BCR Signaling in Immune Responses: Decoding Fate Decisions in Germinal Center vs. Extrafollicular Pathways
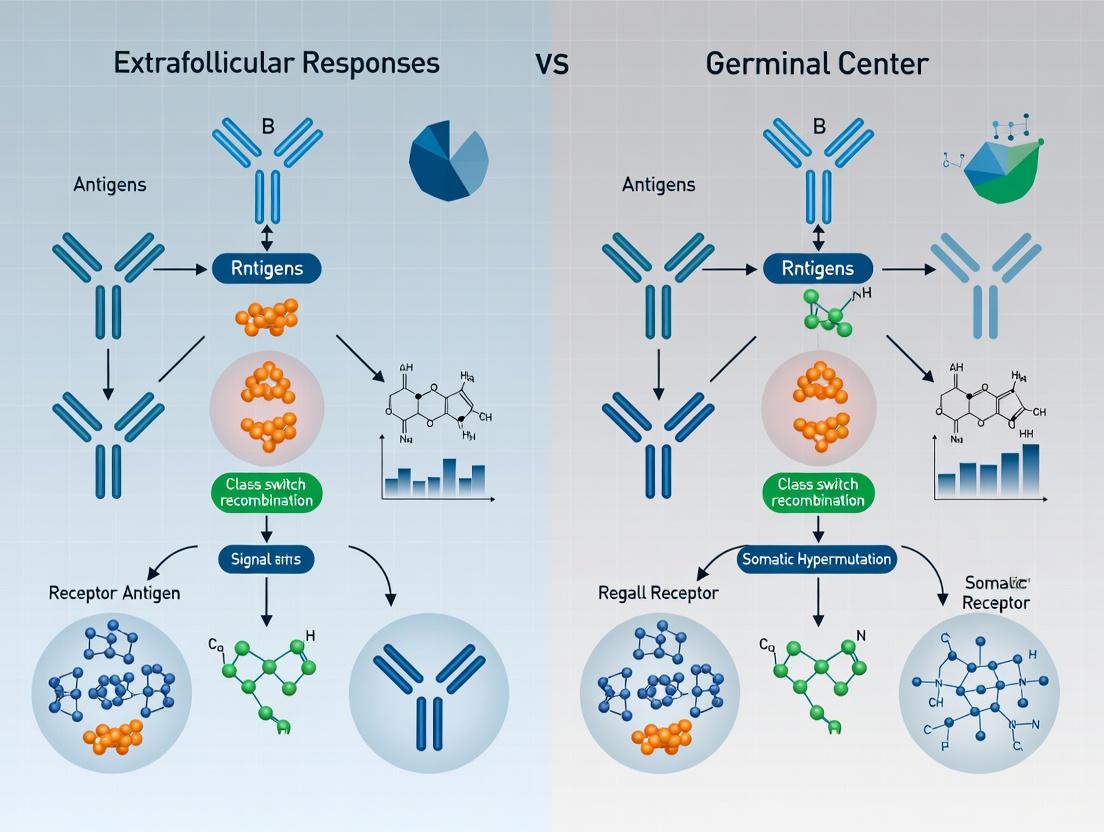
B cell responses to antigen are channeled through two principal pathways: the germinal center (GC) reaction and the extrafollicular (EF) response.
BCR Signaling in Immune Responses: Decoding Fate Decisions in Germinal Center vs. Extrafollicular Pathways
Abstract
B cell responses to antigen are channeled through two principal pathways: the germinal center (GC) reaction and the extrafollicular (EF) response. This article provides a comprehensive analysis for researchers and drug development professionals on the distinct roles of B cell receptors (BCRs) in directing and differentiating these pathways. We explore the foundational biology, where high-affinity BCR engagement often favors the rapid EF plasmablast production, while cyclic BCR revision drives GC affinity maturation. The scope extends to methodologies for dissecting these responses, troubleshooting dysregulation in autoimmunity and cancer, and validating pathway-specific biomarkers for therapeutic intervention. Understanding these BCR-driven mechanisms is critical for advancing vaccine design and targeted therapies for immune-mediated diseases.
The Fundamental Dichotomy: How BCR Signals Initiate Divergent Immune Pathways
Upon encountering antigen, the adaptive immune system mounts a humoral response that can follow one of two principal pathways: the germinal center (GC) reaction or the extrafollicular (EF) response [1] [2]. These parallel processes represent distinct yet sometimes interconnected arms of B cell immunity, each producing terminally differentiated antibody-secreting cells (ASCs) and memory B cells with unique functional characteristics [3]. The GC is a specialized microstructure that develops in secondary lymphoid organs where B cells undergo clonal expansion, somatic hypermutation (SHM), and affinity maturation, eventually differentiating into long-lived plasma cells and memory B cells [1]. In contrast, the EF response occurs outside the B cell follicle in splenic bridging channels, red pulp, and lymph node medullary cords, generating rapid bursts of antibody production through short-lived plasmablasts while also contributing to certain memory B cell subsets [2] [4]. Understanding the fundamental distinctions between these pathways is essential for elucidating protective immunity against pathogens and pathogenic mechanisms in autoimmune diseases, where EF responses are increasingly recognized as key drivers of autoantibody production [1] [3].
Defining the Germinal Center Pathway
Anatomical and Cellular Organization
The germinal center is a highly organized microstructure that forms within secondary lymphoid organs following antigen stimulation [1]. GCs are morphologically divided into two distinct compartments: the dark zone (DZ) and the light zone (LZ) [1]. The DZ is filled with rapidly dividing B cell blasts (centroblasts) that undergo extensive clonal expansion and somatic hypermutation, a process that introduces point mutations into immunoglobulin variable region genes [1]. These mutated B cells then exit the cell cycle and migrate to the LZ, where they become centrocytes that must compete for binding to antigen displayed as immune complexes on follicular dendritic cells (FDCs) [1]. LZ B cells process and present this antigen to a specialized subset of CD4+ T cells called T follicular helper (TFH) cells, which provide critical survival signals through cognate interactions [1]. B cell clones that receive adequate TFH help may differentiate into memory B cells or long-lived plasma cells, or recycle back to the dark zone for further rounds of mutation [1].
Molecular Regulation
GC development and maintenance are regulated by the coordinated expression of specific transcriptional programs and chemokine receptors. The transcription factor Bcl-6 serves as the master regulator of GC formation, suppressing alternative differentiation pathways including plasma cell generation mediated by Blimp-1 [1] [2]. B cell migration between GC compartments is directed by chemokine receptors, with CXCR4 promoting dark zone localization and CXCR5 guiding cells to the light zone [1]. The enzyme activation-induced cytidine deaminase (AID) is essential for both somatic hypermutation and class-switch recombination within the GC, enabling affinity maturation and antibody isotype diversification [1] [2]. The GC reaction is further regulated by a balance between TFH cells that promote GC responses and T follicular regulatory (TFR) cells that limit selection of self-reactive B cells and help terminate GC reactions [1].
Figure 1: Germinal Center Pathway Organization and Cellular Dynamics
Defining the Extrafollicular Pathway
Anatomical Locations and Cellular Participants
The extrafollicular response represents a distinct arm of humoral immunity that occurs outside the B cell follicle [2] [4]. In the spleen, EF responses develop in the bridging channels and red pulp, while in lymph nodes they localize to the medullary cords [2] [3]. These sites are rich in dendritic cells and macrophages that support plasmablast differentiation through factors like BAFF and IL-12 [3]. Unlike the highly structured GC, EF foci consist of clusters of rapidly proliferating B cell blasts and differentiating plasmablasts that express activation markers such as CD44 and CD86 but do not initially express definitive plasma cell markers like CD138 [3]. EF responses can be generated against both T-dependent and T-independent antigens, with the latter involving robust B cell activation through either Toll-like receptor (TLR) signaling or strong, prolonged B cell receptor engagement in the absence of peptide-specific T cell help [3].
Functional Characteristics and Outputs
EF responses are characterized by their rapid kinetics, typically peaking within 4-6 days after antigen exposure [4]. While initially regarded as primarily generating short-lived, low-affinity IgM antibodies, recent evidence demonstrates that EF responses can produce isotype-switched antibodies and memory B cells, including CD11c+ "atypical" B cells (ABCs) [2] [3]. EF-derived B cells can undergo significant clonal expansion and, in the presence of T cell help, can exhibit both class-switch recombination and somatic hypermutation, albeit typically to a lesser extent than GC B cells [3] [4]. The EF pathway serves as a critical source of early protective antibodies during acute infections and has been implicated as a major contributor to pathogenic autoantibody production in systemic autoimmune diseases like lupus [1] [2].
Figure 2: Extrafollicular Pathway Organization and Cellular Dynamics
Comparative Analysis: GC versus EF Pathways
Quantitative Comparison of Pathway Characteristics
Table 1: Key Characteristics of Germinal Center versus Extrafollicular Responses
| Feature | Germinal Center Response | Extrafollicular Response |
|---|---|---|
| Anatomic Location | B cell follicles of secondary lymphoid organs [1] | Splenic bridging channels/red pulp; LN medullary cords [2] [4] |
| Kinetics | Slower onset (peaks ~day 7-10), long-lasting [4] | Rapid onset (peaks ~day 4-6), typically short-lived [4] |
| Key Transcription Factors | Bcl-6 (suppresses Blimp-1) [1] [2] | Blimp-1 (suppresses Bcl-6) [1] [2] |
| B Cell Migration | CXCR5↑, S1PR2↑, EBI2↓ [1] [2] | EBI2↑, CCR7↑, CXCR5↓ [2] |
| Somatic Hypermutation | Extensive [1] | Limited, but can occur [3] [4] |
| Affinity Maturation | Robust, through iterative selection [1] | Limited, primarily through initial BCR affinity [4] |
| T Cell Dependence | Absolutely T-dependent [1] | Can occur with or without T cell help [3] |
| Primary Outputs | Long-lived plasma cells, high-affinity memory B cells [1] | Short-lived plasmablasts, CD11c+ ABCs, early antibodies [2] [3] |
| Role in Autoimmunity | Controversial, may be protective [1] | Major source of pathogenic autoantibodies [1] [3] |
Decision Points in B Cell Fate
The choice between GC and EF differentiation represents a critical fate decision for activated B cells, influenced by multiple factors including B cell subset identity, antigen nature, and inflammatory context [2]. B cell receptor signal strength and affinity play important roles, with some evidence suggesting that higher affinity B cells may preferentially enter the EF pathway during early responses [4]. The presence and quality of T cell help significantly influences this decision, though EF responses can proceed with minimal T cell involvement, particularly when strong innate signals are present [3]. Inflammatory cytokines, particularly IL-12 and IFN-γ, have been identified as key regulators that promote EF responses while suppressing GC formation, effectively acting as molecular switches that direct B cell fate based on infection context [3]. The nature of the antigen itself is also determinative, with T-independent antigens favoring EF responses and protein antigens with CD4 T cell epitopes enabling both pathways [2] [3].
Experimental Methodologies for Pathway Analysis
Genetic Approaches to Dissect Pathway Contributions
Table 2: Key Genetic Models for Studying GC and EF Pathways
| Experimental Approach | Methodology | Application | Key Findings |
|---|---|---|---|
| Genetic GC Ablation | B cell-specific deletion of Bcl-6 or other essential GC factors [1] | Test GC requirement for immune outcomes | Autoimmunity develops despite GC absence; EF pathway sufficient for pathogenesis [1] |
| TFH Manipulation | Use of sanroque mice or other TFH-dysregulated models [1] | Understand T cell help in pathway selection | TFH overactivity correlates with autoimmunity but may function outside GCs [1] |
| Cytokine Manipulation | IL-12/IFN-γ gain or loss of function [3] | Define inflammatory signals in fate decisions | IL-12 enhances EF responses while suppressing GC formation [3] |
| Fate Mapping | Conditional reporters for Bcl-6 or Blimp-1 expression [4] | Track lineage commitment | Many memory B cells originate from GC-independent pathways [4] |
Analytical Techniques for Pathway Characterization
Modern analysis of GC and EF responses employs multidimensional approaches. Immunofluorescence microscopy of lymphoid tissues remains essential for identifying the anatomical location of responses using markers like GL7/CD95 for GCs and Ig/CD138 for EF plasmablasts [4]. Flow cytometry enables detailed phenotypic analysis with key markers including B220+CD38+GL7+ for GC B cells, B220+CD138+ for plasmablasts, and CD11c+T-bet+ for ABCs [4]. Adoptive transfer of traceable antigen-specific B cells (e.g., SWHEL models) allows precise tracking of clonal expansion and differentiation over time [4]. Single-cell RNA sequencing provides comprehensive transcriptional profiles of responding B cells and can be coupled with BCR sequencing to trace clonal relationships and mutation patterns between GC and EF compartments [3] [4]. ELISPOT and in vitro differentiation assays help quantify functional outputs including antibody secretion and memory B cell reactivation potential [4].
Figure 3: Experimental Workflow for GC and EF Pathway Analysis
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying GC and EF Responses
| Reagent/Category | Specific Examples | Experimental Function | Application Notes |
|---|---|---|---|
| Genetic Models | Bcl-6fl/flCD23Cre (GC-deficient) [1] | Ablate GC formation to test pathway necessity | Confirm efficient GC loss by histology/flow cytometry [1] |
| Bcl-6-GFP, Blimp-1-YFP fate reporters [4] | Track lineage commitment in real time | Enable fate mapping of early activation events [4] | |
| Monoclonal Antibodies | Anti-Bcl-6 (transcription factor) [4] | Identify GC B cells by intracellular staining | Requires cell permeabilization; combine with surface markers [4] |
| Anti-GL7, anti-CD95 (FAS) [4] | Surface markers for GC B cells | Standard combination for mouse GC B cell identification [4] | |
| Anti-CD138 (Syndecan-1) [3] | Identify plasmablasts/plasma cells | Marks terminal B cell differentiation [3] | |
| Anti-CD11c, anti-T-bet [4] | Identify ABCs and EF-derived subsets | Characteristic of GC-independent memory populations [4] | |
| Cytokine Reagents | Recombinant IL-12, anti-IL-12 [3] | Manipulate EF-promoting pathways | IL-12 enhances EF while suppressing GC responses [3] |
| Recombinant IL-21, anti-IL-21 [2] | Modulate TFH help | Key cytokine for both GC and EF responses with context-dependent effects [2] | |
| Detection Reagents | ELISA/ELISPOT kits | Quantify antibody-secreting cells | Distinguish isotypes to assess class switching [4] |
| Multiplex immunofluorescence panels | Spatial analysis in tissue sections | Preserve architectural context of responses [4] | |
| Antigen Systems | NP-conjugated antigens (NP-CGG, NP-Ficoll) [3] [4] | Standard T-dependent/T-independent antigens | Well-characterized systems with known kinetics [3] [4] |
| SWHEL transgenic B cells [4] | Traceable hen egg lysozyme-specific B cells | Precise tracking of antigen-specific responses [4] |
The germinal center and extrafollicular pathways represent two fundamentally distinct arms of the humoral immune response, each with unique anatomical organization, regulatory mechanisms, kinetic profiles, and functional outputs. While the GC pathway specializes in generating high-affinity, long-lived immunity through iterative selection, the EF pathway provides rapid antibody production and can contribute to both protective immunity and pathogenic autoantibody responses. The balance between these pathways is dynamically regulated by inflammatory context, antigen nature, and cellular interactions, with cytokines like IL-12 serving as critical molecular switches. Contemporary research has revealed considerable plasticity between these pathways and has challenged earlier simplistic distinctions, particularly regarding the EF pathway's capacity for isotype switching, somatic hypermutation, and memory generation. For research and drug development focused on manipulating humoral immunity, precise understanding of both GC and EF pathways will be essential for developing targeted interventions that either enhance protective responses or suppress pathogenic ones in autoimmunity and other B cell-mediated disorders.
Upon antigen encounter in secondary lymphoid organs, mature naïve B cells face a critical fate decision: to initiate an extrafollicular (EF) response or to enter a germinal center (GC) response. This decision, fundamental to shaping the quality and timing of humoral immunity, is principally guided by the affinity of the B cell receptor (BCR) for its cognate antigen. B cells expressing high-affinity BCRs are preferentially directed toward the EF pathway, rapidly differentiating into antibody-secreting cells (ASCs) that provide early, potent antibody protection. In contrast, B cells bearing lower-affinity BCRs are typically recruited into the GC, where they undergo cyclic rounds of somatic hypermutation (SHM) and selection to achieve affinity maturation [5] [6]. This whitepaper delineates the molecular mechanisms, signaling pathways, and cellular interactions through which BCR affinity serves as a primary determinant in this fate choice, providing a technical resource for researchers and therapeutic developers aiming to manipulate humoral immunity.
The initial BCR-antigen engagement triggers a cascade of biochemical and migratory changes that commit the B cell to either the EF or GC pathway. The table below summarizes the core mechanistic differences induced by high versus low BCR affinity.
Table 1: Core Mechanisms Influenced by BCR Affinity in EF vs. GC Fate Decisions
| Determinant | High-Affinity BCR (EF Fate) | Low-Affinity BCR (GC Fate) |
|---|---|---|
| T-B Interactions | Upregulates PDL1, inhibiting Tfh differentiation; ICOSL expression can be maintained to block pre-Tfh access to bystander B cells [5]. | Promotes T follicular helper (Tfh) cell differentiation via ICOSL upregulation [5]. |
| Chemokine Receptor Expression | High CCR7:CXCR5 ratio, promoting retention at T-B border and migration to EF sites [5]. | Lower CCR7:CXCR5 ratio, facilitating return to the follicle for GC formation [5]. |
| EBI2 (GPR183) Expression | Maintained expression, directing migration to interfollicular and outer follicular regions rich in its ligand (7α,25-OHC) [5] [2]. | Downregulated, enabling escape from EF zones and re-entry into the follicle [5] [2]. |
| Transcriptional Programming | High IRF4 expression represses BCL6, blocking the GC program and promoting an ASC fate [5]. | Lower IRF4 levels permit BCL6 expression, which is essential for establishing and maintaining the GC reaction [5]. |
| Primary Output | Early, high-titer antibody secretion from short-lived plasmablasts [5] [2]. | Affinity-matured memory B cells and long-lived plasma cells [5] [2]. |
Experimental Evidence: Key Studies and Methodologies
The paradigms outlined above are supported by rigorous in vivo and in vitro models. The following section details key experimental approaches and findings that have elucidated the role of BCR affinity in B cell fate determination.
Foundational Affinity Comparison Studies
A seminal study employed Ig heavy-chain transgenic (Tg) mice on a JH knockout background to "freeze" the BCR repertoire and isolate the effect of affinity. Researchers compared the fates of B cells expressing a medium-affinity (B1-8, Ka ~9.64×10⁵ M⁻¹) versus a very low-affinity (V23, Ka <5.0×10⁴ M⁻¹) BCR for the NP antigen [7].
Experimental Protocol:
- Mouse Models: V23 and B1-8 IgH Tg mice crossed onto a JH knockout background.
- Immunization: Intraperitoneal injection with 50 µg NP25-CGG in alum.
- Analysis:
- Histology: Spleen sections stained with PNA-biotin (GC marker) and anti-λ antibody to identify antigen-specific B cells.
- Flow Cytometry: Analysis of GC B cell (B220⁺PNA⁺) formation and apoptosis using caspase glow assays.
- In vivo BrdU Labeling: To measure proliferation rates of GC B cells.
Key Findings: Low-affinity (V23) B cells had an intrinsically higher apoptosis rate within the GC compared to medium-affinity (B1-8) B cells, even in the absence of direct competition. This demonstrated that BCR affinity intrinsically controls B cell survival, a key selective mechanism in the GC [7].
Investigating BCR and Tfh Signal Integration
The interplay between BCR signal strength (Signal 1) and T follicular helper cell support (Signal 2) is critical for GC B cell selection. Subsequent research has refined the model from simple competition to one of integrated signaling.
Experimental Protocol (Limiting T Cell Help):
- An in vivo system was developed to independently deliver BCR-crosslinking antigen or Tfh help to GC B cells.
- Researchers assessed GC B cell expansion and plasmablast differentiation under conditions of limiting T cell help [8].
Key Findings:
- Tfh help alone was sufficient to induce GC B cell expansion and differentiation.
- BCR crosslinking alone could not drive selection.
- When Tfh help was limiting, BCR crosslinking synergistically enhanced GC and plasmablast responses [8]. This established that GC B cells integrate variable inputs from both signals, with BCR signaling augmenting selection particularly when T cell help is suboptimal.
Recent models further propose a three-checkpoint process for GC B cells:
- Checkpoint 1 (DZ to LZ): Tonic BCR signals are required for transition from the Dark Zone to the Light Zone.
- Checkpoint 2 (LZ Survival): Antigen-induced BCR crosslinking provides a survival signal and primes LZ GC B cells to receive Tfh help.
- Checkpoint 3 (Apoptosis Prevention): Strong, sustained BCR signals can induce ROS-mediated apoptosis, which is counteracted by Tfh-derived signals, ensuring only B cells receiving both signals undergo clonal expansion [9].
Quantitative Data: Affinity-Dependent Outcomes
The following tables consolidate quantitative findings from key studies, providing a reference for the magnitude of affinity-dependent effects on B cell responses.
Table 2: Survival and Proliferation Metrics of Low vs. Medium Affinity GC B Cells [7]
| BCR Affinity | Apoptosis Rate in GC | Proliferation Rate (BrdU Incorporation) | GC Competence |
|---|---|---|---|
| Very Low (V23) | High (Intrinsically higher) | Comparable to medium affinity | Poor |
| Medium (B1-8) | Low | Comparable to low affinity | Good |
Table 3: Impact of BCR Affinity on Key Molecular Expression [5]
| BCR Affinity | ICOSL Expression | PDL1 Expression | CCR7:CXCR5 Ratio | IRF4/BCL6 Axis |
|---|---|---|---|---|
| High Affinity | Maintained in vivo | Upregulated | High | High IRF4, represses BCL6 |
| Low Affinity | Upregulated (promotes Tfh) | Not specifically upregulated | Low | Lower IRF4, permits BCL6 expression |
Visualizing B Cell Fate Decisions: Signaling and Migration Pathways
The diagrams below illustrate the critical signaling and migratory pathways that determine B cell fate based on BCR affinity.
B Cell Fate Decision Signaling Network
B Cell Migration and Fate Determination
The Scientist's Toolkit: Key Research Reagents and Models
Table 4: Essential Research Tools for Studying BCR Affinity and B Cell Fate
| Reagent / Model | Key Feature / Function | Application in Research |
|---|---|---|
| MD4 Transgenic Mice | BCRs specific for Hen Egg Lysozyme (HEL) [5]. | Studying high-affinity interactions using HEL and low-affinity variants like DEL [5]. |
| B1-8 & V23 IgH Transgenic Mice | Fixed medium and very low affinity BCRs for NP antigen on JH KO background [7]. | Isolating intrinsic effects of BCR affinity on GC survival and selection without competition [7]. |
| 10E8 BCR Humanized Knock-in Mice | Express heavy chains from HIV bnAb precursors (e.g., 10E8-UCA, 10E8-NGS-04) [10]. | Evaluating recruitment and residency of specific B cell clonotypes in GCs in response to germline-targeting immunogens [10]. |
| 10E8-GT Series Immunogens | Engineered epitope scaffolds with MPER epitopes for increased binding to bnAb precursors [10]. | Priming and shepherding low-affinity precursor B cells through the GC reaction to study affinity maturation pathways [10]. |
| α-ICOSL Blocking Antibody | Inhibits ICOS-ICOSL interactions [5]. | Probing the role of Tfh cell differentiation and help in GC versus EF fate decisions [5]. |
The evidence is conclusive: BCR affinity is a primary determinant directing B cells toward an EF or GC fate. High-affinity engagement promotes rapid EF differentiation through a program of distinct migratory receptor expression (high CCR7, maintained EBI2) and transcriptional regulation (high IRF4, low BCL6). Conversely, lower-affinity engagement fosters a GC fate by permitting BCL6 expression and enabling Tfh cell-dependent cyclic selection. This paradigm is supported by sophisticated in vivo models demonstrating intrinsic, affinity-based survival advantages and the synergistic integration of BCR and Tfh signals.
Future research must further elucidate the molecular switches that fine-tune the initial fate decision and the potential for plasticity between these pathways. Understanding these mechanisms at a deeper level is paramount for the next generation of vaccine design, particularly for pathogens like HIV and influenza where the elicitation of broadly neutralizing antibodies requires guiding B cells through complex affinity maturation pathways [11] [10]. Furthermore, a refined understanding of EF responses, which can generate high-affinity, class-switched antibodies and contribute to autoimmune pathology, opens new avenues for therapeutic intervention in allergic and autoimmune diseases [2] [6] [4].
The adaptive immune system orchestrates precise antibody responses within specialized anatomic niches, primarily secondary lymphoid organs (SLOs) like lymph nodes and spleen. Within these structures, the critical decision for activated B cells to initiate extrafollicular (EF) responses or enter germinal centers (GCs) shapes the quality, breadth, and duration of humoral immunity. This review delineates the architectural and molecular composition of these niches, comparing them with tertiary lymphoid structures (TLSs) that form in non-lymphoid tissues during chronic inflammation. We examine how B cell receptor (BCR) signaling nuances direct cell fate towards rapid EF plasmablast production or GC-based affinity maturation, with implications for vaccine design, autoimmune therapy, and oncology.
Humoral immunity depends on more than just cellular lineage and molecular signals; it is spatially organized within highly specialized microarchitectural niches. These niches—whether the pre-programmed SLOs or the inducible TLSs—provide the structured microenvironment necessary for rare, antigen-specific T and B cells to encounter each other, receive licensing signals, and undergo clonal expansion and differentiation [4]. The BCR plays a gatekeeping role in this process, not only in antigen recognition but also in directing the migratory behavior and subsequent fate decisions of activated B cells. The choice between the EF and GC pathways, occurring in distinct but sometimes adjacent niches, determines whether the immune response will be characterized by rapid, short-lived antibody production or the slower development of high-affinity, long-lived memory [2] [12]. Understanding the anatomy of these niches is therefore fundamental to manipulating immune outcomes in disease.
Secondary Lymphoid Organs: The Primary Niches for B Cell Activation
Architectural Compartments and Key Stromal Cells
SLOs are anatomically structured to maximize the probability of antigen-specific immune cell encounters. The fundamental compartments include:
- B Cell Follicles: Zones rich in B cells, defined by stromal production of the chemokine CXCL13, which attracts CXCR5-expressing B cells [4].
- T Cell Zones: Areas populated by T cells, characterized by production of CCL19 and CCL21, which bind to CCR7 on T cells and some dendritic cells [13].
- Specialized Vasculature: High endothelial venules (HEVs) are specialized blood vessels that facilitate the transmigration of lymphocytes from the blood into the lymphoid tissue [14].
Stromal cells, including follicular dendritic cells (FDCs) and fibroblastic reticular cells, are not passive scaffolds. They are active participants in immunity, providing survival factors, presenting native antigen to B cells (FDCs), and creating chemokine gradients that guide cell movement [14].
The Bone Marrow Niche for B Cell Development
B cell development originates in the bone marrow, a process sustained by specific cellular niches. Early B cell precursors are found in close contact with CXCL12-abundant reticular (CAR) cells [15]. As development progresses from pre-pro-B to pro-B cells, the precursors move away from CAR cells and adjoin IL-7-expressing stromal cells, which are vital for proliferation and differentiation [15] [16]. Mature, naive B cells then egress from the bone marrow to populate SLOs. The bone marrow also serves as a key niche for long-lived plasma cells, which home back to this environment and are maintained by factors including CXCL12 [15].
Table 1: Key Cellular Niches in the Bone Marrow for B Lymphopoiesis
| Niche Cell Type | Key Produced Factors | Role in B Cell Development |
|---|---|---|
| CXCL12hi Reticular (CAR) Cell | CXCL12 [15] [16] | Homing and maintenance of earliest B cell precursors (pre-pro-B) and long-lived plasma cells. |
| IL-7+ Stromal Cell | IL-7 [15] [16] | Supports proliferation and differentiation of pro-B and pre-B cells. |
| Osteoblasts / Pre-Osteoblasts | CXCL12, IL-7, IGF-1, WNT5A [16] | Supports early lymphoid progenitor commitment and pro-B to pre-B cell transition. |
The Germinal Center Niche
Architecture and Function
The GC is a transient but complex niche that forms within B cell follicles upon antigen exposure. It is subdivided into two key microcompartments that facilitate affinity maturation:
- Dark Zone (DZ): A niche dominated by proliferating B cells (centroblasts) that undergo somatic hypermutation (SHM) to introduce point mutations into their BCR variable regions [17].
- Light Zone (LZ): A niche where B cells (centrocytes) interact with a network of FDCs displaying native antigen and with T follicular helper (Tfh) cells. The LZ is the primary site for selection based on antigen binding and T cell help [17].
Continuous cyclic re-entry between the DZ and LZ allows for iterative rounds of mutation and selection, ultimately yielding B cells with BCRs of high affinity for the antigen.
BCR Signaling in GC Selection
The role of BCR signaling in the GC niche extends beyond antigen endocytosis. Recent research using a Bruton’s tyrosine kinase (BTK) drug-resistant mouse model demonstrated that continuous BCR signaling is essential for the survival of LZ B cells and for "priming" them to receive positive selection signals from Tfh cells [17]. This work established that both the signaling and endocytic functions of the BCR are non-redundant requirements for the selection of high-affinity clones within the GC niche.
Tertiary Lymphoid Structures: Ectopic Niches in Inflammation
Formation and Clinical Relevance
Tertiary lymphoid structures are ectopic lymphoid aggregates that form in non-lymphoid tissues (e.g., lung, liver, kidney, tumor margins) in response to persistent antigenic stimulation and chronic inflammation [13] [14] [18]. Unlike SLOs, which form during embryogenesis, TLSs are induced postnatally and lack a capsule, exposing them directly to the inflammatory tissue microenvironment [18]. Their presence is a double-edged sword, with clinical outcomes dependent on context:
- Favorable Associations: In many cancers (e.g., non-small cell lung cancer, melanoma) and infections, the presence of mature TLSs is associated with improved immune control and better patient prognosis [13] [14].
- Detrimental Associations: In autoimmune diseases (e.g., rheumatoid arthritis, lupus), chronic inflammatory diseases, and transplant rejection, TLSs are sites of autoreactive lymphocyte activation and are linked to worse disease outcomes [14] [18].
A Maturation Spectrum
TLSs are not uniform and are thought to mature through a tiered spectrum of organization [13]:
- Early/Immature TLS (Tier 1): Loosely organized perivascular clusters of T and B cells without distinct zoning.
- Intermediate TLS (Tier 2): Defined T cell and B cell areas emerge, with the presence of stromal FDC-like cells and early HEVs.
- Mature TLS (Tier 3): Highly organized structures containing a fully formed GC with Tfh cells, GC B cells, and FDC networks, capable of supporting SHM and class-switching [13].
Table 2: Comparison of Secondary and Tertiary Lymphoid Structures
| Feature | Secondary Lymphoid Organs (SLOs) | Tertiary Lymphoid Structures (TLSs) |
|---|---|---|
| Induction & Timing | Pre-programmed during embryonic development [18] | Induced after birth by chronic inflammation [18] |
| Anatomic Location | Spleen, lymph nodes, gut-associated lymphoid tissue [13] | Non-lymphoid organs (e.g., lung, liver, kidney, tumor site) [13] [18] |
| Capsule | Encapsulated, separating it from the environment [18] | Not encapsulated, directly exposed to tissue microenvironment [18] |
| Key Inducer Cells | Embryonic Lymphoid Tissue Inducer (LTi) cells [14] [18] | Post-natal B cells, Th17 cells, etc., acting as LTi-like cells [14] [18] |
| Functional Role | Generate adaptive immune responses to delivered antigens [18] | Generate adaptive immune responses to locally presented antigens [18] |
| Self-Tolerance | Censoring mechanisms eliminate autoreactive B cells [18] | Autoreactive B cells can survive and produce autoantibodies [18] |
Extrafollicular versus Germinal Center B Cell Responses
Defining the Pathways
Upon antigen encounter in an SLO or TLS, activated B cells face a fate decision, leading to two primary types of responses that occur in distinct anatomic niches:
- Extrafollicular (EF) Response: Activated B cells migrate to the outer edges of the follicle and into EF areas like the splenic bridging channels and medullary cords of LNs. In these niches, they undergo rapid proliferation and differentiate into short-lived plasmablasts that secrete early waves of antibody [2] [4]. This pathway is prominent in responses to both T-dependent and T-independent antigens.
- Germinal Center (GC) Response: Other activated B cells, along with Tfh cells, re-enter the follicle to form a GC. This niche supports the longer processes of SHM and affinity maturation, producing long-lived plasma cells and memory B cells [2] [12].
Molecular Switches and BCR Influence
The commitment to the EF or GC pathway is guided by molecular switches that regulate B cell migration and positioning.
- Migration to EF Sites: Upregulation of the orphan receptor Ebi2 (GPR183) and CCR7, coupled with downregulation of CXCR5, directs activated B cells towards the T-B border and EF zones where their ligands are abundant [2] [4].
- Migration to Follicles/GC: Re-expression of CXCR5 and downregulation of Ebi2 and CCR7, along with upregulation of S1PR2, enables B cells to return to the follicle and initiate the GC reaction [2].
BCR signal strength is a critical factor in this fate decision. B cells receiving stronger BCR and co-stimulatory signals early in the response are competitively favored for entry into both the EF and GC pathways [4] [12]. The EF response can generate isotype-switched, high-affinity antibodies, particularly when derived from B cells with higher initial BCR affinity [4].
Table 3: Characteristics of Extrafollicular and Germinal Center B Cell Responses
| Characteristic | Extrafollicular (EF) Response | Germinal Center (GC) Response |
|---|---|---|
| Anatomic Location | Splenic bridging channels/red pulp, LN medullary cords [4] | B cell follicles within SLOs/TLSs [13] [12] |
| Kinetics | Rapid onset (peaks days 4-6), typically short-lived [4] [12] | Slower onset (peaks ~day 12), can persist for weeks/months [12] |
| Primary Outputs | Short-lived plasmablasts, GC-independent memory B cells [2] [12] | Long-lived plasma cells, memory B cells [12] |
| Somatic Hypermutation | Little to no SHM [4] | Extensive SHM and affinity maturation [17] [12] |
| Key Migratory Receptors | Ebi2 (GPR183) hi, CCR7 hi, CXCR5 lo [2] [4] | CXCR5 hi, S1PR2 hi, Ebi2 lo [2] |
| Metabolic Program | Aerobic glycolysis and oxidative phosphorylation for clonal expansion [12] | Fatty acid oxidation via oxidative phosphorylation for maintenance [12] |
| Pathological Association | Dominant in severe COVID-19 and SLE autoimmunity [19] | Dysregulated in autoimmunity; beneficial in anti-tumor immunity [13] [14] |
Experimental Models and Methodologies
Tracking Antigen Engagement and B Cell Fate
To investigate BCR signaling and selection in the GC niche, Chen et al. developed a sophisticated antigen tracking system [17].
Experimental Protocol: NP-Eα Antigen Tracking [17]
- Antigen Design: A tetrameric tracking antigen (NP-Eα) was created by coupling fluorescently labeled streptavidin (SA-AF647) to the hapten NP and a biotinylated I-Eα peptide.
- Mouse Model: Adoptive transfer of congenically marked B1-8hi B cells (which have a high-affinity BCR for NP) into OVA-primed mice.
- Immunization & Tracking: Mice were boosted with NP-OVA to initiate a GC reaction. The NP-Eα tracker was injected to label B cells engaging antigen in vivo.
- Detection: NP-specific B cells that bound and internalized NP-Eα were identified by AF647 fluorescence. Cells that processed and presented the I-Eα peptide were detected using an antibody (Y-Ae) specific for the resulting pMHC complex.
- Cell Sorting and Analysis: GC B cells were sorted based on tracking status (NP-Eα+ vs. NP-Eα-) and zone (LZ vs. DZ) for subsequent Ig gene sequencing and affinity measurement (bio-layer interferometry).
This methodology allowed researchers to correlate in vivo antigen engagement with BCR mutation status and affinity, revealing that loss of antigen binding was associated with deleterious SHM [17].
Experimental Workflow for Tracking GC B Cell Antigen Engagement
Disrupting BCR Signaling in the GC
To dissect the role of BCR signaling from antigen capture, the same study employed a BTK drug-resistant mouse model. This allowed for the specific, acute inhibition of BCR signaling in an established GC using a BTK inhibitor, which would otherwise be lethal to GC B cells. This experiment demonstrated that BCR signaling is continuously required for LZ B cell survival and their ability to be positively selected by Tfh cells [17].
The Scientist's Toolkit: Key Research Reagents and Models
Table 4: Essential Research Tools for Studying B Cell Niches
| Tool / Reagent | Function/Application | Key Insight Enabled |
|---|---|---|
| NP-Eα Tracking Antigen [17] | Simultaneously track antigen binding (AF647) and presentation (Y-Ae pMHC) in vivo. | Correlated antigen engagement with BCR mutation status and affinity in GC B cells. |
| BTK Drug-Resistant Mouse Model [17] | Enables acute, specific inhibition of BCR signaling in GC B cells without cell death. | Established that BCR signaling per se is necessary for LZ B cell survival and T cell priming. |
| High-Dimensional Flow Cytometry [19] | Deep immunophenotyping of B cell subsets (e.g., DN2, ABCs) in human diseases. | Identified expansion of an EF B cell response signature in severe COVID-19 and SLE. |
| Spatial Transcriptomics / Multi-omics [13] | Resolve gene expression patterns within the intact architecture of SLOs and TLSs. | Revealing cellular heterogeneity and stromal-immune cell crosstalk in situ. |
| CXCL13-Fate Reporting Mice | (Inferred from widespread use) Track and fate-map cells responding to this key follicular chemokine. | Maps cell migration to B cell niches and identifies CXCR5-expressing cells in tissues. |
The anatomic niche in which a B cell is activated is a primary determinant of the immune response's character and outcome. The molecular machinery guiding B cells to EF or GC niches, and the subsequent BCR signaling within those niches, are integrated processes that can be therapeutically targeted. Future research, powered by spatial multi-omics and high-resolution in vivo imaging, will continue to decode the complex cellular and molecular dialogues within TLSs and SLOs. This knowledge is pivotal for developing next-generation vaccines that steer responses towards protective GC reactions, and for therapies that dismantle pathogenic EF or GC responses in autoimmunity, or induce them to fight cancer.
Germinal centers (GCs) are transient, specialized microstructures that form within secondary lymphoid organs following exposure to T-cell-dependent antigens [20] [21]. They serve as the primary site for antibody affinity maturation, a process that enhances the ability of antibodies to neutralize pathogens and is fundamental to effective vaccination and adaptive immunity [22]. GCs are spatially organized into two distinct microanatomical regions: the dark zone (DZ) and the light zone (LZ). This architectural division facilitates the temporal separation of two critical processes: somatic hypermutation (SHM) of immunoglobulin genes in the DZ, and clonal selection based on antigen binding and T-cell help in the LZ [20] [21] [22]. The cyclic migration of B cells between these zones enables the iterative refinement of antibody affinity. This whitepaper delves into the sophisticated spatiotemporal regulation of these mechanisms, framing them within broader B cell biology by contrasting them with the rapid, but less refined, extrafollicular (EF) responses [2].
