at problematic positions in the primer sequence [55]. SAMRS primers maintain efficient annealing to natural DNA targets while dramatically reducing primer-primer interactions. Optimal implementation requires:
- Limiting SAMRS components to 3-5 modifications per primer to maintain efficient extension
- Strategic placement at the 3'-end where dimer initiation occurs
- Validation of SNP discrimination capability, which often exceeds conventional allele-specific PCR [55]
Experimental Optimization Protocols
Reaction Condition Optimization
Even well-designed primers require optimized reaction conditions to prevent artifacts. The following protocol systematically addresses primer-dimer formation:
Protocol 1: PCR Condition Optimization
Primer Concentration Titration
Thermal Cycling Optimization
- Implement a hot-start activation by withholding polymerase activity until temperatures >60°C
- Use commercial hot-start polymerases or physical barriers (wax beads)
- Increase annealing temperature incrementally (1-2°C steps) to determine the maximum temperature that maintains specific amplification [54]
- Extend denaturation times to ensure complete separation of primer dimers between cycles [54]
Additive Optimization for Tricky Templates
- For GC-rich trinucleotide repeats, include DMSO (3-10%) or betaine (0.5-1.5 M) to reduce secondary structure
- Optimize MgCl₂ concentration (1.5-5.0 mM) in 0.5 mM increments
- Consider template-enhanced PCR by adding non-specific DNA (e.g., 100 ng/μL salmon sperm DNA) to compete for mispriming sites
Primer-Dimer Troubleshooting Workflow
The following decision tree provides a systematic approach to diagnose and resolve persistent primer-dimer issues:
Validation and Quality Control
Rigorous validation ensures primer specificity and efficiency:
Protocol 2: Primer-Dimer Assessment
No-Template Control (NTC) Setup
- Include NTC reactions containing all components except template DNA
- Process NTCs alongside experimental samples through entire protocol
- Any amplification in NTC indicates primer-dimer formation [54]
Analytical Electrophoresis
- Use high-resolution gels (4% agarose or polyacrylamide)
- Run gels sufficiently long to separate primer-dimers (<100 bp) from target amplicons
- Identify primer-dimers by their characteristic smeary appearance below 100 bp [54]
qPCR Efficiency Calculation
Research Reagent Solutions
Table 2: Essential Reagents for Optimized PCR
| Reagent Category | Specific Examples | Function & Mechanism | Application Notes |
|---|---|---|---|
| Hot-Start Polymerases | JumpStart Taq, HotStar Plus | Antibody or chemical modification inhibits activity at room temperature; prevents pre-PCR mispriming [61] | Essential for multiplex assays and low-template applications |
| PCR Additives | DMSO (3-10%), Betaine (0.5-1.5 M), Formamide | Reduce secondary structure in template/primers; alter effective Tm [57] | Titrate concentration for specific template types |
| Modified Nucleotides | SAMRS phosphoramidites (Glen Research, ChemGenes) [55] | Enable synthesis of primers with reduced primer-primer interactions | Require specialized synthesis and purification |
| Detection Chemistries | EvaGreen, SYBR Green, TaqMan probes [60] | Enable real-time monitoring of amplification specificity | EvaGreen preferred for high-resolution melt analysis |
| Software Tools | Tm Calculator (nearest-neighbor), OligoAnalyzer | Predict secondary structures, Tm values, and primer interactions | Must incorporate salt correction algorithms [57] |
Optimizing primer design to avoid primer-dimers and mispriming requires a multifaceted approach combining sophisticated in silico design with empirical validation. For challenging targets like trinucleotide repeat regions, strategic implementation of advanced technologies such as SAMRS-modified primers, coupled with rigorous optimization of reaction conditions, enables successful amplification without artifacts. These protocols provide researchers with a comprehensive framework to enhance PCR specificity and efficiency, ultimately supporting accurate genetic analysis and diagnostic development.
Fine-Tuning Additive Concentrations and Denaturation Times
Trinucleotide repeat disorders are a group of human diseases caused by the abnormal expansion of repetitive sequences, primarily affecting the nervous system and occurring during various stages of human development [62]. These conditions, which include Huntington's disease, fragile X syndrome, myotonic dystrophy, Friedreich's ataxia, and various spinocerebellar ataxias, present significant challenges for molecular diagnosis and research due to the unique properties of the repeat regions [62] [63]. The molecular diagnosis of these disorders often relies on genetic tests to identify the expansion of these repeat sequences, with polymerase chain reaction (PCR) serving as a fundamental technique for amplification and analysis [28] [63].
Amplifying Guanine-Cytosine (GC)-rich trinucleotide repeat regions presents particular difficulties in PCR-based methods. These regions are characterized by complex inter- and intra-strand folding, producing secondary structures such as hairpins and loops due to increased hydrogen bonding between neighboring cytosine and guanine bases [28]. These stable secondary structures are resistant to melting during standard denaturation steps and can cause DNA polymerases to stall, resulting in incomplete or non-specific amplification [28]. Since approximately 3% of the human genome is highly GC-rich and 28% of genes are located within these GC-rich regions, optimizing PCR conditions for such challenging templates is essential for reliable genetic analysis [28]. This application note provides detailed protocols and data-driven recommendations for fine-tuning additive concentrations and denaturation parameters to successfully amplify trinucleotide repeat regions.
Experimental Protocols for PCR Optimization
DNA Extraction and Quality Assessment
For genetic testing involving trinucleotide repeats, DNA can be extracted from various sources, including buccal mucosal cells, which provide a non-invasive collection method [28]. A rapid protocol for buccal cell DNA extraction involves the following steps:
- Sample Collection: Participants should refrain from eating and drinking for 30 minutes prior to sample collection. Rinse the mouth and collect buccal cells by rolling an autoclaved cotton bud inside the cheek twenty times on each side [28].
- Cell Lysis: Dip the cotton bud containing buccal cells in a microcentrifuge tube containing 500 μl of lysis buffer (0.1× Gitschiez buffer with 0.5% Triton ×100) [28].
- Protein Digestion: Add Proteinase K to a final concentration of 40 μg/ml, mix well, and incubate at 63°C for 6 minutes [28].
- DNA Precipitation: Add 250 μl of 4.5 M NaCl, mix vigorously, and centrifuge at 13,000× g for 15 minutes. Recover the supernatant and ethanol-precipitate the genomic DNA [28].
- DNA Resuspension: Resuspend the extracted DNA in 20 μl of Tris-EDTA buffer (pH 8.0). Assess concentration and integrity using ethidium bromide-stained 0.8% agarose gel electrophoresis compared against known concentrations of lambda DNA [28].
Primer Design Considerations for Trinucleotide Repeat Regions
Appropriate primer design is essential for successful PCR amplification. When designing primers for trinucleotide repeat regions, follow these guidelines:
- Primer length should be 15–30 nucleotide bases [64].
- Optimal G-C content should range between 40–60% [64].
- The 3' end of primers should contain a G or C residue to increase priming efficiency and prevent "breathing" of ends [64].
- Avoid complementary 3' ends between primer pairs to prevent primer-dimer formation [64].
- Optimal melting temperatures (Tm) for primers range between 52–58°C, with both primers having Tm values within 5°C of each other [64].
- Avoid di-nucleotide repeats or single base runs with more than 4 bases [64].
- Utilize bioinformatics tools such as NCBI Primer-BLAST or Primer3 for primer design and specificity verification [64].
