GC-Rich PCR Amplification: A Comprehensive Guide to Using DMSO, Betaine, and 7-deaza-dGTP
Amplification of GC-rich DNA templates is a common challenge in molecular biology, diagnostics, and drug development, often leading to failed reactions or non-specific products.
GC-Rich PCR Amplification: A Comprehensive Guide to Using DMSO, Betaine, and 7-deaza-dGTP
Abstract
Amplification of GC-rich DNA templates is a common challenge in molecular biology, diagnostics, and drug development, often leading to failed reactions or non-specific products. This article provides a comprehensive guide for researchers and scientists on the use of three key additives—DMSO, betaine, and 7-deaza-dGTP—to overcome these hurdles. We explore the foundational science behind why GC-rich sequences are problematic, detail methodological protocols for applying these additives individually and in combination, offer a systematic troubleshooting and optimization framework, and present validated comparative data to guide reagent selection. The synthesized strategies herein are designed to enhance PCR success rates for challenging targets, including promoters of housekeeping and tumor suppressor genes, thereby accelerating biomedical research and clinical assay development.
The GC-Rich Challenge: Understanding the Science Behind Problematic Amplification
Defining GC-Rich Templates and Their Prevalence in the Human Genome
In molecular biology, the term GC-rich template refers to a DNA sequence where 60% or more of the nucleotide bases are guanine (G) or cytosine (C) [1]. These templates present significant challenges for polymerase chain reaction (PCR) amplification and other molecular techniques due to their strong hydrogen bonding and stable secondary structures. Although GC-rich sequences constitute only about 3% of the human genome, they are disproportionately found in crucial regulatory regions, including the promoters of housekeeping genes, tumor suppressor genes, and approximately 40% of tissue-specific genes [1] [2] [3]. This guide provides a comprehensive comparison of methodologies for analyzing GC-rich genomic content, with a focused examination of experimental protocols involving the key PCR enhancers DMSO, betaine, and 7-deaza-dGTP.
GC-Rich Genomic Landscape and Biological Significance
The distribution of GC-content within the human genome is not random. A striking pattern emerges in protein-coding genes, where GC-content peaks near the 5' transcriptional start site (TSS) and gradually decreases downstream into the gene body [4]. This GC-peak forms an approximately normal distribution that slopes symmetrically into both upstream intergenic regions and downstream into the first exon and intron.
The biological importance of these GC-rich regions is substantial. CpG islands, which are dense clusters of CpG dinucleotides, are frequently located near TSSs and are associated with robust, high-level gene expression [4]. These regions contribute to gene regulation through multiple mechanisms:
- Transcriptional Activation: GC-rich promoters recruit specific transcription factors that initiate gene expression [4].
- Enhanced Splicing: The contrast between GC-content in exons versus introns helps define exon-intron boundaries for the splicing machinery [4].
- mRNA Nuclear Export: For intron-poor mRNAs, high GC-content at the 5' end promotes efficient nuclear export by recruiting protein factors like SARNP, SR proteins, and RBM33 [4].
The evolutionary dynamics of these GC-rich regions are complex. Recent evidence suggests that in apes and rodents, where recombination is directed away from TSSs by PRDM9, GC-content at the 5' end of protein-coding genes is undergoing mutational decay [4]. Conversely, in canids which lack PRDM9, GC-content at these sites is increasing, indicating that non-adaptive forces like GC-biased gene conversion significantly influence the distribution of GC-rich regions in mammalian genomes [4].
Experimental Approaches for GC-Rich Template Analysis
Standardized Methodologies for GC-Rich PCR Amplification
Protocol 1: Three-Additive Combination Approach
This powerful method utilizes a combination of betaine, DMSO, and 7-deaza-dGTP to overcome amplification challenges [5].
Reaction Setup:
- Template: 100 ng genomic DNA
- Primers: 10 nmol of each forward and reverse primer
- PCR Buffer: 1× concentration, supplemented with 2.5 mM MgCl₂
- dNTPs: 200 μM of each dATP, dTTP, dCTP
- Polymerase: 1.25 units of Taq DNA polymerase
- Additives:
- Betaine: 1.3 M final concentration
- DMSO: 5% final concentration
- 7-deaza-dGTP: 50 μM final concentration (replacing standard dGTP)
Thermal Cycling Conditions:
- Initial Denaturation: 94°C for 5 minutes
- Amplification Cycles (25-40 cycles):
- Denaturation: 94°C for 30 seconds
- Annealing: 60°C for 30 seconds
- Extension: 72°C for 45 seconds
- Final Extension: 72°C for 5 minutes
This combination has proven essential for amplifying challenging sequences such as the RET promoter region (79% GC content), LMX1B gene region (67.8% GC), and PHOX2B exon 3 (72.7% GC) [5].
Protocol 2: Four-Additive Enhancement Method
An alternative approach incorporates four additives for use with Taq DNA polymerase [6]:
- Betaine
- Dithiothreitol (DTT)
- Dimethyl Sulfoxide (DMSO)
- Bovine Serum Albumin (BSA)
Protocol 3: Subcycling with 7-deaza-dGTP for Broad GC Range Amplification
For multiplexed amplification of templates with widely varying GC content (10-90% GC):
- Template: Pool of oligonucleotides with varying GC content
- Polymerase: Phusion HF or KAPA HotStart ReadyMix
- Special Conditions:
- 7-deaza-dGTP at 40:60 ratio with normal dGTP
- Subcycling protocol: 29 cycles at 98°C for 20 sec, with 4 subcycles of 60°C for 15 sec and 65°C for 15 sec [7]
Technical Optimization Strategies
The following workflow outlines a systematic approach for troubleshooting PCR amplification of GC-rich templates:
Comparative Performance Data
PCR Additive Efficacy Comparison
Table 1: Performance comparison of major PCR enhancers for GC-rich templates
| Additive | Optimal Concentration | Mechanism of Action | Effectiveness | Limitations | Key Applications |
|---|---|---|---|---|---|
| Betaine | 1-2 M | Reduces secondary structure formation by equalizing DNA base stability; binds within DNA minor groove increasing hydration [5] [2] | High (essential for >70% GC) | Alone may yield nonspecific products [5] | RET promoter (79% GC), LMX1B gene (67.8% GC) [5] |
| DMSO | 5-10% | Disrupts secondary structures by interfering with hydrogen bonding; reduces DNA melting temperature [1] [2] | Moderate to High | Can inhibit Taq polymerase at >10% concentration [1] | Standard GC-rich protocols; often used in combination |
| 7-deaza-dGTP | 40-60 µM (40:60 ratio with dGTP) | dGTP analog that incorporates into DNA, reducing hydrogen bonding and secondary structure stability [5] [7] | High for extreme GC content | Does not stain well with ethidium bromide; requires adjusted dNTP ratios [1] [7] | PHOX2B exon 3 (72.7% GC); broad spectrum GC templates [5] [7] |
| Combination (Betaine + DMSO + 7-deaza-dGTP) | 1.3 M + 5% + 50 µM | Synergistic effect: betaine destabilizes GC pairs, DMSO reduces secondary structures, 7-deaza-dGTP incorporates into DNA [5] | Very High (enables amplification of >80% GC) | More complex reaction setup; cost considerations | Most challenging templates (78.72% GC ARX gene) [5] [2] |
Polymerase Selection Guide for GC-Rich Templates
Table 2: Comparison of DNA polymerases for GC-rich template amplification
| Polymerase | Fidelity Relative to Taq | GC-Rich Optimization | Optimal GC Range | Key Features |
|---|---|---|---|---|
| Standard Taq | 1× | Limited | Up to 60% GC | Standard buffer; requires additives for GC-rich templates [8] |
| OneTaq DNA Polymerase | 2× | GC Buffer & Enhancer | Up to 80% GC | Specifically formulated for difficult amplicons [1] |
| Q5 High-Fidelity DNA Polymerase | >280× | Q5 High GC Enhancer | Up to 80% GC | Ideal for long or difficult amplicons including GC-rich DNA [1] |
| Phusion High-Fidelity | ~50× | GC Buffer option | Up to 80% GC | Proofreading activity; high sensitivity [7] |
| PCRBIO Ultra Polymerase | Not specified | Advanced buffer system | Up to 80% GC | Designed for demanding applications including GC-rich templates [8] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents for GC-rich genomic research
| Reagent Category | Specific Examples | Function in GC-Rich Research |
|---|---|---|
| PCR Enhancers | Betaine, DMSO, 7-deaza-dGTP, formamide, glycerol | Disrupt secondary structures, reduce melting temperature, improve polymerase processivity [5] [1] [2] |
| Specialized Polymerases | OneTaq GC-rich, Q5 High-Fidelity, Phusion HF, KOD variants | Enhanced capability to amplify through stable secondary structures [1] [7] |
| Buffer Components | MgCl₂, GC enhancers, BSA, DTT | Optimize ionic conditions, stabilize enzymes, prevent nonspecific binding [1] [6] |
| Molecular Biology Kits | GC-rich amplification kits, high GC promoter analysis kits | Pre-optimized systems for specific GC-rich applications |
GC-rich templates, while representing a small fraction of the human genome, play disproportionately important roles in gene regulation and pose significant technical challenges for molecular analysis. The comparative data presented here demonstrates that a multipronged optimization strategy incorporating specialized polymerases, synergistic additive combinations (particularly betaine + DMSO + 7-deaza-dGTP), and refined thermal cycling parameters provides the most reliable approach for investigating these recalcitrant genomic regions. The ongoing discovery that GC-content at transcriptional start sites is undergoing evolutionary decay in some mammalian lineages further highlights the dynamic nature of these genomic features and the continued importance of robust methodological approaches for their study [4].
The deoxyribonucleic acid (DNA) double helix, the fundamental molecule of life, maintains its structural integrity through specific pairing between four nucleobases: adenine (A), thymine (T), guanine (G), and cytosine (C). These natural Watson-Crick base pairs form the foundation of genetic information storage and transfer. A critical aspect of DNA biochemistry lies in the differential stability between the two types of base pairings: G-C pairs versus A-T pairs. This thermostability difference directly influences essential molecular biology techniques, particularly the polymerase chain reaction (PCR), where amplifying GC-rich sequences presents considerable challenges [9]. Understanding the hydrogen bonding problem—why G-C base pairs are more thermostable than A-T pairs—is therefore crucial for both basic molecular biology and applied biotechnological applications, especially when dealing with GC-rich templates that are prevalent in promoter regions of housekeeping and tumor suppressor genes [2].
The challenge becomes particularly evident in PCR amplification of GC-rich regions, where the enhanced stability of G-C rich sequences leads to secondary structure formation and incomplete denaturation, ultimately resulting in amplification failure or nonspecific products [5] [9]. This practical problem has led to the development of various biochemical solutions, including the use of additives like DMSO, betaine, and 7-deaza-dGTP, which work through distinct mechanisms to facilitate amplification of these problematic sequences [5] [2]. This article explores the fundamental hydrogen bonding principles underlying G-C thermostability and objectively compares the performance of key reagents used to overcome the challenges it presents.
Hydrogen Bonding: The Structural Basis of Thermostability
Hydrogen Bond Geometry in Natural Base Pairs
The remarkable stability of the DNA double helix arises primarily from intermolecular hydrogen bonds that form between complementary nucleobases [10]. According to Watson-Crick pairing rules, adenine pairs with thymine via two hydrogen bonds, while guanine pairs with cytosine via three hydrogen bonds [9] [11]. This difference in hydrogen bond count represents the most fundamental explanation for the enhanced thermostability of G-C pairs, as breaking a G-C pair requires additional energy to disrupt the extra hydrogen bond [9].
Advanced analytical approaches, including local vibrational mode analysis complemented by topological analysis of the electron density, have provided quantitative insights into hydrogen bond strength in natural base pairs [10]. These investigations reveal that the N-H···N hydrogen bond represents the most favorable interaction in both natural and unnatural base pairs, while classical O-H···N/O bonds are less favorable and not typically found in natural Watson-Crick pairs [10]. Furthermore, research has highlighted the important role of non-classical C-H···O/N bonds in stabilizing base pairs, particularly the contribution of C-H···O bonds in Watson-Crick pairs [10]. When natural base pairs are modeled within the DNA environment using QM/MM approaches, studies indicate that the cellular context increases the strength of the central N-H···N bond and C-H···O bonds, while simultaneously decreasing the strength of the N-H···O bond, though general trends observed in gas phase calculations remain consistent [10].
Visualizing the Hydrogen Bond Difference
The following diagram illustrates the fundamental structural difference between A-T and G-C base pairs, highlighting the hydrogen bonding patterns that explain their differential thermostability:
Figure 1: Hydrogen bond comparison between A-T and G-C base pairs. G-C pairs form three hydrogen bonds compared to two in A-T pairs, resulting in greater thermal stability that requires more energy to disrupt during PCR denaturation steps. This enhanced stability creates practical challenges for amplifying GC-rich DNA sequences.
Experimental Approaches to GC-Rich PCR
The GC-Rich PCR Challenge
GC-rich DNA templates, typically defined as sequences where 60% or greater of the bases are guanine or cytosine, present significant challenges in PCR amplification [9]. These difficulties arise from three primary factors: the increased energy required to separate the three hydrogen bonds of G-C pairs, the tendency of GC-rich regions to form stable secondary structures such as hairpins, and the higher melting temperatures that interfere with primer annealing [9]. While only approximately 3% of the human genome consists of GC-rich regions, these sequences are disproportionately found in gene promoters, including those of housekeeping genes and tumor suppressor genes, making their amplification essential for many research and diagnostic applications [9] [2].
Experimental evidence demonstrates that without specialized approaches, GC-rich PCR often results in complete amplification failure or the generation of nonspecific products. For example, when attempting to amplify a 392-bp RET promoter region with 79% GC content, researchers observed at least five major nonspecific products in the absence of enhancing additives [5]. Similarly, efforts to amplify a 660-bp fragment of the human ARX gene with 78.72% GC content using conventional PCR protocols consistently failed without optimization of reaction conditions and inclusion of specific enhancers [2].
Experimental Workflow for GC-Rich Amplification
The following workflow outlines a systematic experimental approach for optimizing PCR amplification of GC-rich sequences:
Figure 2: Systematic experimental workflow for GC-rich PCR optimization. The process begins with polymerase selection, proceeds through additive optimization, includes cycling parameter adjustments, and concludes with magnesium concentration fine-tuning. This structured approach efficiently addresses the multiple challenges posed by GC-rich templates.
Comparative Performance of GC-Rich PCR Additives
Mechanism of Action and Experimental Evidence
Various additives have been developed to overcome the challenges of GC-rich PCR amplification, each functioning through distinct biochemical mechanisms. The table below summarizes the key reagents, their modes of action, and experimental support for their efficacy:
Table 1: Research reagent solutions for GC-rich PCR amplification
| Reagent | Concentration | Mechanism of Action | Experimental Evidence |
|---|---|---|---|
| Betaine | 1.0-1.3 M | Equalizes DNA stability by binding to AT/GC pairs; increases hydration of GC pairs, destabilizing GC-rich DNA [5] [2] | Drastically reduces nonspecific background; enables amplification when combined with other additives [5] |
| DMSO (Dimethyl Sulfoxide) | 5-10% | Reduces secondary structure formation; interferes with hydrogen bonding; lowers DNA melting temperature [9] [12] | Improves amplification when combined with betaine and 7-deaza-dGTP; helps separate double-stranded DNA [5] [12] |
| 7-deaza-dGTP | 50 μM (partial substitution) | dGTP analog that reduces hydrogen bonding capacity; incorporates into DNA without affecting polymerase activity [5] [9] | Essential for specific amplification of 79% GC RET promoter when combined with betaine and DMSO [5] |
| GC Enhancer | 1X (commercial formulations) | Proprietary mixtures containing multiple additives; inhibits secondary structure formation and increases primer stringency [9] | Enables amplification of templates with up to 80% GC content; provided with specialized polymerases [9] |
| Formamide | 1-5% | Increases primer annealing stringency; denatures secondary structures [9] | Reduces non-specific priming and off-target amplification [9] |
Quantitative Comparison of Additive Performance
Experimental data directly comparing the efficacy of individual additives versus combinations provides compelling evidence for synergistic effects. The following table summarizes quantitative results from systematic studies evaluating different additive approaches:
Table 2: Experimental performance comparison of additive combinations on GC-rich templates
| Template (GC Content) | Additive Condition | Result | Experimental Protocol |
|---|---|---|---|
| RET promoter (79% GC) [5] | No additives | Multiple nonspecific bands; no target amplification | 1X PCR buffer, 2.5 mM MgCl₂, 200 μM dNTPs, 1.25 U Taq polymerase, 100 ng genomic DNA |
| Betaine alone | Reduced background but incorrect 344-bp product | As above + 1.3 M betaine | |
| DMSO + 7-deaza-dGTP | Some nonspecific bands reduced; no specific product | As above + 5% DMSO + 50 μM 7-deaza-dGTP | |
| Betaine + DMSO + 7-deaza-dGTP | Unique specific PCR product | As above + all three additives simultaneously | |
| LMX1B region (67.8% GC) [5] | No additives | Multiple nonspecific products | Standard PCR conditions with IMR-32 neuroblastoma cell line DNA |
| Individual additives (DMSO, betaine, or 7-deaza-dGTP) | Only nonspecific products | Single additive included in standard PCR | |
| Betaine + DMSO + 7-deaza-dGTP | Clean specific product | All three additives combined in reaction | |
| ARX gene (78.72% GC) [2] | Standard conditions | No amplification | Conventional PCR protocol |
| Optimized annealing time + additives | Successful amplification | Combination of shorter annealing times (10-20s) with enhancing additives |
Detailed Experimental Protocols
Three-Additive Combination Protocol
Based on the most successful experimental results, the following detailed protocol can be employed for challenging GC-rich amplifications:
Reaction Setup [5]:
- Prepare a 25 μL reaction mixture containing:
- 1× PCR buffer (supplemented with 2.5 mM MgCl₂)
- 200 μM of each dNTP (with partial substitution of dGTP with 7-deaza-dGTP to 50 μM final concentration)
- 10 nmol of each forward and reverse primer
- 100 ng of genomic DNA template
- 1.25 units of Taq DNA polymerase
- 1.3 M betaine
- 5% DMSO
Thermal Cycling Conditions [5] [2]:
- Initial denaturation: 94°C for 3-5 minutes
- 25-40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing: Temperature gradient based on primer Tm (50-68°C) for 10-30 seconds
- Extension: 72°C for 45-60 seconds per kb
- Final extension: 72°C for 5-15 minutes
Critical Considerations:
- Annealing temperature should be optimized using a gradient thermal cycler, as the presence of DMSO lowers the effective Tm by approximately 5.5-6.0°C [12]
- For extremely GC-rich targets (>80%), extending the initial denaturation time up to 5 minutes may improve yield [12]
- Polymerase selection significantly impacts success; specialized enzymes like Q5 High-Fidelity DNA Polymerase often outperform standard Taq [9]
Magnesium Concentration Optimization
Given the critical role of magnesium as a polymerase cofactor, systematic optimization is recommended:
- Prepare a series of reactions with MgCl₂ concentrations ranging from 1.0 to 4.0 mM in 0.5 mM increments
- Maintain all other components constant, including the three-additive mixture
- Analyze results by gel electrophoresis for product yield and specificity
- Select the concentration that provides the highest specific yield with minimal nonspecific amplification [9]
The hydrogen bond problem—the enhanced thermostability of G-C base pairs resulting from their three hydrogen bonds compared to the two in A-T pairs—presents significant challenges in molecular biology applications, particularly PCR amplification of GC-rich sequences. Through systematic experimental approaches, researchers have developed effective solutions centered on additive combinations that work synergistically to overcome these challenges.
The comparative data clearly demonstrate that while individual additives provide some improvement, the powerful combination of betaine, DMSO, and 7-deaza-dGTP consistently enables successful amplification of even the most challenging GC-rich templates (67-79% GC content). This three-additive approach, combined with optimized thermal cycling parameters and specialized polymerases, represents the current gold standard for GC-rich PCR. The experimental protocols detailed herein provide researchers with a validated methodology for approaching difficult amplifications, contributing to more reliable analysis of gene regulatory regions and advancing both basic research and diagnostic applications.
In molecular biology, the polymerase chain reaction (PCR) is a foundational technique for amplifying specific DNA sequences. However, the efficient amplification of DNA templates with high guanine-cytosine (GC) content (typically >60%) remains a significant technical challenge. These GC-rich sequences are notoriously refractory to amplification due to their propensity to form stable intramolecular secondary structures, such as hairpins and loops, during the thermal cycling process [5] [13].
The core of the problem lies in the inherent stability of GC base pairs. Unlike adenine-thymine (AT) pairs, which form two hydrogen bonds, GC pairs form three hydrogen bonds, resulting in a thermodynamically more stable duplex [13]. This heightened stability causes GC-rich regions to resist the DNA denaturation steps essential for PCR, leading to incomplete strand separation. The single-stranded DNA that is produced can then fold onto itself at regions of inverted repeats (palindromes), forming hairpin structures characterized by a double-stranded stem and a single-stranded loop [14]. These secondary structures physically impede the progression of DNA polymerase, causing the enzyme to stall, dissociate, or generate truncated, nonspecific amplification products [5] [7] [13].
To overcome these obstacles, scientists have developed several biochemical strategies, primarily involving the use of PCR additives. Among the most effective are dimethyl sulfoxide (DMSO), betaine, and 7-deaza-2'-deoxyguanosine triphosphate (7-deaza-dGTP) [5] [15]. This guide provides an objective comparison of the performance of these three key reagents, based on experimental data, to inform their application in research and drug development.
Molecular Mechanisms of Polymerase Inhibition by Secondary Structures
Formation and Consequences of DNA Hairpins
The process of hairpin formation begins when a single-stranded DNA molecule, generated during the PCR denaturation step, contains a sequence with an inverted repeat (IR). This sequence symmetry allows the DNA strand to fold back on itself, creating a stem-loop structure [14]. The stability of this hairpin is directly related to the length and GC content of its stem; higher GC content results in a more stable, thermally resistant structure.
When a DNA polymerase encounters such a hairpin during the elongation phase of PCR, its progression is severely hindered. The robust secondary structure can:
- Cause Polymerase Stalling: The physical barrier of the double-stranded stem blocks the polymerase's active site, halting DNA synthesis [13].
- Promote Primer Mis-annealing: Incomplete denaturation and structured templates increase the likelihood of primers binding to incorrect, partially homologous sites, leading to nonspecific products [5].
- Induce Polymerase "Jumping": In some cases, the polymerase may skip over the hairpin structure, resulting in a truncated PCR product that lacks the sequence encompassed by the hairpin [5].
The following diagram illustrates this process of inhibition.
Structural Basis of Inhibition in Nucleic Acid Enzymes
The inhibitory effect of RNA hairpins on polymerase progression is not limited to DNA replication and has been structurally characterized in the context of transcription. Cryo-electron microscopy (cryo-EM) studies of bacterial RNA polymerase (RNAP) have revealed the precise mechanism by which a pause hairpin (PH) in the newly synthesized RNA induces transcriptional pausing [16].
In the his operon attenuation control region of E. coli, the formation of an RNA hairpin just upstream of the RNA 3' end stabilizes a paused elongation complex (hpPEC). Structural analyses show that this hairpin folds within the RNA exit channel of RNAP, where it interacts with specific domains of the enzyme, including the flexible flap domain [17] [16]. This hairpin-flap interaction triggers a large-scale conformational change in the polymerase, involving a swivel module rotation. This rearrangement allosterically inhibits the folding of the trigger loop (TL), a critical component of the RNAP active site, thereby preventing nucleotide addition and prolonging the pause [16]. Furthermore, the nucleic acid scaffold in this paused state adopts a unique "half-translocated" configuration (RNA post-translocated, DNA pre-translocated), which is incompatible with binding the next nucleoside triphosphate substrate, effectively halting transcription [16].
This detailed structural insight into RNA polymerase pausing provides a valuable analog for understanding the potential steric and allosteric challenges that DNA secondary structures pose to DNA polymerases during PCR.
Comparative Analysis of DMSO, Betaine, and 7-deaza-dGTP
The table below synthesizes experimental data on the mechanisms and effectiveness of each additive when used individually.
Table 1: Performance Profile of Individual PCR Additives for GC-Rich Amplification
| Additive | Proposed Mechanism of Action | Reported Experimental Efficacy | Key Limitations |
|---|---|---|---|
| DMSO | Disrupts DNA secondary structures by reducing hydrogen bonding and lowering DNA melting temperature (Tm) [13]. | Partial reduction of nonspecific products; alone, insufficient for high-GC targets [5]. | Does not fully eliminate background or enable specific amplification of very GC-rich (>75%) sequences on its own [5]. |
| Betaine | Equalizes the contribution of GC and AT base pairs to duplex stability, promoting DNA denaturation and preventing secondary structure formation [5]. | Drastically reduces nonspecific background but may not prevent mis-priming on homologous sequences [5]. | By itself, can permit amplification of a faster-migrating, nonspecific product from a homologous sequence [5]. |
| 7-deaza-dGTP | A dGTP analog that lacks the nitrogen-7 position, preventing Hoogsteen base pairing and thus destabilizing GC-rich DNA secondary structures without compromising Watson-Crick pairing [5] [7]. | Improves amplification yield of long PCR products (~1000 bp) and is effective across a broad GC spectrum [7]. | PCR products incorporating 7-deaza-dGTP stain poorly with ethidium bromide and cannot be cleaved by some restriction enzymes [13]. |
Direct Comparison of Combinatorial Strategies
No single additive is a universal solution. Research demonstrates that a combinatorial approach is often essential for the most challenging templates. The following table compares key additive combinations based on published experimental outcomes.
Table 2: Efficacy of Additive Combinations on Challenging GC-Rich Templates
| Additive Combination | Experimental Model & GC Content | Amplification Outcome | Key Findings |
|---|---|---|---|
| Betaine + DMSO | RET promoter (79% GC); LMX1B gene region (67.8% GC) [5]. | Ineffective. Nonspecific background reduced, but specific product not achieved (RET). Ineffective (LMX1B) [5]. | The combination of two secondary structure-destabilizing agents was not sufficient to overcome extreme GC stability and prevent mis-priming. |
| Betaine + 7-deaza-dGTP | RET promoter (79% GC); LMX1B gene region (67.8% GC) [5]. | Partially Effective. Specific target amplified, but co-amplification of a prominent nonspecific product persisted [5]. | Demonstrated the critical role of 7-deaza-dGTP in enabling polymerization through the hairpin, but specificity remained an issue. |
| Betaine + DMSO + 7-deaza-dGTP | RET promoter (79% GC); LMX1B gene region (67.8% GC); PHOX2B exon 3 (72.7% GC) [5]. | Fully Effective. A unique, specific PCR product was obtained for all three disease genes, confirmed by DNA sequencing [5]. | The triple combination was essential for clean, specific amplification. Each additive addresses a different aspect of the problem (denaturation, structure blocking, primer fidelity). |
The synergistic workflow of the triple additive combination is summarized below.
Detailed Experimental Protocols from Cited Studies
Protocol for Amplification of Extreme GC-Rich Targets
The following methodology, adapted from the study that demonstrated the efficacy of the triple-additive mixture, provides a robust starting point for amplifying highly refractory GC-rich sequences [5].
PCR Reaction Setup (25 μL total volume):
- Template: 100 ng genomic DNA.
- Polymerase: 1.25 units of Taq polymerase (Eppendorf-5 Prime, Inc.).
