Advanced Virtual Screening Strategies for STAT SH2 Domain Inhibitors: From Ultra-Large Libraries to Clinical Translation
This article provides a comprehensive overview of contemporary virtual screening (VS) strategies specifically tailored for identifying inhibitors of STAT protein SH2 domains, challenging targets in oncology drug discovery.
Advanced Virtual Screening Strategies for STAT SH2 Domain Inhibitors: From Ultra-Large Libraries to Clinical Translation
Abstract
This article provides a comprehensive overview of contemporary virtual screening (VS) strategies specifically tailored for identifying inhibitors of STAT protein SH2 domains, challenging targets in oncology drug discovery. It covers foundational concepts of STAT biology and the significance of the SH2 domain in protein-protein interactions. The scope extends to evaluating advanced methodological approaches, including ultra-high-throughput screening of billion-compound libraries, evolutionary algorithms like REvoLd, and AI-driven workflows such as Deep Docking. It also addresses critical troubleshooting aspects, such as overcoming scoring function inaccuracies and managing protein flexibility, and concludes with rigorous validation and comparative analysis of different VS pipelines. Designed for researchers and drug development professionals, this review synthesizes recent advances to guide the effective application of VS in targeting STAT-driven cancers.
Understanding STAT SH2 Domains: Biology, Therapeutic Significance, and Screening Challenges
The Role of STAT3 and STAT5b as Key Oncological Targets in Cancer
The Signal Transducer and Activator of Transcription (STAT) proteins are a family of transcription factors that play central roles in cytokine signaling, growth factor stimulation, and DNA transcription activation [1]. Among the seven STAT family members, STAT3 and STAT5b have been identified as critical drivers of oncogenesis, promoting cancer cell survival, proliferation, and immune evasion [2]. These proteins share a conserved multidomain structure consisting of six functional domains: an N-terminal domain (NTD), coiled-coil domain (CCD), DNA-binding domain (DBD), linker domain (LD), Src Homology 2 (SH2) domain, and transactivation domain (TAD) [1]. The SH2 domain is particularly crucial for STAT function, as it mediates phosphotyrosine recognition and facilitates the receptor recruitment and dimerization that are essential for STAT activation [3] [1].
Persistent activation of STAT3 and STAT5b is a hallmark of numerous malignancies. STAT3 hyperactivation has been documented in neuroblastoma, glioblastoma, osteosarcoma, hepatocellular carcinoma, nasopharyngeal carcinoma, renal cell carcinoma, lung cancer, colorectal cancer, pancreatic cancer, cervical cancer, esophageal cancer, ovarian cancer, and breast cancer [2]. Similarly, STAT5b is associated with breast cancer, colorectal cancer, lung cancer, prostate cancer, and leukemias [1]. The constitutive activation of these transcription factors drives tumorigenesis through multiple mechanisms, including promoting cancer stem cell (CSC) maintenance, epithelial-mesenchymal transition (EMT), drug resistance, and immune suppression [2]. Given their multifaceted roles in tumor biology, the SH2 domains of STAT3 and STAT5b have emerged as promising targets for therapeutic intervention in cancer treatment [3] [1].
Structural and Functional Significance of the SH2 Domain
Unique Features of STAT-type SH2 Domains
The SH2 domain is a modular protein unit that evolved approximately 600 million years ago and is integral to metazoan signal transduction [3]. STAT-type SH2 domains are structurally distinct from Src-type SH2 domains, featuring an α-helix (αB') at the C-terminus compared to the β-sheet found in Src-type domains [3]. This structural distinction has important implications for drug development, as the unique features of STAT-type SH2 domains create potential targeting opportunities not available in other SH2 domain-containing proteins.
All SH2 domains contain conserved structural motifs organized in an αβββα motif, with a central anti-parallel β-sheet (βB-βD strands) flanked by two α-helices (αA and αB) [3]. The β-sheet partitions the SH2 domain into two functionally critical subpockets:
- pY pocket (phosphate-binding pocket): Formed by the αA helix, BC loop, and one face of the central β-sheet, this pocket engages the phosphotyrosine residue of binding partners.
- pY+3 pocket (specificity pocket): Created by the opposite face of the β-sheet along with residues from the αB helix and CD and BC* loops, this pocket determines binding specificity by accommodating residues C-terminal to the phosphotyrosine [3].
The critical role of the SH2 domain in governing STAT transcriptional capacity, combined with its relatively shallow binding surfaces elsewhere on the protein, has made it a primary focus for small molecule inhibitor development [3]. However, STAT SH2 domains exhibit significant flexibility even on sub-microsecond timescales, with the accessible volume of the pY pocket varying dramatically—a crucial consideration for drug discovery efforts [3].
SH2 Domain Mutations in Human Cancers
Genomic sequencing of patient samples has revealed that the SH2 domain represents a hotspot in the mutational landscape of STAT proteins [3]. These mutations can have either activating or deactivating effects on STAT function, underscoring the delicate evolutionary balance of wild-type STAT structural motifs in maintaining precise levels of cellular activity.
Table 1: Disease-Associated Mutations in STAT3 SH2 Domain
| Mutation | Location | Pathology | Type | Effect |
|---|---|---|---|---|
| K591E/M | αA2, pY pocket | AD-HIES | Germline | Loss-of-function |
| S611G/N/I | βB7, pY pocket | AD-HIES | Germline | Loss-of-function |
| S614R | BC3, pY pocket | T-LGLL, NK-LGLL, ALK-ALCL, HSTL | Somatic | Gain-of-function |
| E616G/K | BC5, pY pocket | DLBCL, NKTL | Somatic | Gain-of-function |
| G617E/V/R | BC6, pY pocket | AD-HIES | Germline | Loss-of-function |
As shown in Table 1, specific mutations in the STAT3 SH2 domain are associated with distinct pathological conditions. Loss-of-function mutations (e.g., K591E/M, S611G/N/I, G617E/V/R) are typically germline mutations associated with immunological deficiencies such as autosomal-dominant Hyper IgE Syndrome (AD-HIES), which results from a diminished STAT3-mediated Th17 T-cell response [3]. In contrast, gain-of-function mutations (e.g., S614R, E616G/K) are often somatic mutations linked to various hematologic malignancies, including T-cell large granular lymphocytic leukemia (T-LGLL), natural killer LGLL (NK-LGLL), anaplastic large cell lymphoma (ALK-ALCL), hepatosplenic T-cell lymphoma (HSTL), diffuse large B-cell lymphoma (DLBCL), and natural killer T-cell lymphoma (NKTL) [3].
The functional impact of SH2 domain mutations stems from their effect on critical STAT processes. Conventional STAT activation begins with cytokine or growth-factor interactions with extracellular receptors, stimulating SH2 domain-mediated recruitment of tyrosine kinases and STAT isoforms to receptor cytoplasmic domains [3]. Following phosphorylation, STAT proteins form homo- or heterodimers through reciprocal phosphotyrosine-SH2 domain interactions, leading to nuclear translocation and DNA binding [3]. Mutations that disrupt phosphotyrosine binding or dimerization interface interactions can therefore profoundly alter STAT signaling output, either diminishing or enhancing transcriptional activity depending on the specific residue affected and the nature of the alteration.