Standard PCR Protocol with Optimization Framework
Set up PCR reactions using the following master mix components and cycling conditions as a starting point for optimization:
Reaction Setup:
- Template DNA: 10–150 ng/μl (complexity-dependent) [28] [65]
- PCR Buffer: 1× concentration (supplied with enzyme)
- dNTPs: 200 μM (50 μM of each nucleotide) [64]
- Magnesium Chloride (MgCl₂): 1.5–4.0 mM (optimization required) [28] [64]
- Forward and Reverse Primers: 20–50 pmol each [64]
- DNA Polymerase: 0.5–2.5 units per 50 μl reaction [64]
- PCR Additives: As determined by optimization experiments
- Sterile Distilled Water: To final volume
Basic Thermal Cycling Parameters:
- Initial Denaturation: 94–98°C for 1–3 minutes [47]
- Denaturation: 94–98°C for 30 seconds to 2 minutes [47]
- Annealing: Temperature 3–5°C below primer Tm for 30 seconds to 2 minutes [47]
- Extension: 68–72°C for 1 minute/kb for standard polymerases [47] [65]
- Cycle Number: 25–35 cycles [47]
- Final Extension: 72°C for 5–15 minutes [47]
Quantitative Optimization Data
Additive Concentrations and Combinations
Table 1: PCR Additives for Amplification of GC-Rich Trinucleotide Repeat Regions
| Additive | Concentration Range | Optimal Concentration | Mechanism of Action | Considerations |
|---|---|---|---|---|
| Betaine | 0.5 M to 2.5 M [64] | 1 M [28] | Equalizes template melting temperatures; stabilizes denatured DNA [28] | Often used in combination with DMSO; reduces base composition bias [28] |
| Dimethyl Sulfoxide (DMSO) | 1–10% [64] | 5% [28] | Disrupts base pairing; reduces secondary structure formation [28] | Decreases annealing temperature by 5.5–6.0°C at 10% concentration [47] |
| Formamide | 1.25–10% [64] | 1.25–5% [28] | Denatures DNA; reduces melting temperature | Can inhibit polymerase at higher concentrations |
| 7-deaza-dGTP | 150 μM with 50 μM dGTP [28] | Partial substitution (3:1 ratio with dGTP) [28] | Reduces hydrogen bonding; prevents stable secondary structures [28] | Requires adjustment of annealing temperature; more expensive than standard dNTPs [28] |
| Glycerol | 5–15% [28] | 10–15% [28] | Stabilizes enzymes; lowers melting temperature | Increases enzyme stability but may affect specificity |
| Bovine Serum Albumin (BSA) | 10–100 μg/ml [64] | 10–100 μg/ml [64] | Binds inhibitors; stabilizes enzymes | Particularly useful with impure templates |
Denaturation Parameter Optimization
Table 2: Denaturation Conditions for Challenging Templates
| Template Type | Initial Denaturation | Cycle Denaturation | Additive Enhancement | Rationale |
|---|---|---|---|---|
| Standard DNA | 94–95°C for 1–3 min [47] [65] | 94–95°C for 30–60 sec [65] | Typically not required | Adequate for most templates without complex secondary structure |
| GC-Rich Templates (>65% GC) | 98°C for 2–5 min [65] | 98°C for 10–30 sec [65] | Betaine (1 M) + DMSO (5%) [28] | Higher temperatures needed to melt stable GC structures [28] [65] |
| Long Amplicons (>10 kb) | 94–98°C for 1–3 min [47] | 94–98°C for 20–30 sec [65] | DMSO (2.5–5%) [65] | Shorter denaturation times reduce depurination risk [65] |
| AT-Rich Templates | 94°C for 1–2 min [65] | 94°C for 30–60 sec [65] | Polymerases optimized for GC-rich templates [65] | Lower denaturation temperatures sufficient |
Comprehensive PCR Optimization Results
Table 3: Optimized PCR Conditions for FMR1 CGG Repeat Amplification
| Optimization Method | Reaction Components | Thermal Cycling Parameters | Amplification Success |
|---|---|---|---|
| Standard PCR | 1× PCR buffer, 2 mM MgCl₂, 200 μM dNTPs, 10% DMSO, 0.75 U Taq polymerase [28] | 95°C for 10 min; 25 cycles of: 95°C/1.5 min, 65°C/1 min, 72°C/2 min [28] | Limited for GC-rich templates |
| 7-deaza-dGTP Substitution | 1× PCR buffer, 2 mM MgCl₂, 150 μM deaza-dGTP, 50 μM dGTP, 200 μM other dNTPs, 10% DMSO [28] | 95°C for 10 min; 25 cycles of: 95°C/1.5 min, 65°C/1 min, 72°C/2 min [28] | Improved but variable |
| Single Additive | 1× PCR buffer, 2 mM MgCl₂, 200 μM dNTPs, various concentrations of single additives [28] | 95°C for 10 min; 25 cycles of: 95°C/1.5 min, 65°C/1 min, 72°C/2 min [28] | Moderate improvement |
| Combined Additives | 1× PCR buffer, 1.5 mM MgCl₂, 200 μM dNTPs, 1 M betaine, 5% DMSO, 1 U Taq polymerase [28] | 95°C for 10 min; 25 cycles of: 95°C/1.5 min, 65°C/1 min, 72°C/2 min [28] | Optimal: reproducible amplification |
Visualization of Optimization Pathways
Research Reagent Solutions
Table 4: Essential Research Reagents for Trinucleotide Repeat PCR
| Reagent Category | Specific Products | Function & Application | Optimization Tips |
|---|---|---|---|
| DNA Polymerases | PrimeSTAR GXL DNA Polymerase [65], Takara LA Taq [65], Advantage GC2 Polymerase [65] | High fidelity amplification of GC-rich templates and long repeats | Select polymerases specifically optimized for GC-rich templates [65] |
| PCR Additives | Betaine (1M stock) [28], Molecular biology grade DMSO [28], 7-deaza-dGTP [28] | Disrupt secondary structures; improve amplification efficiency | Use combination approach: 1M betaine + 5% DMSO often most effective [28] |
| Buffer Systems | GC Buffer I/II [65], Magnesium-free buffers with separate MgCl₂ [65] | Provide optimal salt conditions for challenging templates | Adjust MgCl₂ concentration from 1.5-4.0 mM for specific applications [28] [64] |
| Template Preparation Kits | Buccal cell DNA extraction kits [28], High-quality genomic DNA kits | Provide high-quality template without inhibitors | Assess DNA integrity by gel electrophoresis before PCR [28] |
| Quantification Tools | Fluorescent DNA quantification kits, Agarose gel electrophoresis standards | Accurate template quantification for reproducible results | Use same quantification method consistently; avoid spectrophotometry alone |
Discussion and Technical Notes
The optimization of PCR amplification for trinucleotide repeat regions requires systematic adjustment of both additive concentrations and denaturation parameters. The data presented demonstrate that a combination of 1 M betaine and 5% DMSO provides the most consistent improvement for amplifying GC-rich repetitive sequences, as evidenced by successful amplification of the FMR1 CGG repeat region [28]. This combination functions synergistically—betaine stabilizes denatured DNA and equalizes the melting temperature of different DNA sequences, while DMSO disrupts hydrogen bonding and prevents formation of stable secondary structures [28].
Denaturation temperature and duration prove critical for successful amplification of trinucleotide repeat regions. Standard denaturation at 94–95°C may be insufficient for GC-rich templates, which often require higher temperatures of 98°C to completely separate strands [65]. However, balance is essential, as excessive heat exposure can lead to DNA depurination, particularly for longer amplicons [65]. For fragments exceeding 10 kb, shorter denaturation times at higher temperatures are recommended to minimize damage while ensuring complete denaturation [65].
The significant impact of template quality on amplification success cannot be overstated. DNA integrity is particularly crucial for long-range PCR across expanded repeat regions, as damage during extraction or storage can dramatically reduce yield [65]. Template concentration should be optimized for each target, with 30–100 ng of human genomic DNA typically sufficient for single-copy genes [65]. When amplifying from cDNA, the input can be as little as 10 pg RNA equivalent, reflecting the higher abundance of specific transcripts [65].
Researchers should note that the presence of expanded trinucleotide repeats not only presents technical challenges for amplification but may also induce cellular responses to DNA damage. Expanded CAG/CTG repeats have been shown to elicit DNA damage checkpoint responses in model systems, potentially impacting cell proliferation [66]. This biological significance underscores the importance of reliable detection methods for understanding both the molecular diagnosis and pathophysiology of trinucleotide repeat disorders.
Strategies for Handling GC-Rich Sequences and Long Amplicons
The amplification of Guanine-Cytosine (GC)-rich sequences and long amplicons represents a significant technical challenge in molecular biology, particularly in diagnostic and pharmaceutical research. GC-rich templates, typically defined as sequences with over 60% GC content, constitute approximately 3% of the human genome and are frequently located in promoter regions of genes, including housekeeping and tumor suppressor genes [67]. Furthermore, many clinically relevant genetic disorders, such as Fragile X syndrome, Huntington's disease, and myotonic dystrophy, arise from expansions of GC-rich trinucleotide repeat regions, making their reliable amplification crucial for screening and diagnosis [28] [68].
The primary challenges in amplifying these recalcitrant sequences stem from their biophysical properties. The presence of three hydrogen bonds in G-C base pairs, compared to two in A-T pairs, confers greater thermostability and a higher melting temperature [67]. This stability leads to incomplete denaturation during standard PCR cycles. Additionally, GC-rich sequences are highly prone to forming stable intra-strand secondary structures, such as hairpin loops and stem-loops, which can cause DNA polymerases to stall, resulting in truncated or non-specific products [28] [69]. For long amplicons, the challenges are compounded by the increased probability of polymerase dissociation and the accumulation of enzymatic errors, necessitating polymerases with high processivity and fidelity.
Key Optimization Strategies
Successful amplification of GC-rich and long templates requires a multifaceted optimization approach, encompassing reagent selection, buffer composition, and thermal cycling parameters.
Polymerase and Buffer Selection
The choice of DNA polymerase is critical. Standard Taq polymerase often fails with complex templates. Instead, specialized or engineered polymerases are recommended. Hyperthermostable DNA polymerases (e.g., from Pyrococcus species) can withstand higher denaturation temperatures (up to 98°C), which improves strand separation of GC-rich duplexes [69] [29]. For long amplicons, polymerases with high processivity (the number of nucleotides incorporated per binding event) are essential to read through the entire template without dissociating [29]. Furthermore, high-fidelity polymerases, which possess proofreading activity (3'→5' exonuclease), are preferred for long PCR to reduce misincorporation errors in extended products.
Many manufacturers offer master mixes and specialized buffers specifically formulated for difficult templates. These often include GC enhancers that contain a proprietary mix of additives designed to disrupt secondary structures and increase primer stringency [67]. Using these tailored systems can provide a more straightforward and reproducible solution than in-house optimization.
