BN-PAGE vs SDS-PAGE: A Comprehensive Guide to Protein Complex Analysis for Biomedical Research
This article provides a detailed comparison of Blue Native-PAGE (BN-PAGE) and SDS-PAGE, two fundamental electrophoretic techniques for protein analysis.
BN-PAGE vs SDS-PAGE: A Comprehensive Guide to Protein Complex Analysis for Biomedical Research
Abstract
This article provides a detailed comparison of Blue Native-PAGE (BN-PAGE) and SDS-PAGE, two fundamental electrophoretic techniques for protein analysis. Tailored for researchers and drug development professionals, it explores the foundational principles of both methods, with BN-PAGE preserving native protein complexes for functional studies and SDS-PAGE providing denaturing separation for molecular weight determination. The scope extends to practical methodological protocols, troubleshooting for complex samples like membrane proteins, and validation through techniques such as in-gel activity assays and 2D electrophoresis. By synthesizing key operational and application differences, this guide aims to empower scientists in selecting the optimal technique for their specific research goals in structural biology and therapeutic development.
Core Principles: How BN-PAGE and SDS-PAGE Work at the Molecular Level
In the field of protein analysis, electrophoresis stands as a fundamental technique for separating and characterizing complex protein mixtures. The choice between native and denaturing polyacrylamide gel electrophoresis (PAGE) represents a critical decision point that directly impacts experimental outcomes, particularly in the study of protein complexes. While SDS-PAGE has become the default method for determining molecular weight and assessing sample purity, Native PAGE (including Blue Native PAGE) offers unique advantages for preserving functional protein interactions. This guide provides a detailed comparison of these techniques, framed within the context of protein complex analysis, to empower researchers in selecting the optimal approach for their specific research objectives.
Core Principles and Separation Mechanisms
The fundamental distinction between these electrophoretic techniques lies in their treatment of protein structure during separation.
Denaturing SDS-PAGE employs the anionic detergent sodium dodecyl sulfate (SDS) along with reducing agents like dithiothreitol (DTT) or beta-mercaptoethanol to dismantle native protein structures. SDS binds uniformly to polypeptide chains, masking intrinsic charges and conferring a consistent negative charge-to-mass ratio, while reducing agents break disulfide bonds [1] [2]. Heating samples to 70-100°C completes the denaturation process, resulting in linearized proteins that separate based almost exclusively on molecular weight as they migrate through the gel matrix [3] [4].
In contrast, Native PAGE (including BN-PAGE) operates under non-denaturing conditions without SDS or reducing agents [1]. This technique preserves proteins in their folded, functional states, maintaining enzymatic activity, protein-protein interactions, and bound cofactors [5] [3]. Separation depends on a combination of the protein's intrinsic charge, size, and three-dimensional shape, allowing researchers to study quaternary structures and native complexes [6] [4].
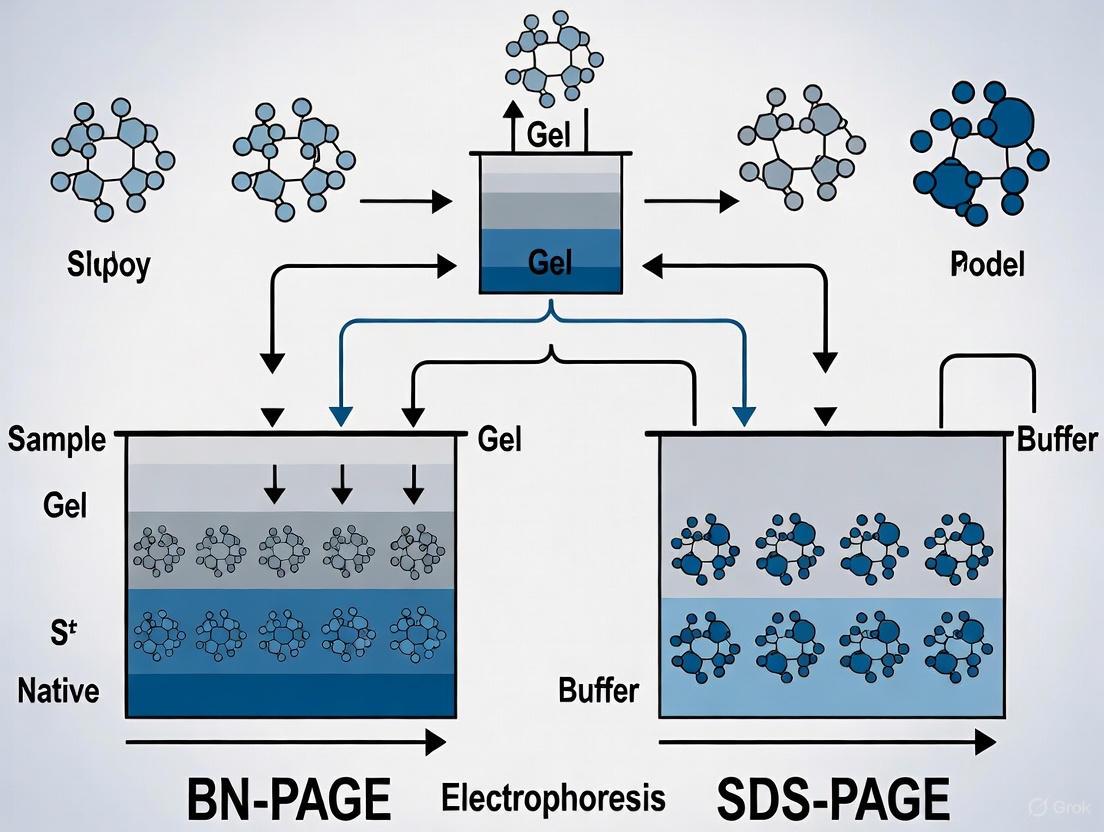
The workflow below illustrates the key procedural differences between these two fundamental methods:
Comparative Analysis: SDS-PAGE vs Native PAGE
The table below summarizes the key technical and application differences between these electrophoretic methods:
| Parameter | SDS-PAGE | Native PAGE |
|---|---|---|
| Gel Conditions | Denaturing [1] [7] | Non-denaturing [1] [7] |
| Key Reagents | SDS, DTT/β-mercaptoethanol [1] [2] | No denaturants; Coomassie in BN-PAGE [1] [8] |
| Sample Preparation | Heating required (70-100°C) [1] [4] | No heating [1] [2] |
| Separation Basis | Molecular weight only [3] [7] | Size, charge, and shape [3] [4] |
| Protein Structure | Denatured, linearized [5] [2] | Native conformation preserved [5] [3] |
| Protein Function | Lost after separation [1] | Retained after separation [1] [4] |
| Protein Recovery | Not recoverable functional [1] [7] | Recoverable functional [1] [7] |
| Primary Applications | Molecular weight determination, western blotting, purity assessment [9] [3] [2] | Protein complexes, oligomeric state, enzymatic activity [9] [5] [3] |
| Typical Running Temperature | Room temperature [1] | 4°C [1] |
Experimental Data and Performance Metrics
Recent research has quantified the performance differences between these techniques, particularly regarding functional preservation. A modified approach called Native SDS-PAGE (NSDS-PAGE) demonstrates the potential for balancing resolution with functional preservation.
Quantitative Comparison of Metal Retention and Enzyme Activity
The following table summarizes experimental data comparing standard SDS-PAGE, BN-PAGE, and NSDS-PAGE in preserving metalloprotein function:
| Method | Zinc Retention (%) | Enzyme Activity Retention (Model Enzymes) | Resolution |
|---|---|---|---|
| Standard SDS-PAGE | 26% [8] | 0/9 active [8] | High [5] [8] |
| BN-PAGE | Not specified | 9/9 active [8] | Lower than SDS-PAGE [8] |
| NSDS-PAGE | 98% [8] | 7/9 active [8] | Comparable to SDS-PAGE [8] |
This data demonstrates that NSDS-PAGE, which eliminates EDTA and reduces SDS concentration in running buffer from 0.1% to 0.0375% while omitting the heating step, achieves near-complete metal retention while maintaining high resolution [8]. This approach represents a valuable hybrid technique for metalloprotein analysis.
Pre-fractionation Efficiency for Protein Complex Analysis
In affinity purification workflows for protein complex isolation, the choice of pre-fractionation method significantly impacts protein identification. When comparing SDS-PAGE and Strong Cation Exchange (SCX) chromatography for pre-fractionating nuclear protein complexes (Bmi-1 and GATA3), SCX consistently identified approximately 3-fold more proteins than SDS-PAGE [10]. This efficiency gap was especially pronounced for the Bmi-1 complex, where the target protein was expressed at low levels [10].
Detailed Experimental Protocols
Sample Preparation: Mix protein sample with 4X LDS sample buffer containing SDS and reducing agent. For standard SDS-PAGE, use 106 mM Tris HCl, 141 mM Tris Base, 0.51 mM EDTA, 2% LDS, 10% glycerol, pH 8.5 [8].
Denaturation: Heat samples at 70-100°C for 10 minutes to complete denaturation [8] [4].
Gel Preparation: Use precast or freshly cast polyacrylamide gels (typically 4-12% or 10% Bis-Tris gels) with SDS incorporated into the gel matrix [8] [4].
Electrophoresis: Load samples and molecular weight markers. Run at constant voltage (200V) for approximately 45 minutes using MOPS SDS running buffer (50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, 0.1% SDS, pH 7.7) at room temperature [8].
Detection: Visualize proteins using Coomassie, silver staining, or transfer to membrane for western blotting [10] [4].
Sample Preparation: Mix protein sample with 4X BN-PAGE sample buffer (50 mM BisTris, 50 mM NaCl, 10% glycerol, 0.001% Ponceau S, pH 7.2). Do not heat [8].
Gel Preparation: Use precast NativePAGE Novex 4-16% Bis-Tris gels or equivalent [8].
Electrophoresis: Load samples and native protein standards. Run at constant voltage (150V) for 90-95 minutes using cathode (50 mM BisTris, 50 mM Tricine, 0.02% Coomassie G-250, pH 6.8) and anode (50 mM BisTris, 50 mM Tricine, pH 6.8) running buffers at room temperature [8].
Detection: Visualize proteins with compatible stains or process for activity assays [8].
Sample Preparation: Mix protein sample with 4X NSDS sample buffer (100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5). Do not heat [8].
Gel Equilibration: Pre-run precast NuPAGE Novex 12% Bis-Tris gels at 200V for 30 minutes in double distilled H₂O to remove storage buffer and unpolymerized acrylamide [8].
Electrophoresis: Run at 200V for 30 minutes using NSDS-PAGE running buffer (50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7) [8].
Research Reagent Solutions
The table below outlines essential materials and their functions for these electrophoretic techniques:
| Reagent/Material | Function | Application |
|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | Denatures proteins, confers negative charge [1] [4] | SDS-PAGE |
| DTT or β-mercaptoethanol | Reduces disulfide bonds [1] [2] | SDS-PAGE |
| Coomassie G-250 | Imparts charge for electrophoresis, staining [8] | BN-PAGE |
| Acrylamide/Bis-acrylamide | Forms porous gel matrix for separation [4] | All PAGE |
| TEMED/Ammonium Persulfate | Catalyzes acrylamide polymerization [4] | All PAGE |
| Tris-based Buffers | Maintains pH during electrophoresis [8] [4] | All PAGE |
| Glycerol | Increases sample density for gel loading [1] [8] | Sample preparation |
| MOPS/Tricine Buffers | Running buffer systems [8] | SDS-PAGE, NSDS-PAGE |
Application Workflows in Protein Complex Research
The diagram below illustrates typical research workflows for analyzing protein complexes using these complementary techniques:
Native and denaturing electrophoresis techniques offer complementary capabilities for protein complex analysis. SDS-PAGE remains the gold standard for determining subunit molecular weight and purity assessment, while Native PAGE (particularly BN-PAGE) excels at preserving functional protein interactions and quaternary structures. The emerging NSDS-PAGE method demonstrates that hybrid approaches can balance resolution with functional preservation, particularly for metalloprotein studies. Researchers should select methodologies based on their specific objectives: SDS-PAGE for analytical separation and molecular weight determination, Native PAGE for functional studies and complex analysis, and SCX chromatography for maximum protein identification in proteomic applications. Understanding these techniques' strengths and limitations enables more informed experimental design in protein complex research and drug development.
In the realm of protein research, particularly in the separation and analysis of protein complexes via techniques like Blue Native PAGE (BN-PAGE) and SDS-PAGE, the choice of detergent is far from a mere technicality. It is a critical decision that dictates the success of an experiment. Detergents, or surfactants, are amphiphilic molecules essential for solubilizing membranes and proteins, but their chemical nature can either preserve or obliterate the delicate structures and functions of biological molecules. Non-ionic detergents, characterized by their uncharged hydrophilic heads, are celebrated for their mild, protein-friendly properties. In contrast, ionic detergents, which carry a distinct electrical charge, are powerful agents for denaturation and complete disruption. This guide provides an objective comparison of these detergent classes, framing their performance within the context of modern protein complex analysis and supporting conclusions with experimental data.
Fundamental Detergent Chemistry and Properties
At the molecular level, the distinction between ionic and non-ionic surfactants is defined by the nature of their hydrophilic (water-attracting) head groups.
- Ionic Surfactants possess a charged hydrophilic head group. When dissolved in water, they dissociate into ions, leading to electrostatic interactions with the solution and other molecules [11]. This class is further divided:
- Anionic Surfactants (e.g., Sodium Dodecyl Sulfate - SDS) have a negatively charged head group [11] [8]. They are renowned for their strong cleaning and foaming power but can be irritating to skin and eyes [11] [12].
- Cationic Surfactants (e.g., Dodecyltrimethylammonium bromide - DTAB) have a positively charged head group [11] [13]. They are often used for their antimicrobial properties and are found in products like fabric softeners and hair conditioners [11].
- Non-Ionic Surfactants, such as Triton X-100 and n-Dodecyl-β-d-maltoside (DDM), feature an uncharged hydrophilic head group [11] [14]. They do not dissociate into ions in water, which makes them generally milder, less irritating, and less disruptive to protein structure and function [11] [15].
The table below summarizes the core differences that arise from these distinct chemical structures.
Table 1: Core Characteristics of Ionic and Non-Ionic Detergents
| Property | Ionic Detergents | Non-Ionic Detergents |
|---|---|---|
| Chemical Structure | Charged head group (positive or negative) [11] | Uncharged head group [11] |
| Protein Interaction | Strong, often denatures proteins [8] [15] | Mild, can preserve native structure/function [14] [8] |
| Foam Production | High [11] | Low [15] |
| Tolerance (Skin/Eyes) | More irritating [11] | Milder, less irritating [11] |
| Hard Water Tolerance | Low (precipitate with ions) [16] | High (effective in hard water) [16] [15] |
| Compatibility | Can interact with charged ingredients [11] | Highly compatible with various ingredients [11] |
A simple method to distinguish between the two classes in the lab is a conductivity test. Since ionic surfactants dissociate into ions, they will conduct electricity in solution, whereas non-ionic surfactants will not significantly alter the solution's conductivity [11].
Detergent Applications in Protein Electrophoresis
The fundamental properties of detergents dictate their roles in key electrophoretic techniques for protein analysis. The choice between BN-PAGE and SDS-PAGE is fundamentally a choice between preserving or destroying native protein structures.
Blue Native PAGE (BN-PAGE): Preserving Complexes with Non-Ionic Agents
The primary goal of BN-PAGE is to separate intact protein complexes in their native, enzymatically active state [14] [8]. To achieve this, non-ionic detergents are the reagents of choice.
- Experimental Protocol for BN-PAGE: A standard protocol involves solubilizing membrane protein complexes with a non-denaturing detergent. The solubilized samples are then mixed with Coomassie Blue G dye, which binds to proteins in a non-stoichiometric way, imparting a negative charge shift to enable migration toward the anode while preserving the complex's structure [14]. Typical detergent concentrations range from 0.5% to 2% final concentration, or a detergent-to-protein ratio from 1:1 to 10:1 [14].
- Key Reagents: The most frequently used non-ionic detergents for BN-PAGE include digitonin, n-dodecyl-β-d-maltoside, and Triton X-100 [14]. Their "mildness" refers to their ability to solubilize membranes without disrupting the critical lipid-protein and protein-protein interactions that hold complexes together [14].
SDS-PAGE: Denaturing Separation with Ionic Agents
In stark contrast, SDS-PAGE aims to separate individual protein subunits based almost exclusively on their molecular weight. This requires the complete denaturation of proteins and the disruption of all non-covalent interactions.
- The Role of Ionic SDS: The anionic detergent Sodium Dodecyl Sulfate (SDS) is the cornerstone of this method. Proteins are denatured by heating in the presence of SDS and a reducing agent. SDS binds uniformly to the polypeptide backbone, with a constant mass ratio, which masks the protein's intrinsic charge and imparts a uniform negative charge [8] [17]. This results in all proteins having a similar charge-to-mass ratio, allowing separation by size as they migrate through a polyacrylamide gel [17].
- Limitation and a Hybrid Approach: A significant limitation of standard SDS-PAGE is the deliberate destruction of functional properties, including enzymatic activity and the presence of non-covalently bound cofactors like metal ions [8]. To address this, a modified method called native SDS-PAGE (NSDS-PAGE) has been developed. This protocol omits the heating step and reduces the SDS concentration in the running buffer (e.g., to 0.0375%), which allows for high-resolution separation while retaining Zn²⁺ in metalloproteins and preserving the activity of many enzymes [8].
Table 2: Detergent Use in Key Electrophoretic Methods
| Electrophoresis Method | Primary Detergent Class | Key Detergents | Objective | Impact on Protein Complexes |
|---|---|---|---|---|
| BN-PAGE | Non-Ionic [14] | Dodecyl maltoside, Digitonin, Triton X-100 [14] | Separate native complexes | Preserves intact structure and function |
| SDS-PAGE | Ionic (Anionic) [8] | Sodium Dodecyl Sulfate (SDS) [8] | Separate denatured subunits | Disassembles complexes into subunits |
| NSDS-PAGE | Ionic (Anionic, reduced) [8] | SDS (at low concentration) [8] | High-resolution native separation | Preserves some metal ions & activity |
Comparative Experimental Data and Emerging Trends
Recent research provides quantitative data on how detergent selection directly impacts experimental outcomes, such as the number of proteins identified and the success of novel reconstitution techniques.
Impact on Proteome Analysis and Identification
A 2025 study investigating detergent screens for bottom-up proteomics on Escherichia coli highlights a clear benefit of using a diverse detergent portfolio. The research employed ionic detergents (SDS and cationic DTAB), a non-ionic detergent (dendritic triglycerol detergent), and related hybrid detergents [13]. The findings were striking:
- Combining proteomics datasets from the different detergents increased the number of unique protein identities (IDs) observed from 1,604 to 2,169 [13].
- This demonstrates that the solubilizing bias of each detergent is unique and that cationic detergents and hybrid detergents, often underrepresented in proteomics, can significantly enhance the observable proteome when used alongside traditional anionic and non-ionic agents [13].
The Rise of Detergent-Free and Hybrid Technologies
The limitations of detergents in preserving the precise native lipid environment have spurred the development of advanced technologies.
- Detergent-Free Reconstitution: A groundbreaking 2025 study published in Nature Communications introduced "DeFrND," a method using engineered membrane-scaffolding peptides (DeFrMSPs) to directly extract membrane proteins from native cell membranes into nanodiscs, completely bypassing the need for detergents [18]. This approach was critical for studying detergent-sensitive complexes like the MalFGK2 transporter, which, when extracted with peptides, maintained its functional coupling, unlike when solubilized with detergents or amphipathic polymers [18].
- Hybrid Detergents: Research is also exploring the potential of ionic/nonionic hybrid detergents, which covalently combine headgroups from both classes. Data suggests their solubilizing properties are not a simple average of the parent detergents and can contribute to increased proteome coverage, as noted above [13].
Table 3: Experimental Performance Data of Detergent Classes
| Experimental Context | Non-Ionic Detergent Performance | Ionic Detergent Performance | Hybrid/Novel Approach |
|---|---|---|---|
| Proteome Coverage (E. coli) | Contributes unique protein IDs [13] | SDS provides high baseline IDs; cationic DTAB adds unique IDs [13] | Combining all data increases unique protein IDs from 1604 to 2169 [13] |
| Membrane Protein Complex Study | Standard for preserving activity in BN-PAGE [14] | Often denatures and inactivates complexes [8] | Detergent-free native nanodiscs (DeFrND) preserve functional coupling better than polymers or detergents [18] |
| Protein Solubilization Bias | Specific bias for certain protein classes [13] [14] | Strong, complementary bias for different protein classes [13] | Hybrid detergents exhibit unique solubilization profiles not seen in canonical detergents [13] |
The Scientist's Toolkit: Essential Reagents for Detergent-Based Research
This table catalogs key reagents and their functions for researchers designing experiments in protein complex analysis.
Table 4: Research Reagent Solutions for Protein Analysis
| Reagent/Solution | Function/Description | Key Example(s) |
|---|---|---|
| Strong Ionic Detergent | Denatures proteins, binds uniformly to impart charge for SDS-PAGE. | Sodium Dodecyl Sulfate (SDS) [8] |
| Mild Non-Ionic Detergent | Solubilizes membranes and proteins while preserving native complexes for BN-PAGE. | n-Dodecyl-β-d-maltoside (DDM), Triton X-100, Digitonin [14] |
| Cationic Detergent | Offers complementary solubilization bias in proteomic screens. | Dodecyltrimethylammonium bromide (DTAB) [13] |
| Hybrid Detergent | Covalently fused ionic/nonionic headgroups for unique solubilization properties. | Ionic/Nonionic hybrid structures [13] |
| Membrane Scaffold Peptide | Enables detergent-free extraction of membrane proteins into native nanodiscs. | Engineered Apolipoprotein-A1 mimetic peptides (DeFrMSPs) [18] |
| Coomassie Dye (G-250) | Imparts charge shift for electrophoretic migration in BN-PAGE without full denaturation. | Coomassie Blue G [14] |
The dichotomy between mild non-ionic and strong ionic detergents is a foundational principle in protein science. Non-ionic detergents are indispensable for the isolation and analysis of intact, functional protein complexes using techniques like BN-PAGE. Conversely, ionic detergents like SDS are powerful tools for deconstructing proteomes into their constituent subunits for precise molecular weight determination via SDS-PAGE. The emerging experimental data unequivocally shows that no single detergent is universally superior. The most advanced proteomic and structural biology research now leverages the complementary strengths of both classes, and even looks beyond them to hybrid and fully detergent-free systems. The optimal experimental outcome hinges on a rational selection of the solubilizing agent, aligned with the fundamental question being asked about the protein or complex under investigation.
In the field of protein analysis, electrophoresis is a foundational technique for separating and characterizing proteins. Two principal methods, Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) and Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), represent fundamentally different approaches defined by how they manage protein state and charge. SDS-PAGE employs denaturing conditions to unfold proteins and impart a uniform negative charge, separating polypeptides primarily by molecular weight. In contrast, BN-PAGE operates under non-denaturing conditions to preserve native protein conformations, multi-subunit complexes, and biological functions, separating complexes based on both size and intrinsic charge [1] [19]. This guide provides an objective comparison of these techniques, focusing on their mechanistic principles, experimental outcomes, and applications in drug development and basic research.
Core Principles: A Tale of Two Techniques
SDS-PAGE: Separation by Molecular Weight
SDS-PAGE is a denaturing electrophoresis technique that separates individual polypeptide chains based almost exclusively on their molecular weight [1]. The anionic detergent sodium dodecyl sulfate (SDS) binds extensively to hydrophobic regions of proteins, disrupting their tertiary structure and unfolding them. This SDS coating confers a uniform negative charge density, meaning all proteins experience similar charge-to-mass ratios. Consequently, when an electric field is applied, separation occurs as proteins migrate through the polyacrylamide gel matrix at rates inversely proportional to their molecular size, with smaller proteins moving faster [1] [8]. The reducing agents present in the buffer, such as DTT or β-mercaptoethanol, break disulfide bonds, ensuring complete denaturation into constituent subunits [20].
BN-PAGE: Separation of Native Complexes by Size and Charge
BN-PAGE is a non-denaturing technique designed to separate intact protein complexes in their functional, folded state [21] [22] [19]. Instead of SDS, the anionic dye Coomassie Brilliant Blue G-250 is used. This dye binds non-covalently to the surface of protein complexes, imparting a negative charge without causing significant dissociation or denaturation [22] [20] [19]. The resulting separation in the polyacrylamide gel depends on both the native molecular mass and the shape of the protein complex, as well as the number of dye molecules bound, which relates to the complex's surface charge [1] [19]. This preserves protein-protein interactions, enzymatic activity, and the binding of non-covalently attached cofactors, including metal ions [8] [22].
The diagram below illustrates the fundamental workflows and outcomes of these two techniques.
Quantitative Comparison: Technical Specifications and Outputs
Direct Technique Comparison
The choice between BN-PAGE and SDS-PAGE has profound implications for the type of data obtained, as summarized in Table 1.
Table 1: Fundamental differences between SDS-PAGE and BN-PAGE
| Criteria | SDS-PAGE | BN-PAGE |
|---|---|---|
| Separation Principle | Molecular weight only [1] | Size, charge, and shape of complex [1] |
| Gel Conditions | Denaturing [1] | Non-denaturing [1] |
| SDS Presence | Present [1] | Absent [1] |
| Buffer Composition | Contains reducing agent (e.g., DTT, BME) [1] | No reducing agent [1] |
| Sample Preparation | Protein samples are heated [1] | Protein samples are not heated [1] |
| Protein Net Charge | Uniformly negative [1] | Can be positive or negative [1] |
| Typely | Room temperature [1] | 4°C [1] |
| Protein State | Denatured, unfolded [1] | Native conformation, folded [1] |
| Protein Function Post-Separation | Lost [1] [8] | Retained [1] [8] |
| Primary Application | Determine molecular weight, check purity/expression [1] | Study structure, subunit composition, and function [1] |
Experimental Data Comparison
The methodological differences lead to distinct functional outcomes, particularly regarding the preservation of enzymatic activity and metal cofactors, which is critical for functional studies.