Experimental Approaches for STAT SH2 Domain Research
Fluorescence Polarization Assay for SH2 Domain Inhibition Screening
Fluorescence polarization (FP) assays provide a robust method for assessing binding interactions between SH2 domains and phosphopeptides, making them invaluable for high-throughput screening of potential inhibitors [4]. The principle behind FP assays relies on the change in rotational mobility that occurs when a small fluorophore-labeled peptide binds to a much larger protein domain. When linearly polarized light excites the fluorophore, only molecules with proper spatial orientation relative to the plane of polarization are excited. The high rotational mobility of unbound peptide results in significant reorientation before emission, producing low polarization. When the peptide binds to the larger SH2 domain, its rotational mobility decreases substantially, resulting in higher polarization of emitted fluorescence [4].
The following protocol outlines a validated FP-based assay for screening STAT4 SH2 domain inhibitors, which can be adapted for STAT3 and STAT5b with appropriate modifications to peptide sequences:
Protocol: FP-Based High-Throughput Screening for SH2 Domain Inhibitors
Reagents and Equipment:
- Purified STAT SH2 domain protein (e.g., STAT3: amino acids 127-722; STAT4: amino acids 136-705; STAT5: corresponding constructs)
- Fluorophore-labeled phosphopeptide probe (e.g., 5-CF-GpYLPQNID for STAT4)
- Black 384-well microplates (Corning)
- Fluorescence plate reader capable of polarization measurements (e.g., Infinite F500, Tecan)
- Assay buffer: 10 mM Tris/HCl, 50 mM NaCl, 1 mM EDTA, 0.1% NP-40 substitute, 2% DMSO, 1 mM DTT, pH 8.0
Procedure:
- Protein Preparation: Express and purify the STAT SH2 domain protein with appropriate tags (e.g., N-terminal MBP and C-terminal 6×His tag). Dialyze against storage buffer (100 mM NaCl, 50 mM Hepes pH 7.5, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1% NP-40 substitute), snap-freeze in liquid nitrogen, and store at -80°C until use [4].
Binding Assay Setup:
- Prepare serial dilutions of the STAT SH2 domain protein in assay buffer.
- Incubate protein solutions for 1 hour at room temperature.
- Add fluorophore-labeled phosphopeptide to a final concentration of 10 nM.
- Incubate for 1 hour at room temperature.
Fluorescence Polarization Measurement:
- Transfer solutions to black 384-well microplates.
- Measure fluorescence polarization using appropriate filters (excitation: 485 nm, emission: 535 nm).
- Calculate normalized FP values by subtracting background polarization (fluorophore-labeled peptide alone).
Inhibition Assays:
- Pre-incubate STAT SH2 domain protein (33 nM) with test compounds or unlabeled competitor peptides for 1 hour.
- Add fluorophore-labeled peptide (10 nM) and incubate for 1 hour.
- Measure fluorescence polarization as above.
- Calculate IC50 values from dose-response curves and convert to inhibition constants (Ki) using the Cheng-Prusoff equation [4].
This assay has demonstrated excellent performance characteristics, with Z'-values of 0.85 ± 0.01 indicating high suitability for high-throughput screening campaigns [4]. For STAT3 and STAT5b, optimal phosphopeptide sequences should be selected based on known SH2 domain binding preferences, such as GpYLPQTV for STAT3 [4].
Virtual Screening Strategies for STAT SH2 Domain Inhibitors
Virtual screening has emerged as a powerful complement to experimental high-throughput screening for identifying STAT SH2 domain inhibitors. Recent advances in ultrahigh-throughput virtual screening (uHTVS) of synthetically accessible compound libraries containing billions of compounds have revolutionized hit identification [1]. The following protocol describes an AI-enhanced virtual screening workflow that has successfully identified inhibitors for STAT3 and STAT5b SH2 domains.
Protocol: AI-Enhanced Virtual Screening for STAT SH2 Domain Inhibitors
Data Sets and Compound Libraries:
- Knowledge-based libraries: Specialized collections such as the OTAVAchemicals SH2 Domain Targeted Library (1,807 compounds) or natural product libraries (∼193,000 compounds)
- Ultralarge screening libraries: Enamine REAL (5.51 billion compounds) or Mcule-in-stock (5.59 million compounds)
- Benchmark set: 117,500 chemically diverse compounds from Mcule-in-stock for validation
Procedure:
- Target Preparation:
- Select appropriate X-ray structure of STAT SH2 domain through retrospective virtual screening.
- Prepare protein structure by adding hydrogen atoms, assigning partial charges, and defining binding site (typically the pY and pY+3 pockets).
Retrospective Validation:
- Compile known active compounds and decoy molecules (e.g., 69 known STAT3 actives from ChEMBL + 959 decoys from DUD-E).
- Perform docking with multiple protein structures and settings.
- Evaluate performance using Area Under the ROC Curve (AUC) and Enrichment Factors (EF) at 1%, 2%, and 5% to select optimal docking parameters.
AI-Enhanced Screening (Deep Docking):
- Train deep learning model on a subset of the ultralarge library (∼100,000 compounds) docked to the STAT SH2 domain.
- Use trained model to predict binding scores for remaining compounds in the library.
- Select top-ranked compounds (typically 0.1-1% of library) for physical docking.
- Cluster docking results and select diverse hits for experimental validation.
Traditional Virtual Screening:
- Perform brute-force docking of knowledge-based libraries (SH2-targeted or natural product collections).
- Apply drug-like filters (Lipinski's Rule of Five, Veber criteria) and remove pan-assay interference compounds (PAINS).
- Select top-ranked compounds for experimental testing.