PCR Additives
Additives are small molecules that modify the nucleic acid environment to facilitate the amplification of challenging sequences. They function primarily by reducing the formation of stable secondary structures or by lowering the melting temperature of DNA. The following table summarizes common additives and their mechanisms of action:
Table 1: Common PCR Additives for GC-Rich and Long Amplicon Amplification
| Additive | Common Concentration | Primary Mechanism of Action | Considerations |
|---|---|---|---|
| Betaine | 1 - 1.5 M | Equalizes the stability of AT and GC base pairs; reduces secondary structure formation [28] [70]. | Often used in combination with DMSO. |
| Dimethyl Sulfoxide (DMSO) | 5 - 10% | Disrupts hydrogen bonding and base stacking, leading to lower DNA melting temperature [28] [29]. | Can reduce Taq polymerase activity; may require adjustment of annealing temperature. |
| Glycerol | 5 - 10% | Lowers template melting temperature and stabilizes the polymerase [67]. | Similar to DMSO in effect. |
| 7-deaza-2'-deoxyguanosine | 150 µM (partial dGTP substitution) | dGTP analog that lacks nitrogen at position 7; reduces Hoogsteen base pairing without disrupting Watson-Crick pairing [67] [28]. | Does not stain well with ethidium bromide; requires adjustment of dNTP ratios. |
| Formamide | 1 - 5% | Denatures DNA strands, aiding in the separation of secondary structures [67]. | Can be inhibitory at higher concentrations. |
Empirical evidence suggests that a combination of additives can be more effective than any single agent. For instance, one optimized protocol for amplifying the GC-rich FMR1 gene used a combination of 1 M betaine and 5% DMSO to achieve reproducible results [28].
Magnesium and Thermal Cycling Optimization
Magnesium ion (Mg²⁺) concentration is a vital cofactor for DNA polymerase activity and must be carefully optimized. While standard PCRs typically use 1.5-2.0 mM MgCl₂, GC-rich amplifications may require deviation from this range. Too much Mg²⁺ can promote non-specific priming, while too little can reduce polymerase activity [67]. A titration experiment using 0.5 mM increments between 1.0 and 4.0 mM is recommended to find the optimal concentration [67].
Thermal cycling parameters also require adjustment:
- Higher Denaturation Temperature: Using 98°C instead of 95°C for denaturation can improve the separation of GC-rich strands [29]. However, the polymerase's thermostability must be confirmed.
- Temperature Gradients and Touchdown PCR: Employing an annealing temperature gradient helps identify the optimal Ta. Touchdown PCR, which starts with an annealing temperature higher than the calculated Tm and gradually decreases it over subsequent cycles, promotes specificity in the early stages of amplification [29].
- Slower Ramp Rates: Reducing the temperature transition speed between cycles (e.g., in "slow-down PCR") can improve the efficiency of primer binding and polymerase initiation on structured templates [69].
Detailed Experimental Protocols
Protocol 1: Amplification of a GC-Rich Trinucleotide Repeat Region
This protocol is adapted from a study that successfully amplified the >80% GC-rich 5' untranslated region of the FMR1 gene, relevant for Fragile X syndrome testing [28].
Research Reagent Solutions Table 2: Essential Reagents for GC-Rich Trinucleotide Repeat Amplification
| Reagent | Function/Justification |
|---|---|
| Thermostable DNA Polymerase (e.g., standard Taq) | The core enzymatic component for DNA synthesis. |
| 10X Standard PCR Buffer | Provides the optimal salt and pH conditions for the polymerase. |
| Betaine (5 M stock) | Primary additive to destabilize secondary structures and equalize base-pair stability. |
| DMSO | Co-additive to further lower DNA melting temperature and disrupt hydrogen bonding. |
| MgCl₂ (25 mM stock) | Essential cofactor for polymerase activity; concentration requires optimization. |
| dNTP Mix | Building blocks for new DNA strand synthesis. |
| Target-Specific Primers | Designed to flank the GC-rich trinucleotide repeat region. |
Methodology:
- Reaction Setup: Prepare a 25 µL reaction mixture containing:
- 1X PCR Buffer
- 1.5 mM MgCl₂ (optimized from a gradient)
- 0.2 mM of each dNTP
- 0.1 µM of each forward and reverse primer
- 1 M Betaine
- 5% DMSO
- 1.0 U of DNA polymerase
- 50 ng of genomic DNA
- Thermal Cycling: Use the following cycling conditions in a thermal cycler:
- Initial Denaturation: 95°C for 5 minutes.
- Amplification (35 cycles):
- Denaturation: 95°C for 45 seconds.
- Annealing: 65°C for 45 seconds (optimize using a gradient).
- Extension: 72°C for 90 seconds.
- Final Extension: 72°C for 7 minutes.
- Hold: 4°C.
Protocol 2: A Multi-Factor Optimization Workflow for GC-Rich Targets
This workflow provides a systematic approach to troubleshoot and optimize PCR for a novel GC-rich target, such as a nicotinic acetylcholine receptor subunit [70].
Data Analysis and Interpretation
For quantitative analysis, particularly in qPCR, ensuring valid data is paramount. PCR efficiency must be calculated when amplifying GC-rich targets, as secondary structures can significantly reduce amplification efficiency, leading to inaccurate quantification [71].
Calculating PCR Efficiency:
- Perform qPCR on a standard curve of serial dilutions (e.g., 1:10, 1:100, 1:1000) of the target template.
- Plot the average Ct value for each dilution against the logarithm of the dilution factor.
- Determine the slope of the resulting linear regression line.
- Calculate PCR efficiency using the formula: Efficiency (%) = (10^(-1/slope) - 1) × 100 [71].
An ideal reaction has an efficiency of 100%, meaning the product doubles every cycle. Acceptable efficiency typically ranges from 90% to 110% [71]. Results outside this range indicate a need for further optimization of the reaction conditions.
Table 3: Troubleshooting Common Issues in GC-Rich and Long Amplicon PCR
| Problem | Possible Cause | Potential Solution |
|---|---|---|
| No Product | Excessive secondary structure; insufficient denaturation; polymerase stalling. | Increase denaturation temperature; use a combination of betaine and DMSO; switch to a high-processivity polymerase. |
| Multiple Bands/Smearing | Non-specific priming; mispriming due to secondary structures. | Optimize Mg²⁺ concentration (often lower it); increase annealing temperature; use touchdown PCR or a hot-start polymerase. |
| Faint Bands/Low Yield | Poor primer annealing; inefficient amplification due to template complexity. | Titrate Mg²⁺ concentration (may need to increase); use PCR enhancers; increase the number of cycles. |
The reliable amplification of GC-rich sequences and long amplicons is achievable through a systematic strategy that addresses their unique physicochemical challenges. The most effective approach involves the synergistic combination of specialized DNA polymerases, destabilizing additives like betaine and DMSO, and carefully optimized buffer and cycling conditions. The protocols and workflows detailed in this application note provide a robust foundation for researchers and drug development professionals to advance their work on genetically complex targets, including those involving trinucleotide repeat regions. Given the target-specific nature of PCR, empirical optimization of these parameters remains essential for success.
Ensuring Accuracy: Validation, Quantification, and Comparative Methodologies
Within the context of advancing research on trinucleotide repeat disorders, the accurate validation of PCR amplification success is a critical step. Amplifying these repetitive genomic regions presents unique challenges due to their high GC content and propensity to form complex secondary structures, such as hairpin loops, which can cause DNA polymerases to dissociate from the template and generate incomplete fragments [16] [72]. These technical difficulties often result in ambiguous data that can compromise experimental outcomes and drug development progress.
This application note provides detailed methodologies for two essential validation techniques—gel electrophoresis and Sanger sequencing—specifically optimized for assessing the amplification of challenging trinucleotide repeat regions. The protocols outlined herein are designed to help researchers overcome the common obstacles associated with these genomic regions, thereby ensuring data reliability for downstream applications including genotyping, cloning, and functional studies relevant to therapeutic development.
The Challenge of Amplifying Repetitive DNA Regions
Trinucleotide repeat expansions (TREs) are implicated in more than 40 neurological disorders, including Huntington's disease, various spinocerebellar ataxias, and fragile X syndrome [72]. The accurate determination of repeat length is crucial for diagnosis, prognosis, and understanding disease mechanisms, as repeat size often correlates with disease severity and age of onset [72].
The primary challenges in amplifying these regions stem from their unique biochemical properties. These sequences typically have high GC content (often exceeding 65-80%) and contain repetitive motifs that form highly stable secondary structures [73] [16]. During PCR amplification, these structures promote polymerase dissociation through a process called "slippage," leading to the generation of truncated fragments that act as megaprimers. These megaprimers then anneal randomly to other sequences, creating a diverse library of undesired artefacts that appear as multiple bands or smearing on electrophoresis gels [16]. Conventional Sanger sequencing methods often fail when encountering these structures, as standard sequencing enzymes have reduced processivity and are easily inhibited by stable hairpin formations [73].
Experimental Protocols
PCR Amplification of Tricky Repeat Regions
The following protocol has been specifically optimized for the amplification of repetitive DNA sequences with high GC content, based on successful amplification of the MaSp1 gene (68.8% GC content) and similar challenging templates [73] [16].
Reaction Setup
- DNA Polymerase: Select a high-fidelity, hot-start enzyme such as TaKaRa LA Taq to minimize nonspecific amplification and enhance processivity [74] [16].
- Primer Design: Design primers with compatible melting temperatures (within 5°C) and approximately 50% GC content. Incorporate a GC clamp at the 3' end (but not exceeding 3 G/C residues) to enhance specific annealing. Avoid stretches of polybases or repeating motifs [74].
- PCR Additives: Include 1-3% DMSO or betaine to facilitate amplification of GC-rich templates by reducing secondary structure formation [74] [73].
- Template Quality: Use highly purified, non-degraded DNA. For formalin-fixed or degraded samples, consider specialized extraction protocols that recover longer DNA fragments [75].