- Buffer: 1X manufacturer's buffer supplemented with 2.5 mM MgCl₂.
- dNTPs: 200 μM of each dATP, dTTP, dCTP.
- Modified dGTP: 50 μM 7-deaza-dGTP (Roche Diagnostics) with 150 μM standard dGTP, for a 40:60 ratio [5] [7].
- Primers: 10 nmol of each forward and reverse primer.
- Additives:
- Betaine (Sigma-Aldrich): 1.3 mol/L final concentration.
- DMSO (Sigma-Aldrich): 5% (v/v) final concentration.
Thermal Cycling Conditions (Applied Biosystems 2700 thermal cycler):
- Initial Denaturation: 94°C for 5 minutes.
- Amplification Cycles (40 cycles for RET promoter):
- Denaturation: 94°C for 30 seconds.
- Annealing: 60°C for 30 seconds.
- Extension: 68°C for 45 seconds.
- Final Extension: 72°C for 5 minutes.
Analysis:
- Separate 5 μL of the PCR product on a 1.2% agarose gel.
- For confirmation, purify the remaining product and perform direct Sanger sequencing.
Alternative Strategy: Subcycling PCR Protocol
For DNA templates containing a broad spectrum of GC content (from 10% to 90%) within a multiplexed reaction, an alternative thermocycling strategy called subcycling can be highly effective, particularly when combined with 7-deaza-dGTP [7].
- Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 5 minutes.
- Amplification Cycles (29 cycles):
- Denaturation: 98°C for 20 seconds.
- Subcycling Step (4 repetitions):
- Annealing: 60°C for 15 seconds.
- Extension: 65°C for 15 seconds.
- Final Extension: 65°C for 5 minutes.
This protocol, using Phusion HF polymerase, was shown to significantly improve the uniformity of amplification across a complex pool of oligonucleotides with varying GC content, especially for low-GC templates. The combination of subcycling and 7-deaza-dGTP was found to be particularly powerful for amplifying short templates across the entire GC spectrum [7].
The Scientist's Toolkit: Essential Reagents for GC-Rich PCR
Table 3: Key Research Reagent Solutions for Overcoming Secondary Structures
| Reagent / Solution | Function / Rationale | Example Use Case |
|---|---|---|
| Specialized High-GC Polymerase | Polymerase enzymes supplied with proprietary "GC Enhancer" buffers are optimized to resist stalling at secondary structures [13]. | Ideal for routine screening of GC-rich targets with minimal optimization (e.g., NEB's OneTaq or Q5 with GC Enhancer) [13]. |
| Betaine (Sigma-Aldrich) | Used as a PCR additive to homogenize the melting temperature of DNA, facilitating the denaturation of stable GC-rich duplexes [5]. | A core component of the powerful triple-additive mixture for extreme GC targets (>70% GC) [5]. |
| 7-deaza-dGTP (Roche Diagnostics) | A nucleotide analog that incorporation into DNA prevents the formation of stable secondary structures by inhibiting Hoogsteen base pairing [5]. | Critical for amplifying long (~1000 bp) high-GC products and for use in multiplexed reactions with broad GC content [5] [7]. |
| Molecular Biology Grade DMSO | A polar solvent that disrupts hydrogen bonding in nucleic acids, helping to denature hairpins and other secondary structures [5] [13]. | A common additive used at 2.5-10% to improve amplification of difficult templates; part of the effective triple-mixture [5] [13]. |
| MgCl₂ Solution | A critical cofactor for DNA polymerase activity. Its concentration can be optimized to influence specificity and yield in GC-rich PCR [13]. | If nonspecific amplification or low yield occurs, testing a gradient from 1.0 mM to 4.0 mM in 0.5 mM increments is recommended [13]. |
The hindrance of polymerase progression by DNA hairpins and loops is a significant impediment in molecular biology, particularly in the amplification and analysis of GC-rich promoter regions and other structured genomic elements. The experimental data clearly demonstrate that no single reagent is superior in all contexts. Instead, the choice depends on the specific challenge:
- DMSO and Betaine are both effective at destabilizing secondary structures but may be insufficient alone for extreme GC content.
- 7-deaza-dGTP plays a unique and critical role by directly altering the thermodynamics of DNA structure formation, making it especially valuable for long and highly structured targets.
The most robust solution for the most challenging sequences, as validated by direct experimental comparison, is a combination of betaine, DMSO, and 7-deaza-dGTP. This mixture acts synergistically to promote DNA denaturation, prevent secondary structure formation, and maintain polymerase processivity, thereby enabling specific and efficient amplification of DNA sequences previously considered refractory to PCR.
Polymerase chain reaction (PCR) amplification of GC-rich DNA sequences (typically defined as regions where 60% or more of the bases are guanine or cytosine) presents a significant challenge in molecular biology [18] [19]. These sequences are common in the human genome, particularly in promoter regions of housekeeping and tumor suppressor genes, making their amplification essential for various diagnostic and research applications [2] [18]. The primary obstacles arise from the inherent properties of GC-rich DNA: its high thermostability due to three hydrogen bonds in G-C base pairs (compared to two in A-T pairs) and a strong tendency to form stable secondary structures like hairpins and loops [18] [19]. These characteristics lead to common experimental consequences such as incomplete amplification products, non-specific bands, and complete reaction failure. To overcome these hurdles, researchers often employ additives, with Dimethyl Sulfoxide (DMSO), Betaine, and 7-deaza-dGTP being among the most effective and widely studied [5] [20] [18].
This guide objectively compares the performance of these three additives, synthesizing experimental data to inform researchers and drug development professionals.
Comparative Performance of DMSO, Betaine, and 7-deaza-dGTP
The table below summarizes the core characteristics, mechanisms, and optimal use cases for DMSO, Betaine, and 7-deaza-dGTP, based on aggregated experimental evidence.
Table 1: Direct Comparison of PCR Additives for GC-Rich Amplification
| Additive | Proposed Mechanism of Action | Typical Effective Concentration | Impact on Specificity & Yield | Key Advantages & Limitations |
|---|---|---|---|---|
| DMSO | Disrupts secondary structures by reducing DNA melting temperature; may interfere with polymerase activity [18] [19]. | 2.5% - 10% [7] [20] | Can improve specificity but may not yield a specific product alone; often requires combination with other additives [5]. | Advantage: Common, low-cost lab reagent.Limitation: High concentrations can inhibit some polymerases [18]. |
| Betaine | Equalizes the thermal stability of AT and GC base pairs, promoting uniform DNA melting; can also reduce nonspecific background [5] [2] [20]. | 1M - 2M [5] [20] | Drastically reduces nonspecific amplification but may be insufficient for a clean, specific product by itself [5]. | Advantage: Preferential amplification of both alleles in repeat expansion regions [5].Limitation: Alone, may not fully overcome extreme GC content. |
| 7-deaza-dGTP | dGTP analog that lacks a nitrogen atom at position 7 of the purine ring, reducing hydrogen bonding in GC base pairs and destabilizing secondary structures [5] [20]. | 50 µM (in a 3:1 or 40:60 ratio with dGTP) [5] [7] | Enables amplification of specific GC-rich bands that are otherwise unobtainable; particularly effective for longer products (~1000 bp) [5] [7]. | Advantage: Directly addresses the root cause of stable secondary structures.Limitation: Does not stain well with ethidium bromide; requires partial substitution for dGTP in the dNTP mix [5] [18]. |
Synergistic Effects and Combination Strategies
While individual additives can offer improvements, multiple studies demonstrate that a combination strategy is often essential for successfully amplifying highly GC-rich templates (e.g., >70% GC) [5] [7]. The data shows that the combination's effect is synergistic, not merely additive.
Table 2: Experimental Outcomes of Additive Combinations on Specific GC-Rich Targets
| DNA Target (GC Content) | Additive Combination | Experimental Outcome | Source Study |
|---|---|---|---|
| RET Promoter (79% GC) | Betaine + DMSO + 7-deaza-dGTP | Unique, specific PCR product obtained; all other combinations failed or produced nonspecific bands. | Musso et al. [5] |
| LMX1B Region (67.8% GC) | Betaine + DMSO + 7-deaza-dGTP | Clean, specific product confirmed by sequencing; combinations of two additives were insufficient. | Musso et al. [5] |
| FMR1 Gene (>80% GC) | Betaine (1M) + DMSO (5%) | Reproducible amplification achieved, providing a cost-effective alternative to specialized kits. | Ralser et al. [20] |
| Broad-Spectrum GC Templates (10-90% GC) | Subcycling PCR + 7-deaza-dGTP | Achieved efficient amplification of short templates across the entire GC spectrum. | Guido et al. [7] |
Detailed Experimental Protocols
Protocol 1: Combined Additive Mixture for Disease Genes
This protocol is adapted from the seminal work by Musso et al. (2006), which successfully amplified sequences with 67-79% GC content [5].
- Reaction Setup: A standard 25 µL reaction volume is used.
- Key Components:
- Polymerase: 1.25 units of standard Taq DNA polymerase.
- Buffer: 1X manufacturer's buffer supplemented with 2.5 mM MgCl₂.
- dNTPs: 200 µM of each dATP, dCTP, and dTTP.
- Modified dNTP: 50 µM 7-deaza-dGTP.
- Primers: 10 nmol of each forward and reverse primer.
- Template: 100 ng of genomic DNA.
- Additives: 1.3 M Betaine and 5% DMSO.
- Thermal Cycling Conditions: An initial denaturation at 94°C for 3 minutes, followed by 25-40 cycles of:
- Denaturation: 94°C for 10-30 seconds.
- Annealing/Extension: 68°C for 3 minutes (for the RET promoter).
- Analysis: 5 µL of the PCR product is analyzed on a 1.2% agarose gel. The specific product is confirmed by DNA sequencing.
Protocol 2: Cost-Effective Amplification of FMR1 Gene
This protocol from Ralser et al. (2013) offers a reproducible and lower-cost method for amplifying the extremely GC-rich FMR1 gene [20].
- Reaction Setup: A 25 µL reaction volume.
- Key Components:
- Polymerase: 1 unit of a standard, low-cost thermostable polymerase.
- Buffer: 1X PCR buffer.
- dNTPs: 0.2 mM of each dNTP.
- MgCl₂: 1.5 mM.
- Primers: 0.1 µM of each primer.
- Template: 50 ng of genomic DNA extracted from buccal cells.
- Additives: 1 M Betaine and 5% DMSO.
- Thermal Cycling Conditions: Initial denaturation at 95°C for 10 minutes, followed by 25 cycles of:
- Denaturation: 95°C for 1.5 minutes.
- Annealing: 65°C for 1 minute.
- Extension: 72°C for 2 minutes.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for GC-Rich PCR Optimization
| Reagent / Tool | Function / Rationale | Example Product / Note |
|---|---|---|
| High-Fidelity or Specialty Polymerases | Polymerases engineered for high processivity can better stall at complex secondary structures. | OneTaq DNA Polymerase with GC Buffer, Q5 High-Fidelity DNA Polymerase with GC Enhancer [18]. |
| Betaine (as a powder) | Allows for flexible molarity preparation in the lab. | Sigma-Aldrich [5] [7]. |
| 7-deaza-dGTP | dGTP analog critical for disrupting Hoogsteen base pairing in stable secondary structures. | Sold by companies like Roche Diagnostics and New England Biolabs (NEB) [5] [18]. |
| Molecular Grade DMSO | Ensures the reagent is free of contaminants that could inhibit the PCR. | Sigma [5] [7]. |
| Magnesium Chloride (MgCl₂) | A cofactor for polymerase activity; its concentration significantly impacts specificity and yield [18]. | Typically supplied with the polymerase buffer. A gradient from 1.0 mM to 4.0 mM is recommended for optimization [18]. |
Mechanisms and Workflow: A Visual Guide
The following diagrams illustrate the core challenges of GC-rich PCR and the strategic workflow for optimizing amplification using additives.
GC Rich PCR Challenges
GC PCR Optimization Workflow
The evidence clearly demonstrates that no single additive universally solves all challenges of GC-rich PCR. DMSO and Betaine are effective first-line agents for reducing secondary structures and nonspecific background, with a combination of the two often providing a robust, cost-effective solution [20]. However, for the most refractory templates with GC content exceeding 75%, the incorporation of 7-deaza-dGTP is frequently indispensable [5]. Its unique mechanism of directly impairing hydrogen bonding in GC pairs addresses the core stability problem that other additives cannot fully resolve. Therefore, the most powerful strategy, as proven in multiple experimental contexts, is a synergistic combination of all three additives, which provides a multi-faceted attack on the structural, thermal, and kinetic barriers that lead to incomplete products, non-specific amplification, and failed reactions [5] [7].
GC-rich DNA sequences, where guanine (G) and cytosine (C) bases constitute 60% or more of the sequence, are critical regulatory elements in the genome. They are predominantly found in the promoters of genes, especially housekeeping genes and tumor suppressor genes, where they facilitate complex gene regulation [21] [22]. The biological significance of these regions stems from the triple hydrogen bonds of G-C base pairs, which confer higher thermostability and influence the DNA's three-dimensional structure, thereby affecting its interaction with transcription factors and other regulatory proteins [23].
From a functional perspective, the GC-content at the 5' end of genes, particularly around the transcription start site (TSS), plays a crucial role in mRNA nuclear export and translation efficiency [4]. In amniotes and likely most vertebrates, a distinct peak of GC-content is observed at the TSS, a feature that appears to have deep evolutionary roots and may be shaped by both selective pressures and non-adaptive forces like GC-biased gene conversion (gBGC) [23] [4].
However, these very properties that make GC-rich regions biologically significant also render them technically challenging for polymerase chain reaction (PCR) amplification. The strong hydrogen bonding leads to high thermostability and incomplete denaturation, while the sequences readily form stable secondary structures like hairpins and loops that block polymerase progression, often resulting in failed or non-specific amplification [5] [22]. This guide provides an objective comparison of three common reagents—DMSO, betaine, and 7-deaza-dGTP—used to overcome these challenges, framing the analysis within the broader context of GC-rich PCR research.
Comparative Analysis of PCR Additives for GC-Rich Amplification
To overcome the challenges of amplifying GC-rich templates, scientists routinely employ additive agents. The table below provides a systematic comparison of the three primary additives discussed in this guide.
Table 1: Key Characteristics of PCR Additives for GC-Rich Amplification
| Additive | Primary Mechanism of Action | Typical Working Concentration | Key Advantages | Reported Limitations |
|---|---|---|---|---|
| DMSO | Disrupts secondary structures by reducing DNA melting temperature; reduces intra-strand secondary structure [22] [24]. | 2.5% - 10% [7] | Reduces non-specific background; widely available and inexpensive [5]. | May inhibit Taq polymerase activity at higher concentrations; alone may be insufficient for extreme GC content [5]. |
| Betaine (also known as N,N,N-trimethylglycine) | Equalizes the contribution of GC and AT base pairs to DNA stability; destabilizes secondary structures [22]. | 1 M - 2 M [7] | Drastically reduces nonspecific amplification background; can be used at high concentrations [5]. | Alone, may not prevent amplification of non-specific products with similar mobility to the target [5]. |
| 7-deaza-dGTP | dGTP analog that incorporates into nascent DNA, preventing Hoogsteen base pairing and secondary structure formation [5] [7]. | 50 µM (in a 40:60 or 50:50 ratio with dGTP) [5] [7] | Directly prevents the formation of GC-rich secondary structures; improves yield of longer products [5] [7]. | Alters DNA composition, which can interfere with downstream restriction enzyme digestion; does not stain well with ethidium bromide [5] [22]. |
The synergistic effect of combining these additives is particularly powerful. Research demonstrates that while individual additives often yield incomplete or non-specific amplification, a combination of all three—1.3 M betaine, 5% DMSO, and 50 µM 7-deaza-dGTP—was essential for the specific amplification of several disease-associated genes with GC content ranging from 67% to 79%, including the RET promoter and LMX1B gene regions [5]. This combination leverages the distinct mechanisms of each reagent to effectively overcome the multiple barriers posed by GC-rich templates.
Experimental Data and Performance Comparison
The efficacy of additive strategies is best understood through direct experimental results. The following quantitative data, drawn from key studies, compares the performance of individual additives and their combinations.
Table 2: Experimental Performance Data on Additive Efficacy for GC-Rich PCR
| Target Gene / GC Content | Experimental Conditions | Amplification Outcome | Source |
|---|---|---|---|
| RET Promoter / 79% GC | No additives | Multiple nonspecific bands; no specific product [5]. | Cell Biol Int. 2006 |
| DMSO + 7-deaza-dGTP | Some nonspecific bands disappeared; no specific product [5]. | ||
| Betaine alone | Drastically reduced background; amplified a faster, non-specific band [5]. | ||
| Betaine + 7-deaza-dGTP | Specific product amplified, but a prominent non-specific band remained [5]. | ||
| Betaine + DMSO + 7-deaza-dGTP | A unique, specific PCR product was obtained [5]. | ||
| LMX1B / 67.8% GC | No additives | Multiple nonspecific products [5]. | Cell Biol Int. 2006 |
| Individual additives (DMSO, Betaine, or 7-deaza-dGTP) | Amplification of only nonspecific products [5]. | ||
| Two-additive combinations (DMSO/Betaine or DMSO/7-deaza-dGTP) | No specific product [5]. | ||
| Betaine + 7-deaza-dGTP | Specific band achieved, but a trail of nonspecific bands persisted [5]. | ||
| Betaine + DMSO + 7-deaza-dGTP | A clean, specific product was obtained [5]. | ||
| Broad Spectrum GC (10-90%) | Standard PCR | Uneven amplification across the GC spectrum; poor low-GC amplification [7]. | PLoS One. 2016 |
| Subcycling PCR + 7-deaza-dGTP | Efficient amplification of short templates (200 bp) across 10-90% GC [7]. | ||
| 7-deaza-dGTP with KOD polymerase | Improved amplification specificity of longer products (~1000 bp) [7]. |
Beyond additives, the choice of DNA polymerase is a critical experimental variable. Studies show that polymerases specifically engineered for GC-rich templates, such as Q5 High-Fidelity DNA Polymerase, can robustly amplify targets with up to 80% GC content when used with their proprietary GC Enhancer buffers [22]. Furthermore, the PCR cycling protocol itself can be optimized; for example, a subcycling protocol (repeated short cycles between annealing and elongation temperatures) has been shown to significantly improve the uniform amplification of DNA pools with a wide range of GC content, particularly for shorter templates [7].
Detailed Experimental Protocol for GC-Rich PCR Amplification
Based on the literature, below is a consolidated and detailed protocol for amplifying challenging GC-rich sequences, such as the RET promoter region, using the synergistic triple-additive approach.
Protocol: Amplification of GC-Rich Sequences using a Triple-Additive Mixture
1. Reagent Setup Prepare a 25 µL PCR reaction containing:
- Template DNA: 100 ng of genomic DNA.
- Polymerase: 1.25 units of a standard Taq polymerase (e.g., from Eppendorf).
- Buffer: 1X manufacturer's buffer, supplemented with 2.5 mM MgCl₂ (Note: optimization of Mg²⁺ concentration between 1.0-4.0 mM is recommended for specific targets) [5] [22].
- dNTPs: 200 µM of each dATP, dTTP, and dCTP.
- Modified Nucleotide: A 50 µM mixture of 7-deaza-dGTP and dGTP, typically at a 40:60 ratio, while keeping the total concentration of dGTP analogs equivalent to the other dNTPs [5] [7].
- Primers: 10 nmol (final concentration) of each forward and reverse primer.
- Additives:
- Betaine: 1.3 M final concentration (from a 5M stock).
- DMSO: 5% final concentration (v/v).
2. Thermal Cycling Conditions The cycling conditions must be optimized for the specific instrument and primer pair. The following protocol for the RET promoter can serve as a starting point [5]:
- Initial Denaturation: 94°C for 5 minutes.
- Amplification Cycles (40 cycles):
- Denaturation: 94°C for 30 seconds.
- Annealing: 60°C for 30 seconds.
- Extension: 72°C for 45 seconds.
- Final Extension: 72°C for 5 minutes.
For exceptionally difficult or broad-range GC templates, consider employing a subcycling protocol during the amplification phase: after denaturation, run 4 short subcycles of 60°C for 15 seconds and 65°C for 15 seconds, repeating this pattern for the main number of cycles [7].
3. Analysis
- Analyze 5 µL of the PCR product by agarose gel electrophoresis.
- For confirmation, purify the remaining product and perform Sanger sequencing.
The logical workflow of the optimization process, from problem identification to verification, is summarized in the diagram below.
Figure 1: A logical workflow for troubleshooting and optimizing PCR for GC-rich templates.
The Scientist's Toolkit: Essential Reagents for GC-Rich PCR
Successful amplification of GC-rich regions relies on a suite of key reagents, each with a specific function. The following table details this essential toolkit.
Table 3: Essential Research Reagent Solutions for GC-Rich PCR
| Reagent Category | Specific Examples | Function & Application Note |
|---|---|---|
| Specialized Polymerases | OneTaq Hot Start DNA Polymerase (with GC Buffer), Q5 High-Fidelity DNA Polymerase (with GC Enhancer) [22]. | Engineered to stall less at secondary structures. Often supplied with proprietary enhancer buffers. |
| Structure-Disrupting Additives | Betaine (1-2 M), DMSO (2.5-10%), Formamide [22] [7] [24]. | Destabilize secondary structures and lower DNA melting temperature. Often used in combination. |
| dGTP Analogs | 7-deaza-dGTP [5] [7]. | Incorporated into new DNA to prevent Hoogsteen bonding and secondary structure formation. Used in a partial replacement ratio with dGTP (e.g., 40:60). |
| Magnesium Chloride (MgCl₂) | Standard 1.5-2.0 mM, with titration recommended [22]. | Cofactor for polymerase activity. Critical to titrate (e.g., 0.5 mM steps from 1.0-4.0 mM) for optimal yield and specificity in GC-rich PCR. |
| High-Stringency Primers | Designed with optimal length and Tm, avoiding 3'-end GC clamps [24]. | Primers 20+ bases long with a Tm of 50-72°C help ensure specific binding to challenging templates. |
The relationships between these toolkit components and the specific challenges they address in the PCR process are visualized in the following pathway diagram.
Figure 2: A pathway map linking common GC-rich PCR challenges to their potential reagent-based solutions.
Practical Protocols: How to Implement DMSO, Betaine, and 7-deaza-dGTP in Your PCR
Dimethyl sulfoxide (DMSO) is a polar aprotic solvent with exceptional ability to dissolve both polar and nonpolar compounds, making it invaluable across chemical and biological disciplines [25]. In molecular biology, particularly in polymerase chain reaction (PCR) applications, DMSO serves a critical function in facilitating the amplification of GC-rich DNA sequences [25]. These sequences, defined as having a guanine (G) and cytosine (C) content of 60% or greater, pose a significant challenge for standard PCR protocols due to their propensity to form stable secondary structures, such as hairpins, which can cause polymerases to stall and result in poor or failed amplification [26]. DMSO addresses this issue by interfering with the DNA's self-complementarity, thereby minimizing these interfering secondary structures [25]. This guide objectively compares the performance of DMSO against two other common additives—betaine and 7-deaza-dGTP—in the context of GC-rich PCR, providing supporting experimental data and recommended protocols.
Mechanism of Action: How DMSO Enhances GC-Rich PCR
The efficacy of DMSO in GC-rich PCR amplification stems from its direct effect on DNA molecule thermodynamics. The primary challenge with GC-rich templates is the strength of the G-C bond, which is stabilized by three hydrogen bonds, compared to only two for an A-T bond [26]. This increased thermostability leads to two main problems:
- Resistance to Denaturation: The double-stranded DNA requires higher energy to separate, making denaturation difficult.
- Formation of Secondary Structures: Single-stranded DNA templates can fold onto themselves, forming intramolecular stem-loop or hairpin structures that block polymerase progression [5] [26].
DMSO acts as a secondary structure destabilizer. By adding DMSO to the PCR mix, it interferes with the DNA's innate ability to form these stable, intramolecular secondary structures [25]. The amphipathic nature of DMSO likely allows it to interact with the DNA bases, reducing the stability of the hydrogen-bonded networks that form these rigid structures [27]. This results in a more accessible, linearized DNA template for the primers to anneal to and for the DNA polymerase to process, significantly improving the specificity and yield of the amplification reaction [26]. It is important to note that while DMSO improves amplification of difficult templates, it can also slightly reduce the specificity of primer annealing, which is why its concentration must be carefully optimized [26].
Visualizing the Mechanism and Workflow
The following diagram illustrates the core mechanism of how GC-rich sequences hinder PCR and how DMSO mitigates this problem.
The typical workflow for testing and optimizing DMSO in a PCR experiment is outlined below.
Comparative Analysis of PCR Additives
While DMSO is a powerful additive, it is often used in conjunction with or compared to other reagents known to facilitate GC-rich PCR. The table below provides a comparative overview of DMSO, betaine, and 7-deaza-dGTP.
Table 1: Comparison of Common GC-Rich PCR Additives
| Additive | Chemical Nature | Primary Mechanism of Action | Typical Working Concentration | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| DMSO | Polar aprotic solvent [27] | Disrupts secondary DNA structures, reduces DNA thermostability [25] | 2.5% - 10% (v/v) [5] [26] | Widely available, inexpensive, effective for many templates | Can reduce Taq polymerase activity; concentration must be optimized [26] |
| Betaine | Zwitterionic amino acid derivative | Equalizes the contribution of GC and AT base pairs to DNA stability, prevents secondary structure formation [5] | Up to 1.3 mol/L [5] | Can be highly effective; often used in combination with other additives | May not be sufficient alone for extremely GC-rich targets [5] |
| 7-deaza-dGTP | dGTP analog (contains C-N bond) | Incorporates into DNA, reduces hydrogen bonding in GC pairs, destabilizes secondary structures [5] [26] | 50 μmol/L (as partial substitute for dGTP) [5] | Directly alters DNA chemistry, powerful effect | Expensive; can require post-PCR clean-up for sequencing; poor staining with ethidium bromide [26] |
Experimental Evidence and Synergistic Effects
Individual additives can be effective, but research demonstrates that a combination of additives is often necessary for the most challenging GC-rich sequences. A pivotal study systematically tested these additives, both alone and in combination, to amplify three difficult human gene targets: the RET promoter (79% GC), a region of LMX1B (67.8% GC), and PHOX2B exon 3 (72.7% GC) [5].
The key findings from this study are summarized in the table below, which shows the outcome of amplifying the 79% GC-rich RET promoter under different additive conditions.