Core Mechanics of the Germinal Center Cycle
The germinal center reaction is a dynamic, cyclic process designed to generate high-affinity antibodies. It relies on the precise coordination of events in two distinct zones.
Dark Zone: Somatic Hypermutation and Proliferation
In the DZ, B cells (centroblasts) undergo rapid proliferation and diversify their B cell receptor (BCR) repertoire through SHM [20] [23]. This process is catalyzed by the enzyme activation-induced cytidine deaminase (AID), which introduces point mutations into the variable regions of immunoglobulin genes at a high rate, approximately 10⁻³ per base pair per cell division [20] [23]. As SHM is a random process, it is significantly more likely to generate deleterious or neutral mutations than affinity-enhancing ones [23]. Following SHM, a critical pre-screening checkpoint occurs within the DZ itself. B cells degrade their pre-existing BCRs and express the newly mutated versions. Cells that acquire crippling mutations and fail to express a functional BCR are identified and triggered to undergo apoptosis, preferentially in the late G1 stage of the cell cycle, preventing them from accumulating and wasting resources [20].
Light Zone: Antigen-Driven Selection
After mutating and proliferating in the DZ, B cells (now called centrocytes) downregulate CXCR4 and migrate to the LZ [20] [21]. Here, they face a competitive selection process based on the affinity of their newly mutated BCRs. The LZ is populated by follicular dendritic cells (FDCs) that display native antigen in the form of immune complexes, and T follicular helper (Tfh) cells that provide essential survival signals [21] [22]. LZ B cells compete to acquire antigen from FDCs. Those with higher-affinity BCRs are more efficient at internalizing antigen, processing it, and presenting it as peptides on MHC-II molecules to Tfh cells [21] [22]. This interaction provides B cells with CD40 and cytokine signals that promote their survival and instruct their subsequent fate. Positively selected B cells induce expression of the transcription factor c-Myc, which licenses them for further expansion and diversification [21].
Cyclic Re-entry and Affinity Maturation
The GC reaction is not a linear pathway. B cells selected in the LZ do not immediately differentiate into output cells; instead, the majority upregulate CXCR4 and re-enter the DZ for further rounds of proliferation and mutation [21] [22]. This process, termed cyclic re-entry, allows for iterative improvement of antibody affinity. With each cycle, B cells bearing affinity-enhancing mutations are preferentially expanded, while those with neutral or deleterious mutations are outcompeted and eliminated. The amount of Tfh cell help a B cell receives in the LZ directly influences the number of divisions it will undergo upon returning to the DZ, creating a feed-forward loop that accelerates affinity maturation [22] [23].
Table 1: Key Characteristics of Germinal Center Zones
| Feature | Dark Zone (DZ) | Light Zone (LZ) |
|---|---|---|
| Primary Functions | Somatic hypermutation (SHM), clonal expansion, BCR pre-screening | Antigen presentation, T follicular helper (Tfh) cell interaction, positive selection, fate decision |
| Key Cell Types | Proliferating B cells (centroblasts) | B cells (centrocytes), Follicular Dendritic Cells (FDCs), Tfh cells |
| Critical Processes | AID-mediated SHM; BCR turnover; apoptosis of cells with non-functional BCRs | Antigen acquisition from FDCs; pMHCII presentation; c-Myc induction |
| Characteristic Markers | High CXCR4, AID | Low CXCR4, high MHC-II, CD40 |
The following diagram illustrates the core germinal center cycle and the checkpoints governing B cell migration and selection.
Advanced Concepts and Recent Discoveries in GC Regulation
Beyond the core cycle, recent research has revealed additional layers of regulation that optimize the affinity maturation process.
Affinity-Dependent Modulation of Somatic Hypermutation
A paradigm-shifting discovery indicates that the SHM rate is not fixed. Instead, it is dynamically regulated in an affinity-dependent manner. High-affinity B cells that receive strong Tfh signals and are programmed for more divisions in the DZ reduce their mutation rate per division [23]. This mechanism protects high-affinity lineages from accumulating deleterious mutations during expansive proliferative bursts, thereby safeguarding their fitness. Computational modeling demonstrates that without this regulation, prolific division would lead to significant generational "backsliding" in affinity due to the random nature of SHM [23].
Metabolic Regulation of Selection
Cell metabolism plays an active role in guiding GC B cell fate. While LZ B cells rely more on glycolysis, DZ B cells undergoing rapid division preferentially utilize oxidative phosphorylation (OXPHOS) and fatty acid oxidation [24]. Enhanced OXPHOS has been directly linked to the efficient positive selection of GC B cell clones that acquire higher-affinity BCRs. Experimentally, impairing OXPHOS reduces positive selection, while enhancing it with drugs like oltipraz promotes affinity maturation [24].
Multiple BCR-Dependent Checkpoints
BCR signaling orchestrates GC transit through several checkpoints. Beyond the classical need for antigen binding in the LZ, a tonic BCR signal is required for DZ B cells to even transition to the LZ (Checkpoint 1) [9]. In the LZ, antigen-induced BCR signaling synergizes with CD40 signaling from Tfh cells to promote survival and c-Myc induction (Checkpoint 2). However, an excessive or sustained BCR signal can be detrimental, inducing reactive oxygen species (ROS)-mediated apoptosis unless counterbalanced by Tfh signals (Checkpoint 3) [9]. This fine-tuning ensures optimal selection.
Table 2: Quantitative Parameters of Somatic Hypermutation and Selection
| Parameter | Value / Probability | Biological Significance |
|---|---|---|
| SHM Rate (Baseline) | ~1 x 10⁻³ per bp/division [20] | Drives antibody diversification |
| Mutation Probability (pₘᵤₜ) per Division | Can vary from ~0.6 to ~0.2 [23] | Affinity-dependent modulation protects high-value clones |
| Probability of an Affinity-Enhancing Mutation | pₑₙₕ ≈ 0.01 [23] | Explains why affinity maturation requires multiple cycles and strong selection |
| Probability of a Lethal Mutation | pₗₑₜ ≈ 0.3 [23] | Highlights the need for rigorous pre-screening and quality control |
Experimental Methods for Investigating GC Dynamics
Studying the spatiotemporal dynamics of GCs requires sophisticated tools that can track cell lineage, division, mutation, and migration over time.
In Vivo Cell Division and Lineage Tracking
A powerful method utilizes H2b-mCherry reporter mice where the fluorescent protein is expressed under a doxycycline (DOX)-sensitive promoter [23]. In this system:
- Protocol: Immunize mice, then administer DOX to turn off the mCherry reporter. As cells divide post-DOX, the mCherry protein is diluted among daughter cells.
- Application: Flow cytometric analysis of mCherry intensity allows researchers to isolate and compare GC B cells that have undergone few ("mCherryʰⁱᵍʰ") versus many ("mCherryˡᵒʷ") divisions. Coupled with single-cell BCR sequencing, this reveals the relationship between division history, mutation load, and clonal expansion [23].
Manipulating Antigen Presentation to Tfh Cells
To dissect the role of Tfh help independently of BCR affinity, researchers use an anti-DEC205 antibody fusion protein [21] [22].
- Protocol: Administer antigen fused to an antibody against DEC-205 (a receptor expressed on GC B cells). This delivers the antigen directly to the B cell's endosomal compartments, bypassing the need for BCR-mediated internalization.
- Application: This method allows for the controlled provision of T cell help to GC B cells regardless of their BCR affinity, demonstrating that the magnitude of help directly determines the extent of subsequent proliferation in the DZ [22].
Metabolic and Genetic Perturbation
Loss-of-function and gain-of-function experiments are critical for establishing mechanism.
- OXPHOS Inhibition: Immunization of mice with B cell-specific deletion of Cox10 (e.g., Cox10fl/fl Aicda+/cre), a gene essential for electron transport chain complex IV assembly, impairs both clonal expansion and positive selection [24].
- OXPHOS Enhancement: Treatment of immunized mice with oltipraz, a drug that increases the oxygen consumption rate (OCR) in B cells, promotes affinity maturation, confirming a causal role for metabolism [24].
The following diagram outlines the workflow for a comprehensive GC B cell analysis experiment that integrates several of these advanced techniques.
The Scientist's Toolkit: Key Research Reagents and Models
Table 3: Essential Reagents and Models for Germinal Center Research
| Tool / Reagent | Function and Application in GC Research |
|---|---|
| H2b-mCherry (or similar) Reporter Mice | Tracks cell division history in vivo by fluorescent protein dilution after doxycycline administration [23]. |
| Anti-DEC205-Antigen Fusion | Delivers antigen directly to GC B cell endosomes, uncoupling Tfh help from BCR affinity to study selection mechanisms [21] [22]. |
| NP-OVA / NP-KLH Antigen | A classic T-cell-dependent model antigen. The NP-specific response is dominated by the VH186.2 gene, allowing precise tracking of affinity-enhancing mutations like W33L [24]. |
| Aicda-Cre Mice | Enables targeted gene deletion specifically in GC B cells, which express Activation-Induced Cytidine Deaminase (AID) [24]. |
| Oltipraz | A pharmacological agent that enhances oxidative phosphorylation (OXPHOS). Used to test the causal role of metabolism in affinity maturation [24]. |
The spatiotemporal orchestration of DZ mutagenesis and LZ selection is a remarkable evolutionary adaptation for producing high-affinity, protective antibodies. The GC cycle, with its iterative rounds of mutation and stringent selection, stands in stark contrast to extrafollicular (EF) responses. EF responses are rapid, occur outside follicles, and generate early-protective antibodies and memory B cells with little to no SHM [2]. While EF responses are crucial for initial pathogen control, the GC is dedicated to long-term immunological refinement and breadth. The discovery of regulated SHM and metabolic control adds new dimensions to our understanding of how GCs optimize this process, effectively solving the theoretical problem of diluting beneficial mutations with neutral or deleterious ones during clonal expansion.
The implications for drug and vaccine development are substantial. Strategies that can modulate the GC cycle—for instance, by temporarily enhancing OXPHOS to boost affinity maturation, or by steering the fate of selected B cells toward long-lived plasma cells—hold great promise for next-generation vaccines against challenging pathogens like HIV and influenza. Furthermore, a deeper understanding of the checkpoints that prevent the emergence of self-reactive B cells in the GC is directly relevant to treating autoimmune diseases. As new spatial omics technologies [25] enable an even more refined mapping of these processes, the next decade will likely yield transformative insights into the spatiotemporal dynamics of antibody evolution.
B cell responses to antigen activation diverge into two principal pathways: the extrafollicular (EF) response and the germinal center (GC) reaction. These pathways are governed by master transcriptional regulators IRF4 and Bcl-6, respectively. This whitepaper delineates the antagonistic molecular switches that commit B cells to either pathway, detailing the transcriptional networks, signaling pathways, and epigenetic modifications involved. Framed within broader B cell receptor (BCR) research, this guide provides researchers and drug development professionals with comprehensive mechanistic insights, quantitative data summaries, and essential methodological protocols for investigating these critical immune determinants.
Upon antigen encounter, naïve B cells face a critical fate decision: migrate to extrafollicular regions and rapidly differentiate into short-lived plasmablasts, or enter germinal centers to undergo affinity maturation and ultimately produce long-lived plasma cells and memory B cells. The EF pathway generates rapid, transient antibody protection, while the GC pathway enables sustained, high-affinity humoral immunity essential for long-term protection and effective vaccination [12]. The molecular master regulators governing this decision are the transcription factors IRF4 and Bcl-6, which function in a mutually antagonistic manner to direct B cell fate toward plasma cell differentiation or GC commitment, respectively [26] [27] [28]. Understanding the precise mechanisms of this regulatory axis provides crucial insights for vaccine development, autoimmune disease treatment, and oncology therapeutics.
Molecular Mechanisms of IRF4 in Extrafollicular Plasma Cell Differentiation
IRF4 as a Transcriptional Switch for Plasma Cell Fate
Interferon Regulatory Factor 4 (IRF4) operates as a crucial determinant in initiating plasma cell differentiation. Studies with conditional knockout mice demonstrate that IRF4 deletion in germinal center B cells results in a complete absence of post-germinal center plasma cells and an inability to differentiate memory B cells into plasma cells [26]. IRF4 functions upstream in a hierarchical transcriptional cascade that includes Blimp-1 (encoded by Prdm1) and XBP-1, essential factors for plasma cell development [26].
The mechanism of IRF4 action involves transcriptional reprogramming of B cell identity. IRF4 directly promotes expression of Prdm1 while simultaneously repressing B cell identity genes such as Pax5, Bach2, and Bcl6 [28]. This dual function facilitates the loss of B cell characteristics and acquisition of plasma cell features, including expanded endoplasmic reticulum capacity and enhanced secretory apparatus [28].
Epigenetic Regulation by H3.3 Histone Variant
Recent findings indicate that histone variant H3.3 plays a critical role in modulating IRF4-driven plasma cell differentiation. During differentiation, H3.3 is markedly downregulated, and its enforced expression impairs plasma cell development by maintaining B cell gene expression programs and preventing the upregulation of plasma cell-associated genes including Irf4, Prdm1, and Xbp1 [28]. Chromatin integration labeling sequencing (ChIL-seq) reveals that H3.3 deposition dynamics at key loci correlate with stage-specific gene expression during differentiation [28].
Table 1: Quantitative Changes in Gene Expression During Plasma Cell Differentiation
| Gene | Function | Expression Change in PC vs. B cell | Regulation by IRF4 |
|---|---|---|---|
| Irf4 | Master regulator of PC differentiation | Upregulated | Auto-regulatory positive feedback |
| Prdm1 | Encodes Blimp-1, represses B-cell genes | Upregulated | Direct transcriptional activation |
| Xbp1 | ER stress response, secretory capacity | Upregulated | Direct/indirect activation |
| Pax5 | B-cell identity maintenance | Downregulated | Direct/indirect repression |
| Bach2 | Antagonizes PC differentiation | Downregulated | Repression |
| Bcl6 | GC program, antagonizes IRF4 | Downregulated | Mutual antagonism |
IRF4 in BCR Signaling and Metabolic Reprogramming
IRF4 expression is induced by BCR and CD40 signaling, creating a positive feedback loop that reinforces plasma cell commitment. In the EF response, strong BCR signals coupled with T cell help or TLR signaling rapidly elevate IRF4, which then cross-antagonizes Bcl-6 to prevent GC entry [12] [29]. Metabolically, EF plasmablasts utilize both glycolysis and oxidative phosphorylation to support their rapid expansion and antibody production, contrasting with the distinct metabolic program of GC B cells [12] [30].
Bcl-6 as the Master Regulator of Germinal Center Commitment
Molecular Functions of Bcl-6 in GC Formation
B-cell lymphoma 6 (Bcl-6) serves as the master transcriptional repressor governing germinal center commitment and development. Genetic studies reveal that the RD2 repression domain of Bcl-6 is specifically required for GC formation, with Bcl-6RD2(MUT) mice exhibiting complete loss of GC structures while maintaining normal extrafollicular responses [27]. This domain recruits histone deacetylases (HDACs) to repress target genes, including trafficking receptors S1pr1 and Gpr183, whose deregulation impairs proper B cell migration and GC organization [27].
Bcl-6 functions by repressing alternative differentiation programs, including plasma cell commitment through direct repression of Prdm1 and memory B cell factors. This repression maintains B cells in a proliferative, mutable state conducive to somatic hypermutation and affinity maturation [27] [31]. The precise level of Bcl-6 expression shortly after antigen engagement determines clonal representation in subsequent GCs, establishing early commitment events that shape the immune response [31].
Metabolic Programming of GC B Cells
GC B cells exhibit a distinct metabolic profile characterized by predominant fatty acid oxidation via oxidative phosphorylation, with minimal glucose or glutamine utilization [12] [30]. This metabolic program supports maintenance rather than expansion, consistent with the constant cell numbers in established GCs despite rapid proliferation. The metabolic regulator mTORC1 plays a critical role in positively selecting GC light zone B cells in a CD40-dependent manner, promoting their migration to the dark zone for further proliferation [30].
Table 2: Quantitative Effects of Bcl-6 Expression Levels on B Cell Fate
| BCL6 Expression Level | GC Formation | EF Response | Clonal Representation in GC | Key Molecular Effects |
|---|---|---|---|---|
| Null (Bcl6-/-) | Absent [27] | Normal [27] | N/A | Failure of GC commitment; inflammatory disease |
| RD2 Domain Mutant | Absent [27] | Normal [27] | N/A | Normal early activation; failed GC coalescence |
| Low (within normal range) | Intact | Intact | Baseline recruitment | Standard progression through pre-GC stages |
| Supraphysiological (upper quartile) | Intact | Intact | Enhanced GC transition [31] | Preferential recruitment through early stages |
The IRF4-Bcl-6 Antagonistic Switch in B Cell Fate Determination
The mutual antagonism between IRF4 and Bcl-6 creates a bistable switch that directs B cells toward either EF plasma cell or GC fates. This reciprocal inhibition occurs at multiple levels:
- Transcriptional repression: Bcl-6 directly binds and represses the Prdm1 promoter, while IRF4 represses Bcl6 expression [28].
- Protein-protein interactions: Both factors interfere with each other's transcriptional activity through direct and indirect mechanisms.
- Epigenetic modifications: Changes in histone variant H3.3 deposition and chromatin accessibility reinforce the chosen differentiation path [28].
The balance of this switch is influenced by signal strength and duration from the BCR, CD40, and cytokine receptors. Strong, sustained signals favor IRF4 induction and plasma cell differentiation, while moderated, cyclic signals promote Bcl-6 expression and GC commitment [12] [29].
Diagram 1: IRF4-BCL6 Molecular Switch in B Cell Fate Determination. Strong BCR and T cell signals promote IRF4 expression, which represses BCL6 and drives plasma cell differentiation via Blimp-1 and H3.3 downregulation. Moderate signals induce BCL6, which represses IRF4 and promotes GC commitment.
Experimental Approaches and Methodologies
Key Experimental Models for Fate Determination Studies
Genetic mouse models have been instrumental in delineating the functions of IRF4 and Bcl-6. Conditional knockout mice with Irf4 deletion in activated B cells demonstrate the absolute requirement for IRF4 in plasma cell generation [26]. Similarly, mice with targeted disruption of the Bcl-6 RD2 domain reveal the essential role of this specific domain in GC formation, while sparing extrafollicular responses [27].
In vitro differentiation systems using LPS stimulation or cytokine cocktails permit controlled investigation of plasma cell differentiation. These systems have revealed the dynamic changes in H3.3 deposition and chromatin accessibility during differentiation [28]. For GC studies, immunization models with T-dependent antigens like NP-CGG allow precise tracking of GC B cell dynamics and molecular analysis.
Quantitative Measurement Techniques
Flow cytometry enables quantification of IRF4 and Bcl-6 protein levels using intracellular staining, allowing correlation of expression levels with differentiation outcomes [31]. Chromatin integration labeling sequencing (ChIL-seq) provides high-resolution mapping of H3.3 deposition dynamics genome-wide [28]. ATAC-seq reveals chromatin accessibility changes during differentiation, while RNA-seq profiles transcriptional changes.
Table 3: Essential Research Reagents for Investigating B Cell Fate Determination
| Reagent/Category | Specific Examples | Research Application | Key Function in Studies |
|---|---|---|---|
| Genetic Models | Irf4-floxed mice [26], Bcl6 RD2 mutant mice [27] | Fate mapping, conditional knockout | Establish requirement of factors in specific pathways |
| Cell Lines | SUDHL-2 (ABC-DLBCL), SUDHL-6 (GCB-DLBCL) [32] | In vitro differentiation studies | Model human B cell differentiation |
| Antibodies | Anti-IRF4 [28], Anti-Bcl6 [27], Anti-Blimp1 [28] | Western blot, flow cytometry, IHC | Protein detection and quantification |
| Inhibitors | Rapamycin (mTOR inhibitor) [30] [32] | Pathway manipulation | Dissect metabolic signaling requirements |
| Stimulation Reagents | LPS [28], anti-CD40 + IL-4/IL-21 | In vitro differentiation | Induce plasma cell or GC-like differentiation |
| Detection Assays | CCK-8 proliferation assay [32], CD38 staining by FCM [32] | Functional assessment | Measure proliferation and differentiation status |
Protocol: Assessing Plasma Cell Differentiation In Vitro
- B Cell Isolation: Isolate naïve B cells from mouse spleen or human peripheral blood using magnetic negative selection (CD43- for mouse).
- Stimulation Culture: Resuspend cells at 1×10^6 cells/mL in RPMI-1640 with 10% FBS and stimulate with:
- LPS at 10-25 μg/mL for mouse cells
- Or CD40L (1 μg/mL) + IL-4 (10 ng/mL) + IL-21 (50 ng/mL) for human cells
- Time Course Sampling: Collect cells at 0, 24, 48, 72, and 96 hours for analysis.
- Differentiation Assessment:
- Flow cytometry: Stain for CD138, CD38, B220, and intracellular IRF4/Blimp1
- Gene expression: qPCR for Irf4, Prdm1, Xbp1, Pax5, Bcl6
- Protein analysis: Western blot for IRF4, Blimp1, Bcl-6, and histone H3.3
- Functional readouts: ELISA for immunoglobulin secretion, CCK-8 for proliferation [28] [32].
Signaling Pathways and Metabolic Regulation
Integrated Signaling Network Controlling B Cell Fate
The B cell fate decision integrates signals from multiple pathways that converge on IRF4 and Bcl-6 expression. The BCR signal strength is a primary determinant, with strong signals promoting IRF4 and weak signals permitting Bcl-6 expression. CD40 signaling provides critical T cell help that can support either pathway depending on timing and context. Cytokine signals (IL-4, IL-21) further modulate the response, particularly for GC commitment and TFH cell help [12] [30].
The mTORC1 pathway serves as a key integrator of metabolic and signaling cues, with activated mTORC1 promoting plasma cell differentiation through IRF4 upregulation [32]. In GC B cells, mTORC1 activity is regulated in a cyclic manner during the dark zone-light zone transition, supporting proliferation while preventing premature differentiation [30].
Diagram 2: Signaling Network Controlling B Cell Fate. Multiple inputs converge on mTORC1, which promotes IRF4 expression and plasma cell fate. BCL6 expression under moderate signaling leads to GC commitment, with mutual antagonism between the two fates.
Metabolic Determinants of Cell Fate
EF plasmablasts utilize both glycolysis and oxidative phosphorylation to support rapid expansion and antibody production, consistent with their role in generating quick antibody responses [12]. In contrast, GC B cells primarily rely on fatty acid oxidation via oxidative phosphorylation, with minimal glycolysis, which may support their unique cyclic pattern of proliferation and selection [12] [30]. Autoreactive B cells show distinct metabolic requirements, with greater dependence on glycolysis compared to antigen-induced GC B cells, suggesting metabolic differences that could be therapeutically targeted [30].
Implications for Disease and Therapeutic Development
Understanding the IRF4-Bcl-6 switch has significant implications for autoimmune disease treatment, vaccine development, and oncology therapeutics. In systemic autoimmunity, pathogenic autoantibodies often arise from extrafollicular responses rather than GC reactions, suggesting therapeutic strategies aimed at suppressing EF responses while preserving GC-mediated protective immunity [1]. In lymphoma, particularly diffuse large B-cell lymphoma (DLBCL), the IRF4-mTORC1 axis drives plasmablast differentiation in the ABC subtype, contributing to autoimmune complications like AIHA [32].
The distinct metabolic requirements of EF, GC, and autoreactive B cells offer opportunities for selective metabolic interventions. Glutaminolysis inhibition suppresses both immunization-induced and autoimmune humoral responses, while glycolysis inhibition preferentially targets autoreactive GC responses [30]. These approaches could enable more precise immunomodulation with fewer global immune effects.
The antagonistic relationship between IRF4 and Bcl-6 represents a fundamental molecular switch directing B cell fate decisions between extrafollicular plasma cell differentiation and germinal center commitment. This regulatory axis integrates BCR signal strength, T cell help, metabolic cues, and epigenetic modifications to determine the quantity, quality, and duration of humoral immune responses. Continued investigation of these pathways will yield critical insights for developing next-generation vaccines, autoimmune therapies, and oncology treatments that precisely modulate B cell fate for therapeutic benefit.
The Role of T Follicular Helper (Tfh) Cells in Providing Cognate Help for Both Pathways
T follicular helper (Tfh) cells are specialized CD4+ T cells essential for adaptive humoral immunity, providing critical cognate help to B cells during both extrafollicular (EF) and germinal center (GC) responses. While GC-Tfh cells are well-established drivers of affinity maturation and long-lived immunity, their roles in supporting the rapidly-activated EF pathway are increasingly recognized. This whitepaper synthesizes current understanding of Tfh cell biology across these divergent B cell response pathways, examining differentiation mechanisms, functional specializations, and molecular requirements. We detail how Tfh cells coordinate with B cells through multi-stage processes involving specific cytokine signals, costimulatory molecules, and transcriptional regulators. Technical protocols for studying these interactions and key research reagents are provided to support ongoing investigations into how modulation of Tfh cell function could advance therapeutic strategies for vaccines, autoimmune diseases, and cancer.
T follicular helper cells represent a distinct lineage of CD4+ T cells specialized in providing help to B cells, forming the cornerstone of T cell-dependent antibody responses. Since the identification of Bcl6 as their lineage-defining transcription factor a decade ago, Tfh cells have been recognized as essential for GC formation—the specialized microanatomical sites where B cells undergo somatic hypermutation and affinity maturation [33] [34]. Beyond this classical role, emerging evidence establishes that Tfh cells also provide crucial help for EF B cell responses, which generate early antibody-secreting cells outside the follicle [3] [4].
The differentiation of Tfh cells is a multi-stage, multi-factorial process distinct from other T helper lineages [33]. Unlike Th1, Th2, or Th17 cells, which can be fully induced by specific cytokine exposures, Tfh cell differentiation requires a coordinated sequence of signals from antigen-presenting cells and B cells across different anatomical locations within secondary lymphoid organs. This complexity enables Tfh cells to dynamically adapt their helper functions to support both the rapid EF response and the protracted GC pathway, making them versatile regulators of humoral immunity with significant implications for vaccine development and treatment of antibody-mediated diseases [3] [34].
Tfh Cell Differentiation: A Multi-Stage Process
Developmental Stages and Key Regulators
Tfh cell differentiation occurs through sequential stages characterized by distinct molecular programs and migratory patterns, as outlined in Table 1. This process begins with initial activation in the T cell zone and progresses through maturation at the T-B border before culminating in the GC reaction [33].
Table 1: Stages of Tfh Cell Differentiation
| Stage | Location | Key Molecular Events | Primary Interacting Cells |
|---|---|---|---|
| Initial Priming | T cell zone | TCR signaling; IL-6/ICOS-induced Bcl6 upregulation; CXCR5 induction; PSGL1/CCR7 downregulation | Dendritic cells |
| Early Tfh Commitment | T-B border & interfollicular zone | Bcl6 stabilization; Enhanced CXCR5 expression; PD-1/ICOS upregulation; IL-21 production | Antigen-specific B cells |
| GC Tfh Maturation | Germinal center light zone | High Bcl6, PD-1, ICOS, CXCR5 expression; SAP expression; IL-21/IL-4 secretion | GC B cells |
The initial priming stage begins when naïve CD4+ T cells are activated by dendritic cells (DCs) in the T cell zone. During this phase, IL-6 signaling synergizes with ICOS costimulation to induce initial expression of Bcl6, the master transcription factor for Tfh lineage commitment [33]. This early Bcl6 expression drives upregulation of CXCR5, the chemokine receptor responsible for homing to B cell follicles, while simultaneously downregulating CCR7 and PSGL1, which otherwise retain T cells in the T cell zone [33].
The second stage occurs at the T-B border and interfollicular zone, where early Tfh cells interact with antigen-specific B cells. Research using fate-mapping strategies has revealed that Tfh cells progress through distinct developmental and functional states, including a progenitor-like stage (Tfh-Prog) and a fully developed effector stage (Tfh-Full) marked by historical IL-21 production [35]. These interactions with B cells are essential for full Tfh maturation, as B cells serve as antigen-presenting cells that provide additional signals through ICOSL and other costimulatory molecules to stabilize the Tfh differentiation program [33] [36].
The third and final stage produces mature GC-Tfh cells, which are characterized by high expression of CXCR5, PD-1, ICOS, Bcl6, Maf, and SAP [33]. These cells provide selective help to GC B cells through serial interactions that promote somatic hypermutation, affinity maturation, and differentiation into memory B cells or long-lived plasma cells.
Transcriptional Regulation of Tfh Differentiation
The Tfh differentiation program is orchestrated by a complex transcriptional network centered around Bcl6, which represses genes associated with alternative T helper lineages while promoting expression of Tfh-associated genes such as CXCR5, PD-1, and ICOS [33]. Other transcription factors including Tcf1, Ascl2, and MAF contribute to establishing the Tfh transcriptional program [35]. Recent single-cell RNA sequencing studies have identified distinct transcriptional states corresponding to different stages of Tfh development, with Tfh-Full cells showing stronger enrichment for core Tfh signatures compared to Tfh-Prog cells [35].
The balance between Tfh differentiation and alternative T cell fates is further regulated by antagonistic relationships with other transcriptional regulators. Blimp1 (encoded by Prdm1) reciprocally represses Bcl6 expression and promotes non-Tfh effector lineages [33]. Similarly, Foxo1 and KLF2 act as negative regulators of Tfh differentiation [35].
Tfh Cells in Germinal Center Responses
Role in GC Initiation and Maintenance
GC-Tfh cells are essential for the formation and maintenance of germinal centers, specialized structures where B cells undergo repeated rounds of proliferation, somatic hypermutation, and selection. Tfh cells provide critical survival and proliferation signals to GC B cells through CD40L-CD40 interactions and cytokine secretion, primarily IL-21 [33] [37]. Without sustained Tfh cell help, GC reactions collapse prematurely, failing to generate high-affinity antibodies and long-lived B cell memory [38].
Recent studies utilizing longitudinal tracking of antigen-specific Tfh cells in non-human primates have demonstrated that functional GC-Tfh subsets correlate with antibody magnitude and quality [38]. Notably, antigen-specific Tfh clones can persist within GCs for over 6 months without signs of exhaustion, maintaining stable gene expression profiles and continuing to provide help to B cells throughout extended GC reactions [38]. This remarkable longevity highlights the importance of Tfh cell persistence for sustaining GC responses, particularly in contexts like HIV vaccination where prolonged affinity maturation is required for the development of broadly neutralizing antibodies.
Molecular Mechanisms of Tfh Help in GCs
Within GCs, Tfh cells provide help through multiple coordinated mechanisms. CD40L-CD40 interactions deliver critical costimulatory signals that promote GC B cell survival, proliferation, and differentiation [37]. Simultaneously, Tfh-derived IL-21 acts as a key growth and differentiation factor for GC B cells, enhancing their proliferation and antibody secretion capacity [37]. The synergy between CD40 signaling and IL-21 reception induces c-Myc and p-S6 in GC B cells, further stimulating their selection within the GC [37].
Recent research has revealed a novel bidirectional help mechanism, with GC B cells providing essential IL-1β signals back to Tfh cells via canonical NLRP3 inflammasome activity [37]. This IL-1β signaling is required for optimal Tfh cell function, including IL-21 production and proper follicular trafficking. Genetic ablation of IL-1β production specifically in B cells results in significant reduction of both GC B cells and Tfh cells following influenza infection, demonstrating the mutual dependence of these cell populations within the GC microenvironment [37].
Tfh Cells in Extrafollicular Responses
EF Responses as Distinct B Cell Activation Pathways
Extrafollicular B cell responses represent an alternative pathway of B cell activation that occurs outside the follicle, typically in the splenic red pulp, medullary cords of lymph nodes, or bridging channels [3] [4]. Unlike GC responses that require days to establish, EF responses generate antibody-secreting cells within days of antigen encounter, providing rapid humoral immunity while the slower GC response develops [3]. While initially regarded as a source of fast but lower-quality antibodies, recent evidence establishes that EF responses constitute an important component of protective humoral immunity, particularly during various viral and bacterial infections [3].
EF responses can occur in response to both T-dependent and T-independent antigens, though T cell help significantly enhances their magnitude and quality [3]. When T cells are present, they greatly enhance the quantity and quality of the EF response, leading to more robust expansion of antigen-experienced B cells, increased isotype switching, and even somatic hypermutation in EF foci [3]. Studies in autoimmune models demonstrate that EF responses can generate pathogenic B cell clones, highlighting their clinical relevance beyond protective immunity [39].
Distinct Nature of Tfh Help in EF Responses
While the full characterization of Tfh cells specialized for EF help remains incomplete, emerging evidence suggests important differences from GC-Tfh cells. EF responses are associated with specific inflammatory contexts, particularly those involving type 1 inflammatory cytokines such as IL-12 and IFNγ, which promote EF responses while suppressing GC formation [3]. This cytokine environment likely shapes the functional properties of Tfh cells participating in EF responses.
The anatomical location of T-B interactions early in immune responses influences the resulting differentiation pathway. Research has identified the interfollicular zone as a site where both GC B cell and Tfh cell differentiation initiates, with antigen-specific T and B cells forming long-lived interactions and upregulating Bcl6 in this location [36]. Notably, in the absence of cognate B cells, Tfh cells can still form and migrate to the follicle, but they fail to maintain characteristic high expression of PD-1, ICOS, and GL7, demonstrating that B cells are required for maintaining the fully differentiated Tfh phenotype but not their initial differentiation or follicular migration [36].
Table 2: Comparison of Tfh Functions in EF vs GC Pathways
| Parameter | Extrafollicular Response | Germinal Center Response |
|---|---|---|
| Timing | Early (days 2-6) | Late (peaks day 8+, can persist months) |
| Primary Signals | IL-12, IFNγ, ICOS | IL-6, IL-21, ICOS, CD40L |
| B Cell Outcomes | Short-lived plasmablasts, early antibody secretion | Memory B cells, long-lived plasma cells |
| Antibody Affinity | Limited affinity maturation | Extensive affinity maturation |
| Key Tfh Cytokines | IL-21 (lower/sustained) | IL-21 (high), IL-4 |
| Location | Medullary cords, red pulp, bridging channels | B cell follicle, GC light zone |
Experimental Approaches for Studying Tfh Cell Help
Key Methodologies and Workflows
Investigating Tfh cell help in both EF and GC pathways requires integrated approaches combining in vivo models, cellular assays, and advanced analytical techniques. The following experimental protocols represent methodologies cited in key studies of Tfh biology:
Influenza Infection Model for GC Tfh Studies:
- Immunize mice with influenza A virus (e.g., PR8 strain)
- Harvest draining lymph nodes and spleen at defined timepoints (days 7-14 for peak GC response)
- Prepare single-cell suspensions and enrich for CD4+ T cells
- Analyze Tfh cells by flow cytometry (CXCR5+PD-1+ICOS+Bcl6+)
- Sort GC-Tfh cells for functional assays or transcriptomic analysis
- For in vivo functional studies, utilize adoptive transfer of antigen-specific T cells (e.g., OT-II) or B cell-specific IL-1β knockout models [37]
Fate-Mapping Tfh Development:
- Use IL-21 fate-mapping mice (Il21Cre × Rosa26Lox-STOP-Lox-YFP)
- Immunize with NP-OVA in appropriate adjuvant
- Analyze Tfh progenitor (YFP-) and Tfh-Full (YFP+) populations by flow cytometry at days 5, 8, and 11 post-immunization
- Sort populations for bulk RNAseq or single-cell transcriptomics
- Assess historical IL-21 production in relation to phenotypic markers and functional capacity [35]
EF Response Analysis in Autoimmunity Models:
- Utilize spontaneous autoimmune models (e.g., lupus-prone mice) or immunization protocols
- Perform serial blood collection and tissue harvesting during disease progression
- Analyze EF foci in spleen sections by immunohistochemistry
- Characterize atypical B cells (CD11c+T-bet+) and antibody-secreting cells by flow cytometry
- Conduct single-cell RNA sequencing of peripheral blood mononuclear cells to identify EF-associated transcriptional signatures [39]
Research Reagent Solutions
Table 3: Essential Research Reagents for Tfh Cell Studies
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Mouse Models | IL-21 fate-mapping mice, Bcl6fl/fl mice, OT-II TCR transgenic mice | Genetic fate-mapping, conditional gene deletion, tracking antigen-specific responses |
| Flow Cytometry Antibodies | Anti-CXCR5, anti-PD-1, anti-ICOS, anti-Bcl6, anti-IL-21 | Identification and characterization of Tfh subsets |
| Cytokine Detection | IL-21 ELISA, IL-4 reporter mice, IL-1β intracellular staining | Quantification of Tfh-derived cytokines and signaling |
| Antigens/Adjuvants | NP-OVA, NP-Ficoll, influenza virus, SARS-CoV-2 spike protein | Immunization models for GC and EF responses |
| Single-Cell Analysis | 10X Genomics platform, CITE-seq antibodies, scRNAseq reagents | High-resolution analysis of Tfh heterogeneity and differentiation states |
Signaling Pathways and Molecular Interactions
The diagram below illustrates the key signaling pathways and cellular interactions involved in Tfh cell differentiation and function across EF and GC pathways:
Diagram 1: Tfh Cell Differentiation and Help in EF and GC Pathways. The diagram illustrates the multi-stage differentiation of Tfh cells from naive CD4+ T cells and their subsequent roles in providing help to B cells through extrafollicular and germinal center pathways. Key signaling molecules and cellular interactions are shown.
Quantitative Analysis of Tfh Cell Responses
Table 4: Quantitative Parameters of Tfh Cell Responses in EF vs GC Pathways
| Response Parameter | Extrafollicular Response | Germinal Center Response | Measurement Techniques |
|---|---|---|---|
| Peak Response Time | Days 2-6 post-immunization | Days 8+ (can persist 6+ months) | Longitudinal flow cytometry, fate mapping |
| Tfh Frequency | Variable (context-dependent) | ~65% of total Tfh at peak [35] | Flow cytometry, scRNAseq |
| IL-21 Production | Lower/sustained | High (defining feature of Tfh-Full) [35] | IL-21 reporter mice, intracellular staining |
| Clonal Persistence | Short-lived (days-weeks) | Long-lived (6+ months without exhaustion) [38] | TCR sequencing, longitudinal tracking |
| B Cell Outcome Ratio | Predominantly plasmablasts | Memory B cells & long-lived plasma cells | ELISPOT, flow cytometry, adoptive transfer |
Discussion and Future Perspectives
The dual role of Tfh cells in supporting both EF and GC pathways highlights their remarkable functional plasticity in adapting to different immunological contexts. Rather than representing a uniform population, Tfh cells exist as a spectrum of differentiation states with distinct functional capacities tailored to the specific needs of EF versus GC responses. The inflammatory environment, particularly cytokines like IL-12 and IL-6, appears to be a critical determinant in steering Tfh cells toward EF- or GC-specialized helper functions [3].