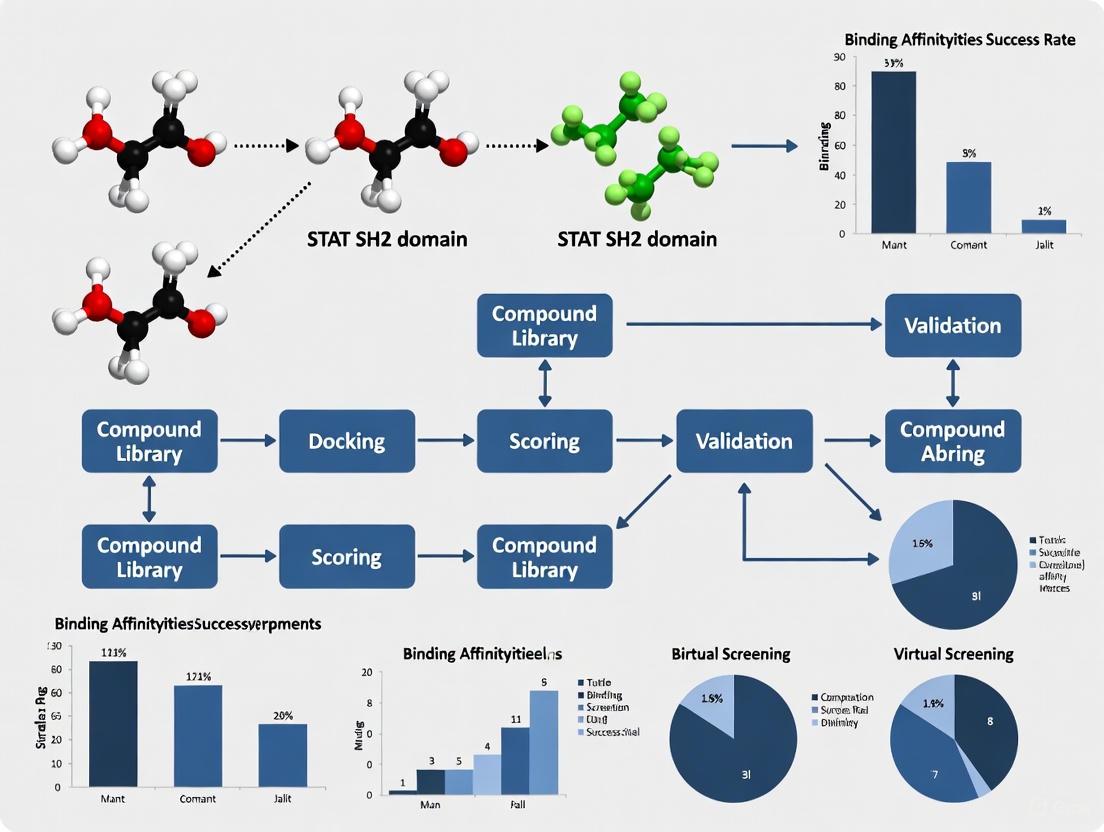
This approach has demonstrated exceptional hit rates, reaching 50.0% for STAT3 SH2 domain and 42.9% for STAT5b SH2 domain in prospective validation studies [1]. The method is particularly valuable for challenging protein-protein interaction targets like STAT SH2 domains, where traditional screening methods often struggle.
Bacterial Peptide Display for SH2 Domain Specificity Profiling
Understanding the sequence specificity of SH2 domain-phosphopeptide interactions is crucial for rational inhibitor design. Bacterial peptide display combined with deep sequencing provides a high-throughput platform for profiling SH2 domain binding specificities [5].
Protocol: Bacterial Peptide Display for SH2 Domain Specificity Profiling
Library Design:
- X5-Y-X5 library: Random 11-residue sequences with central tyrosine (10^6-10^7 diversity)
- pTyr-Var library: Defined sequences spanning 3000 human tyrosine phosphorylation sites plus 5000 variants with disease-associated mutations and natural polymorphisms
Procedure:
- Library Construction:
- Clone peptide libraries into bacterial surface display vector (e.g., eCPX fusion).
- Transform into E. coli cells to create library diversity.
Bait Protein Preparation:
- Generate biotinylated SH2 domains as bait proteins.
Screening:
- Incubate bacterial peptide library with purified tyrosine kinase to phosphorylate tyrosine residues (for kinase specificity profiling).
- For SH2 domain binding screens, use pre-phosphorylated libraries or incorporate phosphotyrosine via genetic code expansion.
- Capture binding cells using avidin-functionalized magnetic beads and biotinylated SH2 domain bait proteins.
- Isolate bound cells and amplify for subsequent rounds of selection.
Deep Sequencing Analysis:
- Extract genomic DNA from input and selected populations.
- Amplify peptide-encoding regions and subject to deep sequencing.
- Calculate enrichment ratios for each peptide sequence between selected and input populations.
- Generate position-specific scoring matrices or sequence logos representing SH2 domain binding preferences.
This method has been successfully applied to quantify the impact of phosphosite-proximal mutations on SH2 domain binding and can be adapted to profile inhibitor specificity across multiple SH2 domains [5].
Research Reagent Solutions for STAT SH2 Domain Studies
Table 2: Essential Research Reagents for STAT SH2 Domain Studies
| Reagent Category | Specific Examples | Application and Function |
|---|---|---|
| Expression Constructs | STAT3 (aa 127-722), STAT4 (aa 136-705), STAT5 SH2 domains | Protein production for biochemical and structural studies |
| Fluorescent Peptide Probes | 5-CF-GpYLPQNID (STAT4), 5-CF-GpYLPQTV (STAT3) | Fluorescence polarization assays to measure binding affinity and inhibition |
| Competitor Peptides | Ac-GpYLPQNID, Ac-pYLPQTV-NH₂ | Positive controls for competition assays and determination of Ki values |
| Virtual Screening Libraries | OTAVAchemicals SH2 Domain Targeted Library, Enamine REAL, Mcule-in-stock | Source compounds for virtual screening campaigns |
| Bacterial Display Libraries | X5-Y-X5 random library, pTyr-Var proteomic library | Profiling SH2 domain binding specificity and sequence requirements |
| Reference Inhibitors | Stattic, SH-4-54 (STAT3); Pimozide (STAT5) | Benchmark compounds for assay validation and comparison |
Signaling Pathways and Experimental Workflows
Discussion and Future Perspectives
The development of targeted therapies against STAT3 and STAT5b represents a promising frontier in cancer treatment. The critical role of the SH2 domain in STAT activation makes it an attractive target for small molecule inhibitors, particularly given that current approaches targeting upstream kinases often lead to feedback activation and drug resistance [2]. However, several challenges remain in translating STAT SH2 domain inhibitors to clinical use.
Key challenges include the shallow, hydrophilic nature of the pY binding pocket, which complicates the design of high-affinity small molecules; the high flexibility of STAT SH2 domains, which adopt multiple conformational states; and the need for isoform selectivity to minimize off-target effects [3] [1]. Despite these hurdles, recent advances in screening technologies and structural biology have created new opportunities for STAT-targeted therapeutics.
Emerging approaches include proteolysis-targeting chimeras (PROTACs) that degrade STAT proteins rather than merely inhibiting them, nanoparticle-based delivery systems to improve bioavailability, and combination therapies that simultaneously target STAT signaling and complementary pathways such as immune checkpoints [2]. The systematic profiling of STAT5B across cancer types has revealed its potential tumor-suppressive role in certain contexts, particularly in lung cancers and hematologic malignancies where high STAT5B expression correlates with favorable prognosis [6]. This context-dependent functionality underscores the importance of patient stratification strategies for STAT-targeted therapies.
The integration of virtual screening with high-throughput experimental validation represents a powerful strategy for accelerating STAT inhibitor discovery. The remarkable hit rates achieved by AI-enhanced virtual screening (50.0% for STAT3 SH2 domain) demonstrate the potential of this approach to identify novel chemical starting points for drug development [1]. As structural information on STAT SH2 domains continues to expand and screening methodologies improve, the prospects for clinically effective STAT3 and STAT5b inhibitors continue to brighten, offering new hope for patients with STAT-driven cancers.