Table 2: Experimental outcomes for protein activity and metal retention
| Experimental Parameter | SDS-PAGE | BN-PAGE | Native SDS-PAGE (NSDS-PAGE) |
|---|---|---|---|
| Retention of Enzyme Activity | All nine model enzymes denatured and inactive [8] | All nine model enzymes active [8] | Seven of nine model enzymes active [8] |
| Zinc (Zn²⁺) Retention in Proteomic Samples | 26% retention [8] | High retention (implied) [8] | 98% retention [8] |
| Suitable for In-Gel Activity Assays | No [8] [22] | Yes [22] [23] | Yes (for most enzymes) [8] |
| Suitable for Western Blotting | Yes, standard method [8] | Yes, requires specific protocols [21] | Yes, requires optimization [8] |
Detailed Experimental Protocols
SDS-PAGE Protocol
The following is a standard denaturing SDS-PAGE protocol based on established methods [8].
- Sample Preparation: Mix the protein sample with an SDS-based sample loading buffer (e.g., containing LDS) and a reducing agent like Dithiothreitol (DTT) [8].
- Denaturation: Heat the mixture at 70°C–95°C for 5–10 minutes to ensure complete denaturation and reduction of disulfide bonds [1] [8].
- Gel Loading: Load the denatured samples into precast or hand-cast polyacrylamide gels. A pre-stained protein molecular weight standard should be loaded in at least one well.
- Electrophoresis: Run the gel at a constant voltage (e.g., 150-200 V) at room temperature using an SDS-containing running buffer (e.g., MOPS-SDS buffer) until the dye front reaches the bottom of the gel [8].
- Post-Electrophoresis Analysis: The gel can be stained (e.g., Coomassie, silver stain) for total protein visualization or used for Western blotting [24].
BN-PAGE Protocol
This BN-PAGE protocol is adapted from methodologies used for mitochondrial complexes [21] [23] and whole cell lysates [25] [26].
- Sample Preparation (Solubilization): Isolate the subcellular fraction (e.g., mitochondria) or whole cells. Solubilize the sample on ice using a mild, non-ionic detergent. Common choices include:
- Clarification: Centrifuge the solubilized lysate at high speed (e.g., 16,000–72,000 x g) for 30 minutes to remove insoluble debris. The supernatant contains the solubilized protein complexes [21].
- Dye Addition: Add Coomassie Blue G-250 dye (e.g., as a 5% solution) to the supernatant to impart charge to the complexes [21].
- Gel Loading: Load the prepared sample into a native gradient gel (e.g., 4-16% or 6-13% acrylamide) without heating [21] [23].
- Electrophoresis: Run the gel under native conditions at 4°C [1]. The cathode buffer (pH ~7.0) contains Coomassie dye, while the anode buffer lacks it. Start electrophoresis at a low voltage (e.g., 150 V) and potentially increase after the dye front has migrated partially into the gel [23].
- Post-Electrophoresis Analysis:
- First Dimension Analysis: Complexes can be visualized by Coomassie staining, immunoblotting with specific antibodies, or in-gel activity assays [22] [23].
- Second Dimension (2D) Analysis: For subunit analysis, a lane from the BN-PAGE gel can be excised, soaked in SDS buffer, and placed on an SDS-PAGE gel. This 2D BN/SDS-PAGE separates complexes in the first dimension and their constituent subunits in the second [25] [23] [20].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of BN-PAGE and SDS-PAGE relies on specific reagents, each with a critical function.
Table 3: Key reagents for protein electrophoresis
| Reagent / Solution | Function | Technique |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Denatures proteins and confers uniform negative charge. | SDS-PAGE [1] |
| Dithiothreitol (DTT) or β-Mercaptoethanol | Reducing agent that breaks disulfide bonds. | SDS-PAGE [1] [20] |
| Coomassie Brilliant Blue G-250 | Imparts negative charge to protein surfaces under native conditions. | BN-PAGE [21] [22] [19] |
| n-Dodecyl-β-D-maltoside (DDM) | Mild non-ionic detergent for solubilizing membrane protein complexes. | BN-PAGE [21] [23] [19] |
| Digitonin | Mild detergent used to preserve labile supercomplexes. | BN-PAGE [19] |
| 6-Aminocaproic Acid | Zwitterionic salt that aids protein solubilization and complex stability. | BN-PAGE [21] [19] |
| Bis-Tris | A buffering agent used to maintain stable pH in native conditions. | BN-PAGE [21] [23] |
Application in Research and Drug Development
The complementary nature of BN-PAGE and SDS-PAGE makes them invaluable across research and development.
Studying Protein Complex Dynamics and Assembly: BN-PAGE is the preferred method for investigating the assembly pathways of multi-subunit complexes, identifying assembly intermediates, and detecting the presence of supercomplexes, such as those in the mitochondrial respiratory chain [22] [19]. This is crucial for understanding diseases arising from defective complex assembly.
Identifying Post-Translational Modifications within Complexes: The 2D BN/SDS-PAGE approach is powerful for identifying specific subunits within a complex that undergo post-translational modifications. For example, it has been used to pinpoint which subunits of mitochondrial complex I are modified by the lipid peroxidation product 4-hydroxynonenal (HNE) in diabetic models [23].
Functional Proteomics and Target Validation: In drug development, confirming a target protein's function and interaction partners is essential. BN-PAGE can validate that a drug candidate does not disrupt essential protein-protein interactions by demonstrating the intactness of complexes after treatment. Furthermore, it allows for in-gel activity assays to test how potential therapeutics affect enzymatic function of native complexes [22].
Analyzing Whole Cellular Lysates: Advancements have shown that with dialysis to remove interfering substances, BN-PAGE can be applied to whole cellular lysates, enabling the study of protein complexes like the proteasome and tumor suppressors (e.g., p53) in a systems biology context [25] [26]. This provides a broader view of cellular interaction networks.
Historical Development and Key Innovators in Electrophoresis
The history of electrophoresis represents a relentless pursuit of analytical precision in biochemical research. From its initial discovery to its sophisticated contemporary applications, electrophoretic techniques have fundamentally transformed our capacity to resolve and characterize biological molecules. This evolution is particularly evident in the development of methods for protein analysis, where the critical dichotomy emerged between denaturing techniques that maximize resolution and native techniques that preserve functional integrity. This guide focuses on the pivotal development of Blue Native PAGE (BN-PAGE) and its comparison with traditional SDS-PAGE, providing researchers with a comprehensive framework for selecting appropriate methodologies based on experimental objectives. The trajectory from early moving-boundary methods to today's high-resolution native gel techniques illustrates how technological innovations have continuously expanded the horizons of proteomic research [27].
Historical Timeline and Key Innovators
The development of electrophoresis spans more than two centuries, marked by foundational discoveries and technological breakthroughs that have progressively enhanced its resolving power and applications.
Table 1: Key Historical Developments in Electrophoresis
| Year | Innovator(s) | Development | Significance |
|---|---|---|---|
| 1807 | Reuß & Strakhov | First observation of electrokinetic phenomena | Discovered clay particle migration in water under electric field [27] |
| 1930s | Arne Tiselius | Moving-boundary electrophoresis apparatus | Enabled analytical separation of chemical mixtures; Nobel Prize (1948) [27] |
| 1950s | Multiple groups | Zone electrophoresis | Introduced solid/gel matrices to separate compounds into discrete zones [27] |
| 1959 | Raymond & Weintraub | Acrylamide gel electrophoresis | Acrylamide introduced as superior supporting medium [27] |
| 1970s | Patrick O'Farrell | Two-dimensional gel electrophoresis | Combined IEF and SDS-PAGE for unprecedented resolution [28] |
| Early 1990s | Schägger & von Jagow | Blue Native PAGE (BN-PAGE) | Enabled separation of native membrane protein complexes [29] [30] |
| 2000s | Multiple groups | Protocol optimizations | Standardized polyacrylamide gels for improved reproducibility [27] |
The earliest roots of electrophoresis trace back to 1807, when Russian professors Peter Ivanovich Strakhov and Ferdinand Frederic Reüss at Moscow University first observed that clay particles dispersed in water would migrate under the influence of a constant electric field—the first documented electrokinetic phenomenon [27]. Throughout the 19th and early 20th centuries, scientists including Johann Wilhelm Hittorf, Walther Nernst, and Friedrich Kohlrausch developed the theoretical and experimental foundations for understanding ion movement in solutions, creating mathematical descriptions of electrochemistry and developing methods for creating moving boundaries of charged particles [27].
The transformative breakthrough for biochemical applications came from Arne Tiselius, who in the 1930s developed the moving-boundary electrophoresis apparatus with support from the Rockefeller Foundation [27]. His method, fully described in his seminal 1937 paper, enabled the analytical separation of chemical mixtures and earned him the Nobel Prize in Chemistry in 1948 [27]. Despite its impact, the Tiselius method could not completely separate electrophoretically similar compounds, leading to the development of zone electrophoresis in the 1940s and 1950s, which used filter paper or gels as supporting media to separate compounds into discrete, stable bands [27].
The introduction of polyacrylamide gels in 1959 by Raymond and Weintraub represented a monumental advance, providing a superior matrix for protein separation [27]. This set the stage for the proteomics revolution when Patrick O'Farrell developed two-dimensional gel electrophoresis in the 1970s, combining isoelectric focusing (IEF) with SDS-PAGE to achieve unprecedented resolution of complex protein mixtures [28]. O'Farrell's innovation emerged from his graduate work at the University of Colorado, where he sought to analyze developmental mutations in Volvox by creating a separation method with dramatically increased resolution [28].
The most significant development for native protein complex analysis came in the early 1990s when Hermann Schägger and Gebhard von Jagow introduced Blue Native PAGE (BN-PAGE), specifically designed to isolate membrane protein complexes in enzymatically active form [29] [30]. This innovation addressed the critical limitation of SDS-PAGE, which denatures proteins and destroys non-covalently bound cofactors [8]. Continuous refinements throughout the 2000s, including standardized polymerization times and optimized protocols, further enhanced the technique's reliability and applications [27] [30].
Diagram 1: Historical development of electrophoresis techniques, showing the parallel development of denaturing and native approaches. The evolution culminated in BN-PAGE as a response to the limitations of fully denaturing methods.
Fundamental Principles: BN-PAGE vs. SDS-PAGE
The core distinction between BN-PAGE and SDS-PAGE lies in their fundamental mechanisms for imparting charge to proteins and their consequent effects on protein structure and complex integrity.
SDS-PAGE: Denaturing Separation
SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) employs the strong ionic detergent SDS, which denatures proteins and binds to them in a ratio of approximately 1.4g SDS per 1g protein [8]. This SDS coating imparts a uniform negative charge proportional to molecular mass, allowing separation primarily by size as proteins migrate through the polyacrylamide gel matrix [8]. The method requires samples to be heated in the presence of SDS and reducing agents to fully denature proteins and break disulfide bonds [8]. While this enables excellent resolution and molecular weight determination, it destroys native protein structure, enzymatic activity, and non-covalent protein-protein interactions [8].
BN-PAGE: Native Complex Separation
BN-PAGE uses the anionic dye Coomassie Blue G-250 to coat proteins with the necessary negative charge for migration toward the anode [31]. Unlike SDS, Coomassie Blue does not denature protein complexes, allowing them to remain intact during electrophoresis [31]. Protein complexes separate according to their size and shape as they migrate through the porosity gradient of the acrylamide gel [31]. The technique employs mild nonionic detergents such as digitonin or n-dodecyl-β-d-maltoside for membrane protein solubilization, which preserve native protein-protein interactions [32] [31]. This maintains enzymatic activity and the integrity of multi-protein complexes, enabling functional studies after separation [29].
NSDS-PAGE: A Hybrid Approach
A modified approach called Native SDS-PAGE (NSDS-PAGE) has been developed to address the limitations of both techniques [8]. This method reduces SDS concentration in running buffers from 0.1% to 0.0375%, eliminates EDTA from buffers, and omits the heating step [8]. Research demonstrates that NSDS-PAGE increases Zn²⁺ retention in proteomic samples from 26% to 98% compared to standard SDS-PAGE, with seven of nine model enzymes retaining activity after separation [8].
Table 2: Comparative Mechanism of BN-PAGE, SDS-PAGE, and NSDS-PAGE
| Parameter | BN-PAGE | SDS-PAGE | NSDS-PAGE |
|---|---|---|---|
| Charge Agent | Coomassie Blue G-250 | SDS (Sodium Dodecyl Sulfate) | Reduced SDS (0.0375%) |
| Protein State | Native | Denatured | Partially Native |
| Complex Integrity | Maintained | Disrupted | Variable Maintenance |
| Molecular Basis | Size & Shape | Molecular Mass | Molecular Mass & Structure |
| Typical Detergents | Digitonin, Dodecyl-β-D-maltoside | SDS, LDS | Reduced SDS |
| Metal Cofactor Retention | High (≥98%) | Low (∼26%) | High (∼98%) |
| Enzyme Activity Post-Electrophoresis | Preserved | Destroyed | Mostly Preserved |
Diagram 2: Decision framework for selecting electrophoresis methods based on research objectives. The choice between BN-PAGE, SDS-PAGE, and hybrid approaches depends primarily on whether native structure preservation or maximum resolution is prioritized.
Comparative Experimental Performance Data
Direct comparative studies provide quantitative evidence of the performance differences between electrophoresis techniques, particularly regarding preservation of enzymatic activity and metal cofactors.
Metal Retention and Enzymatic Activity
Research comparing SDS-PAGE, BN-PAGE, and NSDS-PAGE demonstrates dramatic differences in metal retention capability. When analyzing Zn²⁺ bound in proteomic samples, BN-PAGE and NSDS-PAGE achieved 98% metal retention, compared to only 26% with standard SDS-PAGE [8]. In functional studies with nine model enzymes, including four Zn²⁺ metalloproteins, all nine enzymes retained activity after BN-PAGE separation, while all were denatured during standard SDS-PAGE [8]. NSDS-PAGE showed intermediate performance, with seven of the nine enzymes retaining activity [8].
Resolution and Molecular Weight Range
SDS-PAGE typically provides superior resolution for individual protein subunits, effectively separating proteins differing by less than 2% in molecular mass [8]. BN-PAGE separates protein complexes ranging from 100 kDa to 10 MDa, with the effective range adjustable based on acrylamide gradient specifications [31]. While BN-PAGE has lower absolute resolution than SDS-PAGE, it successfully resolves complexes that would be disrupted in denaturing conditions [8].
Diagnostic Applications in Disease Research
BN-PAGE has proven particularly valuable in diagnosing oxidative phosphorylation (OXPHOS) defects. Research demonstrates that tissues from patients with severe OXPHOS deficiencies show almost complete absence of the corresponding enzyme band after catalytic staining in BN-PAGE gels [29]. In patients with partial deficiencies, a milder decrease in the enzyme band intensity is observed [29]. This application capitalizes on BN-PAGE's ability to maintain enzymatic activity post-separation, allowing direct in-gel activity staining for Complexes I, II, IV, and V [29] [30].
Table 3: Quantitative Performance Comparison of Electrophoresis Techniques
| Performance Metric | BN-PAGE | SDS-PAGE | NSDS-PAGE |
|---|---|---|---|
| Metal Cofactor Retention | 98% [8] | 26% [8] | 98% [8] |
| Enzyme Activity Preservation | 9/9 model enzymes [8] | 0/9 model enzymes [8] | 7/9 model enzymes [8] |
| Molecular Weight Range | 100 kDa - 10 MDa [31] | 5 - 250 kDa | Similar to SDS-PAGE |
| Resolution | Moderate | High | High |
| Protein Complex Integrity | Maintained | Disrupted | Partially Maintained |
| Diagnostic Reliability | High for OXPHOS defects [29] | Not applicable | Limited data |
Detailed Methodological Protocols
BN-PAGE Protocol for Protein Complex Analysis
The standard BN-PAGE protocol involves multiple carefully optimized steps to preserve native protein complexes:
Sample Preparation: Cells or tissues are solubilized using mild nonionic detergents such as digitonin (0.5-1%), n-dodecyl-β-d-maltoside (0.25-1%), or Triton X-100 (0.25-0.5%) in BN-PAGE sample buffer [32] [31]. The choice of detergent significantly impacts complex preservation, with digitonin being preferred for maintaining supercomplex organization [30]. The extraction is typically performed for 30 minutes at 4°C with gentle shaking, followed by centrifugation at 20,000 × g for 30 minutes to remove insoluble material [32].
Gel Electrophoresis: Linear gradient polyacrylamide gels (typically 4-16% acrylamide) are used to achieve optimal separation across a broad molecular weight range [31] [30]. Coomassie Blue G-250 is added to both the samples and the cathode buffer, providing the charge for electrophoretic migration while maintaining protein solubility [31] [30]. Electrophoresis is performed at constant voltage (typically 100-150V) for 90-95 minutes at 4°C to prevent heat denaturation [32] [30].
Detection Methods: Following electrophoresis, multiple detection approaches can be employed. Immunoblotting with specific antibodies enables identification of particular complexes [32]. Mass spectrometry provides comprehensive identification of complex components [25] [33]. In-gel enzymatic activity staining allows direct functional assessment of resolved complexes [29] [30].
Two-Dimensional BN/SDS-PAGE Protocol
For comprehensive analysis of protein complex composition, two-dimensional BN/SDS-PAGE combines native separation in the first dimension with denaturing separation in the second dimension:
First Dimension: Protein complexes are separated by BN-PAGE as described above [25] [33].
Dimension Transfer: Individual lanes from the BN-PAGE gel are excised and incubated in reducing LDS sample buffer containing 50 mM dithiothreitol for 30 minutes [32]. This step denatures the complexes and reduces disulfide bonds.
Second Dimension: The denatured strips are applied to standard SDS-PAGE gels for separation in the second dimension, resolving individual subunits by molecular mass [25] [33]. This approach enables identification of subunit composition within native complexes and has been successfully applied to analyze multi-protein complexes from various sources, including Helicobacter pylori and mitochondrial respiratory complexes [25] [33] [30].
Essential Research Reagents and Materials
Successful electrophoresis requires specific reagents optimized for each methodology. The table below details critical components and their functions.
Table 4: Essential Research Reagents for Electrophoresis Techniques
| Reagent Category | Specific Examples | Function | Technique |
|---|---|---|---|
| Charge-Modifying Agents | Coomassie Blue G-250 | Imparts negative charge, maintains solubility | BN-PAGE [31] |
| SDS (Sodium Dodecyl Sulfate) | Denatures proteins, imparts uniform charge | SDS-PAGE [8] | |
| Mild Detergents | Digitonin, n-Dodecyl-β-D-maltoside | Solubilizes membranes while preserving complexes | BN-PAGE [32] [31] |
| Strong Detergents | SDS, LDS | Complete solubilization and denaturation | SDS-PAGE [8] |
| Buffer Additives | 6-Aminocaproic acid, EDTA | Stabilizes proteins, chelates metals | BN-PAGE/SDS-PAGE [8] [30] |
| Gel Matrix Components | Acrylamide, Bis-acrylamide | Forms porous separation matrix | All Techniques |
| Enzyme Activity Stains | NADH, Nitrotetrazolium Blue | Detects dehydrogenase activity | BN-PAGE [29] |
| Lead nitrate, DAB | Detects cytochrome c oxidase activity | BN-PAGE [29] |
Applications in Contemporary Research
Mitochondrial Research and OXPHOS Analysis
BN-PAGE has become indispensable for mitochondrial research, enabling detailed analysis of oxidative phosphorylation (OXPHOS) complexes and supercomplexes [29] [30]. The technique allows simultaneous assessment of all five OXPHOS complexes, with in-gel activity staining providing functional data complementary to protein abundance measurements [29]. This application has proven particularly valuable for diagnosing mitochondrial disorders, as BN-PAGE can detect specific complex deficiencies in patient tissues including heart, skeletal muscle, liver, and cultured fibroblasts [29].
Viral Membrane Protein Complexes
BN-PAGE has provided crucial insights into the organization of viral envelope glycoprotein complexes. Research on measles virus (MeV) demonstrated that native H complexes exist predominantly as loosely assembled tetramers in purified viral particles [32]. This application of BN-PAGE has helped elucidate the stoichiometric requirements for functional fusion complexes and the molecular mechanisms linking receptor binding with membrane fusion initiation [32].
Bacterial Multi-Protein Complexes
The method has been successfully applied to map protein interaction networks in bacterial systems. Studies of Helicobacter pylori identified 34 different proteins grouped in 13 multi-protein complexes, including both cytoplasmic and membrane complexes [33]. This approach revealed interactions between known pathogenic factors, such as urease with heat shock protein GroEL and CagA protein with DNA gyrase GyrA, providing insights into potential mechanisms of pathogenesis [33].
Protein-Protein Interaction Mapping
BN-PAGE serves as a powerful tool for comprehensive mapping of protein-protein interactions under native conditions. When combined with antibody shift assays, the technique can detect specific protein interactions directly within the gel matrix [25]. This capability has been leveraged to study complex dynamics in response to cellular stimuli, such as the changes in proteasome complexes following γ-interferon stimulation of cells [25].
Limitations and Technical Considerations
BN-PAGE Limitations
Despite its advantages, BN-PAGE presents several technical challenges. The requirement for clean and robust antibodies that recognize proteins in their native conformation can limit immunodetection options, as antibodies raised against denatured antigens may not bind effectively [31]. The Coomassie dye is not completely inert and may disrupt some weak protein-protein interactions, potentially leading to complex dissociation [31]. Additionally, the presence of salts or other solutes in samples can cause protein smearing during electrophoresis, requiring careful sample preparation and desalting [31].
Detection Limitations
While in-gel activity staining provides valuable functional data, its sensitivity varies between complexes. The comparative insensitivity of in-gel Complex IV activity staining and the complete lack of in-gel Complex III activity staining represent notable limitations for comprehensive OXPHOS analysis [30]. In tissues with high background staining, such as liver and cultured skin fibroblasts, evaluation of protein amount by conventional staining becomes difficult, necessitating immunoblotting after BN-PAGE separation [29].
Resolution Constraints
BN-PAGE provides lower absolute resolution compared to SDS-PAGE, which can make distinguishing between similarly sized complexes challenging [8]. This limitation may require troubleshooting of gradient gel parameters or implementation of two-dimensional approaches for adequate separation [31]. In cases where Coomassie dye interferes with analysis, Colorless Native PAGE (CN-PAGE) that lacks the anionic G-250 dye may be preferred [31] [30].
The historical development of electrophoresis reflects an ongoing effort to balance resolution with biological relevance. While SDS-PAGE revolutionized protein biochemistry by providing high-resolution separation of denatured polypeptides, BN-PAGE emerged as a complementary technique that preserves the functional organization of native protein complexes. The methodological comparison presented in this guide demonstrates that technique selection must be guided by specific research objectives: SDS-PAGE for maximum resolution of individual subunits, BN-PAGE for functional analysis of native complexes, and hybrid approaches like NSDS-PAGE for intermediate requirements. As electrophoretic methods continue to evolve, their integration with complementary analytical technologies promises to further expand our understanding of complex biological systems, maintaining their central role in biochemical research and diagnostic applications.
Practical Protocols: From Sample Prep to Specialized Applications
Step-by-Step BN-PAGE Protocol for Membrane Protein Complexes
Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) is a powerful technique designed to separate native protein complexes by molecular weight, preserving their functional integrity and enabling the study of protein-protein interactions. This stands in direct contrast to SDS-PAGE, which denatures proteins into their constituent polypeptides, thereby destroying native complexes and associated functional properties [8]. The core distinction lies in their applications: BN-PAGE is indispensable for analyzing the oligomeric state, stoichiometry, and native molecular weight of protein complexes, particularly those from membranes [34] [31], whereas SDS-PAGE is optimal for analyzing denatured protein subunits.
This guide provides a objective comparison of these techniques, with a detailed, experimentally-validated BN-PAGE protocol for the analysis of membrane protein complexes.
The fundamental difference between these techniques lies in the mechanism by which proteins are prepared for electrophoretic separation.
Table 1: Fundamental Comparison of BN-PAGE and SDS-PAGE
| Feature | BN-PAGE | SDS-PAGE |
|---|---|---|
| Core Principle | Coomassie dye imposes negative charge on native proteins [35] [31]. | SDS denatures proteins and confers a uniform negative charge [8]. |
| Protein State | Native, intact complexes and oligomers [34]. | Denatured, individual polypeptide subunits [8]. |
| Molecular Weight Determination | Native molecular weight of the complex [31]. | Molecular weight of denatured subunits [8]. |
| Ideal Application | Studying protein-protein interactions, oligomeric state, and native complex composition [34] [36]. | Assessing protein purity, expression levels, and subunit composition via western blot [8]. |
| Key Limitation | Potential for mild disruption of some complexes by Coomassie dye; may require native-specific antibodies [31]. | Complete loss of native structure, function, enzymatic activity, and non-covalently bound cofactors [8]. |
A key advantage of BN-PAGE is its utility for membrane proteins. While SDS-PAGE and other solution-based methods like Dynamic Light Scattering (DLS) can be complicated by interference from detergent micelles, BN-PAGE effectively separates the protein-detergent complex from empty micelles, providing a more reliable assessment of monodispersity, a key indicator of sample quality for crystallization [34].
Essential Reagents and Materials
Successful BN-PAGE relies on specific reagents to solubilize and separate protein complexes in their native state.
Table 2: The Scientist's Toolkit: Key Reagents for BN-PAGE
| Reagent | Function | Critical Notes |
|---|---|---|
| Coomassie Blue G-250 | Imparts negative charge to protein surfaces for migration toward anode; prevents aggregation [35] [37]. | Must be the "G" (green) variant. The dye is considered non-denaturing but can disrupt some sensitive interactions [31]. |
| Mild Non-Ionic Detergents | Solubilizes membrane proteins while preserving protein-protein interactions [14]. | n-Dodecyl-β-D-Maltoside (DDM) and Digitonin are most common. Choice is critical for complex stability [34] [37]. |
| 6-Aminocaproic Acid | Zwitterionic salt used in extraction buffers; helps maintain protein integrity and supports solubilization [35]. | Provides a buffering environment at ~pH 7.0 without interfering with electrophoresis [35]. |
| Bis-Tris Buffer System | The standard buffer for BN-PAGE gel and running buffers, typically at pH 7.0 [35]. | Provides stable pH conditions crucial for maintaining native protein states during electrophoresis. |
| Gradient Gels | Separating gel with a gradient of increasing acrylamide concentration (e.g., 3-12%, 4-16%) [35]. | The molecular sieve separates complexes from ~100 kDa up to 10 MDa based on their size and shape [31]. |
Detailed BN-PAGE Experimental Protocol
The following step-by-step protocol is adapted from validated laboratory procedures for the analysis of membrane protein complexes [34] [35] [37].