From a therapeutic perspective, modulating Tfh cell activity represents a promising approach for enhancing vaccine efficacy or treating antibody-mediated diseases. Strategies that promote GC-Tfh responses could improve long-term immunity in vaccines against challenging pathogens like HIV and influenza [38]. Conversely, interventions that selectively inhibit pathogenic EF-Tfh responses without compromising GC reactions could prove beneficial for autoimmune conditions like idiopathic nephrotic syndrome or systemic lupus erythematosus [39].
Future research should focus on better characterizing the molecular signatures distinguishing Tfh cells specialized for EF versus GC help, identifying surface markers that enable selective targeting of these subsets, and understanding how Tfh cell functional states are maintained during prolonged immune responses. The development of more sophisticated fate-mapping tools and single-cell multi-omics approaches will be crucial for these efforts, potentially revealing new opportunities for therapeutic intervention across a range of infectious and immune-mediated diseases.
Tools for Deconvolution: Profiling BCR Repertoires and Cellular Dynamics in Research & Drug Discovery
Single-cell multi-omics technologies represent a transformative approach for dissecting the complexity of B cell responses, enabling the simultaneous measurement of various molecular layers within individual cells. These methodologies allow researchers to capture the multidimensional aspects of single-cell transcriptomes, immune repertoires, and proteomic data in diverse spatiotemporal contexts [40]. The integration of B cell receptor (BCR) sequencing with transcriptomic and proteomic data is particularly valuable for understanding the functional dynamics of B cell responses, especially in distinguishing between the extrafollicular and germinal center pathways that underpin humoral immunity. By concurrently analyzing gene expression profiles, surface protein markers, and clonal BCR sequences from the same single cells, researchers can establish direct links between B cell clonality, cellular identity, functional state, and antigen experience history. This integrated approach provides unprecedented resolution for mapping the developmental trajectories of B cells and identifying the molecular switches that direct their differentiation toward extrafollicular or germinal center fates—critical insights for both vaccine development and therapeutic interventions in autoimmune diseases [12].
Technological Foundations of Single-Cell Multi-Omics
Core Methodologies for Data Generation
The foundation of single-cell multi-omics lies in technologies that enable the capture and analysis of multiple molecular modalities from individual cells. Single-cell RNA sequencing (scRNA-seq) enables the unbiased characterization of gene expression programs, while BCR sequencing (scBCR-seq) effectively delineates the repertoires of B cells, revealing their clonal diversity and antigen exposure history [40]. The integration with single-cell proteomics, exemplified by CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing), enriches the information with protein data that may show both similarities and discrepancies with the transcriptome [40]. These approaches typically utilize microfluidic platforms for partitioning individual cells into nanoliter-scale droplets or wells, where molecular barcoding occurs. Each cell receives a unique cellular barcode, while individual mRNA transcripts and amplified V(D)J segments are labeled with unique molecular identifiers (UMIs) to enable accurate quantification and to distinguish biological signals from technical noise [41]. The simultaneous capture of transcriptomic, proteomic, and BCR data from the same single cell creates a multidimensional dataset that can reveal coordinated changes across molecular layers and provide mechanistic insights into B cell behavior and regulation.
Table 1: Core Single-Cell Multi-Omics Technologies for B Cell Analysis
| Technology | Molecular Target | Key Outputs | Applications in B Cell Biology |
|---|---|---|---|
| scRNA-seq | mRNA transcripts | Gene expression profiles, cell states and subtypes | Identification of B cell subsets (naive, memory, plasma cells), metabolic states, activation pathways |
| scBCR-seq | Rearranged V(D)J segments of immunoglobulin genes | BCR clonotype, V/J gene usage, somatic hypermutation, clonal relationships | Tracing B cell lineages, tracking clonal expansion, assessing affinity maturation, distinguishing naive from memory B cells |
| CITE-seq | Surface proteins | Abundance of specific proteins (e.g., CD markers) | Validation of cell identity, analysis of protein-level expression correlated with transcriptome, identifying rare populations |
| scATAC-seq | Accessible chromatin regions | Epigenetic landscape, regulatory elements, transcription factor binding sites | Understanding gene regulatory mechanisms driving B cell differentiation and function |
Experimental Workflow for Integrated BCR and Multi-Omics Analysis
The standard workflow for generating single-cell multi-omics data with BCR integration involves a coordinated series of wet-lab and computational steps. First, a single-cell suspension is prepared from the tissue of interest (e.g., peripheral blood, lymph nodes, or tumor tissue). The cells are then loaded into a single-cell partitioning system, such as the 10x Genomics Chromium or BD Rhapsody platform, where individual cells are encapsulated in droplets or microwells along with barcoded beads. Inside these partitions, the cells are lysed, and the released RNA, DNA (for V(D)J sequencing), and antibodies bound to oligonucleotide tags (for CITE-seq) hybridize to the barcoded beads. Following reverse transcription and library construction, separate libraries are generated for gene expression, BCR sequences, and surface proteins [41] [40]. These libraries are sequenced simultaneously, and the resulting data undergoes demultiplexing, alignment, and quality control before integration.
Diagram 1: Single-Cell Multi-Omics Workflow. The integrated experimental process from cell preparation to data generation, showing parallel library preparation for different molecular modalities.
Data Integration and Analytical Approaches
Computational Integration of Multi-Modal Data
The analysis of single-cell multi-omics data requires specialized computational approaches to integrate the different molecular modalities captured from the same cells. Tools for analyzing these datasets are primarily written in R and Python, with R-based representative software including Seurat and SingleCellExperiment, while Python-based representative software includes Scanpy and AnnData [40]. The general analysis workflow begins with quality control to filter out doublets, cells with high mitochondrial content, and other low-quality cells. Following data preprocessing, the integration of transcriptomic, proteomic, and BCR data enables the identification of distinct cell populations and states. For B cell analysis specifically, the BCR information can be used to track clonal relationships across different cell subtypes identified through transcriptomics and proteomics. This allows researchers to determine whether particular BCR clonotypes are enriched in certain functional subsets, such as extrafollicular responders versus germinal center B cells [39]. Advanced analytical techniques including trajectory inference, gene regulatory network reconstruction, and cell-cell communication analysis can then be applied to the integrated data to extract biological insights about B cell differentiation and function in various immunological contexts.
Table 2: Key Analytical Tools for Single-Cell Multi-Omics Data Integration
| Tool | Programming Language | Primary Function | Application in B Cell Studies |
|---|---|---|---|
| Seurat | R | Data integration, clustering, visualization, differential expression | Identifying B cell subsets, analyzing gene expression changes across conditions, integrating protein and BCR data |
| Scanpy | Python | Clustering, trajectory inference, differential expression | Large-scale data processing, pseudotime analysis of B cell differentiation |
| SingleCellExperiment | R | Data structure for storing and manipulating single-cell data | Organizing multi-modal B cell data for analysis |
| CellTypist | R/Python | Automated cell type annotation | Rapid classification of B cell subsets using reference datasets |
| Monocle | R | Trajectory inference and pseudotime analysis | Reconstructing B cell differentiation pathways from naive to memory/plasma cells |
BCR-Centered Integration Strategies
The integration of BCR sequencing data with other modalities enables particularly powerful analyses for understanding B cell biology. By combining BCR clonotype information with gene expression profiles, researchers can track how specific B cell clones expand, differentiate, and acquire functional capabilities during immune responses. This approach can reveal whether clonally expanded B cells display distinct transcriptional programs compared to non-expanded B cells, and whether certain clones are preferentially directed toward extrafollicular versus germinal center pathways. In a study of childhood idiopathic nephrotic syndrome, researchers used this integrated approach to demonstrate that an extrafollicular B cell response marked by the expansion of atypical B cells (atBCs) and antibody-secreting cells represented the major immune perturbation [39]. The analysis revealed that these expanded clones exhibited a transcriptional program poised for effector functions, with elevated expression of genes involved in BCR signaling, antibody production, and metabolic pathways supporting plasma cell differentiation. Such integrated analyses provide direct evidence of the functional correlates of clonal expansion and can identify pathogenic B cell subsets in autoimmune and infectious diseases.
Applications in Distinguishing Extrafollicular versus Germinal Center B Cell Responses
Molecular Signatures of B Cell Differentiation Pathways
Single-cell multi-omics approaches have dramatically advanced our understanding of the distinct molecular programs associated with extrafollicular versus germinal center B cell responses. Extrafollicular responses are characterized by the rapid generation of short-lived plasmablasts with lower degrees of somatic hypermutation, while germinal center responses typically produce long-lived plasma cells and memory B cells with higher levels of affinity maturation [12]. Through integrated transcriptomic and proteomic profiling, researchers have identified specific markers associated with each pathway. Extrafollicular B cells often exhibit upregulation of genes involved in inflammatory signaling (e.g., T-bet encoded by TBX21), surface markers such as CD11c, and a metabolic program favoring oxidative phosphorylation and fatty acid oxidation to support rapid differentiation into antibody-secreting cells [39]. In contrast, germinal center B cells maintain expression of markers like BCL6, CD38, and AICDA (activation-induced cytidine deaminase), which is essential for somatic hypermutation and class-switch recombination. The integrated analysis of BCR sequences further reveals differential patterns of somatic hypermutation between these pathways, with germinal center-derived clones typically showing higher levels of V region mutations indicative of affinity maturation.
Diagram 2: B Cell Differentiation Pathways. Key transcriptional and functional differences between extrafollicular and germinal center responses, revealed through single-cell multi-omics.
Case Study: Extrafollicular B Cell Responses in Human Disease
A compelling application of single-cell multi-omics in distinguishing B cell response pathways comes from studies of childhood idiopathic nephrotic syndrome (INS). Using scRNA-seq coupled with BCR analysis and protein validation, researchers demonstrated that active INS is characterized by a pronounced extrafollicular B cell response marked by the expansion of atypical B cells (atBCs) and antibody-secreting cells [39]. The integrated analysis revealed that INS B cells exhibited a distinct transcriptional signature with upregulation of 642 genes compared to healthy controls, including genes involved in BCR signaling (SYK, BTK), B cell activation (CD27, TNFRSF13B), antibody production (IGHG1, IGHG3, IGHA1), and metabolic pathways supporting plasma cell differentiation. Proteomic validation confirmed the expansion of proliferating rituximab-sensitive extrafollicular (CXCR5–) CD21low T-bet+ CD11c+ atBCs and short-lived T-bet+ antibody-secreting cells in INS patients. This study not only identified the extrafollicular origin of humoral immunity in INS but also demonstrated how single-cell multi-omics can reveal previously unrecognized pathogenic B cell subsets in human disease, with important implications for understanding disease mechanisms and developing targeted therapies.
Essential Research Reagents and Technical Considerations
Research Reagent Solutions for B Cell Multi-Omics
Table 3: Essential Research Reagents and Platforms for Single-Cell B Cell Multi-Omics
| Reagent/Platform | Function | Key Features | Application Context |
|---|---|---|---|
| 10x Genomics Chromium | Single-cell partitioning | High-throughput, simultaneous 5' or 3' gene expression and V(D)J profiling | Simultaneous capture of transcriptome and paired BCR sequences from thousands of single B cells |
| BD Rhapsody | Single-cell partitioning | Combinatorial barcoding, compatible with mRNA, protein, and BCR analysis | Targeted analysis of B cell populations with integrated protein and BCR data |
| Parse Evercode assays | Whole transcriptome analysis | Combinatorial barcoding, cost-effective for large sample numbers | Large-scale B cell studies requiring high sample throughput |
| CITE-seq antibodies | Protein detection | Oligonucleotide-tagged antibodies for surface protein quantification | Multiplexed protein marker detection alongside transcriptome and BCR data |
| Cell Hashtag Oligos (HTOs) | Sample multiplexing | Antibody-based sample barcoding for sample pooling | Pooling multiple samples in one run to reduce batch effects and costs |
| VDJ enrichment primers | BCR amplification | Target-specific primers for immunoglobulin loci | Efficient amplification of rearranged V(D)J segments for BCR sequencing |
Technical Considerations and Methodological Challenges
While single-cell multi-omics offers powerful insights into B cell biology, several technical challenges must be addressed in experimental design and data interpretation. The quality of starting material is critical, as cell viability and sample preparation can significantly impact data quality. Methods for single-cell isolation include fluorescence-activated cell sorting (FACS), magnetic-activated cell sorting (MACS), and microfluidic technologies, each with trade-offs in throughput, viability, and cost [41]. Batch effects represent another significant challenge, as technical variations between experiments can confound biological signals. Computational approaches such as Seurat's canonical correlation analysis (CCA), mutual nearest neighbors (MNN), or Harmony can help correct for these effects [40]. For BCR-specific analyses, the sensitivity of V(D)J capture is crucial, as low capture efficiency may miss important clonal relationships. The integration of different data modalities also presents analytical challenges, as each data type has distinct statistical properties and noise structures. Finally, the high cost of single-cell multi-omics and the computational resources required for data analysis and storage remain significant considerations for study design. As noted by bioinformatics experts, "High cost and batch effects remain the major obstacles for large cohort studies on scRNA-seq" [40], though technological advances are gradually addressing these limitations.
Diagram 3: Key Considerations in B Cell Multi-Omics Studies. Critical factors across the experimental and analytical workflow that impact data quality and biological interpretation.
The integration of BCR sequencing with transcriptomic and proteomic data at single-cell resolution represents a powerful approach for elucidating the complexities of B cell biology, particularly in distinguishing the molecular signatures and functional outcomes of extrafollicular versus germinal center responses. As these technologies continue to evolve, they will undoubtedly yield deeper insights into the mechanisms governing B cell differentiation, activation, and pathogenicity across infectious diseases, autoimmunity, and cancer immunotherapy. The comprehensive view provided by single-cell multi-omics will facilitate the identification of novel therapeutic targets and biomarkers, ultimately advancing the development of more precise and effective immunomodulatory treatments.
B-cell receptor repertoire sequencing (BCR-Seq) has emerged as a powerful methodology for interrogating the dynamics of humoral immunity under physiological and pathological conditions. Each B cell possesses a unique BCR generated through somatic recombination of variable (V), diversity (D), and joining (J) gene segments, with the collective entirety of these receptors throughout an organism forming the "BCR repertoire" [42]. Comprehensive analysis of this repertoire provides critical insights into the pathogenesis of immune-mediated diseases and can inform diagnostic and therapeutic strategy development.
A fundamental framework for interpreting BCR repertoire data involves understanding the two primary pathways of B cell activation: the germinal center (GC) response and the extrafollicular (EF) response. Canonical immune responses feature a GC reaction where B cells undergo extensive somatic hypermutation (SHM) and affinity maturation, producing long-lived plasma cells and memory B cells [12]. In contrast, extrafollicular responses involve rapid B cell proliferation and differentiation into short-lived plasmablasts at sites outside follicles, with limited somatic hypermutation and lower affinity antibodies [12] [39]. Recent evidence indicates that in several immune contexts, including autoimmune diseases like systemic lupus erythematosus (SLE) and childhood idiopathic nephrotic syndrome (INS), as well as certain infections, the extrafollicular response becomes dominant, often correlating with pathogenic outcomes [39] [43]. Therefore, BCR repertoire analysis must be contextualized within these distinct differentiation pathways to accurately decipher their functional implications in health and disease.
Methodological Foundations of BCR Repertoire Sequencing
Sequencing Technology Platforms
The choice of sequencing technology profoundly impacts the resolution and type of information that can be extracted from BCR repertoire studies. Each mainstream technology offers distinct advantages and limitations for specific research applications.
Table 1: Comparison of BCR Repertoire Sequencing Technologies
| Technology | Key Features | Advantages | Limitations | Primary Applications |
|---|---|---|---|---|
| Sanger Sequencing | Low-throughput; clones isolated from display libraries | Gold standard for clinical DNA sequencing; high accuracy | Limited throughput; cannot sequence large DNA fragments quickly | B cell/CDR3 spectratyping; detection of BCR-ABL1 mutations [42] |
| Next-Generation Sequencing (NGS) | High-throughput; millions of V(D)J sequences simultaneously | Cost-effective; fast; detailed nucleotide-level examination of repertoires | Limited read length; difficulty recognizing novel chromosomal aberrations | Clonality assessment; minimal residual disease detection; repertoire analysis of BCR IG genes [42] |
| Single-Cell RNA Sequencing | Full-length sequences of IgH and IgL chains from individual cells | Reveals paired heavy/light chains; couples BCR sequence with transcriptomic data | Computationally intensive; higher cost per cell | Reconstruction of immune repertoire; analysis of BCR correlations with cell state [42] [44] |
Template Selection and Experimental Design
The selection of the starting template represents a critical decision point in BCR repertoire study design, as it defines the scope, sensitivity, and interpretability of the resulting data [45].
Genomic DNA (gDNA): Provides a stable template that captures both productive and nonproductive TCR or BCR rearrangements, making it suitable for estimating the total diversity of the immune repertoire. Since a single template is assigned to each cell, gDNA is ideal for clone quantification and analyzing relative clonotype abundance. However, gDNA-based approaches do not provide information on transcriptional activity and may not reflect functional immune responses [45].
RNA/cDNA: Messenger RNA templates directly represent the actively expressed repertoire, focusing on functional clonotypes. This makes RNA optimal for studies aiming to understand the immune system's dynamic responses. While RNA is less stable than gDNA and prone to biases during extraction and reverse transcription, these concerns have diminished with the rising prevalence of single-cell RNA sequencing, which can accurately identify even rare mutations [45].
Complementary DNA (cDNA), synthesized from mRNA, serves as a common template for high-throughput sequencing, retaining the functional relevance of mRNA while offering improved stability for experimental workflows [45].
Bioinformatics Processing Pipeline
The analysis of BCR repertoire sequencing data requires specialized bioinformatics pipelines to transform raw sequencing reads into biologically interpretable results. The typical workflow consists of three major stages [46]:
Pre-processing: This initial stage transforms raw sequencing reads into error-corrected BCR sequences. Key steps include:
- Quality control and read annotation using tools like FastQC
- Primer identification and masking
- Handling of unique molecular identifiers (UMIs) for error correction
- Assembly of paired-end reads
- This stage typically utilizes specialized toolkits such as pRESTO/Change-O [46]
Determination of Population Structure: This stage involves:
- V(D)J assignment using tools like IgBLAST or IMGT HighV-QUEST
- Detection of novel alleles
- Clonal assignment by grouping sequences with the same V and J genes and identical CDR3 lengths
- This stage outputs an IMGT-style clonal table for downstream analysis [46]
Repertoire Analysis: The final stage performs in-depth characterization of repertoire properties through:
- Clonality and diversity assessment (using D20/D50 indices)
- CDR3 spectratyping
- Somatic hypermutation analysis
- Lineage tree construction
- Selection analysis
- Identification of stereotyped or convergent responses [46]
Figure 1: BCR Repertoire Analysis Bioinformatics Workflow. This pipeline transforms raw sequencing reads through three major stages into biologically interpretable repertoire characteristics.
Core Analytical Framework: Clonality, SHM, and VDJ Usage
Clonality Assessment in EF versus GC Responses
Clonality analysis measures the distribution and abundance of B cell clones within a repertoire, providing critical insights into antigen-driven selection and expansion. In healthy individuals, the BCR repertoire typically exhibits high diversity, with a polyclonal distribution where no single clone dominates. During immune responses, antigen-specific B cells undergo clonal expansion, leading to reduced diversity and increased clonality [42].
The pattern of clonal expansion differs markedly between extrafollicular and germinal center responses:
Extrafollicular responses are characterized by focused clonality with limited diversity. These responses generate short-lived plasmablasts through rapid proliferation without extensive diversification. In autoimmune conditions like childhood idiopathic nephrotic syndrome, this manifests as expansion of CD21low CXCR5- T-bet+ CD11c+ atypical B cells (atBCs) and T-bet+ antibody-secreting cells [39]. EF responses can persist for weeks to months, continually replenished by recruitment of new B cell blasts, as seen in chronic infections and spontaneous autoimmunity [12].
Germinal center responses produce diversified clonality within expanded lineages. GC B cells undergo rapid proliferation (3-4 divisions per day) but maintain relatively constant cell numbers after peak expansion, with most division balanced by cell death rather than differentiation [12]. This process generates substantial clonal diversity through somatic hypermutation, creating a repository of variants from which high-affinity clones can be selected.
Table 2: Clonality Patterns in EF versus GC Responses
| Feature | Extrafollicular Response | Germinal Center Response |
|---|---|---|
| Clonal Diversity | Focused, limited diversity | Diversified within expanded lineages |
| Time Course | Rapid onset (days); can persist weeks to months | Slower onset (peaks at ~2 weeks); typically self-limiting |
| B Cell Subsets | Atypical B cells (atBCs), marginal zone-like B cells | Centroblasts, centrocytes, memory B cells |
| Metabolic Program | Aerobic glycolysis, oxidative phosphorylation [12] | Fatty acid oxidation via oxidative phosphorylation [12] |
| Pathological Associations | SLE, childhood INS, acute COVID-19 [12] [39] | Chronic infections, protective vaccine responses |
Somatic Hypermutation Analysis
Somatic hypermutation (SHM) introduces point mutations into the variable regions of immunoglobulin genes at an exceptionally high rate—approximately one mutation per B cell subclone in the relevant locus [42]. This process is primarily associated with germinal center responses, where it works in tandem with affinity-based selection to generate high-affinity antibodies.
Analysis of SHM patterns provides crucial insights into the history of antigen exposure and selection pressure:
Mutation Frequency: Calculated as the total number of mutations in V region genes divided by the number of sequenced bases. GC-derived B cells typically show >5% mutation rates in their variable regions [46].
Mutation Targeting: SHM exhibits intrinsic biases, preferentially targeting certain motifs (e.g., RGYW/WRCY) in the immunoglobulin variable region. Analysis of these patterns can help distinguish between antigen-driven selection and stochastic mutation processes.
Selection Analysis: The ratio of replacement to silent mutations (R/S) in complementarity-determining regions (CDRs) versus framework regions (FWRs) indicates antigen-driven selection. Positive selection is evidenced by higher R/S ratios in CDRs (which directly contact antigen) compared to FWRs (where structural integrity must be maintained) [46].
While SHM is most robust in GC responses, emerging evidence indicates that extrafollicular responses can also support limited somatic hypermutation and affinity maturation. For instance, in Salmonella infection, B cells undergo "promiscuous B cell activation followed by extrafollicular affinity maturation" with measurable SHM [43]. However, EF-derived antibodies generally exhibit lower mutation loads and affinity compared to their GC-derived counterparts.
VDJ Gene Usage Analysis
VDJ gene usage analysis examines the representation of specific variable, diversity, and joining gene segments in the BCR repertoire. The random combinatorial diversity generated by VDJ recombination forms the foundation of BCR diversity, with the theoretical repertoire exceeding 1014 unique receptors [42].
Key aspects of VDJ gene usage analysis include:
Gene Segment Preferences: Identification of overrepresented or underrepresented V, D, and J genes in specific disease states or immune responses.
Public Clonotypes: Detection of identical BCR sequences across different individuals, suggesting common antigen specificities. These shared responses are particularly relevant for vaccine development and understanding stereotyped immune responses in autoimmunity.
Biased Recombination: Assessment of non-stochastic patterns in VDJ rearrangement that may indicate developmental abnormalities or selective pressures.
VDJ usage patterns differ between EF and GC pathways. Extrafollicular responses often show more restricted VDJ usage, reflecting the limited diversification and selection compared to GC responses. For example, in systemic autoimmunity, EF responses frequently exhibit biased VDJ recombination toward autoreactive specificities that have escaped central tolerance [43].
Figure 2: B Cell Differentiation Pathways and Their Association with BCR Features. The extrafollicular and germinal center pathways produce B cells and antibodies with distinct molecular and functional characteristics.
Advanced Integrative Analysis Approaches
Single-Cell Multi-Omic Integration
Recent technological advances enable simultaneous sequencing of BCRs and transcriptomes from single cells, permitting unprecedented resolution in correlating BCR sequence features with cellular states. Tools like Benisse (BCR embedding graphical network informed by scRNA-seq) leverage this multi-modal data to reveal relationships between BCR sequences and B cell transcriptomics [44].
Analysis of 43,938 B cells from 13 scRNA-seq datasets with matched scBCR sequencing demonstrated a positive correlation between BCR sequence similarity and gene expression profile similarity, with B cells in the same clonotype showing much more similar expressions than those from different clonotypes [44]. This integration revealed a gradient of B-cell activation along BCR trajectories and discovered a stronger coupling between BCRs and B-cell gene expression during COVID-19 infections [44].
Lineage Tree Analysis
Lineage tree construction traces the phylogenetic relationships between related B cell clones, visualizing the evolutionary history of an immune response. These trees are built using the pattern of shared and unique somatic mutations across clonally related sequences [46].
BCR lineage analysis has revealed that BCRs can form "a directed pattern of continuous and linear evolution to achieve the highest antigen targeting efficiency," contrasting with the more convergent evolution pattern observed in T-cell receptors [44]. This pattern is particularly evident in HIV-specific B cell lineages, where sequential accumulation of mutations progressively increases antibody breadth and potency over time [44].
Table 3: Key Research Reagent Solutions for BCR Repertoire Studies
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Commercial Library Prep Kits | SMARTer Human BCR IgG/IgM H/κ/λ Profiling Kit (Takara Bio); 10x Genomics Single Cell V(D)J | Targeted amplification of BCR regions for sequencing; single-cell resolution of paired heavy/light chains |
| Primer Sets | Framework-specific primers (FW1, leader region, FW3) | Amplification of specific heavy chain regions; balance between sequence completeness and multiplexing capability [47] |
| Analysis Pipelines | Immcantation (pRESTO/Change-O); MiXCR; ImmuneDB; Cell Ranger (10x) | Processing raw sequencing data to annotated clonal tables; diversity analysis; lineage reconstruction [46] [47] |
| Reference Databases | IMGT/GENE-DB; OGRDB; VDJdb | Germline gene reference; standardized nomenclature; curated receptor specificities [47] |
| Visualization Tools | Immunarch (R); VDJtools (Java/R); scRepertoire (R) | Diversity plots; clonal tracking; repertoire overlap visualization |
BCR repertoire analysis provides a powerful window into the dynamics of adaptive immunity, with distinct patterns emerging from extrafollicular versus germinal center responses. The analytical framework presented here—encompassing clonality, somatic hypermutation, and VDJ usage—enables researchers to decipher these patterns and draw meaningful biological conclusions. As single-cell multi-omic technologies continue to mature and computational methods become more sophisticated, BCR repertoire analysis will play an increasingly central role in understanding immune-mediated diseases, developing targeted therapies, and advancing vaccine design. Contextualizing BCR signatures within the EF/GC paradigm will be essential for accurately interpreting their functional relevance across different immunological contexts.
The adaptive immune response hinges on the activation of B cells, a process initiated by the engagement of the B cell receptor (BCR) with its cognate antigen. Following this encounter, activated B cells can embark on one of two primary differentiation pathways: the extrafollicular (EF) response or the germinal center (GC) response [12] [2]. The EF response is characterized by the rapid generation of short-lived plasmablasts in the splenic bridging channels and lymph node medullary cords, providing a swift, early wave of antibody production [48] [2]. In contrast, the GC response is a more protracted process within B cell follicles, yielding long-lived plasma cells and memory B cells that undergo affinity maturation and somatic hypermutation to provide high-affinity, durable immunity [12]. The precise mechanisms that direct a B cell toward one fate over the other remain a central question in immunology, with profound implications for vaccine design and the treatment of autoimmune diseases and B-cell lymphomas [12] [49] [50].
A significant technical challenge in dissecting these pathways has been the inability to visualize and quantify antigen-specific B cell activation as it occurs within the complex environment of a living organism. Traditional, endpoint histological methods provide only static snapshots, missing the dynamic cellular interactions and signaling events that dictate functional outcomes. This whitepaper details the latest advancements in molecular reporter systems and imaging technologies that are now enabling researchers to track antigen engagement in vivo with unprecedented spatial and temporal resolution. These tools are not only illuminating the fundamental biology of B cell responses but are also critical for the preclinical development of emerging immunotherapies, such as Chimeric Antigen Receptor (CAR) T-cell therapies, where understanding on-target/off-tumor toxicity is paramount [51].
Scientific Context: B Cell Receptor Signaling in Extrafollicular versus Germinal Center Responses
Distinct Pathways and Locations
The divergence between EF and GC responses begins with the initial activation of a naïve B cell. Upon BCR engagement and antigen internalization, the cell migrates to the T-cell–B-cell border, upregulating chemokine receptors like Ebi2 (GPR183) and CCR7 that guide this movement [2]. At this critical juncture, the cell's fate is determined.
- Extrafollicular Response: B cells destined for the EF pathway remain CXCR5-negative and are guided by Ebi2 and CCR7 to EF regions like the splenic bridging channels and lymph node medullary cords [48] [2]. Here, they undergo brisk proliferation and differentiate into Blimp-1+ Bcl6− IRF4hi antibody-secreting cells (ASCs) [2]. This pathway is a major source of early, protective antibodies during acute infections and is also a significant contributor to pathogenic autoantibody production in systemic lupus erythematosus (SLE) [48] [2].
- Germinal Center Response: For entry into the GC pathway, B cells must re-express CXCR5, downregulate Ebi2 and CCR7, and upregulate the master transcriptional regulator Bcl6 [2]. These cells, along with T follicular helper (Tfh) cells, re-enter the follicle to form a GC, a specialized microstructure with dark zones (for somatic hypermutation) and light zones (for affinity-based selection) [48] [12]. The GC is essential for generating high-affinity, class-switched antibodies and memory B cells.
Molecular Switches and Signaling Networks
The decision to pursue an EF or GC fate is influenced by the quality of BCR signaling and T cell help. Dysregulated BCR signaling is a hallmark of B-cell lymphomas, and quantitative modeling has revealed a conserved core signaling network in aggressive lymphomas, featuring intricate feedback and crosstalk, such as a negative crosstalk from p38 to MEK/ERK [49]. Furthermore, expression quantitative trait locus (eQTL) analysis has identified genetic variants that regulate the expression of key BCR signaling components like SYK, VAV2, and PLCG2, potentially influencing disease susceptibility and the nature of the immune response [50].
The following diagram illustrates the key signaling and migratory events in B cell activation and fate determination, integrating the roles of novel reporter systems.
Diagram Title: B Cell Activation and Reporter Integration in EF vs. GC Pathways
Advanced Molecular Reporters for Imaging Antigen Engagement
A new generation of molecular reporters moves beyond constitutive expression to generate signals specifically upon antigen recognition, providing a direct readout of functional engagement.
The SNIPR Inducible Reporter System
A groundbreaking approach for imaging antigen-specific T-cell interactions is the Synthetic Intramembrane Proteolysis Receptor (SNIPR) system, which has direct applicability for studying B-cell responses given shared antigen-recognition domains [51].
- Mechanism: The SNIPR receptor contains an extracellular antigen-recognition domain (e.g., a single-chain variable fragment, scFv), a transmembrane domain from human Notch, and an intracellular transcriptional activator domain (Gal4-VP64). Upon engagement with the target antigen, the receptor undergoes regulated intramembrane proteolysis, releasing the Gal4-VP64 domain. This fragment translocates to the nucleus and drives the expression of a reporter gene cassette [51].
- Key Features: This system is inducible and antigen-dependent. The signal is generated only when the cell encounters its target, making it a powerful tool for mapping productive antigen interactions in vivo, a critical need for safety assessment in CAR T-cell therapy [51].
Diagram Title: Antigen-Dependent SNIPR Reporter System Mechanism
Quantitative Comparison of Reporter Modalities
The choice of reporter gene is crucial and depends on the required sensitivity, depth of imaging, and translational potential. The table below summarizes the key characteristics of commonly used reporter genes.
Table 1: Reporter Genes for In Vivo Immune Cell Tracking
| Reporter Gene | Modality | Signal Trigger | Key Substrate/Probe | Primary Application | Sensitivity & Notes |
|---|---|---|---|---|---|
| Firefly Luciferase | Bioluminescence | ATP + O₂ + Luciferin | D-Luciferin | Preclinical | Very high sensitivity; surface-weighted signal [52]. |
| Renilla/Gaussia Luciferase | Bioluminescence | Coelenterazine | Coelenterazine | Preclinical | Allows for dual imaging with Firefly luciferase [52]. |
| HSV-Thymidine Kinase (HSV-TK) | PET (Positron Emission Tomography) | Phosphorylation of probe | 9-(4-[¹⁸F]fluoro-3-[hydroxymethyl]butyl)guanine ([¹⁸F]FHBG) | Preclinical & Clinical | High spatial resolution; quantifiable; used in SNIPR system [51]. |
| Green/Red Fluorescent Protein (GFP/RFP) | Fluorescence | Light excitation | N/A | Preclinical (in vitro & intravital) | Lower sensitivity for whole-body imaging; ideal for microscopic techniques [52]. |
Detailed Experimental Protocol: Applying the SNIPR System
This protocol outlines the key steps for implementing the SNIPR reporter system to track antigen-specific lymphocyte engagement in vivo, based on the work of Shin et al. [51].
Receptor and Reporter Construct Design
- SNIPR Receptor Engineering: Clone a gene construct encoding, from N- to C-terminus:
- A signal peptide for membrane expression.
- An extracellular scFv domain specific for the target antigen (e.g., anti-HER2, anti-EGFRvIII).
- A truncated CD8α hinge region.
- The transmembrane domain of human Notch1.
- An intracellular domain containing the Gal4-VP64 transcriptional activator.
- Reporter Gene Cassette Design: Clone a separate gene construct where a minimal promoter is upstream of a multiple cloning site, preceded by Gal4 Upstream Activation Sequence (UAS) repeats. Into this site, insert your reporter genes of choice (e.g., HSV-TKSR39 for PET imaging and/or Luciferase for bioluminescence).
Cell Engineering and Validation In Vitro
- Isolation and Activation: Isolate primary human T cells (or relevant B cell lines) from peripheral blood via negative selection. Activate the cells using anti-CD3/CD28 beads and culture in media containing IL-2.
- Viral Transduction: Co-transduce the activated cells with lentiviral vectors carrying the SNIPR receptor and the reporter gene cassette.
- In Vitro Validation:
- Specificity Assay: Co-culture engineered cells with target-positive and target-negative cell lines.
- Flow Cytometry: Assess reporter protein (e.g., GFP) expression.
- Bioluminescence: Measure luciferase activity after adding substrate.
- PET Reporter Assay: Incubate cells with [¹⁸F]FHBG and measure uptake via gamma counting to confirm specific induction only in the presence of the target antigen.
In Vivo Imaging and Analysis
- Animal Model: Establish dual-flank xenograft models in immunodeficient mice by implanting target-antigen-positive tumors on one flank and target-antigen-negative tumors on the contralateral flank.
- Cell Transfer: Adoptively transfer SNIPR-engineered cells into tumor-bearing mice.
- Image Acquisition:
- PET/CT Imaging: At defined time points post-transfer, inject the mouse with [¹⁸F]FHBG. After a suitable uptake period (e.g., 2 hours), anesthetize the animal and acquire static PET/CT images.
- Bioluminescence Imaging (Optional): If a luciferase reporter is included, inject the animal with D-luciferin and acquire optical images.
- Image Analysis: Quantify the PET signal (e.g., as Percentage of Injected Dose per Gram of tissue, %ID/g) in the target-positive and target-negative tumors. A statistically significant higher signal in the antigen-positive tumor indicates specific antigen engagement and reporter induction.
The workflow for this entire process is summarized in the diagram below.
Diagram Title: SNIPR Reporter System Experimental Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Antigen Engagement Studies
| Reagent / Tool | Function / Feature | Example Application |
|---|---|---|
| SNIPR Plasmid System | Modular receptor for antigen-dependent gene induction. | Engineering T or B cells to report specific antigen engagement in vivo [51]. |
| scFv Libraries | Antigen-binding domain for engineered receptors. | Providing target specificity to SNIPR or CAR constructs (e.g., anti-HER2, anti-EGFRvIII) [51]. |
| Multimodality Reporter Constructs | Single construct encoding fluorescent, bioluminescent, and PET reporters. | Enabling cross-validation and tracking of cell location vs. activation state [52]. |
| [¹⁸F]FHBG Radiotracer | PET probe substrate for HSV-TK reporter. | Quantifying the spatial distribution and intensity of reporter gene activation in vivo [51]. |
| Reporter Transgenic Mice | Cell-specific, constitutive reporter expression (e.g., CD2-Firefly Luc). | Tracking bulk cell population dynamics in vivo without adoptive transfer [52]. |
B cell immune responses bifurcate into extrafollicular (EF) and germinal center (GC) pathways, each characterized by distinct metabolic programs essential for their function and output. Activated B cell blasts initiating both pathways utilize high levels of aerobic glycolysis to support rapid clonal expansion. In contrast, germinal center B cells (GCBCs) undergoing affinity maturation fundamentally shift to reliance on oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO). This metabolic switch is governed by B cell receptor (BCR) affinity signals and is critical for the positive selection of high-affinity clones. This whitepaper details the core metabolic distinctions, underlying molecular mechanisms, and essential experimental protocols for investigating immunometabolism in B cell responses, providing a resource for therapeutic development in immunity and autoimmunity.
Upon antigen encounter and T cell help, activated B cells differentiate along one of two primary pathways: the extrafollicular (EF) response or the germinal center (GC) response [12] [2]. The EF response generates early waves of antibody-secreting plasmablasts (PBs), often with minimal somatic hypermutation, and is prominent in acute infections and autoimmune drives [12] [4]. The GC response, a hallmark of T cell-dependent immunity, produces long-lived plasma cells and memory B cells with high-affinity, somatically hypermutated B cell receptors (BCRs) [12] [53].
The functional specialization of these pathways is supported by distinct metabolic programs. EF B cell blasts are characterized by a metabolism that supports rapid biomass generation and proliferation. Conversely, GCBCs adopt a metabolic state that facilitates cyclic re-entry and stringent selection, relying on mitochondrial respiration over glycolysis [12] [54]. Understanding this metabolic divergence is crucial for manipulating immune outcomes, from improving vaccine responses to treating autoimmune diseases where pathogenic EF responses often dominate [12] [55].
Core Metabolic Dichotomy: EF Blasts vs. GC B Cells
The metabolic profiles of EF and GC B cells are tailored to their specific biological roles, with a sharp contrast between the anabolic metabolism of blasts and the oxidative, maintenance-focused metabolism of GCBCs.