The Src Homology 2 (SH2) domain is a structurally conserved protein module of approximately 100 amino acids that plays a fundamental role in intracellular signal transduction by specifically recognizing and binding to phosphotyrosine (pTyr) motifs [7] [8]. Found in over 100 human proteins involved in tyrosine kinase signaling cascades, including kinases, phosphatases, adaptor proteins, and transcription factors, SH2 domains serve as critical "reader" modules that translate tyrosine phosphorylation events into downstream cellular responses [7] [9] [10]. The fundamental importance of SH2 domains is evidenced by their involvement in crucial processes such as cell growth, differentiation, survival, and migration, with dysregulation contributing to various diseases, especially cancer and immunodeficiencies [7] [9] [10].
SH2 domains function within an elaborate pTyr signaling system consisting of three major components: protein tyrosine kinases (PTKs) as "writers" that create the phosphorylation mark, SH2 domains as "readers" that recognize this mark, and protein tyrosine phosphatases (PTPs) as "erasers" that remove the phosphate group to terminate signaling [9] [10]. This sophisticated system allows eukaryotic cells to coordinate complex signaling networks that respond to extracellular stimuli with precise spatial and temporal control.
Table 1: Key Characteristics of SH2 Domains
| Feature | Description | Significance |
|---|---|---|
| Size | ~100 amino acids [7] [8] | Compact modular domain |
| Prevalence | 120 SH2 domains in 110 human proteins [11] [9] | One of the largest families of pTyr readers |
| Structural Motif | Central antiparallel β-sheet flanked by two α-helices (αβββα) [11] [12] | Highly conserved tertiary structure |
| Key Binding Residue | Conserved arginine on βB strand (ArgβB5) [13] [9] [10] | Forms bidentate hydrogen bonds with phosphate moiety |
| Binding Affinity Range | 0.1-10 μM (typical KD values) [9] [10] | Allows transient interactions for dynamic signaling |
Structural Architecture of SH2 Domains
The SH2 domain adopts a highly conserved three-dimensional structure characterized by a central antiparallel β-sheet consisting of three major strands (βB, βC, βD), flanked on both sides by two α-helices (αA and αB) [9] [12]. This core αβββα motif forms a compact globular domain that presents a binding surface for phosphorylated tyrosine residues. The central β-sheet effectively divides the phosphopeptide binding surface into two adjacent binding pockets: the phosphotyrosine-binding pocket (pY pocket) and the specificity pocket (pY+3 pocket) [11] [12]. This structural arrangement allows SH2 domains to recognize their target sequences in an extended conformation perpendicular to the central β-strands.
Molecular Determinants of Phosphotyrosine Recognition
The pY pocket is located in the N-terminal half of the SH2 domain and is primarily responsible for engaging the phosphotyrosine residue. A strictly conserved arginine residue (ArgβB5) from the βB strand serves as the central coordinator for phosphate binding, forming a bidentate salt bridge with two oxygen atoms of the phosphate moiety [9] [12] [10]. Additional positively charged residues, including ArgαA2 and LysβD6 (in many but not all SH2 domains), provide supplementary interactions that stabilize phosphate binding [10]. The remarkable conservation of this arginine across virtually all SH2 domains underscores its fundamental role in pTyr recognition, with mutations at this position typically abolishing phosphopeptide binding capacity [12] [10].
Specificity Determinants and Peptide Recognition
The C-terminal half of the SH2 domain contains the specificity pocket (pY+3 pocket), which engages residues C-terminal to the phosphotyrosine and confers sequence selectivity [9] [10]. This predominantly hydrophobic pocket is formed by the DE, EF, and BG loops, along with elements from βD and αB, and accommodates the side chain of the residue at the pY+3 position [9] [10]. The structural composition and configuration of these loops vary significantly among different SH2 domains, thereby dictating whether a particular domain has specificity for hydrophobic, acidic, or basic residues at the +1, +2, or +3 positions relative to pTyr. This variability in the specificity pocket enables the human complement of SH2 domains to recognize distinct pTyr motifs, thereby ensuring precise signaling specificity within complex cellular networks.
SH2 Domain Functions in Cellular Signaling
SH2 domains mediate critical protein-protein interactions that underlie numerous signaling pathways in metazoans. Their functions can be categorized into several key mechanistic roles:
Recruitment and Assembly of Signaling Complexes
SH2 domains serve as modular adaptors that recruit downstream effector proteins to activated, tyrosine-phosphorylated receptor tyrosine kinases (RTKs) at the plasma membrane [8] [10]. A classic example is the adapter protein Grb2, which uses its SH2 domain to bind specific pTyr sites on activated growth factor receptors, thereby localizing the guanine nucleotide exchange factor SOS to the membrane where it can activate Ras and initiate the MAPK signaling cascade [10]. This recruitment function enables the spatial and temporal assembly of multiprotein signaling complexes in response to extracellular stimuli.
Regulation of Enzymatic Activity
In many signaling proteins, SH2 domains play an allosteric regulatory role that controls catalytic activity. This is particularly well-characterized in the Src family kinases (SFKs), where the SH2 domain mediates intramolecular interactions that maintain the kinase in an autoinhibited state [14] [9]. In SFKs, the SH2 domain binds to a phosphotyrosine motif in the C-terminal tail of the kinase itself, forming a closed conformation that sterically hinders substrate access to the active site [14]. Activation occurs when competitive binding of a higher-affinity external pTyr ligand to the SH2 domain disrupts this intramolecular interaction, resulting in kinase activation [14].
Substrate Targeting and Processive Phosphorylation
For tyrosine kinases, SH2 domains can facilitate substrate recognition and enable processive phosphorylation of multiple sites on target proteins. Active Src family kinases, for instance, use their SH2 domains for intermolecular interactions that allow multisite processive phosphorylation of substrates [14]. This function enhances signaling efficiency and fidelity by ensuring that specific substrates are preferentially phosphorylated by their cognate kinases.
Experimental Approaches for Studying SH2 Interactions
High-Throughput SH2 Profiling
Global analysis of SH2 domain interactions provides comprehensive insights into tyrosine phosphorylation signaling networks. Proteomic binding assays encompassing nearly the full complement of human SH2 domains have been developed to profile the global tyrosine phosphorylation state of cells [15]. These approaches typically employ:
- Large-scale far-western analyses to assess SH2 domain binding to cellular proteins
- Reverse-phase protein arrays to generate comprehensive, quantitative SH2 binding profiles for phosphopeptides, recombinant proteins, and entire proteomes
- Interaction proteomics to identify specific proteins whose tyrosine phosphorylation and SH2 binding are modulated by specific cellular stimuli
These high-throughput methods have been successfully applied to profile adhesion-dependent SH2 interactions in fibroblasts, identifying specific focal adhesion complex proteins whose phosphorylation state and SH2 binding capacity change in response to cell adhesion [15].