Sample Preparation and Solubilization
- Membrane Isolation: Harvest biological material (cells, tissues) and isolate the membrane fraction using differential centrifugation. For cultured cells, wash pellets with phosphate-buffered saline (PBS) and freeze at -80°C until use [35].
- Solubilization: Thaw membrane pellets on ice. Solubilize by adding a non-ionic detergent. The optimal detergent and concentration must be determined empirically [14].
- Common detergents include 1-2% n-Dodecyl-β-D-Maltoside (DDM) for individual complexes or 2-4% Digitonin for labile supercomplexes [14] [37].
- The solubilization buffer should also contain 50 mM NaCl, 10% glycerol, and protease inhibitors [34] [37].
- Incubate the solubilization mixture for 20-30 minutes on ice with gentle agitation.
- Clarification: Remove insoluble material by ultracentrifugation at 100,000 - 138,000 × g for 30-60 minutes at 4°C [34] [35].
- Sample Buffer Preparation: Mix the clarified supernatant with a BN-PAGE sample buffer. A typical 4X concentrated sample buffer contains 100 mM Bis-Tris, 100 mM 6-aminocaproic acid, 40% glycerol, and 0.02% Coomassie Blue G-250 (added from a 5% stock solution in 500 mM 6-aminocaproic acid), pH 7.0 [35] [37].
Gel Electrophoresis
- Gel Casting: While pre-cast gradient gels are commercially available, manual casting of linear gradient gels (e.g., 3-12% or 4-16% acrylamide) is common [35]. The gel buffer is typically 150 mM Bis-Tris, pH 7.0.
- Loading and Running: Load the prepared samples onto the gel. Use native molecular weight markers for calibration.
- The anode buffer (lower chamber) is typically 50 mM Bis-Tris, pH 7.0 [35] [37].
- The cathode buffer (upper chamber) is initially the same as the anode buffer but supplemented with 0.02% Coomassie Blue G-250 (Blue Cathode Buffer) or, for Clear-Native PAGE (CN-PAGE), a mixture of anionic and neutral detergents to replace the dye [35] [37].
- Electrophoresis Conditions: Run the gel at a constant voltage. For mini-gels, start at 100 V for 30-60 minutes until the sample has entered the gel, then continue at 150-200 V for 2-3 hours. The run should be performed at 4°C to maintain complex stability [35] [37].
Downstream Analysis
Following electrophoresis, the blue bands representing native protein complexes can be analyzed by several methods:
- In-Gel Activity Staining: Complexes remain active, allowing histochemical staining for enzymes like those in the oxidative phosphorylation system [35].
- Western Blotting: Transfer complexes to a PVDF membrane for immunodetection. Note that antibodies must recognize the native protein epitopes [31].
- Two-Dimensional Electrophoresis (2D-BN/SDS-PAGE): Excise a lane from the BN-PAGE gel, incubate it in SDS-PAGE sample buffer to denature the complexes, and place it horizontally on top of an SDS-PAGE gel. This resolves the individual protein subunits that make up each native complex, identified by mass spectrometry [36] [38].
BN-PAGE Experimental Workflow
Performance Comparison and Experimental Data
The choice between BN-PAGE and SDS-PAGE has profound implications for experimental outcomes, especially concerning protein function and complex integrity.
Table 3: Experimental Outcomes and Performance Data
| Analysis Parameter | BN-PAGE Performance | SDS-PAGE Performance | Experimental Basis |
|---|---|---|---|
| Metal Cofactor Retention | High retention of non-covalently bound metal ions [8]. | Minimal retention (26% for Zn²⁺) [8]. | ICP-MS analysis of Zn²⁺ in proteome samples [8]. |
| Enzymatic Activity Post-Electrophoresis | High activity retention (9/9 model enzymes active) [8]. | No activity (all enzymes denatured) [8]. | In-gel activity assays for dehydrogenases, phosphatases, etc. [8]. |
| Membrane Protein Monodispersity Assessment | High correlation with crystallization success; effective separation of PDC from micelles [34]. | Not applicable for native state assessment. | Comparison with Dynamic Light Scattering (DLS) and Size Exclusion Chromatography (SEC) [34]. |
| Identification of ATP Synthase Complexes | Successful identification of intact F-type and V-type subcomplexes (~300 kDa) from C. thermocellum [38]. | Would only identify individual subunits (e.g., α, β ~55 kDa). | 2D BN/SDS-PAGE coupled with MALDI-TOF/TOF Mass Spectrometry [38]. |
A modified technique known as NSDS-PAGE demonstrates a potential middle ground, reducing SDS concentration and eliminating heating to retain some native properties while achieving resolution closer to SDS-PAGE [8]. This method retained Zn²⁺ in 98% of proteomic samples and preserved the activity of 7 out of 9 model enzymes [8].
BN-PAGE is an indispensable tool for the functional analysis of native protein complexes, particularly membrane proteins, while SDS-PAGE remains the standard for analytical separation of denatured proteins. The experimental data clearly shows BN-PAGE's superior ability to preserve protein function, complex integrity, and cofactor binding. For researchers studying protein-protein interactions, oligomeric states, and the functional architecture of macromolecular complexes, BN-PAGE provides critical insights that are completely lost in a standard SDS-PAGE analysis. The choice of technique should be decisively guided by the fundamental scientific question: the study of native structure and function requires BN-PAGE, while the analysis of denatured polypeptide composition is the domain of SDS-PAGE.
Standard SDS-PAGE Procedure for Denatured Protein Separation
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) represents the most widely employed technology for obtaining high-resolution analytical separation of complex protein mixtures. [8] This method revolutionized protein biochemistry when introduced in the 1970s and continues to serve as a fundamental technique in laboratories worldwide for assessing protein purity, evaluating expression patterns, and determining molecular mass. [8] [4] The technique's enduring popularity stems from its simplicity, reproducibility, and requirement for only microgram quantities of protein material. [39] [4]
Within the context of protein complex analysis, SDS-PAGE occupies a specific niche focused on denatured polypeptide separation, standing in direct contrast to blue-native PAGE (BN-PAGE), which preserves native protein complexes. This guide objectively examines the standard SDS-PAGE procedure, its experimental parameters, and its performance relative to BN-PAGE for specific applications in pharmaceutical and basic research settings.
Fundamental Principles of SDS-PAGE
Core Mechanism of Separation
SDS-PAGE separates proteins primarily based on molecular weight through a two-part mechanism involving protein denaturation and gel matrix sieving. [39] [4] The ionic detergent sodium dodecyl sulfate (SDS) denatures proteins by wrapping around the polypeptide backbone and conferring a uniform negative charge that overwhelms the protein's intrinsic charge. [4] Under reducing conditions that cleave disulfide bonds, proteins unfold into linear chains with charge proportional to polypeptide length. [39] These SDS-polypeptide complexes then migrate through a porous polyacrylamide gel matrix when an electrical field is applied, with smaller proteins moving faster due to less resistance from the gel matrix. [39] [4]
The polyacrylamide gel serves as a molecular sieve, with its pore size determined by the concentration of acrylamide bis-acrylamide. [4] The gel system typically employs a discontinuous buffer system with a stacking gel that concentrates proteins into a sharp band before they enter the resolving gel, where separation primarily occurs. [39] [4] This combination of charge standardization and molecular sieving enables reliable molecular weight estimation when samples are compared to protein standards of known mass. [40]
Comparative Principle: BN-PAGE
In contrast to denaturing SDS-PAGE, blue-native PAGE separates proteins according to both net charge and mass while maintaining their native structure. [19] [4] BN-PAGE uses mild non-ionic detergents for solubilization and employs Coomassie Blue G250 to impart negative charges to protein complexes without disrupting subunit interactions. [19] [21] This preservation of quaternary structure allows BN-PAGE to separate intact protein complexes and supercomplexes, providing information about protein-protein interactions that is completely lost in SDS-PAGE. [19]
Table 1: Fundamental Separation Principles of SDS-PAGE vs. BN-PAGE
| Parameter | SDS-PAGE | BN-PAGE |
|---|---|---|
| Separation Basis | Polypeptide molecular weight | Native mass, charge, and shape |
| Protein State | Denatured and linearized | Native conformation preserved |
| Detergent Used | Denaturing ionic SDS (0.1-0.5%) | Mild non-ionic (dodecylmaltoside, digitonin, Triton X-100) |
| Charge Source | Bound SDS molecules | Coomassie Blue dye binding |
| Quaternary Structure | Destroyed | Maintained |
| Enzymatic Activity | Lost after separation | Often retained |
Standard SDS-PAGE Protocol
Reagent Preparation
The following research reagent solutions are essential for performing standard SDS-PAGE:
Table 2: Essential Reagents for SDS-PAGE Experiments
| Reagent | Composition/Example | Function |
|---|---|---|
| Acrylamide/Bis Solution | 30% acrylamide, 0.8% bisacrylamide | Gel matrix formation through polymerization |
| SDS-PAGE Sample Buffer | 62.5 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 0.002% Bromophenol blue | Protein denaturation, charge impartation, and density for loading |
| Reducing Agent | β-mercaptoethanol (0.55M) or DTT | Disulfide bond reduction for complete unfolding |
| Running Buffer | 25 mM Tris, 192 mM glycine, 0.1% SDS | Conducts current and maintains SDS saturation |
| Resolving Gel Buffer | 1.5 M Tris-HCl, pH 8.8 | Creates high-pH environment for separation |
| Stacking Gel Buffer | 0.5 M Tris-HCl, pH 6.8 | Creates low-pH environment for protein stacking |
| Polymerization Initiators | APS (ammonium persulfate) and TEMED | Catalyze acrylamide polymerization |
Step-by-Step Experimental Procedure
Gel Preparation: Assemble glass plates with spacers to form a cassette. For a standard 10% resolving gel, combine 7.5 mL of 40% acrylamide solution, 3.9 mL of 1% bisacrylamide, 7.5 mL of 1.5 M Tris-HCl (pH 8.7), water to 30 mL, 0.3 mL of 10% SDS, 0.3 mL of 10% APS, and 0.03 mL TEMED. [4] Pour between plates, overlay with water or isopropanol to ensure even polymerization, and allow to set for 20-30 minutes. Once polymerized, pour a stacking gel (typically 4-5% acrylamide in Tris-HCl, pH 6.8) and insert a comb to form wells. [39]
Sample Preparation: Mix protein samples with SDS-PAGE sample buffer containing a reducing agent such as β-mercaptoethanol at a final concentration of 0.55M. [40] Heat samples at 95°C for 5 minutes (or 70°C for 10 minutes) to ensure complete denaturation. [39] [40] Centrifuge at 15,000 rpm for 1-3 minutes to pellet any debris. [39] [40]
Electrophoresis: Mount the gel cassette in the electrophoresis chamber and fill with running buffer. Load prepared samples and molecular weight markers (5-35 μL per lane). [40] Apply constant voltage (150-200V) until the dye front reaches the bottom of the gel (approximately 45-90 minutes). [8] [40]
Post-Electrophoresis Analysis: Following separation, proteins can be visualized using stains (Coomassie, silver staining), transferred to membranes for western blotting, or excised for mass spectrometric analysis. [4]
Comparative Performance Data: SDS-PAGE vs. BN-PAGE
Quantitative Comparison of Separation Outcomes
Direct comparison studies reveal fundamental performance differences between these techniques. In one systematic analysis, standard SDS-PAGE resulted in only 26% retention of bound Zn²⁺ in metalloproteins, while a modified native SDS-PAGE approach achieved 98% metal retention. [8] Similarly, when model enzymes were separated, all nine underwent complete denaturation and activity loss during standard SDS-PAGE, whereas all nine retained activity in BN-PAGE. [8]
Table 3: Experimental Performance Comparison Between Electrophoresis Methods
| Performance Metric | SDS-PAGE | BN-PAGE | Experimental Context |
|---|---|---|---|
| Metal Ion Retention | 26% | >90% | Zn²⁺ in metalloproteins [8] |
| Enzyme Activity Preservation | 0/9 enzymes active | 9/9 enzymes active | Model enzyme study [8] |
| Resolution of Complex Mixtures | High for polypeptides | Moderate for complexes | Proteomic separation [8] [19] |
| Suitable for Hydrophobic Membrane Proteins | Limited without special detergents | Excellent with optimized solubilization | Membrane protein complexes [14] [19] |
| Molecular Weight Determination | Accurate for polypeptides | Approximate for native complexes | Comparative analysis [8] [19] |
Two-Dimensional Applications: BN-/SDS-PAGE
The complementary strengths of both techniques can be leveraged through two-dimensional electrophoresis. In this approach, protein complexes are first separated by BN-PAGE, then individual lanes are excised, soaked in SDS buffer, and placed atop an SDS-PAGE gel for second-dimension separation. [23] [21] This powerful method resolves both native complexes and their subunit composition, as demonstrated in studies identifying HNE-modified complex I subunits in diabetic kidney mitochondria. [23]
Applications in Research and Drug Development
Appropriate Use Cases for SDS-PAGE
SDS-PAGE remains the method of choice for applications requiring polypeptide-level resolution without need for native structure preservation. These include:
- Molecular Weight Determination: Estimation of protein subunit size relative to standards. [4] [40]
- Purity Assessment: Evaluation of protein preparation homogeneity during purification. [40]
- Expression Analysis: Comparison of protein expression levels across samples. [8]
- Western Blotting: Initial separation for immunodetection of specific proteins. [8] [4]
- Quality Control in Biopharmaceuticals: Purity analysis of biotherapeutic products using CE-SDS (capillary electrophoresis SDS), a related method. [41]
Limitations and Method Constraints
The fundamental limitation of SDS-PAGE lies in its deliberate denaturation of proteins prior to electrophoresis, which destroys functional properties including enzymatic activity, protein binding interactions, and non-covalently bound cofactors. [8] This makes it unsuitable for studying native protein complexes, protein-protein interactions, or enzymatic function after separation. Additionally, very large protein complexes (>500 kDa) may not enter standard gels effectively, while highly acidic or basic proteins may migrate anomalously due to residual charge effects. [40]
The choice between SDS-PAGE and BN-PAGE depends entirely on research objectives. SDS-PAGE provides superior resolution of denatured polypeptides by molecular weight and is ideal for standard protein characterization, purity assessment, and immunoblotting applications. In contrast, BN-PAGE preserves native protein structures and functions, enabling the study of protein complexes, interactions, and enzymatic activities. For comprehensive protein complex analysis, the two-dimensional BN-/SDS-PAGE approach offers the most complete information by combining the strengths of both techniques. Researchers must therefore align their electrophoretic method selection with their specific analytical needs—whether studying polypeptide composition or native protein function.
The analysis of protein complexes, particularly membrane-bound complexes, is a cornerstone of modern biological research and drug development. The choice of detergent for solubilizing these complexes is arguably the most critical factor determining the success of downstream analytical techniques, primarily Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) and denaturing SDS-PAGE. These two techniques serve fundamentally different purposes: BN-PAGE separates intact protein complexes in their native state, preserving enzymatic activity and protein-protein interactions, while SDS-PAGE denatures complexes into individual subunits separated by molecular weight [42] [8]. Within this context, dodecylmaltoside, digitonin, and Triton X-100 have emerged as three of the most widely used detergents, each with distinct properties that influence experimental outcomes. This guide provides a detailed, evidence-based comparison of these detergents to inform method development in protein complex analysis.
Detergent Properties and Performance Comparison
Key Characteristics and Recommended Applications
The table below summarizes the fundamental properties of these three detergents to guide initial selection.
Table 1: Fundamental Properties of Dodecylmaltoside, Digitonin, and Triton X-100
| Property | n-Dodecyl-β-D-Maltoside (DDM) | Digitonin | Triton X-100 |
|---|---|---|---|
| Type | Non-ionic, glycosidic | Non-ionic, steroid-based | Non-ionic, polyoxyethylene |
| Aggregation Number | 78-140 [43] | Mixture (natural product) | 100-155 [44] |
| Critical Micelle Concentration (CMC) | 0.0087% - 0.017% (w/v) [43] | ~0.2% (w/v) | 0.015% [44] |
| Typical Solubilization Concentration | 0.5-2% (w/v) [45] [43] | 1-4% (w/v) [45] [19] | 0.5-2% (w/v) [44] [19] |
| Primary Application in BN-PAGE | Solubilizing individual protein complexes with high activity [45] [19] | Preserving weak interactions and supercomplexes [45] [19] | General solubilization of membrane proteins [19] |
Experimental Performance Data
The functional performance of these detergents varies significantly in experimental settings, as evidenced by the following quantitative data.
Table 2: Experimental Performance Comparison in Protein Complex Studies
| Performance Metric | n-Dodecyl-β-D-Maltoside (DDM) | Digitonin | Triton X-100 |
|---|---|---|---|
| Protein Complex Activity Retention | High (e.g., active GABA receptors purified) [43] | High (e.g., PS synthase activity retained) [45] | Variable (can denature some complexes) [8] |
| Supercomplex Preservation | Low (typically dissociates supercomplexes) [19] | High (preserves respiratory supercomplexes) [19] | Low (typically dissociates supercomplexes) [19] |
| Enzymatic Activity Post-Electrophoresis | 7/9 model enzymes active after NSDS-PAGE [8] | N/A (often used in BN-PAGE where activity is retained) | N/A |
| Metal Cofactor Retention | 98% Zn²⁺ retained in NSDS-PAGE [8] | N/A | 26% Zn²⁺ retained in standard SDS-PAGE [8] |
| Solubilization Efficiency | Robust (>90% of membrane pellets) [43] | Effective for specific complexes [45] | Effective for general membrane proteins [44] |
Experimental Protocols and Workflows
Standard BN-PAGE Protocol with Detergent Optimization
The following workflow outlines a standard BN-PAGE procedure, highlighting critical points for detergent selection and application.
Protocol Details:
- Sample Preparation: Harvest and wash 10x10⁶ cells in ice-cold PBS. Pellet cells by centrifugation at 350g for 5 minutes at 4°C [42].
- Solubilization: Resuspend the cell pellet in 250 µL of ice-cold BN-Lysis Buffer. Incubate on ice for 15 minutes. The key step is adding the selected detergent. Use a detergent-to-protein ratio that must be optimized empirically [19]:
- DDM: Often used at 1-2% (w/v) for solubilizing individual complexes.
- Digitonin: Typically 1-4% (w/v) for preserving supercomplexes.
- Triton X-100: Commonly 0.5-2% (w/v) for general membrane protein solubilization.
- Clarification and Dialysis: Centrifuge the lysate at 13,000g for 15 minutes at 4°C to remove insoluble material. Transfer the supernatant to a tube with a dialysis membrane (10 kDa cutoff) and dialyze against BN-Dialysis Buffer for 6 hours or overnight in the cold room [42]. This step removes salts and small metabolites that can interfere with electrophoresis.
- Gel Preparation and Electrophoresis: Pour a polyacrylamide gradient gel (e.g., 4-15%). Load the dialyzed lysate (1-40 µL) and appropriate native markers into the wells. Run the gel at 4°C, starting at low voltage (e.g., 100V for a minigel) until samples enter the separating gel, then increase to 180V until the dye front reaches the gel bottom [42].
- Analysis: For complexes separated by BN-PAGE, a second dimension SDS-PAGE can be performed. The BN-PAGE gel lane is incubated in SDS sample buffer, briefly heated, and then loaded onto an SDS gel to separate the individual subunits of the complex [42].
Case Study: Solubilizing Candida albicans Cho1 Protein
A 2023 study on solubilizing the transmembrane protein Cho1 from Candida albicans provides a direct comparison of detergent efficacy [45]. The research screened six non-ionic detergents and three styrene maleic acid copolymers. The results demonstrated that:
- Digitonin and DDM solubilized Cho1 effectively and were associated with the highest phosphatidylserine (PS) synthase activity.
- BN-PAGE and immunoblot analysis showed a single band for Cho1 in digitonin- and DDM-solubilized fractions.
- Further analysis via pull-down assays and electron microscopy indicated that the native Cho1 exists as a hexamer, a finding enabled by the gentle solubilization conditions.
This case underscores that while multiple detergents may achieve solubilization, the choice between DDM and digitonin is critical for retaining maximal enzymatic activity.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents required for experiments comparing detergents in protein complex analysis.
Table 3: Essential Reagents for Detergent-Based Protein Complex Analysis
| Reagent/Category | Specific Examples | Function and Importance |
|---|---|---|
| Primary Detergents | n-Dodecyl-β-D-Maltoside (DDM), Digitonin, Triton X-100 | Solubilize membrane proteins while maintaining native protein-protein interactions to varying degrees. The core subject of comparison. |
| Buffers & Salts | BN-Lysis Buffer, BN-Dialysis Buffer, Aminocaproic Acid, NaCl | Maintain pH and ionic strength; low-salt buffers like aminocaproic acid support solubilization without disrupting complexes [19]. |
| Electrophoresis Consumables | Acrylamide/Bis-acrylamide, Coomassie Blue G-250, NativeMark Protein Standard | Form the gel matrix; Coomassie dye provides negative charge for migration in BN-PAGE [42] [31]. |
| Protease Inhibitors | PMSF, Leupeptin, Aprotinin, Pepstatin | Prevent proteolytic degradation of protein complexes during the extraction and solubilization process [45] [43]. |
| Affinity Matrices | Anti-FLAG Agarose, Ni-NTA Agarose, M2 Agarose | Purify tagged protein complexes after solubilization for functional or structural studies [45] [43]. |
| Activity Assay Reagents | GABA (for GABA receptors), CDP-diacylglycerol & Serine (for PS synthase) | Measure the functional integrity of solubilized and purified protein complexes [45] [43]. |
The selection of an appropriate detergent is not a one-size-fits-all process but a strategic decision based on experimental goals. The following diagram summarizes the logical decision process for selecting the optimal detergent.
Summary of Guidelines:
- Choose Digitonin when the experimental goal is to preserve weak interactions and isolate large supercomplexes, as it is the mildest detergent and best maintains these fragile assemblies [19].
- Choose Dodecylmaltoside (DDM) for the solubilization of individual protein complexes where retaining high enzymatic activity and stability is the primary concern. It is the detergent of choice for crystallography pipelines and high-activity purifications [45] [43]. It also shows superior performance in modified Native SDS-PAGE (NSDS-PAGE) for retaining metal cofactors [8].
- Choose Triton X-100 for initial, cost-effective screens or for solubilizing complexes known to be stable in this detergent, acknowledging that it may disrupt some protein-protein interactions and supercomplexes [19].
Ultimately, the optimal detergent and its working concentration should be determined empirically for each specific protein complex and biological source, as the lipid composition of membranes can significantly influence detergent efficacy [19].
Two-dimensional Blue Native/SDS-PAGE (BN/SDS-PAGE) represents a powerful electrophoretic technique that combines native separation of intact protein complexes with denaturing resolution of their individual subunits. This methodology enables researchers to analyze protein complex composition, stoichiometry, assembly states, and interactions under near-physiological conditions. As a cornerstone technique in structural proteomics, it provides critical insights into complexome dynamics that are inaccessible to conventional SDS-PAGE alone. This guide objectively compares BN-PAGE against standard SDS-PAGE approaches, detailing their respective strengths, limitations, and optimal applications in modern protein research and drug development.
The comprehensive analysis of protein complexes remains a fundamental challenge in molecular biology and drug development. Most cellular processes are executed by multi-protein assemblies rather than individual proteins, making the understanding of these complexes crucial for deciphering disease mechanisms and identifying therapeutic targets [19]. While SDS-PAGE has served as the workhorse for protein separation for decades, its denaturing nature destroys protein complexes and eliminates enzymatic activity. Blue Native PAGE (BN-PAGE), developed by Schägger and von Jagow in 1991, emerged to fill this critical technological gap by enabling the separation of intact protein complexes under non-denaturing conditions [30] [32].
The two-dimensional BN/SDS-PAGE technique represents a sophisticated integration of these approaches, where protein complexes are first separated intact according to their size and charge through BN-PAGE, followed by orthogonal separation of constituent subunits by molecular weight using SDS-PAGE [23] [26]. This powerful combination has revolutionized the study of multi-protein complexes, particularly in membrane proteomics, where it has been instrumental in characterizing mitochondrial oxidative phosphorylation (OXPHOS) complexes, respiratory supercomplexes, and their alterations in disease states [30] [46]. The technique preserves protein-protein interactions through the first dimension while providing high-resolution subunit analysis in the second dimension, offering a unique window into the structural and functional organization of cellular machineries.
Fundamental Principles: BN-PAGE vs SDS-PAGE
Core Mechanistic Differences
The fundamental distinction between BN-PAGE and SDS-PAGE lies in their treatment of protein structure during separation. SDS-PAGE employs the ionic detergent sodium dodecyl sulfate (SDS) to denature proteins into linear polypeptides, masking intrinsic charge and enabling separation primarily by molecular weight [1]. In contrast, BN-PAGE utilizes mild non-ionic detergents for solubilization combined with Coomassie Blue G-250 dye, which imparts negative charge to proteins without disrupting their tertiary or quaternary structure [46] [19]. This critical difference preserves native protein interactions and enzymatic activities throughout the electrophoretic process.
In SDS-PAGE, SDS binding to hydrophobic regions of proteins unfolds the tertiary structure and confers a uniform negative charge density, causing migration through the polyacrylamide gel matrix to depend almost exclusively on polypeptide chain length [1]. BN-PAGE operates through a more sophisticated mechanism where the anionic Coomassie dye binds to hydrophobic protein patches, providing the charge necessary for electrophoretic migration while maintaining complex integrity [30] [19]. The dye's binding also enhances the solubility of membrane proteins during electrophoresis, preventing aggregation that would otherwise occur in detergent-free conditions.
Table 1: Fundamental Characteristics of BN-PAGE and SDS-PAGE
| Parameter | BN-PAGE | SDS-PAGE |
|---|---|---|
| Separation Principle | Size, charge, and shape of native complexes | Molecular weight of denatured polypeptides |
| Protein State | Native, folded conformation | Denatured, linearized |
| Detergent System | Mild non-ionic (e.g., DDM, digitonin) | Strong ionic (SDS) |
| Charge Provider | Coomassie Blue G-250 dye | SDS molecules |
| Buffer Additives | Aminocaproic acid, protease inhibitors | Reducing agents (DTT, BME) |
| Temperature | Typically 4°C | Room temperature |
| Protein Function | Retained post-separation | Lost |
Applications and Limitations
The applications of BN-PAGE and SDS-PAGE reflect their fundamental mechanistic differences. SDS-PAGE excels in determining protein molecular weight, assessing purity, and analyzing subunit composition when combined with Western blotting [1]. Its simplicity, reproducibility, and wide adoption make it ideal for routine protein analysis. However, its inability to preserve protein complexes or enzymatic activities represents a significant limitation for functional studies.