Table 1: Comparative Metabolic Profiles of EF Blasts and GC B Cells
| Feature | EF Blasts (Early Activated B Cells) | GC B Cells |
|---|---|---|
| Primary Metabolic Pathway | Aerobic glycolysis & Oxidative Phosphorylation [12] | Oxidative Phosphorylation (OXPHOS) & Fatty Acid Oxidation (FAO) [12] [54] |
| Glucose Utilization | High [12] | Low/Not Detectably Utilized [12] |
| Fatty Acid Metabolism | Not Specified in search results | High uptake and oxidation [12] [54] |
| Oxygen Consumption | High [12] | High [12] |
| Net Biomass Output | High (clonal expansion) [12] | Low/Constant (maintenance) [12] |
| Functional Rationale | Support massive clonal expansion and differentiation [12] | Support survival, cyclic re-entry, and affinity-based selection [12] [54] |
The Metabolic Switch from Activation to the GC
The transition from an activated B cell to a GCBC involves a major metabolic rewiring. Activated B cell blasts, which include precursors for both EF and GC pathways, are large cells that metabolize both glucose and fatty acids, consume oxygen, and undergo aerobic glycolysis [12]. This state supports the generation of new macromolecules necessary for the initial rapid clonal expansion.
Once the GC is established and initial expansion is complete, GCBCs shift their metabolism. They mainly metabolize fatty acids via oxidative phosphorylation and do not detectably utilize glucose or glutamine [12]. This aligns with a state where net cell numbers remain constant, and the metabolic focus shifts from biomass creation to energy for cell cycling and selection processes. A key study demonstrated that OXPHOS is required not only for GCBC clonal expansion but also for the efficient positive selection of high-affinity BCR clones [54].
Molecular Mechanisms and Signaling Pathways
The distinct metabolic programs in EF and GC B cells are orchestrated by a network of transcriptional regulators, signaling pathways, and microenvironmental cues.
Figure 1: Molecular pathways regulating B cell fate and metabolism. High-intensity BCR and CD40 signaling, often from high-affinity antigens, can promote a glycolytic EF fate via HIF-1α and Myc. Concurrently, these signals can also induce Bcl6, which promotes a GC fate by upregulating OXPHOS and the follicle-homing receptor S1PR2.
Transcriptional Control of Metabolism
- Bcl6 and the GC Program: The master GC transcription factor Bcl6 is implicated in promoting the OXPHOS state in GCBCs [12]. Its expression is associated with the suppression of the glycolytic program and the engagement of mitochondrial metabolism.
- HIF-1α and Glycolysis: Hypoxia-inducible factor 1-alpha (HIF-1α) is a key regulator of glycolysis. While the GC was initially thought to be hypoxic, GCBCs do not demonstrate a strong HIF-1α gene expression program, which may contribute to their low glycolytic activity [12] [54]. In contrast, EF blasts likely utilize HIF-1α to sustain their glycolytic metabolism.
- FOXO1: FOXO1 is a critical transcription factor in GC biology, regulating genes involved in cell survival, and its dysregulation is linked to lymphomagenesis [53].
Experimental Protocols for Metabolic Profiling
Investigating B cell metabolism requires a combination of bulk metabolic flux analyses and single-cell technologies that can correlate metabolic state with cell phenotype.
Extracellular Flux Analysis (EFA)
Purpose: To measure real-time glycolytic flux and mitochondrial respiration in live cells [56]. Protocol (Simplified):
- Isolate B cell subsets (e.g., via FACS sorting for naive, activated EF, and GC B cells).
- Seed cells on a specialized microplate pre-coated with a cell adhesion reagent.
- Assay Media: Use a proprietary, serum-free, bicarbonate-buffered assay medium.
- Glycolysis Stress Test:
- Inject Glucose and measure the increase in the Extracellular Acidification Rate (ECAR), indicating glycolysis.
- Inject Oligomycin (ATP synthase inhibitor) and measure the increase in ECAR, indicating glycolytic capacity.
- Inject 2-Deoxy-D-glucose (2-DG), a glycolytic inhibitor, to confirm glycolytic flux.
- Mitochondrial Stress Test:
- Inject Oligomycin and measure the drop in the Oxygen Consumption Rate (OCR), indicating ATP-linked respiration.
- Inject FCCP (mitochondrial uncoupler) and measure the maximal OCR.
- Inject Rotenone & Antimycin A (Complex I & III inhibitors) to measure non-mitochondrial respiration.
Single-Cell Metabolic Regulome Profiling (scMEP) by Spectral Flow Cytometry
Purpose: To simultaneously assess immune phenotype and the expression of key metabolic proteins at single-cell resolution [57] [56]. Protocol (Core Workflow):
- Cell Preparation: Generate single-cell suspensions from spleen or lymph nodes. Include a viability dye (e.g., Zombie NIR).
- Surface Staining: Stain with a panel of antibodies against immune cell markers (e.g., CD19, B220, CD95, GL7, CD38, CD138) to define B cell subsets.
- Intracellular Staining for Metabolic Proteins: Fix and permeabilize cells. Stain intracellularly with a validated antibody panel targeting metabolic pathway regulators.
- Data Acquisition & Analysis: Acquire data on a spectral flow cytometer. Use dimensionality reduction algorithms (e.g., t-SNE, UMAP) to visualize distinct metabolic states of B cell populations.
Figure 2: Experimental workflow for single-cell metabolic profiling of B cells via spectral flow cytometry.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Studying B Cell Metabolism
| Reagent / Tool | Category | Function/Application | Example Use Case |
|---|---|---|---|
| 2-Deoxy-D-Glucose (2-DG) | Glycolysis Inhibitor | Competitive inhibitor of hexokinase, blocks glycolysis. | Short-term in vivo treatment to reduce pathogenic EF/GC B cells in lupus models [55]. |
| Oltipraz | OXPHOS Enhancer | Small molecule that increases oxygen consumption rate (OCR). | In vivo enhancement of OXPHOS to promote affinity maturation in GCs [54]. |
| Seahorse XF Analyzer | Instrument Platform | Measures OCR and ECAR in real-time for metabolic phenotyping. | Functional comparison of mitochondrial respiration and glycolysis in naive vs. activated B cells [56]. |
| AID-Cre-ERT2 Fate Mapping Mice | Genetic Model | Allows indelible labeling of GC-experienced B cells upon tamoxifen administration. | Tracing the fate of GC-derived B cells in recall responses [58]. |
| Anti-Metabolic Protein Antibodies | Detection Reagents | Enable protein-level quantification of metabolic regulators by flow/cyTOF. | Profiling expression of GAPDH, CPT1A, Cytochrome c in B cell subsets via scMEP [57] [56]. |
| LTβR-Ig Fusion Protein | Bioactive Protein | Disrupts GC architecture by blocking lymphotoxin signaling. | Studying the role of persistent GC structures in B cell memory [58]. |
Therapeutic Implications and Future Directions
The metabolic differences between EF and GC B cells present attractive therapeutic targets. In systemic lupus erythematosus (SLE), pathogenic activated B cells and GCBCs show a remarkably high reliance on glucose oxidation for survival [55]. Short-term inhibition of glycolysis with 2-DG selectively reduced these pathogenic subsets and ameliorated disease in lupus-prone mice, highlighting the potential of metabolic intervention [55]. Furthermore, BCMA-expressing GCB cells were found to be particularly dependent on glucose oxidation, suggesting that targeting metabolic vulnerabilities on specific subsets could enhance therapies like CAR-T cells for autoimmune disease [55].
Future work should focus on further elucidating the upstream signals that trigger the metabolic switch, using more sophisticated spatial multi-omics approaches to map metabolism within tissue microenvironments, and developing more specific small molecule modulators of immunometabolism for clinical translation.
The adaptive immune system orchestrates B cell responses through two principal pathways: the germinal center (GC) reaction and the extrafollicular (EF) response. The GC is a specialized microstructure where B cells undergo somatic hypermutation and affinity maturation to produce high-affinity, long-lived immunity [59] [1]. In contrast, the EF response generates a rapid wave of antibody-secreting plasmablasts that provide early but often transient protection [6] [12]. Understanding the molecular switches that direct B cells toward these divergent fates is critical for advancing vaccine design and developing therapies for autoimmune diseases, infections, and B-cell malignancies [6] [12].
This technical guide synthesizes current methodologies for genetically manipulating these pathways in murine models, providing a foundational resource for researchers investigating B cell receptor-mediated immunity within the broader context of humoral immune regulation. We detail specific genetic models, experimental protocols, and analytical frameworks for selectively ablating or modulating GC and EF compartments to delineate their distinct functional contributions to protective and pathogenic immunity.
Biological Foundations of GC and EF Responses
Anatomical and Functional Compartmentalization
GC reactions unfold within secondary lymphoid organs, exhibiting functional compartmentalization into dark zones (DZ) and light zones (LZ). DZ B cells (centroblasts) undergo rapid proliferation and somatic hypermutation, while LZ B cells (centrocytes) engage in affinity-based selection based on signals from follicular dendritic cells and T follicular helper cells [59] [1]. Successful LZ B cells either differentiate into memory B cells or long-lived plasma cells, or recycle to the DZ for further mutation cycles [59].
Conversely, EF responses occur outside follicles in medullary and interfollicular regions, where activated B cells rapidly differentiate into short-lived plasmablasts that secrete early antibodies [60] [12]. While traditionally considered a source of lower-affinity antibodies, recent evidence indicates EF responses can generate protective, pathogen-specific antibodies, particularly during viral infections like influenza [60].
Distinct Molecular Regulators and Metabolic Programs
These pathways employ divergent molecular programs. GC B cells depend on Bcl6, the master transcriptional regulator of GC formation, while EF plasmablasts require Blimp-1 for differentiation [1] [12]. The enzyme activation-induced cytidine deaminase (AID) mediates both somatic hypermutation and class switch recombination in GC B cells [59] [61].
Metabolically, these pathways also differ substantially. Activated B cell blasts in early EF and GC responses utilize both glucose and fatty acids via oxidative phosphorylation and aerobic glycolysis to support biomass accumulation during clonal expansion. In contrast, established GC B cells primarily metabolize fatty acids through oxidative phosphorylation without net biomass increase, consistent with their maintenance programming [12].
Table 1: Key Characteristics of GC and EF Responses
| Feature | Germinal Center (GC) Response | Extrafollicular (EF) Response |
|---|---|---|
| Primary Function | Affinity maturation, long-lived immunity | Rapid antibody production |
| Location | B cell follicles in secondary lymphoid organs | Medullary/extrafollicular regions |
| Key Transcription Factors | Bcl6, IRF8 | Blimp-1, IRF4 |
| Metabolic Program | Fatty acid oxidation, oxidative phosphorylation | Aerobic glycolysis & oxidative phosphorylation |
| Time Course | Peaks at ~2 weeks, can persist months | Emerges within days, often transient |
| Output | Memory B cells, long-lived plasma cells | Short-lived plasmablasts |
| Somatic Hypermutation | Extensive | Limited but possible |
Genetic Models for Pathway Ablation
Germinal Center Ablation Models
Multiple genetic strategies exist for disrupting GC formation. The most direct approach involves Bcl6 deletion using B cell-specific Cre drivers (e.g., Cd19-Cre or Mb1-Cre), as Bcl6 serves as the master regulator for GC development [60]. Similarly, T follicular helper cell ablation through deletion of Bcl6 in CD4+ T cells indirectly disrupts GC formation by eliminating essential help signals [1].
An alternative strategy targets GC-specific signaling molecules. Mice deficient in signaling lymphocytic activation molecule-associated protein (SAP) lack stable T-B cell conjugates necessary for GC initiation. The Mb-1-Cre Bcl6 f/f model represents another effective approach, where Bcl6 is specifically deleted in B cells, preventing GC formation while preserving EF responses, as demonstrated in influenza infection studies [60].
Extrafollicular Response Ablation Models
Targeting EF responses presents greater challenges due to their rapid initiation and dispersed nature. However, several strategic approaches show efficacy:
- IRF4 deficiency: Disrupts plasmablast differentiation, a key EF output
- Blimp-1 deletion: Prevents full plasmablast development
- TLR signaling disruption: Particularly MyD88 deletion, impairs EF response initiation
Research indicates that B cell-intrinsic TLR signaling supports antigen-stimulated B cell survival, clonal expansion, and differentiation toward EF responses through IRF4 induction via NF-κB c-Rel activation [60]. Therefore, mice with B cell-specific MyD88 deletion show impaired EF responses to infections like influenza.
Inducible and Conditional Systems
For temporal control over gene ablation, inducible Cre systems (e.g., hCD20-Cre-ERT2) enable tamoxifen-dependent gene deletion in mature B cells after immunization [61]. This approach is invaluable for studying genes with essential roles in development that would cause embryonic lethality if constitutively deleted.
Table 2: Murine Models for Genetic Ablation of GC and EF Pathways
| Target Pathway | Genetic Model | Key Mechanism | Phenotypic Outcome |
|---|---|---|---|
| GC Ablation | Bcl6-/- (B cell-specific) | Loss of master GC transcription factor | Complete absence of GCs |
| GC Ablation | SAP-/- | Disrupted T-B cell stable interactions | Impaired GC formation |
| GC Ablation | Mb-1-Cre Bcl6 f/f | B cell-specific Bcl6 deletion | Absent GCs, intact EF responses |
| EF Impairment | Irf4-/- | Disrupted plasmablast differentiation | Reduced early antibody secretion |
| EF Impairment | Myd88-/- (B cell-specific) | Impaired TLR signaling | Diminished EF plasmablast formation |
| EF Impairment | Prdm1-/- (Blimp-1 deficient) | Failed plasmablast development | Absent short-lived plasmablasts |
| Dual Modulation | Tfr cell manipulation | Altered GC selection stringency | Modulated GC output, autoimmunity |
Advanced Experimental Approaches
Competitive Adoptive Transfer with CRISPR Editing
Traditional knockout generation is time-consuming and costly. An innovative alternative approach combines ex vivo CRISPR editing with competitive adoptive transfer to rapidly study gene function in GC selection [62].
Diagram: Experimental Workflow for Competitive B Cell Transfer
The protocol involves several key stages:
B Cell Isolation and Editing: Naïve B cells are purified from donor mice (e.g., using B1-8 transgenic models with defined BCR affinities) and transfected with CRISPR-Cas9 ribonucleoproteins targeting the gene of interest via systems like the Neon Transfection System [62].
Competitive Transfer: Edited B cells are mixed with wild-type control B cells of different affinities and adoptively transferred into recipient mice (e.g., 33.C9γ1 mice that cannot mount endogenous responses to the immunizing antigen) [62].
Immunization and Analysis: Recipients are immunized, and GC responses are analyzed by flow cytometry 7-14 days post-immunization. Competitive fitness is assessed by comparing the proportions of edited versus control B cells in GC compartments.
This method was validated by targeting the Fas gene, revealing that FasKO low-affinity B cells accumulated in GCs compared to FasWT low-affinity competitors, though they were still outcompeted by high-affinity B cells, demonstrating a role for Fas in GC selection [62].
Pathway Fate Mapping Using Inflammatory Signals
The decision between EF and GC fates can be manipulated through inflammatory signaling. Research demonstrates that sustained TLR4 stimulation via LPS administration during immunization shifts virus-specific B cells toward EF responses instead of GCs, prompting rapid antibody production [60]. This approach enables researchers to preferentially enhance one pathway over the other without genetic manipulation.
The mechanistic basis involves B cell-intrinsic TLR signaling supporting antigen-stimulated B cell survival and differentiation via IRF4 induction through NF-κB c-Rel activation. This pathway can be harnessed to fate B cells toward EF responses for studying their protective capacity [60].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for GC and EF Response Manipulation
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cre Driver Lines | Cd19-Cre, Mb1-Cre, Cγ1-Cre, hCD20-Cre-ERT2 | Cell/temporal-specific gene deletion |
| CRISPR Components | Cas9 protein, gene-specific gRNAs, Neon Transfection System | Ex vivo gene editing in primary B cells |
| B Cell Media/Stimulation | RPMI-1640 + 10% FCS, CpG (ODN1826), recombinant IL-4 | B cell culture and in vitro activation |
| Flow Cytometry Antibodies | Anti-CD19, CD38, CD24, GL7, CD138, IgD, IgM | Identification of GC/EF B cell subsets |
| Adoptive Transfer Models | B1-8 (B1-8lo/B1-8hi), 33.C9γ1 recipient mice | Competitive GC selection assays |
| TLR Agonists | LPS (TLR4), CpG (TLR9) | Direction of B cells toward EF responses |
| GC Culture System | 40LB cells (CD40L/BAFF-expressing fibroblasts) | In vitro GC differentiation |
Signaling Pathways Governing B Cell Fate Decisions
The decision between GC and EF fates is regulated by integrated signaling networks. Strong BCR signaling coupled with TLR activation promotes IRF4 upregulation, driving plasmablast differentiation and EF commitment [60] [12]. In contrast, moderate BCR signaling with CD40 and cytokine signals (IL-21) promotes Bcl6 expression and GC commitment [59].
Diagram: Molecular Switches in B Cell Fate Decisions
The Fc receptor-like 1 (FcRL1) molecule has emerged as a important regulator of B cell activation. Expressed predominantly on naïve and memory B cells but downregulated in GC B cells, FcRL1 contains ITAM-like motifs that modulate BCR signaling [63]. Upon BCR engagement, FcRL1 co-oligomerizes with BCR within immune synapses, fine-tuning activation thresholds that may influence pathway commitment [63].
Metabolic differences further reinforce these fate decisions, with EF plasmablasts utilizing aerobic glycolysis to support rapid expansion, while GC B cells rely on fatty acid oxidation – metabolic programs aligned with their respective functions of rapid expansion versus sustained mutation and selection [12].
Interpretation and Application of Experimental Results
Analytical Frameworks for Pathway Assessment
When utilizing these ablation models, researchers should employ multiparameter flow cytometry to comprehensively assess both GC and EF compartments. Key GC markers include B220+CD95+GL7+ for murine GC B cells, while EF plasmablasts are typically identified as B220intCD138+ [60]. Histological analysis using immunofluorescence further provides spatial context, revealing GC architecture and EF plasmablast localization.
Functional output assessment should include ELISPOT for antibody-secreting cells from both compartments, serum antibody titers over time, and sequencing of immunoglobulin variable regions to quantify somatic hypermutation – a hallmark of GC responses [60] [12].
Context-Dependent Pathway Contributions
Research reveals that pathway contributions are context-dependent. In systemic autoimmunity models like lupus, EF responses appear dominant for pathogenic autoantibody production, with GC ablation failing to prevent disease [1]. Conversely, during influenza infection, EF responses generate early protective antibodies independently of GCs [60]. These findings highlight the importance of selecting appropriate models for specific research questions.
The competitive adoptive transfer system enables precise quantification of cell-intrinsic fitness defects, allowing researchers to determine whether gene function specifically affects positive selection, clonal expansion, or survival within either pathway [62]. This approach has revealed genes like Fas that specifically regulate GC selection without completely abrogating the response.
Translation to Human Immunity and Therapeutic Development
These murine models provide critical insights for human disease therapy. The discovery that EF responses are prominent in severe COVID-19 and lupus suggests pathway imbalance may underlie disease pathogenesis [1] [12]. Similarly, the success of B cell-depleting therapies in autoimmunity may partly reflect elimination of pathogenic EF responses [64].
Emerging strategies aim to therapeutically modulate pathway balance, such as using TLR agonists to enhance EF responses for rapid protection in vaccines, or developing inhibitors to suppress pathogenic EF responses in autoimmunity [60]. The continued refinement of murine models for pathway-specific manipulation will undoubtedly accelerate these therapeutic advances.
Vaccine adjuvants, long considered mere stimulators of the immune response, are now recognized as master conductors capable of precisely steering B cell fate. The efficacy of modern vaccines, particularly those based on highly purified recombinant antigens, hinges on the inclusion of adjuvants to overcome poor immunogenicity. By strategically engaging innate immune pathways, adjuvants exert profound influence over the fundamental decision point in B cell biology: whether activated B cells pursue extrafollicular (EF) responses that generate rapid but transient protection or commit to germinal center (GC) reactions that yield sophisticated, long-lived immunity [65]. This paradigm shift from simple immune potentiation to precise immunological directioning represents a transformative advance in vaccine science, enabling tailored immune responses optimized for specific pathogens and populations.
The critical balance between EF and GC pathways forms the cornerstone of effective humoral immunity. EF responses provide rapid frontline defense through quickly generated, short-lived plasma cells producing early antibodies, while GC responses enable affinity maturation, class switching, and generation of long-lived plasma cells and memory B cells for sustained protection [12]. The strategic application of adjuvants to bias this fate decision holds particular promise for addressing persistent challenges in vaccinology, including poor responses in immunosenescent populations, inadequate cellular immunity against intracellular pathogens, and the need for broad protection against rapidly evolving viruses.
Molecular Mechanisms: How Adjuvants Influence B Cell Fate Decisions
Fundamental Mechanisms of Adjuvant Action
Adjuvants primarily function through two complementary mechanisms: as delivery systems that enhance antigen presentation or as immunostimulants that directly activate innate immunity [66]. Delivery systems—including emulsions, liposomes, nanoparticles, and mineral salts—prolong antigen bioavailability and facilitate targeted delivery to antigen-presenting cells (APCs) in lymphoid tissues [65]. Immunostimulants, typically pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), engage pattern recognition receptors (PRRs) on APCs to trigger coordinated inflammatory and costimulatory signals [66]. Many modern adjuvant systems combine both approaches to achieve synergistic effects.
The initial encounter between adjuvant and innate immune system creates a cytokine milieu and cellular microenvironment that profoundly influences subsequent B cell fate decisions. Upon activation, APCs upregulate costimulatory molecules (CD40, CD80, CD86) and secrete polarizing cytokines (IL-6, IL-12, IL-21, type I IFNs) that shape the quality and magnitude of helper T cell responses [66]. These primed T cells then provide critical signals to activated B cells, influencing their differentiation toward EF or GC pathways through CD40 engagement, ICOS signaling, and cytokine production [12].
Targeting Pattern Recognition Receptors
Toll-like receptors (TLRs) represent the most extensively exploited PRR class for adjuvant development. TLR4 agonists (e.g., MPL) typically promote Th1-biased responses through MyD88 and TRIF signaling pathways, enhancing CD8+ T cell responses and IgG2 production [65]. TLR9 agonists (CpG oligodeoxynucleotides) trigger robust Th1 and CTL responses via IRF7 and NF-κB activation [66]. TLR7/8 agonists (imidazoquinolines) similarly favor Th1 polarization and strong antibody responses [67]. Intracellular TLRs (TLR3, 7, 8, 9) generally induce type I interferons that promote cross-presentation and Th1 differentiation, while surface TLRs (TLR1, 2, 4, 5, 6) typically drive Th2 or Th17 responses through distinct cytokine profiles [66].
Beyond TLRs, adjuvants targeting NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and C-type lectin receptors (CLRs) offer additional avenues for immune modulation [65]. The specific PRR engagement pattern determines not only the magnitude but also the quality of the resulting immune response, enabling rational adjuvant selection based on desired immunological outcomes.
Table 1: Classification of Vaccine Adjuvants and Their Mechanisms
| Adjuvant Category | Representative Examples | Primary Mechanisms | B Cell Fate Influence |
|---|---|---|---|
| Mineral Salts | Aluminum hydroxide, Aluminum phosphate | Antigen depot, NLRP3 inflammasome activation, enhanced antigen uptake | Favors Th2 responses, limited GC induction |
| Emulsions | MF59, AS03 (α-tocopherol + squalene) | Recruitment of APCs, enhanced antigen uptake, cytokine production | Promotes balanced EF/GC responses; AS03 strongly enhances GC reactions |
| TLR Agonists | MPL (TLR4), CpG (TLR9) | Direct APC activation, costimulatory molecule upregulation, cytokine polarization | TLR4: Th1 bias; TLR9: Strong Th1/CTL, enhanced GC formation |
| Saponin-Based | QS-21, AS01 (MPL+QS-21 liposome) | Direct APC activation, potentially membrane interaction | Strong Th1/CTL, enhanced GC responses and antibody affinity |
| Combination Systems | AS04 (MPL + Alum) | Combined mechanisms: depot + targeted TLR activation | Synergistic effects, enhanced antibody persistence |
Metabolic and Epigenetic Regulation of B Cell Fate
Emerging research reveals that adjuvants exert profound influence over B cell fate through metabolic and epigenetic reprogramming. Activated B cell blasts undergoing clonal expansion utilize both glucose and fatty acids through oxidative phosphorylation and aerobic glycolysis to support biomass generation [12]. In contrast, GC B cells primarily metabolize fatty acids via oxidative phosphorylation, reflecting their maintenance rather than expansion program [12]. Adjuvants that modulate metabolic pathways can therefore directly influence B cell differentiation trajectories.
The STAT5-c-Myc axis has been identified as a critical regulatory node in B cell metabolism following vaccination. Studies comparing SARS-CoV-2 vaccinated individuals to COVID-19 recovered patients revealed elevated expression of pS6, c-Myc, pmTOR, and pSTAT5 in vaccinees, correlating with enhanced BCR signaling and more robust coronavirus-specific immune responses [68]. This metabolic programming creates a permissive environment for GC formation and affinity maturation, demonstrating how adjuvants can architect favorable metabolic conditions for desired B cell fates.
Experimental Evidence: Adjuvant Effects on B Cell Responses
Comparative Adjuvant Studies with Glycoconjugate Vaccines
Systematic comparisons of adjuvant platforms reveal striking differences in their capacity to drive EF versus GC responses. In a comprehensive study evaluating five adjuvants (AS01, AS03, AS04, AS37, and Alum) with Staphylococcus aureus glycoconjugate vaccines, AS03 emerged as particularly potent in enhancing GC reactions [67]. Mice immunized with AS03-adjuvanted vaccines demonstrated significantly higher frequencies of splenic GC B cells, mature memory B cells in lymph nodes and spleen, and long-lived plasma cells in bone marrow compared to other adjuvants [67].
Notably, AS03 induced superior antibody avidity maturation that persisted for at least 25 weeks post-immunization, with effects increasing after each booster dose [67]. This avidity maturation directly correlated with enhanced GC activity, confirming the critical role of GC responses in antibody quality. Interestingly, while all adjuvants enhanced responses to the protein antigen (HlaH35L), profound differences emerged in their effects on polysaccharide antigens, revealing antigen-specific adjuvant preferences [67].
Table 2: Adjuvant Effects on B Cell Responses to Glycoconjugate Vaccines
| Adjuvant | GC B Cell Expansion | Memory B Cell Generation | Bone Marrow Plasma Cells | Antibody Avidity |
|---|---|---|---|---|
| AS01 | Moderate | Moderate | Moderate | Moderate |
| AS03 | High | High | High | High |
| AS04 | Moderate | Moderate | Moderate | Moderate |
| AS37 | Moderate | Moderate | Moderate | Moderate |
| Alum | Low | Low | Low | Low |
| None | Very Low | Very Low | Very Low | Very Low |
Systems Biology Approaches to Adjuvant Evaluation
Advanced analytical technologies have enabled unprecedented resolution in dissecting adjuvant mechanisms. In studies of the heterologous Ad26.ZEBOV, MVA-BN-Filo Ebola vaccine, researchers employed systems immunology approaches to characterize B cell responses with exceptional granularity [69]. Machine learning models trained on blood gene expression signatures accurately predicted antibody response magnitude, demonstrating the predictive power of early immune signatures in forecasting B cell outcomes [69].
Single-cell analyses further identified distinct B cell receptor CDRH3 sequences post-vaccination that resembled known Ebola virus glycoprotein-binding antibodies, providing molecular-level insights into adjuvant-driven repertoire selection [69]. These findings highlight how modern analytical approaches can deconstruct the black box of adjuvant action into defined molecular and cellular events, facilitating more rational adjuvant design and selection.
Experimental Approaches: Methodologies for Evaluating Adjuvant Effects
In Vivo Immunization and Sampling Strategies
Rigorous evaluation of adjuvant effects on B cell fate requires carefully designed immunization and sampling strategies. For comprehensive analysis of both EF and GC responses, staggered sampling timepoints are essential, as EF-derived plasmablasts typically peak at 4-6 days post-immunization while GC responses peak around 2 weeks [12]. The following protocol outlines a standardized approach for comparative adjuvant assessment:
Immunization Protocol:
- Animals: 6-8 week-old female BALB/c mice (n=10-15/group)
- Antigen: Model glycoconjugate vaccine containing Staphylococcus aureus CP5/8 conjugated to tetanus toxoid (2μg each) with HlaH35L toxin (10μg)
- Adjuvant groups: AS01, AS03, AS04, AS37, Alum, and non-adjuvanted controls
- Immunization route: Intramuscular (25μL/leg) at days 0, 28, and 56
- Sample collection: Blood (days 0, 14, 28, 42, 56, 70, 84); Spleen/LNs (days 7, 14, 28, 56, 84); Bone marrow (day 84) [67]
Tissue Processing and Analysis:
- Serum: ELISA for antigen-specific IgG titers and avidity measurements
- Splenocytes/LNs: Flow cytometry for GC B cells (B220+GL7+FAS+), memory B cells (B220+CD38+CD80+CD73+), and plasmablasts (B220intCD138+)
- Bone marrow: ELISpot for long-lived plasma cells
- Histology: Immunofluorescence microscopy of spleen sections for GC enumeration and structure [67]
Molecular and Cellular Characterization Techniques
Advanced molecular techniques provide deep insights into adjuvant-driven B cell differentiation:
Germinal Center B Cell Isolation and Analysis:
- Magnetic enrichment for B220+ cells followed by fluorescence-activated cell sorting (FACS) of GC B cells (B220+GL7+FAS+)
- RNA sequencing for transcriptional profiling of light zone (LZ) versus dark zone (DZ) GC B cells
- B cell receptor sequencing to track clonal expansion and somatic hypermutation [69]
Metabolic Profiling:
- Seahorse XF Analyzer to measure oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)
- 13C-glucose and 13C-glutamine tracing to quantify metabolic flux
- In vivo glucose and fatty acid uptake assays using fluorescent analogs [12]
In vivo Fate Mapping:
- AID-CreERT2 × Rosa26-LSL-YFP mice for tracking GC-derived B cells
- Blimp1-GFP reporters for identifying plasmablast commitment
- Intravenous immunizations with fluorescently-labeled antigens to track antigen-specific B cell responses [2]
Diagram 1: B Cell Fate Decisions Following Antigen Encounter - This diagram illustrates the molecular switches directing B cells toward extrafollicular (red) or germinal center (blue) pathways, highlighting key regulators and cellular outputs.
Table 3: Research Reagent Solutions for Studying Adjuvant Effects
| Reagent/Method | Function/Application | Key Examples |
|---|---|---|
| Adjuvant Systems | Comparative evaluation of adjuvant mechanisms | AS01 (MPL+QS-21 liposome), AS03 (α-tocopherol emulsion), AS04 (MPL+Alum), AS37 (TLR7 agonist+Alum) [67] |
| Flow Cytometry Panels | B cell subset discrimination | GC B cells: B220+GL7+FAS+; Memory B cells: B220+CD38+CD80+CD73+; Plasmablasts: B220intCD138+ [67] |
| Gene Expression Analysis | Transcriptional profiling of B cell responses | Nanostring PanCancer Immune Panel, single-cell RNA sequencing, blood transcriptional modules [69] |
| Metabolic Assays | Assessment of B cell metabolic states | Seahorse XF Analyzer (OCR/ECAR), 13C metabolite tracing, in vivo glucose/fatty acid uptake [12] |
| B Cell Receptor Sequencing | Tracking clonal dynamics and SHM | Single-cell V(D)J sequencing, bulk BCR repertoire analysis, lineage tracing [69] |
| In vivo Modeling | Fate mapping and cellular tracking | AID-CreERT2 × Rosa26-LSL-YFP mice, Blimp1-GFP reporters, intravital microscopy [2] |
Future Perspectives: Next-Generation Adjuvant Design
The frontier of adjuvant science is increasingly focused on precision engineering of immune responses through novel formulation technologies and targeted delivery approaches. Genetic adjuvants—cytokines, chemokines, and immune modulators encoded within DNA, RNA, or viral vectors—represent a promising direction for overcoming limitations of traditional adjuvants, particularly in vulnerable populations [70]. These genetically encoded immune potentiators enable precise spatial and temporal control over immune signaling, potentially overcoming immune senescence and enhancing CD8+ T cell activation in immunocompromised individuals [70].
Systems biology approaches and artificial intelligence are revolutionizing adjuvant development by enabling predictive modeling of adjuvant-immune interactions [65]. Machine learning algorithms trained on high-dimensional immune profiling data can identify early signatures predictive of long-term B cell outcomes, accelerating adjuvant selection and optimization [69]. These computational approaches, combined with single-cell multi-omics, promise to unravel the complex interplay between adjuvant composition, innate sensing, and adaptive immunity, ultimately enabling rational design of adjuvant systems tailored to specific pathogen challenges.
The growing understanding of metabolic checkpoints in B cell differentiation opens new avenues for adjuvant innovation [12] [68]. Small molecule modulators of metabolic pathways such as the STAT5-c-Myc axis may provide complementary approaches to conventional adjuvants for steering B cell fate [68]. Similarly, growing appreciation of epigenetic regulation in B cell memory formation suggests opportunities for epigenetic modulators to enhance vaccine efficacy, particularly in aging populations where epigenetic landscapes may constrain immune responses.
Diagram 2: Comprehensive Workflow for Adjuvant Evaluation - This diagram outlines an integrated experimental pipeline for systematic assessment of adjuvant effects on B cell responses, incorporating multi-platform analyses and computational integration.
As the field advances, the next generation of adjuvants will likely move beyond one-size-fits-all approaches toward precision adjuvants tailored to specific populations, pathogens, and immune challenges. The integration of materials science with immunology promises innovative delivery platforms that control antigen and adjuvant release kinetics, while synthetic biology approaches enable engineering of precisely tuned immune modulators. These advances, coupled with deep learning-based design, will ultimately transform vaccine development, enabling rapid creation of tailored immunization strategies that optimally steer B cell fate for maximal protection.
Dysregulation and Disease: When GC and EF Pathways Go Awry in Autoimmunity and Cancer
Upon antigen encounter, naive follicular B cells face a critical developmental choice: they can either initiate a germinal center (GC) response or rapidly differentiate via the extrafollicular (EF) pathway. The GC pathway generates high-affinity, class-switched antibodies and long-lived memory B cells through somatic hypermutation (SHM) and affinity maturation in a specialized microenvironment. In contrast, the EF pathway produces short-lived antibody-secreting cells (ASCs) that provide rapid but lower-affinity protection against pathogens [71]. The balance between these pathways is crucial for effective immunity while maintaining self-tolerance. However, dysregulation of this balance, particularly through failures in B cell receptor (BCR) checkpoint controls, can lead to the emergence and expansion of autoreactive clones in both pathways, driving autoimmune pathogenesis [71] [72] [1].
The integration of activating and inhibitory signals during the initial 2-3 days post-antigen exposure plays a decisive role in B cell fate determination. Key signaling pathways, especially the PI3K/AKT/mTOR cascade, serve as a central hub that processes signals from the BCR, co-stimulatory molecules, Toll-like receptors (TLRs), cytokines, and inhibitory receptors [71] [73]. The strength and duration of these signals ultimately guide developmental decisions, with dysregulation predisposing individuals to autoimmunity. This whitepaper examines the mechanisms of BCR checkpoint failure in both EF and GC pathways, highlighting their contributions to autoreactive clone emergence and implications for therapeutic intervention.
Signaling Pathways Governing B Cell Fate Decisions
The Central Role of PI3K/AKT/mTOR Signaling
The PI3K/AKT/mTOR pathway integrates multiple signaling inputs to determine B cell fate decisions between GC and EF pathways. As illustrated below, this pathway serves as a central regulatory hub:
This central signaling pathway is regulated by multiple inhibitory mechanisms. FAS (CD95) negatively regulates CD40-mediated PI3K/AKT/mTOR activation in human B cells, serving as a crucial checkpoint against excessive activation [71]. Additional regulators include PTEN, which inhibits PI3K signaling; TRAF3, which limits BAFF-mediated activation; AMPK, which suppresses mTOR activity; and GSK3, which inhibits c-Myc signaling [73]. In autoimmune contexts, these regulatory mechanisms are often compromised, leading to unchecked signaling that promotes autoreactive B cell expansion.
Metabolic Reprogramming in B Cell Fate Determination
Metabolic reprogramming is an essential component of B cell activation and differentiation. Autoreactive B cells in systemic lupus erythematosus (SLE) exhibit heightened glycolytic metabolism and PI3K/AKT/mTOR pathway activity [73]. BAFF (B cell activating factor), which is elevated in SLE patients, promotes glycolysis through Erk1/2 and PI3K/AKT/mTOR signaling, enabling the survival and proliferation of autoreactive B cells that would normally be eliminated [73]. This metabolic dysregulation provides autoreactive clones with the necessary energy and biosynthetic precursors for expansion and differentiation into pathogenic effector cells.
Checkpoint Failures in the Germinal Center Pathway
GC Tolerance Mechanisms
Germinal centers are specialized microstructures where B cells undergo SHM and affinity maturation. This process is inherently risky as random mutations can generate self-reactive clones. To mitigate this danger, GCs incorporate multiple tolerance checkpoints:
- T follicular regulatory (TFR) cells that limit selection and differentiation of self-reactive B cells [1] [43]
- Clonal deletion of overtly self-reactive GC B cells [43]
- Apoptotic mechanisms for removing autoreactive clones [43]
- Receptor editing to rescue moderately self-reactive B cells [43]
The GC microenvironment is organized into dark and light zones that facilitate selective processes. The dark zone hosts proliferating centroblasts that undergo SHM, while the light zone contains follicular dendritic cells (FDCs) that display antigen and T follicular helper (TFH) cells that provide selective signals [1] [43]. B cells with enhanced antigen affinity have a competitive advantage in acquiring TFH cell help, while self-reactive clones are typically eliminated.
GC Checkpoint Failure in Autoimmunity
In autoimmune conditions, GC tolerance mechanisms can fail through multiple mechanisms:
Table: Mechanisms of GC Checkpoint Failure in Autoimmunity
| Failure Mechanism | Consequence | Evidence |
|---|---|---|
| TFR cell deficiency | Reduced suppression of self-reactive B cell selection | TFR cells limit GC selection of self-reactive B cells [1] |
| Excessive TFH cell help | Breach of self-tolerance checkpoints | Roquinsan mice show increased TFH and lupus-like autoimmunity [1] |
| Dysregulated apoptosis | Survival of self-reactive GC B cells | Impaired clearance of autoreactive clones [43] |
| Aberrant AID activity | Increased generation of self-reactive mutations | Elevated SHM in autoimmunity [71] |
Despite these risks, recent evidence challenges the conventional view of GCs as the primary source of pathogenic autoantibodies. Genetic ablation studies demonstrate that GCs are dispensable for systemic autoimmunity in several lupus-prone mouse models [1] [43]. Instead, the extrafollicular pathway appears to play a more dominant role in autoimmune pathogenesis.
Checkpoint Failures in the Extrafollicular Pathway
EF Response Characteristics
The EF pathway enables rapid B cell differentiation into short-lived ASCs, typically producing low-affinity antibodies with few somatic mutations [71]. EF-destined B cells retain EBI2 expression, guiding their migration to extrafollicular sites, while GC-committed B cells downregulate EBI2 and increase CXCR5 expression [71]. EF responses are prominent in certain immunological settings, including antiviral responses, chronic infections, and autoimmunity [71].