Structure-Based Inhibitor Design
The therapeutic potential of targeting SH2 domains has motivated detailed structural studies and inhibitor development efforts. Structure-based drug discovery approaches have been successfully applied to identify potential small-molecule inhibitors for SH2 domains, such as the N-SH2 domain of SHP2 phosphatase [13]. The general methodology includes:
Diagram 1: SH2 inhibitor discovery workflow.
Molecular docking studies followed by molecular dynamics simulations and MM/PBSA calculations have identified promising inhibitor candidates, such as compound CID 60838 (Irinotecan), which showed a binding free energy value of -64.45 kcal/mol and significant interactions with key residues including the critical Arg32 in the N-SH2 domain of SHP2 [13]. These computational approaches provide valuable insights for developing therapeutic compounds that disrupt pathological SH2-mediated interactions in cancer and other diseases.
Table 2: Key Research Reagents for SH2 Domain Studies
| Research Tool | Composition/Type | Research Application | Key Features |
|---|---|---|---|
| Monobodies [14] | Synthetic binding proteins based on fibronectin type III scaffold | Selective inhibition of SFK SH2 domains | Nanomolar affinity, high selectivity, pY-competitive |
| SH2 Superbinder [9] | Engineered SH2 domain with enhanced pY binding | Dominant-negative disruption of pY signaling | Broad pY recognition, altered signaling outcomes |
| SH2db [11] | Database of SH2 domain structures and sequences | Structural bioinformatics and comparative analysis | Generic residue numbering, integrated AlphaFold models |
| Phosphopeptide Libraries [15] | Collections of pY-containing peptides | Specificity profiling and binding studies | Represents natural SH2 binding motifs |
Advanced Binding Reagents: Monobodies
Monobodies are synthetic binding proteins developed from the fibronectin type III domain scaffold that offer exceptional potency and selectivity in targeting SH2 domains [14]. These engineered proteins have been generated for six of the eight Src family kinase (SFK) SH2 domains with nanomolar affinity and strong selectivity for either the SrcA (Yes, Src, Fyn, Fgr) or SrcB (Lck, Lyn, Blk, Hck) subgroups [14]. The application of monobodies includes:
- Dissecting SFK functions in normal development and signaling
- Interfering with aberrant SFK signaling in cancer cells
- Selective perturbation of kinase regulation and downstream signaling
- Intracellular expression for target validation and functional studies
Crystal structures of monobody-SH2 complexes have revealed distinct and only partly overlapping binding modes that rationalize the observed selectivity and enable structure-based mutagenesis to modulate inhibition mode and selectivity [14].
SH2 Domains in Therapeutic Development
SH2 Domains as Drug Targets
The critical roles of SH2 domains in disease processes, particularly in oncology, have made them attractive targets for therapeutic intervention. Disease-associated mutations in SH2 domains have been identified in numerous conditions. For example, gain-of-function mutations in the N-SH2 domain of SHP2 phosphatase that disrupt its autoinhibitory conformation are implicated in Noonan syndrome, LEOPARD syndrome, and juvenile myelomonocytic leukemia [13] [10]. Similarly, mutations in the SH2D1A gene, which encodes the SAP protein (consisting almost exclusively of an SH2 domain), lead to X-linked lymphoproliferative syndrome [10].
Targeting Challenges and Innovative Strategies
Targeting SH2 domains with small molecules has historically been challenging due to the shallow, charged nature of the pY binding pocket and the high conservation among different SH2 domains [11]. However, several innovative approaches have emerged:
- Peptidomimetic compounds that replicate key features of phosphopeptide ligands
- Structure-based design leveraging crystallographic and computational data
- Alternative binding modalities such as monobodies that target unique structural epitopes
- Allosteric inhibitors that exploit regulatory mechanisms rather than direct pY pocket competition
These approaches have yielded promising leads, such as inhibitors developed for the p56lck SH2 domain using molecular docking and in silico scaffold hopping approaches [16]. The resulting compounds showed favorable predicted binding affinities and drug-like properties, suggesting their potential as starting points for antibiotic development given the role of Src family kinases in bacterial invasion [16].
Application to STAT SH2 Domain Inhibitor Screening
Virtual Screening Strategies
The development of inhibitors targeting STAT (Signal Transducer and Activator of Transcription) SH2 domains represents a promising therapeutic approach for cancer and inflammatory diseases. Structure-based virtual screening protocols can be optimized for STAT SH2 domains by incorporating the following key considerations:
- Pocket Selection: Focus on the conserved pY pocket and adjacent specificity determinants that recognize the pY-X-pY motif characteristic of STAT SH2 domains
- Conserved Interactions: Prioritize compounds capable of engaging the critical arginine residue (ArgβB5) and other conserved phosphate-coordinating residues
- Specificity Design: Exploit unique structural features of the STAT SH2 specificity pocket to enhance selectivity over other SH2 domains
Experimental Validation Workflow
Following virtual screening, a tiered experimental approach provides comprehensive characterization of putative STAT SH2 inhibitors:
Diagram 2: STAT inhibitor validation cascade.
This workflow progresses from in vitro binding assays such as surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) to determine affinity and thermodynamics, to cellular functional assays assessing inhibition of STAT phosphorylation, dimerization, nuclear translocation, and target gene expression [10]. Comprehensive selectivity profiling across a panel of SH2 domains ensures specificity for the intended STAT target, minimizing potential off-target effects on other SH2-mediated signaling pathways.
The integration of structural insights, computational screening, and rigorous experimental validation provides a powerful framework for developing next-generation therapeutics that target pathological SH2 interactions in cancer and other diseases, with STAT family transcription factors representing particularly promising targets for this approach.
STAT SH2 Domains as Challenging Protein-Protein Interaction (PPI) Targets
Signal Transducer and Activator of Transcription (STAT) proteins are a family of transcription factors with key roles in cytokine signaling, growth factor stimulation, and DNA transcription activation [1]. Among the seven STAT family members, STAT3 and STAT5b are particularly significant in oncology, as their constitutive activation is directly linked to various human cancers, including leukemias, melanoma, breast cancer, and prostate cancer [1] [17]. STAT proteins share a conserved domain architecture consisting of six domains: the N-terminal domain (NTD), coiled-coil domain (CCD), DNA-binding domain (DBD), linker domain (LD), Src Homology 2 (SH2) domain, and transcription activation domain (TAD) [1].
The SH2 domain is the most critical module for STAT activation and function. This approximately 100-amino-acid domain specifically recognizes phosphotyrosine (pTyr) motifs and mediates STAT dimerization through a reciprocal phosphotyrosine-SH2 interaction [18] [1] [19]. Upon phosphorylation at a conserved tyrosine residue (Y705 in STAT3), two STAT monomers form an active dimer via their SH2 domains, enabling nuclear translocation and DNA binding [20] [17]. This makes the STAT-SH2 domain a compelling target for therapeutic intervention in cancer and other diseases driven by aberrant STAT signaling.