BN-PAGE enables the investigation of protein complex stoichiometry, identification of assembly intermediates, detection of supercomplexes, and in-gel enzyme activity assays [30] [46]. It has become particularly valuable in mitochondrial research, where it facilitates the analysis of OXPHOS complexes and their dysfunction in metabolic diseases [30] [23]. The technique does present challenges, including more complex protocol optimization, potential for complex dissociation during electrophoresis, and interference from Coomassie dye in downstream applications. Clear Native PAGE (CN-PAGE), a variant that replaces Coomassie with mixed detergent micelles, addresses some limitations by eliminating dye interference while maintaining native separation [30].
Table 2: Application-Based Comparison of BN-PAGE and SDS-PAGE
| Analysis Type | BN-PAGE | SDS-PAGE |
|---|---|---|
| Molecular Weight Determination | Indirect, for complexes | Direct, for subunits |
| Complex Stoichiometry | Yes | No |
| Protein-Protein Interactions | Preserved | Disrupted |
| Enzymatic Activity Assays | In-gel possible | Not possible |
| Membrane Protein Analysis | Excellent for complexes | Challenging for hydrophobic proteins |
| Post-Translational Modifications | Limited detection | Good detection |
| Western Blotting Compatibility | Possible with optimization | Standard procedure |
| Mass Spectrometry Compatibility | Requires special processing | Well-established |
The BN/SDS-PAGE Methodology: An Integrated Workflow
Sample Preparation and Solubilization Strategies
Effective sample preparation constitutes the most critical step in BN/SDS-PAGE, requiring careful optimization to preserve labile protein interactions while achieving sufficient solubilization. For mitochondrial complexes, isolation of intact mitochondria precedes solubilization with mild detergents [21] [30]. The choice of detergent profoundly influences which complexes remain intact, with n-dodecyl-β-D-maltoside (DDM) typically preserving individual complexes, while digitonin maintains supercomplex associations [46] [19]. The zwitterionic salt 6-aminocaproic acid is routinely included to support solubilization without interfering with electrophoresis [30].
The detergent-to-protein ratio requires empirical optimization for different sample types, as excessive detergent disrupts native interactions while insufficient detergent yields poor solubilization and sample aggregation [19]. Protease inhibitors (PMSF, leupeptin, pepstatin) are essential additives to prevent protein degradation during the extended procedures [21]. For tissue samples, mechanical homogenization followed by differential centrifugation enables organelle enrichment, reducing sample complexity and enhancing detection of low-abundance complexes [23]. The final solubilized samples are supplemented with Coomassie G-250 (typically 0.02-0.1%) to impart the negative charge required for electrophoretic migration [30] [19].
Figure 1: Two-Dimensional BN/SDS-PAGE Workflow. The integrated procedure begins with native sample preparation, followed by sequential electrophoretic separations that preserve complex information while enabling high-resolution subunit analysis.
First Dimension: Blue Native PAGE
The first dimension BN-PAGE separation employs gradient gels (typically 4-16% acrylamide) to resolve protein complexes across a broad molecular weight range [21] [30]. The gel system utilizes specialized buffers with Bis-Tris and aminocaproic acid at pH 7.0 to maintain neutral conditions throughout electrophoresis [23]. The cathode buffer contains Coomassie G-250 (0.02%) to ensure continuous dye availability during protein migration, while the anode buffer lacks dye to establish the proper electrical field [19].
Electrophoresis conditions must balance separation efficiency with complex preservation, typically running at 4°C to minimize proteolytic activity and complex dissociation [1] [30]. Voltage parameters vary by system, with an initial low voltage (50-100V) to facilitate complex entry into the gel matrix, followed by higher voltages (300-500V) for separation [21] [47]. The progress can be monitored by dye migration, with complexes typically separated within 2-4 hours for mini-gel systems. Post-electrophoresis, complexes can be visualized directly by Coomassie staining, subjected to in-gel activity assays, or processed for second-dimension separation [30] [23].
Second Dimension: SDS-PAGE
The second dimension begins with excising entire lanes from the BN-PAGE gel and equilibrating them in SDS-containing buffer to denature the complexes into constituent subunits [23]. This equilibration step incorporates SDS, reducing agents (DTT or β-mercaptoethanol), and glycerol, thoroughly denaturing proteins and disrupting all non-covalent interactions [1] [23]. The gel strip is then positioned horizontally atop an SDS-polyacrylamide gel, with the original BN-PAGE separation direction perpendicular to the second electrophoretic dimension.
The SDS-PAGE separation follows standard Laemmli protocols, resolving subunits by molecular weight [23]. The resulting two-dimensional pattern provides a comprehensive map where intact complexes from the first dimension are resolved into their subunit components in the second dimension. This orthogonal separation enables correlation of complex molecular weight with subunit composition, identification of shared subunits between complexes, and detection of post-translational modifications that may alter subunit migration [23] [26].
Research Applications and Experimental Data
Mitochondrial Complex Analysis
BN/SDS-PAGE has revolutionized mitochondrial research by enabling comprehensive analysis of oxidative phosphorylation complexes. Aref et al. (2025) validated BN-PAGE protocols for characterizing mitochondrial OXPHOS complexes, demonstrating robust separation of Complexes I-V with preserved enzymatic activities [30]. Their optimized methodology detected as few as 0.4 mg of mitochondrial protein, making it suitable for precious clinical samples. The in-gel activity assays revealed dynamic alterations in Complex I and IV activities in diabetic models, with quantitative data showing approximately 40% reduction in Complex I function compared to controls [30].
Wu et al. (2015) applied BN/SDS-PAGE to identify specific Complex I subunits modified by the lipid peroxidation product 4-hydroxynonenal (HNE) in diabetic kidney mitochondria [23]. Their two-dimensional approach isolated intact Complex I by BN-PAGE before resolving its 45 subunits by SDS-PAGE, enabling precise identification of HNE-modified targets through Western blotting and mass spectrometry. Densitometric quantification revealed statistically significant (p < 0.05) increases in HNE adduct formation on specific subunits, providing mechanistic insights into mitochondrial dysfunction in diabetes [23].
Table 3: Quantitative Analysis of Mitochondrial Complexes by BN/SDS-PAGE
| Complex | Native Mass (kDa) | Subunit Number | In-Gel Activity Method | Detection Limit |
|---|---|---|---|---|
| Complex I | ~1000 | 45 | NADH-dehydrogenase + NBT | 2 μg |
| Complex II | ~140 | 4 | Succinate dehydrogenase | 1 μg |
| Complex III | ~250 | 11 | No reliable method | N/A |
| Complex IV | ~200 | 13 | Cytochrome c oxidase | 5 μg |
| Complex V | ~600 | 18 | ATP hydrolysis | 10 μg |
Supercomplex and Assembly Intermediate Resolution
The detergent selection during sample preparation dramatically influences complex resolution, enabling investigation of different organizational states. Camacho-Carvajal et al. (2004) established that BN/SDS-PAGE could resolve multi-protein complexes from whole cellular lysates, extending the technique beyond purified organelles [26]. Their methodology employed dialysis to adjust cellular lysates to BN-PAGE-compatible buffers, successfully separating proteasomal complexes and demonstrating interferon-induced complex dynamics.
The technique has been particularly valuable in resolving respiratory chain supercomplexes, known as "respirasomes." When digitonin solubilization precedes BN-PAGE, stable associations between Complexes I, III, and IV remain intact, revealing higher-order organizational states not observable with DDM solubilization [46] [19]. These structural insights have fundamentally altered understanding of mitochondrial respiratory chain organization, shifting from the "fluid state" model of freely diffusing individual complexes to the "solid state" model of structured supercomplex assemblies [46].
Essential Reagents and Research Solutions
Successful BN/SDS-PAGE requires carefully selected reagents optimized for native protein separation. The following table details critical components and their functions in the experimental workflow.
Table 4: Essential Research Reagents for BN/SDS-PAGE
| Reagent Category | Specific Examples | Function | Concentration Range |
|---|---|---|---|
| Mild Detergents | n-Dodecyl-β-D-maltoside (DDM), Digitonin, Triton X-100 | Solubilize membranes while preserving protein complexes | 0.5-2% (w/v) |
| Charge Providers | Coomassie Blue G-250 | Impart negative charge for electrophoretic migration | 0.02-0.1% |
| Solubilization Enhancers | 6-Aminocaproic acid | Support membrane protein solubilization | 0.5-0.75 M |
| Buffer Components | Bis-Tris, Tricine | Maintain neutral pH environment | 50-75 mM |
| Protease Inhibitors | PMSF, Leupeptin, Pepstatin A | Prevent protein degradation during processing | 1 mM, 1 μg/mL |
| Reducing Agents | Dithiothreitol (DTT), β-mercaptoethanol | Reduce disulfide bonds in second dimension | 50-100 mM |
| Gel Matrix | Acrylamide/Bis-acrylamide | Form porous separation matrix | 4-16% gradient |
Advanced Integration with Mass Spectrometry
The combination of BN/SDS-PAGE with mass spectrometry has dramatically enhanced its analytical power for comprehensive complexome profiling. Cryo-slicing BN-MS represents a technological advancement where BN-PAGE gel lanes are sectioned into sub-millimeter slices using a cryo-microtome before LC-MS/MS analysis [47]. This approach achieved exceptional resolution, discriminating complexes with <5% molecular weight difference and generating abundance-mass profiles for 774 mitochondrial proteins from rat brain [47].
Recent innovations like PEPPI-MS (Passively Eluting Proteins from Polyacrylamide Gels as Intact species for MS) address the historical challenge of efficiently recovering proteins from gel matrices [48]. This methodology uses Coomassie Brilliant Blue as an extraction enhancer, achieving 68% recovery efficiency for proteins below 100 kDa, finally enabling effective integration of gel-based separation with top-down proteomics [48]. These advances establish BN/SDS-PAGE as a foundational separation platform for structural proteomics, particularly valuable for characterizing membrane protein complexes that remain challenging for alternative approaches.
Figure 2: BN/SDS-PAGE Integration with Mass Spectrometry. Two primary pathways enable proteomic analysis following electrophoretic separation: traditional in-gel digestion for bottom-up approaches and intact protein recovery for top-down proteomics.
Comparative Technical Performance and Research Considerations
Decision Framework for Technique Selection
The choice between BN-PAGE, SDS-PAGE, or their two-dimensional combination depends on specific research questions and sample characteristics. SDS-PAGE remains the appropriate choice for routine protein analysis, molecular weight determination, and studies focusing on individual protein subunits without concern for native structure [1]. Its simplicity, reproducibility, and compatibility with downstream applications like Western blotting make it ideal for quality control and initial characterization.
BN-PAGE provides unique capabilities for functional studies requiring preserved protein interactions, enzymatic activities, or complex stoichiometry [30] [46]. It is particularly valuable for investigating assembly disorders, detecting supercomplexes, and analyzing membrane protein complexes. The two-dimensional BN/SDS-PAGE approach becomes essential when both complex-level and subunit-level information are required, such as identifying specific modified subunits within multi-protein complexes or characterizing complex composition changes in disease states [23] [26].
Limitations and Alternative Approaches
While powerful, BN/SDS-PAGE presents notable limitations. The technique has limited dynamic range, with abundant complexes potentially masking less prevalent species [19]. The Coomassie dye can interfere with certain downstream applications, though CN-PAGE provides an alternative for such cases [30]. Protein complexes may partially dissociate during electrophoresis, and the requirement for empirical optimization of solubilization conditions adds complexity [46] [19].
Emerging alternatives like native mass spectrometry, cross-linking MS, and cryo-EM offer complementary approaches for studying protein complexes [48]. However, BN/SDS-PAGE maintains advantages in accessibility, throughput, and ability to directly link separation with functional enzymatic assays. The technique continues to evolve through integration with advanced MS methods, establishing it as a cornerstone methodology in the structural proteomics toolkit for researchers and drug development professionals.
Respiratory Chain Complexes and Snake Venom Proteomics
The analysis of protein complexes is a cornerstone of modern molecular biology, particularly in specialized fields such as snake venom proteomics and respiratory chain research. Two primary electrophoretic techniques—Blue Native PAGE (BN-PAGE) and Sodium Dodecyl Sulfate-PAGE (SDS-PAGE)—offer complementary approaches for characterizing these complexes. BN-PAGE preserves native protein structures and multi-subunit interactions, enabling the study of functionally active complexes [8]. In contrast, SDS-PAGE denatures proteins into their constituent polypeptides, providing high-resolution separation based primarily on molecular mass [8]. This methodological comparison is especially relevant for snake venom research, where protein complexes often exhibit synergistic toxicity that far exceeds the potency of individual components [49]. Understanding the capabilities and limitations of each technique empowers researchers to select appropriate analytical strategies based on their specific research objectives, whether focused on structural characterization, functional activity, or compositional analysis of complex protein mixtures.
Theoretical Foundation: BN-PAGE vs. SDS-PAGE
Principles and Mechanisms
Blue Native PAGE (BN-PAGE) employs the dye Coomassie G-250, which imparts a negative charge to protein complexes without disrupting their native structure. This allows separation based on both mass and charge under non-denaturing conditions, preserving protein-protein interactions, enzymatic activity, and bound cofactors such as metal ions [8]. The technique is particularly valuable for studying multi-protein assemblies like respiratory chain complexes, where functional integrity is essential for analysis.
Sodium Dodecyl Sulfate-PAGE (SDS-PAGE) utilizes the anionic detergent SDS to denature proteins and impart a uniform negative charge proportional to their molecular mass. This denaturation eliminates higher-order structure, separating proteins primarily by molecular weight while destroying most functional properties, including enzymatic activity and non-covalently bound metal ions [8]. The method provides excellent resolution of individual polypeptide chains but cannot preserve native complexes.
Technical Specifications and Buffer Composition
Table 1: Comparative Buffer Compositions for Electrophoretic Techniques
| Component | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Sample Buffer | 2% LDS, 0.51 mM EDTA, 10% glycerol | 50 mM BisTris, 50 mM NaCl, 10% glycerol, 0.001% Ponceau S | 10% glycerol, 0.01875% Coomassie G-250, 0.00625% Phenol Red |
| Running Buffer | 0.1% SDS, 1 mM EDTA, MOPS/Tris | BisTris/Tricine with cathode and anode buffers | 0.0375% SDS, MOPS/Tris |
| Protein State | Denatured | Native | Native |
| Metal Retention | 26% (Zn²⁺) | High | 98% (Zn²⁺) |
| Enzyme Activity | Destroyed | Preserved | Preserved (7 of 9 tested) |
A modified approach called Native SDS-PAGE (NSDS-PAGE) represents a hybrid technique that reduces SDS concentration and eliminates EDTA and heating steps. This modification maintains excellent protein resolution while significantly improving retention of native properties, including metal ions (98% Zn²⁺ retention versus 26% in standard SDS-PAGE) and enzymatic activity in most tested proteins [8].
Application Case Study: Snake Venom Proteomics
Experimental Workflow for Venom Analysis
The following diagram illustrates a comprehensive proteomic workflow for analyzing snake venom protein complexes, integrating both BN-PAGE and SDS-PAGE techniques:
Key Findings in Snake Venom Research
Research on snake venom complexes has revealed critical insights into their structure-function relationships. In Crotalus durissus terrificus venom, the crotoxin complex exemplifies the pharmacological advantage of protein complexes. This heterodimeric complex consists of a basic phospholipase A₂ (PLA₂) subunit with weak neurotoxicity and an acidic non-enzymatic subunit (crotapotin) that acts as a chaperone [49]. The complex exhibits neurotoxicity one order of magnitude greater than the isolated PLA₂ subunit, as the acidic component directs the enzyme to specific presynaptic targets while preventing non-specific binding [49]. Interestingly, the complex must dissociate upon binding to synaptic membranes to exert its toxic effect, as covalently linked subunits show complete loss of lethal potency [49].
Similar synergistic interactions have been documented in other venom systems. The recent discovery of Brownitoxin-1 in Micrurus ephippifer venom reveals a complex between phospholipase A₂ and three-finger toxins (3FTx) that produces enhanced lethality not observed with individual components [50]. Proteomic analyses indicate that while 3FTx constitute 54% and PLA₂ 29% of this venom, only a small subset of these proteins exhibits toxicity, suggesting specialized functions for many venom components [50].
Comparative Experimental Data
Performance Metrics in Venom Proteomics
Table 2: Technique Performance in Snake Venom Protein Complex Analysis
| Parameter | BN-PAGE | SDS-PAGE | Integrated Approach |
|---|---|---|---|
| Complex Preservation | Excellent (Crotoxin, β-Bungarotoxin) | Poor (Dissociates complexes) | Complementary data |
| Protein Family Identification | Moderate (Limited resolution) | Excellent (svMPs, svSPs, PLA₂s) | Comprehensive coverage |
| Toxic Activity Correlation | High (Native structure maintained) | Low (Denatured proteins) | Functional insights |
| Synergistic Toxin Detection | Yes (Brownitoxin-1 complex) | No | Reveals cooperative effects |
| Metal Cofactor Retention | Preserved (Zn²⁺, Ca²⁺) | Lost during denaturation | Context-dependent |
| Dynamic Range | Limited by complex stability | Excellent for individual proteins | Enhanced coverage |
Quantitative proteomic analysis of Crotalus atrox and C. oreganus helleri venoms using these techniques reveals distinct compositional profiles. In crude venoms, metalloproteinases (svMPs) constitute 31% and 24% of C. atrox and C. o. helleri venoms respectively, while serine proteases (svSPs) represent 21% and 14% [51]. However, analysis of snake venom extracellular vesicles (svEVs) shows significant enrichment of L-amino acid oxidase (17% in C. atrox, 13% in C. o. helleri) and ecto-5'-nucleotidase (12% in C. atrox, 9% in C. o. helleri), highlighting the importance of technique selection for specific venom components [51].
Cardiotoxicity Mechanisms Revealed by Proteomics
A proteomic investigation of Crotalus durissus terrificus envenomation in mouse models demonstrated significant alterations in cardiac tissue proteins following venom injection. Researchers identified changes in over 1300 cardiac proteins, with noteworthy modulation in proteins associated with cytochrome P450 pathways, lipid metabolism, acute phase inflammation, immune response, and heat shock proteins [52]. Concurrent downregulation occurred in mitochondrial electron transport, NADH metabolism, TCA cycle, and oxidative reduction pathways [51]. These findings illustrate how venom components disrupt cardiac function through coordinated effects on multiple physiological systems, mechanisms that can only be fully elucidated through techniques that preserve functional protein interactions.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Protein Complex Analysis
| Reagent/Category | Specific Examples | Function in Analysis |
|---|---|---|
| Electrophoresis Systems | Invitrogen NuPAGE Novex, NativePAGE Novex | High-resolution protein separation under denaturing or native conditions |
| Detection Antibodies | AzureSpectra IR700, IR800 fluorescent secondaries | Multiplex detection of multiple proteins simultaneously on the same blot |
| Protein Stains | Coomassie G-250, Ponceau S, Reversible Protein Stain | Visualization of proteins while maintaining activity (native) or high sensitivity |
| Proteomic Analysis | Trypsin, LC-MS/MS systems, EVtrap | Protein digestion, identification, and quantification of complex mixtures |
| Specialized Buffers | MOPS/Tris, BisTris/Tricine, Anode/Cathode buffers | Maintaining specific pH and conditions for native or denatured separation |
| Membrane Types | Nitrocellulose, PVDF | Protein immobilization for subsequent antibody probing |
Fluorescent Western blotting has emerged as particularly valuable for quantitative analysis of protein complexes. Unlike chemiluminescent detection with variable enzyme-substrate kinetics, fluorophore-conjugated secondary antibodies provide consistent light emission directly proportional to protein abundance [53]. This enables multiplex detection of multiple proteins on the same blot, allowing simultaneous analysis of target proteins and loading controls even when proteins are close in molecular weight [53]. Near-infrared fluorescent antibodies (e.g., IR700, IR800) offer additional advantages due to reduced membrane autofluorescence and higher signal-to-noise ratios [53].
The comparative analysis of BN-PAGE and SDS-PAGE demonstrates that these techniques provide complementary rather than competing information for protein complex characterization. BN-PAGE excels in preserving native structures and functional interactions, making it indispensable for studying biologically active complexes like those found in snake venoms. SDS-PAGE offers superior resolution of individual components and remains the gold standard for compositional analysis. The emerging approach of NSDS-PAGE represents a promising intermediate, balancing resolution with preservation of certain native properties. For comprehensive understanding of complex biological systems like snake venoms, integrated methodologies that leverage the strengths of multiple techniques will continue to provide the most insightful results, particularly as advanced detection methods like multiplex fluorescent Western blotting enhance quantitative accuracy. These technical capabilities are especially critical for developing targeted therapies for envenomation and exploring the therapeutic potential of venom components themselves.
Solving Common Challenges in Native and Denaturing Gel Electrophoresis
In the study of proteins, particularly multi-subunit complexes, the initial solubilization step is critical and largely determines the success of downstream structural and functional analyses. Two primary electrophoretic techniques—Blue Native PAGE (BN-PAGE) and SDS-PAGE—offer complementary approaches for separating proteins, yet they start from fundamentally different principles. BN-PAGE preserves protein complexes in their native, functional state, allowing researchers to study intact assemblies, their stoichiometry, and interactions [21]. In contrast, standard SDS-PAGE denatures proteins into their constituent polypeptides, providing information on molecular weights but destroying higher-order structure and function [8]. The choice between these techniques dictates the required solubilization strategy, with detergent selection and buffer conditions being paramount for success. This guide objectively compares the performance of these approaches, focusing on the critical parameters of detergent-to-protein ratios and buffer composition that enable optimal protein complex analysis for research and drug development applications.
Fundamental Principles and Comparative Mechanics
BN-PAGE: Preserving Native Complexes
Blue Native PAGE was developed specifically to isolate membrane protein complexes in an enzymatically active form. The technique relies on the binding of the anionic dye Coomassie Blue G-250 to protein complexes, which imparts a negative charge without disrupting intrinsic protein-protein interactions. This charge allows migration in a polyacrylamide gel under native conditions. The resulting separation reveals information about the size, abundance, and subunit composition of complexes, and can even detect assembly intermediates [21]. The method typically involves a first-dimension BN-PAGE separation, which can be followed by a second-dimension denaturing SDS-PAGE to resolve the individual subunits of each complex, providing a powerful two-dimensional analysis system.
SDS-PAGE: Denaturing Separation
Traditional SDS-PAGE is a denaturing technique that involves heating proteins in the presence of the ionic detergent Sodium Dodecyl Sulfate (SDS) and a reducing agent. SDS binds cooperatively to polypeptides, unfolding them and conferring a uniform negative charge density. This process masks the proteins' intrinsic charge and allows separation based almost exclusively on molecular mass [8]. While excellent for determining purity and molecular weight, this denaturation destroys native complexes, quaternary structure, and enzymatic activity. A modified approach called Native SDS-PAGE (NSDS-PAGE) reduces the denaturing conditions by removing SDS and EDTA from the sample buffer and omitting the heating step, allowing some proteins to retain function and bound metal ions [8].
Comparative Workflow Diagram
The following diagram illustrates the key procedural differences between BN-PAGE and SDS-PAGE, highlighting how the choice of detergent and buffer conditions dictates the analytical outcome:
Detergent Mechanisms and Solubilization Strategies
The Role of Detergents in Membrane Protein Solubilization
Detergents are amphipathic molecules essential for manipulating hydrophobic-hydrophilic interactions in biological samples. In protein research, they serve to lyse cells, solubilize membrane proteins and lipids, and control protein crystallization [54]. Detergents form thermodynamically stable colloidal aggregates called micelles above a specific concentration known as the Critical Micelle Concentration (CMC). The number of detergent monomers per micelle is called the aggregation number [54].
For membrane protein solubilization, detergent monomers partition into the membrane bilayer. As detergent concentration increases, membranes undergo progressive stages of solubilization:
- At low detergent:lipid molar ratios (0.1:1 to 1:1), the lipid bilayer remains largely intact, but selective extraction of some membrane proteins occurs.
- At a ~2:1 ratio, solubilization begins, resulting in mixed micelles containing phospholipid-detergent, detergent-protein, and lipid-detergent-protein complexes.
- At a ~10:1 ratio, all native membrane lipid:protein interactions are effectively exchanged for detergent:protein interactions [54].
Selecting Appropriate Detergents
The choice of detergent fundamentally determines the success of protein complex preservation:
For BN-PAGE: Mild, non-denaturing detergents are essential. n-dodecyl-β-D-maltoside is frequently used, as it effectively solubilizes membranes without disrupting protein-protein interactions [21]. Other non-ionic (Triton X-100, Digitonin) or zwitterionic (CHAPS) detergents may also be used.
For SDS-PAGE: The strongly denaturing anionic detergent SDS is used, which completely disrupts membranes and denatures proteins by breaking protein-protein interactions [54].
Table: Properties of Common Detergents Used in Protein Solubilization
Detergent Type CMC (mM) Aggregation Number Applications and Considerations n-dodecyl-β-D-maltoside Non-ionic 0.24 140 BN-PAGE: Effective for solubilizing membrane complexes with minimal denaturation [21] SDS Anionic 6-8 62 SDS-PAGE: Strong denaturation, ideal for molecular weight determination [54] Triton X-100 Non-ionic 0.24 140 Mild lysis: Isolating cytoplasmic and membrane proteins in native state [54] CHAPS Zwitterionic 8-10 10 Mild lysis: Useful for protein isolation while maintaining functionality [54] Octyl-glucoside Non-ionic 23-24 27 High CMC: Easily removable by dialysis [54]
Experimental Protocols for Optimal Solubilization
BN-PAGE Protocol for Mitochondrial Complexes
This protocol, based on the method by Schägger and von Jagow, is optimized for the analysis of mitochondrial protein complexes [21]:
Step 1: Sample Preparation
- Resuspend 0.4 mg of sedimented mitochondria in 40 μL of Buffer A (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris/HCl, pH 7.0) containing protease inhibitors (e.g., 1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin).
- Add 7.5 μL of 10% n-dodecyl-β-D-maltoside (final concentration of approximately 1.6%).
- Mix and incubate for 30 minutes on ice.
- Centrifuge at 72,000 x g for 30 minutes at 4°C to remove insoluble material.
- Collect the supernatant and add 2.5 μL of 5% Coomassie Blue G in 0.5 M aminocaproic acid.