Thermocycling Conditions The thermal profile is crucial for successful amplification of repetitive sequences. The optimized parameters are summarized in Table 1.
Table 1: Optimized Thermocycling Conditions for Repetitive DNA Amplification
Step Temperature Duration Cycles Notes Initial Denaturation 98°C 2 minutes 1 Ensures complete template denaturation Denaturation 98°C 20 seconds 30-35 Higher temperature is key for complex templates [16] Annealing 55-65°C 30 seconds 30-35 Temperature gradient recommended for new targets Extension 68°C 1 minute/kb 30-35 Suitable for fragments up to 3kb Final Extension 72°C 10 minutes 1 Ensures complete extension of all products
Gel Electrophoresis for Amplicon Validation
Following PCR, analyze the amplification products to verify specificity and yield before proceeding to sequencing.
Procedure
- Prepare a 1-2% agarose gel in 1X TAE or TBE buffer, incorporating a fluorescent nucleic acid stain.
- Mix 5 μL of PCR product with an appropriate loading dye.
- Load the mixture alongside a DNA molecular weight marker.
- Run the gel at 5-10 V/cm until adequate separation is achieved.
- Visualize the gel under UV transillumination.
Interpretation of Results A successful amplification for Sanger sequencing should yield a single, sharp band corresponding to the expected product size [74]. Multiple bands indicate non-specific amplification or sequence duplications, while smearing suggests primer-dimer formation or significant non-specific products [74] [16]. If multiple bands are present, gel purification of the specific band of interest is required before sequencing. Column purification or enzymatic cleanup will not isolate the desired product if multiple PCR products are present [74].
Sanger Sequencing of Amplified Products
Standard Sanger sequencing protocols often fail when confronted with the stable secondary structures of trinucleotide repeats. The following workflow (Figure 1) and optimized protocol address these challenges.
Figure 1: Workflow for Sanger sequencing of challenging repetitive DNA regions, highlighting key technical obstacles and mitigation strategies.
PCR Product Purification Cleanup is essential to remove excess primers, dNTPs, and enzyme from the PCR reaction, as these can interfere with the sequencing reaction. Several methods are available:
- Enzymatic Purification: Treatment with shrimp alkaline phosphatase (SAP) and Exonuclease I (Exo I) effectively degrades remaining nucleotides and single-stranded DNA primers [74].
- Column-Based Purification: Effective for removing salts and other impurities [75].
- Gel Purification: Required if multiple bands are present on the initial agarose gel to isolate the specific amplicon of interest [74].
Sequencing Primer Design
- For large projects, consider using universal-tailed PCR primers (e.g., M13 tails) during the initial amplification. This standardizes the sequencing step, as the same universal primer can be used for all samples [74].
- Ensure primers are specific, have minimal secondary structure, and are located 150-350 bp from the repetitive region for optimal results [73].
Cycle Sequencing Reaction Standard sequencing kits often fail when encountering stable secondary structures. Specialized methods, such as the one developed by GENEWIZ for AAV-ITR regions (which share structural similarities with trinucleotide repeats), can successfully sequence through these obstacles [73]. Key modifications include:
- Using specialized polymerases with higher processivity.
- Optimizing reaction components to overcome thermodynamic barriers.
- If standard kits must be used, additives like DMSO may offer some improvement, though they may not be sufficient for extremely difficult structures [73].
Troubleshooting Sequencing Failures If sequencing fails or the signal is lost within the repetitive region:
- Sequence from Both Ends: If the initial sequencing direction fails (e.g., through a poly-G tract), sequencing from the opposite direction (through the complementary poly-C tract) can often yield successful results [73].
- Verify Template Quality: Ensure the submitted DNA is pure, concentrated correctly (Fig. 1A), and represents a single, specific amplicon [75].
- Consider Alternative Technologies: For very long repeat expansions that exceed the capabilities of Sanger sequencing (typically 800-1000 bp), long-read sequencing technologies like PacBio or Oxford Nanopore may be necessary [72].
The Scientist's Toolkit
Table 2: Essential Research Reagents for Amplifying and Sequencing Challenging Repeats
| Reagent / Solution | Function / Application | Considerations |
|---|---|---|
| Hot-Start DNA Polymerase | Reduces non-specific amplification by remaining inactive until high temperatures are reached [74]. | Essential for GC-rich and repetitive templates. |
| Proofreading High-Fidelity Enzyme | Provides accurate DNA synthesis for longer amplicons; reduces error rate [16]. | Critical for cloning applications. |
| DMSO / Betaine | PCR additives that destabilize secondary structures, facilitating amplification of GC-rich templates [74] [16]. | Typical working concentration: 1-5%. |
| dNTP Mix | Building blocks for DNA synthesis. | Use balanced, high-quality dNTPs. |
| MgCl₂ Solution | Cofactor for DNA polymerase; concentration is critical for enzyme activity and fidelity [74]. | Concentration often needs optimization (1.5-3.0 mM). |
| Shrimp Alkaline Phosphatase (SAP) & Exonuclease I | Enzymatic cleanup of PCR products; degrades unused dNTPs and primers prior to sequencing [74]. | More convenient than column-based cleanups for high-throughput work. |
| Specialized Sequencing Kits | Formulated with polymerases and chemistry to overcome difficult templates like hairpins [73]. | Required for sequencing through stable secondary structures. |
| Gel Extraction Kits | Purification of specific amplicons from agarose gels after electrophoresis [74]. | Necessary when PCR yields multiple products. |
Data Analysis and Quality Assessment
Interpreting Chromatograms
High-quality Sanger sequencing data is characterized by evenly spaced, single peaks with low background noise. When sequencing trinucleotide repeats, several anomalies may appear:
- Signal Degradation: A sudden drop in signal intensity or complete termination within the repeat region indicates polymerase stalling due to secondary structure [73].
- Overlapping Peaks: Multiple peaks at a single position can suggest heterogeneous amplification or the presence of both normal and expanded alleles in the sample.
- Baseline Noise: Increased noise following a structured region may result from non-templated nucleotide additions or polymerase slippage [74] [72].
For reliable data, manually inspect chromatograms rather than relying solely on base-called sequences, particularly in regions surrounding the repeat expansion. Software such as Phred can assign quality scores to each base call; establish a minimum quality threshold (e.g., QV ≥ 30) for reliable sequence data [75].
Confirming Repeat Size
While Sanger sequencing can determine the exact sequence of shorter repeats, accurately sizing long expansions remains challenging. If the repeat length exceeds the sequencing read length or causes complete signal loss, supplemental techniques are required:
- Fragment Analysis by Capillary Electrophoresis: A highly accurate method for determining the size of fluorescently labeled PCR products, ideal for genotyping known repeat expansions [72].
- Long-Read Sequencing (PacBio/Nanopore): These technologies can span long, repetitive stretches, enabling the characterization of expansions that are "unsequenceable" by Sanger methods. Computational tools like RepeatHMM have been developed to estimate repeat counts from this data [72].
- Southern Blotting: A traditional but reliable method for detecting large expansions, though it is labor-intensive and provides limited sequence information [72].
The reliable amplification and sequencing of trinucleotide repeat regions demand carefully optimized protocols that address their unique biochemical challenges. By implementing the specialized methods for PCR, gel electrophoresis, and Sanger sequencing outlined in this application note, researchers can significantly improve the success rate of their experiments. Robust validation of amplification products is a foundational step in generating high-quality data for research into the mechanisms and therapeutic interventions for repeat expansion disorders. As these technologies evolve, the integration of long-read sequencing platforms with sophisticated computational tools will further enhance our ability to interrogate these clinically significant genomic regions.
Utilizing qPCR for Sensitive Detection and Absolute Quantification
The accurate detection and absolute quantification of specific DNA sequences are fundamental to advancing molecular diagnostics and therapeutic development. This is particularly critical when working with challenging genomic targets, such as trinucleotide repeat regions, which are implicated in a range of neurological disorders and cancers [76]. These repetitive sequences are prone to forming complex secondary structures that impede reliable amplification and quantification using standard PCR methods [76] [77].
Quantitative PCR (qPCR) provides a powerful platform for sensitive detection, but its application to these difficult templates requires careful optimization of reaction components and conditions. The integration of PCR additives and specialized reagents is essential to overcome the technical barriers presented by high GC content and stable non-B DNA structures [8] [77]. This protocol details optimized methodologies for applying qPCR to achieve sensitive and absolute quantification of trinucleotide repeat regions, with specific emphasis on addressing amplification challenges through strategic reagent selection and condition optimization.
Key Research Reagent Solutions
The following reagents are essential for successfully amplifying and quantifying challenging trinucleotide repeat regions via qPCR. Their functions and optimal usage are summarized in the table below.
Table 1: Essential Research Reagents for Amplifying Challenging Trinucleotide Repeats
| Reagent Category | Specific Examples | Function & Rationale |
|---|---|---|
| Specialized DNA Polymerases | Platinum SuperFi DNA Polymerase, Phusion High-Fidelity DNA Polymerase [8] | Proofreading activity and enhanced processivity for accurate amplification of structured DNA. |
| PCR Additives | Betaine (1M), DMSO (5%) [8] [77] | Destabilize secondary structures (e.g., hairpins) by reducing melting temperature, improving primer access and enzyme efficiency. |
| MgCl₂ Optimization | Magnesium Chloride (MgCl₂) [14] | Cofactor for DNA polymerase; fine-tuning concentration (e.g., 3 mM) is critical for enzyme processivity and yield. |
| Optimized Primer Systems | TaqMan Assays with exon-spanning designs [78] [79] | Fluorogenic probes provide target-specific quantification; designing across exon junctions prevents genomic DNA amplification. |
Optimized Experimental Protocols
Primer and Probe Design for Repetitive Regions
Careful assay design is the most critical step for success. Adhere to the following guidelines to ensure specificity and efficiency:
- Primer Length and Tm: Design primers between 18–30 bases in length. Aim for a melting temperature (Tm) of 60–64°C, with forward and reverse primers within 2°C of each other [79].