Table 1: Key Characteristics of STAT SH2 Domains
| Feature | Description | Functional Significance |
|---|---|---|
| Size | ~100 amino acids [18] | Compact structural domain |
| Primary Function | Binds phosphotyrosine (pTyr) motifs [18] | Mediates specific protein-protein interactions |
| Structural Motif | Central antiparallel β-sheet flanked by α-helices (αβββα) [21] | Highly conserved fold |
| Key Binding Residue | Arginine at βB5 position in FLVR motif [18] | Essential for phosphotyrosine recognition |
| STAT Dimerization | Reciprocal pTyr-SH2 interaction between STAT monomers [17] | Critical for STAT activation and nuclear translocation |
Structural Biology of STAT SH2 Domains
Architecture and Classification
SH2 domains adopt a conserved three-dimensional structure described as a "sandwich" consisting of a central antiparallel β-sheet flanked by two α-helices [18]. The basic structural organization follows an αA-βB-βC-βD-αB pattern, with most SH2 domains containing additional secondary structural elements [18] [19]. The N-terminal region is highly conserved and contains a deep pocket within the βB strand that binds the phosphate moiety of phosphotyrosine [18].
STAT SH2 domains belong to a distinct structural subclass characterized by the absence of βE and βF strands found in Src-type SH2 domains [21] [19]. Instead, STAT-type SH2 domains feature a split αB helix, an adaptation believed to facilitate the dimerization required for STAT transcriptional function [19]. This structural divergence reflects the evolutionary ancestry of STAT SH2 domains, which predate animal multicellularity and represent one of the most ancient functional SH2 domain templates [21].
Molecular Recognition Mechanism
The SH2 domain recognizes phosphorylated tyrosine residues through a "two-pronged plug" mechanism involving two adjacent binding sites [22] [23]:
- Phosphotyrosine (pTyr) binding pocket: A deep basic pocket that coordinates the phosphate moiety of phosphotyrosine through critical hydrogen bonds and salt bridges.
- Specificity pocket: Adjacent to the pTyr pocket, this region recognizes amino acid residues C-terminal to the phosphotyrosine, typically with preference for specific residues at the +3 position.
The pTyr binding pocket contains a highly conserved arginine residue at position βB5 (part of the "FLVR" motif) that directly coordinates the phosphate group through a salt bridge [18] [23]. Mutation of this arginine reduces binding affinity by up to 1000-fold, demonstrating its critical role in phosphotyrosine recognition [23]. Additional conserved basic residues at positions αA2 and βD6 further contribute to phosphate coordination [23].
The STAT3 SH2 domain binding pocket can be divided into three sub-pockets designated pY+X (hydrophobic side), pY+0 (binds pY705), and pY+1 (binds L706) [17]. Key residues involved in ligand binding include Arg609, Glu594, Lys591, Ser636, Ser611, Val637, Tyr657, Gln644, Thr640, Glu638, and Trp623 [17].
Challenges in Targeting STAT SH2 Domains
Molecular and Cellular Barriers
Targeting STAT SH2 domains for therapeutic intervention presents several formidable challenges:
- Charge and bioavailability: Phosphotyrosine and its isosteres contain multiple negative charges, resulting in poor cytosolic penetration and bioavailability [20].
- Rapid dephosphorylation: Phosphotyrosine residues are rapidly hydrolyzed in the cytosol by protein tyrosine phosphatases (PTPs), limiting the stability of phosphopeptide-based inhibitors [20].
- Solvent-exposed PPI interface: The protein-protein interaction interface of STAT SH2 domains is large and solvent-exposed, making it difficult to target with small molecules [1].
- Specificity challenges: The high sequence conservation among human SH2 domains (approximately 120 domains across 110 proteins) poses significant challenges for achieving selective inhibition [14].
Experimental Hurdles in Inhibitor Development
Despite extensive efforts to develop STAT3 SH2 domain inhibitors, many promising candidates have failed to demonstrate efficacy in cellular models. Research has shown that peptides combining STAT3-specific binding sequences with difluorophosphonomethyl phenylalanine (F2Pmp) as a phosphatase-stable phosphotyrosine mimetic and cell-penetrating peptides (CPPs) for enhanced delivery still showed no STAT3 inhibitory activity in cells, despite substantial cytosolic delivery and stability [20]. This highlights the delicate balance required between target affinity, resistance to degradation, and cytosolic penetration for effective SH2 domain inhibitors.
Computational Approaches for STAT SH2 Inhibitor Discovery
Virtual Screening Methodologies
Computational screening has emerged as a powerful strategy for identifying STAT SH2 domain inhibitors, particularly given the challenges of targeting protein-protein interactions. Current approaches include:
- Ultrahigh-throughput virtual screening (uHTVS): AI-assisted screening of ultralarge (10⁸+ compounds) synthetically accessible libraries [1].
- Deep Docking: Machine learning-based workflow that reduces computational cost by using iterative deep learning to prioritize compounds for docking [1].
- Multi-level precision docking: Hierarchical screening using High-Throughput Virtual Screening (HTVS), Standard Precision (SP), and Extra Precision (XP) modes [17].
- Molecular Mechanics/Generalized Born Surface Area (MM-GBSA): Calculations to determine binding free energy and prioritize hits [17].
These computational methods have demonstrated remarkable success, with Deep Docking achieving hit rates as high as 50.0% for STAT3 SH2 domain inhibitors in prospective screens [1].