Key B cell subsets associated with EF responses include:
- CD21low B cells (also termed atypical memory cells or age-associated B cells)
- CD11c+ T-bet+ B cells
- Double-negative (DN) 2 and DN3 B cells (based on CD21 and CD11c expression) [71]
In SLE patients, DN2 and DN3 B cells constitute a major population of antigen-specific B cells after SARS-CoV-2 vaccination, highlighting the importance of EF populations in human immune responses [71].
EF Checkpoint Failures and Autoreactivity
The EF pathway possesses intrinsically weaker tolerance checkpoints compared to GCs, making it particularly susceptible to autoreactive B cell emergence:
Table: EF Checkpoint Deficiencies in Autoimmunity
| Checkpoint Deficiency | Consequence | Autoimmune Association |
|---|---|---|
| Reduced stringency of negative selection | Survival of autoreactive B cells | Expansion of CD21low B cells in SLE [71] |
| TLR signaling dominance | T cell-independent activation | EF ASC differentiation driven by TLR signaling [71] |
| Altered T cell help requirements | Broader B cell activation | EF TFH cells drive B cell maturation outside GCs [71] |
| Diminished SHM regulation | Altered antibody specificity | SHM observed at EF sites in FAS-deficient mice [71] |
EF responses can generate class-switched and somatically mutated autoantibodies despite their extragerminal center location. In FAS-deficient MRL-lpr mice, EF responses show mutation rates comparable to peaking GC responses [71]. Similarly, in severe SARS-CoV-2 infection, GC formation is suppressed while EF pathways expand, generating self-reactive antiviral IgG1 ASCs with detectable SHM [71].
The inflammatory milieu in autoimmunity further promotes EF responses. Conditions that suppress GC formation while enhancing EF differentiation create an environment where naive B cells with germline-encoded autoreactivity are continuously recruited into the EF pathway [71]. This is particularly evident in SLE, where chronic inflammation favors EF responses over regulated GC reactions.
Experimental Models and Methodologies
Key Research Models and Approaches
The investigation of BCR checkpoint failures employs diverse experimental models and methodologies:
Table: Experimental Models for Studying BCR Checkpoint Failures
| Model/Approach | Application | Key Findings |
|---|---|---|
| Lupus-prone mouse models (MRL-lpr, NZB/W) | Study spontaneous GC formation and autoantibody production | Demonstrated GC-independent autoimmunity [1] [43] |
| BAFF transgenic mice | Investigate B cell survival and tolerance | BAFF overexpression rescues autoreactive B cells [73] |
| FAS-deficient models | Study apoptotic checkpoint failure | Revealed SHM in EF responses [71] |
| BCR repertoire analysis | Characterize autoreactive clones | Identified IGHV4-34 bias in SLE [74] |
| Single-cell RNA/TCR-seq | Resolution of B cell heterogeneity | Revealed clonal expansion in autoimmunity [74] |
Essential Research Reagents and Tools
The following research reagents are critical for investigating BCR checkpoint failures:
Table: Essential Research Reagents for B Cell Checkpoint Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Animal Models | FAS-deficient mice, BAFF-transgenic mice, Lyn-deficient mice | In vivo study of checkpoint failures [71] [73] |
| Flow Cytometry Antibodies | Anti-CD21, CD11c, CD38, CD27, T-bet, CXCR5, CD95(FAS) | B cell subset identification [71] |
| Signaling Inhibitors | PI3K inhibitors, mTOR inhibitors (rapamycin), BTK inhibitors | Pathway manipulation studies [72] [73] |
| Cytokines/Biologicals | Recombinant BAFF, IFN-α, IL-21, anti-CD40, TLR ligands | B cell stimulation and differentiation [71] [73] |
| Detection Reagents | Phospho-specific antibodies (pAKT, pS6), cell viability dyes | Signaling and survival assessment [72] [73] |
The experimental workflow for analyzing B cell fate decisions and checkpoint failures typically involves B cell isolation and stimulation, followed by multidimensional analysis of differentiation outcomes and signaling events:
BCR Repertoire Abnormalities in Autoimmunity
High-throughput BCR sequencing has revealed distinct repertoire abnormalities across autoimmune diseases, providing insights into checkpoint failures:
Table: BCR Repertoire Abnormalities in Autoimmune Diseases
| Disease | Key BCR Repertoire Abnormalities | Clinical Significance |
|---|---|---|
| Systemic Lupus Erythematosus (SLE) | Increased IGHV4-34 usage; Longer CDR3 in memory B cells; Elevated autoreactive clones in unswitched memory B cells | Strong association with disease activity [74] |
| Rheumatoid Arthritis (RA) | Skewed IGHV gene usage; Clonal expansion in synovial B cells; Altered antibody glycosylation | Linked to ACPA production [74] |
| Systemic Sclerosis (SSc) | Altered IGHV gene usage; Shorter CDR3 length; Increased SHM in expanded clones | Distinct patterns in SSc-PAH [74] |
| Sjögren's Syndrome (SS) | Skewed IGHV gene usage; Accumulation of self-reactive naive B cells; Increased non-synonymous CDR mutations | Suggests early checkpoint failure [74] |
The unswitched memory B (USM B) cell subset appears particularly important in SLE pathogenesis. USM B cells from SLE patients show increased frequencies of autoreactive clones that strongly correlate with disease activity [74]. These cells, which normally produce natural IgM with protective functions, may undergo aberrant activation in autoimmunity, contributing to disease pathogenesis.
Therapeutic Implications and Future Directions
Targeting B Cell Signaling and Metabolism
Understanding BCR checkpoint failures has enabled development of targeted therapies:
- BAFF inhibition (belimumab, ianalumab): Reduces B cell survival and differentiation [73]
- BTK inhibitors: Suppress BCR signaling pathway activation [72]
- mTOR inhibition (rapamycin): Modulates metabolic reprogramming [73]
- Metabolic modulators (metformin, 2-deoxy-D-glucose): Target aberrant B cell metabolism [73]
Combination approaches that simultaneously target multiple checkpoints may yield enhanced efficacy. For example, combining PI3Kδ inhibitors with BAFF blockade addresses both activation and survival signals in autoreactive B cells.
Emerging Therapeutic Concepts
Novel therapeutic strategies are emerging from recent research insights:
- TIM-1 targeting: TIM-1 functions as a B cell immune checkpoint; its blockade enhances B cell antigen presentation and amplifies anti-tumor and potentially anti-autoreactive responses [75]
- TLS modulation: Tertiary lymphoid structures in autoimmune tissues may be targeted to disrupt pathological immune organization [76] [77]
- BCR-directed therapies: Chimeric antigen receptor (CAR)-T cells targeting B cell subsets show promise in eliminating pathogenic B cell populations [77]
Future research directions should focus on elucidating the metabolic-epigenetic cross-talk that determines B cell fate decisions, developing subset-specific therapeutics that spare protective immunity, and leveraging spatial multi-omics to resolve functional heterogeneity within GC and EF responses. The integration of BCR repertoire analysis with clinical parameters may enable personalized approaches for autoimmune disease treatment.
BCR checkpoint failures in both GC and EF pathways contribute significantly to the emergence of autoreactive clones in autoimmunity. While GCs were historically considered the primary source of high-affinity autoantibodies, recent evidence highlights the dominant role of EF responses in systemic autoimmunity. The EF pathway's intrinsic tolerance vulnerabilities, combined with inflammatory signals that favor EF over GC commitment, create an environment permissive for autoreactive B cell expansion and differentiation.
The PI3K/AKT/mTOR pathway serves as a central integrator of signals that determine B cell fate, with dysregulation leading to altered GC-EF balance and breakdown of self-tolerance. Metabolic reprogramming further supports the survival and pathogenicity of autoreactive clones in both pathways. Emerging therapeutic strategies that target these checkpoint failures hold promise for restoring immune tolerance while preserving protective immunity in autoimmune diseases.
Systemic Lupus Erythematosus (SLE) is a chronic, systemic autoimmune disease characterized by a loss of immune tolerance, production of pathogenic autoantibodies, and multi-organ damage [78]. A critical aspect of SLE pathogenesis involves the breakdown of B cell tolerance, leading to the generation of autoreactive plasma cells (PCs) [78] [79]. While these PCs can originate from two primary pathways—the germinal center (GC) and the extrafollicular (EF) response—emerging evidence underscores the dominance of the EF pathway in driving severe, pathogenic autoimmunity in a significant subset of SLE patients [2] [79] [80]. This whitepaper details the mechanisms, identification, and therapeutic targeting of EF-dominated responses within the broader context of B cell receptor (BCR) signaling research.
EF vs. GC B Cell Responses: A Comparative Analysis
B cell activation upon encountering antigen can diverge into two major pathways, each with distinct anatomical locations, outcomes, and roles in SLE [2] [79].
Table 1: Key Characteristics of Germinal Center and Extrafollicular B Cell Responses
| Feature | Germinal Center (GC) Response | Extrafollicular (EF) Response |
|---|---|---|
| Anatomic Location | B cell follicles within Secondary Lymphoid Organs (SLOs) [2] | Splenic bridging channels, red pulp, and lymph node medullary cords (outside follicles) [2] |
| Primary Output | Long-lived plasma cells, high-affinity memory B cells [79] | Short-lived plasmablasts, early-protective antibodies; in SLE, pathogenic autoantibodies [2] [79] |
| Somatic Hypermutation (SHM) | Extensive, leading to affinity maturation [2] | Limited to none [2] |
| Class Switch Recombination (CSR) | Yes [2] | Yes, can occur in EF locations [79] |
| Role in SLE | Can contribute to autoimmunity [79] | Major driver of pathogenic autoantibody production, correlates with disease [2] [80] |
| BCR Affinity Selection | Stringent, based on high affinity [2] | Permissive, allows activation of low-affinity and polyreactive B cells [2] |
The EF response serves as a rapid reaction system, generating a wave of antibody-secreting cells (ASCs) within days of infection or immunization [2]. However, in the context of SLE and chronic inflammation, this evolutionarily conserved pathway is co-opted to support the expansion of autoreactive B cells and the production of pathogenic antibodies, notably against nucleic acid-associated antigens [2] [80].
Mechanisms of EF Response Dominance in SLE Pathogenesis
The predominance of the EF pathway in SLE is driven by a confluence of genetic, signaling, and microenvironmental factors that skew B cell fate toward extrafollicular differentiation.
Key Signaling Pathways and Molecular Triggers
Several interconnected signaling modules are critical for initiating and sustaining EF responses in SLE.
Figure 1: Key Signaling Pathways Promoting EF Responses in SLE
BCR and Endosomal TLR Synergy
A cardinal feature of SLE is the synergistic signaling between the BCR and endosomal Toll-like Receptors (TLRs), particularly TLR7 and TLR9 [80].
- Mechanism: Autoreactive B cells internalize nuclear self-antigens (e.g., RNA-associated or DNA-associated immune complexes) via their BCR. The nucleic acid components are then delivered to endosomes where they engage TLR7 (for RNA) or TLR9 (for DNA) [80].
- Synergy: Co-engagement of the BCR and TLRs leads to a potent, synergistic activation signal primarily mediated through the adaptor protein MyD88, driving robust B cell proliferation, class-switch recombination (CSR), and differentiation [80].
- Pathogenic Imbalance: In SLE, TLR7 signaling is strongly pathogenic. Its overactivity (e.g., due to gene duplication or increased expression) promotes EF responses and spontaneous GC formation. Surprisingly, TLR9 can play a protective role by constraining TLR7-driven autoimmunity, despite being required for anti-dsDNA antibody production [80].
Cytokine Networks
The cytokine milieu in SLE patients strongly favors EF plasmablast differentiation.
- Type I Interferons (IFN-I): A hallmark of SLE, IFN-I directly promotes B cell activation and PC differentiation. It enhances TLR7 expression and signaling, creating a feed-forward loop that amplifies autoimmunity [81] [79].
- BAFF (B-cell Activating Factor): Overexpressed in SLE, BAFF supports the survival of autoreactive B cells that have escaped central tolerance, thereby expanding the pool of cells available for EF activation [78]. BAFF can also signal through the TACI receptor via MyD88 to promote CSR [79].
- IL-6 and IL-21: These cytokines provide critical pro-differentiation signals for B cells. IL-21, in particular, is a potent driver of PC generation in both T-dependent and T-independent contexts [79].
B Cell Intrinsic and Microenvironmental Drivers
- BCR Affinity and Signal Strength: The EF pathway is notably permissive, allowing B cells with a wide range of BCR affinities, including low-affinity and polyreactive clones, to become activated. This contrasts with the GC, which imposes stringent affinity-based selection [2].
- Metabolic Reprogramming: EF B cells undergo rapid proliferation, which requires a shift toward aerobic glycolysis and increased nutrient uptake. This metabolic state is distinct from that of GC B cells and may present a unique therapeutic vulnerability.
- Chemotactic Cues: The orphan receptor Ebi2 (GPR183) is critical for positioning activated B cells at the outer follicular and EF zones. Its ligand, 7α,25-dihydroxycholesterol, is abundant in these regions and promotes B cell retention and cluster formation, establishing the EF niche [2].
Experimental Models and Methodologies for Studying EF Responses
Investigating EF-dominated responses requires a combination of sophisticated animal models and precise analytical techniques.
Key Murine Models of SLE
Table 2: Murine Models for Studying EF-Dominated Responses in SLE
| Model | Genetic Background / Induction Method | Key Features Related to EF Response |
|---|---|---|
| MRL/Mp-lpr/lpr | Spontaneous mutation in Fas gene [82] | Robust spontaneous EF response; production of pathogenic autoantibodies; severe lupus-like disease with lymphoproliferation [82] |
| TLR7 Transgenic | Overexpression of TLR7 [80] | Dominant EF response; spontaneous GC formation; autoantibody production; rescued by TLR7 deletion [80] |
| BAFF Transgenic | Overexpression of BAFF [78] | Polyclonal B cell activation; rescues autoreactive B cells from deletion; SLE-like disease with immune complex deposition [78] |
| Chronic Graft-vs-Host Disease | Transfer of parental splenocytes into F1 hybrid [2] | Inducible model; leads to rapid expansion of EF plasmablasts and production of pathogenic antibodies [2] |
Core Methodological Toolkit for Analysis
A multi-faceted approach is essential to definitively identify and characterize EF responses.
Table 3: Research Reagent Solutions for Analyzing EF Responses
| Research Reagent / Tool | Function and Application in EF Research |
|---|---|
| Anti-CD138 (Syndecan-1) Antibody | Identifies and isolates plasmablasts and plasma cells via flow cytometry or immunohistochemistry [79] |
| 9G4 Anti-VH4-34 Antibody | Detects a human B cell repertoire with inherent self-reactivity common in SLE; labels EF-derived pathogenic clones [78] |
| Ebi2 (GPR183) Antagonists | Inhibits B cell migration to EF zones; used to probe the requirement for Ebi2 in EF response formation [2] |
| TLR7 Agonists (e.g., Imiquimod) & Antagonists | To experimentally enhance or inhibit TLR7 signaling and assess its impact on EF plasmablast generation [80] |
| AID-Reporter Mice (e.g., Aicda-GFP) | Tracks B cells undergoing class-switch recombination and somatic hypermutation, processes active in both EF and GC pathways [2] |
| MyD88 Inhibitors | Blocks signaling downstream of TLR7/9 and TACI; used to dissect the role of this key pathway in EF PC differentiation [79] [80] |
Detailed Protocol 1: Tracking Antigen-Specific EF Responses In Vivo
This protocol is used to dissect the dynamics of an EF response to a known lupus-associated antigen.
- Immunization: Administer a T-dependent antigen like sheep red blood cells (SRBCs) or a defined RNA-associated autoantigen mixed with an adjuvant (e.g., incomplete Freund's adjuvant) intraperitoneally to SLE-prone mice (e.g., MRL/lpr) and controls [2].
- Tissue Harvest and Processing: At peak of the EF response (days 4-7), harvest spleen and lymph nodes. Create single-cell suspensions and enrich for CD19+ B cells using magnetic-activated cell sorting (MACS) [2].
- Flow Cytometric Identification of EF Plasmablasts: Stain cells with the following antibody panel and analyze by flow cytometry:
- CD19+ B220+ CD138+: Identifies total plasmablasts [79].
- CXCR5lo/neg Ebi2hi: Phenotype consistent with EF localization (vs. GC B cells which are CXCR5hi Ebi2lo) [2].
- Blimp-1 (Prdm1) GFP Reporter: High expression denotes commitment to the PC lineage [2].
- Antigen-specific BCR staining: Use labeled antigen (e.g., nucleosomes, dsDNA) to identify autoreactive clones [78].
- Functional Validation – ELISPOT: To confirm antibody secretion, perform an enzyme-linked immunospot (ELISPOT) assay on sorted EF-phenotype (CD138+ CXCR5neg) B cells, using plates coated with the immunizing antigen or general anti-Ig to detect total ASCs [79].
Detailed Protocol 2: Single-Cell BCR Sequencing of EF-Derived Plasmablasts
This protocol defines the clonal ancestry and level of somatic hypermutation of EF-derived autoreactive B cells.
- Single-Cell Sorting: Using a fluorescence-activated cell sorter (FACS), individually sort live, CD19+ CD138+ CXCR5neg cells from the spleens of lupus mice or human SLE patient samples into 96-well plates pre-loaded with cell lysis buffer [78].
- Reverse Transcription and Amplification: Perform reverse transcription using primers specific for the constant regions of immunoglobulin heavy and light chains. Subsequently, amplify the variable regions of the BCR genes using multiplex PCR [78].
- Sequencing and Bioinformatics Analysis: Subject amplicons to next-generation sequencing. Analyze the data using tools like IMGT/HighV-QUEST to determine:
Figure 2: Experimental Workflow for Isolating and Characterizing EF B Cells
Implications for Therapeutic Development and Future Research
The recognition of EF-dominated responses as a key driver of SLE pathogenesis opens novel avenues for targeted therapeutic intervention.
- Targeting Key Signaling Hubs: Inhibition of TLR7 or its downstream signaling components (e.g., MyD88) represents a promising strategy to specifically dampen the pathogenic EF axis without completely ablating protective immunity [80]. The protective role of TLR9 suggests that therapeutic modulation, rather than broad TLR inhibition, may be optimal.
- Disrupting the EF Niche: Chemokine receptors like Ebi2 (GPR183) are required for B cell localization to EF sites. Small molecule inhibitors of Ebi2 could disrupt the formation of pathogenic EF foci, providing a novel mechanistic treatment [2].
- BCR-Directed Therapies: The permissive nature of EF BCR signaling allows for the activation of low-affinity, polyreactive clones. Therapies that enforce stricter BCR signaling thresholds could potentially limit the initial activation of these aberrant EF B cells.
- Biomarker-Driven Patient Stratification: A critical future direction is the development of biomarkers to identify patients with EF-dominated disease. This could include a serum antibody signature (e.g., specific autoantibodies with low SHM), increased frequencies of circulating CD11c+ atypical B cells, or a specific blood transcriptional signature. Such stratification will enable precision medicine, directing EF-targeted therapies to the patients most likely to benefit [2] [79].
In conclusion, the EF pathway is a major contributor to the pathogenic plasma cell pool in SLE, driven by synergistic BCR/TLR signaling, a permissive cytokine environment, and distinct metabolic and migratory cues. Focusing research and drug development on this pathway offers a promising route to more effective and specific treatments for this complex autoimmune disease.
The B cell receptor (BCR) repertoire represents the totality of BCRs within an individual, reflecting the state of the immune system through its diversity, clonal composition, and sequence features [83]. In autoimmune diseases, this repertoire undergoes characteristic perturbations that provide critical insights into disease mechanisms. Skewed BCR repertoires emerge from breakdowns in central and peripheral tolerance checkpoints, leading to the expansion of autoreactive clones that drive pathology across conditions like systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systemic sclerosis (SSc) [83] [74]. These abnormalities manifest most prominently in two key response pathways: the extrafollicular (EF) response, which generates rapid, often lower-affinity antibody production, and the germinal center (GC) response, which facilitates affinity maturation and long-lived immunity [12] [84]. The distinct repertoire features associated with each pathway offer valuable biomarkers for disease activity and potential targets for therapeutic intervention.
Advanced high-throughput sequencing technologies now enable detailed characterization of these repertoire abnormalities at unprecedented resolution, revealing disease-specific signatures in VDJ gene usage, complementarity-determining region 3 (CDR3) composition, and clonal selection patterns [42]. This technical guide synthesizes current understanding of skewed BCR repertoires in autoimmunity, with particular emphasis on their relationship to EF versus GC responses, and provides methodologies for researchers investigating B cell-driven autoimmunity.
BCR Repertoire Fundamentals and Analytical Frameworks
Molecular Architecture of the BCR Repertoire
BCRs are heterodimeric proteins composed of two identical heavy chains and two identical light chains, each containing variable and constant regions [83] [42]. The tremendous diversity of BCRs arises through several molecular mechanisms:
- V(D)J recombination: Random combinatorial rearrangement of variable (V), diversity (D), and joining (J) gene segments for heavy chains, and V and J segments for light chains [42]
- Junctional diversity: Random nucleotide deletion and insertion at segment junctions [83]
- Somatic hypermutation (SHM): Introduction of point mutations in variable regions during B cell activation [83]
- Class switch recombination (CSR): Changing the constant region of the heavy chain to alter effector functions [83]
The variable regions of both heavy and light chains contain three complementarity-determining regions (CDR1, CDR2, and CDR3) that form the antigen-binding site. CDR3 demonstrates the greatest diversity and plays the most critical role in determining antigen specificity [83] [42]. The collection of all BCRs constitutes the BCR repertoire, which serves as a dynamic record of immune history and current status.
Sequencing Technologies for BCR Repertoire Analysis
Each sequencing technology offers distinct advantages for BCR repertoire characterization, with choice dependent on research goals, budget, and required resolution [42].
Table 1: BCR Repertoire Sequencing Technologies
| Technology | Throughput | Read Length | Key Applications | Major Limitations |
|---|---|---|---|---|
| Sanger Sequencing | Low | Long (~800 bp) | Clonal validation, CDR3 spectratyping | Low throughput, cannot capture full repertoire diversity |
| Next-Generation Sequencing (NGS) | High | Short (50-300 bp) | Comprehensive repertoire analysis, diversity assessment, clonality tracking | PCR amplification biases, short reads may not capture full VDJ regions |
| Single-Cell RNA Sequencing | Medium | Full-length | Paired heavy and light chain analysis, B cell phenotyping with transcriptomics | High cost, specialized bioinformatics requirements |
| Third-Generation Sequencing | High | Very long (>10,000 bp) | Full-length repertoire without assembly, isoform detection | Higher error rates, expensive instrumentation |
Abnormal BCR Repertoire Features in Autoimmune Diseases
Disease-Specific Skewing in VDJ Gene Usage
Autoimmune diseases demonstrate characteristic biases in V, D, and J gene segment usage, suggesting antigen-driven selection or defective tolerance mechanisms [83] [74] [85].
Table 2: VDJ Gene Usage Abnormalities in Autoimmune Diseases
| Disease | VDJ Gene Abnormalities | CDR3 Features | Other Repertoire Characteristics |
|---|---|---|---|
| Systemic Lupus Erythematosus (SLE) | Increased usage of IGHV4 family, especially IGHV4-34 with 9G4 idiotype [83] [74] | Longer CDR3 in class-switched memory B cells and plasmablasts; shorter in naïve B cells [83] [74] | Over-representation of IgA and IgE isotypes; association with interferon signature [83] |
| Rheumatoid Arthritis (RA) | Increased IGHV4 family usage; IGHV1-69 polymorphisms associated with disease susceptibility [83] [85] | Conflicting reports (longer or unchanged CDR3 length) [83] | Clonal expansion in synovial tissues; importance of BCR glycosylation [83] |
| Systemic Sclerosis (SSc) | Generally biased IGHV usage; reduced IGHV2-5 in SSc-PAH [83] | Significantly shortened CDR3 length in PBMCs [83] | Increased SHM in expanded clones; more diverse clonotypes [83] |
| Sjögren's Syndrome (SS) | Altered IGHV gene usage [83] | No significant length change; increased non-synonymous mutations in CDR regions of memory B cells [83] | Accumulation of self-reactive clones in naïve B cells; impaired early tolerance [83] |
| ANCA-Associated Vasculitis (AAV) | No significant differences detected [83] | No significant differences in clonal expansion or diversification [83] | Limited pathogenic ANCA clones in PBMC may explain lack of detectable differences [83] |
In SLE, the IGHV4-34 gene segment is particularly noteworthy because its germline-encoded sequence has intrinsic self-reactivity and is typically eliminated from memory B cell compartments through negative selection in healthy individuals [83] [74]. The increased presence of IGHV4-34 clones, especially those bearing the 9G4 idiotype, in SLE patients strongly correlates with disease activity and represents a failure of tolerance mechanisms [74]. Longitudinal analyses reveal that IGHV4-34 usage increases during disease flares, further supporting its pathogenic role [83].
CDR3 Length and Chemical Property Alterations
CDR3 regions demonstrate characteristic length distributions and biochemical properties across autoimmune conditions, reflecting distinct selection pressures:
SLE: CDR3 length abnormalities differ by B cell subset, with longer CDR3 in class-switched memory B cells and plasmablasts suggesting breakdown of peripheral tolerance checkpoints that normally eliminate autoreactive clones with long CDR3s [83]. Conversely, shorter CDR3 in naïve B cells correlates with interferon signatures, potentially indicating altered B cell development in the bone marrow [83] [74].
Systemic Sclerosis: Significantly shortened CDR3 regions in peripheral blood mononuclear cells may reflect unique antigenic pressures or selection processes [83].
Rheumatoid Arthritis: Reports on CDR3 length are conflicting, with some studies showing elongation and others no significant change, possibly due to differences in patient populations or analyzed B cell subsets [83].
The chemical properties of CDR3 regions, including hydrophobicity and charge, also influence autoreactivity, with certain profiles promoting self-reactivity through interactions with specific autoantigens [83].
Figure 1: Molecular Abnormalities Driving Autoreactive BCR Repertoires. Skewed VDJ usage, altered CDR3 features, and increased somatic hypermutation collectively contribute to BCR autoreactivity in autoimmune diseases.
Extrafollicular Versus Germinal Center Responses in Autoimmunity
Distinct Functional and Metabolic Programs
B cell responses to antigen proceed through either extrafollicular (EF) or germinal center (GC) pathways, each with distinct characteristics, locations, and functional outputs [12]:
Extrafollicular Responses: Generate rapid plasmablast differentiation and antibody production at EF sites in the bridging channels of the spleen or medulla of lymph nodes, typically within 2-4 days post-antigen exposure [12]. EF responses are characterized by substantial clonal expansion but limited affinity maturation, though some SHM can occur [12]. These responses are prominent in certain infections and autoimmunity, particularly in SLE where they persist chronically [12] [19].
Germinal Center Responses: Develop later (peaking around 2 weeks post-immunization) within B cell follicles and support extensive SHM, affinity maturation, and class switch recombination [12]. GC responses generate long-lived plasma cells that migrate to bone marrow and memory B cells that provide lasting immunity [12].
These two response types employ different metabolic programs reflective of their distinct functions. EF-activated B cell blasts are large cells that utilize both glucose and fatty acids through aerobic glycolysis and oxidative phosphorylation to support clonal expansion [12]. In contrast, GC B cells primarily metabolize fatty acids via oxidative phosphorylation with minimal glucose utilization, consistent with their role in maintaining rather than expanding cell numbers [12].
BCR Affinity Directs Response Pathway Selection
BCR affinity for antigen plays a determining role in directing B cells toward EF or GC pathways [84]. Higher affinity BCRs preferentially enter EF responses, while lower affinity BCRs are directed toward GC responses [84]. This differentiation is mediated through several mechanisms:
Costimulatory molecule expression: High-affinity B cells upregulate PDL1 and downregulate ICOSL, inhibiting T follicular helper (Tfh) differentiation and promoting EF responses [84]. Low-affinity B cells maintain ICOSL expression, promoting Tfh differentiation necessary for GC responses [84].
Chemokine receptor expression: BCR affinity influences CCR7:CXCR5 ratios, with high-affinity B cells expressing higher ratios that maintain positioning at the follicular periphery near EF sites [84]. The chemokine receptor EBI2 directs B cells toward EF regions through response to its ligand 7α,25-dihydroxycholesterol present in interfollicular areas [84].
T-B cell interactions: The duration and quality of interactions between B cells and CD4+ T cells at the T-B border determine subsequent B cell fate, with stable interactions promoting GC formation and transient interactions favoring EF differentiation [84].
Figure 2: BCR Affinity Directs EF vs. GC Fate. High-affinity BCRs promote extrafollicular responses through PDL1 upregulation, while low-affinity BCRs foster germinal center formation via ICOSL expression.
Pathogenic EF Activation in Severe Autoimmunity
In autoimmune contexts, particularly SLE, there is a marked shift toward EF responses that contributes to disease pathogenesis [12] [19]. This pathogenic EF activation shares features with EF responses observed in severe COVID-19, where it correlates with robust antibody-secreting cell expansion and high concentrations of neutralizing antibodies but also with severe inflammation and poor clinical outcomes [19].
Key cellular mediators of pathogenic EF responses include:
- Activated naïve B cells (aN): CD11c+ T-bet+ B cells that differentiate into effector populations [19]
- Double-negative 2 (DN2) B cells: IgD-CD27- CXCR5-CD21- cells that are expanded in active SLE and severe COVID-19 and correlate with disease activity [19]
- Plasmablasts/antibody-secreting cells: Short-lived progenitors that generate pathogenic autoantibodies [12] [19]
This EF bias in autoimmunity is driven by cytokine milieus rich in IFN-γ and IL-21, which promote T-bet expression and TLR7-dependent B cell activation, creating a positive feedback loop that sustains autoantibody production [19].
Experimental Approaches for BCR Repertoire Analysis
Sample Processing and B Cell Subset Isolation
Comprehensive BCR repertoire analysis requires careful sample processing and often involves sorting specific B cell subsets to identify subset-specific abnormalities [83] [74]:
PBMC isolation: Collect peripheral blood in anticoagulant tubes and isolate mononuclear cells via density gradient centrifugation within 24 hours of collection [19]
B cell enrichment: Use negative selection kits to isolate total B cells, minimizing activation during processing
Fluorescence-activated cell sorting (FACS): Sort defined B cell subsets using surface markers:
- Naïve B cells: CD19+CD27-IgD+
- Unswitched memory B cells (USM B): CD19+CD27+IgD+
- Class-switched memory B cells: CD19+CD27+IgD-
- Double-negative B cells: CD19+IgD-CD27-
- Plasmablasts: CD19+CD27+CD38+
RNA extraction: Use high-quality RNA extraction methods with DNase treatment to eliminate genomic DNA contamination
Library Preparation and Sequencing Strategies
BCR repertoire sequencing library preparation involves targeted amplification of rearranged VDJ regions:
Multiplex PCR approaches: Use V gene family-specific primers or consensus primers targeting framework regions combined with constant region primers [42]
Unique molecular identifiers (UMIs): Incorporate UMIs during reverse transcription to correct for PCR amplification biases and enable accurate clonal quantification [42]
Single-cell BCR sequencing: Use emulsion-based partitioning or plate-based methods to pair heavy and light chains, preserving native pairings [42]
Full-length versus target-enriched sequencing: Choose between amplifying full-length VDJ regions or targeting specific regions like CDR3 based on research objectives
Bioinformatics Analysis Pipelines
BCR repertoire data analysis requires specialized bioinformatics tools and pipelines:
Preprocessing and quality control: Filter low-quality reads, remove sequencing adapters, and correct errors using UMIs
VDJ assignment: Align sequences to reference V, D, and J genes using tools like IMGT/HighV-QUEST, IgBLAST, or partis
Clonotype definition: Group sequences into clonotypes based on shared V and J genes and identical CDR3 amino acid sequences
Repertoire metrics calculation:
- Clonality: Measure dominance of expanded clones (high clonality indicates few dominant clones)
- Diversity: Calculate Shannon entropy or Hill numbers to quantify repertoire richness and evenness
- SHM analysis: Identify mutations by comparison to germline V gene sequences
- CDR3 property analysis: Calculate length, hydrophobicity, and charge distributions
Statistical analysis: Compare repertoires between groups using differential abundance analysis, network analysis, and multivariate methods
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for BCR Repertoire Studies
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| B Cell Surface Markers | Anti-CD19, CD20, CD27, IgD, CD38, CD21, CXCR5, CD11c, T-bet | Flow cytometry, cell sorting to isolate B cell subsets | Panel design critical for identifying rare populations like DN2 B cells |
| Cytokines & Activation Reagents | IFN-γ, IL-21, IL-6, anti-IgM, CD40L, TLR agonists (e.g., R848) | In vitro B cell stimulation to model EF vs. GC differentiation | Concentration and timing determine differentiation outcomes |
| Sequencing Library Prep Kits | SMARTer Human BCR Kit, NEBNext Immune Sequencing Kit | BCR repertoire library construction | Consider UMI incorporation to reduce PCR biases |
| Bioinformatics Tools | IMGT/HighV-QUEST, IgBLAST, MiXCR, Change-O, ALICE | BCR sequence analysis, clonotype identification, mutation analysis | Pipeline integration essential for reproducible analysis |
| Animal Models | Lyn-/- mice, TLR7 transgenic, SLE-prone mice (e.g., MRL/lpr) | In vivo studies of autoreactive B cell development | Model selection depends on specific research questions |
Skewed BCR repertoires in autoimmune diseases reflect fundamental breakdowns in B cell tolerance and distinct biases toward extrafollicular versus germinal center response pathways. The characteristic abnormalities in VDJ gene usage, CDR3 features, and clonal expansion patterns provide both insights into disease mechanisms and potential biomarkers for diagnosis, stratification, and therapeutic monitoring.
Future research directions will likely focus on single-cell multi-omics approaches that simultaneously capture BCR sequences, transcriptomes, and epigenomic states to comprehensively define pathogenic B cell states. The integration of BCR repertoire analysis with clinical outcomes in therapeutic trials will help validate repertoire features as predictive biomarkers. Finally, advancing our understanding of how BCR signaling and antigen engagement drive pathogenic EF responses may reveal novel therapeutic targets for restoring immune tolerance in autoimmune diseases.
Technical advances in long-read sequencing, microfluidics, and computational analysis will continue to enhance resolution and decrease costs, making comprehensive BCR repertoire analysis increasingly accessible for both basic research and clinical applications.
TLR and Inflammatory Signals as B Cell Fate-Switches in Infection and Autoimmunity
The adaptive immune response possesses a remarkable capacity to generate highly specific antibodies through distinct effector pathways. For decades, the germinal center (GC) reaction has been regarded as the primary engine of affinity-matured, long-lived humoral immunity, while the extrafollicular (EF) response was often characterized as a transient producer of low-affinity antibodies. Emerging research now fundamentally challenges this dichotomy, revealing that innate immune signals, particularly through Toll-like receptors (TLRs), serve as critical fate-determinants that direct B cells toward one pathway or the other [12] [80] [60]. This paradigm shift reframes our understanding of B cell biology, positioning TLR and inflammatory signaling not merely as adjuvants but as master regulators of B cell fate decisions with profound implications for infectious disease control, autoimmunity pathogenesis, and therapeutic development.
The context of B cell receptor (BCR) engagement remains crucial—where antigen affinity and co-stimulation provide essential cues—but intrinsic TLR signals now emerge as the decisive factor tipping the balance between EF and GC commitment. This review synthesizes recent advances demonstrating how TLR-mediated inflammation directs B cell differentiation, examining the molecular mechanisms, functional consequences, and therapeutic potential of targeting these fate-switches in both protective immunity and pathological autoimmunity.
Molecular Mechanisms of TLR Signaling in B Cell Fate Decisions
TLR Expression and Signaling Pathways in B Cells
B cells express a diverse repertoire of Toll-like receptors, though the specific TLR profile varies between subsets and maturation states. Human memory B cells constitutively express TLR2, TLR6, TLR7, TLR9, and TLR10, while naïve B cells require pre-stimulation through the BCR to upregulate significant TLR levels [86]. Murine B cells generally express TLR2, TLR3, TLR4, TLR7, and TLR9 constitutively, with marginal zone B cells showing particularly high expression [86]. This differential expression pattern provides the first layer of fate-determination, privileging certain B cell subsets for TLR-mediated activation.
All TLRs except TLR3 utilize the adaptor molecule MyD88 for downstream signal transduction, initiating a cascade that leads to activation of the transcription factors NF-κB and AP-1 [86]. TLR3 signals via the TRIF adaptor, while TLR4 employs both MyD88 and TRIF pathways [87]. The intracellular TLRs (TLR3, TLR7, TLR8, TLR9) that recognize nucleic acids are localized to endosomal membranes, a strategic compartmentalization that prevents aberrant activation by self-nucleic acids under normal conditions [87]. Upon TLR stimulation, B cells undergo dramatic changes: they increase in size, proliferate, upregulate activation markers (CD80, CD86, CD25, CD69, MHC II), and produce cytokines including IL-6, IL-10, IL-12, TNFα, and IFNs [86].
Diagram: TLR Signaling Directs B Cell Fate Decisions. Strong TLR signaling via MyD88 activates NF-κB, leading to IRF4 upregulation and extrafollicular commitment. TLR4/TLR9 engagement can also promote T-bet expression, further reinforcing the EF pathway. In contrast, weak TLR signals combined with BCR engagement favor germinal center formation.
Synergistic BCR-TLR Engagement
A critical mechanism underlying TLR-mediated fate specification is the synergistic engagement of TLRs with the B cell receptor. This synergy is particularly evident in autoreactive B cells, where immune complexes containing nucleic acids can simultaneously engage the BCR (via the antigen component) and TLRs (via the nucleic acid component) [86] [80]. Seminal studies using AM14 transgenic mice, which bear BCRs specific for self-IgG (rheumatoid factor), demonstrated that effective activation of these autoreactive B cells required dual engagement of both the BCR and TLR7 or TLR9 [86]. This cooperative signaling creates a positive feedback loop wherein BCR-mediated internalization delivers TLR ligands to endosomal compartments, enhancing TLR activation, which in turn lowers the threshold for BCR signaling.
The functional outcome of BCR-TLR synergy depends on the integration of signal strength and timing. Strong, simultaneous engagement of both receptors drives rapid IRF4 upregulation, a master regulator of plasma cell differentiation that commits B cells to the extrafollicular pathway [60]. In contrast, weaker or temporally segregated signals favor the GC fate, allowing for the sustained proliferation and mutation required for affinity maturation. This signal integration occurs through shared downstream elements, including the PI3K-AKT pathway and NF-κB activation, which are amplified when both receptors are engaged [88].