- GC Content and Clamp: Maintain a GC content between 40–60%. Include a GC clamp at the 3' end (a G or C base) to enhance binding stability but avoid runs of four or more consecutive G residues [13] [79].
- Specificity Checks: Utilize tools like NCBI BLAST to ensure primer uniqueness and avoid off-target binding. Screen for self-dimers, hairpins, and heterodimers using analysis tools (e.g., IDT OligoAnalyzer), rejecting designs with ΔG values more stable than -9.0 kcal/mol [79].
- Amplicon Length and Location: Keep amplicons short (70–150 bp) for optimal amplification efficiency. When working with cDNA, design assays to span an exon-exon junction to preclude amplification of contaminating genomic DNA [79].
Sample Preparation and Quality Control
The sensitivity of qPCR demands high-quality input material.
- Nucleic Acid Extraction: Use extraction methods that yield pure DNA/RNA with an A260/A280 ratio of ~1.8. Impurities like EDTA can inhibit polymerase activity [14].
- DNAse Treatment: For gene expression studies (qRT-PCR), treat RNA samples with DNase I to remove genomic DNA contamination [80] [79].
- Quality Assessment: Use agarose gel electrophoresis and spectrophotometry to confirm RNA integrity and purity prior to cDNA synthesis [8] [80].
qPCR Master Mix Optimization for GC-Rich Repeats
This protocol is optimized for challenging, structure-prone trinucleotide repeats.
Table 2: Optimized qPCR Reaction Setup
| Component | Final Concentration | Notes |
|---|---|---|
| Specialized High-Fidelity Master Mix | 1X | Use a buffer system formulated for GC-rich content. |
| Forward & Reverse Primers | 10–30 pM each | Requires empirical testing for optimal specificity [14]. |
| TaqMan Probe | 100–200 nM | Tm should be 5–10°C higher than the primers [79]. |
| Betaine | 1 M | Final concentration; crucial additive for destabilizing secondary structures [8] [77]. |
| DMSO | 2–5% (v/v) | Can be used in combination with betaine, but requires testing [8]. |
| MgCl₂ | 3 mM | Adjust based on polymerase requirements; a critical cofactor [79]. |
| Template DNA | 1–50 ng | Keep consistent across samples for absolute quantification. |
| Nuclease-Free Water | To volume | - |
Procedure:
- Prepare Master Mix: Combine all components (except template) in a master mix to minimize pipetting error.
- Aliquot and Add Template: Dispense the master mix into reaction wells and then add the template DNA.
- qPCR Cycling: Use the following optimized cycling conditions on a real-time PCR instrument:
- Initial Denaturation: 98°C for 2 minutes (or as recommended for the polymerase).
- Amplification (40–45 cycles):
- Denature: 98°C for 10 seconds.
- Anneal/Extend: 62–68°C for 30–60 seconds (data acquisition for TaqMan).
- Data Collection: Acquire fluorescence data during the annealing/extension step of every cycle.
Standard Curve for Absolute Quantification
For absolute quantification, a standard curve of known copy numbers is essential.
- Standard Preparation: Use a linearized plasmid or a synthetic gBlock containing the target sequence. Perform a serial dilution (e.g., 10^7 to 10^1 copies) in the same background as the sample (e.g., carrier DNA) to create a standard curve.
- Data Analysis: The qPCR software will plot the Cq values against the log of the starting quantity for each standard. The resulting curve allows for the interpolation of the absolute copy number in unknown samples based on their Cq values.
Workflow Visualization
The following diagram illustrates the complete optimized workflow for the sensitive detection and quantification of trinucleotide repeats, from sample preparation to data analysis.
Data Analysis and MIQE Compliance
To ensure the reliability and reproducibility of qPCR data, adherence to the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines is mandatory [78] [81].
- Assay Validation: Report the amplification efficiency (90–105%), correlation coefficient (R² > 0.980), and the dynamic range of the standard curve [80].
- Technical Replication: Perform reactions in at least triplicate to measure technical variability. The intra-assay coefficient of variation (CV) should ideally be <5% [80].
- Specificity: Confirm a single, specific amplification product through melt curve analysis (if using SYBR Green) or through the use of sequence-specific probes.
- Data Reporting: When publishing, provide the assay sequence (e.g., Assay ID or amplicon context sequence) as required by MIQE guidelines to ensure full transparency [78].
Troubleshooting Common Issues
Table 3: Troubleshooting Guide for qPCR Amplification of Trinucleotide Repeats
| Problem | Potential Cause | Solution |
|---|---|---|
| No Amplification | Excessive secondary structure; inefficient polymerase; low primer Tm. | Increase betaine/DMSO concentration; use a specialized polymerase; lower annealing temperature gradientally. |
| Non-Specific Bands/High Background | Primers annealing non-specifically; low annealing temperature; contaminations. | Increase annealing temperature; optimize primer concentration; use hot-start polymerase; ensure clean template. |
| Poor Replicate Consistency | Pipetting errors; inadequate master mix mixing; low quality template. | Always prepare a master mix; vortex and centrifuge reagents; check template purity and integrity. |
| Low Amplification Efficiency | Inhibitors in sample; suboptimal Mg²⁺ concentration; poor probe design. | Purify template; titrate MgCl₂ concentration (1–5 mM); re-design and validate probe. |
Polymerase chain reaction (PCR) technologies represent foundational tools in molecular biology, with specific formats offering distinct advantages for particular applications. This application note provides a detailed comparative analysis of standard PCR, nested PCR, and long-range PCR methodologies, with specific focus on their utility in amplifying challenging trinucleotide repeat regions. Such regions, particularly those with high GC content, present significant amplification obstacles due to their propensity for forming complex secondary structures that impede DNA polymerase progression [8] [11]. The performance characteristics, optimal applications, and detailed protocols for each method are critically examined to guide researchers in selecting and optimizing the most appropriate technique for their experimental needs, particularly within the context of genetic disorder research and drug development.
Performance Comparison and Quantitative Data
The three PCR techniques exhibit markedly different performance characteristics in sensitivity, specificity, and amplicon size capacity. The table below summarizes key comparative data derived from clinical and research studies.
Table 1: Quantitative Performance Comparison of Standard, Nested, and Long-Range PCR
| Parameter | Standard PCR | Nested PCR | Long-Range PCR |
|---|---|---|---|
| Detection Sensitivity | 45-63.9% [82] [83] | 86-100% [82] [83] | Not explicitly quantified |
| Specificity | 100% [82] [83] | 73-100% [82] [83] | High (with high-fidelity enzymes) |
| Practical Amplicon Size | <5 kb [29] | Typically short (second amplicon) | >5 kb [29] |
| Key Advantages | Simplicity, speed, good specificity | Exceptional sensitivity, enhanced specificity for low-abundance targets | Amplification of large genomic fragments |
| Major Limitations | Lower sensitivity for rare targets | Contamination risk from two rounds of amplification | Challenges with complex secondary structures |
| Optimal Applications | Routine amplification of abundant targets | Pathogen detection, low-copy number genes, degraded samples | Genome walking, sequencing large inserts, complex repeat analysis |
The data reveal a clear sensitivity gradient, with nested PCR demonstrating superior detection capabilities for low-abundance targets. In a prospective study detecting V. vulnificus in blood samples, nested PCR (86% sensitivity) significantly outperformed standard PCR (45% sensitivity) [82]. Similarly, for detecting Severe Fever with Thrombocytopenia Syndrome virus (SFTSV), nested PCR maintained a 70% detection rate up to 40 days post-symptom onset, far exceeding standard PCR which showed markedly reduced sensitivity after just 7 days [83].
Addressing the Challenge of Trinucleotide Repeat Amplification
Amplification of trinucleotide repeat regions (TNRs), such as the CTG/CCTG repeats implicated in myotonic dystrophies (DMs), presents particular challenges for standard PCR protocols. These regions are characterized by high GC content and strong secondary structure formation, which hinder polymerase processivity and lead to amplification failure [8] [11].
Optimization Strategies for GC-Rich TNRs:
- PCR Additives: Incorporating co-solvents such as dimethyl sulfoxide (DMSO) or betaine is crucial for reducing secondary structure formation by lowering the melting temperature of GC-rich duplexes, thus facilitating strand separation and primer annealing [8] [29].
- Polymerase Selection: Using highly processive DNA polymerases or specialized enzyme blends is essential for synthesizing through long, repetitive, and structured DNA templates [29] [11].
- Thermal Cycling Modifications: Employing higher denaturation temperatures (e.g., 98°C) and longer extension times helps ensure complete template denaturation and sufficient time for polymerase to navigate through difficult regions [29] [11].