Table 2: Performance of Virtual Screening Approaches Against STAT SH2 Domains
| Screening Approach | Compound Library | Hit Rate | Key Advantages |
|---|---|---|---|
| Deep Docking [1] | Enamine REAL (5.51B compounds) | 50.0% (STAT3) | Exceptional hit rates; feasible without supercomputers |
| Economic Deep Docking [1] | Mcule-in-stock (5.59M compounds) | 42.9% (STAT5b) | Cost-effective; only ~120,000 compounds actually docked |
| Knowledge-Based Screening [1] | OTAVA SH2 Targeted Library (1,807 compounds) | Not specified | Focused on compounds with predicted SH2 domain affinity |
| Natural Product Screening [1] [17] | Natural product libraries (193,757 compounds) | Not specified | Leverages inherent bioactivity and structural diversity |
Structure-Based Drug Design Protocols
Protocol 1: Molecular Docking and Virtual Screening Workflow
This protocol outlines a comprehensive computational approach for identifying STAT SH2 domain inhibitors through virtual screening [17]:
Protein Preparation
- Retrieve STAT3 SH2 domain structure from PDB (e.g., 6NJS, resolution 2.70 Å)
- Process structure using Protein Preparation Wizard (Schrödinger)
- Add hydrogen atoms, fill missing side chains, assign bond orders
- Optimize hydrogen bonding network and minimize energy using OPLS3e force field
Ligand Library Preparation
- Retrieve natural compounds from ZINC15 database (182,455 compounds)
- Prepare 3D structures with LigPrep (Schrödinger)
- Generate ionization states at pH 7.4 ± 0.5
- Apply OPLS3e force field for energy minimization
Receptor Grid Generation
- Define binding site using co-crystallized ligand coordinates
- Set grid box dimensions: X:13.22, Y:56.39, Z:0.27 (length: 20 Å)
- Validate grid by redocking native ligand (RMSD < 2.0 Å)
Hierarchical Docking Protocol
- Step 1: High-Throughput Virtual Screening (HTVS) of entire library
- Step 2: Standard Precision (SP) docking of top ~30% compounds from HTVS
- Step 3: Extra Precision (XP) docking of top-scoring compounds (cut-off: -6.5 kcal/mol)
Binding Affinity Assessment
- Perform MM-GBSA calculations on top hits
- Calculate binding free energy using OPLS3e force field and VSGB solvent model
- Prioritize compounds with most favorable ΔG binding values
Pharmacokinetic Property Prediction
- Analyze drug-like properties using QikProp
- Evaluate adherence to Lipinski's rule of five and Veber criteria
- Assess absorption, distribution, metabolism, and excretion (ADME) properties
Protocol 2: AI-Enhanced Ultrahigh-Throughput Virtual Screening
For screening billion-compound libraries, AI-enhanced approaches provide computational efficiency [1]:
Library Selection
- Obtain synthetically accessible compound library (e.g., Enamine REAL, 5.51 billion compounds)
- Apply Lipinski's rule of five and Veber criteria filters
- Remove pan-assay interference compounds (PAINS)
Benchmark Set Preparation
- Select diverse subset of compounds (e.g., 117,500 compounds) using RDKit Diversity Picker
- Include known actives from ChEMBL and decoy molecules from DUD-E database
Deep Docking Implementation
- Perform initial docking on benchmark set to generate training data
- Train deep neural network to predict docking scores based on chemical features
- Apply trained model to prioritize compounds from full library for docking
- Iteratively refine model based on docking results
Validation and Hit Identification
- Dock top-prioritized compounds (typically 1-5% of full library)
- Select compounds with best docking scores for experimental validation
- Confirm binding through secondary assays (SPR, ITC, FP)
Experimental Validation and Characterization
Biochemical Assay Protocols
Protocol 3: Fluorescence Polarization (FP) Binding Assay
This protocol enables quantitative measurement of inhibitor binding to STAT SH2 domains [20]:
Reagent Preparation
- Express and purify recombinant STAT3 SH2 domain protein
- Prepare fluorescein-labeled phosphopeptide tracer (e.g., Flu-G(pTyr)LPQTV-NH₂)
- Serially dilute test compounds in assay buffer (PBS, pH 7.4, 0.01% Triton X-100)
Assay Setup
- Prepare reaction mixtures in 384-well black plates:
- Constant tracer concentration (5-10 nM)
- Varying STAT3 SH2 domain concentrations (0-100 μM for Kd determination)
- Or constant protein with varying inhibitor concentrations (for IC50 determination)
- Include controls: blank (tracer only), full binding (tracer + protein), competition (unlabeled reference peptide)
- Prepare reaction mixtures in 384-well black plates:
Measurement and Data Analysis
- Incubate plates for 60 minutes at room temperature in the dark
- Measure fluorescence polarization using plate reader (λex = 485 nm, λem = 535 nm)
- Calculate normalized fluorescence polarization values
- Fit data to appropriate binding models to determine Kd or IC50 values
Protocol 4: Cell-Based STAT3 Transcriptional Reporter Assay
This protocol assesses functional inhibition of STAT3 signaling in cellular models [20]:
Cell Line Preparation
- Maintain U3A fibrosarcoma cells (STAT1-deficient) or other STAT3-responsive cells
- Culture in DMEM with 10% FBS, penicillin/streptomycin at 37°C, 5% CO₂
Reporter Construct Transfection
- Transfect cells with STAT3-responsive luciferase reporter (e.g., pLucTKS3)
- Include constitutive Renilla luciferase control for normalization
- Use appropriate transfection reagent (e.g., lipofectamine)
Compound Treatment and Stimulation
- Pre-treat cells with test compounds (0-25 μM) for 1-2 hours
- Stimulate with IL-6 (50 ng/mL) or oncostatin M (10 ng/mL) for 6-8 hours
- Include controls: unstimulated, stimulated without inhibitor, reference inhibitor
Luciferase Activity Measurement
- Lyse cells and measure firefly and Renilla luciferase activities
- Calculate normalized luciferase activity (firefly/Renilla ratio)
- Express results as percentage inhibition compared to stimulated control
Advanced Targeting Strategies
Emerging strategies for targeting STAT SH2 domains include:
- Non-peptidic small molecules: Development of compounds with reduced charge and improved pharmacokinetic properties [20] [1].
- Protein-based inhibitors: Engineered monobodies and other binding proteins that achieve high affinity and selectivity [14].
- Lipid-binding pocket targeting: Exploitation of SH2 domain-lipid interactions for allosteric modulation [18] [19].
- Multivalent inhibitors: Compounds that simultaneously target multiple STAT domains or interaction interfaces.
Research Reagent Solutions
Table 3: Essential Research Reagents for STAT SH2 Domain Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Recombinant Proteins | STAT3 SH2 domain (expressed and purified) [20] | Binding assays, structural studies, screening |
| Peptide Inhibitors | Ac-G(pTyr)LPQTV-NH₂ (gp130-derived) [20] | High-affinity positive control for binding studies |
| Phosphotyrosine Mimetics | F2Pmp (difluorophosphonomethyl phenylalanine) [20] | Phosphatase-stable pTyr replacement in peptide inhibitors |
| Cell-Penetrating Peptides | CPP12 (cyclo(FφR₄) improved version) [20] | Enhanced cytosolic delivery of peptide inhibitors |
| Chemical Libraries | OTAVA SH2 Domain Targeted Library [1] | Knowledge-based screening focused on SH2 domains |
| Natural Product Libraries | Zinc15 Natural Product Collection [17] | Screening of structurally diverse natural compounds |
| Reporter Cell Lines | U3A fibrosarcoma STAT3 reporter cells [20] | Functional assessment of STAT3 pathway inhibition |
| Reference Inhibitors | Stattic, SD-36 [17] | Benchmark compounds for validation experiments |
STAT SH2 domains represent challenging but therapeutically valuable targets in oncology and inflammatory diseases. Their critical role in STAT activation through dimerization, combined with the difficulties in targeting large, solvent-exposed PPI interfaces, has driven the development of sophisticated computational and experimental approaches. The integration of AI-enhanced virtual screening with rigorous biochemical and cellular validation provides a powerful framework for identifying novel STAT SH2 domain inhibitors with improved potency, selectivity, and drug-like properties. As our understanding of SH2 domain biology and chemical targeting continues to advance, these approaches hold significant promise for delivering new therapeutic agents that disrupt aberrant STAT signaling in human disease.
In modern drug discovery, the concept of "chemical space" represents the multidimensional universe of all possible organic compounds. Navigating this vast space efficiently is crucial for identifying hit compounds against therapeutic targets. This application note examines two complementary strategies for exploring chemical space in the context of virtual screening (VS) for STAT SH2 domain inhibitors: the use of ultra-large make-on-demand libraries and the application of smaller, focused sets guided by prior knowledge [1]. STAT proteins, especially STAT3 and STAT5b, are compelling oncological targets due to their roles in cancer cell survival and proliferation, with their Src Homology 2 (SH2) domains being particularly critical for function [24] [1]. The strategic definition of the chemical space to be screened significantly influences the success rate, cost, and efficiency of discovering novel inhibitors.