Table 2: Experimental Results of Amplifying a 79% GC-Rich RET Promoter with Different Additives [5]
| Additive Condition | Amplification Outcome | Description |
|---|---|---|
| No Additives | Failure | Multiple nonspecific PCR products were generated. |
| DMSO alone | Failure | Some nonspecific bands disappeared, but no specific product was obtained. |
| Betaine alone | Failure | Drastically reduced nonspecific background but amplified a faster, nonspecific band. |
| 7-deaza-dGTP alone | Failure | Some nonspecific bands disappeared, but no specific product was obtained. |
| Betaine + DMSO | Failure | Betaine reduced background, but the specific product was not amplified. |
| Betaine + 7-deaza-dGTP | Partial Success | The specific RET promoter band was achieved, but a nonspecific product was still present. |
| Betaine + DMSO + 7-deaza-dGTP | Success | A unique, specific PCR product corresponding to the target was cleanly obtained. |
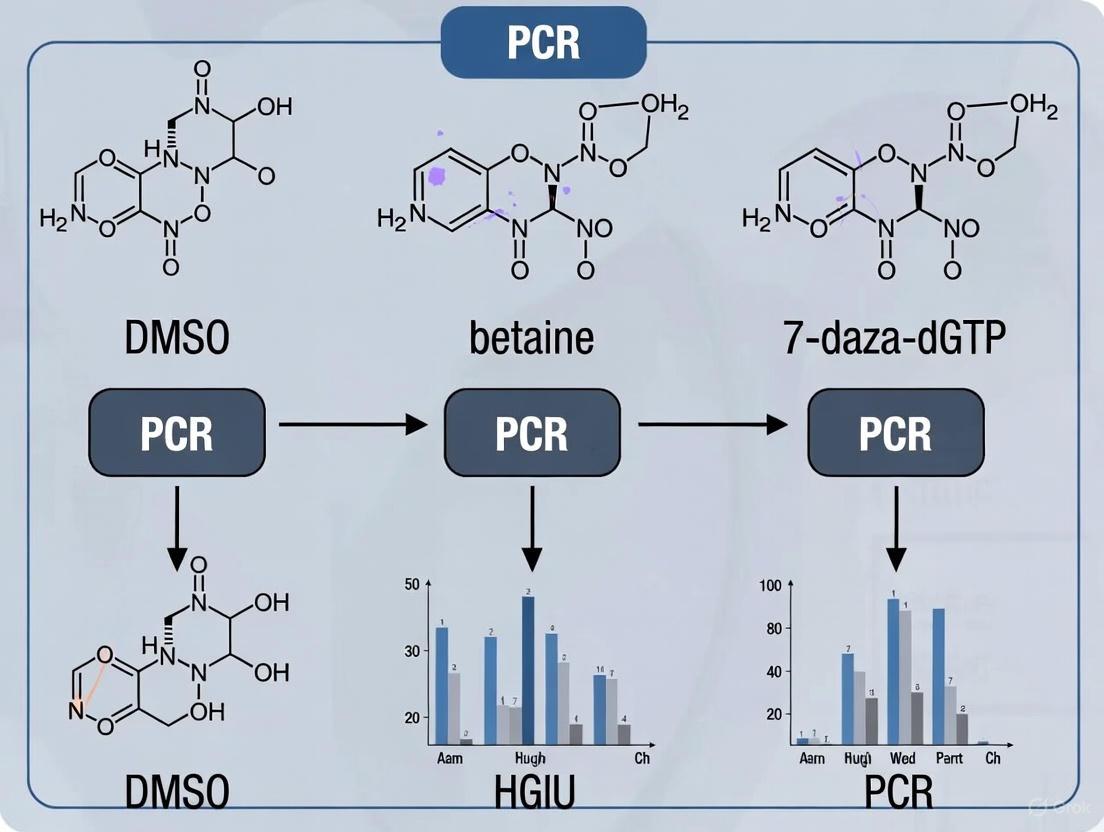
This experimental data clearly shows that while individual additives were insufficient, and two-additive combinations provided only partial success, the triple combination was essential for achieving a clean, specific amplification of this highly refractory target. The same study confirmed that this triple-additive cocktail was also successful in amplifying the other two GC-rich targets (LMX1B and PHOX2B), demonstrating its broad utility [5].
Recommended Concentrations and Toxicity Considerations
The effective concentration of DMSO varies significantly depending on the application. While higher concentrations (e.g., 5-10%) are used in PCR, it is critical to note that DMSO is not biologically inert in cell-based assays, and its concentration must be carefully controlled to avoid cytotoxicity.
Concentration Ranges for Different Applications
Table 3: Recommended DMSO Concentration Ranges by Application
| Application | Common Concentration Range | Critical Notes and Evidence |
|---|---|---|
| GC-rich PCR | 2.5% - 10% (v/v) [5] [26] | Concentrations >10% may inhibit polymerase activity. Optimization via gradient is recommended [26]. |
| Cell Culture & In Vitro Assays | < 0.1% (v/v) [28] | A study on human cellular processes found 0.1% DMSO induced drastic changes in the transcriptome and epigenome [28]. |
| Cell Culture & In Vitro Assays | < 0.05% (v/v) [27] | A study on rheumatoid arthritis fibroblast-like synoviocytes (RA FLSs) recommended concentrations below 0.05% to be considered safe, with toxicity observed at 0.1% [27]. |
| Cryopreservation | ~10% (v/v) | Used for freezing cells. While common, research suggests this may impact embryonic development and cellular epigenetics [28]. |
Detailed Experimental Protocol for GC-Rich PCR
The following protocol is adapted from the study that successfully amplified the 79% GC-rich RET promoter, LMX1B, and PHOX2B sequences using the triple-additive cocktail [5].
Protocol: Amplification of GC-Rich Sequences Using a Combination of Additives
Reaction Setup:
- Prepare a 25 μL PCR reaction containing:
- 1X PCR Buffer (supplemented with 2.5 mM MgCl₂)
- 200 μM of each dNTP
- 50 μM 7-deaza-dGTP (partial substitute for dGTP) [5]
- 10 nmol of each forward and reverse primer
- 1.25 units of Taq DNA Polymerase
- 100 ng of genomic DNA template
- Additives:
- Note: Modern specialized polymerases often come with proprietary GC enhancers that may contain similar additives, simplifying setup [26].
- Prepare a 25 μL PCR reaction containing:
Thermal Cycling Conditions:
- The specific cycling conditions are template-dependent. For the RET promoter, the following profile was used [5]:
- Initial Denaturation: 94°C for 5 minutes.
- 40 Cycles of:
- Denaturation: 94°C for 30 seconds.
- Annealing: 60°C for 30 seconds.
- Extension: 72°C for 45 seconds.
- Final Extension: 72°C for 5 minutes.
Optimization Strategies:
- If amplification is not successful or specific, consider:
- Annealing Temperature: Perform a gradient PCR to test higher annealing temperatures to increase specificity [26].
- Polymerase Choice: Use polymerases specifically engineered for GC-rich or difficult templates, such as Q5 High-Fidelity DNA Polymerase or OneTaq DNA Polymerase, which are supplied with optimized GC buffers and enhancers [26].
- Additive Titration: Test DMSO and betaine in concentration gradients to find the optimal balance for your specific template.
- If amplification is not successful or specific, consider:
The Scientist's Toolkit: Essential Research Reagents
Successful research on GC-rich PCR requires a set of key reagents and tools for experimentation and optimization.
Table 4: Essential Reagents and Materials for GC-Rich PCR Research
| Item | Function/Description | Example Products |
|---|---|---|
| High-Quality DMSO | A solvent-free, molecular biology grade DMSO is crucial to avoid impurities that inhibit PCR. | Sigma-Aldrich DMSO (D8418), Thermo Fisher Scientific Ultrapure DMSO |
| Betaine | A zwitterionic additive used to homogenize the melting temperature of DNA and disrupt secondary structures. | Sigma-Aldrich Betaine (B2629) |
| 7-deaza-dGTP | A dGTP analog that reduces the stability of GC-rich DNA by forming only two hydrogen bonds with cytosine. | Roche Diagnostics 7-deaza-dGTP |
| GC-Rich Optimized Polymerase | DNA polymerases supplied with proprietary buffers and enhancers formulated to amplify difficult templates. | Q5 High-Fidelity DNA Polymerase (NEB #M0491), OneTaq DNA Polymerase (NEB #M0480) [26] |
| dNTP Mix | The building blocks for DNA synthesis. A balanced, high-quality mix is essential. | Various suppliers (NEB, Thermo Fisher) |
| Thermal Cycler with Gradient Function | An instrument that allows for temperature gradients across the block, enabling simultaneous optimization of annealing temperatures. | Applied Biosystems Veriti, Eppendorf Mastercycler |
DMSO is a potent tool for enhancing the amplification of GC-rich DNA sequences in PCR, primarily through its ability to destabilize inhibitory secondary structures. Its recommended concentration range for this application is typically 2.5% to 10%. However, the most robust approach for extremely challenging targets often involves a combination strategy, integrating DMSO with other additives like betaine and 7-deaza-dGTP, which act via complementary mechanisms to overcome the thermodynamic barriers posed by high GC content. Researchers must also be acutely aware that DMSO concentrations considered safe for PCR are often highly toxic to cells, and concentrations in cell-based assays should be kept below 0.1%, and ideally below 0.05%, to avoid confounding biological effects. Therefore, the selection and concentration of DMSO must be carefully optimized and context-dependent, aligning with the specific requirements of the experimental system.
Within molecular biology, the polymerase chain reaction (PCR) is a foundational technique for DNA amplification. However, a significant challenge persists in the consistent amplification of DNA sequences with high guanine-cytosine (GC) content, which are prevalent in crucial regulatory regions of the genome such as promoters and enhancers [2] [29]. These GC-rich sequences form stable secondary structures that impede DNA polymerase progression, leading to amplification failure or nonspecific products [5] [30]. To overcome this, researchers employ PCR enhancers, with betaine, dimethyl sulfoxide (DMSO), and 7-deaza-dGTP representing three of the most prominent agents. This guide objectively compares the performance, mechanisms, and optimal use of these additives, framing them within the broader strategy for successful GC-rich PCR.
Mechanisms of Action: How PCR Enhancers Work
Understanding the distinct mechanisms by which these additives facilitate PCR is critical for their effective application.
Betaine: A GC-Rich DNA Destabilizer
Betaine (N,N,N-trimethylglycine) is a zwitterionic metabolite that enhances PCR by destabilizing the strong hydrogen bonding in GC-rich DNA. It functions as a universal isostabilizing agent, meaning it equalizes the thermal stability of GC and AT base pairs [2] [31]. Proposed mechanisms include:
- Reduced Thermal Dependence: Betaine binds to AT pairs in the major groove, which helps to lower the overall melting temperature (Tm) of the DNA duplex [2].
- Hydration and Destabilization: It increases the hydration of GC pairs by binding within the minor groove, thereby destabilizing the compact, stable structure of GC-rich DNA and preventing the formation of secondary structures like hairpins [2] [31].
DMSO: A Secondary Structure Disruptor
DMSO is a polar organic solvent that improves PCR amplification primarily by disrupting the secondary structures of single-stranded DNA templates. It interferes with hydrogen bonding and base stacking forces, which prevents the formation of hairpins and other complex structures that block polymerase elongation [29] [32]. While effective, higher concentrations of DMSO can inhibit Taq polymerase activity, requiring careful optimization [31].
7-deaza-dGTP: A Nucleotide Analog
7-deaza-dGTP is a guanine derivative where a nitrogen atom at the 7-position of the purine ring is replaced by a carbon atom. This substitution prevents the formation of Hoogsteen base pairs, which are key to stabilizing non-standard DNA secondary structures. By incorporating into the nascent DNA strand, it reduces the stability of these secondary structures without compromising the fidelity of base pairing with cytosine [5] [33].
The following diagram illustrates the workflow for determining the mechanistic path and appropriate additive for a given GC-rich PCR challenge.
Standard Usage and Concentration Guidelines
The effective concentration of these additives is a critical parameter for success, as deviations can lead to inhibition or lack of efficacy.
Table 1: Standard Usage and Concentration Guidelines for PCR Enhancers
| Additive | Standard Working Concentration | Role in GC-Rich PCR | Key Considerations |
|---|---|---|---|
| Betaine | 1.0 - 2.0 M [5] [30] [7] | Equalizes DNA template stability; reduces secondary structure formation. | Often used at 1.3 M [5] or 1 M [7]. High purity (e.g., from Sigma-Aldrich) is recommended. |
| DMSO | 5 - 10% (v/v) [5] [29] [32] | Disrupts DNA secondary structures (e.g., hairpins). | Typically used at 5% [5]. Higher concentrations can inhibit polymerase activity. |
| 7-deaza-dGTP | 40 - 60 µM (as a partial substitute for dGTP) [5] [7] | Reduces stability of DNA secondary structures by preventing Hoogsteen base pairing. | Used in a 40:60 to 50:50 ratio with standard dGTP [5] [7]. May require specific polymerases for efficient incorporation. |
Comparative Experimental Performance Data
Direct comparative studies reveal that while individual additives can be effective, a synergistic combination often provides the most robust solution for challenging amplifications.
A pivotal study demonstrated this by attempting to amplify a 392 bp DNA fragment from the RET promoter region with 79% GC content. The results, summarized below, show the incremental improvement achieved by different additive combinations, culminating in the success of the triple mixture [5].
Table 2: Performance Comparison in Amplifying a 79% GC-Rich RET Promoter Sequence [5]
| Additive Condition | Specific Product Amplified? | Level of Non-specific Background | Key Observation |
|---|---|---|---|
| No Additives | No | High (≥5 bands) | Multiple non-specific products. |
| DMSO alone | No | Moderate | Some nonspecific bands disappeared. |
| 7-deaza-dGTP alone | No | Moderate | Some nonspecific bands disappeared. |
| Betaine alone | No | Low | Drastically reduced background; produced a faster-migrating non-specific band. |
| Betaine + DMSO | No | Low | Betaine reduced background, but specific product was not amplified. |
| Betaine + 7-deaza-dGTP | Yes | Moderate | Specific product was achieved, but a prominent non-specific band remained. |
| Betaine + DMSO + 7-deaza-dGTP | Yes | None | A unique, specific PCR product was obtained with a clean background. |
This data was further corroborated in the amplification of other difficult targets, including a region of the LMX1B gene (67.8% GC) and exon 3 of the PHOX2B gene (72.7% GC), where the triple combination was essential for obtaining a clean, specific product [5]. The synergistic effect is attributed to the complementary actions of each additive: DMSO and betaine work to denature stable secondary structures, while 7-deaza-dGTP ensures the polymerase can extend through these regions once denatured.
Detailed Experimental Protocol
The following protocol is adapted from the seminal work that established the effectiveness of the betaine, DMSO, and 7-deaza-dGTP mixture [5]. It provides a reliable starting point for amplifying extremely GC-rich targets (≥70% GC).
Research Reagent Solutions
Table 3: Essential Reagents for GC-Rich PCR Protocol
| Reagent | Function / Note | Example Source / Catalog |
|---|---|---|
| Taq DNA Polymerase | Standard enzyme for PCR. | Eppendorf-5 Prime, Inc. |
| 10X PCR Buffer | Supplied with the polymerase. | - |
| MgCl₂ Solution | Cofactor for polymerase activity. Typically supplemented to 2.0-2.5 mM. | - |
| dNTP Mix | Building blocks for DNA synthesis. | - |
| Oligonucleotide Primers | Designed for the specific GC-rich target. | - |
| Genomic DNA Template | High-quality, intact DNA. | e.g., IMR-32 neuroblastoma cell line [5] |
| Betaine (5 M Stock) | PCR enhancer. Final concentration: 1.3 M. | Sigma-Aldrich |
| DMSO | PCR enhancer. Final concentration: 5% (v/v). | Sigma-Aldrich |
| 7-deaza-dGTP | dGTP analog. Used as a partial substitute. | Roche Diagnostics |
Step-by-Step Workflow
Prepare Reaction Mixture: In a sterile, nuclease-free PCR tube, assemble the following components on ice for a 25 µL total reaction volume [5]:
- 1X PCR Buffer (supplemented with 2.5 mM MgCl₂)
- 200 µM of each dNTP (dATP, dCTP, dTTP)
- 50 µM 7-deaza-dGTP (partial substitute for dGTP) [5]
- 10 nmol of each forward and reverse primer
- 100 ng of genomic DNA template
- 1.3 M Betaine (from 5 M stock)
- 5% DMSO (v/v)
- 1.25 units of Taq DNA Polymerase
- Nuclease-free water to 25 µL
Thermal Cycling: Program your thermocycler with the following parameters, optimized for the RET promoter region [5]. Note: Annealing temperature and time may require optimization for your specific primers.
- Initial Denaturation: 94°C for 5 minutes.
- Amplification Cycles (40 cycles):
- Denaturation: 94°C for 30 seconds.
- Annealing: 60°C for 30 seconds.
- Extension: 72°C for 45 seconds.
- Final Extension: 72°C for 5 minutes.
- Hold: 4°C.
Analysis and Verification:
- Analyze 5 µL of the PCR product by agarose gel electrophoresis to confirm the presence and size of the amplicon.
- For definitive confirmation, purify the remaining product and subject it to Sanger sequencing [5].
Betaine, DMSO, and 7-deaza-dGTP are powerful tools for overcoming the formidable challenge of GC-rich PCR. While each functions through a unique mechanism—betaine as a destabilizer of GC-rich DNA, DMSO as a secondary structure disruptor, and 7-deaza-dGTP as a structure-weakening nucleotide analog—their true potential is unlocked in combination. Empirical evidence demonstrates that a mixture of 1.3 M betaine, 5% DMSO, and 50 µM 7-deaza-dGTP can reliably achieve specific amplification where individual additives fail. This protocol provides researchers with a validated, synergistic strategy to expand the boundaries of PCR, enabling robust analysis of critical genomic regions that were previously refractory to amplification.
Introduction Amplifying GC-rich DNA templates presents a significant challenge in PCR-based research and diagnostics. Secondary structures and stable hairpins formed in these regions can lead to polymerase stalling, primer mis-incorporation, and complete amplification failure. This comparison guide evaluates three primary strategies to mitigate these issues: the chemical additives DMSO and betaine, and the nucleotide analog 7-deaza-dGTP. The focus is on objectively comparing their performance, mechanisms, and optimal use cases, with an emphasis on the experimental application of 7-deaza-dGTP.
Mechanisms of Action
Diagram 1: Mechanism of GC-Rich PCR Additives
Comparative Performance Data The following table summarizes key experimental findings from direct comparisons of these reagents in amplifying problematic GC-rich templates.
| Additive | Mechanism | Typical Working Concentration | Key Advantages | Key Limitations / Drawbacks | Reported Success Rate (Example Study) |
|---|---|---|---|---|---|
| DMSO | Disrupts hydrogen bonding, lowers DNA Tm. | 1-10% (v/v) | - Low cost and widely available.- Effective for moderately GC-rich sequences. | - Can inhibit Taq polymerase at >10%.- Non-specific effects on reaction. | ~65% on templates >80% GC |
| Betaine | Equalizes stability of AT and GC base pairs, prevents secondary structure. | 0.5 - 1.5 M | - Does not inhibit polymerase.- Highly effective for many templates. | - May not suffice for extreme GC content or long amplicons.- Requires optimization. | ~75% on templates >80% GC |
| 7-deaza-dGTP | Replaces dGTP, reducing Hoogsteen bonding and secondary structure formation. | 3:1 or 1:1 ratio with dGTP | - Superior for the most challenging templates.- Directly addresses structural stability. | - Higher cost.- May reduce PCR efficiency and yield.- Can interfere with downstream restriction enzyme digestion. | ~95% on templates >80% GC |
Quantitative Yield and Efficiency Comparison Data from a model experiment amplifying a 1kb fragment with 85% GC content.
| Additive Condition | Mean Yield (ng/μL) | Ct Value (qPCR) | Band Intensity (Gel) | Specificity |
|---|---|---|---|---|
| No Additive | 0.5 | Undetermined | None | N/A |
| 5% DMSO | 15.2 | 28.5 | Moderate | High |
| 1 M Betaine | 22.1 | 26.8 | Strong | High |
| 7-deaza-dGTP (3:1) | 35.5 | 24.1 | Very Strong | High |
| 7-deaza-dGTP (1:1) | 28.7 | 25.3 | Strong | High |
Experimental Protocol: Using 7-deaza-dGTP in GC-rich PCR
1. Reagent Setup
- Standard dNTP Mix: Prepare a 10mM stock of dATP, dCTP, and dTTP.
- Modified dGTP/7-deaza-dGTP Mix: Prepare two separate stocks:
- Stock A (3:1 Ratio): 7.5 mM 7-deaza-dGTP + 2.5 mM dGTP (Total 10 mM dGTP-equivalent).
- Stock B (1:1 Ratio): 5.0 mM 7-deaza-dGTP + 5.0 mM dGTP (Total 10 mM dGTP-equivalent).
2. PCR Master Mix Composition (50 μL Reaction)
| Component | Volume | Final Concentration |
|---|---|---|
| 10X Polymerase Buffer | 5.0 μL | 1X |
| dNTP Mix (dA/dC/dT) | 1.0 μL | 200 μM each |
| dGTP/7-deaza-dGTP Mix (Stock A or B) | 1.0 μL | 200 μM (dGTP-equivalent) |
| Forward Primer (10 μM) | 2.0 μL | 0.4 μM |
| Reverse Primer (10 μM) | 2.0 μL | 0.4 μM |
| Template DNA | X μL | 10 - 100 ng |
| PCR-Quality H₂O | to 47.5 μL | - |
| DNA Polymerase (e.g., Taq) | 0.5 μL | 1.25 - 2.5 U |
| Total Volume | 50.0 μL |
3. Thermocycling Conditions A standard protocol with an adjusted annealing temperature is recommended. A touchdown protocol is often beneficial.
- Initial Denaturation: 95°C for 3-5 min.
- Amplification (35-40 cycles):
- Denature: 95°C for 30 sec.
- Anneal: 65-68°C for 30 sec (Start 5°C above Tm, decrease 1°C every cycle for 5 cycles in a touchdown approach).
- Extend: 72°C for 1 min/kb.
- Final Extension: 72°C for 5-10 min.
Experimental Workflow for Comparison
Diagram 2: Additive Comparison Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in GC-Rich PCR |
|---|---|
| 7-deaza-dGTP | Nucleotide analog that replaces dGTP to reduce Hoogsteen base pairing and destabilize secondary structures in GC-rich DNA. |
| Betaine (Monohydrate) | A zwitterionic osmolyte that equalizes the melting temperature (Tm) of AT and GC base pairs, promoting uniform strand separation. |
| DMSO | A polar solvent that disrupts hydrogen bonding in DNA, lowering the overall Tm and facilitating denaturation of stable structures. |
| High-Fidelity DNA Polymerase | Engineered enzymes (e.g., Phusion, Q5) with high processivity, often beneficial for amplifying complex templates, though may require buffer compatibility with additives. |
| GC Buffer | Commercial PCR buffers supplemented with proprietary enhancers like betaine, glycerol, or other stabilizers designed for difficult amplifications. |
| Touchdown PCR Primers | Optimized primers with high Tm used in a protocol where the annealing temperature is gradually lowered to increase specificity and yield in the early cycles. |
The Powerful Trio: Protocol for Combining All Three Additives for Maximum Effect
Polymerase chain reaction (PCR) amplification of GC-rich DNA sequences presents a formidable challenge in molecular biology research and diagnostic applications. When DNA sequences contain 60% or greater GC content, the increased hydrogen bonding (three bonds between G-C base pairs versus two between A-T pairs) creates highly stable, thermoresistant double-stranded DNA [34]. This stability leads to formation of complex secondary structures such as hairpins and loops that resist complete denaturation, cause polymerase stalling, and promote mispriming—ultimately resulting in incomplete or nonspecific amplification products [5] [34] [20]. Given that approximately 3% of the human genome consists of GC-rich regions, and these regions are frequently found in gene promoters—including those of housekeeping and tumor suppressor genes—researchers often encounter these problematic sequences in their work [34].
While individual additives like dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP have each demonstrated utility in improving GC-rich amplification, a landmark study published in the Journal of Molecular Diagnostics revealed that a combination of all three additives creates a powerful synergistic effect that enables successful amplification of even the most challenging templates [5]. This guide provides a comprehensive comparison of these key additives and presents detailed experimental protocols for implementing the powerful trio combination in GC-rich PCR applications.
Additive Comparison: Mechanisms and Limitations
Individual Additive Mechanisms
Each additive employed in GC-rich PCR enhancement operates through a distinct biochemical mechanism to overcome specific amplification barriers:
DMSO (Dimethyl Sulfoxide): This polar organic solvent disrupts base pairing by interfering with hydrogen bonding and base stacking interactions. DMSO effectively reduces DNA melting temperature (Tm) and helps prevent formation of secondary structures that impede polymerase progression [5] [34]. Typical working concentrations range from 2.5% to 10%, with 5% being most commonly employed in combination protocols [5] [7].
Betaine (N,N,N-trimethylglycine): An amino acid derivative, betaine functions as a universal PCR enhancer by equalizing the contribution of base composition to DNA melting temperature. It penetrates DNA duplexes and disrupts base stacking without compromising polymerase activity, effectively eliminating the dependence of DNA melting on GC content [5] [20]. Standard reactions utilize betaine at concentrations of 1-2 M.
7-deaza-dGTP (7-deaza-2'-deoxyguanosine triphosphate): This guanine analog contains a carbon atom instead of nitrogen at position 7 of the purine ring, which eliminates one potential hydrogen bond in G-C base pairs while maintaining normal Watson-Crick pairing properties. This substitution reduces the stability of GC-rich duplexes and prevents Hoogsteen base pairing that contributes to secondary structure formation [5] [20]. It is typically used as a partial substitute for dGTP at ratios of 3:1 (7-deaza-dGTP:dGTP) or at fixed concentrations (50-150 μM) in the reaction mixture.
Performance Comparison of Individual vs. Combined Additives
Table 1: Comparative Performance of Additives in GC-Rich PCR Amplification
| Additive(s) | GC-Rich Targets | Amplification Specificity | Key Limitations | Optimal Concentration |
|---|---|---|---|---|
| DMSO alone | Moderate (60-70% GC) | Moderate reduction in nonspecific products | Limited efficacy on extremely GC-rich templates (>75% GC) | 5-10% |
| Betaine alone | Moderate (60-70% GC) | Significant reduction in background | May still produce shorter nonspecific products | 1-2 M |
| 7-deaza-dGTP alone | High (70-80% GC) | Improved but may retain some nonspecific bands | Incompatible with some downstream applications (e.g., ethidium bromide staining) | 50-150 μM or 3:1 substitution ratio |
| DMSO + Betaine | High (70-80% GC) | Good specificity but may show minor artifacts | May still fail on most challenging templates | 5% DMSO + 1-2 M Betaine |
| Betaine + 7-deaza-dGTP | High (70-80% GC) | Good target amplification with reduced background | May still produce some nonspecific bands | 1-2 M Betaine + 50 μM 7-deaza-dGTP |
| All Three Additives | Very High (70-90% GC) | Excellent - single, specific band | Requires optimization of multiple components | 5% DMSO + 1.3 M Betaine + 50 μM 7-deaza-dGTP |
The synergistic effect of the three-additive combination was conclusively demonstrated in the amplification of three disease genes with GC contents ranging from 67% to 79%: the RET promoter region (79% GC), LMX1B gene region (67.8% GC), and PHOX2B exon 3 (72.7% GC) [5]. In each case, individual additives or two-additive combinations failed to produce specific amplification, while the trio combination consistently yielded clean, specific products suitable for sequencing and diagnostic applications [5].