Table 2: Research Reagent Solutions for Challenging PCR Applications
| Reagent Category | Specific Example | Function in PCR | Application Context |
|---|---|---|---|
| Specialized Polymerases | Platinum SuperFi DNA Polymerase [8] | High-fidelity amplification of complex templates | GC-rich gene amplification |
| DyNAzyme EXT DNA Polymerase [11] | Processive enzyme for long amplicons | CTG/CCTG-repeat expansion detection | |
| PCR Additives | DMSO (5-10%) [8] [29] | Disrupts secondary structures, lowers Tm | GC-rich PCR, trinucleotide repeats |
| Betaine (1 M) [8] | Equalizes Tm of AT and GC base pairs | GC-rich PCR | |
| Hot-Start Enzymes | Platinum II Taq Hot-Start [29] | Inhibits activity until heated, reduces primer-dimer formation | Multiplex PCR, enhances specificity |
| Enzyme Blends | Taq + High-Fidelity Enzyme Blend [29] | Combines processivity with accuracy | Long-range PCR |
Detailed Experimental Protocols
Optimization PCR for Trinucleotide Repeat Expansions
This protocol is adapted from methodologies successfully used for detecting CTG/CCTG-repeat expansions in myotonic dystrophies [11].
Reaction Setup:
- Template DNA: 50 ng of human genomic DNA
- Primers: 10 pmol each of forward and reverse primers flanking the repeat region
- PCR Buffer: 1X supplied buffer with Mg²⁺
- dNTPs: 200 μmol/L each
- DNA Polymerase: 1.6 units of a high-processivity enzyme (e.g., DyNAzyme EXT)
- Additives: Include 5% DMSO or 1M betaine for GC-rich templates
- Total Reaction Volume: 25 μL
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 5 minutes
- Amplification (30 cycles):
- Denaturation: 95°C for 45 seconds
- Annealing: 66°C for 8 seconds (optimized for primer Tm)
- Extension: 78°C for 3 minutes (extended for long amplicons)
- Final Extension: 72°C for 10 minutes
- Hold: 4°C
Analysis:
- Separate PCR products on 2.5% agarose gels
- Visualize with ethidium bromide or SYBR Safe staining
- For precise sizing, purify products and perform fragment analysis or sequencing
Nested PCR for Enhanced Sensitivity
This protocol is designed for maximum detection sensitivity, particularly useful for low-abundance targets or problematic templates [82] [83].
First Round PCR:
- Reaction Components: As described in section 4.1, using outer primer set
- Thermal Cycling: 25-30 cycles using optimized annealing temperature for outer primers
- Product: Use 1-2 μL of first-round product as template for second round
Second Round PCR:
- Primers: Nested primer set binding internal to first amplicon
- Reaction Components: Identical to first round except for primers
- Thermal Cycling: 25-30 cycles using optimized annealing temperature for nested primers
Critical Considerations:
- Physically separate pre- and post-amplification areas to prevent contamination
- Use dedicated pipettes and aerosol-resistant tips
- Include negative controls (no template and no amplification controls) in both rounds
Diagram 1: Nested PCR enhances sensitivity by re-amplifying a primary PCR product with internal primers.
Long-Range PCR for Large Fragments
This protocol enables amplification of targets >5 kb, particularly useful for spanning large trinucleotide repeat expansions [29].
Reaction Setup:
- Template DNA: 50-100 ng of high-quality genomic DNA
- Primers: 10-20 pmol each of forward and reverse primers
- PCR Buffer: 1X supplied buffer (often specialized for long-range)
- dNTPs: 200-500 μmol/L each (higher concentration for long products)
- Polymerase Blend: Combination of high-processivity and proofreading enzymes
- Additives: Include GC enhancers as needed (DMSO, betaine)
- Total Reaction Volume: 50 μL
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 2 minutes
- Amplification (30-35 cycles):
- Denaturation: 98°C for 10-20 seconds (higher temperature for GC-rich templates)
- Annealing: 60-68°C for 15-30 seconds (primer-specific)
- Extension: 68°C for 1 minute per kb (e.g., 6 minutes for 6 kb product)
- Final Extension: 72°C for 10 minutes
- Hold: 4°C
Analysis:
- Analyze products on 0.8-1.0% agarose gels for large fragments
- Use appropriate DNA size standards for accurate sizing
- Southern blotting may be required for verification of very large expansions
Method Selection Workflow
Selecting the appropriate PCR method requires careful consideration of experimental goals and template characteristics. The following decision pathway provides guidance for method selection.
Diagram 2: A workflow to guide the selection of the most appropriate PCR method based on experimental requirements.
Standard PCR, nested PCR, and long-range PCR each occupy distinct niches in molecular biology research. Standard PCR offers simplicity and reliability for routine applications, while nested PCR provides exceptional sensitivity for challenging detection scenarios. Long-range PCR enables amplification of large genomic regions inaccessible to other methods. For the specific challenge of amplifying trinucleotide repeat regions - a crucial capability in genetic disorder research and drug development - protocol optimization incorporating specialized polymerases, PCR additives, and tailored thermal cycling parameters is essential. The comprehensive performance data and detailed protocols provided herein equip researchers to select and implement the most appropriate PCR strategy for their specific experimental requirements in amplifying these challenging genomic targets.
Assessing Fidelity and Error Rates for Downstream Applications
The polymerase chain reaction (PCR) has become an indispensable tool for molecular diagnosis and genetic research, particularly in the analysis of trinucleotide repeat disorders such as fragile X syndrome (FMR1), Huntington disease, and myotonic dystrophy [28]. However, the accurate amplification of Guanine-Cytosine (GC)-rich trinucleotide repeat regions presents substantial technical challenges due to their propensity to form stable secondary structures that impede DNA polymerase progression [28] [70]. These regions are not only difficult to amplify but also require exceptional replication fidelity to ensure that downstream applications—including clinical diagnostics, cloning, and next-generation sequencing—produce reliable, actionable data. The fidelity of DNA replication, defined as the accuracy with which a DNA polymerase copies a template sequence, becomes paramount when the amplified products are used for diagnostic purposes or scientific conclusions that may affect patient care or therapeutic development [84].
Error rates in PCR amplification can significantly impact the interpretation of results, especially when assessing repeat lengths in trinucleotide repeat disorders where small variations can differentiate between healthy, premutation, and full mutation alleles [68] [11]. Researchers and clinicians must therefore carefully consider both the biochemical optimization of PCR conditions for challenging templates and the intrinsic error rate of the enzymatic systems employed. This application note provides a comprehensive framework for assessing fidelity and optimizing amplification protocols specifically for trinucleotide repeat regions, with particular emphasis on practical considerations for downstream applications in research and drug development.
Polymerase Fidelity: Mechanisms and Measurement
Mechanisms of Polymerase Fidelity
DNA polymerase fidelity is maintained through a multi-step process that ensures the correct nucleotide is incorporated during DNA synthesis. The primary mechanisms include:
- Geometric Selection: The polymerase active site is shaped to accommodate correct Watson-Crick base pairs, aligning catalytic groups for efficient incorporation of complementary nucleotides. Incorrect nucleotides create suboptimal architecture that slows incorporation, increasing the opportunity for dissociation before the polymerase proceeds [84].
- Proofreading Activity (3´→5´ Exonuclease): Many high-fidelity DNA polymerases contain a dedicated exonuclease domain that recognizes and excises misincorporated nucleotides from the 3' end of the growing DNA strand before further extension. This proofreading function can decrease error rates by up to 125-fold, as demonstrated by comparisons between exonuclease-proficient and deficient versions of Deep Vent DNA Polymerase [84].
Quantitative Measurement of Fidelity
Polymerase error rates have been quantified using several methodological approaches, each with distinct advantages and limitations:
Table 1: Methods for Assessing DNA Polymerase Fidelity
| Method | Principle | Detection Limit | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Blue/White Colony Screening | Functional assay based on loss-of-function mutations in lacZ gene causing white colonies | ~1.4 × 10⁻⁶ errors/base [84] | High-throughput; cost-effective for large sample numbers | Only detects mutations in specific regions affecting gene function; limited sequence context |
| Sanger Sequencing | Direct sequencing of cloned PCR products | ~1 × 10⁻⁶ errors/base [84] | Identifies all mutation types within sequenced region; no special reagents required | Lower throughput than screening methods; cost-prohibitive for extensive sequencing |
| Next-Generation Sequencing (Illumina) | High-throughput sequencing with molecular barcoding | ~1 × 10⁻⁶ errors/base [84] | Extremely high throughput; comprehensive mutation profiling | Requires intermediary amplification step; background noise from library preparation |
| PacBio SMRT Sequencing | Single-molecule real-time sequencing without amplification | ~9.6 × 10⁻⁸ errors/base [84] | No amplification bias; detects all error types; lowest background | Higher cost per sample; specialized equipment required |
The evolution of sequencing technologies has progressively lowered the detection limits for polymerase errors, enabling more accurate fidelity assessments of high-fidelity enzymes. Single-molecule real-time (SMRT) sequencing currently offers the most sensitive approach, with a background error rate of approximately 9.6 × 10⁻⁸ errors/base, making it suitable for quantifying the fidelity of proofreading polymerases [84].
Comparative Fidelity Analysis of DNA Polymerases
Direct comparisons of polymerase fidelity across multiple studies reveal significant variation in error rates among enzymes commonly used in PCR applications. Taq DNA polymerase serves as a common reference point, with fidelity measurements ranging from 1.5 × 10⁻⁴ to 3.0 × 10⁻⁵ errors per base per doubling [84] [20]. This variation highlights the importance of standardized measurement conditions when comparing polymerase performance.