Step 2: Native Gel Electrophoresis (1st Dimension)
- Prepare a linear gradient native acrylamide gel (e.g., 6-13%) using a gradient former.
- Load 5-20 μL of the prepared sample per well.
- Run electrophoresis using specific anode and cathode buffers (see Table 2) at 150 V for approximately 2 hours at 4°C until the dye front approaches the gel bottom.
Step 3: Second Dimension Electrophoresis (Optional)
- Excise a lane from the first-dimension BN-PAGE gel.
- Soak the gel strip in SDS-PAGE denaturing buffer (containing SDS and dithiothreitol) for 30-60 minutes.
- Place the strip horizontally on a second SDS-PAGE gel (e.g., 10-20% acrylamide) and proceed with standard denaturing electrophoresis.
NSDS-PAGE Protocol for Semi-Native Separation
This modified protocol from the search results allows for high-resolution separation with retention of some native properties [8]:
Step 1: Sample Preparation
- Mix 7.5 μL of protein sample with 2.5 μL of 4X NSDS-PAGE sample buffer (100 mM Tris HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5).
- Do not heat the sample.
Step 2: Gel Pre-Run and Electrophoresis
- Pre-run a precast NuPAGE Novex 12% Bis-Tris mini-gel at 200V for 30 minutes in double-distilled H₂O to remove storage buffer and unpolymerized acrylamide.
- Load the prepared samples alongside molecular weight standards.
- Perform electrophoresis at a constant voltage (200V) for approximately 45 minutes using NSDS-PAGE running buffer (50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7).
Buffer Formulations for Electrophoretic Techniques
The composition of electrophoresis buffers is a critical factor for successful protein separation. The table below compares key buffer recipes for BN-PAGE, SDS-PAGE, and NSDS-PAGE:
- Table: Buffer Compositions for Different PAGE Methodologies
Buffer Type Components Purpose and Characteristics BN-PAGE Cathode Buffer [21] 50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0 Provides the anionic dye for continuous charge transfer during native electrophoresis. BN-PAGE Anode Buffer [21] 50 mM Bis-Tris, pH 7.0 Completes the electrical circuit in the lower buffer chamber. SDS-PAGE Running Buffer [8] 50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, 0.1% SDS, pH 7.7 Maintains denaturing conditions and uniform charge during separation. NSDS-PAGE Running Buffer [8] 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 Reduced SDS concentration (vs. standard SDS-PAGE) helps preserve some native protein features. SDS-PAGE Denaturing Buffer [21] 10% Glycerol, 2% SDS, 50 mM Tris, 0.002% Bromophenol Blue, 50 mM DTT, pH 6.8 Used to denature samples for 2D-SDS-PAGE; SDS unfolds proteins, DTT reduces disulfide bonds.
Performance Comparison and Experimental Data
Functional Retention and Metal Binding Analysis
Comparative research reveals significant differences in the ability of various electrophoretic methods to preserve protein function. One study systematically tested the retention of Zn²⁺ bound to proteins and the enzymatic activity of nine model enzymes after different electrophoretic separations [8]:
- Metal Retention: After standard SDS-PAGE, only 26% of Zn²⁺ remained bound to proteins. In contrast, 98% retention was achieved with NSDS-PAGE, comparable to the high retention seen with BN-PAGE [8].
- Enzymatic Activity: All nine model enzymes tested were denatured and inactivated during standard SDS-PAGE. However, seven of the nine enzymes retained activity after NSDS-PAGE, while all nine remained active after BN-PAGE [8].
This data demonstrates that NSDS-PAGE offers a valuable intermediate approach, providing higher resolution than BN-PAGE while preserving significantly more functional characteristics than standard SDS-PAGE.
Solubilization Efficiency and Complex Recovery
The efficiency of initial solubilization directly impacts the yield and quality of protein complexes recovered for analysis:
- Detergent-to-Protein Ratio: For membrane proteins, the solubilization buffer must contain sufficient detergent to provide more than one micelle per membrane protein molecule to ensure individual protein molecules are isolated in separate micelles [54]. The optimal ratio depends on the CMC, aggregation number, and the nature of both the membrane and detergent.
- BN-PAGE on Whole Cellular Lysates: Research demonstrates that with appropriate sample preparation, including dialysis, BN-PAGE can be successfully applied to whole cellular lysates, not just purified mitochondria. This enables the study of complex dynamics, such as changes in the eukaryotic proteasome after γ interferon stimulation, directly from complex mixtures [25].
Essential Research Reagent Solutions
Successful protein complex analysis requires a carefully selected toolkit of reagents. The following table details key materials and their functions:
- Table: Essential Reagents for Protein Solubilization and Electrophoresis
Research Reagent Function and Application n-dodecyl-β-D-maltoside Non-ionic detergent for mild solubilization of membrane protein complexes in BN-PAGE [21]. Coomassie Blue G-250 Anionic dye that binds to protein complexes, providing charge for electrophoresis without denaturation [21]. Protease Inhibitor Cocktail Prevents proteolytic degradation during sample preparation (e.g., PMSF, leupeptin, pepstatin) [21]. 6-aminocaproic acid & Bis-Tris Key buffer components in BN-PAGE that maintain stable pH and a non-denaturing environment [21]. SDS (Sodium Dodecyl Sulfate) Strong anionic detergent that denatures proteins and confers uniform charge for SDS-PAGE [54]. Dithiothreitol (DTT) or β-mercaptoethanol Reducing agents that break disulfide bonds in proteins for complete denaturation in SDS-PAGE. CHAPS Detergent Zwitterionic detergent useful as a mild alternative for cell lysis and protein isolation [54]. PVDF Membrane Preferred membrane for electroblotting BN-PAGE gels due to superior protein binding and handling properties [21].
The choice between BN-PAGE and SDS-PAGE, and the subsequent optimization of solubilization conditions, must be driven by the specific research goals. The experimental data and protocols presented provide a clear framework for this decision:
For studies aimed at understanding intact protein complexes, their native molecular weights, interactions, and assembly states, BN-PAGE with mild detergents like n-dodecyl-β-D-maltoside is the unequivocal choice. The protocol requires careful optimization of detergent-to-protein ratios, typically using detergents at concentrations well above their CMC.
For standard molecular weight determination, purity assessment, or analysis of polypeptide composition, denaturing SDS-PAGE remains the gold standard due to its excellent resolution and reproducibility.
For projects requiring a balance between high resolution and the preservation of certain native properties, such as metal binding or enzymatic activity, NSDS-PAGE offers a viable intermediate approach, as demonstrated by its high rates of metal retention and functional preservation.
Ultimately, the comparative performance of these techniques underscores a fundamental trade-off in protein biochemistry: the balance between structural resolution and functional preservation. By understanding and strategically applying the principles of detergent-based solubilization outlined in this guide, researchers can effectively tailor their experimental design to answer specific biological questions in basic research and drug development.
Pre-fractionation Strategies for Low-Abundance Complexes
The comprehensive analysis of protein complexes, particularly those of low abundance, represents a significant challenge in proteomics and drug development research. Due to the immense dynamic range of protein concentrations in biological samples, low-abundance complexes are often masked by highly abundant proteins during analytical procedures [55]. Pre-fractionation strategies prior to detailed analysis are therefore not merely beneficial but essential for revealing these elusive targets that may have crucial physiological and pathological roles [56].
The central dilemma in protein complex analysis lies in the balance between resolution and native state preservation. While standard SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) offers high resolution separation based on molecular mass, it does so at the cost of denaturing protein complexes and destroying functional properties, including non-covalently bound cofactors and metal ions [8]. In contrast, BN-PAGE (Blue Native-PAGE) preserves native interactions and enzymatic activities but traditionally falls short of the resolution power achieved by denaturing techniques [8] [19]. This methodological divide frames the ongoing challenge for researchers seeking to characterize low-abundance complexes with both structural and functional fidelity.
This guide objectively compares the performance of these orthogonal approaches, their hybrid applications, and supporting experimental data to inform strategic decisions in protein complex analysis. By understanding the relative strengths and limitations of each method, researchers can design fractionation workflows that maximize sensitivity for detecting and characterizing the least abundant components of the proteome.
Technical Comparison of BN-PAGE and SDS-PAGE
The selection between BN-PAGE and SDS-PAGE involves fundamental trade-offs between preservation of native states and achieving high resolution separation. The table below summarizes the core characteristics and performance metrics of these techniques.
Table 1: Performance Comparison of BN-PAGE and SDS-PAGE for Protein Complex Analysis
| Feature | BN-PAGE | SDS-PAGE |
|---|---|---|
| Separation Principle | Size & charge via Coomassie binding [19] | Molecular mass via SDS binding [8] |
| Native Structure Preservation | Yes - maintains enzymatic activity and protein interactions [8] [25] | No - denatures proteins and disrupts complexes [8] |
| Resolution | Lower for individual proteins, higher for intact complexes [8] | High for individual polypeptide separation [8] |
| Compatibility with Low-Abundance Targets | Requires pre-fractionation/enrichment for low-abundance complexes [19] | GeLC-MS/MS provides good sensitivity; 1-D SDS-PAGE yielded highest identifications in one study [56] |
| Metal Cofactor Retention | Excellent (e.g., 98% Zn²⁺ retention demonstrated) [8] | Poor (e.g., 26% Zn²⁺ retention demonstrated) [8] |
| Enzymatic Activity Recovery | High (7 of 9 model enzymes remained active) [8] | None (all 9 model enzymes denatured) [8] |
| Typical Applications | Analysis of multiprotein complexes, supercomplexes, assembly intermediates [25] [21] | Molecular weight determination, purity assessment, immunoblotting [8] |
| Optimal Pre-fractionation Strategies | Cellular/organelle pre-fractionation, affinity chromatography [19] | GeLC-MS/MS, preparative SDS-PAGE [56] |
The experimental data clearly demonstrates that BN-PAGE and SDS-PAGE serve complementary roles in protein complex analysis. BN-PAGE excels in preserving functional properties, with studies showing 98% retention of bound Zn²⁺ ions and maintenance of enzymatic activity in 7 of 9 model enzymes [8]. This makes it indispensable for studying metalloproteins and functional complexes. Conversely, SDS-PAGE provides superior resolution for separating individual polypeptides, with 1-D SDS-PAGE demonstrating the highest number of protein identifications in comparative proteomic profiling [56].
For low-abundance targets specifically, BN-PAGE typically requires additional pre-fractionation steps such as organelle isolation or affinity enrichment to reduce sample complexity and improve detection sensitivity [19]. SDS-PAGE-based approaches like GeLC-MS/MS (where the gel lane is sliced into multiple fractions followed by in-gel digestion and LC-MS/MS analysis) can provide excellent proteome coverage but sacrifice information about native complexes and interactions [56].
Advanced Technical Applications and Workflow Integration
Two-Dimensional Electrophoresis Approaches
The integration of BN-PAGE and SDS-PAGE in two-dimensional (2D) electrophoresis represents a powerful strategy that combines the strengths of both techniques. In 2D BN-/SDS-PAGE, protein complexes are first separated under native conditions using BN-PAGE, followed by denaturing separation of their individual subunits in the second dimension using SDS-PAGE [57]. This approach allows researchers to simultaneously determine the size and subunit composition of multiprotein complexes while maintaining information about their native interactions [25].
This technique has proven particularly valuable for studying signaling complexes, as demonstrated in research on SLP family adaptor proteins in immune cells. Using 2D BN-/SDS-PAGE, researchers identified that in non-stimulated T cells, all SLP-76 proteins exist in a ~400 kDa complex with the adaptor protein Gads, whereas in B cells, SLP-65 exists in both a 180 kDa complex and monomeric form [57]. Importantly, the study revealed that upon antigen stimulation, only the complexed form of SLP-65 was phosphorylated, highlighting the functional significance of complex formation that would be undetectable using fully denaturing methods [57].
Table 2: Detection of Low-Abundance Proteins: Enhanced Western Blotting Strategies
| Strategy | Specific Technique | Benefit for Low-Abundance Targets |
|---|---|---|
| Sample Preparation | Subcellular fractionation; protease/phosphatase inhibitors; ultrasonic disruption [58] | Enriches target proteins; prevents degradation; improves nuclear protein release |
| Gel Electrophoresis | Bis-Tris gels (neutral pH); Tris-Acetate gels (high MW); Tricine gels (low MW) [59] | Preserves protein integrity; better resolution for extreme MW proteins |
| Membrane Transfer | PVDF membranes; optimized wet/semi-dry/dry transfer [58] | Higher protein binding capacity; improved transfer efficiency |
| Immunodetection | High-ab concentration; low-blocker concentration; high-sensitivity chemiluminescent substrates [59] | Enhances signal-to-noise ratio; maximizes detection sensitivity |
Integration with Mass Spectrometry
The combination of gel-based separation with mass spectrometry has dramatically enhanced the detection and characterization of low-abundance protein complexes. The development of PEPPI-MS (Passively Eluting Proteins from Polyacrylamide Gels as Intact Species for MS) has addressed the long-standing challenge of efficiently recovering proteins from gel matrices, with mean recovery rates of 68% for proteins below 100 kDa [48]. This technical advancement has enabled the application of GeLC-MS workflows to top-down proteomics, facilitating the large-scale analysis of intact proteoforms [48].
For structural proteomics, the integration of PAGE with techniques like native MS, cross-linking MS (XL-MS), and top-down MS has created new opportunities for comprehensive protein complex characterization [48]. These approaches allow researchers to obtain structural information while leveraging the exceptional separation power of gel-based methods to reduce sample complexity and enhance detection of low-abundance components that would otherwise be masked in direct LC-MS analyses.
Experimental Protocols for Enhanced Detection
BN-PAGE for Multiprotein Complex Analysis
The following protocol, adapted from established methodologies [21], enables the separation of native protein complexes from mitochondrial preparations or whole cell lysates:
Sample Preparation:
- Isolate mitochondria or whole cells and resuspend in 0.75 M 6-aminocaproic acid, 50 mM Bis-Tris, pH 7.0 [21].
- Add n-dodecyl-β-D-maltopyranoside to 1-2% final concentration for membrane solubilization. For soluble complexes, milder detergents like digitonin may be preferable [19].
- Incubate on ice for 30 minutes, then centrifuge at 72,000 × g for 30 minutes at 4°C [21].
- Collect supernatant and add Coomassie Blue G to 0.25% final concentration along with protease inhibitors (e.g., 1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin) [21].
Gel Electrophoresis:
- Prepare a native gradient gel (e.g., 6-13% acrylamide) in 50 mM Bis-Tris, 0.75 M 6-aminocaproic acid, pH 7.0 [21].
- Use a stacking gel (typically 3-4% acrylamide) in the same buffer system.
- Load samples and run with cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0) and anode buffer (50 mM Bis-Tris, pH 7.0) [21].
- Run at 100-150 V for approximately 2 hours at 4°C until the dye front approaches the gel bottom [21].
Downstream Processing:
- For complex analysis, proceed to western blotting using PVDF membranes with Tris-Glycine transfer buffer [21].
- For subunit resolution, incubate gel strips in SDS denaturing buffer (2% SDS, 50 mM DTT) for 30 minutes, then perform second dimension SDS-PAGE [21].
Enhanced Western Blotting for Low-Abundance Proteins
This protocol includes optimizations specifically for detecting low-abundance targets [58]:
Sample Preparation:
- Use fresh or protease-inhibited samples. For secreted proteins, add Brefeldin A (BFA) before cell lysis to prevent secretion [58].
- Lysate cells in RIPA or specialized buffer with protease/phosphatase inhibitors.
- Employ ultrasonication (3-second pulses, 10-second intervals, 5-15 cycles) to disrupt cells completely, especially for nuclear proteins [58].
- Determine protein concentration and use 5× loading buffer to minimize sample dilution.
- Load 50-100 μg protein per lane using 1.5 mm thick gels to increase capacity [58].
Electrophoresis and Transfer:
- Use appropriate gel chemistry: Bis-Tris gels (neutral pH) for most proteins, Tris-Acetate for high molecular weight targets (>300 kDa), or Tricine gels for low molecular weight targets (<30 kDa) [59].
- Transfer to PVDF membranes (pre-wetted in methanol) using wet, semi-dry, or dry transfer systems [58].
- Verify transfer efficiency with Ponceau S staining [58].
Immunodetection:
- Block with 5% non-fat milk or BSA for 1 hour at room temperature. Reduced blocking time or concentration may enhance signal for some targets [58].
- Incubate with primary antibody at higher than standard concentration (e.g., 2-5× typical dilution) overnight at 4°C [58].
- Use high-affinity HRP-conjugated secondary antibodies with high-sensitivity chemiluminescent substrates (e.g., SuperSignal West Atto) for maximal detection sensitivity [59].
- Avoid sodium azide in detection systems as it inhibits HRP activity [58].
Visualizing Methodologies and Workflows
The following diagrams illustrate key experimental workflows and technical relationships for pre-fractionation strategies targeting low-abundance protein complexes.
BN-PAGE vs SDS-PAGE Workflows
Essential Research Reagent Solutions
Successful analysis of low-abundance protein complexes requires carefully selected reagents optimized for specific methodological approaches.
Table 3: Essential Research Reagents for Protein Complex Analysis
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Solubilization Detergents | n-Dodecyl-β-D-maltoside, Digitonin, Triton X-100 [19] | Mild non-ionic detergents for native complex solubilization in BN-PAGE |
| Specialized Gels | Bis-Tris gels (neutral pH), Tris-Acetate gels, Tricine gels [59] | Optimized separation for different molecular weight ranges; preserve protein integrity |
| Staining Dyes | Coomassie Blue G-250 [21] | Imparts negative charge to proteins in BN-PAGE; reversible staining |
| Protease Inhibitors | PMSF, Leupeptin, Pepstatin [21] | Prevent protein degradation during extraction and processing |
| Membranes for Blotting | PVDF membranes [58] | High protein-binding capacity for low-abundance target detection |
| Detection Substrates | SuperSignal West Atto Ultimate Sensitivity Substrate [59] | High-sensitivity chemiluminescent detection for low-abundance proteins |
Concluding Analysis and Strategic Recommendations
The comparative analysis of pre-fractionation strategies for low-abundance protein complexes reveals that method selection must be guided by specific research objectives. BN-PAGE emerges as the superior approach when maintaining native structure, enzymatic activity, or metal cofactor associations is paramount [8]. Its application in studying respiratory chain supercomplexes and signaling assemblies demonstrates unique capabilities for functional analysis that denaturing methods cannot provide [19]. However, its limitations in resolution and sensitivity for low-abundance targets necessitate complementary approaches.
SDS-PAGE-based strategies, particularly GeLC-MS/MS, provide exceptional proteomic coverage and sensitivity for identifying low-abundance proteins, with studies showing that 1-D SDS-PAGE yielded the highest number of protein identifications in comparative profiling [56]. The development of improved protein recovery methods like PEPPI-MS, which achieves 68% recovery efficiency for proteins below 100 kDa, has further enhanced the utility of SDS-PAGE for top-down proteomics [48].
For the most challenging targets, hybrid approaches such as 2D BN-/SDS-PAGE offer a powerful compromise, enabling initial separation of intact complexes followed by high-resolution analysis of their subunits [57]. This strategy has proven valuable for characterizing signaling complexes in immunology research, revealing stimulus-dependent phosphorylation events specifically associated with complexed forms of adaptor proteins [57].
The optimal pre-fractionation strategy for low-abundance complexes often involves orthogonal separation techniques tailored to the specific biological question. For comprehensive analysis, researchers may implement sequential fractionation at both protein and peptide levels, leveraging the complementary strengths of BN-PAGE for native complex preservation and SDS-PAGE for maximum resolution and proteomic depth [56]. As mass spectrometry technologies continue to advance in sensitivity, the integration of these gel-based separation methods with structural MS approaches promises to further expand our ability to characterize the most elusive components of the cellular proteome.
In the rigorous field of protein analysis, the choice of electrophoresis technique and parameters directly determines the resolution, accuracy, and reliability of experimental data. For researchers and drug development professionals investigating protein complexes, the strategic use of gradient gels provides a significant technical advantage over fixed-concentration gels by enabling the analysis of a broader spectrum of protein sizes within a single experiment [60]. This guide provides a detailed comparative analysis of two foundational electrophoretic techniques: Blue Native PAGE (BN-PAGE), which preserves native protein complexes for functional studies, and SDS-PAGE, which denatures proteins to separate subunits by molecular weight [4]. The selection between these methods is not merely procedural but strategic, influencing everything from the initial experimental design to the interpretation of complex biological mechanisms. Within this framework, gradient gels serve as a powerful tool that enhances both techniques, offering superior resolution across a wide molecular weight range and producing sharper protein bands that are critical for publication-quality data and accurate quantification [60]. By objectively comparing the performance of these systems and detailing the experimental parameters that optimize their resolving power, this guide aims to equip scientists with the knowledge needed to maximize the yield of information from precious protein samples.
Fundamental Principles: BN-PAGE vs. SDS-PAGE
The core distinction between BN-PAGE and SDS-PAGE lies in the state of the protein during separation—native or denatured—which in turn dictates the type of biological information that can be obtained.
Blue Native PAGE (BN-PAGE) is a high-resolution technique specifically designed for the separation of intact, native protein complexes. First developed by Schägger and von Jagow in the 1990s, it uses the mild, nonionic detergent n-dodecyl-β-d-maltoside for membrane protein solubilization and the anionic dye Coomassie Blue G-250 to impose a negative charge shift on the proteins [35] [21]. This charge shift facilitates electrophoretic migration toward the anode while keeping hydrophobic proteins soluble without dissociating the complexes. A key advantage of BN-PAGE is its ability to maintain the enzymatic activity of separated complexes, allowing for subsequent functional assays and in-gel activity staining [35] [23]. When the even milder detergent digitonin is used, BN-PAGE can resolve higher-order structures such as respiratory chain supercomplexes (respirasomes), providing insights into functional protein interactions within biological membranes [35].
In contrast, SDS-PAGE is a denaturing technique that separates individual protein subunits primarily by molecular weight. The ionic detergent sodium dodecyl sulfate (SDS) denatures proteins and binds to the polypeptide backbone in a constant mass ratio, conferring a uniform negative charge that overwhelms the protein's intrinsic charge [4]. Consequently, separation is based almost entirely on polypeptide size due to the molecular sieving effect of the polyacrylamide matrix, with smaller proteins migrating faster than larger ones [4]. This makes SDS-PAGE ideal for determining protein purity, estimating molecular weight, and analyzing subunit composition when combined with reducing agents to break disulfide bonds.
Table 1: Core Principles and Applications of BN-PAGE vs. SDS-PAGE
| Parameter | BN-PAGE (Native) | SDS-PAGE (Denaturing) |
|---|---|---|
| Separation Principle | Size, charge, and native shape of complexes [4] | Molecular weight of polypeptide subunits [4] |
| Protein State | Native, intact complexes | Denatured, linearized subunits |
| Key Reagents | Coomassie Blue G-250, n-dodecyl-β-d-maltoside, digitonin [35] [21] | SDS, β-mercaptoethanol or DTT [4] |
| Primary Applications | Studying oligomeric state, supercomplex formation, in-gel enzyme activity [35] [23] | Determining protein size, purity, and subunit composition [4] |
| Typical Gel Type | Linear gradient (e.g., 4-16%) [35] | Single percentage or gradient (e.g., 4-20%) [60] |
Technical Comparison: Resolution and Performance Data
The resolving power of an electrophoretic system is quantified by its ability to distinguish proteins of similar size and its effective separation range. Gradient gels enhance both BN-PAGE and SDS-PAGE by creating a pore size gradient that linearly decreases from the top to the bottom of the gel.
Performance Advantages of Gradient Gels
In practice, gradient gels offer three key performance benefits over fixed-concentration gels:
- Broad Dynamic Range: A single gradient gel (e.g., 4-20%) can effectively resolve proteins from 10-200 kDa, a range that would otherwise require running multiple single-percentage gels (e.g., 8%, 12%, and 15%) [60].
- Sharper Banding: As a protein migrates, its leading edge encounters progressively smaller pores and slows down, while the trailing edge continues to move forward. This "piling up" effect compresses the protein into a sharper, more discrete band [60].
- Enhanced Resolution of Similar-Sized Proteins: The sharpening effect and extended run time possible with gradient gels increase the physical distance between bands of similar molecular weights, making it easier to distinguish them during analysis [60].
Table 2: Quantitative Comparison of Electrophoresis Systems and Their Resolving Power
| System | Effective Separation Range | Optimal Gradient for Broad Separation | Key Performance Metric |
|---|---|---|---|
| BN-PAGE | ~100 kDa to several MDa (complexes) [35] | 3–12% or 4–16% linear gradient [35] | Resolution of individual OXPHOS complexes and supercomplexes [35] |
| SDS-PAGE | 4-250 kDa (polypeptides) [60] | 4–20% or 8–16% linear gradient [60] | Sharp, discrete bands for proteins from 10-100 kDa on a single gel [60] |
| 2D BN/SDS-PAGE | Combines both BN and SDS separation ranges | First dimension: 3–12% BN; Second: 10-20% SDS [21] [23] | Identification of modified subunits within a complex (e.g., HNE-modified Complex I subunits) [23] |
Experimental Validation Data
Independent validation of a streamlined BN-PAGE protocol demonstrates its robust performance. Using a linear gradient gel system (e.g., 4-16%), this protocol successfully resolves the five oxidative phosphorylation (OXPHOS) complexes from human cell lines and patient fibroblasts [35]. The in-gel activity staining for Complexes I, II, IV, and V remains functional after electrophoresis, confirming the preservation of enzymatic activity, a hallmark of successful native electrophoresis [35]. Furthermore, the protocol shows a high dynamic range, detecting assembly intermediates and pathological defects in models of monogenetic mitochondrial disorders [35].
Experimental Protocols
Protocol 1: BN-PAGE for Analysis of Mitochondrial Complexes
This protocol is adapted for the analysis of small patient samples and uses a simplified extraction procedure [35] [21].
Sample Preparation
- Isolate mitochondria from cells or tissue. While whole-cell extracts can be used, mitochondrial isolation yields a stronger signal [21].
- Resuspend 0.4 mg of mitochondrial pellet in 40 µL of Buffer A (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris, pH 7.0, plus protease inhibitors) [21].
- Add 7.5 µL of 10% n-dodecyl-β-D-maltoside (DDM). Mix and incubate on ice for 30-60 minutes [35] [21].
- Centrifuge at 16,000–72,000 x g for 30 minutes at 4°C to remove insolubilized material [21].