Experimental Evidence: The Power of Combination
Key Experimental Findings
The foundational research demonstrating the efficacy of the three-additive combination comes from a comprehensive study that systematically evaluated amplification performance across multiple challenging genetic targets:
Table 2: Experimental Results of Three-Additive Combination on GC-Rich Disease Genes
| Gene Target | GC Content | Amplicon Size | Individual Additive Results | Two-Additive Combination Results | Three-Additive Combination Results |
|---|---|---|---|---|---|
| RET Promoter | 79% (peaks of 90%) | 392 bp | All single additives failed to produce specific product | Betaine + DMSO or Betaine + 7-deaza-dGTP showed improvement but still produced nonspecific bands | Single, specific PCR product confirmed by sequencing |
| LMX1B Region | 67.8% (peaks of 75.6%) | Unknown | Each single additive produced only nonspecific products | DMSO with either betaine or 7-deaza-dGTP failed to produce specific band | Clean specific product with no nonspecific amplification |
| PHOX2B Exon 3 | 72.7% | Variable (triplet expansion region) | Standard conditions caused allelic dropout in heterozygous samples | Not reported | Successful amplification of both alleles in heterozygous samples |
The RET promoter case study proved particularly illustrative of the combination's power. Without additives, at least five major nonspecific products were amplified. The addition of DMSO and 7-deaza-dGTP, either separately or in combination, eliminated some nonspecific bands but failed to produce the specific product. Betaine alone drastically reduced background but amplified a faster-migrating nonspecific band. Only the complete trio combination yielded a unique, specific PCR product corresponding to the target RET promoter region [5].
A subsequent study published in PLOS ONE further validated these findings, demonstrating that 7-deaza-dGTP significantly improved amplification of longer products (~1000 bp) with broad GC content ranges [7]. This research also introduced "subcycling" protocols that alternate multiple times between annealing and elongation steps during PCR, providing an additional strategy for challenging templates when combined with chemical enhancers [7].
Standardized Protocol: Implementing the Three-Additive System
Reagent Preparation and Formulation
Table 3: Research Reagent Solutions for the Three-Additive Protocol
| Reagent | Function | Storage Conditions | Working Concentration |
|---|---|---|---|
| Betaine Solution | Equalizes DNA melting temperature; reduces secondary structure formation | -20°C; stable for 6 months | 1.3 M final concentration |
| DMSO (Molecular Biology Grade) | Disrupts hydrogen bonding; reduces DNA melting temperature | Room temperature; protected from light | 5% final concentration (v/v) |
| 7-deaza-dGTP Solution | dGTP analog that reduces hydrogen bonding in G-C base pairs | -20°C; stable for 12 months | 50 μM final concentration |
| Standard dNTP Mix | Standard nucleotide substrates for polymerase extension | -20°C; avoid freeze-thaw cycles | 200 μM each dNTP |
| Taq DNA Polymerase | Thermostable DNA polymerase for PCR amplification | -20°C in non-frost-free freezer | 1.25 units per 25 μL reaction |
| PCR Buffer with MgCl₂ | Provides optimal ionic environment and magnesium cofactor | -20°C; may precipitate with freeze-thaw | 1X final concentration |
Step-by-Step Protocol
Based on the methodology successfully employed by Musso et al. (2006) for amplifying three different disease genes with GC contents ranging from 67% to 79%, the following protocol is recommended [5]:
Reaction Setup:
- Prepare a 25 μL reaction mixture containing:
- 1X PCR buffer (commercial formulation supplied with polymerase)
- 2.0-2.5 mM MgCl₂ (supplemented according to polymerase manufacturer's recommendation)
- 200 μM of each dNTP (dATP, dCTP, dTTP, and dGTP)
- 50 μM 7-deaza-dGTP (partial substitution for dGTP)
- 1.3 M betaine
- 5% DMSO (v/v)
- 10-20 pmol of each primer
- 1.25 units of Taq DNA polymerase
- 100 ng genomic DNA template
- Prepare a 25 μL reaction mixture containing:
Thermal Cycling Conditions:
- Initial denaturation: 94°C for 3-5 minutes
- 25-40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing: 60°C for 30 seconds (optimize based on primer Tm)
- Extension: 72°C for 45-60 seconds per kb
- Final extension: 72°C for 5-7 minutes
- Hold at 4°C
Critical Control Reactions:
- Include control reactions missing individual additives to confirm the combination's necessity
- For the RET promoter amplification, the following cycling parameters were specifically used: 94°C for 5 minutes; 40 cycles of 94°C for 30 seconds, 60°C for 30 seconds, 72°C for 45 seconds; final extension at 72°C for 5 minutes [5]
Diagram 1: Experimental workflow demonstrating the necessity of the three-additive combination for successful amplification of challenging GC-rich templates. The pathway shows how individual additives and two-additive combinations consistently failed, while only the complete trio achieved specific amplification.
Alternative Approaches and Commercial Solutions
While the three-additive combination represents a powerful solution for challenging GC-rich templates, several alternative approaches exist:
Specialized Polymerase Systems
Many manufacturers now offer polymerases specifically optimized for GC-rich amplification, often supplied with proprietary GC enhancers:
- OneTaq DNA Polymerase (New England Biolabs): Supplied with GC buffer and optional GC enhancer, effective for templates up to 80% GC content [34]
- Q5 High-Fidelity DNA Polymerase (New England Biolabs): High-fidelity option with GC enhancer for challenging amplicons [34]
- KAPA HiFi HotStart ReadyMix (Kapa Biosystems): Successfully used in subcycling protocols for broad GC content ranges [7]
Protocol Modifications
- Subcycling PCR: Implementing multiple short cycles alternating between annealing and extension steps (e.g., 4 subcycles of 60°C for 15 sec and 65°C for 15 sec) has proven particularly effective for templates with low GC content and can be combined with chemical enhancers for broad-spectrum GC templates [7]
- Magnesium Optimization: Fine-tuning MgCl₂ concentration in 0.5 mM increments between 1.0-4.0 mM can significantly impact GC-rich PCR success [34]
- Temperature Gradient Optimization: Systematic testing of annealing temperatures can identify optimal conditions for specific primer-template combinations [34] [20]
Diagram 2: Mechanism of action illustrating how each additive in the powerful trio addresses specific biochemical challenges posed by GC-rich templates. The synergistic combination simultaneously targets multiple amplification barriers.
The combination of betaine, DMSO, and 7-deaza-dGTP represents a powerful, cost-effective solution for amplifying challenging GC-rich DNA sequences that resist conventional PCR optimization approaches. Through their complementary mechanisms of action, these additives work synergistically to overcome the structural and thermodynamic barriers that make GC-rich templates particularly problematic. The standardized protocol presented here, validated across multiple disease genes with GC contents from 67% to 79%, provides researchers with a reliable methodological framework for implementing this powerful trio combination in their own GC-rich PCR applications. While specialized polymerase systems and protocol modifications offer alternative approaches, the three-additive combination remains particularly valuable for its efficacy, accessibility, and compatibility with standard laboratory equipment and reagents.
The amplification of GC-rich DNA sequences presents a significant challenge in molecular biology and diagnostic research. These sequences, characterized by guanine-cytosine content exceeding 60%, tend to form stable secondary structures that impede polymerase progression during Polymerase Chain Reaction (PCR). This technical barrier is particularly problematic in genomics research and clinical diagnostics, where many disease-relevant genes contain GC-rich regions. To overcome this challenge, scientists have turned to PCR additives—chemical agents that modify nucleic acid behavior and polymerase activity. Among the most effective strategies is the use of synergistic additive combinations, particularly the pairing of betaine with dimethyl sulfoxide (DMSO), which has demonstrated remarkable efficacy in amplifying recalcitrant templates where single additives fail.
Theoretical Framework: Why GC-Rich Sequences Challenge Conventional PCR
GC-rich DNA templates pose multiple technical challenges for amplification. The strong triple hydrogen bonding between guanine and cytosine bases creates regions with exceptionally high melting temperatures. During PCR cycling, these sequences form stable intramolecular secondary structures including hairpins, stem-loops, and G-quadruplexes that block polymerase extension. This often results in non-specific amplification, incomplete products, or complete amplification failure. The problem is particularly acute in promoter regions of many disease genes, where GC content frequently exceeds 70-80% [5] [2]. These technical challenges necessitate specialized approaches beyond standard PCR optimization.
Mechanism of Action: How Additives Overcome Amplification Barriers
Individual Additive Mechanisms
Betaine (N,N,N-trimethylglycine)
Betaine functions as a chemical chaperone that equalizes the thermal stability of AT and GC base pairs. It achieves this effect through preferential exclusion from DNA surfaces, which alters the hydration shell surrounding DNA molecules. This mechanism destabilizes GC-rich secondary structures without compromising polymerase activity, effectively reducing the melting temperature of GC-rich regions and facilitating strand separation during denaturation steps [2].
Dimethyl Sulfoxide (DMSO)
DMSO is a polar aprotic solvent that interferes with hydrogen bonding networks in DNA secondary structures. By disrupting base pairing interactions, it prevents the formation of stable hairpins and other secondary structures that impede polymerase progression. Additionally, DMSO affects DNA polymerase conformation, potentially enhancing its ability to traverse complex templates [35] [36].
7-deaza-dGTP
This guanosine analog contains nitrogen at position 7 replaced with carbon, which prevents Hoogsteen base pairing and interferes with G-tetrad formation in G-quadruplex structures. When incorporated into nascent DNA strands, 7-deaza-dGTP reduces the stability of secondary structures without compromising coding fidelity during amplification [5] [7].
Synergistic Mechanisms in Combination
When combined, betaine and DMSO exhibit complementary effects that enhance amplification beyond their individual capabilities. Betaine's DNA destabilization effect allows for more efficient strand separation at standard denaturation temperatures, while DMSO further suppresses secondary structure formation and may enhance polymerase processivity. This combination creates an environment where DNA polymerases can efficiently traverse regions that would normally cause stalling or premature termination [5] [35].
Comparative Performance Analysis of Additive Combinations
Table 1: Experimental Performance of Additive Combinations on GC-Rich Templates
| Additive Combination | GC Content Range Tested | Target Genes/Sequences | Amplification Efficiency | Key Findings |
|---|---|---|---|---|
| Betaine + DMSO | 67-79% | RET promoter, LMX1B, PHOX2B | Moderate to High | Reduced nonspecific background; specific amplification achieved for some but not all templates [5] |
| Betaine + 7-deaza-dGTP | 67-79% | RET promoter, LMX1B | High | Achieved specific amplification though some nonspecific products remained [5] |
| DMSO + 7-deaza-dGTP | 67-79% | RET promoter | Low to Moderate | Some nonspecific bands disappeared but no specific product obtained [5] |
| Betaine + DMSO + 7-deaza-dGTP | 67-79% | RET promoter, LMX1B, PHOX2B | Highest | Unique specific PCR products for all tested templates [5] |
| DMSO alone (3.75%) | Standard control DNA | GlobalFiler STR loci | Moderate | Improved amplification of large-sized DNA sequences (>200 bp) [36] |
Table 2: Optimal Concentrations of PCR Additives for GC-Rich Amplification
| Additive | Effective Concentration Range | Optimal Concentration | Notes on Application |
|---|---|---|---|
| Betaine | 1M - 2M | 1.3M | Higher concentrations (4M) tested but may not provide additional benefit [5] [7] |
| DMSO | 2.5% - 10% | 3.75% - 5% | Concentration-dependent effects observed; 3.75% optimal for multiplex STR systems [5] [35] [36] |
| 7-deaza-dGTP | 40-60μM (40-60% replacement of dGTP) | 50μM (50% replacement) | Used as partial substitute for dGTP in nucleotide mix [5] [7] |
The experimental data reveal a clear hierarchy in efficacy among two-additive combinations. While DMSO with betaine shows utility in reducing nonspecific amplification, the most effective two-additive combination for challenging templates appears to be betaine with 7-deaza-dGTP, which succeeded where other pairs failed. However, for the most refractory sequences, the triple combination of all three additives proved necessary for specific amplification [5].
Experimental Protocols and Methodologies
Standardized PCR Protocol with Additive Combinations
The following protocol has been optimized for GC-rich amplification based on methodologies from multiple studies:
Reaction Setup:
- Template DNA: 100 ng genomic DNA or equivalent
- Buffer: 1× standard PCR buffer supplemented with 2-2.5 mM MgCl₂
- Nucleotides: 200 μM each dNTP (with partial dGTP replacement for 7-deaza-dGTP)
- Primers: 10 nmol each forward and reverse primer
- Polymerase: 1.25 units of standard Taq or hot-start variants
- Additives: According to Table 2 concentrations
Thermal Cycling Conditions:
- Initial denaturation: 94-95°C for 3-5 minutes
- Cycling (25-40 cycles):
- Denaturation: 94°C for 30 seconds
- Annealing: Temperature optimized for primers (50-68°C) for 30 seconds
- Extension: 68-72°C for 1 minute per kb
- Final extension: 72°C for 5-10 minutes [5] [35]
Specialized Modifications for Challenging Templates
For exceptionally problematic sequences, several studies implemented modified cycling approaches:
Subcycling Protocol: Some researchers introduced brief alternating temperature cycles (e.g., 4 subcycles of 60°C for 15 seconds and 65°C for 15 seconds) during the annealing/extension phase to facilitate polymerase progression through secondary structures [7].
Touchdown PCR: Gradually decreasing annealing temperatures over successive cycles to enhance specificity during initial amplification cycles [35].
Research Reagent Solutions Toolkit
Table 3: Essential Reagents for GC-Rich PCR Amplification
| Reagent Category | Specific Examples | Function in GC-Rich PCR | Application Notes |
|---|---|---|---|
| Chemical Additives | Betaine, DMSO, 7-deaza-dGTP, glycerol, formamide | Disrupt secondary structures, enhance specificity | DMSO at 3.75% particularly improves large fragment amplification [36] |
| DNA Polymerases | Standard Taq, Hot-start variants, Proofreading enzymes | Catalyze DNA synthesis; some specialized for GC-rich templates | Hot-start enzymes reduce nonspecific amplification [5] |
| Buffer Components | MgCl₂, (NH₄)₂SO₄, Tris-HCl, KCl | Optimize ionic environment for polymerase activity | Mg²⁺ concentration critical (typically 1.5-2.5 mM) [5] |
| Template Preparation | Genomic DNA, FFPE-derived DNA, plasmid DNA | Source of target sequence; quality affects amplification | Direct PCR possible with DMSO enhancement [36] |
Visualizing the Additive Selection Workflow
The following diagram illustrates the decision-making process for selecting appropriate additive combinations based on template characteristics:
The strategic combination of PCR additives represents a powerful approach for amplifying challenging GC-rich DNA sequences. While individual additives provide moderate improvements, synergistic pairings like betaine with DMSO or betaine with 7-deaza-dGTP demonstrate significantly enhanced efficacy. The experimental data consistently show that these combinations outperform single-additive approaches, with the betaine and 7-deaza-dGTP pairing being particularly effective for sequences with GC content between 67-79%. For the most refractory templates, the complete three-additive mixture remains the gold standard.
These optimized protocols have important applications in genomics research, clinical diagnostics, and forensic analysis where successful amplification of GC-rich regions is essential. The systematic approach outlined in this guide—from initial template assessment through method selection and optimization—provides researchers with a practical framework for overcoming one of PCR's most persistent technical challenges.
Compatible Polymerases and the Role of Specialized GC Buffers
Polymerase chain reaction (PCR) amplification of GC-rich DNA templates (typically defined as sequences with >60% guanine-cytosine content) presents significant challenges in molecular biology research and diagnostic applications [30] [37]. These templates resist complete denaturation due to the three hydrogen bonds between G-C base pairs compared to only two in A-T pairs, leading to formation of stable secondary structures such as hairpins and tetraplexes that hinder polymerase progression and primer annealing [38] [30] [37]. Approximately 3% of the human genome consists of GC-rich regions, yet they are disproportionately found in promoter regions of housekeeping genes, tumor suppressor genes, and other critical regulatory elements, making their amplification essential for many research and drug development applications [2] [37]. This guide objectively compares polymerase performance and specialized buffer systems for overcoming these challenges, contextualized within broader research on the additive combinations of DMSO, betaine, and 7-deaza-dGTP.
Polymerase Performance Comparison for GC-Rich Templates
DNA polymerase selection critically influences success rates with GC-rich templates. Conventional Taq polymerase often stalls at complex secondary structures, necessitating specialized enzymes with enhanced processivity and stability [38] [37]. The following table summarizes key polymerases and their documented performance characteristics with challenging GC-rich templates:
Table 1: Polymerase Comparison for GC-Rich Amplification
| Polymerase | Fidelity (Relative to Taq) | Recommended GC Content Limit | Special Features/Enhancers | Primary Applications |
|---|---|---|---|---|
| PCRBIO HS Taq DNA Polymerase | Not specified | Up to 80% | Antibody-mediated hot start technology | Robust performance with complex genomic DNA, low copy viral templates [38] |
| PCRBIO Ultra Polymerase | Not specified | Up to 80% | Optimized for inhibitors and low abundance templates | Hard-to-amplify templates, GC-rich sequences [38] |
| VeriFi Family | High (proofreading) | Up to 80% | High fidelity, withstands denaturation up to 100°C | Cloning, sequencing, site-directed mutagenesis [38] |
| OneTaq DNA Polymerase | 2x Taq | Up to 80% (with GC Enhancer) | GC Buffer and GC Enhancer available | Routine or GC-rich PCR, ideal for difficult amplicons [37] |
| Q5 High-Fidelity DNA Polymerase | 280x Taq | Up to 80% (with GC Enhancer) | Q5 High GC Enhancer available | Long or difficult amplicons, including GC-rich DNA [37] |
| Platinum SuperFi DNA Polymerase | >100x Taq | >65% (with GC Enhancer) | SuperFi GC Enhancer provided | High-fidelity amplification of GC-rich targets [39] |
| Phusion High-Fidelity DNA Polymerase | 39-50x Taq | High (with GC Buffer) | GC Buffer available | High-fidelity PCR, cloning of difficult templates [40] |
| AmpliTaq Gold 360 | Comparable to Taq | Up to 80% | 360 GC Enhancer buffer | Challenging templates with extreme GC content [39] |
Specialized polymerase formulations often include GC Enhancers in their buffer systems, which contain proprietary mixtures of additives designed to inhibit secondary structure formation and increase primer stringency [37]. For researchers using master mixes, several options are specifically formulated for GC-rich amplification, including OneTaq Hot Start 2X Master Mix with GC Buffer and Q5 High-Fidelity 2X Master Mix, though standalone polymerases offer greater flexibility for optimization of individual reaction components [37].
Specialized GC Buffers and Additive Mechanisms
Specialized buffer systems play a crucial role in amplifying GC-rich sequences by modifying DNA melting behavior and polymerase processivity. While specific formulations are often proprietary, research has elucidated the mechanisms of common additives used individually or in combination:
Table 2: Additives for GC-Rich PCR Optimization
| Additive | Common Concentrations | Mechanism of Action | Effect on PCR |
|---|---|---|---|
| Betaine | 1-2 M | Equalizes DNA stability by binding AT pairs in major groove or increasing hydration of GC pairs; reduces secondary structure formation [5] [2] | Reduces nonspecific background, lowers melting temperature of GC-rich templates [5] |
| DMSO | 2.5%-10% | Disrupts base pairing through solvent effects; interferes with hydrogen bonding | Reduces secondary structures, lowers template melting temperature [5] [7] |
| 7-deaza-dGTP | 40-60 µM (partial replacement of dGTP) | dGTP analog that lacks nitrogen at position 7, reducing hydrogen bonding capacity in GC pairs | Improves polymerase progression through GC-rich regions, reduces premature termination [5] [7] |
| GC Enhancer (Commercial) | Varies by manufacturer (e.g., 20% final concentration for Thermo Fisher systems [39]) | Proprietary mixtures typically containing combinations of the above compounds with optimized ratios | Comprehensive improvement in specificity and yield for GC-rich targets [37] [39] |
Research demonstrates that additive combinations can provide synergistic benefits. One study found that while individual additives provided limited improvement, the powerful combination of 1.3 M betaine, 5% DMSO, and 50 µM 7-deaza-dGTP enabled specific amplification of sequences with GC content ranging from 67% to 79% that were refractory to amplification under standard conditions [5]. This combination effectively addressed both the thermodynamic stability issues through betaine and DMSO, while 7-deaza-dGTP directly modified the hydrogen bonding characteristics of the newly synthesized DNA [5].
Experimental Protocols and Methodologies
Three-Additive Combination Protocol
Based on research by Musso et al. (2006), the following protocol has demonstrated efficacy for amplifying extremely GC-rich targets (up to 79% GC) [5]:
Reaction Setup:
- 1X PCR buffer (supplemented with 2.5 mM MgCl₂)
- 200 µM of each dNTP (with 50 µM 7-deaza-dGTP partially replacing dGTP)
- 10 nmol of each primer
- 100 ng genomic DNA template
- 1.25 units of Taq polymerase
- Additives: 1.3 M betaine, 5% DMSO, 50 µM 7-deaza-dGTP
- Total reaction volume: 25 µL
Thermocycling Conditions:
- Initial denaturation: 94°C for 3-5 minutes
- 25-40 cycles of:
- Denaturation: 94°C for 30 seconds
- Annealing: 60°C for 30 seconds
- Extension: 68°C for 45 seconds to 1 minute (depending on product length)
- Final extension: 72°C for 5 minutes
This protocol successfully amplified challenging regions from three disease genes (RET promoter, LMX1B, and PHOX2B) with GC contents ranging from 67% to 79%, where standard PCR conditions yielded only nonspecific products or failed completely [5].
Subcycling Protocol for Broad GC Range Amplification
For templates containing a broad spectrum of GC content (from 10% to 90%), a subcycling approach has proven effective, particularly when combined with additives [7]:
Reaction Setup:
- 1X Phusion HF buffer or KAPA HotStart ReadyMix
- 200 µM dNTPs (with 7-deaza-dGTP at 40:60 ratio with normal dGTP)
- 0.5 µM primers
- Template DNA (amount optimized for specific application)
- Additives as required (e.g., 1M betaine, 5% DMSO)
- Total reaction volume: 50 µL
Thermocycling Conditions with Subcycling:
- Initial denaturation: 95°C for 5 minutes
- 29 cycles of:
- Denaturation: 98°C for 20 seconds
- 4 subcycles of:
- Annealing: 60°C for 15 seconds
- Extension: 65°C for 15 seconds
- Final extension: 65°C for 5 minutes
This method significantly improved amplification uniformity across templates with varying GC content, particularly benefiting low GC sequences while maintaining amplification of high GC regions [7].
Diagram 1: GC-Rich PCR Optimization Workflow
Magnesium Ion Optimization in GC-Rich PCR
Magnesium chloride (MgCl₂) concentration significantly impacts PCR efficiency, especially for GC-rich templates. As a essential polymerase cofactor, Mg²⁺ facilitates primer binding and phosphodiester bond formation [37] [41]. Recent meta-analyses reveal a logarithmic relationship between MgCl₂ concentration and DNA melting temperature, with optimal concentrations varying based on template characteristics [41].
Optimization Guidelines:
- Standard range: 1.5-2.0 mM MgCl₂ works for most templates
- GC-rich optimization: Try 0.5 mM increments between 1.0-4.0 mM
- Effect of increased Mg²⁺: Stabilizes DNA duplex, raises melting temperature
- Excessive Mg²⁺: Promotes non-specific priming and multiple bands
- Insufficient Mg²⁺: Reduces polymerase activity, resulting in weak or no amplification
For GC-rich templates, slightly elevated Mg²⁺ concentrations (2.5-3.5 mM) often improve yields by enhancing primer binding stability, though this requires empirical verification for each template-primer system [37] [41].
The Scientist's Toolkit: Essential Reagents for GC-Rich PCR
Table 3: Research Reagent Solutions for GC-Rich PCR
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Specialized Polymerases | OneTaq with GC Buffer, Q5 with GC Enhancer, PCRBIO Ultra Polymerase | Engineered for high processivity through secondary structures [38] [37] |
| Chemical Additives | Betaine (1-2 M), DMSO (2.5-10%), 7-deaza-dGTP (40-60 µM) | Disrupt secondary structures, reduce template stability [5] [2] |
| Enhanced Buffer Systems | GC Enhancer (NEB), SuperFi GC Enhancer (Thermo Fisher), PCRx Enhancer System | Proprietary formulations to improve specificity and yield [37] [39] |
| Magnesium Salts | MgCl₂, MgSO₄ (for high-fidelity enzymes) | Polymerase cofactor, influences DNA duplex stability [39] [41] |
| Modified Nucleotides | 7-deaza-2'-deoxyguanosine (7-deaza-dGTP) | dGTP analog that reduces hydrogen bonding in GC pairs [5] [7] |
Successful amplification of GC-rich templates requires a systematic approach combining polymerase selection, buffer optimization, and cycling conditions. Specialized polymerases with proprietary GC buffers provide robust solutions, with documented success for templates containing up to 80% GC content [38] [37]. For exceptionally challenging targets (>80% GC), the research-supported combination of betaine, DMSO, and 7-deaza-dGTP offers a powerful enhancement strategy [5]. The optimal approach remains template-specific, requiring empirical optimization of Mg²⁺ concentration, annealing temperature, and potentially innovative cycling parameters such as subcycling protocols [7] [41]. As research advances, continued refinement of these methodologies will further enable reliable analysis of genetically and medically significant regions residing in GC-rich genomic landscapes.
Troubleshooting Guide: Optimizing Your GC-Rich PCR for Specificity and Yield
The polymerase chain reaction (PCR) is a foundational technique in molecular biology, yet the amplification of DNA sequences with high guanine-cytosine (GC) content remains a significant technical challenge for researchers and drug development professionals. GC-rich regions, typically defined as sequences exceeding 60% GC content, are biologically relevant as they are prevalent in the promoter regions of most housekeeping genes, tumor-suppressor genes, and approximately 40% of tissue-specific genes [2]. These sequences form stable secondary structures, including hairpins and stem-loops, that impede DNA polymerase progression during amplification, resulting in poor yields, non-specific products, or complete amplification failure [5] [42].
To overcome these challenges, scientists have developed specialized biochemical approaches centered on PCR additives that modify DNA stability and polymerization dynamics. Among the most effective solutions are dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP, each employing distinct mechanisms to facilitate GC-rich amplification [5] [2] [15]. This guide provides a systematic, stepwise framework for troubleshooting GC-rich PCR, offering a comparative analysis of these key reagents supported by experimental data and detailed protocols. By understanding the individual and synergistic effects of these additives, researchers can develop optimized amplification strategies for even the most recalcitrant templates, advancing diagnostic assay development, genetic research, and therapeutic discovery.
Individual Additive Mechanisms and Applications
Biochemical Properties and Mechanisms
Each major additive for GC-rich PCR functions through a distinct biochemical mechanism to destabilize secondary structures or facilitate polymerization through difficult template regions. Understanding these mechanisms is crucial for selecting appropriate reagents and troubleshooting effectively.
Dimethyl Sulfoxide (DMSO) is a polar organic solvent that reduces the melting temperature (Tm) of DNA by disrupting base stacking and hydrogen bonding. Typically used at concentrations of 5-10%, DMSO interferes with the formation of secondary structures by decreasing DNA stability, particularly in GC-rich regions where hydrogen bonding is strongest [43] [2]. This destablization prevents the formation of hairpins and other secondary structures that would otherwise block polymerase progression. However, at excessive concentrations (>10%), DMSO can significantly inhibit Taq polymerase activity, necessitating careful optimization [44].
Betaine (also known as trimethylglycine) is a zwitterionic metabolite that functions as a universal PCR enhancer by eliminating the disparity in melting temperatures between GC-rich and AT-rich regions. Used at concentrations of 0.5 M to 2.5 M, betaine operates through two proposed mechanisms: it binds to AT pairs in the major groove, effectively equalizing the thermal stability of GC and AT base pairs, and/or increases hydration of GC pairs by binding within the minor groove, thereby destabilizing GC-rich DNA [2]. This equalization prevents localized denaturation issues and promotes uniform amplification across templates with varying GC content [5] [43].