TLR-Mediated Fate Switching in Infection and Autoimmunity
Determinants of EF versus GC Fate Decisions
The decision between EF and GC pathways is not random but governed by specific molecular cues that integrate antigenic and inflammatory signals. TLR-mediated inflammation has emerged as a primary determinant of this fate choice, with the strength and quality of TLR signals directing B cell differentiation. During influenza infection, TLR signals drive antigen-specific B cells toward the EF pathway through IRF4 induction via NF-κB c-Rel activation [60]. This inflammatory programming results in rapid plasmablast differentiation and antibody production at EF sites, providing crucial early protection.
The metabolic reprogramming of activated B cells further reinforces these fate decisions. EF B cells and GC B cells utilize distinct metabolic programs: activated B cell blasts supporting clonal expansion are large cells that metabolize both glucose and fatty acids through oxidative phosphorylation and aerobic glycolysis, while GC B cells primarily utilize fatty acids via oxidative phosphorylation with minimal glucose consumption [12]. This metabolic specialization aligns with their functional requirements—biomass generation for EF expansion versus maintenance signaling for GC selection.
Table 1: Characteristics of Extrafollicular Versus Germinal Center B Cell Responses
| Parameter | Extrafollicular Response | Germinal Center Response |
|---|---|---|
| Timing | Early (peaks 4-7 days) [12] [60] | Late (peaks ~2 weeks, persists) [12] |
| Location | Medullary cords, red pulp, bridging channels [4] | Follicular structures with dark/light zones [12] |
| Antibody Affinity | Can be high (germline-encoded) [60] [4] | High (affinity-matured) [12] |
| Somatic Hypermutation | Minimal to none [4] | Extensive [12] |
| Duration | Short-lived (days to weeks) [12] | Long-lived (months to years) [12] |
| Key Transcriptional Regulators | IRF4, T-bet [60] | Bcl6 [12] |
| Metabolic Program | Aerobic glycolysis + oxidative phosphorylation [12] | Fatty acid oxidation (oxidative phosphorylation) [12] |
| TLR Dependence | TLR7/MyD88/IRF4 axis [80] [60] | Context-dependent TLR involvement [87] |
TLR Signals in Autoimmunity: Breaking Tolerance
In autoimmune contexts, particularly systemic lupus erythematosus (SLE), TLR signaling plays a pathogenic role by breaking B cell tolerance. The recognition of self-nucleic acids by TLR7 and TLR9 activates autoreactive B cells that have escaped central tolerance mechanisms [86] [80]. Surprisingly, despite their structural similarity and common signaling adapters, TLR7 and TLR9 play opposing roles in lupus pathogenesis. TLR7 deficiency markedly decreases autoantibody production and disease symptoms, while TLR9 deficiency exacerbates disease despite reducing anti-dsDNA antibodies [80]. This paradox suggests that TLR9 may exert protective effects by limiting TLR7-driven pathology.
The Y-linked autoimmune accelerator (Yaa) locus, which contains a duplication of the TLR7 gene, provides compelling genetic evidence for TLR7's central role in autoimmunity [86]. Male mice with the Yaa mutation develop more severe lupus-like disease, associated with a B cell-intrinsic bias toward production of pathogenic nucleolar autoantibodies [86]. In humans, TLR7 gene duplication and polymorphisms are associated with increased susceptibility to SLE [80], and the escape of TLR7 from X-chromosome inactivation may contribute to the female predominance of this disease [80].
Diagram: TLR-Mediated Loss of B Cell Tolerance in Autoimmunity. Nucleic acid-containing immune complexes simultaneously engage the BCR and endosomal TLRs in autoreactive B cells, leading to loss of tolerance. TLR7 and TLR9 have opposing effects on disease pathogenesis despite similar signaling mechanisms, with TLR7 driving pathology and TLR9 exerting protective functions.
Experimental Approaches and Methodologies
Key Research Models and Reagent Solutions
Investigating TLR-mediated B cell fate decisions requires specialized experimental approaches that can dissect complex cell-intrinsic and extrinsic mechanisms. The table below summarizes critical research tools and their applications in this field.
Table 2: Essential Research Reagents and Models for Studying TLR in B Cell Fate Decisions
| Reagent/Model | Key Features | Experimental Applications | References |
|---|---|---|---|
| AM14 Transgenic Mice | BCR transgenic specific for self-IgG2a (rheumatoid factor) | Demonstrating dual BCR/TLR activation by nucleic acid-containing immune complexes | [86] |
| Mb-1-Cre Bcl6 f/f Mice | B cell-specific Bcl6 deletion, unable to form GCs | Studying EF responses independent of GC formation | [60] |
| Conditional MyD88 KO Mice | Cell-type specific deletion of MyD88 signaling | Dissecting B cell-intrinsic vs. extrinsic TLR signaling | [87] |
| Yaa (TLR7 Duplication) Mice | Enhanced TLR7 signaling due to gene duplication | Modeling TLR7-driven autoimmunity | [86] [80] |
| Virus-like Particles (VLPs) | Antigen presentation with controlled TLR ligand incorporation | Studying B cell-intrinsic TLR signaling in GC responses | [87] |
| Flow Cytometry Panels | EF PBs (CD19loCD45RloCD24+CD38lo), GC B cells (CD45RhiCD19hiCD24hiCD38med) | Identifying and isolating B cell subsets from lymphoid tissues | [60] |
Protocol: Assessing B Cell Fate Decisions In Vivo
To evaluate TLR-mediated B cell fate switching during immune responses, researchers can employ the following methodology adapted from key studies [60]:
Step 1: Immunization/Infection Models
- For EF-biased responses: Use intranasal influenza infection (e.g., A/PR8 strain, 100-500 PFU) or immunization with antigens conjugated to TLR ligands (e.g., CpG-DNA for TLR9, R848 for TLR7)
- For GC-biased responses: Use subcutaneous immunization with protein antigens in alum adjuvant without strong TLR ligands
Step 2: Tissue Processing and Cell Isolation
- Harvest draining lymph nodes or spleen at multiple time points (days 4-7 for EF responses, days 10-14 for GC responses)
- Prepare single-cell suspensions using mechanical dissociation followed by enzymatic treatment (e.g., collagenase D, 1 mg/mL, 30 min at 37°C)
- Enrich B cells using magnetic negative selection kits (>90% purity recommended)
Step 3: Flow Cytometric Analysis
- Stain cells with viability dye and antibodies against:
- EF plasmablasts: CD19, CD45R (B220), CD24, CD38, CD138, IRF4
- GC B cells: CD19, CD45R, CD95, GL7, Bcl6, IRF8
- Antigen-specific B cells: Fluorescently-labeled antigens (e.g., HA for influenza)
- Include intracellular staining for transcription factors (requires fixation/permeabilization)
- Analyze using flow cytometer capable of detecting ≥12 parameters
Step 4: Functional Assessment
- Sort EF plasmablasts and GC B cells using FACs (>98% purity)
- Measure antigen-specific antibody secretion by ELISPOT
- Analyze V-region mutations by single-cell sequencing
- Evaluate metabolic profiles using Seahorse Analyzer (glycolysis vs. oxidative phosphorylation)
Step 5: B Cell-Intrinsic TLR Requirement
- Use mixed bone marrow chimeras with WT and TLR-deficient B cells
- Alternatively, employ B cell-specific conditional knockout mice
- Track competitive fitness and fate decisions of TLR-sufficient versus deficient B cells
This protocol enables comprehensive assessment of how TLR signals influence the EF versus GC fate decision, providing insights into both protective immunity and autoimmune pathogenesis.
Therapeutic Implications and Future Directions
The emerging understanding of TLRs as B cell fate-switches opens promising therapeutic avenues for autoimmune diseases, vaccine design, and immunodeficiencies. In SLE, targeted inhibition of TLR7 and/or enhancement of TLR9 signaling represents a potential strategy to rebalance the aberrant B cell response [80]. For vaccine development, incorporating specific TLR ligands could strategically bias responses toward rapid EF protection or sustained GC immunity depending on the pathogen [60]. The demonstrated capacity of sustained TLR4 stimulation to shift virus-specific B cells toward protective EF responses [60] offers particularly exciting possibilities for vaccines against rapidly-acting pathogens.
Future research must address several critical questions: How do different TLRs generate distinct fate decisions despite shared signaling adapters? What epigenetic modifications reinforce TLR-directed B cell fates? Can metabolic interventions selectively modulate EF versus GC responses? Answering these questions will not only advance fundamental immunology but also enable precision manipulation of B cell responses for therapeutic benefit.
The positioning of TLR and inflammatory signals as central regulators of the EF-GC fate decision represents a paradigm shift in B cell biology with far-reaching implications. As our understanding of these fate-switching mechanisms grows, so too does our capacity to harness them for combating disease and promoting health.
The germinal center (GC) is a transient and dynamic microenvironment within secondary lymphoid organs where B cells undergo rapid proliferation, somatic hypermutation (SHM) of their immunoglobulin genes, and class-switch recombination to generate high-affinity antibodies [89]. This process, essential for adaptive immunity, requires B cells to temporarily adopt several phenotypes that are hallmarks of cancer, including massive proliferation, genotoxic stress, and evasion of apoptosis [89]. Consequently, the GC reaction is a double-edged sword; while it enables effective humoral immunity, it also creates a permissive environment for malignant transformation. Diffuse large B-cell lymphoma (DLBCL), the most common form of non-Hodgkin lymphoma, originates predominantly from GC B-cells or their immediate descendants [89] [90]. The disease is notorious for its clinical and biological heterogeneity, with approximately 30-40% of patients failing to be cured by standard chemoimmunotherapy [89]. Understanding how the normal biological processes of the GC—specifically the delicate balance between proliferation and apoptosis—become dysregulated is fundamental to unraveling the pathogenesis of DLBCL and developing novel therapeutic strategies.
Normal Germinal Center Biology and Its Inherent Risks
The Germinal Center Reaction: A Multi-Compartmental Process
The GC is functionally divided into two main compartments: the dark zone (DZ) and the light zone (LZ).
- Dark Zone: Here, GC B-cells (centroblasts) undergo rapid clonal expansion and SHM, an process mediated by activation-induced cytidine deaminase (AID) that introduces point mutations into immunoglobulin variable region genes [89] [91].
- Light Zone: Post-replicative B-cells (centrocytes) migrate here to be selected based on the affinity of their B-cell receptor (BCR) for antigen displayed on follicular dendritic cells. Successful interaction with T-follicular helper (TFH) cells allows high-affinity B-cells to either differentiate into memory B cells or plasma cells, or to recycle back to the dark zone for further rounds of mutation and selection [89].
Table 1: Key Transcription Factors in GC B-Cell Fate Decisions
| Transcription Factor | Role in Normal GC Biology | Effect in GC B-Cells |
|---|---|---|
| BCL6 | Master regulator of the GC reaction [91] | Establishes the GC phenotype; represses DNA damage checkpoint and differentiation genes [89] |
| MYC | Induced early in GC initiation and upon T-cell help for re-entry [89] | Licenses cell proliferation and metabolic reprogramming |
| IRF4 | Induced by strong BCR signaling [91] | Promotes plasma cell differentiation; represses BCL6 [92] |
| BLIMP1 (PRDM1) | Master regulator of plasma cell differentiation [91] | Terminally represses the GC program and initiates the antibody-secreting program |
Inherent Vulnerabilities to Malignant Transformation
The GC reaction inherently places B-cells at a high risk for malignant transformation. Several physiological processes contribute to this vulnerability [89]:
- Genome Instability: The activities of AID and error-prone DNA polymerase eta introduce DNA point mutations not only in immunoglobulin genes but also in off-target loci, including oncogenes and tumor suppressors.
- Resistance to DNA Damage: To accommodate SHM, GC B-cells downregulate key DNA damage response proteins like ATR, CHEK1, and TP53, creating a permissive environment for the accumulation of genetic alterations.
- Attenuation of Apoptosis: The default fate for GC B-cells that fail positive selection is apoptosis. However, the GC environment is rich in survival signals, and premature apoptosis is actively repressed by factors like BCL6.
- Metabolic Reprogramming: GC B-cells undergo dynamic metabolic shifts to support their rapid proliferation and bioenergetic demands, relying primarily on fatty acid oxidation and oxidative phosphorylation [12].
These inherent risks mean that the majority of healthy individuals harbor premalignant clones of mutant B-cells [89]. The transformation to overt lymphoma occurs when somatic mutations lock these cells into a perpetual state of proliferation and survival, blocking their normal exit from the GC via differentiation or death.
Dysregulated Apoptosis and Proliferation in DLBCL Pathogenesis
The dominant biological theme in GC-derived lymphomas is the acquisition of mutations that enhance the oncogenic functions of BCL6 or overcome its tumor-suppressive effects, thereby locking B-cells in a proliferative, death-resistant state [89].
Genetic Lesions Disrupting Key Cellular Processes
DLBCL is characterized by a high burden of somatic mutations that disrupt epigenetic regulation, signal transduction, and immune surveillance [90] [93]. Many of these mutations converge on pathways controlling cell cycle and apoptosis.
Table 2: Common Genetic Alterations in DLBCL and Their Functional Consequences
| Gene/Pathway | Frequency/Subtype | Functional Consequence in DLomagenesis |
|---|---|---|
| BCL2 Translocations | Common in GCB-DLBCL [90] | Inhibits mitochondrial apoptosis; promotes cell survival |
| MYC Translocations | Common in BL and High-Grade B-cell Lymphoma [89] | Drives cell cycle progression and metabolic reprogramming |
| TP53 Mutations | ~20% of DLBCLs [93] | Abrogates cell cycle checkpoints and DNA damage-induced apoptosis |
| CARD11 Mutations | ~10% of ABC-DLBCL [92] | Constitutively activates NF-κB, promoting proliferation and survival |
| CD79A/B Mutations | ~20% of ABC-DLBCL [92] | Enhances chronic active BCR signaling, leading to NF-κB activation |
| MYD88 L265P Mutation | ~30% of ABC-DLBCL [92] [94] | Activates NF-κB and JAK-STAT signaling via TLR pathways |
| EP300 and CREBBP Mutations | ~40% of DLBCLs [90] | Disrupts histone acetylation and epigenetic control of differentiation |
The Central Role of BCL6 and Epigenetic Modifiers
The transcription factor BCL6 is a central oncoprotein in GC-derived lymphomas. It promotes proliferation and survival by repressing genes involved in DNA damage sensing (e.g., ATR, p53), cell cycle arrest (e.g., CDKN1A/p21), and terminal differentiation (e.g., PRDM1/BLIMP1) [89]. In normal GC B-cells, this repression is transient. In DLBCL, however, genetic lesions can sustain BCL6 activity or its downstream effects. For instance, recurrent mutations in epigenetic regulators like EZH2, CREBBP, and EP300 prevent the proper expression of genes required for GC exit and differentiation, effectively trapping cells in a proliferative state [90]. Mutations in TP53 further exacerbate genomic instability by removing a critical fail-safe mechanism against uncontrolled proliferation [93].
Constitutive Signaling through the BCR and NF-κB Pathways
A pivotal pathway dysregulated in DLBCL, particularly in the ABC subtype, is the B-cell receptor (BCR) signaling pathway. Approximately 30-40% of ABC-DLBCLs rely on chronic active BCR signaling for survival [92] [95]. This signaling is characterized by constitutive activation of the CARD11-BCL10-MALT1 (CBM) complex, which leads to sustained activation of the NF-κB pathway [92]. NF-κB is a potent anti-apoptotic and pro-proliferative transcription factor. Its activation creates a self-reinforcing loop by inducing the expression of IRF4, which in turn supports the survival of ABC-DLBCL cells and represses a toxic interferon response [92]. This "addiction" to NF-κB signaling makes ABC-DLBCL particularly vulnerable to inhibitors of the BCR signaling pathway, such as ibrutinib (a BTK inhibitor) [92].
Diagram 1: Oncogenic BCR signaling in ABC-DLBCL. Contrasts transient signaling in normal B-cells with constitutive NF-κB-driven survival in lymphoma.
Experimental Approaches for Investigating DLBCL Pathogenesis
Key Methodologies and Workflows
Research into the molecular basis of DLBCL relies on a combination of genomic, biochemical, and functional techniques.
Genomic Characterization and Cell-of-Origin Classification:
- Gene Expression Profiling (GEP): RNA from tumor biopsies is subjected to microarray or RNA-sequencing. A gene expression signature is compared to normal GC B-cells and in vitro activated B-cells to classify the tumor as ABC, GCB, or Unclassified [94].
- Next-Generation Sequencing (NGS): Whole-exome or whole-genome sequencing of tumor and matched normal DNA is performed to identify somatic mutations, copy number alterations, and structural variants [94]. This has enabled more refined molecular subtyping (e.g., MCD, BN2, N1, EZB) based on co-occurring genetic alterations [94].
Functional Validation of Genetic Lesions:
- RNA Interference (RNAi) Screens: Lentiviral shRNA libraries are used to perform loss-of-function screens in DLBCL cell lines. This approach identified the essential role of the CBM complex and NF-κB signaling in ABC-DLBCL survival [92].
- In Vitro and In Vivo Modeling: Oncogenes (e.g., mutant CARD11) or tumor suppressors (e.g., TP53) are overexpressed or knocked out in GC B-cell models. The functional impact is assessed using proliferation assays (e.g., CFSE dilution), apoptosis assays (e.g., Annexin V staining), and transcriptomic analysis. For in vivo validation, murine models, including xenografts of human DLBCL cell lines in immunodeficient mice, are used to study tumor growth and response to targeted therapies [92].
Diagram 2: Integrated experimental workflow for DLBCL research, from genomic discovery to therapeutic testing.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents and Models for DLBCL Research
| Reagent/Model | Type | Key Application | Rationale |
|---|---|---|---|
| DLBCL Cell Lines (e.g., TMD8, OCI-Ly3, SU-DHL-4) | In vitro model | Functional studies; drug screening | Represent molecular subtypes (ABC/GCB); genetically characterized [92] |
| Ibrutinib | Small molecule inhibitor | Target validation; combination therapy | Inhibits Bruton's Tyrosine Kinase (BTK); selectively toxic for ABC-DLBCL [92] |
| shRNA Libraries | Genetic tool | Loss-of-function screens | Identifies essential genes and pathways for lymphoma survival (e.g., CBM complex) [92] |
| Aicda-Cre; Bcl-6flx/flx Mice | Genetically engineered mouse model (GEMM) | Study GC biology and lymphomagenesis | Enables cell-type-specific deletion of Bcl6 in GC B-cells to dissect its role [96] |
| Anti-BCL6 Inhibitors (e.g., FX1) | Small molecule inhibitor | Target validation | Disrupts BCL6 corepressor binding; reverses transcriptional repression [89] |
Molecular Classification and Therapeutic Implications
The historical classification of DLBCL into ABC and GCB subtypes has provided prognostic value but limited therapeutic impact. Recent comprehensive genetic analyses have revealed at least four distinct molecular subtypes that transcend the ABC/GCB dichotomy, each with unique therapeutic vulnerabilities [94]:
- MCD/C5: Characterized by MYD88L265P and CD79B mutations, often ABC. Associated with extra-nodal involvement and sensitivity to BTK inhibitors.
- BN2/C1: Defined by BCL6 fusions and NOTCH2 mutations, often ABC or Unclassified. Features a favorable prognosis.
- N1/C3: Driven by NOTCH1 mutations and often classified as ABC. Shows similarity to T-cell acute lymphoblastic leukemia.
- EZB/C2: Characterized by EZH2 mutations and BCL2 translocations, typically GCB. May be sensitive to EZH2 and BCL2 inhibitors.
This refined classification provides a roadmap for precision medicine in DLBCL. For instance, patients with MCD/C5 tumors, which are addicted to chronic active BCR signaling, are prime candidates for BTK inhibitor therapy. Conversely, patients with EZB/C2 tumors harboring EZH2 mutations may benefit from EZH2 inhibitors [94]. The future of DLBCL therapy lies in moving beyond the one-size-fits-all R-CHOP regimen towards biomarker-driven, combination therapies that target the specific genetic lesions responsible for dysregulated apoptosis and proliferation in each molecular subset.
The humoral immune response orchestrates B cell activation through two primary pathways: the rapid, inflammatory extrafollicular (EF) response and the slower, more refined germinal center (GC) reaction. While both are essential for effective immunity, the inappropriate engagement or dysregulation of either pathway can lead to autoimmune pathology. Emerging research reveals that pathogenic B cells in systemic lupus erythematosus (SLE), idiopathic nephrotic syndrome, and other autoimmune conditions exhibit distinct preferences for these differentiation routes. This technical review examines cutting-edge strategies for the selective depletion of pathogenic EF or GC B cell populations, focusing on molecular signatures, signaling pathways, and innovative therapeutic modalities that enable precise targeting while preserving protective immunity. We synthesize experimental data and clinical evidence to provide a framework for the next generation of B-cell-directed therapeutics.
B cell responses to antigenic challenge bifurcate early after activation into extrafollicular (EF) and germinal center (GC) pathways, each generating functionally distinct effector populations [97]. The EF pathway produces rapidly differentiated antibody-secreting cells (ASCs) with limited somatic hypermutation, typically within days of antigen encounter. These short-lived plasmablasts provide immediate but often lower-affinity antibody protection. In contrast, the GC pathway involves extensive somatic hypermutation, affinity maturation, and class switching over weeks, resulting in long-lived plasma cells and memory B cells that provide high-affinity, durable immunity [97] [98].
Table 1: Key Characteristics of EF and GC B Cell Pathways
| Feature | Extrafollicular (EF) Response | Germinal Center (GC) Response |
|---|---|---|
| Timing | Early (2-5 days post-activation) | Delayed (1-2 weeks post-activation) |
| Location | Bridging channels, medullary cords | Follicular structures in lymphoid organs |
| Primary Output | Short-lived plasmablasts | Long-lived plasma cells, memory B cells |
| Somatic Hypermutation | Limited | Extensive |
| Affinity Maturation | Minimal | Robust |
| B Cell Phenotype | CD21low, T-bet+, CD11c+ [39] | BCL-6+, GL7+ |
| Key Chemokine Receptors | EBI2 (persistent), CXCR4 [97] | CXCR5, EBI2 (lost) |
| Role in Autoimmunity | SLE, idiopathic nephrotic syndrome [97] [39] | Rheumatoid arthritis, certain SLE aspects |
The balance between these pathways is carefully regulated, but in autoimmune disease, this balance is disrupted. In SLE, for instance, autoreactive B cells preferentially differentiate through the EF pathway, generating bursts of pathogenic ASCs [97]. Similarly, in childhood idiopathic nephrotic syndrome, a pronounced EF signature characterizes the B cell response [39]. Understanding the molecular determinants of these fate decisions provides the foundation for selective therapeutic intervention.
Molecular Signatures and Signaling Pathways
Distinct Transcriptional and Surface Profiles
Pathogenic EF B cells exhibit a characteristic transcriptional and surface profile that distinguishes them from their GC counterparts. Single-cell RNA sequencing of blood from children with active idiopathic nephrotic syndrome revealed a B cell transcriptional program poised for effector functions, with elevated expression of genes involved in B cell receptor (BCR) signaling (SYK, BTK), activation (CD27, TNFRSF13B/TACI), and antibody production (IGHG1, IGHG3, MZB1) [39]. Flow cytometry analyses further identified the expansion of CD21low CXCR5– T-bet+ CD11c+ atypical B cells (atBCs) and T-bet+ antibody-secreting cells (ASCs) in this condition [39].
The commitment to the EF versus GC pathway is determined early after B cell activation, guided by integrated signaling from the BCR, Toll-like receptors (TLRs), and cytokines [99]. The PI3K/AKT/mTOR pathway plays a central role in this fate decision, with its intensity and duration influencing whether B cells undergo GC maturation or rapid EF differentiation [99].
Key Signaling Pathways Regulating B Cell Fate
The B cell receptor (BCR) is a complex structure composed of membrane immunoglobulin (mIg) and associated Igα/Igβ (CD79a/CD79b) heterodimers that transduce intracellular signals [100] [101]. Upon antigen binding, BCR signaling activates three major pathways: the PLC-γ2 pathway, the PI3K pathway, and the MAPK pathway [101]. The integration of these signals, along with input from other receptors such as CD40, IL-21R, and BAFF-R, determines the outcome of B cell activation [100].
Figure 1: BCR Signaling Pathways and Fate Decisions. B cell receptor activation triggers multiple signaling cascades whose intensity and duration help determine differentiation toward extrafollicular (EF) or germinal center (GC) fates. The PLC-γ2, PI3K/AKT/mTOR, and MAPK pathways integrate with other signals to influence cell fate, with high PI3K/AKT/mTOR signaling and IRF4 induction favoring the EF pathway [99] [101].
Recent work highlights that non-apoptotic FAS signaling controls mTOR activation and extrafollicular maturation in human B cells [99]. Furthermore, B cell-intrinsic TLR signaling supports antigen-stimulated B cell survival, clonal expansion, and differentiation via induction of IRF4 through activation of NF-κB c-Rel, directing cells toward the EF pathway [60]. This explains why influenza infection, but not immunization with virus particles in alum, rapidly induces protective EF responses through TLR-mediated mechanisms [60].
Experimental Models and Methodologies
Tracking and Depleting Pathogenic B Cell Populations
Table 2: Key Research Reagents for Studying EF and GC B Cell Biology
| Reagent/Category | Specific Example | Function/Application |
|---|---|---|
| Surface Markers for Flow Cytometry | CD21, CD23, CXCR5, T-bet, CD11c, CD19, CD38, CD24, CD138, IgD, IgM, GL7 | Identification and isolation of EF (CD21low CXCR5– T-bet+ CD11c+) and GC (GL7+ CD38low) B cell subsets [97] [39] |
| Genetic Models | Mb-1-Cre Bcl6 f/f mice | Enables selective ablation of GC formation to study EF responses in isolation [60] |
| Therapeutic Modalities | Anti-CD20 mAbs (Rituximab), Seldegs (Selective Degradation), CAR-T cells, Bispecific TCEs | Depletion of B cell populations via different mechanisms (effector function, targeted delivery to liver, T-cell mediated killing) [102] [103] |
| Signaling Agonists/Antagonists | TLR ligands (LPS, CpG), PI3K/AKT/mTOR inhibitors, SYK inhibitors | Manipulation of key signaling pathways to study B cell fate decisions [99] [60] |
| Tracking & Detection | Recombinant HA probes, ELISPOT for antigen-specific ASCs, scRNA-seq | Detection of antigen-specific B cells and ASCs, transcriptional profiling [39] [60] |
Detailed Experimental Protocol: Seldeg-Mediated Depletion of Autoantibodies
The selective degradation (Seldeg) strategy represents a novel approach for precise targeting of pathogenic antibodies. The following protocol outlines key steps for developing and testing Seldegs, based on the work of Sun et al. (2020) [102]:
Seldeg Construct Design and Generation:
- Create a fusion protein containing the target autoantigen (e.g., myelin oligodendrocyte glycoprotein/MOG) linked to an Fc domain with mutated FcγR binding (G236R/L428R) to prevent effector function.
- Incorporate a targeting component (e.g., C2A domain of synaptotagmin 1 for phosphatidylserine binding or Fc domain for FcRn binding) to direct the complex to specific cellular compartments.
- Introduce knobs-into-holes and electrostatic steering mutations in the CH3 domains to enable monomeric display of the antigen.
In Vitro Validation:
- Validate binding specificity using ELISA to confirm autoantigen-antibody interaction and cell-based assays with PS-exposing cells (e.g., 2H11, RAW264.7) for PS-targeting Seldegs.
- Assess internalization and lysosomal trafficking using flow cytometry and fluorescence microscopy with pulse-chase experiments and lysosomal markers (e.g., dextran).
In Vivo Efficacy Testing:
- Utilize established disease models (e.g., experimental autoimmune encephalomyelitis/EAE for MOG-specific antibodies).
- Administer Seldeg intravenously and monitor disease progression and autoantibody titers compared to control Fc fusions.
- Quantify antigen-specific antibody levels using ELISA and examine cellular localization in tissues (e.g., liver) via immunohistochemistry.
This approach demonstrated that MOG-Seldeg-PS could selectively clear MOG-specific antibodies without affecting antibodies of other specificities, ameliorating disease in an antibody-mediated EAE exacerbation model [102].
Current and Emerging Therapeutic Strategies
Broad Spectrum B Cell Depletion
Conventional B cell depletion therapies (BCDTs), particularly anti-CD20 monoclonal antibodies like rituximab, have demonstrated efficacy in various autoimmune diseases [103]. These antibodies primarily deplete B cells through effector mechanisms including antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) [103]. However, they exhibit limitations:
- Incomplete depletion of certain B cell subsets, particularly tissue-resident B cells and those with low CD20 expression [103]
- Variable efficacy across autoimmune conditions (e.g., effective in rheumatoid arthritis but not in SLE) [103]
- Failure to target long-lived plasma cells, which lack CD20 expression [103]
- Broad immunosuppression rather than antigen-specific targeting [102]
Selective Targeting Approaches
Next-generation strategies aim for greater precision in targeting pathogenic B cell populations:
1. Antigen-Specific Depletion (Seldegs): As described previously, Seldegs use antigen-Fc fusion proteins to selectively bind and eliminate autoantibodies of a single specificity while preserving protective antibodies [102]. This approach directly targets the pathogenic humoral component without broad B cell depletion.
2. Bispecific T-Cell Engagers (TCEs): These antibodies simultaneously target CD3 on T cells and B-cell surface antigens (e.g., CD20, CD19), redirecting T cell cytotoxicity toward pathogenic B cells. This enables more complete B cell depletion, including of rituximab-resistant subsets [103].
3. Chimeric Antigen Receptor T (CAR-T) Cells: Autologous T cells engineered to express chimeric antigen receptors against B cell surface markers (e.g., CD19) have shown remarkable efficacy in refractory autoimmune diseases, achieving deep B cell depletion and sustained remission [103].
4. Signaling Pathway Inhibitors: Small molecule inhibitors targeting key signaling nodes in B cell activation (e.g., BTK, SYK, PI3K) offer the potential to modulate B cell function without complete depletion [99] [101].
Figure 2: Mechanisms of Action of B Cell-Targeting Therapies. Traditional anti-CD20 monoclonal antibodies (mAbs) deplete B cells through effector functions like complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP). Next-generation approaches include Seldegs that target autoantibodies to liver cells (Kupffer cells and liver sinusoidal endothelial cells/LSECs), bispecific T-cell engagers (TCEs) and CAR-T cells that employ T-cell mediated cytotoxicity, and signaling inhibitors that modulate BCR activation and survival pathways [102] [103].
The strategic targeting of pathogenic B cells based on their developmental origin represents a paradigm shift in the treatment of antibody-mediated autoimmune diseases. The accumulating evidence for distinct roles of EF and GC pathways across different conditions suggests that future therapies must be tailored to the specific immunopathology of each disease. In SLE and idiopathic nephrotic syndrome, where EF responses appear dominant, interventions targeting the unique molecular signatures of these cells (e.g., T-bet, CD11c) or their survival signals (PI3K/AKT/mTOR) may offer enhanced efficacy with reduced off-target effects [97] [99] [39].
Future work should focus on several key areas:
- Biomarker Development: Identifying reliable biomarkers to classify patients based on their dominant B cell pathway (EF vs. GC) for precision targeting.
- Combination Strategies: Exploring sequential or simultaneous targeting of both EF and GC pathways in refractory cases.
- Temporal Dynamics: Understanding how B cell responses evolve during disease progression to optimize intervention timing.
- Safety Refinements: Enhancing the specificity of novel therapies like Seldegs, TCEs, and CAR-T cells to minimize unintended immune consequences.
The integration of advanced sequencing technologies with functional immune profiling will enable increasingly precise delineation of pathogenic B cell subsets, paving the way for a new era of targeted B cell therapeutics in autoimmune disease.
Comparative Analysis and Functional Validation: Defining Pathway-Specific Outputs and Biomarkers
The adaptive immune system employs two principal pathways to launch an antibody response: the extrafollicular (EF) response and the germinal center (GC) response. These pathways represent a fundamental fate decision for activated B cells, initiating distinct molecular programs that ultimately dictate the functional profile of the resulting antibodies. Within the context of B cell receptor (BCR) mediated research, understanding this dichotomy is crucial, as the choice between EF and GC pathways determines the kinetics, quality, and durability of humoral immunity [2] [6]. The EF response is characterized by the rapid generation of short-lived plasmablasts in the splenic bridging channels and lymph node medullary cords, outside the confines of B cell follicles. In contrast, the GC response is a slower, more structured process within follicles where B cells undergo extensive proliferation and somatic hypermutation under the guidance of T follicular helper (TFH) cells [2] [1]. This review provides a head-to-head comparison of the antibody output from these competing pathways, focusing on affinity, durability, and protective capacity, to inform research and therapeutic development.
Functional Output: A Comparative Analysis
The functional divergence of EF and GC responses is evident in the distinct characteristics of the antibodies they produce. The table below summarizes the key differences in their functional output.
Table 1: Functional Output of Extrafollicular vs. Germinal Center Responses
| Feature | Extrafollicular (EF) Response | Germinal Center (GC) Response |
|---|---|---|
| Kinetics | Rapid onset; plasmablasts appear within days of antigen encounter [2] | Delayed onset; peaks over weeks to months [2] |
| Antibody Affinity | Germline-encoded or low-to-intermediate affinity; limited somatic hypermutation (SHM) [2] | High affinity; undergoes extensive SHM and affinity maturation [1] |
| Somatic Hypermutation | Minimal to absent [2] | Extensive, a hallmark of the GC reaction [1] |
| Class-Switching | Yes, can generate IgM and class-switched antibodies (e.g., IgG) [2] [1] | Yes, generates a variety of class-switched antibodies [1] |
| Durability | Short-lived; generates short-lived plasmablasts [1] | Long-lived; generates long-lived plasma cells and memory B cells [1] [6] |
| Primary Protective Role | Critical for early control of acute infections [2] [60] | Foundation of long-term protective immunity and recall responses [1] [6] |
| Pathogenic Role | Implicated in generating pathogenic autoantibodies in systemic autoimmunity (e.g., SLE) [2] [1] | Traditionally suspected, but recent evidence suggests it may be more tightly regulated and even protective in autoimmunity [1] |
Molecular Switches and Signaling Pathways Governing B Cell Fate
The decision between the EF and GC pathways is governed by a network of molecular signals that integrate BCR affinity, co-stimulation, and inflammatory cues.
Key Molecular Determinants
- BCR Signal Strength: Strong BCR-affinity for antigen can drive rapid proliferation and differentiation towards the EF pathway, while lower affinity interactions may favor GC responses [60].
- Transcription Factors: The balance between Bcl-6 (pro-GC) and IRF4/Blimp-1 (pro-EF/Plasmablast) is a critical switch. Bcl-6 suppresses the plasmablast program, allowing for GC formation, while IRF4, induced by strong BCR and TLR signaling, promotes EF differentiation [2] [60].
- Chemokine Receptors: Following activation, the differential expression of homing receptors directs B cell migration. EBI2 (GPR183) expression promotes retention in the EF regions, supporting the EF response. Conversely, downregulation of EBI2 and upregulation of CXCR5 and S1PR2 are required for B cells to re-enter the follicle and form a GC [2] [1].
- Inflammatory Cues: Toll-like receptor (TLR) signaling can act as a fate-determinant. B cell-intrinsic TLR signaling supports survival, clonal expansion, and IRF4 induction via NF-κB c-Rel, skewing responses towards the protective EF pathway during viral infection [60].
- T Cell Help: While both pathways can be T cell-dependent, the quality of help differs. EF responses can be supported by early T cell help, while GC responses are absolutely dependent on a sustained interaction with T follicular helper (TFH) cells [2] [1].
The following diagram illustrates the key signaling pathways and molecular switches that direct B cell fate.
Diagram 1: Molecular switches direct B cell fate. The diagram summarizes integrated signals from BCR strength, TLRs, and T cell help that converge on transcription factors (IRF4/Bcl-6) and chemokine receptors (EBI2/CXCR5) to direct B cells towards EF or GC fates [2] [1] [60].
Investigating the Pathways: Key Experimental Models and Protocols
Elucidating the distinct contributions of EF and GC responses requires specialized experimental models and methodologies designed to isolate and probe each pathway.
Genetic and Pharmacological Models
Table 2: Experimental Models for Discerning EF and GC Responses
| Model / Approach | Mechanism | Utility in Pathway Analysis |
|---|---|---|
| Mb1-Cre Bcl6f/f mice | B cell-specific ablation of Bcl-6, a master transcription factor required for GC formation [60]. | Allows for the study of EF responses in the complete absence of GCs, confirming the GC-independence of EF-derived antibodies [60]. |
| EBI2 Manipulation | Genetic deletion or pharmacological antagonism of the EBI2 receptor. | Impairs B cell migration to EF zones, thereby curtailing EF responses without directly affecting GC initiation [2]. |
| CD4 T Cell Depletion | Use of depleting antibodies (e.g., anti-CD4) to remove T cell help. | Helps distinguish T-dependent (both EF and GC can be affected) from T-independent responses (primarily EF). The use of specific anti-TFH agents can more selectively target GCs. |
| TLR Ligand Administration | Co-administration of antigens with ligands like LPS (TLR4 agonist) during immunization [60]. | Acts as a B cell fate-determinant; sustained TLR4 signaling can shift the balance towards EF responses, enabling study of TLR-driven EF antibody production. |
Key Methodologies for Functional Analysis
- Cell Sorting and ELISpot: Isolation of specific B cell populations (e.g., EF plasmablasts: CD19loCD45RloCD138+; GC B cells: CD19hiCD45RhiGL7+) via fluorescence-activated cell sorting (FACS), followed by Enzyme-Linked Immunospot (ELISpot) to quantify antigen-specific antibody-secreting cells from each compartment [60].
- Sequencing for Somatic Hypermutation (SHM): Amplification and sequencing of immunoglobulin variable region genes from sorted EF plasmablasts versus GC B cells. A high frequency of nucleotide mutations is characteristic of a GC origin, while their absence suggests an EF origin [2] [1].
- Adoptive Transfer and Challenge Models: Transfer of sorted, antigen-specific B cell populations from each pathway into naïve or recipient mice, followed by pathogen challenge. This directly tests the protective capacity and durability of the antibody response generated by each pathway [60].
The following diagram outlines a typical experimental workflow for comparing EF and GC responses.
Diagram 2: Workflow for analyzing EF and GC responses. The experimental pipeline from immunization to downstream analysis, highlighting the isolation of pure B cell populations and the assays used to determine their functional output [60].
The Scientist's Toolkit: Essential Research Reagents
Research into B cell fate decisions relies on a specific toolkit of reagents and models. The following table details key resources for investigating EF and GC pathways.