- Collect the supernatant and add 2.5 µL of 5% Coomassie Blue G solution [21].
Gel Casting and Electrophoresis
- Cast a native gradient gel. A 6–13% linear gradient gel is highly recommended for resolving OXPHOS complexes [21]. Use a gradient mixer to pour the gel with a low-percentage solution (e.g., 6%) and a high-percentage solution (e.g., 13%) [35] [21].
- Load 5–20 µL of prepared sample per well.
- Run the gel using a cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G, pH 7.0) and an anode buffer (50 mM Bis-Tris, pH 7.0) [21] [23].
- Conduct electrophoresis at 100-150 V for the first phase. Once the dye front has migrated about one-third into the gel, replace the cathode buffer with a version without Coomassie dye to reduce interference with downstream activity stains. Continue running at 200 V until completion [23].
Downstream Analysis
- Western Blotting: Transfer proteins to a PVDF membrane for immunodetection with antibodies against specific complex subunits [21].
- In-Gel Activity Staining: Incubate the gel in specific substrate solutions to visualize active complexes (e.g., NADH and Nitro Blue Tetrazolium for Complex I activity) [35] [23].
- Two-Dimensional Electrophoresis (2D-BN/SDS-PAGE): Excise a lane from the BN-PAGE gel, soak it in SDS buffer, and place it on a second-dimension SDS-PAGE gel to separate the individual subunits of the complexes [23].
Protocol 2: 2D BN/SDS-PAGE for Identifying Protein Modifications
This method is ideal for identifying post-translational modifications within specific complexes, such as HNE-modified subunits of mitochondrial Complex I [23].
First Dimension (BN-PAGE)
- Solubilize mitochondrial membranes in BN-PAGE sample buffer (1% DDM, 0.75 M aminocaproic acid, 75 mM Bis-Tris, pH 7.0) [23].
- Separate the solubilized complexes on a non-gradient (e.g., 6%) or gradient BN-PAGE gel as described in Protocol 1.
- After electrophoresis, cut out the band corresponding to the intact Complex I, identified by a Coomassie blue stain or in-gel activity stain [23].
Second Dimension (SDS-PAGE)
- Equilibrate the BN-PAGE gel strip in SDS denaturing buffer (2% SDS, 5% 2-mercaptoethanol, 62.5 mM Tris-HCl, pH 6.8, 10% glycerol) for 20 minutes [23].
- Place the strip onto a 10% SDS-PAGE gel.
- Run the second-dimension SDS-PAGE according to standard protocols.
- Transfer proteins to a membrane for Western blotting with anti-HNE antibodies or stain the gel with Coomassie or silver to visualize all subunits [23].
- Excise HNE-positive bands from a parallel stained gel for protein identification by mass spectrometry [23].
Visualization of Workflows
The following diagrams illustrate the logical flow of the two primary electrophoretic methods discussed, highlighting their divergent paths and applications.
Diagram 1: A workflow diagram comparing BN-PAGE and SDS-PAGE analysis paths. BN-PAGE preserves native complexes for functional studies and second-dimension analysis, while SDS-PAGE provides straightforward separation of denatured subunits.
Diagram 2: A detailed workflow for two-dimensional BN/SDS-PAGE analysis. The process begins with native complex separation, followed by denaturing separation of subunits, enabling the identification of specific proteins and their modifications within a complex.
Essential Research Reagent Solutions
The following table catalogues the key reagents required for successful BN-PAGE and gradient gel electrophoresis, along with their critical functions.
Table 3: Essential Reagents for Protein Complex Analysis by BN-PAGE
| Reagent | Function | Application Notes |
|---|---|---|
| n-Dodecyl-β-D-maltoside (DDM) | Mild, nonionic detergent for solubilizing membrane proteins without disrupting complex integrity [35] [21]. | Preferred for resolving individual OXPHOS complexes. Concentration typically 1% [23]. |
| Digitonin | Very mild, nonionic detergent used to preserve supercomplex assemblies [35]. | Used for analyzing respirasomes (Complex I/III/IV supercomplexes) [35]. |
| Coomassie Blue G-250 | Anionic dye that binds hydrophobic protein surfaces, imparting negative charge for migration and preventing aggregation [35] [21]. | Added to sample and cathode buffer. Critical for BN-PAGE separation [35]. |
| 6-Aminocaproic Acid | Zwitterionic salt that supports solubilization and acts as a protease inhibitor [35]. | Creates a zero net charge environment at pH 7.0, preventing complex dissociation [35]. |
| Bis-Tris | Buffering agent used in gels and running buffers at pH 7.0 [21] [23]. | Maintains a neutral pH compatible with native protein structures [21]. |
| Linear Gradient Gels (e.g., 4-16%) | Polyacrylamide gels with a continuous concentration gradient for separating a wide size range of complexes [35] [60]. | Provides high resolution of complexes from ~100 kDa to several MDa [35]. |
| Acrylamide/Bis-Acrylamide | Matrix-forming monomers that create the sieving network for electrophoretic separation [4]. | The ratio and total concentration determine gel pore size and resolution [4]. |
Preserving Enzyme Activity and Complex Integrity During Processing
In molecular biology and drug development, the method chosen for protein separation can fundamentally determine the success of downstream analyses. When research objectives require the study of functional protein complexes—with intact enzymatic activity and preserved subunit interactions—the choice of electrophoretic technique becomes paramount. Blue Native PAGE (BN-PAGE) and Sodium Dodecyl Sulfate-PAGE (SDS-PAGE) represent two fundamentally different approaches to protein separation, with divergent outcomes for protein complex integrity [1].
This comparison guide examines the core distinctions between these techniques, focusing specifically on their capabilities for preserving enzyme activity and native complex structure. We present experimental data and detailed methodologies to help researchers select the optimal approach for applications ranging from mitochondrial complex analysis to therapeutic protein characterization.
Fundamental Principles and Comparative Analysis
Core Mechanism and Design Philosophy
SDS-PAGE operates as a denaturing technique that deliberately dismantles protein complexes into individual polypeptide subunits. The anionic detergent SDS binds extensively to proteins in a ratio of approximately 1.4g SDS per 1g protein, unfolding the tertiary structure and masking the protein's intrinsic charge [1] [8]. This results in separation based almost exclusively on molecular weight, as all proteins migrate toward the anode with charge-to-mass ratios that are nearly identical [1].
In contrast, BN-PAGE employs a non-denaturing approach designed specifically to preserve supramolecular protein organizations. The key differentiator is the use of Coomassie Blue G-250 dye, which binds to protein complexes without causing dissociation [22]. This dye imparts a negative charge to the complexes, enabling electrophoretic migration while maintaining native protein-protein interactions, bound cofactors, and enzymatic functionality [21] [61].
Direct Technical Comparison
Table 1: Comprehensive Comparison Between BN-PAGE and SDS-PAGE
| Analysis Criteria | BN-PAGE | SDS-PAGE |
|---|---|---|
| Separation Basis | Size, charge, and shape of native complexes [1] | Molecular weight only [1] |
| Gel Conditions | Non-denaturing [1] | Denaturing [1] |
| Detergent System | Non-ionic or mild detergents (e.g., DDM, digitonin) [14] | Ionic detergent SDS with reducing agents [1] |
| Sample Preparation | No heating step [1] | Heating at 70-100°C [1] [8] |
| Protein State | Native conformation, folded [1] | Denatured, unfolded [1] |
| Enzyme Activity | Preserved post-separation [1] [8] | Destroyed [1] [8] |
| Protein Complex Integrity | Maintains intact complexes and quaternary structure [61] [22] | Dissociates into individual subunits [1] |
| Metal Cofactor Retention | Retains bound metal ions [8] | Loss of non-covalently bound metals [8] |
| Protein Recovery | Functional proteins can be recovered [1] | Proteins cannot be recovered in functional form [1] |
| Molecular Weight Range | 10 kDa - 10 MDa [62] | Typically <500 kDa |
| Primary Applications | Studying protein-protein interactions, complex composition and assembly, functional assays [1] [22] | Molecular weight determination, purity assessment, protein expression analysis [1] |
Experimental Evidence: Functional Preservation Data
Quantitative Analysis of Functional Retention
The critical advantage of BN-PAGE for functional studies is demonstrated through direct experimental measurements of enzyme activity and metal retention post-electrophoresis.
Table 2: Experimental Measurements of Functional Preservation
| Experimental Parameter | BN-PAGE | Native SDS-PAGE | Standard SDS-PAGE |
|---|---|---|---|
| Zn²⁺ Retention in Proteomic Samples | Not explicitly measured | 98% [8] | 26% [8] |
| Enzyme Activity Retention (Model Zn²⁺ Proteins) | All nine enzymes active [8] | Seven of nine enzymes active [8] | No enzyme activity [8] |
| Complex I Activity (Mitochondrial) | Retained, measurable via in-gel assay [23] [22] | Not applicable | Not retained |
| Membrane Protein Complex Integrity | Preserved, including supercomplexes [14] [62] | Partially preserved | Fully disrupted |
Case Study: Mitochondrial Complex I Analysis
Research investigating HNE (4-hydroxynonenal) modifications to mitochondrial complex I subunits demonstrates the practical application of BN-PAGE for functional studies. The technique enabled isolation of intact complex I from rat kidney mitochondria, followed by successful in-gel activity staining using NADH and nitro blue tetrazolium [23]. This preservation of enzymatic function allowed researchers to directly correlate complex I activity with specific oxidative modifications in diabetic models—something impossible with SDS-PAGE due to complete complex dissociation and loss of function [23].
Methodological Guide: BN-PAGE Experimental Protocol
Standard BN-PAGE Workflow
The following detailed protocol has been adapted from established methodologies for mitochondrial complex analysis [21] [23]:
Key Reagent Solutions for BN-PAGE
Table 3: Essential Research Reagents for Native Electrophoresis
| Reagent Solution | Composition | Function in Protocol |
|---|---|---|
| Solubilization Buffer | 0.75 M aminocaproic acid, 50 mM Bis-Tris, pH 7.0 [23] | Maintains native pH while providing ionic environment |
| Non-Ionic Detergent | 1% n-dodecyl-β-D-maltoside (DDM) [23] or digitonin [62] | Solubilizes membrane proteins while preserving complexes |
| Coomassie Dye Solution | 0.02% Coomassie Blue G-250 in cathode buffer [21] | Imparts charge shift for migration without denaturation |
| BN-PAGE Cathode Buffer | 50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G-250, pH 7.0 [21] | Upper buffer for electrophoretic separation |
| BN-PAGE Anode Buffer | 50 mM Bis-Tris, pH 7.0 [21] | Lower buffer for completing circuit |
| In-Gel Activity Stain | 0.1 mg/ml NADH, 0.2 mg/ml NBT in phosphate buffer [23] | Detects enzymatic activity of complex I |
| Second Dimension Equilibration Buffer | 5% 2-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.8 [23] | Denatures complexes for second dimension |
Critical Optimization Parameters
Successful implementation of BN-PAGE requires careful attention to several key parameters:
Detergent Selection and Concentration: The choice of detergent significantly impacts complex preservation. For mitochondrial complexes, dodecyl maltoside (1-2%) effectively solubilizes individual complexes, while digitonin (1-2%) can preserve supercomplex organizations [14] [62]. Detergent-to-protein ratios typically range from 1:1 to 3:1 (w/w) [14].
Sample Preparation Considerations: Mitochondrial isolation should be performed in isotonic buffers (e.g., 250 mM sucrose, 20 mM sodium phosphate) to preserve organelle integrity [62]. Protease inhibitors (PMSF, leupeptin, pepstatin) are essential to prevent degradation during solubilization [21].
Electrophoresis Conditions: BN-PAGE is typically performed at 4°C to maintain protein stability, using constant voltage (150-200V) until the dye front migrates completely through the gel [1] [23].
Advanced Application: Two-Dimensional BN/SDS-PAGE
For comprehensive analysis of protein complex composition, two-dimensional separation combines the strengths of both techniques:
In this approach, protein complexes are first separated by BN-PAGE, then individual gel lanes are excised, denatured in SDS buffer, and placed perpendicularly on an SDS-PAGE gel [61] [23] [63]. This enables researchers to correlate intact complexes with their subunit composition, revealing which proteins co-migrate in native conditions [63].
The choice between BN-PAGE and SDS-PAGE fundamentally depends on research objectives. SDS-PAGE remains the superior choice for applications requiring precise molecular weight determination, assessment of protein purity, or expression level analysis where preservation of native structure is unnecessary [1] [64].
For studies investigating protein-protein interactions, enzymatic function, complex assembly states, or metalloprotein characterization, BN-PAGE provides indispensable capabilities for maintaining native structure and function [1] [22]. The development of related techniques like Native SDS-PAGE (reduced SDS concentration without heating) offers intermediate options that preserve some functional aspects while maintaining high resolution [8].
In drug development contexts where understanding therapeutic protein interactions or assessing complex integrity is crucial, BN-PAGE provides critical functional data that complements the structural information obtained from SDS-PAGE [64]. By implementing the appropriate technique—or strategically combining them in two-dimensional approaches—researchers can obtain comprehensive insights into both the structural and functional aspects of protein systems.
Within the diverse toolkit for protein analysis, polyacrylamide gel electrophoresis (PAGE) is a fundamental technique. While SDS-PAGE separates denatured proteins by molecular weight and Blue Native PAGE (BN-PAGE) resolves native complexes by size, Clear Native PAGE (CN-PAGE) emerges as a critical alternative when preserving enzymatic function for in-gel activity assays is the primary goal [1] [65]. This guide provides an objective comparison of these techniques, focusing on the experimental data that establishes CN-PAGE's unique value in studying active protein complexes, particularly in mitochondrial research and membrane protein biochemistry.
Technical Comparison: SDS-PAGE, BN-PAGE, and CN-PAGE
The core distinction between these techniques lies in their treatment of protein structure and the resulting impact on functionality.
- SDS-PAGE employs the ionic detergent sodium dodecyl sulfate (SDS) and a reducing agent to denature proteins into linear chains, masking their intrinsic charge. Separation is based almost exclusively on molecular weight, but biological activity is lost [1] [5].
- BN-PAGE uses the mild detergent n-dodecyl-β-D-maltoside (DDM) and the anionic dye Coomassie Brilliant Blue G-250. The dye binds to proteins, conferring a negative charge shift that allows separation by size and native shape. While it preserves protein complexes and some function, the bound Coomassie dye can inhibit enzymatic activity and interfere with fluorescence detection [65] [30] [66].
- CN-PAGE is a "milder" variant. It forgoes Coomassie dye, instead relying on mixtures of anionic and neutral detergents in the cathode buffer to impose a charge shift. This results in a clear gel, eliminating dye-related interference and making it the superior choice for subsequent in-gel enzyme activity staining and fluorescence studies [65] [30].
Table 1: Core Characteristics and Applications of PAGE Techniques.
| Feature | SDS-PAGE | BN-PAGE | CN-PAGE |
|---|---|---|---|
| Protein State | Denatured / Unfolded [1] | Native / Folded [1] | Native / Folded [65] |
| Key Reagent | SDS & Reducing Agent (DTT/BME) [1] | Coomassie Blue G-250 Dye [30] | Sodium Deoxycholate (DOC) or mixed detergents [30] [67] |
| Separation Basis | Molecular Weight [1] | Size & Shape (Native Mass) [1] | Size, Shape & Intrinsic Charge [65] |
| Protein Function | Lost [1] | Often Retained, but can be inhibited [30] | Optimally Preserved [65] |
| Primary Application | Molecular weight determination, purity checks [1] | Analysis of protein complexes, assembly pathways [30] | In-gel activity assays, functional studies, fluorescence detection [65] [30] |
CN-PAGE in Practice: Key Advantages and Experimental Evidence
Enhanced Resolution of Enzymatic Activity
The most cited advantage of CN-PAGE is its compatibility with sensitive in-gel enzyme activity staining. Research on mitochondrial oxidative phosphorylation (OXPHOS) complexes validates this. A 2025 protocol paper states that a "key advantage of CN-PAGE is the absence of residual blue dye interference during downstream in-gel enzyme activity staining" [30]. This is crucial for detecting the catalytic activity of complexes like ATP synthase (Complex V), which can be masked in BN-PAGE. The same study developed a simple enhancement step for in-gel Complex V activity staining that "markedly improves sensitivity" [30] [35].
Furthermore, a comparative study highlighted that CN-PAGE enabled the identification of "enzymatically active oligomeric states of mitochondrial ATP synthase previously not detected using BN-PAGE" [65]. This demonstrates CN-PAGE's ability to reveal functional states of complexes that are labile or inhibited by the conditions of other methods.
Preservation of Labile Supramolecular Assemblies
CN-PAGE excels at stabilizing weak protein interactions that might dissociate under other conditions. The combination of the mild detergent digitonin for membrane solubilization with CN-PAGE has been shown to "retain labile supramolecular assemblies of membrane protein complexes that are dissociated under the conditions of BN-PAGE" [65]. This makes CN-PAGE particularly valuable for studying higher-order structures like respiratory chain supercomplexes (respirasomes).
Compatibility with Fluorescence and Structural Biology
The transparency of CN-PAGE gels makes it ideal for techniques requiring fluorescence detection. For instance, CN-PAGE has been used to screen for optimal purification and crystallization conditions of the membrane protein A2A adenosine receptor (A2AAR) fused to a red fluorescent protein (RFP) [66]. This "mCBB CN-PAGE" method allowed researchers to rapidly screen detergents and additives without time-consuming protein purification.
More recently, CN-PAGE has been integrated into cryo-electron microscopy (cryo-EM) workflows. A novel CN-PAGE-based method was developed for preparing ultra-large protein complexes like the photosystem I–light-harvesting complex I (PSI–LHCI) supercomplex. This approach allowed direct grid preparation after electroelution from the gel, achieving high-resolution (2.18 Å) structure determination without additional chromatographic purification [67].
Table 2: Experimental Evidence Showcasing CN-PAGE Advantages.
| Research Context | Experimental Finding | Implication |
|---|---|---|
| Mitochondrial OXPHOS Complexes [65] [30] | CN-PAGE identified active ATP synthase oligomers not seen with BN-PAGE; no dye interference in activity stains. | Superior for functional enzymology and detecting labile active states. |
| GPCR Crystallization (A2AAR) [66] | Enabled fluorescence-based screening of fusion protein stability and optimal conditions directly from crude extracts. | Accelerates pre-crystallization screening for difficult membrane proteins. |
| Cryo-EM Sample Prep [67] | PSI–LHCI supercomplex was isolated via CN-PAGE and used for high-resolution (2.18 Å) structure determination. | Provides a rapid, high-resolution purification path for structural biology. |
Experimental Protocols: A Workflow for CN-PAGE
The following workflow is adapted from established protocols for analyzing mitochondrial complexes [30] [35].
Sample Preparation
- Isolation: Isolate mitochondria or membrane fractions from tissues or cell lines (e.g., HEK293T, HeLa) using standard differential centrifugation.
- Solubilization: Solubilize the membrane pellet using a mild non-ionic detergent. For individual complexes, use n-dodecyl-β-D-maltoside (DDM). For supramolecular assemblies like respirasomes, use digitonin.
- Clarification: Remove insoluble material by high-speed centrifugation (e.g., 100,000 × g for 30 minutes at 4°C). The supernatant contains the solubilized native protein complexes.
CN-PAGE Electrophoresis
- Gel Casting: Prepare a native polyacrylamide gradient gel (e.g., 3–12% or 4–16% acrylamide). A bis-tris buffer system at pH 7.0 is standard.
- Sample Loading: Mix the solubilized sample with a loading buffer that lacks Coomassie dye (in contrast to BN-PAGE) but contains glycerol for density.
- Electrophoresis Run: Use an anode buffer (e.g., bis-tris, pH 7.0) and a cathode buffer containing sodium deoxycholate (DOC) or other anionic detergents to provide the necessary charge shift [30] [67]. Run the gel at low temperatures (e.g., 4°C) to maintain complex stability.
Downstream Applications
- In-Gel Activity Staining: Immediately after electrophoresis, incubate the gel in specific substrate buffers to visualize active enzymes.
- Two-Dimensional Electrophoresis (2D-CN/SDS-PAGE): Excise a lane from the CN-PAGE gel, incubate it in SDS-PAGE sample buffer, and place it on a second denaturing gel. This separates the individual subunits of each native complex [30] [68].
- Western Blotting: Transfer proteins from the CN-PAGE gel to a membrane for immunodetection with specific antibodies.
CN-PAGE Experimental Workflow
The Scientist's Toolkit: Essential Reagents for CN-PAGE
Table 3: Key Research Reagent Solutions for CN-PAGE.
| Reagent | Function in CN-PAGE |
|---|---|
| n-Dodecyl-β-D-maltoside (DDM) | Mild, non-ionic detergent for solubilizing individual membrane protein complexes while preserving their native state [30]. |
| Digitonin | Very mild, non-ionic detergent used to solubilize membranes more gently, preserving labile supramolecular assemblies like respiratory supercomplexes [65] [30]. |
| Sodium Deoxycholate (DOC) | Anionic detergent used in the cathode buffer to impose a charge shift on proteins, enabling migration without Coomassie dye [30] [67]. |
| 6-Aminocaproic Acid | Zwitterionic salt added to extraction buffers; provides ionic strength but does not interfere with electrophoresis, helping to stabilize complexes [30]. |
| Bis-Tris Buffer | The preferred buffering agent (at pH 7.0) for native gel and running buffer systems due to its compatibility with native protein structures [30]. |
CN-PAGE is not a replacement for BN-PAGE or SDS-PAGE but a powerful complementary technique. Its defining value is in functional proteomics where the research question revolves around enzymatic activity, labile protein interactions, or fluorescence-based detection. The experimental evidence is clear: for researchers studying the functional architecture of mitochondrial complexes, screening membrane proteins for structural studies, or isolating supercomplexes for cryo-EM, CN-PAGE provides an efficient and often essential path to reliable and interpretable results.
Technique Validation and Direct Comparison for Informed Method Selection
In-Gel Enzyme Activity Assays for Functional Validation
For researchers and drug development professionals, validating protein function is a critical step that often follows initial separation and identification. In-gel enzyme activity assays provide a powerful tool for this functional validation, allowing direct visualization of enzymatic activity within the polyacrylamide gel matrix after electrophoresis. The fundamental choice between Blue Native-PAGE (BN-PAGE) and SDS-PAGE separation techniques profoundly impacts the success of these functional assays, as it determines whether proteins retain their native conformation and catalytic capabilities.
SDS-PAGE, introduced by Ulrich K. Laemmli, employs sodium dodecyl sulfate to denature proteins, rendering them suitable for molecular weight determination but functionally inactive [1]. In contrast, BN-PAGE, developed by Hermann Schägger in the 1990s, preserves protein complexes in their native state using non-denaturing conditions and the charge-conferring Coomassie blue G-250 dye [35] [21]. This preservation enables researchers to directly link separation with functional assessment, making BN-PAGE indispensable for studying intact multi-protein complexes like the mitochondrial oxidative phosphorylation (OXPHOS) system [35]. This guide objectively compares these techniques, providing experimental data and protocols to inform method selection for protein complex analysis.
Fundamental Principles: Separation Mechanisms and Their Functional Implications
SDS-PAGE: A Denaturing Approach for Molecular Weight Determination
SDS-PAGE operates on the principle of complete protein denaturation. The technique uses sodium dodecyl sulfate (SDS), an anionic detergent that binds to hydrophobic regions of proteins in a constant ratio, approximately 1.4 grams of SDS per gram of protein [1]. This binding unfolds the tertiary structure, masks the protein's intrinsic charge, and imparts a uniform negative charge density. Consequently, separation occurs primarily based on molecular weight as proteins migrate through the polyacrylamide gel matrix under an electric field. The addition of reducing agents like DTT or β-mercaptoethanol further breaks disulfide bonds, ensuring complete denaturation [1]. While this method provides excellent resolution for molecular weight determination, the denatured proteins lose their biological activity, making SDS-PAGE unsuitable for direct in-gel enzyme activity assays.
BN-PAGE: A Native Approach for Functional Complex Analysis
BN-PAGE preserves protein structure and function through several key mechanisms. The technique utilizes mild non-ionic detergents like n-dodecyl-β-D-maltoside for membrane protein solubilization, which disrupts lipid bilayers without dissociating protein complexes [35] [21]. Coomassie blue G-250 dye plays a dual role: it binds to hydrophobic protein surfaces, imparting a negative charge shift that facilitates electrophoretic migration toward the anode, and it helps prevent protein aggregation during separation [35] [21]. The procedure is conducted in bis-tris buffer systems at pH 7.0, maintaining a physiological pH that preserves complex integrity [21]. This gentle treatment allows the OXPHOS complexes to remain catalytically active after separation, enabling direct in-gel activity detection [35].
Clear Native PAGE (CN-PAGE): A Variant for Enhanced Activity Staining
A variant of BN-PAGE, Clear Native PAGE (CN-PAGE), replaces Coomassie blue with mixtures of anionic and neutral detergents in the cathode buffer to induce the necessary charge shift [35]. A key advantage of CN-PAGE is the absence of residual blue dye interference during downstream in-gel enzyme activity staining, which can be crucial for sensitive detection methods [35]. However, the resolution of CN-PAGE is generally lower compared to standard BN-PAGE [35].
Table 1: Core Principle Comparison Between SDS-PAGE and BN-PAGE
| Parameter | SDS-PAGE | BN-PAGE |
|---|---|---|
| Separation Principle | Molecular weight | Size, charge, and shape of native complexes |
| Protein State | Denatured and linearized | Native, folded conformation |
| Detergent Used | Ionic (SDS) | Mild non-ionic (e.g., n-dodecyl-β-D-maltoside) |
| Key Additive | Reducing agents (DTT, BME) | Coomassie Blue G-250 |
| Functional Preservation | No enzymatic activity retained | Enzymatic activity preserved |
| Typical Buffer pH | Basic (pH ~8.3-8.8) | Physiological (pH 7.0) |
Technical Comparison: Resolution, Applications, and Limitations
Separation Characteristics and Data Output
The separation characteristics of SDS-PAGE and BN-PAGE differ significantly in their underlying mechanisms and data output. SDS-PAGE provides high-resolution separation of polypeptide chains based primarily on molecular weight, with smaller proteins migrating faster through the gel matrix [1]. Protein sizing is achieved by comparing migration distances against known molecular weight standards, with denatured proteins typically separated in the range of 5-250 kDa [1]. In contrast, BN-PAGE separates intact protein complexes ranging from approximately 100 kDa to 10 MDa, with migration influenced by both complex size and the number of Coomassie dye molecules bound to hydrophobic surfaces [35] [63]. This technique can resolve not only individual complexes but also higher-order supercomplexes, such as the respiratory chain respirasomes comprising Complexes I, III, and IV [35].