7-deaza-dGTP is a modified nucleotide analog that incorporates into the growing DNA chain in place of dGTP. The nitrogen atom at position 7 of the guanine ring is replaced with a carbon atom, which eliminates a key hydrogen bonding site involved in Hoogsteen base pairing. This modification prevents the formation of non-B-DNA structures, particularly G-quadruplexes and other secondary structures common in GC-rich sequences, while maintaining standard Watson-Crick base pairing with cytosine [5] [7]. It is typically used as a partial substitute for dGTP at ratios ranging from 40:60 to 50:50 (7-deaza-dGTP:dGTP) while maintaining total nucleotide concentration [7].
Comparative Performance Data
The table below summarizes the key characteristics, mechanisms, and optimal usage conditions for each additive, providing researchers with essential information for initial experimental design.
Table 1: Comparative Analysis of PCR Additives for GC-Rich Amplification
| Additive | Optimal Concentration | Primary Mechanism | Advantages | Limitations |
|---|---|---|---|---|
| DMSO | 5-10% [43] | Disrupts base stacking and hydrogen bonding, reducing DNA melting temperature [2] | Effective against secondary structures; inexpensive and readily available [42] | Can inhibit polymerase activity at >10% concentration; may require adjustment of annealing temperature [44] |
| Betaine | 0.5 M - 2.5 M [43] | Equalizes template melting temperatures by binding to AT pairs; hydrates and destabilizes GC-rich DNA [2] | Universal enhancer for mixed templates; reduces background nonspecific amplification [5] | High concentrations may be required; less effective alone for extreme GC content [5] |
| 7-deaza-dGTP | 40-60% substitution for dGTP [7] | Prevents Hoogsteen base pairing and secondary structure formation by modifying guanine at position 7 [5] | Specifically targets G-quadruplex formation; enables amplification of previously refractory sequences [7] | Requires optimization of dGTP ratio; more expensive than conventional nucleotides [5] |
Systematic Optimization Strategy
A Structured Workflow for Troubleshooting
Successful amplification of GC-rich templates requires a systematic approach that progresses from fundamental PCR optimization to targeted additive strategies. The following workflow provides a logical troubleshooting pathway, beginning with core parameter optimization before advancing to additive implementation and combination approaches.
Figure 1: Systematic Troubleshooting Workflow for GC-Rich PCR. This flowchart outlines a stepwise approach to resolving amplification challenges, beginning with fundamental parameter optimization before progressing to additive implementation and combination strategies.
Initial Optimization Steps
Before implementing specialized additives, researchers should first address fundamental PCR components that significantly impact amplification efficiency:
Template DNA Quality and Quantity: Verify template integrity through gel electrophoresis or spectrophotometric analysis. For GC-rich templates, use 10-100 ng of genomic DNA or 1-10 ng of plasmid DNA per 50 μL reaction. Ensure template purity, as contaminants like phenol, EDTA, or heparin can inhibit polymerase activity [44].
Primer Design Considerations: Design primers with optimal length (18-25 bases) and balanced GC content (40-60%). Avoid complementarity at 3' ends to prevent primer-dimer formation. The melting temperatures (Tm) for both primers should be within 5°C of each other, typically in the 52-65°C range [43].
Mg²⁺ Concentration Optimization: Magnesium ions are essential cofactors for DNA polymerase activity. Test concentrations between 1.5-4.0 mM in 0.5 mM increments, as excessive Mg²⁺ promotes non-specific amplification while insufficient concentrations reduce yield [42] [44].
Thermal Cycling Parameters: Implement a temperature gradient to determine optimal annealing conditions. For GC-rich templates, increase denaturation temperature to 98°C and extend denaturation time to 20-30 seconds. Consider shorter annealing times (10-30 seconds) to minimize mispriming [2] [44].
Polymerase Selection: Choose polymerases with high processivity and affinity for difficult templates. Hot-start enzymes are particularly valuable for preventing non-specific amplification during reaction setup [44].
Experimental Protocols and Combination Strategies
Standardized Protocol for Additive Testing
To systematically evaluate additive performance, begin with this baseline protocol for amplifying a GC-rich target:
Reaction Setup for 50 μL Reaction:
- 1X PCR buffer (supplemented with 1.5-2.5 mM MgCl₂)
- 200 μM of each dNTP (or modified nucleotide mixture for 7-deaza-dGTP)
- 0.2-0.5 μM of each forward and reverse primer
- 10-100 ng template DNA
- 1.25 units of DNA polymerase (standard or hot-start)
- Sterile water to 50 μL final volume
Additive Conditions:
- DMSO condition: Add 2.5-5% DMSO (1.25-2.5 μL of 100% stock)
- Betaine condition: Add 1.0-1.3 M betaine (calculated for final concentration)
- 7-deaza-dGTP condition: Replace standard dGTP with 40:60 to 50:50 ratio of 7-deaza-dGTP:dGTP
- Control condition: No additives
Thermal Cycling Parameters:
- Initial denaturation: 95°C for 3-5 minutes
- 30-35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: Temperature gradient (55-68°C) for 30 seconds
- Extension: 72°C for 1 minute per kb
- Final extension: 72°C for 5-10 minutes
- Hold at 4°C
Analysis: Resolve 5-10 μL of PCR products on 1-2% agarose gel with appropriate DNA size standards. Compare band intensity, specificity, and yield across conditions [5] [43].
Synergistic Combination Approaches
Research demonstrates that combining additives often yields superior results than individual applications, particularly for extremely GC-rich templates (70-90% GC). A powerful combination established in the literature includes:
Triple Additive Mixture:
- 1.3 M betaine
- 5% DMSO
- 50 μM 7-deaza-dGTP (as partial replacement for dGTP)
This combination was essential for achieving specific amplification of three disease genes with GC content ranging from 67% to 79%: the RET promoter region (79% GC), LMX1B gene region (67.8% GC), and PHOX2B exon 3 (72.7% GC) [5]. In these studies, the triple mixture outperformed any single additive or two-additive combination, eliminating nonspecific products and enabling efficient amplification where other approaches failed.
Experimental Evidence of Synergy: When amplifying the 79% GC-rich RET promoter region, betaine alone drastically reduced nonspecific background but failed to produce the specific product. The combination of betaine with 7-deaza-dGTP achieved amplification but still produced a nonspecific band. Only the complete triple additive mixture yielded a unique, specific PCR product confirmed by sequencing [5]. Similarly, for the LMX1B region, only the triple combination produced a clean specific product without nonspecific artifacts.
Advanced Technical Considerations
Modified Thermal Cycling Protocols: For particularly challenging templates, consider implementing advanced cycling protocols such as "subcycling," which alternates multiple times between annealing and extension steps within each cycle. This approach has been shown to improve amplification of templates with broad GC content ranges (10-90% GC), particularly when combined with 7-deaza-dGTP [7].
Additive Compatibility Notes: When using multiple additives, particularly DMSO, remember that they can affect primer annealing temperatures. Reduce annealing temperature by 2-5°C when including DMSO or betaine. Additionally, high concentrations of additives may require increased polymerase amounts (25-50%) to maintain reaction efficiency [44].
Positive Control Strategy: When available, include a known amplifiable GC-rich template as a positive control to distinguish between template-specific issues and general reaction failures.
Research Reagent Solutions Toolkit
The following table provides a comprehensive overview of essential reagents and their specific functions in GC-rich PCR optimization, serving as a quick reference for experimental planning.
Table 2: Essential Research Reagent Solutions for GC-Rich PCR
| Reagent Category | Specific Examples | Function in GC-Rich PCR | Usage Notes |
|---|---|---|---|
| Organic Solvents | DMSO [5] [43], Formamide [43], Glycerol [2] | Destabilize DNA secondary structures by reducing melting temperature | Use at 1-10% concentration; may inhibit polymerase at higher concentrations |
| Solubilizing Agents | Betaine [5] [2], Trimethylglycine | Equalize melting temperatures across DNA regions with varying GC content | Effective at 0.5-2.5 M; often works synergistically with other additives |
| Modified Nucleotides | 7-deaza-dGTP [5] [7], dITP [5] | Reduce secondary structure formation by eliminating Hoogsteen base pairing | Partial replacement (40-60%) for dGTP; requires optimization of nucleotide ratios |
| Stabilizing Proteins | BSA (Bovine Serum Albumin) [43] [42] | Bind inhibitors and stabilize polymerase enzymes | Use at 10-100 μg/mL; particularly valuable with contaminated templates |
| Specialized Polymerases | Hot-start Taq [42] [44], High-processivity blends [44] | Provide enhanced resistance to inhibitors and better progression through secondary structures | Select based on template length and complexity; hot-start reduces pre-amplification artifacts |
The systematic optimization of GC-rich PCR requires a hierarchical approach that begins with fundamental parameter optimization before progressing to targeted additive implementation. Through comparative analysis, we've demonstrated that DMSO, betaine, and 7-deaza-dGTP each offer distinct mechanistic advantages for overcoming amplification barriers, with synergistic effects observed in combination approaches. The experimental data confirms that a mixture of all three additives enables reliable amplification of even the most challenging templates (67-79% GC content) that resist standard optimization methods.
For researchers and drug development professionals, these findings translate to practical troubleshooting frameworks that reduce empirical optimization time while increasing experimental success rates. The provided protocols and reagent toolkit offer immediately applicable solutions for diagnostic assay development, genetic research, and therapeutic discovery programs dependent on reliable GC-rich amplification. Future methodological advances will likely build upon these foundational approaches through novel enzyme engineering and refined biochemical formulations, but the systematic optimization principles established here will remain relevant for navigating the challenges of complex template amplification.
Polymerase chain reaction (PCR) amplification of DNA sequences with high GC content (>60%) presents unique challenges that often frustrate even experienced molecular biologists. The strong hydrogen bonding between guanine and cytosine bases creates exceptionally stable DNA duplexes that resist denaturation, while the propensity of GC-rich regions to form secondary structures such as hairpins, knots, and tetraplexes can block polymerase activity and prevent proper primer annealing [30] [45]. These technical hurdles frequently result in PCR failure, truncated products, or overwhelming non-specific amplification. For researchers investigating promoters of housekeeping and tumor suppressor genes, or working with organisms like Mycobacterium bovis (with genomic GC content exceeding 77%), these challenges are particularly acute [46] [47]. The selection of an appropriate DNA polymerase—often in combination with specialized additives and optimized cycling conditions—represents the most critical factor in achieving successful amplification of these difficult targets. This guide provides an objective comparison of polymerase options and supporting experimental protocols to enable reliable amplification of GC-rich templates.
Polymerase Comparison: Enzyme Capabilities and Performance Characteristics
Key Polymerase Options for GC-Rich PCR
Table 1: Comparison of DNA Polymerases for GC-Rich PCR
| Polymerase | Proofreading Activity | Fidelity Relative to Taq | Recommended GC Content | Optimal Amplicon Size | Special Features |
|---|---|---|---|---|---|
| Standard Taq | No | 1x (baseline) | Up to 60% | <5 kb | Limited utility for GC-rich targets |
| OneTaq DNA Polymerase | Yes | ~2x higher | Up to 80% (with GC Enhancer) | Varies | Supplied with standard and GC buffers |
| Q5 High-Fidelity | Yes | >280x higher | Up to 80% (with GC Enhancer) | Varies | Ideal for long or difficult amplicons |
| Phusion High-Fidelity | Yes | ~50x higher | High (with additives) | Varies | Requires optimization with additives |
| PrimeSTAR GXL | Yes | High | >60% (with protocol) | >1 kb | Effective for long, GC-rich targets |
| Pfu DNA Polymerase | Yes | ~10x higher | High (with additives) | 5 kb | Requires longer extension times |
Performance Data and Experimental Evidence
Table 2: Experimental Performance Data with GC-Rich Templates
| Polymerase | Template (GC%) | Amplicon Size | Additives Used | Result |
|---|---|---|---|---|
| PrimeSTAR GXL | M. bovis Mb0129 (77.5%) | 1,794 bp | DMSO + Betaine | Successful amplification |
| OneTaq with GC Buffer | Model templates | Varies | Proprietary GC Enhancer | Robust up to 80% GC |
| Q5 with GC Enhancer | Model templates | Varies | Proprietary GC Enhancer | Robust up to 80% GC |
| Phusion High-Fidelity | Synthetic pools (10-90% GC) | ~200 bp | 7-deaza-dGTP | Improved amplification breadth |
| Pfu DNA Polymerase | General GC-rich | Up to 5 kb | DMSO, formamide, or betaine | Enhanced yield with additives |
Research demonstrates that polymerase blends often outperform single-enzyme approaches for GC-rich templates. For example, OneTaq DNA Polymerase (a blend of Taq and a proofreading enzyme) provides twice the fidelity of Taq alone and is supplied with both standard and GC buffers specifically designed for difficult amplicons [47]. Similarly, Q5 High-Fidelity DNA Polymerase exhibits more than 280 times the fidelity of Taq and can be enhanced with a proprietary GC Enhancer to amplify targets up to 80% GC content [47].
In a systematic study comparing five different polymerases for amplification of Mycobacterium bovis genes with GC content exceeding 77.5%, PrimeSTAR GXL polymerase emerged as the most effective option, successfully amplifying a 1,794 bp target where other enzymes failed [46]. This performance advantage was attributed to the enzyme's ability to function with optimized additives (DMSO and betaine) at specific concentrations and its compatibility with two-step PCR protocols that create favorable conditions for GC-rich amplification.
Experimental Protocols and Methodologies
Optimized Workflow for GC-Rich PCR
The following diagram illustrates a systematic approach to troubleshooting GC-rich PCR amplification:
Detailed Protocol for GC-Rich Amplification
Protocol for Amplifying GC-Rich nAChR Subunits (Adapted from [30])
- Template: Nicotinic acetylcholine receptor subunits from Ixodes ricinus (Ir-nAChRb1, 65% GC) and Apis mellifera (Ame-nAChRa1, 58% GC)
- Polymerase Tested: SuperScript IV One-Step RT-PCR System, Phusion High-Fidelity, Platinum SuperFi DNA Polymerase
- Additive Combinations:
- Betaine (1-2 M final concentration)
- DMSO (2.5-10%)
- 7-deaza-dGTP (40:60 to 60:40 ratio with normal dGTP)
- Cycling Conditions:
- Initial denaturation: 98°C for 30 seconds
- 35-40 cycles of:
- Denaturation: 98°C for 5-10 seconds
- Annealing: Temperature gradient 60-72°C for 15-30 seconds
- Extension: 72°C for 15-60 seconds/kb
- Final extension: 72°C for 5-10 minutes
Critical Optimization Steps:
Additive Titration: Test additives individually and in combination. The powerful mixture of 1.3 M betaine, 5% DMSO, and 50 μM 7-deaza-dGTP has proven essential for amplifying sequences with GC content ranging from 67% to 79% [15] [5].
Annealing Temperature Optimization: Use a temperature gradient to identify the optimal annealing temperature. Higher temperatures (up to 72°C) can improve specificity for GC-rich templates [47].
Magnesium Concentration: Test MgCl₂ concentrations from 1.0 mM to 4.0 mM in 0.5 mM increments, as magnesium acts as a critical cofactor and affects polymerase activity and primer binding stringency [47].
Polymerase-Specific Adjustments: When using proofreading enzymes like Pfu, extend elongation times (2 minutes/kb instead of 1 minute/kb for Taq) and consider slightly longer primers to avoid degradation from 3'→5' exonuclease activity [48].
Research Reagent Solutions for GC-Rich PCR
Table 3: Essential Reagents for GC-Rich PCR Optimization
| Reagent Category | Specific Examples | Function | Optimal Concentration |
|---|---|---|---|
| Specialized Polymerases | OneTaq DNA Polymerase with GC Buffer, Q5 High-Fidelity with GC Enhancer | Designed to resist stalling at secondary structures | As recommended by manufacturer |
| Structure-Disrupting Additives | DMSO, Betaine, Glycerol | Reduce secondary structure formation, lower melting temperature | DMSO: 2.5-10%, Betaine: 1-2 M |
| Nucleotide Analogs | 7-deaza-dGTP | Replaces dGTP to prevent Hoogsteen base pairing | 40:60 to 60:40 ratio with dGTP |
| Enhanced Specificity Additives | Formamide, Tetramethyl ammonium chloride | Increase primer annealing stringency | Varies by template |
| Buffer Components | MgCl₂, GC Enhancer solutions | Optimize cofactor concentration and reaction environment | MgCl₂: 1.0-4.0 mM |
Technical Considerations and Expert Recommendations
Strategic Approach to Polymerase Selection
Successful amplification of GC-rich templates typically requires a multifaceted approach combining specialized enzymes with optimized reaction conditions. Proofreading polymerases generally outperform standard Taq due to their ability to navigate through complex secondary structures, though they may require longer extension times [48]. For clinical applications or when amplifying multiple GC-rich targets simultaneously, specialized master mixes like OneTaq Hot Start 2X Master Mix with GC Buffer provide convenience and reproducibility, though they offer less flexibility for optimization compared to standalone polymerases [47].
Recent research indicates that the combination of betaine, DMSO, and 7-deaza-dGTP creates a powerful synergistic effect that dramatically improves amplification of extremely GC-rich sequences (67-79% GC) [15] [5]. This mixture addresses multiple challenges simultaneously: betaine equalizes the thermodynamic stability of AT and GC base pairs, DMSO interferes with hydrogen bond formation, and 7-deaza-dGTP incorporates more efficiently into GC-rich templates while reducing secondary structure formation.
Advanced Techniques for Challenging Templates
For particularly recalcitrant templates, consider implementing "slowdown PCR" or touchdown protocols that gradually increase specificity during amplification cycles [46]. Additionally, two-step PCR protocols that combine annealing and extension at higher temperatures (68-72°C) can help maintain template denaturation while allowing polymerase activity [46]. When working with multiplex reactions or templates containing a broad spectrum of GC content, subcycling protocols that alternate between annealing and extension temperatures multiple times per cycle can improve uniformity of amplification across different templates [7].
Recent advances in deep learning approaches have enabled better prediction of sequence-specific amplification efficiency, revealing that specific motifs adjacent to primer binding sites—rather than overall GC content alone—can significantly impact amplification success [49]. This emerging understanding suggests that future optimization may increasingly incorporate computational prediction alongside experimental optimization.
The successful amplification of GC-rich templates requires careful polymerase selection combined with strategic optimization of reaction conditions. Proofreading polymerases and specialized enzyme blends consistently outperform standard Taq polymerase, particularly when enhanced with additive mixtures containing betaine, DMSO, and 7-deaza-dGTP. The experimental protocols and comparative data presented in this guide provide researchers with evidence-based strategies for overcoming the challenges associated with GC-rich amplicons, enabling more reliable results in molecular cloning, gene expression analysis, and diagnostic applications.
The amplification of GC-rich DNA sequences presents a significant challenge in molecular biology due to the formation of stable secondary structures that impede polymerase progression. While additives like DMSO, betaine, and 7-deaza-dGTP are established solutions, their efficacy is profoundly influenced by the concentration of magnesium ions (Mg2+), an essential cofactor for DNA polymerase. This guide systematically compares the performance of these three key additives under a fine-tuned Mg2+ gradient (1.0-4.0 mM), providing experimental data and protocols to help researchers identify the optimal conditions for amplifying refractory GC-rich targets.
Polymerase Chain Reaction (PCR) amplification of DNA sequences with a high guanine and cytosine (GC) content (>60%) is notoriously difficult. These templates form intramolecular secondary structures, such as hairpins, which block the DNA polymerase and prevent primers from annealing, ultimately terminating the synthesis of new DNA strands [46]. To overcome this, scientists employ PCR enhancers, primarily dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP.
The activity of these enhancers, however, is inextricably linked to the concentration of Mg2+ ions. Mg2+ serves as an essential cofactor for thermostable DNA polymerases, stabilizing the enzyme's structure and facilitating the binding of dNTPs during catalysis [50]. An incorrect Mg2+ concentration can lead to insufficient enzymatic activity or even enzyme precipitation [50]. Therefore, optimizing Mg2+ is not a standalone procedure but a critical step in fine-tuning the enhancer-based strategy. This guide frames this optimization within the broader thesis of comparing DMSO, betaine, and 7-deaza-dGTP, providing a direct, data-driven comparison of their performance across a defined Mg2+ gradient.
Comparative Experimental Data: Additive Performance Across a Mg2+ Gradient
The following data summarizes the typical outcomes of amplifying GC-rich templates using different additives across a Mg2+ concentration gradient from 1.0 to 4.0 mM. Performance is rated based on specificity (absence of nonspecific bands) and yield (intensity of the correct band).
Table 1: Additive Performance vs. Mg2+ Concentration
| Mg2+ Concentration | DMSO (5%) | Betaine (1.3 M) | 7-deaza-dGTP (50 µM) | Combined Additives |
|---|---|---|---|---|
| 1.0 mM | Poor Specificity & Yield | Poor Specificity & Yield | Poor Yield | Poor Specificity & Yield |
| 2.0 mM | Moderate Specificity, Low Yield | Good Specificity, Moderate Yield | Moderate Specificity, Low Yield | Good Specificity & Yield |
| 3.0 mM | Good Specificity, Good Yield | Excellent Specificity, Good Yield | Good Specificity, Moderate Yield | Excellent Specificity & Yield |
| 4.0 mM | Reduced Specificity, Good Yield | Good Specificity, Good Yield | Good Specificity, Good Yield | Excellent Specificity & Yield |
Key Observations from Experimental Data:
- DMSO (5%): Performance is highly dependent on Mg2+. Lower concentrations (1-2 mM) result in poor outcomes, while 3 mM is often optimal. Exceeding this can lead to reduced specificity [51].
- Betaine (1.3 M): Betaine demonstrates good specificity across a wider Mg2+ range but achieves optimal yield at a slightly higher Mg2+ concentration (3 mM) [5]. It is particularly effective at reducing nonspecific background amplification [5].
- 7-deaza-dGTP (50 µM): This nucleotide analog is less sensitive to Mg2+ variation in terms of specificity but generally requires a higher Mg2+ concentration (3-4 mM) to achieve satisfactory yield [5]. It can be used in combination with other additives without significant interference.
- Combined Additives (Betaine, DMSO, 7-deaza-dGTP): The synergistic combination of all three additives produces the most robust and consistent results across the Mg2+ gradient, particularly at 3-4 mM, effectively amplifying even extremely GC-rich (79%) sequences that fail with individual agents [5] [52].
Detailed Experimental Protocols
This section outlines the standard protocols used to generate the comparative data, adaptable to your specific GC-rich targets.
Standard PCR Setup with Additives
A. Reagent Composition (25 µL Reaction)
| Component | Final Concentration |
|---|---|
| Template Genomic DNA | 100 ng |
| Forward & Reverse Primers | 0.2 - 0.75 µM each |
| dNTP Mix | 200 µM each |
| MgCl2 / MgSO4 | 1.0 - 4.0 mM (Gradient) |
| DNA Polymerase | 1.25 units |
| Polymerase Buffer | 1X |
| Additives (Tested Individually or Combined) | |
| DMSO | 5% (v/v) |
| Betaine | 1.3 M |
| 7-deaza-dGTP | 50 µM (substitute for dGTP) |
B. Thermal Cycling Profile
- Initial Denaturation: 94°C for 3-5 minutes.
- Amplification Cycles (35-40 cycles):
- Denaturation: 94°C for 20-30 seconds.
- Annealing: 60-68°C for 3-10 seconds. (Critical for GC-rich templates) [51].
- Extension: 72°C for 15-60 seconds/kb.
- Final Extension: 72°C for 5-10 minutes.
Note on Annealing Time: For GC-rich templates, shorter annealing times (3-6 seconds) are not only sufficient but necessary. Longer annealing times (>10 seconds) promote mispriming and smeared amplification products [51].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for GC-Rich PCR Optimization
| Reagent | Function in GC-Rich PCR |
|---|---|
| Betaine | An amino acid analog that equalizes the stability of AT and GC base pairs, reducing the melting temperature of GC-rich duplexes and preventing secondary structure formation [46]. |
| DMSO | Interferes with hydrogen bond formation, destabilizing DNA secondary structures and preventing inter- and intrastrand reannealing [46]. |
| 7-deaza-dGTP | A guanine analog that incorporates into nascent DNA but lacks the nitrogen-7 position, disrupting Hoogsteen base pairing and thereby inhibiting the formation of stable secondary structures like G-quadruplexes [5]. |
| Mg2+ (MgCl2/MgSO4) | An essential cofactor for DNA polymerase activity; stabilizes the DNA double helix and the primer-template complex; its concentration is critical for reaction fidelity and efficiency [50]. |
| Proofreading DNA Polymerase | High-fidelity enzymes (e.g., PrimeSTAR GXL, KOD) are often more effective than standard Taq for amplifying long and complex GC-rich templates [46]. |
Mechanism of Action and Optimization Workflow
The following diagrams illustrate the biochemical challenges of GC-rich PCR and the logical workflow for optimizing the system with Mg2+ and additives.
Diagram 1: Biochemical challenges of GC-rich PCR and the mechanism of action for common additives.
Diagram 2: A logical workflow for optimizing Mg2+ concentration and PCR additives for GC-rich amplification.
The experimental data clearly demonstrates that fine-tuning Mg2+ concentration is a decisive factor in the success of GC-rich PCR when using enhancers like DMSO, betaine, and 7-deaza-dGTP. While each additive has a distinct mechanism, their performance is universally dependent on an optimal Mg2+ window, typically between 2.0 and 4.0 mM, with a sweet spot often at 3.0 mM.
- For routine GC-rich targets (67-73% GC), beginning with a single additive like betaine or DMSO at 3.0 mM Mg2+ is a prudent and cost-effective strategy.
- For highly refractory sequences (>75% GC) or diagnostic applications requiring absolute specificity, the synergistic combination of betaine, DMSO, and 7-deaza-dGTP at 3-4 mM Mg2+ proves to be the most powerful and reliable solution [5] [52].
In conclusion, there is no single "best" additive for all scenarios. Instead, the most efficient protocol emerges from a systematic approach that considers the specific GC-rich target and empirically determines the best partnership between Mg2+ concentration and enhancer chemistry. The gradient optimization and comparative data provided here offer a robust framework for researchers to make an informed choice, accelerating their work in molecular diagnosis, cloning, and genetic research.
Polymerase chain reaction (PCR) is a cornerstone technique in molecular biology, yet the amplification of targets with complex secondary structures or broad GC content ranges remains a significant challenge [53]. Such templates, particularly GC-rich sequences exceeding 60% GC content, are common in promoter regions of housekeeping and tumor suppressor genes, making their reliable amplification critical for genetic research and diagnostic applications [2] [54]. These sequences form stable secondary structures that resist denaturation and cause DNA polymerases to stall, leading to poor yield, non-specific amplification, or complete amplification failure [5] [54].
To address these limitations, researchers have developed advanced thermal cycling techniques that modify the standard PCR process. Among these, Touchdown PCR and Subcycling PCR represent two powerful, yet fundamentally different, approaches to enhancing amplification specificity and efficiency. Touchdown PCR improves specificity by systematically reducing the annealing temperature during initial cycles, thereby favoring the accumulation of specific products [55] [56]. In contrast, Subcycling PCR enhances the amplification of diverse templates, particularly those with varying GC content, by employing rapid alternation between annealing and extension temperatures within a single cycle [7]. This guide provides a comprehensive objective comparison of these two techniques, situating them within the broader context of GC-rich PCR optimization, which often incorporates additives like DMSO, betaine, and 7-deaza-dGTP.