Table 3: Research Reagent Solutions for B Cell Fate Studies
| Reagent / Model | Function / Application |
|---|---|
| Anti-CXCR5 Antibody | Used to block or identify follicular homing; critical for validating B cell localization. |
| Anti-Bcl-6 Antibody | Essential for intracellular staining to identify and isolate GC B cells by flow cytometry. |
| Anti-IRF4 Antibody | Used for intracellular staining to identify B cells committed to the plasmablast differentiation pathway. |
| Anti-GL7 Antibody | A canonical surface marker for staining and sorting GC B cells in mouse models. |
| Anti-CD138 (Syndecan-1) Antibody | Surface marker for identifying and sorting plasmablasts and plasma cells. |
| LPS (TLR4 Agonist) | Used as an adjuvant in immunization studies to investigate the role of inflammatory signals in driving EF responses [60]. |
| Bcl6-floxed (Bcl6f/f) Mice | When crossed with appropriate Cre-drivers (e.g., Mb1-Cre, CD23-Cre), enables cell-type specific knockout of Bcl6 to ablate GC formation [60]. |
| Recombinant HA Protein | Recombinant hemagglutinin from pathogens like influenza, often fluorescently tagged, is used to identify and sort antigen-specific B cells by flow cytometry [60]. |
The head-to-head comparison of EF and GC responses reveals a sophisticated division of labor in humoral immunity. The EF pathway serves as a rapid-response unit, generating early, protective antibodies that are crucial for controlling acute infections, albeit with limited durability. The GC pathway is the specialist, producing high-affinity, long-lived antibodies that form the basis of sustained immunity and memory. The balance between these pathways is influenced by BCR signal strength, T cell help, and critically, inflammatory cues via TLR signaling [60].
From a therapeutic perspective, this dichotomy has profound implications. Vaccine design may be optimized by incorporating adjuvants that strategically skew responses towards one pathway—towards EF for rapid protection during outbreaks, or towards GC for long-term immunity [6] [60]. In autoimmune disease, the emerging paradigm that pathogenic autoantibodies may arise predominantly from the less-stringently regulated EF response [1] opens new avenues for targeted therapies that spare the protective GC pathway. Future research, leveraging the experimental tools and models detailed herein, will continue to dissect the molecular nuances of B cell fate, enabling more precise manipulation of the immune system for human health.
B cell receptor (BCR) signaling is a fundamental process in adaptive immunity, directing the affinity maturation of antibodies within germinal centers (GCs). This whitepaper delineates the critical role of Bruton's tyrosine kinase (BTK)-mediated BCR signaling in the GC light zone (LZ), a process essential for B cell survival and priming for T follicular helper (Tfh) cell selection. Beyond the established paradigm of the BCR as merely an endocytic receptor for antigen capture, emerging evidence confirms that BCR signaling per se is indispensable for positive selection. This document synthesizes recent experimental data, provides detailed methodologies for key validation experiments, and presents a curated toolkit for ongoing research. Understanding these mechanisms is paramount for advancing therapeutic strategies in vaccine development and managing B-cell malignancies.
Germinal centers (GCs) are transient, specialized microanatomical structures that form in secondary lymphoid organs during T cell-dependent immune responses. They are the primary sites where B cells undergo clonal expansion, somatic hypermutation (SHM), and affinity maturation, ultimately producing high-affinity antibodies and long-lived memory B cells [17] [9]. The GC is spatially organized into two distinct compartments: the dark zone (DZ), where B cells proliferate and undergo SHM, and the light zone (LZ), where a critical selection process occurs [17] [30].
The classical model of GC selection posits that LZ B cells compete for cognate antigen displayed as immune complexes on the surface of follicular dendritic cells (FDCs). B cells with higher BCR affinity more efficiently endocytose, process, and present this antigen as peptide-Major Histocompatibility Complex (pMHC) complexes to T follicular helper (Tfh) cells. The subsequent "help" in the form of CD40 signaling and cytokines promotes the positive selection of these high-affinity B cells [17]. Within this framework, the BCR's role has been largely viewed as that of an antigen-capture device. However, the BCR is a dual-purpose receptor, also functioning as a potent signal transducer. The precise contribution of this signaling function to GC B cell selection has remained a central and only recently resolved question in immunology [17] [9] [104].
The Central Role of BTK in BCR Signaling and Germinal Center Biology
BTK in the BCR Signaling Cascade
Bruton's tyrosine kinase (BTK) is a non-receptor tyrosine kinase of the Tec family, critically positioned in the BCR signaling pathway. Upon BCR engagement by antigen, BTK is recruited to the cell membrane and activated by upstream kinases like SYK. A primary substrate of activated BTK is phospholipase C gamma 2 (PLCγ2). BTK phosphorylates PLCγ2, triggering a downstream signaling cascade that generates inositol triphosphate (IP3) and diacylglycerol (DAG). These second messengers drive calcium flux and activate protein kinase C (PKC) and NF-κB pathways, collectively regulating B cell survival, proliferation, and metabolism [105].
A Delicate Balance: The Dual Nature of BCR Signaling in GCs
BCR signaling within the GC must be precisely tuned. While insufficient signaling leads to failure of selection, evidence indicates that excessive BCR signaling can be equally detrimental. Studies have shown that enhanced BCR signaling, without adequate co-stimulation, can inflict early plasmablast and GC B cell death [106]. This hyper-signaling can induce reactive oxygen species (ROS)-mediated apoptosis, a fate that can only be counteracted by synergistic signals from Tfh cells [9]. This establishes a critical balance between BCR and Tfh cell help signals as a fundamental checkpoint for successful affinity maturation.
Experimental Validation of BTK-Mediated BCR Signaling in GC LZ Selection
Recent pioneering research has provided direct experimental validation for the essential role of BTK-mediated BCR signaling in the GC LZ. The key findings from these investigations are summarized in the table below.
Table 1: Key Experimental Findings on BTK in GC LZ Selection
| Experimental System | Key Finding | Functional Consequence |
|---|---|---|
| BTK drug-resistant mouse model [17] [104] | BCR signaling is necessary for LZ B cell survival. | Prevents apoptosis during the antigen-sensing phase. |
| NP-Eα antigen tracking system [17] | BCR signaling primes LZ B cells for T cell help. | Enhances synergy with CD40 signaling for positive selection. |
| BTK inhibition [17] | Disrupts positive selection of GC B cells. | Impairs affinity maturation and high-affinity antibody production. |
| SHP-1 deletion model (Cγ1Cre/wt Ptpn6fl/fl*) [106] | Unchecked BCR signaling induces GC B cell apoptosis. | Highlights the necessity for balanced signaling intensity. |
Critical Experimental Protocols and Methodologies
In Vivo Antigen Binding and Presentation Tracking
Objective: To identify and isolate GC B cells that are actively engaging with cognate antigen in vivo and presenting it to T cells [17].
Protocol Details:
- Reagent Generation: A tetrameric antigen tracker (NP-Eα) is constructed by coupling fluorescently labeled streptavidin (SA-AF647) to the hapten 4-hydroxy-3-nitrophenylacetyl (NP) and biotinylated I-E52–73 (Eα) peptide.
- Mouse Model: Adoptive transfer of congenically marked B1-8hi B cells (which carry a high-affinity NP-specific BCR when paired with a lambda light chain) into ovalbumin (OVA)-primed recipient mice.
- Immunization & Tracking: Mice are boosted with NP-OVA to induce a GC reaction. The NP-Eα tracker is introduced intravenously.
- Flow Cytometry Analysis: GC B cells are analyzed for:
- AF647 fluorescence: Indicating NP-Eα binding and internalization.
- Y-Ae antibody staining: Detecting presentation of the Eα peptide on MHC-II.
- Cell Sorting and Sequencing: NP-Eα+ and NP-Eα− GC B cells from both LZ and DZ are sorted for subsequent Ig gene sequencing and affinity measurements.
This protocol allows for the direct correlation of a B cell's antigen-binding status in vivo with its mutational landscape and functional capacity.
Genetic Validation of BTK Signaling Dependency
Objective: To definitively establish that BCR signaling, specifically via BTK, is required for LZ B cell selection, independent of the BCR's antigen capture function [17] [104].
Protocol Details:
- Drug-Resistant BTK Mouse Model: A knock-in mouse model is generated harboring a point mutation in BTK (C481F) that confers resistance to the inhibitory effects of the BTK inhibitor ibrutinib.
- Adoptive Transfer and Inhibition: BTK drug-resistant (or wild-type control) B cells are transferred into mice and a GC reaction is initiated. Mice are then treated with ibrutinib.
- Functional Readouts:
- Survival: Apoptosis in LZ B cells is measured via flow cytometry (e.g., Annexin V staining). Ibrutinib treatment in wild-type settings induces apoptosis, which is rescued in BTK drug-resistant B cells.
- Priming for T cell Help: The synergistic upregulation of activation markers (e.g., c-MYC) in response to combined BCR and CD40 stimulation is assessed. BTK inhibition ablates this priming effect.
- Positive Selection: The efficiency of GC B cell cyclic re-entry (LZ to DZ transition) and the output of high-affinity plasma cells are quantified with and without BTK inhibition.
This genetic approach uncouples BTK's signaling function from other BCR roles and provides causal evidence for its necessity.
Signaling Pathway and Experimental Workflow Visualization
The following diagrams illustrate the core signaling pathway and the key experimental workflow used to validate its function.
Diagram 1: BTK-dependent BCR signaling pathway for LZ survival.
Diagram 2: Experimental workflow for validating BTK role.
The Scientist's Toolkit: Key Research Reagents and Models
To investigate BCR signaling and BTK function in GCs, researchers rely on a suite of specialized reagents and model systems.
Table 2: Essential Research Reagents for Investigating BTK in GC Biology
| Reagent / Model | Function and Utility | Key Application |
|---|---|---|
| NP-Eα Tracking System [17] | Fluorescent tetramer that simultaneously tracks antigen binding (AF647) and peptide-MHC presentation (Y-Ae staining). | Directly identifies GC B cells actively engaged in antigen acquisition and presentation in vivo. |
| BTK Drug-Resistant Mouse Model [17] [104] | Knock-in model (e.g., BTK-C481F) that allows for specific inhibition of endogenous BTK while leaving the genetically engineered BTK functional. | Causally validates the role of BTK kinase activity in BCR signaling, independent of pharmacological side effects. |
| B1-8hi Knock-In Mouse [17] | B cells from this model carry a rearranged heavy chain that, with a lambda light chain, produces a defined high-affinity anti-NP BCR. | Provides a synchronized, antigen-specific B cell population to study GC responses and affinity maturation. |
| SHP-1 Deletion Model [106] (Cγ1Cre/wt Ptpn6fl/fl) | Enables rapid deletion of the negative regulator SHP-1 in activated B cells, leading to enhanced BCR signaling. | Studies the consequences of hyper-active BCR signaling and the importance of signaling balance. |
| BTK Inhibitors (Ibrutinib, Acalabrutinib) [107] [105] | Small molecule inhibitors that covalently bind to C481 of BTK, blocking its enzymatic activity. | Tool compounds for acute and specific inhibition of BTK to probe its function in GC B cell selection. |
The paradigm of GC selection has been expanded. The BCR is not merely a passive portal for antigen entry but an active signaling entity whose output, mediated by BTK, is a non-negotiable prerequisite for positive selection. BTK-dependent BCR signaling provides a critical survival signal to LZ B cells and primes their molecular machinery to optimally receive and integrate Tfh cell help. This dual function ensures that B cells with functional, high-affinity BCRs are selectively expanded. The experimental validation of this mechanism, facilitated by sophisticated antigen-tracking systems and genetic models, deepens our fundamental understanding of adaptive immunity. Furthermore, it provides a refined framework for therapeutic intervention, guiding the development of next-generation BTK inhibitors for autoimmune diseases and strategies to enhance vaccine efficacy by modulating GC output.
The adaptive immune system relies on immunological memory to provide long-term protection against re-infection. This memory is fundamentally anchored by two key cell types: long-lived plasma cells (LLPCs), which secrete protective antibodies for years or even a lifetime, and memory B cells (MBCs), which stand ready to mount a rapid and robust response upon antigen re-encounter [108] [109]. The generation of these cells occurs through two primary, competitive pathways: the germinal center (GC) reaction and the extrafollicular (EF) response [4] [2]. Understanding the distinct contributions of these pathways is crucial for rational vaccine design and for developing therapies targeting pathogenic antibody responses in autoimmunity. This review delineates the mechanisms, outputs, and functional significance of GC and EF responses in establishing the two pillars of B cell memory, framed within the context of B cell receptor (BCR)-mediated fate decisions.
Germinal Center (GC) Pathway
The germinal center is a transient, specialized microstructure that forms within the follicles of secondary lymphoid organs after antigen exposure. It is the cornerstone of the T cell-dependent humoral immune response, responsible for refining antibody quality and establishing high-quality memory [110] [111].
- Objective: To generate high-affinity, class-switched antibodies and to create a diverse repertoire of B cells capable of recognizing antigenic variants.
- Location: B cell follicles within secondary lymphoid organs (e.g., spleen, lymph nodes) [4].
- Kinetics: A slower process, peaking days after initial infection or immunization and capable of persisting for months, continually producing output cells [4] [109].
Mechanistic Steps and Signaling
The GC pathway is a multi-stage process driven by precise cellular interactions and molecular signals:
- Initial B Cell Activation and Migration: Naïve B cells are activated by cognate antigen in the follicles. This triggers upregulation of transcription factors and chemokine receptors, causing them to migrate to the T-cell zone border [2] [112].
- Cognate T-B Cell Interaction: At the T-B border, antigen-specific B cells present processed antigen via MHC class II to cognate T follicular helper (Tfh) cells. This interaction, mediated by CD40-CD40L binding and cytokine signaling (e.g., IL-21), provides critical survival and activation signals [113] [112].
- Germinal Center Formation and Cyclic Re-entry: Successfully activated B cells downregulate Ebi2 and CCR7, re-express CXCR5 and S1PR2, and upregulate Bcl-6. This allows them, along with Tfh cells, to re-enter the follicle and form a GC [2]. Within the GC, B cells cycle between the dark zone, where they undergo rapid proliferation and somatic hypermutation (SHM) of their immunoglobulin genes, and the light zone, where they are selected based on the affinity of their BCR for antigen displayed on follicular dendritic cells (FDCs) [110] [112].
- Differentiation into Output Cells: High-affinity B cells receive positive selection signals from Tfh cells and can then exit the GC as either memory B cells or long-lived plasma cells [109]. The transcription factor IRF4 plays a critical role in this fate decision, with prolonged expression driving plasma cell differentiation [109].
Table 1: Key Molecular Signals in the GC Pathway
| Molecule | Role in GC Pathway | Cellular Source |
|---|---|---|
| CD40L / CD40 | Critical co-stimulatory signal for B cell activation, survival, and GC initiation [113] [112]. | Tfh cells / B cells |
| CXCR5 / CXCL13 | Homing of B and Tfh cells to the follicle [4] [2]. | B cells, Tfh / Stromal cells |
| BCL-6 | Master transcriptional regulator of the GC B cell and Tfh cell fate [2]. | B cells, Tfh cells |
| IL-21 | Key cytokine promoting B cell proliferation, class-switch recombination, and differentiation [2]. | Tfh cells |
| AID (AICDA) | Enzyme essential for somatic hypermutation and class-switch recombination [2]. | B cells |
Output Cells and Their Characteristics
The GC is the primary source of the most durable components of humoral memory.
- Long-Lived Plasma Cells (LLPCs): These are non-proliferative, terminally differentiated cells that migrate to survival niches, primarily in the bone marrow. They constitutively secrete high-affinity, class-switched antibodies (e.g., IgG, IgA) without requiring antigen persistence, providing sustained serological memory [108] [109]. Their longevity is governed by both intrinsic fitness and extrinsic factors from the niche, including cytokines (e.g., APRIL, IL-6) and contact with stromal cells [108].
- Memory B Cells (MBCs): GC-derived MBCs are quiescent, long-lived cells that express somatically hypermutated, class-switched BCRs. They recirculate and can respond rapidly upon re-exposure to antigen by differentiating into antibody-secreting plasma cells or re-initiating GC reactions [110] [109]. They often express surface markers like CD27 in humans [113] [112].
Extrafollicular (EF) Pathway
The extrafollicular response represents an evolutionary older, rapid-reaction pathway that occurs outside the B cell follicle [4] [2].
- Objective: To quickly generate a wave of effector cells for early pathogen control.
- Location: Splenic bridging channels and red pulp; medullary cords of lymph nodes [4] [2].
- Kinetics: A fast process, with foci of proliferating cells appearing within days of antigen encounter, but typically being short-lived [4].
Mechanistic Steps and Signaling
The EF pathway shares initial activation steps with the GC pathway but diverges early.
- Initial Activation and Divergent Migration: Like GC-precursors, naïve B cells are activated by antigen and migrate to the T-B border. However, B cells destined for the EF pathway maintain high expression of Ebi2 and CCR7, directing their migration to the outer follicular regions and interfollicular zones, rather than back into the follicle [2].
- T Cell Help and Clonal Expansion: In the EF foci, B cells can receive help from activated T cells that are not necessarily canonical Tfh cells. This help drives robust clonal expansion and class-switch recombination, but typically with little to no somatic hypermutation [4] [2].
- Rapid Differentiation: In these EF sites, activated B cells rapidly differentiate into plasmablasts under the influence of transcription factors like BLIMP-1 and IRF4 [2].
Table 2: Key Molecular Signals in the EF Pathway
| Molecule | Role in EF Pathway | Cellular Source |
|---|---|---|
| Ebi2 (GPR183) | Critical for guiding activated B cells to EF zones; its sustained expression is a hallmark of the EF fate [2]. | B cells |
| CCR7 / CCL19,21 | Promotes B cell migration towards T cell-rich zones at the follicular border [2]. | B cells / Stromal cells |
| CD40L / CD40 | Required for T cell-dependent EF responses, though the quality of help may differ from GCs [113]. | T cells / B cells |
| IL-6, BAFF, APRIL | Survival and differentiation factors for plasmablasts and plasma cells in EF niches [108]. | Myeloid cells, Stromal cells |
| AID (AICDA) | Supports class-switch recombination in EF responses, but SHM is minimal [2]. | B cells |
Output Cells and Their Characteristics
The EF response generates a first line of defense, but its outputs are functionally distinct from those of the GC.
- Short-Lived Plasma Cells/Plasmablasts: The primary output of the EF response is a large number of antibody-secreting cells. These cells are often short-lived (days to weeks) and produce antibodies that can be isotype-switched but generally possess a lower average affinity than GC-derived antibodies due to the absence of affinity maturation [4] [2]. They are crucial for controlling early pathogen replication.
- Memory B Cells: The EF pathway can also generate GC-independent memory B cells [2] [109]. These MBCs often carry an unmutated or minimally mutated BCR and can include populations of IgM+ memory B cells and "atypical" memory B cells (often identified by markers like CD11c and T-bet in mice) [4] [2]. Their reactivation properties and long-term maintenance are areas of active investigation.
Comparative Analysis: GC vs. EF Pathways
Table 3: Comprehensive Comparison of GC and EF Pathways and Their Contributions to Immunological Memory
| Feature | Germinal Center (GC) Pathway | Extrafollicular (EF) Pathway |
|---|---|---|
| Kinetics | Slow (peaks over weeks), sustained | Rapid (peaks within days), transient |
| Primary Location | B cell follicles of SLOs | Bridging channels, red pulp, medullary cords of SLOs |
| Dependence on T Cells | Strictly T cell-dependent (Tfh) | Can be T cell-dependent or T cell-independent |
| Somatic Hypermutation | Extensive, leading to affinity maturation | Minimal to none |
| Class-Switch Recombination | Yes, extensive | Yes, but spectrum may be different |
| Primary Outputs | Long-lived plasma cells, high-affinity memory B cells | Short-lived plasmablasts, GC-independent memory B cells (e.g., IgM+, atypical) |
| Antibody Affinity | High affinity | Low to moderate affinity |
| Role in Immune Memory | Long-term serological memory via bone marrow LLPCs; recall responses via high-quality MBCs | Early protective immunity; potential role in recall via GC-independent MBCs |
| Key Regulators | BCL-6, Tfh cells, CD40/CD40L, AID (SHM+CSR) | Ebi2, BLIMP-1, IRF4, AID (primarily CSR) |
The following diagram illustrates the key decision points and divergent differentiation pathways for B cells following antigen activation:
Experimental Approaches for Delineating GC and EF Responses
Key Methodologies and Workflows
To dissect the relative contributions of GC and EF pathways, researchers employ a combination of techniques.
1. Cell Tracking and Fate Mapping:
- Method: Use of congenic mouse models (e.g., CD45.1 vs. CD45.2) or fluorescent reporter mice (e.g., for AID or Blimp-1) to track the fate of transferred or endogenous antigen-specific B cells over time.
- Workflow: Immunize recipient mice → Harvest organs at multiple time points → Analyze by flow cytometry or microscopy for cell phenotype (e.g., GL7+ for GC B cells, intracellular Ig for plasmablasts) and location (via immunohistochemistry) [4] [109].
- Key Insight: This approach can identify early-appearing plasmablasts in EF zones and later-appearing, high-affinity B cells in GCs.
2. Genetic and Pharmacological Perturbations:
- Method: Using knockout mice or administering blocking antibodies to disrupt specific signals.
- Sample Protocol: Treat mice with anti-CD40L antibody to block T cell help → Immunize → Measure GC formation (e.g., by flow cytometry for GL7+ FAS+ B cells) and EF responses (e.g., by ELISpot for early antibody-secreting cells) [113] [112]. This abolishes most GCs and T-dependent EF responses, highlighting the requirement for CD40 signaling.
3. Analysis of BCR Repertoire and Affinity:
- Method: Single-cell sorting of MBCs or plasma cells based on surface markers (e.g., CD38+ CD27+ for human MBCs) followed by single-cell BCR sequencing.
- Workflow: Sort single B cells → Perform RT-PCR to amplify Ig heavy and light chain V(D)J genes → Clone into expression vectors → Produce recombinant monoclonal antibodies → Test antigen binding and affinity [111] [113].
- Key Insight: B cells derived from EF responses typically show few somatic mutations, while GC-derived B cells are heavily mutated and have higher affinity, as demonstrated in studies of SARS-CoV-2 booster vaccinations [111].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Studying B Cell Memory Pathways
| Reagent / Tool | Function/Application | Specific Example / Target |
|---|---|---|
| Anti-CD40L Antibody | Blocks CD40-CD40L interaction; inhibits GC formation and T-dependent B cell responses [113]. | MR1 clone (in mice) |
| BCR Transgenic Models | Provides a source of B cells with known antigen specificity for tracking and functional assays. | MD4 (anti-HEL) mice |
| Congenic Markers | Allows for tracking of adoptively transferred cells in vivo via flow cytometry. | CD45.1, CD45.2, Thy1.1 |
| AID Reporter Mice | Identifies and tracks cells that have undergone or are undergoing class-switch recombination and somatic hypermutation [2]. | Aicda-Cre x Rosa26-LSL-YFP |
| Recombinant Cytokines | Used in in vitro B cell cultures to promote differentiation towards plasma cell or GC B cell fates. | IL-21, IL-4, IL-6 [108] [2] |
| Flow Cytometry Panels | Phenotypic identification of B cell subsets based on surface and intracellular markers. | Anti-B220, CD138, GL7, CD95, CD38, CD27, CXCR5, T-bet, CD11c [4] [112] |
| ELISpot / FLUOSpot | Quantifies antigen-specific antibody-secreting cells (ASC) at the single-cell level from tissues. | Kits for IgG, IgA, IgM |
The germinal center and extrafollicular pathways represent two distinct yet interconnected strategies for establishing B cell memory. The EF response acts as a rapid deployment force, generating quick, short-lived antibody protection and potentially seeding a layer of GC-independent memory. In contrast, the GC reaction is a specialized training ground, meticulously crafting high-affinity antibodies and generating the elite, long-lived plasma cells and memory B cells that constitute durable, recall-ready immunological memory [109]. The balance between these pathways is influenced by the nature of the antigen, the strength of BCR signaling, and the quality of T cell help. A deep understanding of their distinct contributions and regulatory mechanisms is paramount for advancing vaccine science, where the goal is to steer immune responses toward optimal, long-lasting protection, and for developing targeted therapies for antibody-mediated autoimmune diseases.
In B cell immunology, deciphering the molecular signatures that define cell state, activation history, and functional capacity is paramount for both basic research and therapeutic development. This is particularly critical when studying the divergent germinal center (GC) and extrafollicular (EF) responses, two fundamental pathways that generate long-lived humoral immunity but are also implicated in autoimmune pathology and B-cell malignancies [4] [2]. The EF response, initially defined by the anatomical location of rapidly proliferating B cells and plasmablasts in the splenic bridging channels and lymph node medullary cords, serves as a rapid source of early, protective antibodies [4] [2]. Conversely, the GC response is a more protracted process within B cell follicles that yields antibodies with high affinity and diverse isotypes through somatic hypermutation and class-switch recombination [2].
A precise understanding of these pathways requires biomarkers that report on pathway activity at multiple levels: the cell surface phenotype, dynamic intracellular signaling events, and systemic serological signatures. This guide provides a technical framework for identifying and applying these biomarkers, with a specific focus on their utility in delineating EF and GC B cell responses. We synthesize current methodologies, present quantitative biomarker data, and provide standardized experimental protocols and visualization tools to equip researchers and drug development professionals with the necessary resources to advance this complex field.
Surface Phenotypes: Defining B Cell Identity and Fate
Surface phenotypes provide a accessible means to identify, isolate, and characterize B cells at different stages of differentiation and activation. The markers expressed on a B cell's surface can reveal its origin, its potential fate, and its functional history.
Core Surface Markers for B Cell Identification and Subsetting
The table below summarizes key surface markers essential for identifying broad B cell lineages and subsets.
Table 1: Core B Cell Surface Markers for Identification and Isolation
| Marker | Expression Profile | Function / Significance | References |
|---|---|---|---|
| CD19 | Pan-B cell marker (immature, mature, activated) | Component of the BCR co-receptor complex; amplifies BCR signaling. | [29] |
| CD20 | Mature B cells | Function not fully defined; target for therapeutic depletion (e.g., Rituximab). | [114] |
| CD79A/B | Pan-B cell marker (as part of BCR) | Igα/Igβ heterodimer; essential for BCR expression and signal transduction. | [101] [114] |
| IgM / IgD | Naïve mature B cells | Antigen-binding component of the BCR; naïve B cells co-express mIgM and mIgD. | [101] [29] |
| CD40 | Activated B cells, memory B cells | Costimulatory receptor; interaction with CD154 on T cells is critical for B cell activation and GC formation. | [29] |
| CD44 | Variably expressed; inversely correlated with cell stiffness in stromal cells. | Adhesion molecule; in some cell types, CD44lo phenotype correlates with higher elastic modulus (stiffness). | [115] |
Surface Phenotypes in Extrafollicular versus Germinal Center Responses
The decision between an EF and GC fate is reflected in distinct surface marker profiles. Following antigen activation, B cells reposition to the outer follicular edges, a process guided by upregulated Ebi2 (GPR183) and CCR7, and downregulated CXCR5 [2]. The sustained downregulation of CXCR5 and maintenance of Ebi2 and CCR7 expression favor the EF pathway, directing B cells to interfollicular and outer T cell zones where they differentiate into plasmablasts [2].
A significant challenge in the field is the association of certain phenotypes, like CD11c+ T-bet+ B cells (often called Age-associated B cells or ABCs), with EF responses, particularly in settings of chronic infection or autoimmunity [4] [2]. However, these cells are often identified in contexts where GCs are absent, and their definitive classification as EF-derived requires direct evidence of their anatomical origin and developmental pathway [4]. Therefore, while surface markers are powerful tools, they should be interpreted with caution and in conjunction with other data.
Table 2: Surface Markers Associated with EF and GC Pathways
| Phenotype/Marker | Expression in EF Response | Expression in GC Response | Notes and Technical Considerations |
|---|---|---|---|
| CXCR5 | Downregulated | Re-expressed and high | Critical for homing back into the B cell follicle for GC formation. |
| Ebi2 (GPR183) | Upregulated | Downregulated | Guides activated B cells to outer follicular zones; critical for establishing EF responses. |
| CCR7 | Upregulated | Downregulated | Promotes movement towards T cell zones. |
| S1PR2 | Not specified | Upregulated | Promotes B cell retention in the GC follicle. |
| CD11c & T-bet | Often associated (e.g., in ABCs) | Not typical | Considered a marker for "atypical" memory B cells often generated independently of GCs; context-dependent. |
| CD38 & CD138 | High on EF-derived plasmablasts | High on GC-derived plasmablasts | Markers for antibody-secreting cells (ASCs); cannot distinguish EF vs. GC origin alone. |
The following diagram illustrates the key surface receptor changes that guide B cell trafficking and fate decisions post-activation.
Intracellular Signaling: Interrogating BCR Activity and Functional Output
B cell receptor signaling is the cornerstone of B cell activation, survival, and proliferation. Measuring the activity of intracellular signaling pathways provides a direct readout of cellular function and can reveal heterogeneity with prognostic and therapeutic implications.
The Core BCR Signaling Cascade
Upon antigen binding, the BCR aggregates and its ITAM motifs on the Igα/Igβ heterodimers are phosphorylated by Src-family kinases (e.g., Lyn) [101] [114]. This leads to the recruitment and activation of Syk, which nucleates the formation of a multi-protein "signalosome" [29] [114]. Key downstream pathways include:
- PLC-γ2 Pathway: Activated PLC-γ2 generates IP₃ and DAG. IP₃ induces calcium release from the ER, activating transcription factors like NFAT, while DAG activates PKCβ and the NF-κB pathway, promoting cell survival and proliferation [101].
- PI3K Pathway: PI3K generates PIP₃, recruiting and activating kinases like Akt, which drives metabolic reprogramming and survival [101].
- MAPK Pathway: Regulates cell proliferation and differentiation [101].
The following diagram details this core signaling network.
Profiling Signaling Activity: Phospho-Flow Cytometry
Phospho-specific flow cytometry is a powerful single-cell technique for quantifying the phosphorylation state of signaling proteins downstream of the BCR, allowing for the functional profiling of heterogeneous B cell populations.
Table 3: Key BCR Signaling Proteins for Phospho-Flow Analysis
| Signaling Protein | Role in BCR Signaling | Phosphorylation Site(s) | Functional Significance |
|---|---|---|---|
| Syk | Proximal tyrosine kinase; nucleates signalosome. | Tyr-352 | Indicator of immediate early BCR activation. |
| BTK | Tyrosine kinase critical for PLC-γ2 activation. | Tyr-223, Tyr-551 | Key node in BCR signaling; target of inhibitors (Ibrutinib). |
| PLC-γ2 | Enzyme generating second messengers IP₃ and DAG. | Tyr-753, Tyr-759 | Central hub for downstream pathway bifurcation. |
| AKT | Serine/threonine kinase; regulates survival and metabolism. | Ser-473 | Readout of PI3K pathway activity. |
| ERK1/2 | MAP kinases; regulate proliferation and differentiation. | Thr202/Tyr204 | Readout of MAPK pathway activity. |
| p38 | MAP kinase; responds to stress and inflammation. | Thr180/Tyr182 | Alternative MAPK pathway readout. |
| NF-κB p65 | Transcription factor; promotes survival and inflammation. | Ser-536 | Indicator of canonical NF-κB pathway activation. |
| STAT5 | Transcription factor; involved in cell growth and survival. | Tyr-694 | Can be activated by cytokine signals alongside BCR. |
Experimental Protocol: BCR Signaling Profiling via Phospho-Flow Cytometry
This protocol is adapted from studies interrogating BCR signaling in primary Mantle Cell Lymphoma (MCL) samples [116].
Key Research Reagents:
- Fresh or viably frozen PBMCs from patients or healthy donors.
- Stimulation Media: Serum-free RPMI 1640.
- BCR Crosslinking Agent: Goat F(ab')₂ anti-human IgM (or anti-IgG/IgD as controls). A common working concentration is 10-20 µg/mL.
- Fixation Buffer: Phosflow Lyse/Fix Buffer (BD Biosciences).
- Permeabilization Buffer: Phosflow Perm Buffer III (BD Biosciences).
- Antibody Panel: Conjugated antibodies against surface markers (e.g., CD19, CD20) and intracellular phospho-proteins (e.g., pSYK, pBTK, pPLCγ2, pAKT, pERK, pp38, pNF-κB p65, pSTAT5).
- Flow Cytometer: High-parameter flow cytometer capable of detecting 8+ colors.
Workflow:
- Cell Preparation: Restore viably frozen PBMCs and rest overnight in complete media. Aliquot 0.5-1x10⁶ cells per condition into tubes.
- Stimulation: Pre-warm cells and stimulation media to 37°C. Stimulate cells with anti-IgM for a predetermined time course (e.g., 0, 5, 15 minutes). Include an unstimulated (media only) control.
- Fixation and Permeabilization: Immediately at the end of the stimulation period, add pre-warmed Fixation Buffer to each tube, vortex, and incubate for 10-15 minutes at 37°C. Centrifuge, wash with PBS, and then permeabilize cells with ice-cold Permeabilization Buffer for 30 minutes on ice.
- Staining: Centrifuge and resuspend cell pellets in staining buffer. Add the pre-titrated antibody cocktail and incubate for 30-60 minutes at room temperature in the dark.
- Acquisition and Analysis: Wash cells, resuspend in PBS, and acquire data on the flow cytometer. Analyze data by first gating on live, single CD19+ B cells. Calculate the median fluorescence intensity (MFI) of each phospho-protein in stimulated and unstimulated conditions. The signaling response can be expressed as a fold-change (MFI-stimulated / MFI-unstimulated) or as a difference (ΔMFI).
Clinical and Research Applications of Signaling Biomarkers
Quantifying BCR signaling activity has direct clinical utility. For example, in MCL, unsupervised clustering of basal and anti-IgM-induced phosphorylation profiles identified a patient subgroup with hyperactive BCR signaling that was associated with significantly shorter progression-free and overall survival [116]. Furthermore, constitutive activation of AKT was predictive of inferior response to the BTK inhibitor ibrutinib, highlighting the predictive value of these functional biomarkers [116].
Serological Signatures: Systemic Indicators of Immune Status
Serological biomarkers, measured in blood plasma or serum, provide a systemic readout of immune activation, tolerance breakdown, and tissue damage. They are crucial for diagnosing disease, monitoring activity, and predicting response to therapy.
Autoantibodies: RF and ACPA in Rheumatoid Arthritis
In rheumatoid arthritis (RA), the presence of rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) is a defining serological feature used to classify patients as having seropositive or seronegative RA [117]. This distinction is clinically meaningful: over the last two decades, long-term outcomes have improved significantly in seropositive RA but not in seronegative RA, suggesting potentially different underlying pathobiologies [117]. Seronegative RA may represent a more heterogeneous group that could be further stratified using synovial tissue signatures [117].
Soluble Inflammatory Mediators and Complement
Beyond autoantibodies, a broad array of soluble proteins can serve as biomarkers. In Long COVID, for instance, a plasma biomarker signature linking breathlessness was identified, centered on proteins like CCL3, CD40, IKBKG, IL-18, and IRAK1, and involved dysregulated pathways associated with apoptosis, lung injury, and platelet activation [118].
The complement system is another rich source of serological biomarkers. Analysis can include quantification of individual components (e.g., C3, C4, C1q), activation products (e.g., C3a, C5a, sC5b-9), and functional activity of the different pathways (classical, lectin, alternative) [119]. These measures are indicated for diagnosing complement deficiencies, disorders with aberrant complement activation (e.g., SLE, aHUS, C3G), and for monitoring patients on complement-regulating drugs [119].
Table 4: Key Serological Biomarker Categories and Examples
| Biomarker Category | Examples | Measurement Technique | Research/Clinical Context |
|---|---|---|---|
| Pathognomonic Autoantibodies | RF, ACPA, anti-dsDNA, ANA | ELISA, Multiplex Immunoassay | Diagnosis and classification of autoimmune diseases (e.g., RA, SLE). |
| Soluble Inflammatory Mediators | CCL3, IL-18, IL-6, TNF-α | Ultrasensitive immunoassays (e.g., SIMOA), Multiplex Cytokine Panels | Identifying inflammatory endotypes (e.g., in Long COVID [118]). |
| Complement Components | C3, C4, C1-INH | Nephelometry, Turbidimetry, ELISA | Screening for deficiencies; monitoring disease activity in SLE. |
| Complement Activation Products | C3a, C5a, sC5b-9, Bb, C4d | ELISA (requires careful sample processing) | Specific indicators of in vivo complement activation. |
| Functional Complement Assays | CH50 (Total), AH50 (Alternative) | Hemolytic assays, ELISA-based functional kits | Assessing the functional integrity of the entire complement cascade. |
| Neutralizing Antibodies | SARS-CoV-2 neutralizing Ab | Plaque Reduction Neutralization Test (PRNT) | Assessing quality of humoral response (e.g., lower in Long COVID vs. healthy convalescence [118]). |
The Scientist's Toolkit: Essential Research Reagents
This table consolidates key reagents and their applications for studying biomarkers of B cell pathway activity.
Table 5: Research Reagent Solutions for B Cell Pathway Analysis
| Reagent / Tool | Function / Application | Specific Example / Target |
|---|---|---|
| Anti-human Immunoglobulin | BCR crosslinking for stimulation assays. | Goat F(ab')₂ anti-human IgM / IgG / IgD. |
| Phospho-Specific Antibodies | Detecting activated signaling proteins by flow cytometry or WB. | pSYK, pBTK, pPLCγ2, pAKT, pERK. |
| Fluorescent Cell Barcoding Dyes | Multiplexing samples for phospho-flow, reducing technical variability. | Pacific Orange, CellTrace Violet. |
| BTK Inhibitors | Inhibiting BCR signaling; research and therapeutic tool. | Ibrutinib, Acalabrutinib. |
| Cytokine/Chemokine Detection Kits | Quantifying soluble serological biomarkers. | CCL3, IL-18, IL-6 ELISA or Multiplex Kits. |
| Complement Assay Kits | Measuring complement components, activation products, or function. | Wieslab Complement System Screen, sC5b-9 ELISA. |
| Cell Surface Staining Antibodies | Defining B cell subsets and activation states by flow cytometry. | Anti-CD19, CD27, CD38, IgD, CXCR5, CCR7. |
| Transcription Factor Staining Kits | Intracellular staining for T-bet, Bcl-6, Ki-67, etc. | FoxP3 / Transcription Factor Staining Buffer Set. |
The integrated analysis of surface phenotypes, intracellular signaling, and serological signatures provides a powerful, multi-dimensional view of B cell pathway activity. As research continues to dissect the nuances of extrafollicular and germinal center responses, a standardized approach to biomarker identification and application—as outlined in this guide—will be essential. The experimental frameworks and tools provided here offer a foundation for researchers to uncover novel biology, stratify patient populations, and ultimately contribute to the development of more targeted and effective immunotherapies.
The germinal center (GC) reaction is a critical process in adaptive immunity, enabling B cells to produce high-affinity, class-switched antibodies. Traditional research has relied heavily on animal models, which often fail to fully recapitulate human immune responses. This whitepaper examines the pivotal role of advanced three-dimensional human lymphoid models in bridging the translational gap between animal studies and human immunology. By providing a detailed technical guide on the development, validation, and application of these 3D architectures within the context of B cell receptor-mediated responses, we highlight how these systems offer unprecedented insights into the distinct pathways of extrafollicular versus germinal center B cell differentiation. The integration of these models promises to accelerate therapeutic development for immunodeficiencies, autoimmune diseases, and vaccine design.