Applications in Protein Research and Drug Development
The applications of these techniques diverge according to their fundamental principles:
SDS-PAGE Applications: Determining protein purity and homogeneity; verifying molecular weights; assessing expression levels; checking proteolytic degradation patterns; preparing for western blotting in denatured conditions [1].
BN-PAGE Applications: Studying native protein-protein interactions; analyzing assembly pathways of multi-subunit complexes; identifying structural composition of membrane protein complexes; monitoring complex dynamics under different physiological or pathological conditions; direct in-gel enzyme activity assays [35] [63].
For drug development professionals, BN-PAGE offers particular value in investigating how therapeutic compounds affect the integrity and function of protein complexes, especially in metabolic pathways and membrane receptor complexes.
Limitations and Technical Considerations
Both techniques present specific limitations that researchers must consider:
SDS-PAGE Limitations:
- Complete loss of biological activity due to denaturation [1]
- Inability to assess native complex formation or subunit interactions
- Potential for artifacts from incomplete denaturation or reduction
- Limited information about native protein structure or function
BN-PAGE Limitations:
- Comparative insensitivity of in-gel Complex IV activity staining [35]
- Lack of established in-gel Complex III activity staining methods [35]
- Potential for protein aggregation without optimal detergent conditions
- More complex protocol requiring optimization for different sample types
- Interference of Coomassie dye with certain downstream applications (mitigated by CN-PAGE) [35]
Table 2: Performance Comparison for Functional Analysis
| Performance Metric | SDS-PAGE | BN-PAGE |
|---|---|---|
| Molecular Weight Determination | Excellent (denatured) | Possible (native complexes) |
| Complex Stoichiometry Analysis | Not possible | Excellent |
| In-Gel Enzyme Activity Detection | Not possible | Excellent (for many enzymes) |
| Protein-Protein Interaction Studies | Not possible | Excellent |
| Membrane Protein Analysis | Poor (often requires denaturation) | Excellent |
| Sample Throughput | High (standardized protocols) | Medium (requires optimization) |
| Quantitative Potential | Good (with proper standards) | Good (semi-quantitative) |
| Compatibility with Mass Spectrometry | Excellent | Good (after complex extraction) |
Experimental Protocols for In-Gel Enzyme Activity Detection
BN-PAGE Protocol for OXPHOS Complex Analysis
The following protocol, validated by research published in 2025, outlines the steps for analyzing functionally active OXPHOS complexes [35]:
Sample Preparation:
- Isolate mitochondria from cells or tissues by differential centrifugation.
- Resuspend mitochondrial pellet (0.4 mg) in 40 μL of buffer A (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris/HCl, pH 7.0) containing protease inhibitors (1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin) [21].
- Add 7.5 μL of 10% n-dodecyl-β-D-maltoside and mix thoroughly.
- Incubate on ice for 30 minutes for membrane solubilization.
- Centrifuge at 72,000 × g for 30 minutes at 4°C to remove insoluble material.
- Collect supernatant and add 2.5 μL of 5% Coomassie blue G in 0.5 M aminocaproic acid [21].
Gel Electrophoresis:
- Prepare a linear gradient native polyacrylamide gel (e.g., 4-16% or 3-12%) using a gradient maker [35]. Commercial pre-cast gels (e.g., Thermo Fisher Scientific NativePAGE Bis-Tris system) can also be used.
- Load samples (5-20 μL) into wells using a stacking gel system [21].
- Run electrophoresis at 150 V for approximately 2 hours at 4°C using anode (50 mM Bis-Tris, pH 7.0) and cathode (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie blue G, pH 7.0) buffers until the blue dye front approaches the gel bottom [21].
In-Gel Activity Assays:
- Complex I (NADH Dehydrogenase): Incubate gel in reaction buffer containing NADH and nitrotetrazolium blue, which forms a purple formazan precipitate upon reduction [35].
- Complex II (Succinate Dehydrogenase): Use buffer containing succinate, phenazine methosulfate, and nitrotetrazolium blue [35].
- Complex IV (Cytochrome c Oxidase): Incubate in buffer containing cytochrome c and diaminobenzidine, which produces a brown precipitate upon oxidation [35].
- Complex V (ATP Synthase): Use an enhancement step for marked sensitivity improvement; assay based on lead phosphate precipitation from ATP hydrolysis [35].
For CN-PAGE, follow similar steps but replace Coomassie blue in the cathode buffer with mixtures of anionic and neutral detergents to avoid dye interference with activity staining [35].
Protocol for Antioxidant Enzyme Activity Staining in Native Gels
Research published in 2022 demonstrates this application for ecotoxicological studies [69]:
Sample Preparation:
- Homogenize tissues in ice-cold buffer (0.5 M sucrose, 0.15 M KCl, 0.02 M Tris-HCl pH 8.0, 1 mM PMSF, 5 mM DTT).
- Centrifuge at 20,000 × g for 20 minutes at 4°C.
- Collect supernatant (SN20) for enzyme assays.
- Determine protein concentration using Bradford assay.
Native Electrophoresis:
- Use pre-cast polyacrylamide gels (7.5% for CAT, 12% for SOD and GPX) under non-denaturing conditions [69].
- Dilute protein samples (25-100 μg) 1:1 with native loading buffer (0.05 M Tris-HCl pH 6.8, 50% glycerol, 0.05% bromophenol blue).
- Run electrophoresis at 40 mA for 90 minutes at 4°C using Tris-glycine running buffer (0.05 M Tris-HCl pH 8.3, 0.3 M glycine, 0.002 M EDTA) without pre-electrophoresis [69].
Activity Staining:
- Catalase (CAT): Wash gel and incubate with reaction mixture containing hydrogen peroxide, then add ferric chloride and potassium ferricyanide (III) to detect remaining H₂O₂ [69].
- Superoxide Dismutase (SOD): Incubate gel in nitroblue tetrazolium in the dark, then expose to riboflavin under light to generate superoxide anions, appearing as light bands on a purple background [69].
- Glutathione Peroxidase (GPX): Incubate with glutathione, glutathione reductase, NADPH, and cumene hydroperoxide, monitoring NADPH oxidation [69].
BN-PAGE Workflow for Functional Validation
Research Reagent Solutions for Native Gel Electrophoresis
Successful in-gel enzyme activity assays require specific reagents optimized for preserving protein function:
Table 3: Essential Reagents for Native Gel Electrophoresis
| Reagent | Function | Example Formulation |
|---|---|---|
| Mild Detergents | Solubilize membrane proteins while preserving complex integrity | n-dodecyl-β-D-maltoside (1-2%), digitonin (for supercomplex preservation) [35] [21] |
| Charge-Shift Dyes | Impart negative charge for electrophoretic migration | Coomassie Blue G-250 (0.02-0.05% in cathode buffer) [35] [21] |
| Protease Inhibitors | Prevent protein degradation during extraction | PMSF (1 mM), leupeptin (1 μg/mL), pepstatin (1 μg/mL) [21] |
| Native Buffers | Maintain physiological pH conditions | Bis-Tris (50 mM, pH 7.0), 6-aminocaproic acid (0.75 M) [21] |
| Electrophoresis Buffers | Provide ionic environment for native separation | Anode: 50 mM Bis-Tris (pH 7.0); Cathode: 50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue (pH 7.0) [21] |
| Activity Assay Reagents | Detect specific enzymatic activities in gel | NADH/NTB (Complex I), succinate/PMS/NTB (Complex II), cytochrome c/DAB (Complex IV) [35] |
Two-Dimensional BN/SDS-PAGE for Comprehensive Complex Analysis
The combination of BN-PAGE with SDS-PAGE creates a powerful two-dimensional system for comprehensive protein complex analysis [63]. In this approach, protein complexes are first separated under native conditions using BN-PAGE. Subsequently, the entire gel lane is excised, incubated in SDS buffer to denature the complexes, and placed horizontally on a second SDS-PAGE gel for separation of individual subunits [21] [63].
This technique provides distinct insights: monomeric proteins typically migrate diagonally in the 2D separation, while components of the same protein complex align vertically in the second dimension [63]. Multiple spots in a horizontal line indicate a protein present in various distinct complexes [63]. The method has been successfully adapted for analyzing multi-protein complexes from whole cellular lysates, expanding its applicability beyond purified organelles [25] [26]. When combined with mass spectrometry, 2D BN/SDS-PAGE offers a holistic view of protein complex composition, stoichiometry, and relative abundance, making it invaluable for studying dynamic regulation of protein interactions in response to cellular stimuli [63].
2D BN/SDS-PAGE Workflow
The choice between SDS-PAGE and BN-PAGE for in-gel enzyme activity assays depends entirely on research objectives. SDS-PAGE remains the gold standard for determining molecular weights, assessing protein purity, and analyzing denatured proteins, but cannot preserve enzymatic function. BN-PAGE and its variant CN-PAGE provide unparalleled capability for studying native protein complexes, their interactions, and their enzymatic activities directly within the gel matrix.
For researchers focusing on functional validation, BN-PAGE offers the unique advantage of correlating protein separation with catalytic activity, enabling studies of complex assembly, dynamics, and responses to physiological or pharmacological perturbations. The ongoing refinement of BN-PAGE protocols, including enhanced sensitivity for in-gel activity staining and adaptation for whole-cell lysates, continues to expand its applications in basic research and drug development. As proteomics moves toward more integrative approaches, BN-PAGE stands as an essential tool in the researcher's arsenal for bridging the gap between protein identification and functional characterization.
Western Blot and Mass Spectrometry for Complex Identification
In the study of protein complexes, particularly within the framework of BN-PAGE (Blue Native Polyacrylamide Gel Electrophoresis) and SDS-PAGE research, Western blotting and mass spectrometry (MS) represent two fundamental, yet distinct, analytical approaches. Protein complexes organize and maintain cellular and organelle functions across all levels of complexity in time and space, governing processes from cell development and division to respiration and photosynthesis [36]. While Western blotting provides highly specific, antibody-based detection that confirms the presence and relative abundance of specific protein subunits, mass spectrometry offers an unbiased, high-sensitivity tool for discovering novel complex components and characterizing post-translational modifications. This guide objectively compares their performance in identifying protein complexes, supported by experimental data and detailed methodologies, to inform researchers and drug development professionals in selecting the optimal technique for their specific analytical needs.
Technical Comparison: Core Principles and Capabilities
Fundamental Mechanisms and Applications
Western Blotting operates through size-based separation of proteins via gel electrophoresis (typically SDS-PAGE or BN-PAGE), transfer to a membrane, and subsequent immunodetection using target-specific antibodies. This method is particularly valuable for targeted protein validation, enabling researchers to confirm the presence of specific known proteins within a complex and obtain information about their molecular weight and relative abundance [70] [71]. When combined with BN-PAGE, Western blotting can analyze mitochondrial protein complexes in their native state, determining size, abundance, and subunit composition while preserving protein-protein interactions [21].
Mass Spectrometry identifies proteins by measuring the mass-to-charge ratios of peptide ions derived from proteolytically digested samples. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) has emerged as a powerful tool to quickly and efficiently identify proteins in biological samples, making it particularly suited for discovery-oriented proteomics [72]. In protein complex analysis, MS is typically coupled with purification techniques such as affinity enrichment (AE-MS) or tandem affinity purification (TAP-tagging), enabling comprehensive characterization of complex composition without requiring purification to homogeneity [73].
Direct Performance Comparison
Table 1: Comparative Performance of Western Blotting and Mass Spectrometry for Protein Complex Analysis
| Parameter | Western Blotting | Mass Spectrometry |
|---|---|---|
| Proteome Coverage | Low (1-10 proteins per blot) [70] | High (1000+ proteins per experiment) [72] [73] |
| Throughput | Low (hours to days for a few targets) [70] | Medium to High (hours for sample preparation, days for analysis) [72] |
| Sensitivity | Moderate (nanogram range) | High (femtomole to attomole range) [72] |
| Specificity | Dependent on antibody quality [74] | Based on mass accuracy and sequence information [72] |
| Quantification Capability | Semi-quantitative with normalization [71] | Highly quantitative with stable isotopes or label-free methods [72] [73] |
| Ability to Detect Novel Components | No (requires prior knowledge) | Yes (unbiased identification) [72] |
| Sample Requirements | Can use electroblotted proteins from BN-PAGE [21] | Often requires cleaner samples, compatible with BN-PAGE gel spots [63] |
| Multiplexing Capacity | Low to moderate (limited by antibody availability and detection channels) [70] | High (can identify thousands of proteins simultaneously) [72] |
| Instrumentation Cost | Low to moderate [70] | High [70] |
| Technical Expertise Required | Moderate [70] | High [72] |
Table 2: Advantages and Limitations for Protein Complex Analysis
| Aspect | Western Blotting | Mass Spectrometry |
|---|---|---|
| Key Advantages | - Easily customizable protocols [70]- Direct compatibility with BN-PAGE [21]- Low equipment costs [70]- Can observe changes in protein size [70] | - Unbiased protein identification [72]- High sensitivity and dynamic range [72]- Comprehensive characterization of complexes [73]- Can detect post-translational modifications [75] |
| Major Limitations | - Limited proteome coverage [70]- Antibody-dependent with potential cross-reactivity [74]- Difficult quantification [71]- Low throughput [70] | - High instrumentation cost [70]- Complex data analysis [72]- Can miss low-abundance proteins in complex mixtures [73]- Requires proteolytic digestion [72] |
Experimental Protocols for Protein Complex Analysis
BN-PAGE with Western Blot Detection
Blue Native PAGE preserves protein complexes in their native state during separation, making it ideal for studying multisubunit enzymes and protein interactions [21]. The protocol below describes the integration of BN-PAGE with Western blotting for complex analysis:
Sample Preparation:
- Isolate mitochondria from cells or tissues (0.4 mg recommended) [21]
- Resuspend in 40 μL buffer containing 0.75 M aminocaproic acid and 50 mM Bis-Tris (pH 7.0)
- Add 7.5 μL of 10% n-dodecyl-β-D-maltopyranoside detergent
- Mix and incubate for 30 minutes on ice
- Centrifuge at 72,000 × g for 30 minutes and collect supernatant
- Add 2.5 μL of 5% Coomassie blue G solution and protease inhibitors [21]
BN-PAGE Electrophoresis:
- Prepare a linear gradient native acrylamide gel (e.g., 6-13%)
- Load 5-20 μL of prepared samples into wells
- Run electrophoresis at 150 V for approximately 2 hours using specific anode and cathode buffers [21]
- Transfer proteins to PVDF membrane using fully submerged system at 150 mA for 1.5 hours [21]
- Proceed with standard immunodetection using target-specific antibodies [21]
Mass Spectrometry-Based Workflow
For mass spectrometry analysis of protein complexes, two primary approaches have been developed: Affinity Enrichment Mass Spectrometry (AE-MS) and Tandem Affinity Purification (TAP-tagging) coupled with MS.
Affinity Enrichment Mass Spectrometry (AE-MS) Protocol:
- Perform single-step affinity enrichment of tagged proteins (e.g., GFP-tagged) and their interactors [73]
- Process samples using single-run, intensity-based label-free quantitative LC-MS/MS analysis [73]
- Normalize data using the approximately 2000 background binders typically present in each pull-down [73]
- Identify interacting proteins by comparison across multiple tagged strains rather than a single control [73]
- Validate potential interactors through intensity profiles across all samples [73]
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Analysis:
- Digest purified protein complexes proteolytically (typically with trypsin) [72]
- Separate peptides using reversed-phase capillary liquid chromatography (RPLC) [72]
- Elute peptides with increasing organic solvent (acetonitrile) directly into the mass spectrometer via electrospray ionization [72]
- Measure mass/charge ratios of peptides (typically 200-2000 m/z range) [72]
- Select peptides for fragmentation to generate MS/MS spectra [72]
- Identify proteins by comparing acquired MS/MS spectra to theoretical spectra in protein databases [72]
Figure 1: Integrated Workflow for Protein Complex Analysis Using BN-PAGE with Western Blot or Mass Spectrometry
Essential Research Reagents and Materials
Successful protein complex analysis requires specific reagents optimized for each methodology. The table below details essential materials for both Western blotting and mass spectrometry workflows in the context of BN-PAGE research.
Table 3: Essential Research Reagents for Protein Complex Analysis
| Reagent/Material | Function/Purpose | Application |
|---|---|---|
| n-dodecyl-β-D-maltopyranoside | Mild detergent for solubilizing membrane protein complexes while preserving interactions [21] | BN-PAGE sample preparation |
| Coomassie Blue G | Imparts negative charge to proteins for native electrophoresis without denaturation [21] | BN-PAGE first dimension |
| 6-aminocaproic acid | Provides ionic strength and minimizes protein aggregation during electrophoresis [21] | BN-PAGE buffer system |
| PVDF membrane | High protein binding capacity and mechanical strength for immunodetection [21] | Western blotting |
| Primary antibodies | Target-specific detection of protein complex subunits | Western blotting |
| Trypsin | Proteolytic enzyme for digesting proteins into identifiable peptides [72] | Mass spectrometry |
| C18 reversed-phase columns | Separate peptides based on hydrophobicity before MS analysis [72] | LC-MS/MS |
| Tandem affinity tags (TAP) | Enable two-step purification to reduce non-specific binders [72] | MS complex purification |
| Nitrocellulose membranes | Protein immobilization for subsequent analysis; can be dissolved for BARN-MS method [75] | Western blotting / BARN-MS |
Comparative Analysis of Supporting Data
Validation and Reproducibility Considerations
A critical consideration in protein complex analysis is the validation of results. While it is common practice to validate mass spectrometry findings with Western blotting, this approach has significant limitations. Research has shown that Western blotting is not always a robust validation technique due to differences in antibody affinities and specificities [74]. In some cases, proteins identified by mass spectrometry show opposite trends when analyzed by Western blot, creating uncertainty about which result to trust [74].
Mass spectrometry offers advantages in quantification through stable isotopic labeling or label-free quantitative methods, providing more reliable quantitative data across multiple samples [72] [73]. However, Western blotting remains widely accepted for confirming the presence of specific proteins, particularly when antibodies are well-validated.
Compatibility with BN-PAGE for Complexome Profiling
Both techniques can be effectively integrated with BN-PAGE for comprehensive complexome analysis:
Western Blotting with BN-PAGE:
- Enables immunodetection of specific subunits after native separation [21]
- Can determine subunit composition and stoichiometry within complexes [36]
- Compatible with two-dimensional BN/SDS-PAGE for higher resolution [63]
Mass Spectrometry with BN-PAGE:
- Protein spots from BN-PAGE gels can be excised and identified by MS [63]
- BARN (Blotting And Removal of Nitrocellulose) method allows MS analysis of electroblotted proteins [75]
- Provides comprehensive identification of complex components without prior knowledge [63]
Western blotting and mass spectrometry offer complementary strengths for protein complex identification within BN-PAGE research. Western blotting remains the method of choice for targeted validation of specific complex subunits, particularly when antibodies are available and well-characterized. Its compatibility with BN-PAGE, relatively low cost, and ease of customization make it ideal for hypothesis-driven research focused on known protein components.
Conversely, mass spectrometry excels in discovery-oriented applications, enabling unbiased identification of novel complex components and comprehensive characterization of complex composition. While requiring more sophisticated instrumentation and expertise, MS provides superior proteome coverage, sensitivity, and quantitative accuracy, particularly when coupled with advanced purification strategies like AE-MS or TAP-tagging.
The optimal choice depends on research objectives: Western blotting for confirming suspected complex memberships, and mass spectrometry for exploring unknown complex compositions or characterizing complex dynamics under different physiological conditions. For the most comprehensive analysis, many researchers employ both techniques in a complementary workflow, using MS for initial discovery and Western blotting for subsequent validation and targeted studies.
In the field of protein analysis, polyacrylamide gel electrophoresis (PAGE) serves as a fundamental methodology for separating and characterizing proteins and protein complexes. Two principal techniques, Blue Native PAGE (BN-PAGE) and Sodium Dodecyl Sulfate PAGE (SDS-PAGE), offer complementary approaches with distinct advantages and limitations. SDS-PAGE, a denaturing technique, separates individual polypeptide chains based primarily on molecular mass [4]. In contrast, BN-PAGE is a non-denaturing technique that resolves intact protein complexes according to their native molecular weight, size, and charge [1] [35]. This comparative guide objectively analyzes the performance of these two techniques, focusing on their separation criteria, protein recovery potential, and applications within protein complex research, providing researchers with a framework for selecting the appropriate method for their experimental objectives.
Comparative Analysis: BN-PAGE vs. SDS-PAGE
The following table provides a detailed comparison of the key parameters for BN-PAGE and SDS-PAGE, summarizing their fundamental differences in separation criteria, protein recovery, and applications.
Table 1: Comprehensive comparison of BN-PAGE and SDS-PAGE
| Criteria | BN-PAGE | SDS-PAGE |
|---|---|---|
| Separation Principle | Size, net charge, and 3D shape of native complexes [1] [4] | Molecular weight of denatured polypeptide subunits [1] [4] |
| Gel Conditions | Non-denaturing, non-reducing [1] | Denaturing and reducing (SDS present) [1] |
| Sample Preparation | Not heated; solubilized with mild detergents [1] [35] | Heated with SDS and reducing agents [1] |
| Protein State | Native, folded conformation; functionality retained [1] [35] | Denatured, linearized; functionality lost [1] |
| Primary Applications | Studying structure, subunit composition, function, and supercomplexes [1] [35] | Determining molecular weight, checking protein expression, and purity analysis [1] |
| Protein Recovery & Post-Separation Analysis | Proteins can be recovered in functional form for activity assays [1] | Recovered proteins are denatured; used for mass spectrometry, sequencing [76] [48] |
| Typical Detergent | Digitonin, Dodecyl-β-D-maltoside [35] [31] | Sodium Dodecyl Sulfate (SDS) [1] |
| Key Reagent | Coomassie Blue G-250 (imparts charge) [35] [31] | SDS (denatures and imparts charge) [1] |
| Running Temperature | Often run at 4°C to maintain stability [1] | Typically run at room temperature [1] |
Experimental Protocols for Key Applications
Protocol for BN-PAGE to Resolve Mitochondrial OXPHOS Complexes
This protocol, adapted from validated methodologies, is used for analyzing the assembly and native molecular weight of oxidative phosphorylation (OXPHOS) complexes [77] [35].
- 1. Sample Preparation: Isolate mitochondria via Percoll gradient centrifugation. Resuspend the mitochondrial pellet (fresh or frozen) in a solubilization buffer containing 50 mM Bis-Tris (pH 7.0), 1% n-dodecyl-β-D-maltoside, and 750 mM ε-amino-N-caproic acid. Keep the suspension on ice for 1 hour with occasional vortexing. Clarify the lysate by centrifugation at 20,000 × g for 30 minutes [77].
- 2. Sample Loading: Mix the supernatant with a concentrated BN-PAGE loading buffer containing 0.75 M ε-amino-N-caproic acid and 3% Serva Blue G-250 (w/v) [77] [35].
- 3. Gel Electrophoresis: Cast a non-gradient or linear gradient (e.g., 3.5-12.5%) polyacrylamide gel. Load the sample (20-30 μg of protein) and run the gel at 150 V using a blue cathode buffer (50 mM Tricine, 15 mM Bis-Tris, pH 7.0, 0.02% Serva Blue G-250). Once the dye front has migrated one-third into the gel, replace the cathode buffer with a clear cathode buffer (without the blue dye) and continue electrophoresis at 200 V until completion [77] [35].
- 4. Downstream Analysis:
- In-Gel Activity Staining: Incubate the gel strip in specific substrate solutions to detect enzymatic activity of complexes. For example, Complex I activity can be visualized by incubation with NADH and nitroblue tetrazolium (NBT), which produces a formazan precipitate [77] [35].
- Second Dimension SDS-PAGE: For subunit analysis, equilibrate a BN-PAGE gel strip in a solution containing SDS and reducing agent (e.g., 2-mercaptoethanol), then place it onto an SDS-PAGE gel for orthogonal separation [77] [35].
Protocol for Protein Recovery from SDS-PAGE Gels
Recovering proteins from SDS-PAGE gels is often necessary for identification and further analysis. The following passive diffusion method is widely used.
- 1. Localization and Excision: Following electrophoresis, stain the gel with Coomassie Brilliant Blue and destain until protein bands are visible. Excise the band of interest with a clean, sharp blade, minimizing excess gel [76] [78].
- 2. Passive Diffusion Elution: Place the gel piece in a microcentrifuge tube and crush it with a small pestle. Add an elution buffer (e.g., 50 mM Tris-HCl, pH 8.0, containing 0.1% SDS). Incubate the crushed gel fragments on a rotator for 4-16 hours, depending on the protein size, to allow the protein to diffuse out [76].
- 3. SDS Removal and Concentration: Separate the eluted protein solution from the gel pieces by centrifugation. Remove SDS and concentrate the protein by acetone precipitation. Resuspend the purified protein in an appropriate buffer for downstream applications like mass spectrometry [76] [78].
Workflow Visualization
The following diagram illustrates the key decision points and procedural steps involved in choosing and executing BN-PAGE or SDS-PAGE for protein analysis.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of BN-PAGE and SDS-PAGE relies on specific reagents, each with a critical function. The table below details these key components.