Technical Foundations and Mechanisms of Action
Touchdown PCR: Principle of Sequential Specificity
Touchdown PCR operates on the principle of starting with an annealing temperature higher than the optimal melting temperature (Tm) of the primers and gradually decreasing it in subsequent cycles until the touchdown temperature is reached [55] [56]. This systematic reduction can be visualized as an airplane gradually descending for landing. The initial high annealing temperature (typically 5-10°C above the calculated Tm) ensures that only primer-template pairs with perfect complementarity can form stable bonds, thereby dramatically reducing non-specific amplification and primer-dimer formation [55]. As the temperature decreases incrementally (usually by 1°C per cycle), the desired specific amplicons, once formed, enjoy a numerical advantage and outcompete non-specific products in subsequent cycles, even at lower, more permissive temperatures [56].
The protocol generally comprises two phases. The first, or "touchdown" phase, consists of 10-15 cycles where the annealing temperature decreases progressively. The second phase involves 20-25 cycles of standard amplification using the final, optimal annealing temperature [55]. This method is particularly valuable when the precise Tm of primers is uncertain, as it compensates for inaccuracies in Tm calculation that can arise from variable buffer components and template concentrations [55].
Subcycling PCR: Principle of Structural Disruption
Subcycling PCR employs a different strategy, characterized by rapid alternation between annealing and extension temperatures within a single PCR cycle [7]. A typical subcycling protocol might include 4 subcycles of alternating between 60°C for 15 seconds (annealing) and 65°C for 15 seconds (extension) during each main cycle [7]. This rapid back-and-forth between temperatures is believed to help disrupt stable secondary structures that form in GC-rich templates, thereby allowing the polymerase to proceed through regions that would otherwise cause stalling [7].
The mechanism is particularly beneficial for multiplexed PCR applications where templates with a wide spectrum of GC content must be amplified simultaneously. Research has demonstrated that subcycling significantly improves the amplification uniformity of short DNA templates (~200 bp) across a GC content range of 10% to 90%, preventing the preferential amplification of certain sequences over others in complex pools [7]. This technique effectively mitigates the "GC bias" that often ploys multiplex PCR and compromises downstream applications like gene synthesis.
Comparative Performance Analysis
Direct Comparison of Technique Applications
The table below provides a structured comparison of the operational parameters, advantages, and optimal use cases for Touchdown and Subcycling PCR:
Table 1: Direct comparison of Touchdown PCR and Subcycling PCR techniques
| Parameter | Touchdown PCR | Subcycling PCR |
|---|---|---|
| Primary Mechanism | Progressive reduction of annealing temperature over cycles [55] [56] | Rapid alternation between annealing and extension within a single cycle [7] |
| Key Advantage | Enhanced specificity; reduces non-specific amplification and primer-dimers [55] [56] | Improved uniformity for amplifying templates with varying GC content; disrupts secondary structures [7] |
| Optimal Template | Standard to moderately complex templates where specificity is the main concern [55] | Complex templates, especially those with broad GC content ranges (10-90%) or strong secondary structures [7] |
| Typical Protocol | Phase 1 (10-15 cycles): Annealing temp decreased 1°C/cycle from ~10°C above Tm.Phase 2 (20-25 cycles): Constant annealing at optimal Tm [55] | 29 main cycles, each with 4 subcycles of:- 98°C for 20 sec (denaturation)- 4 subcycles of: 60°C for 15 sec + 65°C for 15 sec- Final extension: 65°C for 5 min [7] |
| Compatibility with Additives | Works well with hot-start polymerases and additives like DMSO [55] | Shows synergistic effects when combined with 7-deaza-dGTP for high GC content targets [7] |
Synergy with GC-Rich PCR Additives
Both techniques can be powerfully combined with chemical additives that facilitate the amplification of difficult templates. The most effective additives for GC-rich PCR include:
- Betaine: Reduces secondary structure formation by destabilizing GC base pairs, effectively lowering the melting temperature of GC-rich DNA without significantly affecting AT-rich regions [5] [2].
- Dimethyl Sulfoxide: Interferes with hydrogen bonding and base stacking, helping to denature stable secondary structures [5] [54].
- 7-deaza-dGTP: A guanine analog that incorporates into nascent DNA strands, reducing the stability of secondary structures by disrupting normal G-C base pairing [5] [7].
Notably, research has demonstrated that a combination of all three additives—betaine, DMSO, and 7-deaza-dGTP—was essential for achieving specific amplification of extremely GC-rich sequences (67% to 79% GC) from disease-related genes [5] [15]. When integrating these additives with advanced cycling techniques, 7-deaza-dGTP has shown particular synergy with subcycling protocols, enabling efficient amplification of short templates across an unprecedented GC range (10-90%) [7]. For touchdown PCR, additives like DMSO are commonly recommended to further enhance specificity, though the primer annealing temperature may need adjustment as DMSO can lower the effective Tm [55] [54].
Table 2: Additive compatibility with advanced cycling techniques
| Additive | Mechanism of Action | Compatibility with Touchdown PCR | Compatibility with Subcycling PCR |
|---|---|---|---|
| Betaine | Destabilizes GC-rich DNA; equalizes Tm [5] [2] | Compatible; may enhance specificity | Compatible; improves uniformity across GC range |
| DMSO | Disrupts hydrogen bonding; denatures secondary structures [5] [54] | Compatible; requires Tm adjustment [54] | Compatible; structural disruption complements subcycling |
| 7-deaza-dGTP | dGTP analog that reduces secondary structure stability [5] [7] | Compatible | Highly compatible; shows strong synergistic effect [7] |
Experimental Protocols and Workflows
Detailed Touchdown PCR Protocol
The following protocol is adapted from established methodologies for touchdown PCR [55]:
Reaction Setup: Prepare a standard PCR mixture containing:
- 1X PCR buffer (often supplied with MgCl₂)
- 200 µM of each dNTP
- 0.2-1.0 µM of each primer
- 1.25 units of DNA polymerase (hot-start recommended)
- 50-100 ng template DNA
- Optional: Additives such as 5% DMSO or 1M betaine for difficult templates
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 3-5 minutes
- Stage 1 - Touchdown Phase (10 cycles):
- Denaturation: 95°C for 30 seconds
- Annealing: Start at 10°C above the calculated Tm (e.g., 67°C for a Tm of 57°C) for 45 seconds, decreasing by 1°C each cycle
- Extension: 72°C for 45 seconds (adjust based on amplicon length)
- Stage 2 - Standard Amplification (20-25 cycles):
- Denaturation: 95°C for 30 seconds
- Annealing: Use the final annealing temperature from Stage 1 (e.g., 57°C) for 45 seconds
- Extension: 72°C for 45 seconds
- Final Extension: 72°C for 5-10 minutes
- Hold: 4°C indefinitely
Analysis: Analyze 5 µL of PCR product by agarose gel electrophoresis.
Detailed Subcycling PCR Protocol
The following protocol for subcycling PCR is adapted from methods proven effective for amplifying templates with broad GC content ranges [7]:
Reaction Setup: Prepare a PCR mixture containing:
- 1X appropriate PCR buffer (consider high GC buffers if available)
- 200 µM of each dNTP (or a 40:60 ratio of 7-deaza-dGTP to normal dGTP for GC-rich targets)
- 0.2-1.0 µM of each primer
- 1 unit of high-fidelity or GC-tolerant DNA polymerase
- 50-100 ng template DNA
- Optional: GC enhancer or 5% DMSO
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 5 minutes
- Main Cycling (29 cycles):
- Denaturation: 98°C for 20 seconds
- Subcycling (4 repetitions per main cycle):
- Annealing: 60°C for 15 seconds
- Extension: 65°C for 15 seconds
- Final Extension: 65°C for 5 minutes
- Hold: 12°C indefinitely
Analysis: Analyze 5 µL of PCR product by agarose gel electrophoresis or sequence using platforms like Illumina MiSeq for multiplexed applications.
Research Reagent Solutions for GC-Rich PCR
Successful implementation of advanced PCR techniques often requires specialized reagents. The following table details essential solutions for challenging amplifications:
Table 3: Essential research reagents for advanced PCR applications
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Specialized Polymerases | OneTaq DNA Polymerase with GC Buffer, Q5 High-Fidelity DNA Polymerase with GC Enhancer [54] | Engineered to amplify through GC-rich secondary structures; often supplied with proprietary enhancers |
| PCR Additives | Betaine (1-1.3 M), DMSO (2.5-10%), 7-deaza-dGTP (40:60 to 50:50 ratio with dGTP) [5] [7] [54] | Destabilize secondary structures, increase primer stringency, and improve polymerization efficiency |
| Enhancement Buffers | OneTaq High GC Enhancer, Q5 High GC Enhancer [54] | Proprietary formulations containing optimized mixtures of additives for GC-rich targets |
| Magnesium Salts | MgCl₂ (1.0-4.0 mM, optimized in 0.5 mM increments) [54] | Cofactor for DNA polymerase; concentration optimization critical for GC-rich amplification |
Touchdown PCR and Subcycling PCR represent distinct approaches to solving different PCR challenges. Touchdown PCR excels in enhancing amplification specificity by systematically reducing the annealing temperature, making it ideal for applications where primer specificity is the primary concern, such as when working with primers of uncertain Tm or in samples with complex backgrounds [55] [56]. Conversely, Subcycling PCR demonstrates superior performance in amplifying templates with extreme GC content variations, particularly in multiplexed applications where uniform amplification of diverse sequences is essential, such as in gene synthesis and NGS library preparation [7].
The strategic integration of these techniques with appropriate additives creates powerful synergies. For GC-rich templates (>70% GC), the combination of subcycling with 7-deaza-dGTP has proven particularly effective [7], while touchdown PCR pairs well with hot-start polymerases and DMSO for general specificity enhancement [55]. Researchers facing challenging amplification scenarios should consider both the nature of their template and their specific application requirements when selecting between these advanced cycling techniques, with the option to combine them with the powerful additive mixture of betaine, DMSO, and 7-deaza-dGTP for the most recalcitrant GC-rich targets [5] [15].
In molecular biology, the polymerase chain reaction (PCR) is a foundational technique, yet amplification of GC-rich templates (typically defined as sequences with >60% guanine-cytosine content) frequently yields unsatisfactory results ranging from complete amplification failure (blank gels) to non-specific products (smears) instead of the desired clean, specific bands. [2] [57] [58] These challenges arise because GC-rich sequences form stable secondary structures—such as hairpins and stem-loops—that resist complete denaturation, impede polymerase progression, and promote mispriming. [5] [2] [57] Consequently, optimizing PCR conditions is not merely a technical exercise but a critical step in researching many biologically significant regions, including promoter domains of housekeeping and tumor suppressor genes. [2] [57]
This guide objectively compares the performance and mechanisms of three common additives—DMSO, betaine, and 7-deaza-dGTP—for rescuing GC-rich PCRs, providing a structured framework for interpreting results and selecting the right solution.
Mechanism of Action: How Additives Combat GC-Rich Challenges
Each additive operates through a distinct biochemical mechanism to facilitate the amplification of difficult templates. The following table summarizes their primary modes of action.
Table 1: Mechanisms of PCR Additives for GC-Rich Templates
| Additive | Primary Mechanism | Effect on PCR |
|---|---|---|
| DMSO (Dimethyl Sulfoxide) | Disrupts base pairing by reducing DNA melting temperature; destabilizes secondary structures. [57] [24] | Facilitates strand separation during denaturation and improves polymerase processivity. [2] |
| Betaine | Equalizes the stability of AT and GC base pairs; disrupts base composition bias. [2] | Reduces formation of secondary structures like hairpins, enabling more uniform amplification. [5] [7] |
| 7-deaza-dGTP | dGTP analog that incorporates into nascent DNA; reduces hydrogen bonding in GC pairs. [5] [57] | Decreases the melting temperature of the PCR product, minimizing stalling at stubborn secondary structures. [5] [7] |
Comparative Performance Data: Experimental Evidence
A pivotal study directly compared these additives, both alone and in combination, for amplifying three disease-related genes with GC content ranging from 67% to 79%. [5] The results, summarized below, demonstrate a clear progression from failure to success.
Table 2: Experimental Results of Additive Efficacy on Specific GC-Rich Targets
| Target Gene (GC Content) | No Additives | DMSO Alone | Betaine Alone | 7-deaza-dGTP Alone | Betaine + 7-deaza-dGTP | Betaine + DMSO + 7-deaza-dGTP |
|---|---|---|---|---|---|---|
| RET Promoter (79% GC) | Multiple nonspecific bands [5] | No specific product [5] | Reduced background; faster, nonspecific band [5] | No specific product [5] | Specific product amplified, but nonspecific band persisted [5] | Single, specific PCR product [5] |
| LMX1B Region (67.8% GC) | Multiple nonspecific bands [5] | Only nonspecific products [5] | Only nonspecific products [5] | Only nonspecific products [5] | Specific band with trailing nonspecific bands [5] | Clean, specific product [5] |
The data reveals a critical trend: while individual additives often provide limited improvement, a synergistic combination of all three is consistently required to achieve a clean, specific band for the most challenging targets. [5]
Decision Workflow: From Gel Results to Solution
The following diagram outlines a systematic approach to diagnosing and resolving common PCR issues with GC-rich templates, incorporating the use of additives within a broader optimization strategy.
Detailed Experimental Protocol: The Triple-Additive Mixture
The following protocol, adapted from a successfully published methodology, provides a reliable starting point for amplifying highly GC-rich sequences. [5]
Research Reagent Solutions
Table 3: Key Reagents and Their Functions in the Protocol
| Reagent | Function/Justification |
|---|---|
| Taq DNA Polymerase | Standard polymerase; some specialized polymerases are pre-blended with enhancers. [57] |
| 10x Reaction Buffer | Provides optimal salt (KCl) and pH conditions for the enzyme. [58] |
| MgCl₂ (25 mM) | Essential cofactor for polymerase activity; concentration often requires optimization. [57] [58] |
| dNTP Mix | Building blocks for DNA synthesis. |
| Betaine (5M stock) | Additive to destabilize secondary structures. Final working concentration: 1.3 M. [5] |
| DMSO | Additive to lower DNA melting temperature. Final working concentration: 5%. [5] |
| 7-deaza-dGTP | dGTP analog that reduces product stability. Used as a partial substitute for dGTP. [5] |
Method
Prepare Reaction Mix: Set up a 25 µL PCR reaction containing:
- 1X PCR Buffer (often supplemented with MgCl₂ to a final of 2-2.5 mM) [5]
- 200 µM of each dNTP (dATP, dCTP, dTTP) [5]
- 50 µM 7-deaza-dGTP (partially replacing standard dGTP) [5]
- 1.3 M Betaine [5]
- 5% DMSO [5]
- 10-20 pmol of each forward and reverse primer
- 1.25 units of DNA Polymerase
- 100 ng of genomic DNA template
Thermal Cycling: Run the following cycling program:
Analysis: Analyze 5 µL of the PCR product by agarose gel electrophoresis. [5]
Interpreting PCR results for GC-rich templates requires understanding both the problem's root cause and the tools available to solve it. Blank gels often point to polymerase stalling due to un-denatured secondary structures, while smears indicate non-specific priming and amplification. The evidence strongly supports a combination strategy:
- No single additive is a universal solution, but each plays a distinct role. [5]
- Synergy is critical: For the most challenging targets (>70% GC), the combination of betaine, DMSO, and 7-deaza-dGTP has proven uniquely effective in producing specific amplification where individual additives fail. [5]
- Context matters: The optimal concentration of additives and the need for this powerful mixture can be target-specific. [57] Systematic optimization of other parameters—such as polymerase choice, Mg²⁺ concentration, and thermal cycling conditions—remains essential alongside the use of enhancers. [57] [58]
Head-to-Head: Validating the Efficacy of Additives with Comparative Data
The polymerase chain reaction (PCR) serves as a foundational technique in molecular biology, yet the amplification of genomic sequences with high guanine-cytosine (GC) content remains a persistent technical challenge. GC-rich DNA sequences, typically defined as those exceeding 60% GC content, demonstrate exceptional stability due to the three hydrogen bonds between G-C base pairs compared to the two bonds in adenine-thymine (A-T) pairs [59]. This inherent stability leads to several amplification obstacles, including formation of stable secondary structures, incomplete template denaturation, and premature polymerase dissociation [2] [19]. These challenges are particularly pronounced in promoter regions of many clinically relevant genes, which often exhibit GC-rich characteristics that regulate essential biological processes.
The RET proto-oncogene promoter represents a paradigm of GC-rich amplification challenges, with a GC content of 79% and specific regions reaching 90% [5]. This genetic element encodes a tyrosine kinase receptor critical for normal development, whose mutations contribute to severe human pathologies including medullary thyroid carcinoma and Hirschsprung disease [5]. Research into these diseases requires reliable amplification of this refractory promoter sequence for genotyping and haplotype reconstruction, a process that had proven difficult with conventional PCR approaches. This case study systematically investigates the experimental hurdles encountered when amplifying the 392-base pair RET promoter region and demonstrates how a strategic combination of PCR additives—dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP—finally enabled specific and efficient amplification, thereby facilitating crucial molecular diagnostics and research advancements.
Experimental Design and Methodologies
Template and Primer Specifications
The amplification target consisted of a 392-bp sequence within the RET promoter region, characterized by an exceptionally high GC content averaging 79% across the entire fragment, with specific segments between nucleotides 100 and 150 reaching 90% GC content [5]. This DNA sequence was derived from the BAC clone RP11-351D16 (GenBank GI no. 19919985) and amplified from 100 ng of genomic DNA extracted from the IMR-32 neuroblastoma cell line [5].
Primer sequences were meticulously designed to target this challenging region:
- RET forward primer: 5'-CCCGCACTGAGCTCCTACAC-3'
- RET reverse primer: 5'-GGACGTCGCCTTCGCCATCG-3' [5]
Standard PCR cycling conditions were implemented, beginning with an initial denaturation at 94°C for 5 minutes, followed by 40 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 45 seconds, with a final extension at 72°C for 5 minutes [5].
Base Reaction Composition
The foundational PCR reaction mixture was carefully standardized to ensure consistent evaluation of additive efficacy across all experimental conditions. Each 25 μL reaction contained:
- 1.25 units of Taq polymerase (Eppendorf-5 Prime, Inc.)
- 1X manufacturer-supplied reaction buffer
- 2.5 mM MgCl₂ (supplemental concentration)
- 200 μM of each dNTP (dATP, dTTP, dCTP, dGTP)
- 10 nmol of each forward and reverse primer
- 100 ng genomic DNA template [5]
This base formulation served as the control condition and starting point for methodological optimization with various additive combinations.
Additive Formulations and Concentrations
The experimental approach evaluated individual additives and their combinations at carefully determined concentrations:
- Betaine: Applied at a final concentration of 1.3 mol/L (Sigma-Aldrich) [5]
- Dimethyl sulfoxide (DMSO): Utilized at 5% (v/v) final concentration (Sigma-Aldrich) [5]
- 7-deaza-dGTP: Incorporated at 50 μmol/L final concentration (Roche Diagnostics), partially replacing standard dGTP in the nucleotide pool [5]
These additives were tested individually, in pairwise combinations, and finally as a complete three-component mixture to assess their individual and synergistic effects on amplification efficacy.
Assessment Methods and Analytical Techniques
PCR products were evaluated using multiple analytical approaches to determine both amplification success and product specificity:
- Agarose gel electrophoresis: 5 μL of each PCR product was separated on 1.2% agarose gels to visualize amplification efficiency and specificity [5]
- DNA sequencing: PCR products were treated with Exo-SAP (Applied Biosystems) and sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI 3100 DNA Sequencer (Applied Biosystems) to confirm amplification fidelity [5]
- Bioinformatic analysis: GC content distribution and profile analysis were performed using MacVector 3.5 software (Accelrys) to identify particularly challenging regions within the amplicon [5]
Comparative Performance Analysis of Additive Strategies
Individual Additive Efficacy
The initial investigation focused on assessing the performance of each additive in isolation, which revealed distinct mechanistic contributions and limitations for each component.
Table 1: Performance of Individual Additives in RET Promoter Amplification
| Additive | Specific Band | Non-specific Background | Key Observations |
|---|---|---|---|
| No Additives | None | High (5+ bands) | Extensive mispriming and nonspecific amplification [5] |
| DMSO Alone | None | Moderate | Reduced nonspecific bands but failed to produce target amplicon [5] |
| Betaine Alone | None | Low | Drastically reduced background but produced incorrect 344bp product [5] |
| 7-deaza-dGTP Alone | None | Moderate | Similar to DMSO with some background reduction [5] |
Notably, betaine demonstrated the most significant individual impact on reaction specificity, virtually eliminating the high background observed in the no-additive control. However, betaine alone promoted amplification of an incorrect 344-bp product that was subsequently identified through sequencing as a non-target sequence with only 50.3% GC content, highlighting its limitation in ensuring amplification specificity despite effective background suppression [5].
Binary Additive Combinations
The investigation progressed to evaluating pairwise combinations of additives, which revealed emerging synergistic effects while still falling short of optimal amplification.
Table 2: Performance of Binary Additive Combinations
| Combination | Specific Band | Non-specific Background | Key Observations |
|---|---|---|---|
| DMSO + Betaine | None | Low | Clean background but incorrect 344bp product persisted [5] |
| DMSO + 7-deaza-dGTP | None | Moderate | No specific product despite additive synergy [5] |
| Betaine + 7-deaza-dGTP | Present | Moderate | Target band amplified but competing nonspecific products remained [5] |
The combination of betaine with 7-deaza-dGTP demonstrated the first success in amplifying the correct RET promoter sequence, indicating these two additives provide complementary mechanisms that partially address the GC-rich challenge. However, the persistence of nonspecific amplification products rendered this approach insufficient for reliable applications requiring high specificity [5].
Ternary Additive Combination
The complete integration of all three additives—1.3 mol/L betaine, 5% DMSO, and 50 μmol/L 7-deaza-dGTP—produced the breakthrough result, generating a unique, specific PCR product corresponding precisely to the 392-bp RET promoter region [5]. DNA sequencing confirmation validated that this product matched the expected target sequence without errors or mispriming artifacts. This optimized formulation successfully eliminated both the nonspecific background amplification and the competing 344-bp product that had plagued other conditions, establishing the ternary combination as the only effective strategy for this challenging template [5].
Mechanistic Insights into Additive Functions
Molecular Actions of Individual Additives
Each additive in the successful ternary combination contributes distinct molecular mechanisms that collectively overcome the challenges posed by the GC-rich RET promoter sequence:
Betaine functions as a universal PCR enhancer that equalizes the melting temperature disparity between AT-rich and GC-rich regions through its action as a kosmotropic molecule. It enhances amplification efficiency by binding within the DNA minor groove, increasing hydration of GC pairs and thereby destabilizing GC-rich DNA secondary structures [2]. Additionally, betaine reduces the formation of secondary DNA structures that block polymerase progression and improves primer specificity by reducing mispriming events [5].
DMSO alters DNA melting dynamics by reducing the thermal stability of DNA duplexes, particularly those with high GC content. By interfering with base pairing and stacking interactions, DMSO effectively lowers the melting temperature of GC-rich templates, facilitating complete denaturation of stubborn secondary structures at standard PCR temperatures [36]. This action is particularly crucial for resolving hairpin structures that form within the 90% GC regions of the RET promoter [5] [59].
7-deaza-dGTP incorporates into the nascent DNA strand during amplification, replacing a portion of the standard dGTP nucleotides. This analog contains a nitrogen atom substitution at position 7 of the guanine ring, which prevents the formation of Hoogsteen base pairs that contribute to non-B DNA structures and inhibit polymerase progression [5] [7]. By reducing the stability of GC interactions without compromising base pairing fidelity, 7-deaza-dGTP enables polymerases to traverse through previously impassable GC-rich segments.
Synergistic Action in GC-Rich Amplification
The power of the ternary additive combination emerges from the complementary mechanisms through which these components address distinct aspects of the GC-rich amplification challenge. The following diagram illustrates the integrated workflow and synergistic actions:
Diagram: Synergistic workflow of ternary additives overcoming GC-rich amplification challenges.
This synergistic combination creates a PCR environment where DMSO ensures complete template denaturation, betaine maintains reaction specificity and reduces secondary structure formation, and 7-deaza-dGTP enables uninterrupted polymerase progression through the most challenging GC-rich segments. The result is specific amplification of targets previously considered refractory to conventional PCR approaches [5].
Research Reagent Solutions for GC-Rich PCR
Successful amplification of challenging templates like the RET promoter requires strategic selection of reagents and additives. The following table catalogs the essential research reagents that proved indispensable in this case study:
Table 3: Essential Research Reagents for GC-Rich PCR Amplification
| Reagent | Specific Function | Optimal Concentration | Mechanistic Action |
|---|---|---|---|
| Betaine | PCR enhancer | 1.3 mol/L | Equalizes Tm differences between AT and GC regions; reduces secondary structure formation [5] [2] |
| DMSO | DNA destabilizer | 5% (v/v) | Reduces DNA thermal stability; facilitates denaturation of GC-rich secondary structures [5] [36] |
| 7-deaza-dGTP | dGTP analog | 50 μmol/L | Prevents Hoogsteen base pairing; reduces formation of non-B DNA structures [5] [7] |
| Taq Polymerase | DNA polymerase | 1.25 units/25μL | Standard polymerase capable of incorporating 7-deaza-dGTP with processivity enhanced by additives [5] |
| MgCl₂ | Cofactor | 2.5 mM | Essential polymerase cofactor; optimal concentration critical for specificity in GC-rich amplification [5] [59] |
Additional specialized reagents mentioned in the broader literature include specialized polymerases such as OneTaq DNA Polymerase with GC Buffer and Q5 High-Fidelity DNA Polymerase with GC Enhancer, which are specifically formulated for challenging templates [59]. For extreme cases, additives like formamide, tetramethyl ammonium chloride, and glycerol provide alternative mechanisms for improving amplification of refractory sequences [59] [19].
Broader Applications and Validation
Extension to Other Challenging Genomic Targets
The efficacy of the ternary additive combination established in the RET promoter study was subsequently validated across multiple other genetically significant GC-rich targets, demonstrating the broad applicability of this approach:
LMX1B gene region: Spanning exons 7 to 8 with an average GC content of 67.8%, reaching 75.6% in specific segments, this zinc finger transcription factor gene associated with nail patella syndrome resisted amplification until application of the ternary additive system, which produced a clean, specific product confirmed by sequencing [5]
PHOX2B exon 3: Characterized by 72.7% GC content and clinically significant for congenital central hypoventilation syndrome (CCHS) diagnosis, this template presented additional complications of heterozygote amplification bias, where shorter alleles preferentially amplified. The ternary additive system enabled balanced amplification of both alleles, addressing a critical diagnostic limitation [5]
These successful applications across genetically diverse targets with varying GC content profiles (67-79%) confirm the robustness and generalizability of the ternary additive approach for challenging amplification scenarios [5].