Table 2: Error Rates of Common DNA Polymerases
| DNA Polymerase | Proofreading Activity | Substitution Rate (errors/base/doubling) | Accuracy (bases/error) | Fidelity Relative to Taq |
|---|---|---|---|---|
| Taq | No | 1.5 × 10⁻⁴ (± 0.2 × 10⁻⁴) | 6,456 | 1× [84] |
| Deep Vent (exo-) | No | 5.0 × 10⁻⁴ (± 0.1 × 10⁻⁴) | 2,020 | 0.3× [84] |
| KOD Hot Start | Yes | 1.2 × 10⁻⁵ (± 0.2 × 10⁻⁵) | 82,303 | 12× [84] |
| PrimeSTAR GXL | Yes | 8.4 × 10⁻⁶ (± 1.1 × 10⁻⁶) | 118,467 | 18× [84] |
| Pfu | Yes | 5.1 × 10⁻⁶ (± 1.1 × 10⁻⁶) | 195,275 | 30× [84] |
| Deep Vent | Yes | 4.0 × 10⁻⁶ (± 2.0 × 10⁻⁶) | 251,129 | 44× [84] |
| Phusion Hot Start | Yes | 3.9 × 10⁻⁶ (± 0.7 × 10⁻⁶) | 255,118 | 39× [84] |
| Q5 High-Fidelity | Yes | 5.3 × 10⁻⁷ (± 0.9 × 10⁻⁺⁷) | 1,870,763 | 280× [84] |
The data reveal that proofreading enzymes generally exhibit significantly enhanced fidelity compared to non-proofreading polymerases. Q5 High-Fidelity DNA Polymerase demonstrates the highest fidelity among commercially available enzymes, with an error rate of approximately 5.3 × 10⁻⁷ errors per base per doubling—approximately 280-fold more accurate than Taq polymerase [84]. This exceptional accuracy makes it particularly suitable for applications requiring minimal errors, such as cloning of trinucleotide repeat regions or diagnostic applications where sequence integrity is paramount.
Experimental Protocols for Amplifying GC-Rich Trinucleotide Repeat Regions
Optimized PCR Protocol for FMR1 CGG Repeat Amplification
The following protocol has been specifically optimized for amplifying the GC-rich 5' untranslated region of the FMR1 gene, which contains more than 80% GC content [28]:
Reaction Setup:
- Template DNA: 50 ng genomic DNA extracted from buccal cells or peripheral blood
- PCR Buffer: 1× concentration (compatible with selected polymerase)
- MgCl₂: 1.5 mM (optimized range: 1-4 mM)
- dNTPs: 0.2 mM each
- Primers: 0.1 μM each (FMR1-specific primers c and f as described in Fu et al., 1991)
- DNA Polymerase: 1 U high-fidelity enzyme with proofreading activity (e.g., Q5, Pfu, or Phusion)
- Additives: 1 M betaine + 5% DMSO
- Reaction Volume: 25 μL
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 5-10 minutes
- 30-35 cycles of:
- Denaturation: 95°C for 45 seconds
- Annealing: 66°C for 8 seconds (optimize based on primer Tm)
- Extension: 72°C for 3 minutes (adjust based on product length)
- Final Extension: 72°C for 10 minutes
- Hold: 4°C
This protocol leverages the synergistic effect of betaine and DMSO in reducing secondary structure formation in GC-rich templates. Betaine functions as a destabilizing agent that equalizes the thermodynamic stability of GC and AT base pairs, while DMSO assists in DNA denaturation by interfering with hydrogen bonding [28].
Alternative Additives for Challenging Amplifications
When standard DMSO and betaine combinations prove insufficient, researchers may consider alternative additives:
- Ethylene Glycol (1.075 M final concentration): Successfully amplified 87% of 104 challenging GC-rich human genomic amplicons in comparative studies [85].
- 1,2-Propanediol (0.816 M final concentration): Demonstrated superior performance to betaine alone, amplifying 90% of tested GC-rich targets [85].
- 7-deaza-dGTP: A substrate analog that can be partially substituted for dGTP (typically at 150 μM with 50 μM dGTP) to reduce hydrogen bonding between guanine and cytosine bases without disrupting normal Watson-Crick base pairing [28].
Protocol for Myotonic Dystrophy CTG/CCTG Repeat Analysis
For amplification of CTG repeats in the DMPK gene (myotonic dystrophy type 1) and CCTG repeats in the ZNF9 gene (myotonic dystrophy type 2), the following optimized protocol has been successfully implemented [11]:
Reaction Components:
- Template DNA: 50 ng from peripheral blood
- PCR Buffer: 1× concentration
- dNTPs: 200 μmol/L each
- DNA Polymerase: 1.6 units of DyNAzyme EXT DNA polymerase
- Primers: 10 pmol each (DMPK or ZNF9 specific)
- Additives: Based on initial optimization (DMSO, betaine, or combinations)
Thermal Cycling Parameters for DMPK:
- Initial Denaturation: 95°C for 5 minutes
- 30 cycles of:
- Denaturation: 95°C for 45 seconds
- Annealing: 66°C for 8 seconds
- Extension: 78°C for 3 minutes
- Final Extension: 72°C for 10 minutes
This protocol has successfully amplified CTG repeats ranging from 53 to 683 repeats (average 535) in diagnostic settings, demonstrating its utility for clinical applications [11].
The Scientist's Toolkit: Essential Reagents for Trinucleotide Repeat PCR
Table 3: Essential Research Reagents for Amplifying Trinucleotide Repeat Regions
| Reagent Category | Specific Examples | Optimal Concentration | Function in PCR |
|---|---|---|---|
| High-Fidelity DNA Polymerases | Q5, Phusion, Pfu, KOD | 1-2 units/50 μL reaction | Accurate nucleotide incorporation with proofreading capability; reduced error rates |
| PCR Additives | Betaine | 1-2 M | Equalizes template stability; disrupts secondary structures |
| DMSO | 5-10% | Reduces DNA melting temperature; prevents secondary structure formation | |
| Ethylene Glycol | 1.075 M | Alternative destabilizing agent with potential superior performance to betaine for some targets | |
| 1,2-Propanediol | 0.816 M | Enhanced amplification efficiency for challenging GC-rich templates | |
| Modified Nucleotides | 7-deaza-dGTP | 150 μM (with 50 μM dGTP) | Reduces hydrogen bonding in GC-rich regions without disrupting Watson-Crick pairing |
| Buffer Components | MgCl₂ | 1.5-4 mM | Essential cofactor for DNA polymerase activity; concentration requires optimization |
| dNTPs | 0.2 mM each | Balanced nucleotide concentrations ensure faithful incorporation |
Visualization of Experimental Workflows
Fidelity Assessment Methodology
Trinucleotide Repeat Amplification Optimization
The accurate amplification of trinucleotide repeat regions requires a multifaceted approach that addresses both the biochemical challenges of GC-rich templates and the fundamental fidelity requirements for downstream applications. Based on current evidence, the following recommendations emerge:
Polymerase Selection: For diagnostic applications or cloning where sequence integrity is paramount, select high-fidelity DNA polymerases with proofreading activity such as Q5, Phusion, or Pfu, which offer error rates 30-280 times lower than Taq polymerase [84].
Additive Strategy: Implement a systematic approach to PCR additives, beginning with DMSO and betaine combinations, then exploring alternatives like ethylene glycol or 1,2-propanediol for particularly challenging templates [28] [85].
Validation Methods: Employ appropriate fidelity assessment methods based on application requirements, with SMRT sequencing providing the most sensitive detection for rigorous characterization of error rates [84].
Template-Specific Optimization: Recognize that optimal conditions vary significantly between trinucleotide repeat targets; protocols require empirical optimization for each specific application, particularly when moving between different repeat regions [70] [11].
By implementing these evidence-based strategies, researchers and clinicians can significantly improve the reliability of trinucleotide repeat amplification, ensuring that downstream applications—from basic research to clinical diagnostics—are built upon a foundation of accurate genetic information.
Conclusion
The reliable amplification of trinucleotide repeat regions is no longer an insurmountable obstacle but a manageable challenge through a systematic approach. By understanding the unique biochemistry of TNRs, meticulously optimizing PCR components with strategic additives, and employing rigorous validation, researchers can achieve specific and high-fidelity amplification. These advancements are crucial for propelling research in neurodegenerative diseases, enabling accurate genotyping, and facilitating the development of novel therapeutic strategies, including those exploring DNA repair pathways and CRISPR-based interventions. Future directions will likely see further refinement of polymerase enzymes and additive cocktails specifically designed for unstable repeats, deepening our grasp of TNR biology and its clinical implications.