B cell responses to antigenic challenge follow two primary trajectories: the extrafollicular (EF) response and the germinal center (GC) response. The EF pathway generates a rapid burst of short-lived plasma cells producing initial, often lower-affinity antibodies, serving as a first line of defense [12]. In contrast, the GC response is a more protracted process within specialized microenvironments in secondary lymphoid organs, supporting somatic hypermutation, affinity maturation, class-switch recombination, and generation of long-lived plasma cells and memory B cells [43]. The balance between these pathways has profound implications for protective immunity and autoimmune pathogenesis, with evidence suggesting that in systemic autoimmunity, dysregulated EF responses can drive disease independently of GCs [43].
A critical challenge in delineating human B cell biology has been the limitations of existing models. Animal models, while invaluable, face translation challenges due to species-specific differences in immune system organization and function [120]. Conventional 2D in vitro cultures fail to recapitulate the complex three-dimensional microenvironment essential for GC formation and function [120]. This whitepaper details how 3D human GC architectures overcome these limitations, providing validated, human-based systems for studying B cell receptor function in determining EF versus GC fate decisions.
Fundamentals of 3D Germinal Center Architecture
The germinal center is not merely a collection of cells but a highly organized specialized microstructure within secondary lymphoid organs. It is morphologically and functionally divided into two compartments: a dark zone filled with proliferating centroblasts undergoing somatic hypermutation, and a light zone where B cells interact with follicular helper T cells and follicular dendritic cells to undergo selection based on antigen affinity [43]. This spatial organization is essential for the GC function, enabling the dynamic shuttling of B cells between zones for repeated rounds of mutation and selection.
The natural GC microenvironment provides signals to residing B-cells through both cell-cell interactions and cell-matrix interactions, which are crucial for differentiation, proliferation, and cellular functions [120]. The extracellular matrix in lymphoid tissues contains proteins such as collagens, fibronectin, laminin, and vitronectin, which form stromal networks that guide cell migration and interaction [120]. Synthetic recreation of this complex architecture requires careful consideration of both cellular composition and biochemical and biophysical properties of the supporting scaffold.
3D Genome Architecture in B Cell Fate
Recent research has revealed that three-dimensional genome organization plays a fundamental regulatory role in B cell development and function. Studies utilizing Hi-C chromosome conformation capture have identified distinct chromatin compartments:
- A-type compartments associated with active, open chromatin with higher transcription activity.
- B-type compartments associated with compact, closed chromatin with lower gene density.
- Intermediate (I-type) compartments enriched in poised and polycomb-repressed chromatin [121].
During normal human B cell differentiation, approximately 28% of genomic compartments change, involving widespread chromatin activation from naive to germinal center B cells and reversal upon maturation into memory B cells [121]. These structural changes directly impact key transcriptional regulators; for instance, during ageing, the Ebf1 gene locus—critical for B cell commitment—switches from compartment A to B, correlating with reduced expression and impaired B lymphopoiesis [122]. Such findings underscore that 3D chromatin organization is not merely a consequence but a major driver of B cell phenotypes.
Development of 3D Human Lymphoid Models
Hydrogel Scaffold Design and Optimization
The foundation of a synthetic 3D human lymphoid model is a tunable hydrogel scaffold that mimics the native extracellular matrix. The PEG-4MAL hydrogel system has emerged as a leading platform due to its synthetic origin, which eliminates batch-to-batch variability associated with natural matrices like Matrigel, and its capacity for precise biochemical functionalization [120].
Table 1: Optimal Composition of PEG-4MAL Hydrogel for 3D Human B Cell Culture
| Component | Optimal Concentration/Type | Function |
|---|---|---|
| PEG-4MAL Macromer | 5.0% | Forms the foundational hydrogel network, providing structural support and defining mechanical properties. |
| RGD Peptide | 2.0 mM | Functionalizes the gel by presenting cell-adhesion motifs that ligate integrins on B cells, facilitating survival and cluster formation. |
| Supporting Cells | CD40L cells, human tonsil-derived lymphoid stromal cells | Provide critical co-stimulatory signals (CD40) and stromal support mimicking the GC niche. |
| Cytokine Supplementation | IL-2, IL-4, IL-6, IL-10, IL-15, IL-21, IFN-α, APRIL | Replace soluble factors naturally produced by GC T cells, dendritic cells, and myeloid cells. |
The optimization process involves systematically varying hydrogel stiffness (through polymer concentration) and biofunctionalization (through adhesive peptide density) to identify the composition that best supports B-cell viability, proliferation, and differentiation. The 5.0% PEG-4MAL, 2.0 mM RGD-peptide composition was found to significantly increase plasmablast and plasma cell numbers as well as antibody production, with less B-cell death compared to 2D cultures [120].
Experimental Workflow for Model Establishment
The process of establishing a 3D human GC model follows a structured workflow from cell isolation to functional analysis, as illustrated below.
Critical Culture Parameters and Protocols
Cell Source and Isolation:
- Source: Human peripheral blood mononuclear cells are an accessible source of primary B-cells.
- Isolation: CD19+ B-cells are typically isolated using magnetic bead-based positive selection.
- Supporting Cells: The model is supported by irradiated murine fibroblast L-cells expressing human CD40L (CD40LCs) to mimic T-cell help, and human tonsil-derived lymphoid stromal cells to recreate the stromal niche [120].
Cytokine Cocktail Regimen: A timed cytokine addition protocol is critical for mimicking the evolving GC microenvironment and guiding B cell fate. The regimen is typically stratified into phases:
Table 2: Phased Cytokine Cocktail for Driving Human B Cell Differentiation in 3D Culture
| Culture Phase | Cytokine Mix 2 (CK2) Composition | Biological Function |
|---|---|---|
| Day 0-4 | CPG ODN, IL-2, IL-4, IL-10, IL-15, IL-21 | Initial activation and proliferation; TLR engagement (CPG) and T-cell like signaling. |
| Day 4-7 | IL-2, IL-4, IL-6, IL-10, IL-15, IL-21 | Support continued proliferation and early differentiation. |
| Day 7-11 | IL-4, IL-6, IL-15, IFN-α, IL-21 | Promote GC maintenance and plasma cell differentiation. |
| Day 11-14 | IL-4, IL-6, IL-21, APRIL | Final maturation and support for antibody secretion. |
This phased cytokine approach, particularly the use of cytokine mix 2 (CK2), was identified as superior in supporting B-cell survival, proliferation, and differentiation [120].
Validation of 3D GC Models Against Human Tissue Benchmarks
A critical step in establishing the fidelity of any in vitro model is its validation against in vivo benchmarks. 3D human GC models must be rigorously evaluated to ensure they recapitulate key features of physiological germinal center reactions.
Phenotypic and Functional Validation Readouts
Table 3: Key Validation Metrics for 3D Human Germinal Center Models
| Validation Category | Specific Readout | Benchmark from Physiological GCs | Performance in 3D Model |
|---|---|---|---|
| Survival & Proliferation | Viability assays, CFSE dilution | Sustained B-cell proliferation in GCs. | Significantly improved vs. 2D cultures; reduced B-cell death [120]. |
| Differentiation | Flow cytometry for CD19+CD38+ plasmablasts and CD19+CD38hi plasma cells. | Generation of antibody-secreting cells. | Significantly increased plasmablast and plasma cell numbers [120]. |
| Antibody Production | ELISA for total and antigen-specific IgG/IgA. | High-rate antibody secretion. | Significantly increased antibody production [120]. |
| Class-Switch Recombination | Flow cytometry for surface IgD (naive), IgG, and IgA. | Isotype switching from IgM/IgD to IgG, IgA, etc. | Observed class switching of naive CD19+IgD+ B-cells toward IgG+ and IgA+ B-cells [120]. |
| Microstructure Formation | Microscopy (confocal, histology) for cluster formation. | Formation of distinct GC-like structures with dark and light zones. | Formation of large B-cell clusters indicating GC-like structures [120]. |
Molecular and Genomic Validation
Advanced genomic techniques provide a deeper layer of validation by comparing the molecular states of in vitro models with primary human cells.
- Single-Cell and Spatial Genomics: These technologies enable direct comparison of the diversity of GC B cell states between the 3D model and human lymphoid tissue, providing an unbiased assessment of how well the model recapitulates in vivo transcriptional signatures [123].
- Epigenomic Landscaping: Mapping histone modifications (H3K27ac, H3K4me3, H3K27me3) and chromatin accessibility in the model and comparing them to datasets from primary human B cell subsets (naive, GC, memory, plasma cells) validates the establishment of correct enhancer and promoter activities [121].
- 3D Genome Profiling: Techniques like Hi-C can assess whether the in vitro model recapitulates key aspects of 3D genome architecture, such as compartment shifts and TAD strengths, observed during normal human B cell differentiation [121].
The Scientist's Toolkit: Research Reagent Solutions
The successful implementation of a 3D human GC model relies on a suite of essential research reagents and materials.
Table 4: Essential Research Reagents for 3D Human Germinal Center Modeling
| Reagent/Material | Function/Description | Example Application |
|---|---|---|
| PEG-4MAL Macromer | A 4-armed, maleimide-functionalized polyethylene glycol polymer that forms a tunable, bio-inert hydrogel scaffold via cross-linking. | Serves as the synthetic, defined extracellular matrix for the 3D culture [120]. |
| RGD Peptide | A cyclic or linear peptide containing the Arg-Gly-Asp sequence that ligates to cell surface integrins. | Functionalizes the PEG-4MAL gel to enable cell adhesion and signaling [120]. |
| CD40 Ligand (CD40L) Expressing Cells | Irradiated murine fibroblast L-cells stably expressing human CD40L. | Provides critical CD40 signaling, mimicking T follicular helper cell co-stimulation for B cell activation and survival [120]. |
| Recombinant Human Cytokines | A panel of purified cytokines including IL-2, IL-4, IL-6, IL-10, IL-15, IL-21, IFN-α, and APRIL. | Used in timed cocktails to drive B cell activation, proliferation, differentiation, and class switching [120]. |
| CpG ODN (Oligodeoxynucleotide) | A Toll-like receptor 9 (TLR9) agonist that acts as a synthetic analog of microbial DNA. | Used as a stimulus to activate B cells, mimicking antigenic trigger [120]. |
Signaling Pathways Governing B Cell Fate in 3D Architectures
The decision between the extrafollicular and germinal center fates is governed by an intricate interplay of signaling pathways and transcriptional networks, which are exquisitely sensitive to the 3D microenvironment. The model successfully recapitulates key signaling axes critical for GC biology.
The 3D model promotes a GC-like fate by enhancing signaling through the CD40 and BCR pathways, supported by integrin-mediated adhesion. The specific cytokine milieu, particularly including IL-21, is crucial for tipping the balance away from the Blimp-1-driven extrafollicular pathway and toward the Bcl-6-dominated GC program [120] [43]. This results in the hallmark outputs of a GC reaction: somatic hypermutation, class-switch recombination, and the formation of large, proliferating B-cell clusters, which are all robustly observed in the optimized 3D system [120].
The development of robust 3D human GC architectures represents a paradigm shift in the study of B cell biology. These models successfully bridge the critical gap between animal studies and human immunology by providing a defined, tunable, and human-based system that recapitulates key aspects of the germinal center reaction, including B cell survival, proliferation, differentiation, class-switching, and microstructure formation. Framed within the broader context of B cell fate decisions, these models serve as powerful tools to dissect the molecular mechanisms that direct B cells toward extrafollicular or germinal center pathways—a distinction with fundamental implications for vaccine design, autoimmune disease therapy, and cancer treatment.
Future advancements will likely focus on increasing model complexity through the incorporation of additional immune cell types, such as T follicular helper and regulatory cells, to better mimic the cellular crosstalk of the GC. Furthermore, the integration of patient-derived cells holds immense promise for personalized immunology, enabling the study of individual disease pathogenesis and tailored drug screening. As these models continue to be refined and validated, they will undoubtedly deepen our understanding of human immunity and accelerate the development of next-generation immunotherapeutics.
The humoral immune response, orchestrated by B lymphocytes, is a cornerstone of adaptive immunity and a critical target for therapeutic intervention. Upon antigen encounter, activated B cells can track towards one of two principal fates: the extrafollicular (EF) response or the germinal center (GC) response [12]. The EF pathway generates a rapid wave of short-lived plasmablasts producing early, often lower-affinity antibodies, serving as a first line of defense. In contrast, the GC reaction is a highly organized, multi-week process within secondary lymphoid organs that yields high-affinity, class-switched antibodies and durable memory B cells and long-lived plasma cells [12] [124] [125]. The molecular switches that direct a B cell toward one pathway over the other are now recognized as crucial determinants of immune efficacy, with profound implications for vaccine design, autoimmune disease therapy, and oncology [12] [19]. This whitepaper delineates an integrative modeling framework that leverages our growing understanding of B cell receptor (BCR) signaling in these divergent pathways to predict therapeutic outcomes and innovate clinical trial design.
Core Biology: Delineating Extrafollicular and Germinal Center Pathways
A detailed comparison of the EF and GC pathways is essential for building predictive biological models. The functional outputs, kinetics, and regulatory mechanisms of these responses are fundamentally distinct.
Table 1: Key Characteristics of Extrafollicular vs. Germinal Center B Cell Responses
| Feature | Extrafollicular (EF) Response | Germinal Center (GC) Response |
|---|---|---|
| Primary Location | Extrafollicular areas of spleen/lymph nodes [12] | B cell follicles of secondary lymphoid organs [124] [125] |
| Kinetics & Duration | Rapid onset (days); can be short-lived or persistent for weeks/months [12] | Slower onset (peaks ~2 weeks); can persist for months [12] [124] |
| Key Effector Cells | Short-lived plasmablasts [12] | Long-lived plasma cells, memory B cells [12] [124] |
| Antibody Affinity & Class | Early, often lower-affinity antibodies; can undergo some maturation [12] | High-affinity, class-switched antibodies (IgG, IgA, IgE) [124] [126] |
| Somatic Hypermutation (SHM) | Can occur but is not the primary feature [12] | Hallmark process; iterative cycles of SHM and selection [124] [125] |
| Metabolic Program | Aerobic glycolysis and oxidative phosphorylation for clonal expansion [12] | Fatty acid oxidation via oxidative phosphorylation for maintenance [12] |
| Pathological Association | Dominant in severe COVID-19 and autoimmune diseases like SLE [12] [19] | Dysregulation linked to autoimmunity and B-cell lymphomas [124] [101] |
The Germinal Center Reaction
The GC is a transient, specialized microstructure that serves as a factory for affinity-matured B cells [124]. It is divided into two anatomically and functionally distinct zones:
- The Dark Zone (DZ): Here, B cells (centroblasts) undergo rapid proliferation and somatic hypermutation (SHM) of their immunoglobulin variable region genes, a process catalyzed by activation-induced cytidine deaminase (AID) [124] [125]. This introduces random point mutations, generating a diverse repertoire of B cell clones with varied affinity for the antigen.
- The Light Zone (LZ): B cells (centrocytes) in the LZ are selected based on antigen affinity. They interact with antigen presented as immune complexes on follicular dendritic cells (FDCs) and receive survival signals from T follicular helper (Tfh) cells [124] [125]. High-affinity B cells are positively selected and can either re-enter the DZ for further rounds of mutation, or exit the GC as memory B cells or long-lived plasma cells [125].
The Extrafollicular Response
Conversely, the EF response is characterized by the activation and differentiation of B cells at sites outside the follicle, leading to a rapid burst of antibody-secreting plasmablasts [12]. While historically viewed as a source of low-affinity, "first-wave" antibodies, it is now appreciated that EF responses can support significant affinity maturation and the production of a substantial fraction of memory B cells, even before GCs are formed [12]. A key insight is that in certain infections and autoimmune conditions, EF responses can become dominant and persistent, actively suppressing the formation of GCs. For instance, in severe COVID-19, a robust, EF-derived B cell response correlates with high titers of neutralizing antibodies but also with heightened inflammation and poor clinical outcomes, mirroring patterns seen in systemic lupus erythematosus (SLE) [19].
Quantitative Data Synthesis for Model Inputs
Integrative models require robust, quantitative data. The following table synthesizes key molecular and cellular metrics that can be used to parameterize models predicting B cell fate and therapeutic efficacy.
Table 2: Quantitative Biomarkers and Signaling Metrics for B Cell Fate Prediction
| Parameter | Quantitative Readout / Example | Experimental Method | Interpretation in Therapeutic Context |
|---|---|---|---|
| BCR Signaling Strength | Calcium flux intensity; Phospho-Syk, Phospho-BLNK levels [101] | Phospho-flow cytometry, Western Blot, Calcium flux assays [101] | Strong, chronic signal may favor EF fate; regulated signal for GC entry. |
| EF-associated B Cells | Frequency of CD11c+ T-bet+ DN2 B cells (e.g., >15% of B cells in severe COVID-19) [19] | High-dimensional flow cytometry (24+ markers) [19] | Biomarker for pathogenic, EF-skewed response (e.g., in autoimmunity, severe infection). |
| GC B Cell Proliferation | Division rate: Cell cycles as short as 5-6 hours for centroblasts [125] | In vivo BrdU/EdU labeling, CFSE dilution [124] | Metric of GC activity and health; suppressed in chronic EF-dominant responses. |
| Tfh Cell Help | IL-21 secretion; CD40L expression [125] | Cytokine secretion assay, FCM for surface CD40L | Essential for GC B cell selection and survival; key node for therapeutic modulation. |
| Plasmablast Output | ASC frequency: Millions of PB at peak in EF/GC early stages [12] | ELISpot, FCM (CD138+ CD38+), scRNA-seq [127] [19] | Measure of acute humoral output; persistent EF ASCs indicate dysregulation. |
| SHM Load | Mutation frequency in Ig V-region genes (e.g., via NGS) [124] | Single-cell BCR sequencing (scBCR-seq) [127] | Indicator of GC activity and affinity maturation history. |
Molecular Mechanisms and Signaling Pathways
The decision between EF and GC fates is governed by integrated signals from the BCR, co-receptors, and cytokines.
BCR Signaling and Downstream Pathways
The BCR complex, comprising membrane-bound immunoglobulin (mIg) and the Igα/Igβ (CD79a/b) heterodimer, initiates signaling upon antigen engagement. The phosphorylation of the Immunoreceptor Tyrosine-Based Activation Motifs (ITAMs) within Igα/Igβ by Src-family kinases (e.g., Lyn) recruits and activates Syk, triggering three major signaling cascades [101]:
- The PLC-γ2 Pathway: Leads to PKCβ and NF-κB activation, plus calcium-NFAT signaling, promoting survival and proliferation.
- The PI3K Pathway: Crucial for metabolic reprogramming and cell growth.
- The MAPK Pathway: Regulates cellular differentiation and proliferation.
The strength and duration of these signals, influenced by antigen affinity and avidity, are a primary determinant of initial B cell fate [101].
Key Transcriptional Regulators
- BCL-6: The master regulator of the GC B cell fate. It represses a plasmablast differentiation program and allows for proliferation and tolerance of the DNA damage inherent in SHM [125].
- IRF4: Different levels of IRF4 expression can guide B cell fate. Lower levels are involved in GC initiation, while high levels drive plasmablast differentiation, a key output of both EF and GC responses [125].
- T-bet (TBX21): A transcription factor highly expressed in EF-derived B cells, particularly in responses to viruses and in autoimmunity. It is associated with a CD11c+ B cell phenotype and is inducible by IFN-γ and IL-21 [19].
The following diagram illustrates the core signaling and transcriptional network governing B cell activation and fate decisions.
Experimental Protocols for Core Assays
High-Dimensional Phenotyping of B Cell Populations
Objective: To comprehensively identify and quantify EF-associated and GC B cell subsets in human peripheral blood or murine lymphoid tissues.
Detailed Methodology:
- Sample Preparation: Isolate PBMCs from whole blood or prepare single-cell suspensions from spleen/lymph nodes.
- Staining Panel Design: A 24+ color flow cytometry panel is required. Critical markers include:
- Lineage/Negative Selection: CD19, CD3, CD14, CD16 (Live/Dead fixable dye).
- EF-associated Populations: CD11c, T-bet, CD21 (lo), CD27 (to define DN2: IgD- CD27- CD21- CD11c+ T-bet+) [19].
- GC-associated Populations: CXCR4 (DZ), CD86 (LZ), CD83 (LZ), BCL-6 (intracellular), GL7/Fas (GC) [124] [125].
- Activation & Homing: CD38, CD138 (ASCs), CXCR5, HLA-DR, CD95.
- Acquisition and Analysis: Acquire data on a high-parameter flow cytometer. Use UMAP or t-SNE for dimensionality reduction and visual exploration of population heterogeneity. Perform manual gating based on established subset definitions for quantitative comparisons [19].
Single-Cell RNA Sequencing with BCR Repertoire Analysis
Objective: To simultaneously analyze transcriptional states and the clonal lineage of B cells, linking specificity and function.
Detailed Methodology:
- Single-Cell Partitioning: Load a single-cell suspension into a platform (e.g., 10x Genomics) to partition individual cells into nanoliter-scale droplets with barcoded beads.
- Library Construction: Generate two libraries from each cell: a whole transcriptome library (for gene expression) and a V(D)J-enriched library for BCR heavy and light chains [127].
- Bioinformatic Analysis:
- Clustering and Annotation: Process gene expression data (alignment, quantification, normalization). Use graph-based clustering and marker gene expression (e.g., MS4A1 for B cells, XBP1 for plasma cells) to annotate cell types.
- BCR Analysis: Assemble BCR sequences from V(D)J data. Identify clonally related cells (sharing the same VDJ rearrangement) and track their distribution across different cell clusters (e.g., EF vs. GC). Calculate SHM frequency per clone [127].
Integrative Workflow for Therapeutic Prediction
The following diagram outlines a holistic workflow, from patient stratification to clinical trial design, integrating the biological insights and experimental methods described above.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for B Cell Fate Research
| Reagent / Technology | Function/Application | Specific Examples |
|---|---|---|
| High-Parameter Flow Cytometry Panels | Deep immunophenotyping to identify rare and novel B cell subsets (e.g., DN2, GC subsets). | Antibodies against CD19, CD11c, T-bet, CD21, CXCR5, CXCR4, CD86, BCL-6, CD38, CD138 [19]. |
| Single-Cell Multi-Omic Platforms | Simultaneous analysis of transcriptome and BCR repertoire from the same cell. | 10x Genomics Single Cell Immune Profiling, BD Rhapsody with BCR solution [127]. |
| Phospho-Specific Antibodies | Interrogation of BCR signaling pathway activation status. | Anti-phospho-Syk, anti-phospho-BLNK, anti-phospho-AKT for flow cytometry/Western blot [101]. |
| Genetically Engineered Mouse Models | In vivo functional validation of targets in a physiological context. | Conditional Bcl6 KO, Irf4 KO, Cd19-Cre models for fate-mapping [125] [126]. |
| Quantitative Systems Pharmacology (QSP) Tools | Computational modeling to integrate multi-scale data and predict drug effects. | Platforms for building PK/PD models incorporating B cell biology, biomarker data, and clinical outcomes [128]. |
Clinical Trial Renovation and Biomarker Application
The ultimate translation of bench insights requires a paradigm shift in clinical trial design. The traditional "one-size-fits-all" approach is inadequate for therapies targeting specific immune pathways. Biomarker-driven, adaptive trial designs are essential [128].
- Patient Stratification: Using the biomarkers and signatures detailed in Table 2, clinical trial populations can be stratified into "EF-high" and "GC-high" groups. For instance, in an autoimmune disease trial, a drug designed to suppress pathogenic EF responses (e.g., an anti-IL-21 or T-bet inhibitor) would be most effectively tested in the "EF-high" stratum, where its effect is most likely to be detected [19].
- Endpoint Selection: Trials should move beyond generic clinical scores to include deep immunological endpoints. These include monitoring the frequency of pathogenic DN2 B cells, tracking the SHM landscape of the B cell repertoire, and measuring the restoration of a balanced GC response. This provides mechanistic proof-of-concept and can serve as early indicators of efficacy [128] [19].
- Leveraging Real-World Data (RWD): RWD from biobanks and large-scale genomic initiatives (e.g., UK Biobank, Tohoku Medical Megabank) can be mined to validate the association of EF/GC signatures with disease manifestations and outcomes, further strengthening trial rationale and patient selection strategies [128].
The bifurcation of B cell responses into EF and GC pathways represents a fundamental axis of immune regulation. The integrative models presented here—synthesizing quantitative biology, advanced experimental protocols, and computational workflows—provide a powerful roadmap for therapeutic prediction. By moving from a descriptive to a predictive understanding of B cell fate, we can design smarter, biomarker-driven clinical trials that deliver the right immunomodulatory therapy to the right patient, ultimately improving outcomes in infection, autoimmunity, and cancer.
Conclusion
The decision for a B cell to embark on an extrafollicular or germinal center journey is a pivotal event in adaptive immunity, masterfully orchestrated by integrated BCR intrinsic and extrinsic signals. The EF pathway serves as a rapid deployment force, generating swift, often high-affinity antibody protection, while the GC is a specialized training ground for precision, yielding affinity-matured, long-lived immunity. Crucially, the dysregulation of either pathway underpins pathologies ranging from autoimmune diseases to B cell malignancies. Future research must focus on integrative human studies to fully map the dynamics of tumor-infiltrating B cells and to rationally design next-generation therapeutics and vaccines that can precisely manipulate these fundamental B cell fate decisions for improved patient outcomes.
References
- 1. Germinal Center vs Extrafollicular Responses in Systemic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7616122/]
- 2. Extrafollicular B cell responses – is one tent big enough? [https://pmc.ncbi.nlm.nih.gov/articles/PMC12632143/]
- 3. Coordinated Regulation of Extrafollicular B Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11986407/]
- 4. A roadmap for defining “extrafollicular” B cell responses [https://www.sciencedirect.com/science/article/abs/pii/S107476132500370X]
- 5. BCR Affinity Influences T-B Interactions and B Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8350505/]
- 6. Germinal Center and Extrafollicular B Cell Responses in ... [https://www.sciencedirect.com/science/article/pii/S1074761320304933]
- 7. Taking Advantage: High Affinity B cells in the Germinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4106706/]
- 8. B Cell Receptor Crosslinking Augments Germinal Center ... [https://www.sciencedirect.com/science/article/pii/S2211124718316310]
- 9. Review BCR signaling in germinal center B cell selection [https://www.sciencedirect.com/science/article/abs/pii/S1471490624001807]
- 10. Affinity gaps among B cells in germinal centers drive the ... [https://www.nature.com/articles/s41590-024-01844-7]
- 11. A paradigm shift in simulating affinity maturation to elicit ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12259674/]
- 12. Germinal Center and Extrafollicular B Cell Responses in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7748291/]
- 13. Tertiary Lymphoid Structures Across Organs - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12463167/]
- 14. Tertiary lymphoid structures in diseases: immune ... - Nature [https://www.nature.com/articles/s41392-024-01947-5]
- 15. Microenvironmental niches in the bone marrow required for B - cell ... [https://www.nature.com/articles/nri1780?error=cookies_not_supported]
- 16. The Bone Marrow Microenvironment in B - Cell Development and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9102980/]
- 17. B cell receptor signaling in germinal centers prolongs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10424567/]
- 18. The roles of tertiary lymphoid structures in chronic diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC10092939/]
- 19. Extrafollicular B cell responses correlate with neutralizing ... [https://www.nature.com/articles/s41590-020-00814-z]
- 20. Germinal Center B Cells Replace Their Antigen Receptors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6162340/]
- 21. Positive Selection in the Light Zone of Germinal Centers [https://pmc.ncbi.nlm.nih.gov/articles/PMC8044421/]
- 22. Germinal center reaction: antigen affinity and presentation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4174395/]
- 23. Regulated somatic hypermutation enhances antibody ... [https://www.nature.com/articles/s41586-025-08728-2]
- 24. A new perspective is needed for positive selection of ... [https://www.nature.com/articles/s41423-021-00823-4]
- 25. Spatiotemporal omics for biology and medicine [https://www.sciencedirect.com/science/article/pii/S0092867424008341]
- 26. Transcription factor IRF4 controls plasma cell ... - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/16767092/]
- 27. The BCL6 RD2 domain governs commitment of activated B ... [https://pubmed.ncbi.nlm.nih.gov/25176650/]
- 28. Plasma cell differentiation is regulated by the expression of ... [https://www.nature.com/articles/s41467-024-49375-x]
- 29. B-Cell Markers, Signaling Pathways & Immune Outcomes [https://blog.cellsignal.com/b-cell-basics-what-a-disease-researcher-needs-to-know]
- 30. Immune metabolism regulation of the germinal center ... [https://www.nature.com/articles/s12276-020-0392-2]
- 31. Article The Amount of BCL6 in B Cells Shortly after Antigen ... [https://www.sciencedirect.com/science/article/pii/S2211124720300188]
- 32. Role of IRF4 in mediating plasmablast differentiation in ... [https://link.springer.com/article/10.1007/s00277-025-06273-6]
- 33. T follicular helper cell differentiation, function, and roles in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4223692/]
- 34. T Follicular Helper Cell Biology: A Decade of Discovery ... [https://www.sciencedirect.com/science/article/pii/S1074761319301918]
- 35. Stepwise differentiation of follicular helper T cells reveals ... [https://www.nature.com/articles/s41467-023-43427-4]
- 36. Germinal center B cell and T follicular helper cell development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3280079/]
- 37. Germinal Center B cells provide essential IL-1β signals to TFH ... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1013404]
- 38. Highly functional and prolonged germinal center T follicular ... [https://www.sciencedirect.com/science/article/abs/pii/S1074761325004625]
- 39. The extrafollicular B cell response is a hallmark of ... [https://www.nature.com/articles/s41467-023-43504-8]
- 40. Single-cell sequencing to multi-omics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11438019/]
- 41. Single-cell multi-omics in cancer immunotherapy: from tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376342/]
- 42. B-cell receptor repertoire sequencing: Deeper digging into ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9494237/]
- 43. Germinal center versus extrafollicular responses in ... [https://www.sciencedirect.com/science/article/abs/pii/S0065277624000221]
- 44. Interpreting the B-cell receptor repertoire with single- ... [https://www.nature.com/articles/s42256-022-00492-6]
- 45. Comprehensive Analysis of TCR and BCR Repertoires [https://pmc.ncbi.nlm.nih.gov/articles/PMC11853700/]
- 46. Practical guidelines for B-cell receptor repertoire sequencing ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-015-0243-2]
- 47. BCR/TCR Sequencing Repertoire Profiling [https://www.med.upenn.edu/humanimmunologycore/bcr-tcr-sequencing.html]
- 48. Antigen presentation by B cells enables epitope spreading ... [https://www.nature.com/articles/s41467-023-42541-7]
- 49. Quantitative modeling of signaling in aggressive B cell ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012488]
- 50. Genetic regulation of B cell receptor signaling pathway [https://www.sciencedirect.com/science/article/abs/pii/S1476927124001762]
- 51. Antigen-Dependent Inducible T-Cell Reporter System for PET ... [https://jnm.snmjournals.org/content/64/1/137]
- 52. Reporter Gene Imaging of Immune Responses to Cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC3326719/]
- 53. Editorial: Molecular and cellular control of B cell responses [https://pmc.ncbi.nlm.nih.gov/articles/PMC10831292/]
- 54. A new perspective is needed for positive selection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8803856/]
- 55. Glucose oxidation-dependent survival of activated B cells ... [https://www.sciencedirect.com/science/article/pii/S258900422301564X]
- 56. Systems-based approaches to study immunometabolism [https://www.nature.com/articles/s41423-021-00783-9]
- 57. Deep metabolic profiling of immune cells by spectral flow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12268576/]
- 58. B cell intrinsic and extrinsic factors impacting memory ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.873886/full]
- 59. Mouse Models of Germinal Center Derived B-Cell Lymphomas [https://pmc.ncbi.nlm.nih.gov/articles/PMC8387591/]
- 60. Toll-like receptor mediated inflammation directs B cells ... [https://www.nature.com/articles/s41467-023-39734-5]
- 61. Murine models of germinal center derived-lymphomas - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5449224/]
- 62. In vivo analysis of CRISPR-edited germinal center murine ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1473760/full]
- 63. FcRL1, a New B-Cell-Activating Co-Receptor - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12250417/]
- 64. The half-century odyssey of regulatory B cells: from Breg ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1681082/full]
- 65. Advances in vaccine adjuvant development and future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12180328/]
- 66. Vaccine adjuvants: mechanisms and platforms [https://www.nature.com/articles/s41392-023-01557-7]
- 67. Distinct effects of adjuvants on B cell responses to protein ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1574941/full]
- 68. STAT5-c-Myc-axis regulates B cell metabolism in ... [https://www.sciencedirect.com/science/article/pii/S1995820X25000963]
- 69. Prediction and characterisation of the human B cell ... [https://www.nature.com/articles/s41467-025-61571-x]
- 70. Genetic adjuvants: A paradigm shift in vaccine ... [https://www.sciencedirect.com/science/article/pii/S2162253125000903]
- 71. Signaling Activation and Modulation in Extrafollicular B Cell... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11803499/]
- 72. B Cell Signaling and Activation in Autoimmunity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9914404/]
- 73. B cell metabolism in autoimmune diseases [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1232820/full]
- 74. B cell receptor repertoire abnormalities in autoimmune ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1326823/full]
- 75. B-cell immune checkpoint TIM-1: a potential target for ... [https://www.nature.com/articles/s41392-023-01643-w]
- 76. B cells and the coordination of immune checkpoint inhibitor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11029261/]
- 77. The dual immunomodulatory role of B cells in tumorigenesis [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1649812/full]
- 78. Organ-based characterization of B cells in patients with ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1509033/full]
- 79. Plasma Cell Differentiation Pathways in Systemic Lupus ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2018.00427/full]
- 80. Toll-like receptor signalling in B cells during systemic lupus ... [https://www.nature.com/articles/s41584-020-00544-4]
- 81. Immunopathogenesis of Systemic Lupus Erythematosus [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641968/]
- 82. New Mechanisms and Therapeutic Targets in Systemic Lupus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146670/]
- 83. B cell receptor repertoire abnormalities in autoimmune disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC10867955/]
- 84. BCR Affinity Influences T-B Interactions and B Cell ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.703918/full]
- 85. Biased Usage of V/D/J Genes and Clonal Diversity in IgG ... [https://pubmed.ncbi.nlm.nih.gov/40905528/]
- 86. B Cell Autonomous TLR Signaling and Autoimmunity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2763483/]
- 87. TLR signaling in B-cell development and activation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4003046/]
- 88. Mechanisms of B Cell Receptor Activation and Responses to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7352865/]
- 89. Germinal center‐derived lymphomas: The darkest side of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6518944/]
- 90. Genetic and epigenetic determinants of diffuse large B-cell ... [https://www.nature.com/articles/s41408-020-00389-w]
- 91. Role of Specific B-Cell Receptor Antigens in ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2020.604685/full]
- 92. B cell Receptor Signaling in Diffuse Large B cell Lymphoma [https://pmc.ncbi.nlm.nih.gov/articles/PMC4374122/]
- 93. Genetics of diffuse large B-cell lymphoma [https://www.sciencedirect.com/science/article/pii/S0006497120322084]
- 94. Molecular classification and therapeutics in diffuse large B ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2023.1124360/full]
- 95. Targeting B-cell receptor and PI3K signaling in diffuse ... [https://www.sciencedirect.com/science/article/pii/S0006497121014117]
- 96. The extrafollicular response is sufficient to drive initiation of ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.1021370/full]
- 97. Extrafollicular responses in humans and SLE - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6422038/]
- 98. The Diversity and Molecular Evolution of B-Cell Receptors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4839220/]
- 99. Signaling Activation and Modulation in Extrafollicular B ... [https://pubmed.ncbi.nlm.nih.gov/39917832/]
- 100. B Cell Receptor Signaling [https://www.cellsignal.com/pathways/b-cell-receptor-signaling?srsltid=AfmBOorFzt2zzGnstqNvuYOetYrzXM7im3Zh_MXBrNQt3e80Cv4Zvcz7]
- 101. The regulators of BCR signaling during B cell activation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8975005/]
- 102. Selective Depletion of Antigen-Specific Antibodies for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7934575/]
- 103. Cutting-edge approaches to B-cell depletion in ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1454747/full]
- 104. B cell receptor signaling in germinal centers prolongs survival ... [https://pubmed.ncbi.nlm.nih.gov/36882061/]
- 105. Inhibitors targeting Bruton's tyrosine kinase in cancers [https://www.nature.com/articles/s41375-020-01072-6]
- 106. Article Enhanced BCR signaling inflicts early plasmablast ... [https://www.sciencedirect.com/science/article/pii/S2589004221000067]
- 107. Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies [https://pubmed.ncbi.nlm.nih.gov/34905129/]
- 108. The Maintenance of Memory Plasma Cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6464033/]
- 109. B cell memory: building two walls of protection against ... [https://www.nature.com/articles/s41577-019-0244-2]
- 110. Immunological memory - Immunobiology - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK27158/]
- 111. Germinal center-mediated broadening of B cell responses ... [https://pubmed.ncbi.nlm.nih.gov/41071904/]
- 112. Memory B cell [https://en.wikipedia.org/wiki/Memory_B_cell]
- 113. Memory B Cell - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/memory-b-cell]
- 114. B-Cell Receptor - an overview [https://www.sciencedirect.com/topics/neuroscience/b-cell-receptor]
- 115. Discovery of surface biomarkers for cell mechanophenotype ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9239330/]
- 116. B-cell receptor signaling activity identifies patients with ... [https://www.nature.com/articles/s41598-024-55728-9]
- 117. Clinical Phenotypes, Serological Biomarkers, and Synovial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11083059/]
- 118. Identification of soluble biomarkers that associate with ... [https://www.nature.com/articles/s41590-025-02135-5]
- 119. Interpretation of Serological Complement Biomarkers in ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2018.02237/full]
- 120. A synthetic human 3D in vitro lymphoid model enhancing B ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9794978/]
- 121. Dynamics of genome architecture and chromatin function ... [https://www.nature.com/articles/s41467-020-20849-y]
- 122. Three-dimensional chromatin reorganization regulates B ... [https://www.nature.com/articles/s41556-024-01424-9]
- 123. Mapping and modelling human B cell maturation in the ... [https://www.sciencedirect.com/science/article/pii/S0952791524000189]
- 124. GERMINAL CENTER B CELL DYNAMICS - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5123673/]
- 125. Germinal center [https://en.wikipedia.org/wiki/Germinal_center]
- 126. Review The unique biology of germinal center B cells [https://www.sciencedirect.com/science/article/pii/S1074761321003010]
- 127. Integrating single-cell RNA and T cell/B cell receptor ... [https://www.nature.com/articles/s41590-024-02059-6]
- 128. A State-of-the-Art Roadmap for Biomarker-Driven Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9143954/]