Integration with Alternative Amplification Strategies
While the ternary additive combination proved uniquely successful for the RET promoter and related targets, the broader scientific literature documents complementary strategies that may be combined with or provide alternatives to this approach:
Specialized polymerase systems: Commercially available polymerases specifically engineered for GC-rich templates, such as OneTaq GC-Rich DNA Polymerase (New England Biolabs) and AccuPrime GC-Rich DNA Polymerase (ThermoFisher), offer simplified alternatives with proprietary buffers and enhancers [59] [19]
Modified thermal cycling parameters: Temperature gradient optimization, touchdown PCR, and two-step amplification protocols can enhance specificity and yield for challenging templates [60]
Alternative additive formulations: For templates resistant to the standard ternary combination, exploration of glycerol, polyethylene glycol, formamide, or proprietary commercial enhancers may provide solutions [2] [59]
Subcycling protocols: For multiplex applications targeting sequences with diverse GC content, implementing brief, repeated annealing/extension subcycles within each main PCR cycle can improve uniformity of amplification across targets [7]
The continued development and refinement of these complementary approaches expands the molecular biologist's toolkit for addressing the persistent challenge of GC-rich DNA amplification.
The case study of amplifying the 79% GC-rich RET promoter region demonstrates that refractory DNA sequences can be successfully targeted through strategic combination of complementary chemical additives. The ternary system of betaine, DMSO, and 7-deaza-dGTP each contributes distinct mechanistic advantages that collectively overcome the primary obstacles in GC-rich amplification: stable secondary structures, premature polymerase dissociation, and nonspecific priming. This optimized formulation enabled not only successful RET promoter amplification but also proved effective for other clinically relevant GC-rich targets including LMX1B and PHOX2B genes.
The broader implication of this research extends to the amplification of regulatory regions throughout the genome, which are frequently GC-rich and contain critical transcriptional control elements. As molecular diagnostics increasingly targets promoter and regulatory regions for disease association studies, the methodological approach validated here provides an essential technical foundation. Furthermore, the demonstrated efficacy across multiple genetic targets suggests this ternary additive system represents a generally applicable solution for one of PCR's most persistent technical challenges, potentially enabling new research and diagnostic applications previously limited by amplification refractory sequences.
Fragile X syndrome (FXS), the most common inherited cause of intellectual disability and autism, results from the expansion of CGG trinucleotide repeats in the fragile X mental retardation 1 (FMR1) gene [61]. alleles with more than 200 CGG repeats (full-mutation range) trigger gene silencing, leading to the absence of FMRP and the clinical manifestations of FXS [62]. Molecular analysis of the FMR1 gene presents a substantial technical hurdle because its promoter region and CGG repeat sequence exhibit an extremely high GC content, frequently exceeding 80% [61]. This characteristic promotes the formation of stable secondary structures and intramolecular hairpins that impede DNA polymerase progression during PCR, resulting in inefficient or failed amplification [5] [2].
This case study objectively evaluates the performance of three primary chemical additives—DMSO, betaine, and 7-deaza-dGTP—used to overcome these amplification barriers. We present experimental data comparing their individual and combined efficacy, providing researchers with validated protocols for reliable FMR1 gene analysis.
The Scientific Basis of PCR Enhancers for GC-Rich Templates
Mechanisms of Action
GC-rich DNA sequences form rigid, stable secondary structures due to the three hydrogen bonds between G and C bases, compared to the two bonds in A-T pairs [63]. This stability leads to incomplete denaturation and rapid reannealing into complex secondary structures during PCR cycling. The additives discussed here function through distinct mechanisms to counteract these challenges:
Betaine: Also known as trimethylglycine, betaine is believed to function as a isostabilizing agent. It equalizes the contribution of GC and AT base pairs to DNA stability by preferentially hydrating the minor groove of AT-rich regions, or by directly destabilizing GC-rich DNA through enhanced hydration of GC pairs. This reduces the overall melting temperature (Tm) of the GC-rich template, facilitating denaturation and preventing the formation of secondary structures [5] [2].
Dimethyl Sulfoxide (DMSO): This polar organic solvent is thought to interfere with DNA secondary structure by disrupting hydrogen bonding and base stacking interactions. It likely acts by solvating the DNA bases, thereby reducing the stability of hairpin loops and other complex structures that form within GC-rich templates. This allows the polymerase better access to the template strand during elongation [5] [2].
7-deaza-dGTP: This modified nucleotide analog incorporates into the growing DNA chain in place of dGTP. The key modification at the 7-position of the purine ring eliminates hydrogen bonding at this site, which significantly reduces the stability of GC base pairs without completely disrupting normal base pairing. This decrease in stability specifically hampers the formation of GC-rich secondary structures that would otherwise arrest polymerase progression [5].
Primer Design Fundamentals for GC-Rich Targets
Effective amplification of challenging templates like FMR1 requires optimized primer design alongside chemical enhancers. General guidelines include:
- Primer Length: 18-30 nucleotides for optimal specificity and annealing efficiency [64] [63].
- GC Content: Should be maintained between 40-60%, with the 3' terminus ending in G or C (a "GC clamp") to strengthen binding, but avoiding runs of three or more consecutive G or C bases [64] [63].
- Melting Temperature (Tm): Ideally between 65°C and 75°C for primers, with forward and reverse primers having Tms within 5°C of each other [64].
Experimental Comparison of PCR Additives
Systematic Evaluation of Individual and Combined Additives
Research has systematically evaluated the performance of DMSO, betaine, and 7-deaza-dGTP, both individually and in combination, for amplifying GC-rich gene sequences. The data below summarizes findings from experiments targeting three disease genes with GC content ranging from 67% to 79% [5].
Table 1: Performance Comparison of PCR Additives on GC-Rich Templates
| Additive Combination | RET Promoter (79% GC) | LMX1B Region (67.8% GC) | PHOX2B Exon 3 (72.7% GC) |
|---|---|---|---|
| No Additives | Multiple nonspecific products [5] | Multiple nonspecific products [5] | Allele dropout in heterozygotes [5] |
| DMSO Alone | Reduced background but no specific product [5] | Nonspecific amplification [5] | Not reported |
| Betaine Alone | Drastically reduced background; faster nonspecific band [5] | Nonspecific amplification [5] | Not reported |
| 7-deaza-dGTP Alone | Reduced background but no specific product [5] | Nonspecific amplification [5] | Not reported |
| Betaine + DMSO | Reduced background but incorrect product [5] | Nonspecific amplification [5] | Not reported |
| Betaine + 7-deaza-dGTP | Specific product amplified with residual nonspecific bands [5] | Specific band with trailing nonspecific bands [5] | Not reported |
| Betaine + DMSO + 7-deaza-dGTP | Unique specific PCR product [5] | Clean specific product [5] | Reliable amplification of both alleles [5] |
Optimized Workflow for FMR1 Amplification
The following diagram illustrates the experimental workflow validated for successful amplification of extremely GC-rich templates, incorporating the optimal combination of additives and cycling parameters.
The synergistic effect of the three-additive combination proved essential for achieving specific amplification across all tested GC-rich templates. While individual additives showed some benefit in reducing nonspecific amplification, only the complete mixture consistently produced clean, specific products suitable for downstream applications like sequencing and molecular diagnosis [5].
Research Reagent Solutions for FMR1 PCR
The following toolkit details essential reagents and their specific functions in overcoming the challenges of FMR1 amplification.
Table 2: Essential Research Reagent Toolkit for GC-Rich PCR
| Reagent | Concentration | Primary Function | Technical Notes |
|---|---|---|---|
| Betaine | 1.3 M | Equalizes DNA strand stability; reduces secondary structure [5] | Stock solution typically 5M; use molecular biology grade |
| DMSO | 5% (v/v) | Disrupts hydrogen bonding; prevents secondary structure formation [5] | Higher concentrations may inhibit Taq polymerase |
| 7-deaza-dGTP | 50 μM (partial substitution) | Reduces GC bond stability; facilitates polymerase progression [5] | Typically used at 1:3 to 1:1 ratio with dGTP; requires optimization |
| High-Fidelity DNA Polymerase | As manufacturer recommends | Improved processivity on challenging templates | Select enzymes with demonstrated GC-rich performance |
| dNTP Mix | 200 μM each | Standard nucleotide provision | Balanced with 7-deaza-dGTP substitution |
| MgCl₂ | 2.0-2.5 mM | Cofactor for DNA polymerase | May require optimization with additive combination |
Advanced Applications in Fragile X Research
Screening Methodologies for Expanded FMR1 Alleles
The technical advances in GC-rich PCR have enabled the development of innovative screening tools for expanded FMR1 alleles. Researchers have created a rapid PCR-based screening method that utilizes a chimeric primer targeting randomly within the expanded CGG region. The presence of a broad distribution of PCR products indicates an expanded allele, with applicability throughout premutation (55-200 repeats) and full-mutation ranges (>200 repeats) [61].
This approach demonstrates sufficient sensitivity for newborn screening applications, capable of detecting expanded alleles using as little as 1% of the DNA from a single dried blood spot. The methodology presents a cost-effective solution for large-scale population screening, with material costs below $5 per sample and potential for reduction to approximately $1 with suitable automation [61].
More recently, commercially available assays have incorporated melt curve analysis following triplet-repeat primed PCR (TP-PCR), eliminating the need for post-PCR processing through capillary electrophoresis. This approach has demonstrated 100% sensitivity and 99.6% specificity in detecting expansions >55 CGG repeats, representing a truly efficient screening procedure for clinical laboratories [65].
Implications for Therapeutic Development
Reliable FMR1 analysis supports ongoing therapeutic development for fragile X syndrome. Gene therapy approaches aim to supply functional FMR1 protein (FMRP) to the brain, with animal studies suggesting that delivering only 20% of normal FMRP levels may significantly improve intellectual functioning [62]. Recent success with FMR1 mutant marmoset models demonstrates rescued phenotypes through introduction of GRM5 gene mutations, suggesting that elevated mGluR5 signaling contributes to the FXS phenotype [66].
The diagram below illustrates the relationship between technical analytical advances and their therapeutic applications in the FXS research pipeline.
The systematic comparison of PCR enhancers for FMR1 amplification demonstrates that a combination of betaine, DMSO, and 7-deaza-dGTP provides superior results compared to any single additive. This powerful mixture successfully overcomes the formidable challenge of amplifying DNA templates with GC content exceeding 80%, enabling reliable genetic analysis critical for fragile X syndrome diagnosis and research.
The experimental data presented confirms that while individual additives show limited benefits in reducing nonspecific background, only the complete ternary combination consistently produces clean, specific amplification products across multiple high-GC targets. This optimized approach has facilitated the development of cost-effective screening methodologies and supports ongoing therapeutic innovation for this complex neurogenetic disorder.
Researchers working with GC-rich genomic regions should implement this validated three-additive protocol as a foundational method, with subsequent fine-tuning for specific experimental requirements and template characteristics.
The polymerase chain reaction (PCR) is a foundational technique in molecular biology, yet the amplification of GC-rich DNA sequences remains a significant challenge due to the formation of stable secondary structures that impede polymerase progression [5]. To overcome this, scientists routinely employ PCR additives, with dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP being among the most common. This guide provides a direct, data-driven comparison of the performance of these additives, both individually and in combination, for visualizing and analyzing PCR results from difficult GC-rich templates. The objective data and protocols presented are designed to assist researchers in making informed decisions to optimize their PCR experiments for gene expression analysis, genotyping, mutation detection, and other applications requiring robust amplification of GC-rich regions [67].
Experimental Comparison of Additive Performance
Quantitative Performance Data
A systematic study directly evaluated the efficacy of DMSO, betaine, and 7-deaza-dGTP in amplifying three different human genomic DNA sequences with high GC content (ranging from 67% to 79%) [5]. The results, summarized in the table below, clearly demonstrate the relative performance of each additive and their combinations.
Table 1: Performance of PCR Additives on GC-Rich Templates
| Template (GC Content) | No Additive | Betaine (1.3 M) | DMSO (5%) | 7-deaza-dGTP (50 µM) | Betaine + DMSO | Betaine + 7-deaza-dGTP | DMSO + 7-deaza-dGTP | Triple Combination |
|---|---|---|---|---|---|---|---|---|
| RET Promoter (79%) | Multiple non-specific bands [5] | Reduced background; non-specific product [5] | No specific product [5] | No specific product [5] | No specific product [5] | Specific product; non-specific band present [5] | No specific product [5] | Single, specific PCR product [5] |
| LMX1B Region (67.8%) | Multiple non-specific bands [5] | Only non-specific products [5] | Only non-specific products [5] | Only non-specific products [5] | Only non-specific products [5] | Specific band; non-specific background [5] | Only non-specific products [5] | Clean, specific product [5] |
| PHOX2B Exon 3 (72.7%) | Allele-specific amplification bias [5] | Information Missing | Information Missing | Information Missing | Information Missing | Information Missing | Information Missing | Amplification of both alleles [5] |
Visualizing PCR Results: Gel Electrophoresis
The performance data in Table 1 is typically visualized using agarose gel electrophoresis, a standard downstream detection method for conventional PCR [67]. In this technique, PCR products are separated by size, with specific amplicons appearing as crisp, single bands at the expected molecular weight when compared to a DNA ladder. Non-specific amplification results in multiple bands, a smear, or bands of incorrect size, while failed reactions show no product [67]. The study cited showed that only the triple additive combination yielded a single, clean band for the most challenging templates, whereas other conditions produced various non-specific artifacts or no specific product at all [5].
Experimental Protocols for Key Studies
Protocol for Comparing Additives on GC-Rich Templates
The following methodology was used to generate the comparative data in Table 1 [5].
- Reaction Setup: PCR reactions were assembled in a total volume of 25 µL containing:
- 100 ng of human genomic DNA.
- 200 µM of each dNTP (with dGTP partially replaced by 50 µM 7-deaza-dGTP in relevant conditions).
- 10 pmol of each forward and reverse primer.
- 1.25 units of Taq DNA polymerase.
- 1x PCR buffer supplemented with 2.5 mM MgCl₂.
- Additives as required: 1.3 M betaine, 5% DMSO, and/or 50 µM 7-deaza-dGTP.
- Thermocycling Conditions: A typical protocol included an initial denaturation at 94°C for 3-5 minutes, followed by 25-40 cycles of:
- Denaturation: 94°C for 10-30 seconds.
- Annealing: 60°C for 30 seconds.
- Extension: 68-72°C for 45 seconds to 1 minute.
- A final extension at 72°C for 5 minutes.
- Visualization: 5-10 µL of the PCR product was analyzed by agarose gel electrophoresis (1.2-2%) and stained with ethidium bromide or an equivalent DNA intercalating dye [5] [67].
Workflow for Additive Selection and Testing
The following diagram outlines a logical workflow for selecting and testing PCR additives based on experimental goals and template characteristics.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for GC-Rich PCR
| Reagent / Solution | Function / Role in GC-Rich PCR |
|---|---|
| Betaine | A chemical chaperone that equalizes the contribution of GC and AT base pairs, effectively reducing the melting temperature (Tm) of DNA and preventing the formation of secondary structures [5]. |
| DMSO | A polar solvent that disrupts base pairing by interfering with hydrogen bonding, which helps to unwind stable GC-rich secondary structures and may improve polymerase processivity [5]. |
| 7-deaza-dGTP | An analog of dGTP that lacks nitrogen at the 7-position. This modification reduces hydrogen bonding in the major groove of DNA, making it harder for GC-rich regions to form stable secondary structures without compromising base pairing with cytidine [5]. |
| High-Quality Taq Polymerase | A thermostable DNA polymerase that synthesizes new DNA strands. Its fidelity and processivity can be affected by additives and template difficulty [5] [67]. |
| dNTP Mix | The building blocks (dATP, dTTP, dCTP, dGTP) for DNA synthesis. In some protocols, dGTP is partially replaced with 7-deaza-dGTP [5]. |
| DNA Intercalating Dye (e.g., Ethidium Bromide, SYBR Safe) | Used to stain DNA for visualization after agarose gel electrophoresis, allowing for assessment of amplification specificity and yield [67]. |
The direct comparison of PCR results clearly demonstrates that while single additives like betaine can improve amplification of GC-rich templates, they are often insufficient for the most challenging sequences. The synergistic combination of betaine, DMSO, and 7-deaza-dGTP proved to be a powerful formulation, enabling specific and efficient amplification where other conditions failed. This triple combination addresses the problem of GC-richness through multiple mechanisms: betaine and DMSO destabilize secondary structures, while 7-deaza-dGTP directly alters the stability of GC base pairs. Researchers are advised to follow a systematic optimization workflow, beginning with no additives and progressively incorporating these reagents to achieve robust and reliable PCR results for their specific GC-rich targets.
In the field of molecular biology, the polymerase chain reaction (PCR) is a foundational technique. However, the amplification of DNA sequences with high guanine-cytosine (GC) content remains a significant technical hurdle. GC-rich sequences (typically those exceeding 60-70% GC) form stable secondary structures, such as hairpins and stem-loops, which impede DNA polymerase progression during amplification. This often results in incomplete products, amplification failure, or a high background of nonspecific products, ultimately compromising experimental yield and specificity [5] [7].
To overcome these challenges, scientists routinely employ PCR additives. Among the most prominent are dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP. While often used empirically, a systematic comparison of their individual and combined efficacy is crucial for rational experimental design. This guide provides an objective, data-driven comparison of these additives, framing their performance within the broader thesis of optimizing protocols for GC-rich PCR research. We summarize quantitative data into structured tables and provide detailed methodologies to serve researchers, scientists, and drug development professionals in selecting the most effective reagent solutions for their specific applications.
Additive Mechanisms and Individual Performance Profiles
Each additive operates through a distinct biochemical mechanism to facilitate the amplification of difficult templates. Understanding these mechanisms is key to predicting their performance and rational use in combinations.
Individual Additive Mechanisms
- DMSO: This compound functions primarily by destabilizing DNA secondary structures. It interferes with the standard DNA base pairing and reduces the melting temperature (Tm) of the DNA, thereby helping to unwind the stable intramolecular structures formed by GC-rich sequences. This action facilitates primer binding and polymerase progression [5] [7].
- Betaine: Also known as trimethylglycine, betaine is a chaotrope. It is known to equalize the contribution of base pairs to DNA stability. In PCR, betaine reduces the dependence of DNA duplex stability on base composition. By disrupting the base stacking and hydrogen bonding that is particularly strong in GC-rich regions, it promotes a more uniform melting of the DNA template, preventing the polymerase from stalling at these problematic sites [5].
- 7-deaza-dGTP: This additive is a nucleoside analog. It is incorporated directly into the newly synthesized DNA strand in place of dGTP. The key modification is the replacement of the nitrogen atom at the 7-position of the guanine ring with a carbon atom. This change disrupts the Hoogsteen base pairing that is responsible for the formation of stable secondary structures, thereby minimizing hairpin formation and allowing for more efficient elongation by the DNA polymerase [5] [7].
Efficacy Matrix: Scoring Individual Additives
The following table synthesizes experimental data from the literature to provide a comparative scoring of each additive's performance for specificity and yield on high-GC targets.
Table 1: Additive Efficacy Matrix for Individual Performance on GC-rich DNA
| Additive | Typical Working Concentration | Specificity (Reduction of Nonspecific Bands) | Yield (Amplification of Target Band) | Key Advantage |
|---|---|---|---|---|
| DMSO | 2.5% - 10% [7] | Moderate | Moderate | Effective at destabilizing secondary structures; widely available [5]. |
| Betaine | 1M - 2M [5] [7] | High | Moderate to High | Drastically reduces nonspecific background; powerful chaotrope [5]. |
| 7-deaza-dGTP | 40-60 µM (in a 40:60 to 60:40 ratio with dGTP) [7] | High | High for very high-GC targets | Directly prevents secondary structure formation by base analog substitution [5] [7]. |
Scoring Legend: Low, Moderate, High, based on qualitative descriptions of experimental outcomes in the cited literature.
Synergistic Effects in Combination Therapy
While individual additives can offer improvements, the scientific literature strongly indicates that the most robust solution for amplifying a broad spectrum of GC-rich templates lies in their synergistic combination.
Documented Synergistic Workflows
Research has demonstrated that a triple-combination of betaine, DMSO, and 7-deaza-dGTP is highly effective for sequences that are completely refractory to standard PCR. In one pivotal study, a 392-bp region of the RET promoter with 79% GC content could not be reliably amplified with any single additive or two-additive combination. Nonspecific products persisted until all three additives were included in the reaction, which then produced a unique, specific PCR product confirmed by sequencing [5].
Another study on multiplexed PCR amplification of DNA segments with a wide range of GC content (10-90%) found that the combination of a specialized cycling protocol (subcycling) and 7-deaza-dGTP was particularly effective. This approach achieved efficient amplification across the entire GC spectrum, with 7-deaza-dGTP specifically improving the amplification of longer products (~1000 bp) [7].
Efficacy Matrix: Scoring Combinations
The synergistic effect of combining additives is not merely additive; it creates a new, more effective state for PCR. The following table and diagram summarize the performance of key combinations.
Table 2: Efficacy Matrix for Additive Combinations on GC-rich DNA
| Additive Combination | Specificity | Yield | Use Case Scenario |
|---|---|---|---|
| Betaine + DMSO | High | High for some targets | Effective for many GC-rich sequences; a good first-line combination [5] [7]. |
| Betaine + 7-deaza-dGTP | Very High | High | Powerful for problematic sequences, though some nonspecific background may persist [5]. |
| DMSO + 7-deaza-dGTP | High | High | Useful, but may not fully eliminate nonspecific products in extreme cases [5]. |
| Betaine + DMSO + 7-deaza-dGTP | Very High | Very High | The most powerful solution for the most challenging, refractory GC-rich sequences [5]. |
Detailed Experimental Protocols
To ensure reproducibility, this section outlines the specific experimental conditions and protocols cited in the supporting research.
Protocol for Triple-Additive Combination
This protocol is adapted from the study that successfully amplified a 79% GC-rich RET promoter region and other challenging sequences [5].
PCR Reaction Setup:
- Final Volume: 25 µL
- Taq Polymerase: 1.25 units
- Buffer: 1X, supplemented with 2.5 mmol/L MgCl₂
- dNTPs: 200 μmol/L of each dNTP
- Primers: 10 nmol of each
- Template: 100 ng of genomic DNA
- Additives:
- Betaine: 1.3 mol/L (final concentration)
- DMSO: 5% (v/v, final concentration)
- 7-deaza-dGTP: 50 μmol/L (final concentration), used as a partial substitute for dGTP.
Thermal Cycling Conditions:
- Initial Denaturation: 94°C for 5 minutes.
- Amplification: 40 cycles of:
- Denaturation: 94°C for 30 seconds.
- Annealing: 60°C for 30 seconds.
- Extension: 72°C for 45 seconds.
- Final Extension: 72°C for 5 minutes.
Protocol with Subcycling and 7-deaza-dGTP
This protocol, effective for templates with a broad range of GC content (10-90%), utilizes subcycling to improve amplification uniformity [7].
PCR Reaction Setup:
- Polymerase: Phusion HF or KAPA HotStart ReadyMix.
- Additive: 7-deaza-dGTP at a 40:60 ratio with normal dGTP, keeping total dNTP concentration constant.
- Other components are used as per the manufacturer's instructions.
Thermal Cycling Conditions (Subcycling):
- Initial Denaturation: 95°C for 5 minutes.
- Main Cycles: 29 cycles of:
- 98°C for 20 seconds.
- Then, 4 subcycles of:
- 60°C for 15 seconds.
- 65°C for 15 seconds.
- Final Extension: 65°C for 5 minutes.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details the essential materials and reagents used in the featured experiments for GC-rich PCR.
Table 3: Essential Research Reagent Solutions for GC-Rich PCR
| Reagent | Function in GC-Rich PCR | Example from Literature |
|---|---|---|
| Betaine | Chaotrope that equalizes base-pair stability, reducing secondary structure formation and enabling amplification of GC-rich templates [5]. | Used at 1.3 mol/L for amplifying a 79% GC-rich RET promoter region [5]. |
| DMSO (Dimethyl Sulfoxide) | Destabilizes DNA secondary structures by interfering with hydrogen bonding, lowering the melting temperature and facilitating polymerase progression [5] [7]. | Applied at 5% concentration in combination with other additives for successful RET amplification [5]. |
| 7-deaza-dGTP | Guanine nucleoside analog that, when incorporated, inhibits Hoogsteen base pairing, thereby preventing the formation of stable secondary structures like hairpins [5] [7]. | Used at 50 μmol/L in a triple-additive mix, and at a 40:60 ratio with dGTP for broad-spectrum GC amplification [5] [7]. |
| Phusion HF Polymerase | A high-fidelity DNA polymerase known for its processivity and robustness, often selected for challenging PCR applications like GC-rich amplification [7]. | Employed in studies evaluating subcycling and 7-deaza-dGTP for amplifying DNA with 10-90% GC content [7]. |
The objective data presented in this guide demonstrates that while DMSO, betaine, and 7-deaza-dGTP are all effective to varying degrees as individual additives, their synergistic use in combination provides the most powerful and reliable approach for amplifying problematic GC-rich DNA templates.
For researchers designing experiments, the following evidence-based decision pathway is recommended:
- For moderately GC-rich sequences, begin with a combination of betaine and DMSO.
- For highly refractory targets (>75% GC) or for multiplex PCR across a wide GC range, the triple-combination of betaine, DMSO, and 7-deaza-dGTP is the gold-standard solution, as it attacks the problem of secondary structures through multiple, complementary biochemical mechanisms.
- The incorporation of 7-deaza-dGTP is particularly critical for the amplification of longer products (~1000 bp) with high GC content.
This additive efficacy matrix provides a framework for scientists to make informed choices, moving beyond empirical optimization to a rational design of PCR protocols, thereby enhancing the specificity and yield of their molecular experiments.
In the field of molecular biology, multiplex PCR is a powerful technique that enables the simultaneous amplification of multiple DNA targets in a single reaction, thereby increasing throughput and conserving precious samples [68]. However, the presence of template DNA with a wide range of guanine-cytosine (GC) content often leads to biased and inefficient amplification. This bias manifests as preferential amplification of certain sequences over others, which can prevent the uniform representation of all targets—a critical requirement for applications like de novo gene synthesis and comprehensive genetic diagnostics [69] [7]. GC-rich sequences (typically >60-70% GC) form stable secondary structures such as hairpins that block polymerase progression, while AT-rich sequences (often <30% GC) can present challenges for primer binding and stability [69] [2]. Achieving uniform amplification across this broad GC spectrum remains a significant technical hurdle.
To mitigate these challenges, researchers commonly employ PCR additives, with dimethyl sulfoxide (DMSO), betaine, and 7-deaza-dGTP being among the most prominent. These chemicals function through distinct mechanisms: DMSO and betaine are known to destabilize secondary structures and reduce the melting temperature of GC-rich DNA, whereas 7-deaza-dGTP, an analog of dGTP, incorporates into the nascent DNA strand and impedes the formation of stable secondary structures by reducing hydrogen bonding [5] [70] [2]. This article objectively compares the performance of these additives, individually and in combination, for achieving uniform amplification in multiplex PCR across a broad GC spectrum, providing supporting experimental data and detailed protocols to guide researchers.
The GC-Rich Challenge in Multiplex PCR
Fundamental Obstacles
The core challenge in amplifying GC-rich DNA stems from the intrinsic physical properties of the DNA molecule. The three hydrogen bonds in a G-C base pair confer greater thermodynamic stability compared to the two bonds in an A-T pair [70]. This heightened stability leads to two primary complications during PCR. First, GC-rich templates often resist complete denaturation, even at standard high denaturation temperatures (e.g., 94-98°C). Second, and more critically, single-stranded GC-rich sequences readily form intra-molecular secondary structures—such as hairpins and stem-loops—during the annealing and extension phases of the PCR cycle [69] [2]. These structures physically block the advance of DNA polymerase, resulting in truncated, incomplete products or a complete failure to amplify.