Table 2: Essential reagents for BN-PAGE and SDS-PAGE
| Reagent | Function | Technique |
|---|---|---|
| Coomassie Blue G-250 | Binds to hydrophobic protein surfaces, imparting a negative charge shift for migration without denaturation [35] [31]. | BN-PAGE |
| n-Dodecyl-β-D-Maltoside (DDM) | A mild, non-ionic detergent for solubilizing membrane proteins while preserving protein-protein interactions [35]. | BN-PAGE |
| Digitonin | A very mild detergent used to solubilize labile supercomplexes and megacomplexes for high-level structural analysis [37] [35]. | BN-PAGE |
| 6-Aminocaproic Acid | A zwitterionic salt that improves solubilization of membrane proteins and enhances resolution during electrophoresis [77] [35]. | BN-PAGE |
| Sodium Dodecyl Sulfate (SDS) | A strong ionic detergent that denatures proteins and binds uniformly to polypeptides, masking their native charge [1] [4]. | SDS-PAGE |
| Dithiothreitol (DTT) / β-Mercaptoethanol | Reducing agents that break disulfide bonds within and between protein subunits, ensuring complete denaturation [1]. | SDS-PAGE |
| Acrylamide/Bis-Acrylamide | Monomer and cross-linker that polymerize to form the porous gel matrix, which sieves proteins during electrophoresis [4]. | BN-PAGE & SDS-PAGE |
| Coomassie Brilliant Blue R-250 | A protein stain used for post-electrophoretic visualization of protein bands in fixed gels [76] [48]. | Downstream Analysis |
In the field of protein analysis, researchers often face a fundamental trade-off: preserving native protein functionality versus achieving accurate molecular weight separation. Two core electrophoretic techniques, Blue Native PAGE (BN-PAGE) and Sodium Dodecyl Sulfate-PAGE (SDS-PAGE), represent opposing ends of this spectrum. This guide provides an objective comparison of these techniques, focusing on their performance in protein complex analysis for research and drug development applications. Understanding these methodologies is crucial for studying multi-protein complexes, which are fundamental to most biological processes and represent important therapeutic targets [25].
Fundamental Principles and Comparative Analysis
Core Mechanistic Differences
The primary distinction between these techniques lies in their treatment of protein structure.
BN-PAGE utilizes the mild, nonionic detergent n-dodecyl-β-d-maltoside and the anionic dye Coomassie Blue G-250 to solubilize and separate protein complexes under native conditions. The dye binds to hydrophobic protein surfaces, imparting a negative charge shift that drives electrophoretic migration while maintaining complex integrity, enzymatic activity, and bound cofactors (e.g., metal ions) [35] [21]. This allows the study of protein-protein interactions, oligomeric states, and supercomplexes like the mitochondrial respirasome [35].
SDS-PAGE, the standard denaturing method, uses the ionic detergent SDS and often a heating step to unfold proteins, destroy functional properties, and impart a uniform negative charge. Separation is based almost exclusively on polypeptide chain mass, destroying quaternary, tertiary, and secondary structures, along with any non-covalently bound cofactors [8].
Quantitative Performance Comparison
The table below summarizes the key performance characteristics of each method, illustrating the core trade-off between functionality and resolution.
Table 1: Direct Comparison of BN-PAGE and SDS-PAGE Characteristics
| Characteristic | BN-PAGE | SDS-PAGE |
|---|---|---|
| Protein State | Native, intact complexes & supercomplexes [35] [68] | Denatured, individual polypeptide chains [8] |
| Functionality Preservation | Retains enzymatic activity and bound metal ions [8] [35] | Destroys enzymatic activity and releases metal ions [8] |
| Separation Basis | Size and shape of native complexes [68] | Molecular mass of individual subunits [8] |
| Molecular Weight Determination | Less accurate for mass; can inform on oligomeric state [65] | Highly accurate for subunit mass [8] |
| Key Advantage | Studies functional protein interactions and active enzymes [8] [35] | High-resolution separation and precise mass estimation [8] |
| Primary Limitation | Lower resolution of complex mixtures; Coomassie dye can interfere with some assays [8] [65] | Loss of all native structural and functional information [8] |
Supporting experimental data demonstrates the tangible impact of these differences. In a study comparing the techniques, retention of natively bound Zn²⁺ in proteomic samples was 98% with BN-PAGE versus only 26% with standard SDS-PAGE [8]. Furthermore, when nine model enzymes were analyzed, all nine remained active after BN-PAGE, whereas all were denatured during SDS-PAGE [8].
Experimental Protocols for Key Applications
Standard BN-PAGE Protocol for Protein Complexes
This protocol, adapted from established methodologies [35] [21], is designed for analyzing intact protein complexes from isolated mitochondria.
Sample Preparation:
- Isolate mitochondria from cells or tissues via differential centrifugation.
- Resuspend mitochondrial pellet (e.g., 0.4 mg) in 40 µL of ice-cold solubilization buffer (0.75 M 6-aminocaproic acid, 50 mM Bis-Tris, pH 7.0) containing protease inhibitors (e.g., 1 mM PMSF).
- Add 7.5 µL of 10% n-dodecyl-β-d-maltoside (or digitonin for studying supercomplexes [35]) to solubilize membrane proteins. Mix and incubate on ice for 30 minutes.
- Centrifuge at ≥16,000 x g for 30 minutes at 4°C to remove insoluble debris.
- Collect the supernatant and add 2.5 µL of 5% Coomassie Blue G-250 in 0.5 M aminocaproic acid [21].
Gel Electrophoresis:
- Cast a native linear gradient gel (e.g., 4-16% or 6-13% acrylamide) using a gradient maker. A low-percentage stacking gel is poured on top after the gradient sets [21].
- Prepare anode (50 mM Bis-Tris, pH 7.0) and cathode (50 mM Tricine, 15 mM Bis-Tris, 0.02% Coomassie Blue G-250, pH 7.0) running buffers [21].
- Load the prepared samples (5-20 µL) into the wells.
- Run electrophoresis at a constant voltage (e.g., 150 V) for approximately 2 hours, or until the dye front approaches the gel bottom [21].
Downstream Analysis:
- For in-gel activity assays, incub the gel in specific substrate buffers to detect active enzymes (e.g., ATP hydrolysis for Complex V) [35].
- For subunit analysis, a lane can be excised, soaked in SDS buffer, and run on a second-dimension SDS-PAGE [68] [21].
- Proteins can also be transferred to a PVDF membrane for western blotting [21].
Modified SDS-PAGE for Partial Functionality Preservation
A hybrid method, termed Native SDS-PAGE (NSDS-PAGE), has been developed to bridge the gap between the two techniques. It aims to offer higher resolution than BN-PAGE while retaining more functionality than standard SDS-PAGE [8].
Protocol Modifications from Standard SDS-PAGE [8]:
- Sample Buffer: Remove SDS and EDTA. Use Tris-HCl, Tris base, glycerol, and trace amounts of Coomassie G-250 and Phenol Red.
- Sample Prep: Omit the heating step. Mix sample with the modified buffer and load without denaturation.
- Running Buffer: Reduce SDS concentration from 0.1% to 0.0375% and delete EDTA.
This method resulted in a dramatic increase in Zn²⁺ retention (from 26% to 98%) and allowed seven out of nine tested enzymes to retain activity after electrophoresis, showcasing its utility for specific applications [8].
Workflow and Decision Pathway
The following diagram illustrates the logical decision process for selecting and applying the appropriate electrophoretic method based on research goals.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of these techniques requires specific reagents. The following table lists key solutions and their critical functions within the protocols.
Table 2: Essential Research Reagent Solutions for Native and Denaturing Electrophoresis
| Reagent Solution | Critical Function | BN-PAGE | SDS-PAGE |
|---|---|---|---|
| n-Dodecyl-β-D-maltoside | Mild, nonionic detergent for solubilizing membrane proteins without disrupting complex integrity [35] [21]. | Essential | Not Used |
| Coomassie Blue G-250 | Anionic dye that binds protein surfaces, providing charge for migration and preventing aggregation [35] [68]. | Essential (in cathode buffer & sample) | Not Used (or trace in NSDS-PAGE [8]) |
| 6-Aminocaproic Acid | Zwitterionic salt; stabilizes proteins and complexes during extraction, improves resolution [35]. | Essential | Not Used |
| SDS (Sodium Dodecyl Sulfate) | Ionic detergent that denatures proteins, masks intrinsic charge, and confers uniform charge-to-mass ratio [8]. | Not Used | Essential |
| Digitonin | Very mild, nonionic detergent used to preserve labile supramolecular assemblies (e.g., respiratory supercomplexes) [65] [35]. | Optional (for specific applications) | Not Used |
| Bis-Tris Buffers | Common buffering system for native gels; provides stable pH environment compatible with downstream assays [35] [21]. | Standard | Optional (e.g., NuPAGE system [8]) |
Advanced Considerations and Complementary Techniques
Clear-Native PAGE (CN-PAGE)
CN-PAGE is a variant that replaces Coomassie dye in the cathode buffer with mixtures of anionic and neutral detergents [35]. Its key advantage is the absence of dye interference with downstream applications like fluorescence resonance energy transfer (FRET) or sensitive in-gel activity assays [65] [35]. It is considered a milder technique than BN-PAGE and can sometimes retain labile supramolecular assemblies that dissociate under standard BN-PAGE conditions [65]. However, its resolution is generally lower than BN-PAGE, and molecular weight estimation is less straightforward because migration depends on both protein size and intrinsic charge [65].
Two-Dimensional Electrophoresis (BN/SDS-PAGE)
A powerful approach combines the strengths of both methods. In the first dimension, protein complexes are separated by BN-PAGE. Then, a single lane is excised, soaked in SDS buffer to denature the complexes, and placed on a standard SDS-PAGE gel for the second dimension separation. This resolves the individual protein subunits that make up each complex [68]. This 2D technique has been successfully used, for instance, to analyze the complex protein composition of snake venoms and to study the subunit makeup of mitochondrial complexes [35] [68].
The choice between BN-PAGE and SDS-PAGE is not a matter of one technique being superior to the other, but rather which is appropriate for the specific biological question. BN-PAGE is the method of choice for investigating the structure, function, and interactions of native protein complexes, offering unparalleled preservation of enzymatic activity and protein-metal partnerships. In contrast, SDS-PAGE provides a high-resolution, denaturing workhorse for analyzing protein purity, expression, and subunit molecular weight. For the most comprehensive analysis, researchers can leverage both techniques sequentially in a two-dimensional approach. Furthermore, modified methods like NSDS-PAGE and CN-PAGE offer valuable intermediate options, allowing scientists to fine-tune the balance between high resolution and the preservation of functional protein states in their research and drug development workflows.
Integrating with Computational Methods for Structural Insights
Understanding the native architecture of protein complexes is fundamental to structural biology and drug development. Two primary polyacrylamide gel electrophoresis (PAGE) techniques facilitate this understanding: Blue Native PAGE (BN-PAGE) and Sodium Dodecyl Sulfate PAGE (SDS-PAGE). These methods serve complementary roles in analyzing protein composition and organization. SDS-PAGE employs the ionic detergent SDS to denature proteins into their constituent polypeptides, separating them by molecular mass with high resolution [46] [8]. In contrast, BN-PAGE utilizes mild non-ionic detergents and the dye Coomassie Blue G-250 to separate intact protein complexes under native conditions, preserving functional properties and multi-subunit interactions [62] [46] [21]. This guide provides an objective comparison of their performance, supported by experimental data, to inform researchers selecting the optimal technique for integrating structural insights with computational methods.
Fundamental Principles and Separation Characteristics
The core distinction between these techniques lies in their treatment of protein structure. SDS-PAGE deliberately denatures proteins, masking intrinsic charge and enabling mass-based separation [8]. BN-PAGE maintains native conformation by using Coomassie dye, which binds to hydrophobic protein surfaces to impart a negative charge shift without disrupting complex integrity [62] [30]. A variant called clear-native PAGE (CN-PAGE) replaces Coomassie with mixed anionic detergent micelles, eliminating dye interference for downstream activity assays [62] [30]. High-resolution CN-PAGE further refines this approach for analyzing fluorescently labeled proteins or conducting in-gel catalytic assays [62].
Table 1: Fundamental Characteristics of BN-PAGE and SDS-PAGE
| Characteristic | BN-PAGE | SDS-PAGE |
|---|---|---|
| Protein State | Native, intact complexes | Denatured, individual subunits |
| Separation Basis | Hydrodynamic size & shape | Molecular mass |
| Mass Range | 10 kDa - 10 MDa [62] | Limited to smaller subunits |
| Charge Agent | Coomassie Blue G-250 or mixed detergents [62] [30] | Sodium dodecyl sulfate (SDS) |
| Functional Retention | Enzymatic activity, bound cofactors, metal ions [8] [30] | No functional retention |
| Membrane Protein Suitability | Excellent with appropriate markers [62] | Limited due to hydrophobicity issues |
Table 2: Quantitative Performance Comparison
| Performance Metric | BN-PAGE | SDS-PAGE | Experimental Support |
|---|---|---|---|
| Zn²⁺ Retention | ~98% [8] | ~26% [8] | Metal analysis of LLC-PK1 proteome |
| Enzyme Activity Retention | 9/9 model enzymes [8] | 0/9 model enzymes [8] | In-gel activity assays |
| Resolution | Moderate for complexes | High for individual subunits | Comparative gel analysis |
| Suitable for Supercomplex Analysis | Yes (with digitonin) [46] [30] | No | 2D separation experiments |
Experimental Protocols for Protein Complex Analysis
BN-PAGE Protocol for OXPHOS Complexes
Sample Preparation from Mitochondria:
- Resuspend 0.4 mg of sedimented mitochondria in 40 μL buffer containing 0.75 M aminocaproic acid and 50 mM Bis-Tris (pH 7.0) [21].
- Add 7.5 μL of 10% n-dodecyl-β-D-maltopyranoside (DDM) for individual complexes or digitonin for supercomplex preservation [46] [21] [30].
- Mix and incubate for 30 minutes on ice, then centrifuge at 72,000 × g for 30 minutes [21].
- Collect supernatant and add 2.5 μL of 5% Coomassie Blue G-250 in 0.5 M aminocaproic acid [21].
Gel Electrophoresis:
- Prepare linear acrylamide gradient gels (e.g., 6-13%) using a gradient mixer [62] [21].
- Load 5-20 μL samples into wells and run at 150V for approximately 2 hours or until dye front approaches gel bottom [21].
- Maintain temperature at 4°C throughout electrophoresis [42].
Downstream Applications:
- For subunit analysis: Excise BN-PAGE lane, incubate in SDS buffer, and perform second-dimension SDS-PAGE [36] [21] [42].
- For activity assays: Use CN-PAGE variant and perform in-gel staining for Complex I, II, IV, or V activity [30].
- For immunodetection: Transfer to PVDF membrane using Tris-Glycine buffer with 10% methanol [21].
SDS-PAGE and NSDS-PAGE Protocols
Standard Denaturing SDS-PAGE:
- Mix protein sample with LDS loading buffer containing SDS and reducing agent [8].
- Heat at 70°C for 10 minutes to denature proteins [8].
- Load onto Bis-Tris gel and run with MOPS-SDS running buffer at 200V for 45 minutes [8].
Native SDS-PAGE (NSDS-PAGE) Modification:
- Omit SDS and EDTA from sample buffer and do not heat samples [8].
- Reduce SDS concentration in running buffer to 0.0375% and remove EDTA [8].
- Add 0.01875% Coomassie G-250 to sample buffer to facilitate charge shift without denaturation [8].
Research Reagent Solutions
Table 3: Essential Reagents for Native Electrophoresis
| Reagent | Function | Example Applications |
|---|---|---|
| n-Dodecylmaltoside | Mild non-ionic detergent for solubilizing individual protein complexes | Mitochondrial complexes I-V analysis [46] [30] |
| Digitonin | Mild detergent for preserving supercomplexes | Respiratory chain supercomplexes [62] [46] |
| Coomassie Blue G-250 | Charge-conferring dye for protein migration in BN-PAGE | Standard BN-PAGE for all membrane proteins [62] [21] |
| 6-Aminocaproic Acid | Zwitterionic salt for solubilization support | Membrane protein extraction without denaturation [30] |
| Triton X-100 | Alternative non-ionic detergent for complex solubilization | Whole cellular lysate analysis [62] [46] |
| Bis-Tris Buffer System | pH stabilization for native conditions | Maintaining pH 7.0 during BN-PAGE [21] [30] |
Workflow Visualization
Figure 1: Comparative workflow for protein complex analysis.
Figure 2: Decision framework for technique selection.
BN-PAGE and SDS-PAGE offer complementary capabilities for protein complex analysis with distinct advantages for specific research objectives. BN-PAGE excels in preserving native protein interactions, maintaining enzymatic activity, and characterizing supercomplex organizations, making it ideal for functional and structural studies. SDS-PAGE provides superior resolution for subunit analysis and molecular weight determination but sacrifices native conformation. The emerging NSDS-PAGE technique offers a promising intermediate approach, balancing resolution with native state preservation. For researchers integrating electrophoretic data with computational structural methods, BN-PAGE provides critical information on native complex architecture. In contrast, SDS-PAGE remains invaluable for validating subunit composition and purity. Selection between these techniques should be guided by the specific structural insights required, with BN-PAGE illuminating native architecture and SDS-PAGE defining constituent components.
Conclusion
BN-PAGE and SDS-PAGE serve distinct, complementary roles in protein analysis. BN-PAGE is unparalleled for studying native protein complexes, their interactions, and enzymatic functions, making it ideal for investigating respiratory chains, supercomplexes, and metabolic pathways. SDS-PAGE remains the gold standard for determining molecular weights, checking protein purity, and expression levels under denaturing conditions. The choice between these techniques should be driven by the research question: study structure-function relationships in native complexes with BN-PAGE, or analyze denatured polypeptides with SDS-PAGE. Future directions include tighter integration with cutting-edge computational predictions from tools like AlphaFold for a multi-faceted understanding of protein interactions, accelerating drug discovery and the mechanistic study of diseases linked to complex dysfunction.
References
- 1. Differences between SDS PAGE and Native PAGE [https://www.geeksforgeeks.org/biology/differences-between-sds-page-and-native-page/]
- 2. Native or Denaturing Gel - Which Is For You? [https://advansta.com/not-all-gels-are-created-equal-native-or-denaturing-which-is-for-you/]
- 3. Should I Use A Native Or Denaturing Gel? [https://info.gbiosciences.com/blog/should-i-use-a-native-or-denaturing-gel]
- 4. Overview of Protein Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 5. How Is Native PAGE Different from SDS-PAGE? [https://synapse.patsnap.com/article/how-is-native-page-different-from-sds-page]
- 6. What is the difference between denaturing and non- ... [https://www.aatbio.com/resources/faq-frequently-asked-questions/What-is-the-difference-between-denaturing-and-non-denaturing-native-gels]
- 7. SDS PAGE vs Native PAGE [https://byjus.com/biology/difference-between-sds-page-and-native-page/]
- 8. Native SDS-PAGE: High Resolution Electrophoretic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4517606/]
- 9. Native Versus Denaturing Gels [https://bitesizebio.com/27332/native-versus-denaturing-gels/]
- 10. Comparison of Strong Cation Exchange and SDS/PAGE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3707127/]
- 11. Ionic vs Non-Ionic Surfactants: What Are the Differences? [https://surfactant.alfa-chemistry.com/ionic-vs-non-ionic-surfactants-what-are-the-differences.html]
- 12. Ionic vs. Non-Ionic Surfactants: What Are the Differences? [https://www.chemicalproductsokc.com/ionic-vs-non-ionic-surfactants-what-are-the-differences/]
- 13. Detergent Screening With Hybrid Detergents Increases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329390/]
- 14. Solubilization of membrane protein complexes for blue ... [https://www.sciencedirect.com/science/article/abs/pii/S1874391908000729]
- 15. Non-Ionic Detergent: Benefits, Applications & Comparison ... [https://texauxchemicals.com/non-ionic-detergent/]
- 16. Which Is Better Ionic or Non-ionic Surfactant [https://www.sancolo.com/news/which-is-better-ionic-or-non-ionic-surfactant.html]
- 17. Gel electrophoresis of proteins [https://grokipedia.com/page/Gel_electrophoresis_of_proteins]
- 18. DeFrND: detergent-free reconstitution into native ... [https://www.nature.com/articles/s41467-025-63275-8]
- 19. Blue-native PAGE in plants: a tool in analysis of protein ... [https://plantmethods.biomedcentral.com/articles/10.1186/1746-4811-1-11]
- 20. Principle of 2D Blue Native/SDS-PAGE Protein Complex ... [https://www.mtoz-biolabs.com/principle-of-2d-blue-native-sds-page-protein-complex-analysis.html]
- 21. Blue native electrophoresis protocol [https://www.abcam.com/en-us/technical-resources/protocols/blue-native-electrophoresis]
- 22. Blue Native electrophoresis to study mitochondrial and ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202302000385]
- 23. Two dimensional blue native/SDS-PAGE to identify ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2015.00098/full]
- 24. Protein Gel and Imaging Analysis [https://www.creative-proteomics.com/services/protein-gel-and-imaging-analysis.htm]
- 25. Two-dimensional Blue Native/SDS Gel Electrophoresis of ... [https://www.sciencedirect.com/science/article/pii/S1535947620334228]
- 26. Two-dimensional Blue native/SDS gel electrophoresis of ... [https://pubmed.ncbi.nlm.nih.gov/14665681/]
- 27. History of electrophoresis [https://en.wikipedia.org/wiki/History_of_electrophoresis]
- 28. The pre-omics era: The early days of two-dimensional gels [https://pmc.ncbi.nlm.nih.gov/articles/PMC2731566/]
- 29. Blue Native Polyacrylamide Gel Electrophoresis [https://www.nature.com/articles/pr2001236]
- 30. and clear-native polyacrylamide gel electrophoresis protocols ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0332065]
- 31. Principles of Blue Native-PAGE [https://blog.benchsci.com/principles-of-blue-native-page]
- 32. Blue Native PAGE and Biomolecular Complementation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2976385/]
- 33. Two-dimensional Blue Native/SDS Gel Electrophoresis of ... [https://www.sciencedirect.com/science/article/pii/S1535947620314109]
- 34. The use of blue native PAGE in the evaluation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2648668/]
- 35. Validation of blue- and clear-native polyacrylamide gel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445495/]
- 36. How to analyze protein complexes by 2D blue native SDS- ... [https://pubmed.ncbi.nlm.nih.gov/17893852/]
- 37. Separation of Thylakoid Protein Complexes with Two- ... [https://bio-protocol.org/en/bpdetail?id=2899&type=0]
- 38. A Blue Native-PAGE analysis of membrane protein complexes ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-11-22]
- 39. The principle and method of polyacrylamide gel ... [https://www.mblbio.com/bio/g/support/method/sds-page.html]
- 40. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOorde5SrwyMUdOT3ix0YB_Aa5hXF5mvKbVsXl8jVvCyFM607AucC]
- 41. Increasing the Throughput of CE-SDS Experiments for ... [https://chemrxiv.org/engage/chemrxiv/article-details/68127ee6927d1c2e6672faaa]
- 42. Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3339838/]
- 43. Dodecyl Maltopyranoside Enabled Purification of Active ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4349990/]
- 44. Triton X-100 - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/triton-x-100]
- 45. Solubilization, purification, and characterization of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10248529/]
- 46. Blue-native PAGE in plants: a tool in analysis of protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1308860/]
- 47. Cryo-slicing Blue Native-Mass Spectrometry (csBN-MS), a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4739680/]
- 48. In-depth structural proteomics integrating mass ... [https://www.frontiersin.org/journals/analytical-science/articles/10.3389/frans.2022.1107183/full]
- 49. Protein complexes in snake venom - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11115964/]
- 50. Integrative transcriptomic, proteomic, biochemical and ... [https://www.sciencedirect.com/science/article/abs/pii/S1874391925000430]
- 51. Proteomic Identification and Quantification of Snake Venom ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8473211/]
- 52. Proteomic analysis reveals rattlesnake venom modulation ... [https://www.sciencedirect.com/science/article/abs/pii/S1874391922000537]
- 53. Multiplex Fluorescent Western Blots [https://azurebiosystems.com/western-blotting-applications/multiplex-fluorescent-western-blots/]
- 54. Detergents for Cell Lysis and Protein Extraction [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/detergents-cell-lysis-protein-extraction.html]
- 55. Strategies for revealing lower abundance proteins in two- ... [https://pubmed.ncbi.nlm.nih.gov/15652797/]
- 56. Comparison of in-gel protein separation techniques ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4234072/]
- 57. Two dimensional Blue Native-/SDS-PAGE analysis of SLP ... [https://www.sciencedirect.com/science/article/abs/pii/S0165247805003500]
- 58. Western blot protocol for low abundance proteins [https://www.abcam.com/en-us/technical-resources/protocols/western-blot-protocol-for-low-abundance-proteins]
- 59. Detecting Low Abundance Proteins in Western Blotting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/detecting-low-abundance-proteins-in-western-blotting.html]
- 60. A Guide to Gradient Gels: The Why's and How's [https://bitesizebio.com/47184/gradient-gels/]
- 61. Mechanism of 2D Blue Native/SDS-PAGE Protein Complex ... [https://www.mtoz-biolabs.com/mechanism-of-2d-blue-native-sds-page-protein-complex-analysis.html]
- 62. Mass Estimation of Native Proteins by Blue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2953912/]
- 63. 2D Blue Native/SDS-PAGE for Complex Analysis [https://www.creative-proteomics.com/services/2d-blue-native-sds-page-for-complex-analysis.htm]
- 64. Implementing a Dual Approach to Protein Characterization [https://www.biopharminternational.com/view/implementing-dual-approach-protein-characterization]
- 65. Advantages and limitations of clear-native PAGE [https://pubmed.ncbi.nlm.nih.gov/16220535/]
- 66. An efficient screening method for purifying and crystallizing ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269718301106]
- 67. An efficient clear-native PAGE–based workflow for ... [https://sciety-discovery.elifesciences.org/articles/by?article_doi=10.21203/rs.3.rs-8130027/v1]
- 68. Two-Dimensional Blue Native/SDS Polyacrylamide Gel ... [https://www.mdpi.com/2072-6651/14/10/661]
- 69. In-Gel Assay to Evaluate Antioxidant Enzyme Response to ... [https://www.mdpi.com/2076-3417/12/6/2760]
- 70. Traditional protein analysis methods - Western blotting ... [https://www.nautilus.bio/blog/traditional-approaches-to-study-proteins-antibodies-and-other-affinity-reagents/]
- 71. Guide to western blot quantification [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-quantification]
- 72. Advances in protein complex analysis using mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1665575/]
- 73. Accurate Protein Complex Retrieval by Affinity Enrichment ... [https://www.sciencedirect.com/science/article/pii/S1535947620316674]
- 74. Mass spectrometry versus western blotting for validation [https://biology.stackexchange.com/questions/26028/mass-spectrometry-versus-western-blotting-for-validation]
- 75. Analysis of Electroblotted Proteins by Mass Spectrometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC2667373/]
- 76. Extraction of proteins from gels-A brief review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7295094/]
- 77. Resolving mitochondrial protein complexes using non-gradient blue... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2795571/]
- 78. How to Isolate and Purify Protein Bands After SDS-PAGE [https://www.mtoz-biolabs.com/how-to-isolate-and-purify-protein-bands-after-sds-page.html]