References
- 1. Trinucleotide Repeats: A Structural Perspective - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3687200/]
- 2. Frustration between preferred states of complementary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10191799/]
- 3. 聚合酶链反应: [https://www.sigmaaldrich.com/HK/zh/technical-documents/technical-article/genomics/pcr/polymerase-chain-reaction?srsltid=AfmBOopQW-Z6-5vVGWHBfX1E0i3IvFtZ93zG_5hGHbN4MnZYh-8vWcrE]
- 4. pcr实验组分详细-pcr反应体系 - 赛默飞 [https://www.thermofisher.cn/cn/zh/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 5. 生物标志物开发和测试: 精准医疗之路 [https://www.labcorp.com/content/labcorp/zh/ch/biopharma/central-labs/modalities/biomarkers.html]
- 6. 核苷酸重复障碍中CRISPR-Cas系统的组合物和使用方法 [https://patents.google.com/patent/CN106029880A/zh]
- 7. 科普| 生物标志物及其在临床研究中的应用 [http://www.innoclinic.com.cn/Shownews.asp?ID=529&BigClass=%B9%AB%CB%BE%B6%AF%CC%AC]
- 8. Optimizing PCR amplification of GC-rich nicotinic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12145843/]
- 9. Enhancement of DNA amplification efficiency by using ... [https://www.bocsci.com/blog/enhancement-of-dna-amplification-efficiency-by-using-pcr-additives/?srsltid=AfmBOopar__YspPXlkYgim5nN_ozZShawXaa5abgmYH3Eu3f-xdTfxDz]
- 10. Predicting sequence-specific amplification efficiency in ... [https://www.nature.com/articles/s41467-025-64221-4]
- 11. Optimization PCR for Detection CTG/CCTG-Repeat ... [https://www.annclinlabsci.org/content/45/5/502.full]
- 12. Primer design guide - 5 tips for best PCR results [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 13. PCR Primer Design Tips - Behind the Bench [https://www.thermofisher.com/blog/behindthebench/pcr-primer-design-tips/]
- 14. 10 Strategies to Achieve Excellent PCR Amplification [https://geneticeducation.co.in/10-strategies-to-achieve-excellent-pcr-amplification/]
- 15. Replication and Expansion of Trinucleotide Repeats in Yeast [https://pmc.ncbi.nlm.nih.gov/articles/PMC141142/]
- 16. Improving the PCR protocol to amplify a repetitive DNA ... [https://www.geneticsmr.org/articles/improving-the-pcr-protocol-to-amplify-a-repetitive-dna-sequence-7440.html]
- 17. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOoo9CSqEfEAKEbcOY3Eqq5a8XR7e4PDDyb97HeKD9jGespoIDwS2]
- 18. SMRT Sequencing Detects Clinically Significant Repeat ... [https://www.pacb.com/rare-disease/detecting-clinically-significant-repeat-changes-in-triplet-expansion-disorders/]
- 19. PCR Setup—Six Critical Components to Consider [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 20. Error Rate Comparison during Polymerase Chain Reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4150459/]
- 21. Increased frequency of repeat expansion mutations across ... [https://www.nature.com/articles/s41591-024-03190-5]
- 22. Mechanism of trinucleotide repeat expansion by MutSβ- ... [https://www.nature.com/articles/s41467-025-64485-w]
- 23. REV1 inhibition enhances trinucleotide repeat mutagenesis [https://pubmed.ncbi.nlm.nih.gov/41000971/]
- 24. Base editing of trinucleotide repeats that cause Huntington's ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12165863/]
- 25. Identification and characterization of repeat expansions in ... [https://www.sciencedirect.com/science/article/pii/S0035378724004831]
- 26. Somatic mutation and selection at population scale [https://www.nature.com/articles/s41586-025-09584-w]
- 27. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOooOzNHuWsH0QPyzABzW7JEXuInPxjs4IzMi04rI5pHbK-uZ4smW]
- 28. Polymerase chain reaction optimization for amplification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3722634/]
- 29. PCR Methods - Top Ten Strategies [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 30. Development of a Simple Direct and Hot-Start PCR Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10379934/]
- 31. Archaeal dUTPase enhances PCR amplifications with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC117351/]
- 32. PCR amplification of a triple-repeat genetic target directly from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4411754/]
- 33. Methods to Study Trinucleotide Repeat Instability Induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6800170/]
- 34. Counting CAG repeats in the Huntington's disease gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC134256/]
- 35. Betaine, Dimethyl Sulfoxide, and 7-Deaza-dGTP, a Powerful ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1876170/]
- 36. Role of DMSO in PCR [https://geneticeducation.co.in/role-of-dmso-in-pcr/]
- 37. DMSO and Betaine Greatly Improve Amplification of GC-Rich ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0011024]
- 38. Betaine improves the PCR amplification of GC-rich DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC146979/]
- 39. Bovine serum albumin further enhances the effects of organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3466135/]
- 40. Why Add BSA to PCR Mixes? The Science Behind ... [https://synapse.patsnap.com/article/why-add-bsa-to-pcr-mixes-the-science-behind-stabilization]
- 41. Emerging drivers of DNA repeat expansions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12493176/]
- 42. of temperature, Effects + Mg and mismatches on... 2 concentration [https://pubmed.ncbi.nlm.nih.gov/10325409/]
- 43. conditions | PCR annealing specificity | Primer buffers PCR [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/introduction/pcr-conditions]
- 44. A Critical Balance: dNTPs and the Maintenance of Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5333046/]
- 45. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 46. A Comprehensive Triple-Repeat Primed PCR and a Long ... [https://www.sciencedirect.com/science/article/pii/S1525157822001349]
- 47. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 48. PCR Protocols & Primer Design Guide | Boster Bio [https://www.bosterbio.com/protocol-and-troubleshooting/pcr-protocol?srsltid=AfmBOoppBGBrC1Iz8FlIdOVCdsk3ogFlBJm49U_KgjhFrnA9GHEC8ZDG]
- 49. Engineered helicase replaces thermocycler in DNA ... [https://www.nature.com/articles/s41467-022-34076-0]
- 50. Troubleshooting your PCR [https://www.takarabio.com/learning-centers/pcr/faq/troubleshooting?srsltid=AfmBOoqWP-xPLS6MHwtVM72CeSRXtwEwOFAjInmpuNqzIVpYJOO0tpYn]
- 51. Repeat Expansion and Somatic Instability in TCF4 in Patients ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12347246/]
- 52. High Speed/Throughput Sizing of Expanded Repeats [https://www.myotonic.org/high-speedthroughput-sizing-expanded-repeats]
- 53. Correcting PCR amplification errors in unique molecular ... [https://www.nature.com/articles/s41592-024-02168-y]
- 54. Troubleshooting primer dimer in PCR [https://www.minipcr.com/primer-dimer-pcr/]
- 55. Eliminating primer dimers and improving SNP detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7200914/]
- 56. PCR Optimization: Strategies To Minimize Primer Dimer [https://genemod.net/blog/pcr-optimization]
- 57. 2025 Oligonucleotide Design Guide: Complete User Manual [https://oligopool.com/resources/user-guide]
- 58. How can I avoid primer-dimer formation during PCR ... [https://www.qiagen.com/us/resources/faq/544]
- 59. Analysis of qPCR Data: From PCR Efficiency to Absolute ... [https://www.preprints.org/manuscript/202510.2485]
- 60. rtpcr: a package for statistical analysis and graphical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530193/]
- 61. Prevention of pre-PCR mis-priming and primer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC312262/]
- 62. Trinucleotide Repeat Disorders - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK559254/]
- 63. Trinucleotide Repeat - an overview [https://www.sciencedirect.com/topics/neuroscience/trinucleotide-repeat]
- 64. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 65. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOooJs2apeDPNdUw_1znapo-6FXiJUTZEz_JuSRRYhFKbhJL0zPrw]
- 66. Expanded CAG/CTG Repeat DNA Induces a Checkpoint ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1001339]
- 67. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoooyaB3MBSaz55WIyS3vytXrvdxl9latogjIbnpJ_OLs52AJuRp]
- 68. Strategies for amplification of trinucleotide repeats [https://www.sciencedirect.com/science/article/abs/pii/S108485929670022X]
- 69. 5 easy tips to address problems amplifying GC-rich regions [https://bitesizebio.com/24002/problems-amplifying-gc-rich-regions-problem-solved/]
- 70. Optimizing PCR amplification of GC-rich nicotinic ... [https://www.sciencedirect.com/science/article/pii/S2405580825001396]
- 71. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOooQuZmhaD_gZbyMara_Sf82GhUSaaEO63igwPbEWbtank-ZdVY4]
- 72. Interrogating the “unsequenceable” genomic trinucleotide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5514472/]
- 73. 金唯智新型技术测通AAV-ITR区域 [https://www.genewiz.com.cn/public/resources/zxzx/0029]
- 74. PCR for Sanger Sequencing [https://www.thermofisher.com/us/en/home/life-science/sequencing/sanger-sequencing/sanger-dna-sequencing/pcr-sanger-sequencing.html]
- 75. Guidelines for Sanger sequencing and molecular assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7649556/]
- 76. Fast and accurate quantification of double-strand breaks in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12377901/]
- 77. Strategies for amplification of trinucleotide repeats [https://ohiostate.elsevierpure.com/en/publications/strategies-for-amplification-of-trinucleotide-repeats-optimizatio/]
- 78. MIQE Guidelines | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/why-choose-taqman-assays/publish-real-time-pcr-results.html]
- 79. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 80. A sensitive and reproducible qRT-PCR assay detects ... [https://www.nature.com/articles/s41598-023-29786-4]
- 81. MIQE 2.0 and the Urgent Need to Rethink qPCR Standards [https://www.mdpi.com/1422-0067/26/11/4975]
- 82. Comparison of Conventional, Nested, and Real-Time PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2546753/]
- 83. Comparison of RT-PCR, RT-nested PCRs, and real-time ... [https://www.nature.com/articles/s41598-021-96066-4]
- 84. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOop83TaK790Ql0Z9crCVvZNiNY6jRFHLSkxoQ-t3G4Y3ws3aHZuz]
- 85. Better Than Betaine: PCR Additives That Actually Work [https://bitesizebio.com/2592/better-than-betaine-pcr-additives-that-actually-work/]