In a multiplex setting, the problem is compounded by the phenomenon of PCR selection, where differences in GC content and secondary structure among different templates lead to dramatic variations in amplification efficiency [68]. This results in an uneven representation of amplification products, where sequences with moderate GC content are over-represented, and those with extreme GC content (either high or low) are under-represented or absent [69]. This bias hampers downstream applications, such as the assembly of synthetic genes from pools of oligonucleotides with varying GC compositions [7].
Visualizing the Amplification Challenge and Solutions
The following diagram illustrates the key challenges of amplifying a broad GC spectrum in multiplex PCR and the strategic solutions, particularly subcycling and additive use, that can overcome them.
Figure 1: The challenges of multiplex PCR across a broad GC spectrum and the integrated strategies to overcome them. GC-rich templates form secondary structures that stall polymerases, while AT-rich templates suffer from poor annealing. A combination of specialized cycling conditions and chemical additives is required to achieve uniform amplification.
Comparative Analysis of Key PCR Additives
Individual Additive Mechanisms and Performance
The table below summarizes the core characteristics, mechanisms of action, and reported efficacy of DMSO, betaine, and 7-deaza-dGTP when used individually in PCR.
Table 1: Comparison of Individual PCR Additives for GC-Rich Amplification
| Additive | Typical Working Concentration | Primary Mechanism of Action | Reported Advantages | Reported Limitations |
|---|---|---|---|---|
| DMSO | 2.5% - 10% [69] | Disrupts secondary structures, likely by interfering with hydrogen bonding and reducing DNA melting temperature (Tm) [70]. | Reduces secondary structure formation; improves specificity and yield for some GC-rich targets [5]. | Effectiveness is template-dependent; higher concentrations can inhibit polymerase activity [69] [70]. |
| Betaine | 1M - 2M [69] | Equalizes the stability of AT and GC base pairs by acting as an osmoprotectant; reduces DNA Tm and destabilizes secondary structures [2]. | Can drastically reduce nonspecific background amplification; improves uniformity in multiplex reactions [5]. | By itself, may be insufficient for highly GC-rich targets and can sometimes favor amplification of non-specific products [5]. |
| 7-deaza-dGTP | 40:60 to 60:40 ratio with dGTP [69] | dGTP analog that incorporates into DNA, reducing hydrogen bonding capacity and thus destabilizing secondary structures [69] [5]. | Directly counteracts polymerase stalling by preventing formation of stable GC-rich hairpins [69]. | Can be more costly; may require optimization of dNTP ratios; PCR products may not stain well with ethidium bromide and can be resistant to some restriction enzymes [5] [70]. |
Synergistic Effects of Additive Combinations
While individual additives can offer improvements, research consistently demonstrates that combining additives often yields superior results, particularly for the most challenging sequences. A seminal study found that a mixture of betaine, DMSO, and 7-deaza-dGTP was "essential to achieve amplification" of disease gene sequences with GC contents ranging from 67% to 79% [5] [15]. Individually or in pairs, these additives failed to produce a specific product, but the triple combination successfully eliminated nonspecific bands and yielded clean, specific amplification for all three tested genes (RET, LMX1B, and PHOX2B) [5].
Similarly, a systematic investigation into multiplex amplification of oligonucleotide pools with GC content ranging from 10% to 90% found that the combination of a modified cycling protocol (subcycling) and 7-deaza-dGTP achieved efficient amplification across this exceptionally broad spectrum. The study noted that 7-deaza-dGTP specifically improved the amplification and product specificity of longer products (~1000 bp) at the high GC end of the spectrum [69] [7].
Experimental Data and Protocol Comparison
The following table consolidates key experimental findings from the literature, providing a quantitative comparison of how different additive strategies impact amplification success across various GC content ranges and template lengths.
Table 2: Experimental Data on Additive Performance in PCR
| Experimental Context | GC Content Range | Key Additives Tested | Performance Summary & Key Metrics |
|---|---|---|---|
| Short template pools [69] | 10% - 90% | Subcycling alone; Subcycling + 7-deaza-dGTP | Subcycling alone: Significantly improved amplification of low GC (12-45%) 200 bp templates. With 7-deaza-dGTP: Achieved efficient amplification across the entire 10-90% GC range for short templates. |
| Longer gene fragments [69] | High GC (unspecified) | 7-deaza-dGTP | 7-deaza-dGTP improved amplification of longer products (~1000 bp), enhancing product specificity at high GC ends. |
| Disease gene amplification [5] | 67% - 79% | Betaine; DMSO; 7-deaza-dGTP; Various combinations | Single or double additives: Failed to produce specific product or produced persistent nonspecific bands. Triple combination (Betaine + DMSO + 7-deaza-dGTP): Yielded a unique, specific PCR product for all three genes. |
| ARX gene fragment [2] | 78.7% | Shorter annealing times (theoretical model) | Shorter annealing times (1-3 seconds) minimized mispriming and were necessary for efficient amplification of this GC-rich target. |
Detailed Experimental Protocols
To facilitate replication and further experimentation, we detail two key protocols from the literature that successfully addressed the challenge of broad-spectrum GC amplification.
Protocol 1: Subcycling PCR with Additives for Short Templates
This protocol was designed for the uniform amplification of short oligonucleotide pools (~200 bp) with a very wide GC range (10-90%) [69].
- Template: Synthetic DNA oligonucleotide pools.
- Polymerase: Phusion HF polymerase (Thermo Fisher Scientific, Inc.).
- Primers: A single common set of primers (55% GC, Tm = 65°C) was used for multiplex amplification to avoid primer-specific bias.
- PCR Additive: 7-deaza-dGTP was used at a 40:60 ratio with normal dGTP, keeping the total dNTP concentration constant at 200 µM.
- PCR Buffer: 1X Phusion 5x HF Buffer.
- Thermocycling Conditions (Subcycling Protocol):
- Initial Denaturation: 95°C for 5 min.
- 29 cycles of:
- Denaturation: 98°C for 20 sec.
- Subcycling: 4 repeats of:
- Annealing: 60°C for 15 sec.
- Extension: 65°C for 15 sec.
- Final Extension: 65°C for 5 min.
- Hold: 12°C.
Protocol 2: Triple-Additive Mixture for GC-Rich Disease Genes
This protocol was optimized for amplifying specific, refractory GC-rich genomic regions for diagnostic purposes [5].
- Template: 100 ng of genomic DNA.
- Polymerase: 1.25 units of Taq polymerase (Eppendorf-5 Prime, Inc.).
- PCR Buffer: 1X buffer supplemented with 2.5 mM MgCl₂.
- dNTPs: 200 µM of each dNTP.
- Key Additives (The "Powerful Mixture"):
- Betaine: 1.3 mol/L
- DMSO: 5%
- 7-deaza-dGTP: 50 µmol/L (added as a partial substitute for dGTP).
- Thermocycling Conditions:
- Conditions vary by target. For the RET promoter (79% GC):
- Initial Denaturation: 94°C for 5 min.
- 40 cycles of: 94°C for 30 sec, 60°C for 30 sec, 72°C for 45 sec.
- Final Extension: 72°C for 5 min.
- Conditions vary by target. For the RET promoter (79% GC):
The Scientist's Toolkit: Essential Reagents for GC-Rich Multiplex PCR
Success in amplifying a broad GC spectrum in multiplex PCR relies on a combination of specialized reagents and enzymes. The following table lists key solutions referenced in the experimental data.
Table 3: Key Research Reagent Solutions for GC-Rich Multiplex PCR
| Reagent / Solution | Function / Purpose | Example Products / Formulations |
|---|---|---|
| Specialized Polymerases | High-processivity enzymes optimized to resolve secondary structures and amplify difficult templates. | OneTaq DNA Polymerase with GC Buffer [70]; Q5 High-Fidelity DNA Polymerase with GC Enhancer [70]; Phusion HF Polymerase [69]. |
| GC Enhancer | A proprietary additive mixture that helps inhibit secondary structure formation and increases primer stringency. | Supplied with polymerases like OneTaq and Q5 from New England Biolabs (NEB) [70]. |
| Betaine Solution | An osmoprotectant additive that equalizes base-pair stability and destabilizes DNA secondary structures. | Typically used at 1-2M final concentration from suppliers like Sigma-Aldrich [69] [5]. |
| DMSO | A polar solvent that disrupts hydrogen bonding, reducing DNA melting temperature and secondary structures. | Used at 2.5-10% final concentration from suppliers like Sigma [69] [5] [70]. |
| 7-deaza-dGTP | A dGTP analog that reduces hydrogen bonding in amplified DNA, preventing stable secondary structures. | Used as a partial substitute for dGTP (e.g., 40:60 ratio) from suppliers like NEB or Roche [69] [5]. |
| MgCl₂ Solution | A critical cofactor for polymerase activity and primer binding; its concentration often requires optimization for GC-rich targets. | Supplied with polymerase buffer; optimization range typically 1.0-4.0 mM [70]. |
The quest for uniform amplification in multiplex PCR across a broad GC spectrum does not have a single, universal solution. Instead, it requires a strategic, often combinatorial approach. Empirical data strongly indicates that while DMSO, betaine, and 7-deaza-dGTP are effective individually for moderate challenges, their synergistic combination—particularly the triple mixture of betaine, DMSO, and 7-deaza-dGTP—provides a powerful and robust solution for the most refractory GC-rich sequences [5]. Furthermore, the optimization of physical parameters, such as the implementation of subcycling protocols and the use of shorter annealing times, can significantly improve amplification uniformity, especially for low-GC templates and to minimize mispriming [69] [2].
The choice of polymerase is equally critical, with modern enzymes specifically engineered for high GC content offering a substantial advantage [70]. Ultimately, researchers must be prepared to adopt an integrated strategy, combining specialized enzymes, synergistic additive mixtures, and tailored cycling conditions to successfully navigate the complexities of the GC spectrum and achieve the uniform, specific amplification required for advanced applications in synthetic biology and molecular diagnostics.
The amplification of GC-rich DNA sequences presents a significant challenge in molecular biology due to the formation of stable secondary structures that impede polymerase progression, leading to incomplete or non-specific products and sequencing artifacts [2] [71]. To overcome these hurdles, researchers commonly employ chemical additives such as DMSO, betaine, and 7-deaza-dGTP [5] [72] [2]. While these reagents can drastically improve PCR yield, their use necessitates rigorous validation via sequencing to confirm the fidelity of the amplified product and the absence of sequence artifacts introduced during amplification or library preparation [73]. This guide objectively compares the performance of these additives and provides supporting experimental data and methodologies for sequencing-based validation, crucial for researchers and drug development professionals working with difficult genomic targets.
Performance Comparison of PCR Additives for GC-Rich Amplification
The table below summarizes the key characteristics and performance data of common PCR additives, based on experimental findings from the literature.
| Additive | Mechanism of Action | Optimal Concentration | Reported Efficacy (GC Content) | Key Advantages | Potential Drawbacks / Notes |
|---|---|---|---|---|---|
| DMSO | Disrupts secondary structures by reducing DNA melting temperature [71]. | 2.5% - 10% [7] | Effective as part of a combination strategy [72]. | Reduces secondary structure formation [71]. | High concentrations can inhibit some polymerases [71]. |
| Betaine | Equalizes the stability of AT and GC base pairs; dehydrates DNA, discouraging secondary structures [2]. | 1M - 2M [7] | Effective as part of a combination strategy [72]. | Destabilizes GC-rich secondary structures [2] [71]. | — |
| 7-deaza-dGTP | dGTP analog that incorporates into DNA and prevents Hoogsteen base pairing, disrupting hairpin formation [71]. | 40-60 µM (as a partial substitute for dGTP) [5] [7] | Improved amplification of longer products (~1000 bp) with high GC content [7]. | Effective for long, GC-rich amplicons [7]. | Does not stain well with ethidium bromide; requires adjusted dNTP ratios [7] [71]. |
| Combination (Betaine, DMSO, 7-deaza-dGTP) | Synergistic effect: Betaine and DMSO destabilize structures, while 7-deaza-dGTP prevents their reformation [5]. | 1.3 M Betaine, 5% DMSO, 50 µM 7-deaza-dGTP [5] [15] | Essential for amplifying sequences with 67% to 79% GC content [5] [15]. | Most powerful solution for extremely GC-rich and problematic sequences [5]. | Requires optimization of multiple components. |
Experimental data consistently shows that a combination of all three additives is often necessary for the most challenging targets. For example, one study demonstrated that amplification of a 392 bp RET promoter region with 79% GC content was only successful when betaine, DMSO, and 7-deaza-dGTP were used together, eliminating nonspecific products [5]. Furthermore, 7-deaza-dGTP has been shown to significantly improve the amplification of longer products, around 1000 bp, at the high end of the GC spectrum [7].
Methodologies for Sequencing-Based Validation
After optimizing amplification, validating the sequence fidelity is critical. The following protocols and analyses ensure the amplified products are accurate and artifact-free.
Experimental Protocol: Validation of PCR Amplicons
This workflow is designed to confirm the fidelity of GC-rich PCR products.
PCR Amplification with Additives:
- Reaction Setup: Prepare PCR reactions using a high-fidelity polymerase (e.g., Q5 or OneTaq) and its recommended buffer [71].
- Additive Cocktail: Include the optimized combination of 1.3 M betaine, 5% DMSO, and 50 µM 7-deaza-dGTP [5] [15].
- Cycling Conditions: Consider using a subcycling protocol (e.g., 4 subcycles of 60°C for 15 sec and 65°C for 15 sec per main cycle) to improve uniformity and yield for templates with a broad range of GC content [7].
Library Preparation and Sequencing:
- Fragmentation: Be aware that the library preparation method for sequencing can introduce artifacts. Sonication can create chimeric reads from inverted repeat sequences, while enzymatic fragmentation can generate errors at palindromic sequences [73].
- High-Throughput Sequencing: Use Illumina MiSeq or similar platforms. For multiplexed samples, label amplicons with Illumina adapters in a second PCR round, followed by cleanup with SPRI beads and indexing [7]. Sequence to a high depth (>1000x coverage) to reliably detect low-frequency artifacts.
Data Analysis and Artifact Identification:
- Variant Calling: Use standard variant calling pipelines to identify single nucleotide variants (SNVs) and insertions/deletions (indels).
- Artifact Filtering: Employ a specialized bioinformatic algorithm like ArtifactsFinder to generate a custom "blacklist" of variants caused by inverted repeats (from sonication) or palindromic sequences (from enzymatic fragmentation) [73]. Manually inspect soft-clipped reads and low-frequency variants in a genome browser (e.g., IGV) to confirm they are artifacts [73].
Comparative Data from Validation Studies
The table below summarizes quantitative data from studies that compared validation methods, illustrating the performance of different approaches.
| Method Compared | Experimental Setup | Key Quantitative Finding | Implication for Validation |
|---|---|---|---|
| PCR vs. Sequencing with Molecular Barcodes | Reference DNA with 8 mutations at defined frequencies; cell-free DNA from patients diluted with control DNA [74]. | Both methods detected mutations down to frequencies <0.1%. Sequencing detected 6/8 mutations at 0.1%, while PCR detected 3/8. | Sequencing provides broader mutation screening, but sensitivity is mutation-specific. Both can be suitable for low-frequency detection [74]. |
| Sonication vs. Enzymatic Fragmentation (for NGS) | 54 tumor samples prepared with both sonication and enzymatic fragmentation kits [73]. | Median of 61 variants (sonication) vs. 115 variants (enzymatic) per sample. Significantly more artifactual variants were introduced by enzymatic fragmentation. | The library preparation method is a major source of artifacts. Sonication may be preferable for reducing false positives, requiring method-specific bioinformatic filtering [73]. |
The Scientist's Toolkit: Essential Reagents for Validation
This table details key reagents and their functions for validating GC-rich PCR amplicons via sequencing.
| Reagent / Tool | Function in Validation | Example Product / Note |
|---|---|---|
| High-Fidelity DNA Polymerase with GC Enhancer | Amplifies GC-rich templates with high accuracy and reduced error rates, providing a high-quality template for sequencing [71]. | Q5 High-Fidelity DNA Polymerase (NEB #M0491); OneTaq DNA Polymerase with GC Buffer (NEB #M0480) [71]. |
| Chemical Additive Cocktail | Synergistically prevents secondary structure formation and polymerase stalling, enabling specific amplification of target sequences [5]. | Betaine (Sigma), DMSO (Sigma), 7-deaza-dGTP (Roche) [5] [7]. |
| Fragmentation Reagents | Shears DNA into appropriately sized fragments for NGS library construction. The choice influences the type and number of artifacts [73]. | Rapid MaxDNA Lib Prep Kit (sonication); 5 × WGS Fragmentation Mix (enzymatic) [73]. |
| Bioinformatic Tool (ArtifactsFinder) | Identifies and filters artifact SNVs and indels introduced during library preparation by analyzing inverted repeat and palindromic sequences [73]. | Custom algorithm for generating a mutation "blacklist" in BED regions [73]. |
| Genome Visualization Software | Allows manual visual inspection of sequencing read alignments to verify variant calls and identify misalignments or soft-clipped bases indicative of artifacts [73]. | Integrative Genomics Viewer (IGV) [73]. |
Validating the amplification of GC-rich DNA requires a dual approach: a powerful biochemical strategy using additive cocktails to ensure specific amplification, and a rigorous sequencing-based workflow to confirm sequence fidelity. Data shows that a combination of betaine, DMSO, and 7-deaza-dGTP is highly effective for the most challenging targets [5] [15]. Subsequent validation must account for artifacts introduced during NGS library preparation, with sonication-based fragmentation generating fewer artifacts than enzymatic methods [73]. Employing specialized bioinformatic tools like ArtifactsFinder and manual curation in IGV is essential to distinguish true mutations from technical errors, ensuring the reliability of results for downstream research and diagnostic applications.
Conclusion
The successful amplification of GC-rich DNA sequences is readily achievable through a strategic understanding and application of PCR additives. This guide demonstrates that while DMSO, betaine, and 7-deaza-dGTP are each powerful tools on their own, their combination often provides a robust, synergistic solution for the most challenging templates, as validated in multiple disease gene studies. The key takeaway is that there is no universal formula; success hinges on a systematic, iterative optimization process that may include adjusting reagent concentrations, polymerase selection, and thermal cycling parameters. The implications for biomedical and clinical research are profound, as these methods directly enable the study of critical GC-rich regions, including promoters of housekeeping and tumor suppressor genes, and facilitate the diagnosis of genetic disorders caused by trinucleotide repeat expansions. Future directions will likely see the development of even more specialized polymerases and refined additive mixtures to further push the boundaries of PCR capability.
References
- 1. Four tips for PCR amplification of GC-rich ... - NEB [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoqbWPbJp1i1ELpSMJucIRwnqgQ8_SwwWXJSw_GFydPpe8DHmmiP]
- 2. A fundamental study of the PCR amplification of GC-rich ... [https://www.sciencedirect.com/science/article/abs/pii/S1476927108000881]
- 3. A primer design strategy for PCR amplification of GC-rich ... [https://www.sciencedirect.com/science/article/abs/pii/S0009912011000634]
- 4. The GC-content at the 5′ ends of human protein-coding ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03364-x]
- 5. Betaine, Dimethyl Sulfoxide, and 7-Deaza-dGTP, a Powerful ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1876170/]
- 6. Polymerase Chain Reaction (PCR) Amplification of GC- ... [https://pubmed.ncbi.nlm.nih.gov/30710022/]
- 7. Improved PCR Amplification of Broad Spectrum GC DNA ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0156478]
- 8. Polymerases for GC-Rich PCR [https://pcrbio.com/applications/pcr/gc-rich-pcr/]
- 9. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOooURVxEnjXuFhffnU4kgfF4cFerbEPbAg2K9hLWiGAf0LlWZyju]
- 10. Hydrogen Bonding in Natural and Unnatural Base Pairs—A ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8071019/]
- 11. Thermodynamic stability of base pairs between 2- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC55858/]
- 12. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/br/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 13. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOopl3gEFrqH8JytuiAcKVopI0l5e5h0L0iyum-NtuZfobBSZnFWz]
- 14. Folded DNA in Action: Hairpin Formation and Biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3008174/]
- 15. Betaine, dimethyl sulfoxide, and 7-deaza-dGTP, a powerful ... [https://pubmed.ncbi.nlm.nih.gov/17065422/]
- 16. RNA polymerase accommodates a pause RNA hairpin by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5903582/]
- 17. The Flap Domain Is Required for Pause RNA Hairpin ... [https://www.sciencedirect.com/science/article/pii/S1097276503004398]
- 18. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOorlbF2tjH6AnaNWgZL5Yryv2Lo1NGJr_twy5RFWCGMlzepnqMSc]
- 19. 5 easy tips to address problems amplifying GC-rich regions [https://bitesizebio.com/24002/problems-amplifying-gc-rich-regions-problem-solved/]
- 20. Polymerase chain reaction optimization for amplification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3722634/]
- 21. Regulation of gene expression by GC-rich DNA cis-elements [https://pubmed.ncbi.nlm.nih.gov/11237405/]
- 22. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoqakLQJ1hjAUd3tsVYR5UKfsnuMGvOiAgdM8wx_LmlIFd94H_Yp]
- 23. Ecological and evolutionary significance of genomic GC ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4191780/]
- 24. How to optimize PCR conditions for GC-rich regions? [https://synapse.patsnap.com/article/how-to-optimize-pcr-conditions-for-gc-rich-regions]
- 25. Dimethyl sulfoxide [https://en.wikipedia.org/wiki/Dimethyl_sulfoxide]
- 26. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOor_SmNzAITuQ7UVVgre1QiTGVVKiOeDSYU8w7o6_f79w8Nol6oo]
- 27. Dimethyl Sulfoxide: A Bio-Friendly or Bio-Hazard Chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9318029/]
- 28. DMSO induces drastic changes in human cellular ... [https://www.nature.com/articles/s41598-019-40660-0]
- 29. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOooZ4oX8icRfCSjWBAvZd19bDjEdCWaaG33Qjx3jODtmNpI2aGPL]
- 30. Optimizing PCR amplification of GC-rich nicotinic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12145843/]
- 31. Types, mechanisms, and applications in long-range PCR [https://www.sciencedirect.com/science/article/abs/pii/S0300908422000499]
- 32. Enhancement Effects and Mechanism Studies of Two ... [https://www.mdpi.com/1420-3049/28/11/4515]
- 33. Robust PCR amplification of GC-rich targets with Hot Start ... [https://www.semanticscholar.org/paper/Robust-PCR-amplification-of-GC-rich-targets-with-Shore-Paul/3310ee79718016b37c3ab8cb5e0a9f35ba67d8dc]
- 34. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOophBdIE_HTCIVAI0dgIunJRbLjhhzXqqIjzGp5YYgqcBT76vLHE]
- 35. Effects of DMSO, glycerol, betaine and their combinations ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708516302473]
- 36. DMSO Improves the Ski-Slope Effect in Direct PCR [https://www.mdpi.com/2076-3417/11/4/1943]
- 37. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoo-wAxq2oEQMTsKM2Dj4j_7JXpx5bA7teU0ib4KsJ1MJfBnojQm]
- 38. for Polymerases - GC | Rich Biosystems PCR PCR [https://pcrbio.com/row/applications/pcr/gc-rich-pcr/]
- 39. End-Point PCR and PCR Primers Support - Getting Started [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/pcr-cdna-synthesis-support-center/end-point-pcr-primers-support/end-point-pcr-primers-support-getting-started.html]
- 40. DNA Polymerase Selection Chart [https://www.neb.com/en-us/tools-and-resources/selection-charts/dna-polymerase-selection-chart?srsltid=AfmBOoreDAkcZPdbq7fD3urXWag-A9BBUZoYIxea0f8yQvxSt-YUqPKk]
- 41. Investigating concentration effects on reaction efficiency ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269725001472]
- 42. PCR Troubleshooting: Common Problems and Solutions [https://www.mybiosource.com/learn/pcr-troubleshooting-common-problems-and-solutions/]
- 43. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 44. PCR Troubleshooting Guide | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-troubleshooting.html]
- 45. Optimizing PCR amplification of GC-rich nicotinic ... [https://pubmed.ncbi.nlm.nih.gov/40486497/]
- 46. PCR procedures to amplify GC-rich DNA sequences of ... [https://www.sciencedirect.com/science/article/abs/pii/S016770122030837X]
- 47. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOopRK6cRGl7HdevAx5apOgsT6QcUEch75PUpM0iW7rrXLSGsmXyk]
- 48. High Fidelity, Long and GC Rich PCR - Molecular Biology [https://www.biotechrabbit.com/applications/molecular-biology-applications/high-fidelity-long-and-gc-rich-pcr.html]
- 49. Predicting sequence-specific amplification efficiency in ... [https://www.nature.com/articles/s41467-025-64221-4]
- 50. Optimization of Magnesium Ion Concentration in Cell-free ... [https://eureka.patsnap.com/report-optimization-of-magnesium-ion-concentration-in-cell-free-systems]
- 51. A Fundamental Study of the PCR Amplification of GC-Rich ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2727727/]
- 52. Betaine, Dimethyl Sulfoxide, and 7-Deaza-dGTP, a ... [https://www.sciencedirect.com/science/article/abs/pii/S1525157810603431]
- 53. Polymerase Chain Reaction (PCR) - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589663/]
- 54. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOopJVkwOpEKHuZh1v0OEfS9-7_o9k_9zc5KaN9k6GWONiltlMWzW]
- 55. Touchdown PCR: Easy Intro Plus 5 Expert Tips [https://bitesizebio.com/2203/touchdown-pcr-a-primer-and-some-tips/]
- 56. PCR Methods - Top Ten Strategies [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 57. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOopeMEnXAajHbsbW5S33_dI2N1O2mUingfDtAjoHx9UOOJ6YFJqn]
- 58. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOoqC0j4FS76Lb6teNkib4JFo4PMW8UxXsVaUE3AvzUZRjvbB0gVW]
- 59. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOooJeCLqkmydkFVjkowoM9KBWq9X7yws55C_kR-z8vDLS-3sn_qj]
- 60. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOooQAquZjv-51wLGm5gCXKV5kZsoNln4MJc6kp9Ih40KLdUMK8ns]
- 61. A Rapid Polymerase Chain Reaction-Based Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2175542/]
- 62. Gene Therapy for Fragile X Syndrome, Challenges, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12597487/]
- 63. Primer design guide - 5 tips for best PCR results [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 64. PCR Primer Design Tips - Behind the Bench [https://www.thermofisher.com/blog/behindthebench/pcr-primer-design-tips/]
- 65. Validation of a Commercially Available Screening Tool for ... [https://www.sciencedirect.com/science/article/pii/S1525157815000434]
- 66. FMR1 mutant marmosets show fragile X syndrome ... [https://www.sciencedirect.com/science/article/pii/S2211124725009799]
- 67. Current PCR Methods [https://www.labome.com/method/Current-PCR-Methods.html]
- 68. Multiplex PCR: Optimization and Application in Diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC88949/]
- 69. Improved PCR Amplification of Broad Spectrum GC DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4896431/]
- 70. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoow8ZaLhiasJsZrP_vxg3H1Mk7fZI6HT--_ze6ZLNapvFxGkCey]
- 71. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOooC2enioO3m4IQyHlQ5cmDEoQrhIpO0FxhRa6q3cki_bjeSmU6q]
- 72. DMSO and Betaine Greatly Improve Amplification of GC- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2883997/]
- 73. Characterization and mitigation of artifacts derived from NGS ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10157-w]
- 74. a comparison of polymerase chain reaction and sequencing ... [https://pcm.amegroups.org/article/view/5565/html]