The table below summarizes key characteristics of different types of chemical libraries used in virtual screening, illustrating the trade-offs between scale and focus.
Table 1: Comparison of Chemical Libraries for Virtual Screening
| Library Name | Type | Approximate Size | Key Characteristics | Example Use Case |
|---|---|---|---|---|
| Enamine REAL Space [25] | Make-on-Demand | 78.1 billion compounds | Synthetically accessible via validated protocols; "on-the-fly" generation via synthons [25]. | Ultra-large virtual screening for novel chemotypes [1]. |
| Mcule-in-stock [1] | Commercial In-Stock | 5.59 million compounds | Readily purchasable; complies with drug-like rules [1]. | Benchmarking and economic screening workflows [1]. |
| Otava SH2 Domain Library [1] | Focused/Targeted | 1,807 compounds | Designed using pharmacophore models for SH2 domains [1]. | Knowledge-based screening for difficult PPI targets like STAT SH2 [1]. |
| Natural Product Library [1] | Focused/Natural | ~190,000 compounds | Contains natural products and natural product-like compounds [1]. | Identifying complex, 3D-like hits against PPI interfaces [1]. |
Experimental Protocols for Virtual Screening
Protocol: AI-Accelerated Ultra-Large Library Screening (e.g., Deep Docking)
This protocol is designed for screening billion-compound libraries against a target protein like the STAT3 SH2 domain [1].
- Objective: To efficiently identify hit candidates from an ultra-large chemical space (e.g., Enamine REAL) using an iterative machine learning process to reduce computational cost.
- Materials:
- Procedure:
- Step 1: Preparation of a Benchmark Set. A diverse subset (e.g., 117,500 compounds) is selected from the full library using a diversity-picking algorithm [1].
- Step 2: Initial Docking and Model Training. The benchmark set is docked into the target's binding site. The docking scores are used to train a deep learning model to predict the scores of unscreened compounds [1].
- Step 3: Iterative Screening and Model Retraining. The trained model predicts scores for a larger portion of the library. The top-predicted compounds (e.g., 5-10%) are docked, and their results are used to retrain and improve the model. This process repeats for several iterations [1].
- Step 4: Final Hit Selection. After the final iteration, the top-ranked compounds from the docking of the filtered set are selected for further experimental validation.
- Expected Outcome: A significant reduction in the number of compounds requiring physics-based docking (e.g., from billions to ~120,000) while achieving high hit rates (up to 50% reported for STAT3) [1].
Protocol: Knowledge-Based Screening with Focused Sets
This protocol leverages smaller, targeted libraries for a more direct route to potential hits [1].
- Objective: To rapidly identify hit compounds using libraries pre-enriched for specific target classes, such as SH2 domains.
- Materials:
- Procedure:
- Step 1: Library Curation. Acquire and prepare the focused library. Filter out pan-assay interference compounds (PAINS) [1].
- Step 2: Structure-Based Pharmacophore Modeling (Optional). Generate a receptor-based pharmacophore model using a known inhibitor-bound crystal structure (e.g., PDB: 6CMR for SHP2, a related PTP). The model should identify critical features like Hydrogen Bond Acceptors (HBA), Donors (HBD), and Hydrophobic (HYP) regions [24].
- Step 3: Pharmacophore-Based Screening. Screen the focused library against the pharmacophore model to identify compounds that match the essential feature set [24].
- Step 4: Molecular Docking. Dock the top compounds from the pharmacophore screen (or the entire pre-filtered library) into the STAT SH2 domain binding site for precise pose prediction and scoring.
- Step 5: Binding Stability Assessment. Subject the top-ranking docked complexes to molecular dynamics (MD) simulations (e.g., 500 ns) and calculate binding free energies (e.g., via MM/PBSA) to assess stability and interaction strength [24].
- Expected Outcome: Identification of a smaller set of high-quality hits with a high likelihood of activity, validated by computational simulations.
Visualizing Virtual Screening Workflows
The following diagrams, generated using Graphviz, illustrate the logical flow of the two primary screening strategies discussed.
Diagram 1: AI-Accelerated Ultra-Large Screening
Diagram 2: Knowledge-Based Focused Screening
The Scientist's Toolkit: Essential Research Reagents & Solutions
The table below lists key resources for conducting virtual screening campaigns for STAT SH2 domain inhibitors.
Table 2: Key Research Reagent Solutions for STAT SH2 Inhibitor Screening
| Tool / Resource | Type | Function in Research | Example / Provider |
|---|---|---|---|
| Make-on-Demand Libraries | Chemical Database | Provides access to billions of novel, synthetically accessible compounds for ultra-large screening. | Enamine REAL Space [25] |
| Focused/Targeted Libraries | Chemical Database | Offers pre-selected compounds designed for specific target classes, increasing hit probability. | Otava SH2 Domain Library [1] |
| Structure-Based Pharmacophore Modeling | Computational Software | Identifies and maps essential interaction features from a protein-ligand complex to guide screening. | Discovery Studio [24] |
| Deep Docking Workflow | AI-Accelerated Tool | Dramatically reduces computational cost of screening billion-compound libraries using iterative ML. | Custom or published protocol [1] |
| Molecular Dynamics Software | Simulation Software | Assesses the stability and binding mechanics of protein-ligand complexes over time. | GROMACS, AMBER, Desmond [24] |
| Targeted Compound Database | Information Database | Curates known actives, decoys, and bioactivity data for benchmarking and validation. | ChEMBL, DUD-E [1] |
Cutting-Edge Virtual Screening Methodologies for Ultra-Large Libraries
The discovery of inhibitors for Src Homology 2 (SH2) domains represents a significant challenge and opportunity in modern drug discovery, particularly for targets like STAT (Signal Transducer and Activator of Transcription) proteins implicated in oncology and inflammatory diseases. SH2 domains are approximately 100 amino acid protein modules that specifically recognize and bind to phosphotyrosine (pY) motifs, playing a crucial role in intracellular signal transduction [19]. The STAT3 and STAT5b SH2 domains, in particular, are clinically relevant oncological targets because their inhibition can cause cancer-derived cells to undergo growth arrest or apoptosis while leaving healthy cells largely unaffected [1].
Traditional virtual screening approaches face insurmountable computational challenges when applied to ultralarge chemical libraries that now exceed billions of "make-on-demand" compounds. While conventional docking can process millions of compounds, screening billion-molecule libraries would require years of computational time, creating a critical bottleneck in drug discovery pipelines [26]. Deep Docking (DD) has emerged as an artificial intelligence-powered solution to this challenge, accelerating virtual screening by up to 50-fold through the integration of quantitative structure-activity relationship (QSAR) deep learning models with conventional docking programs [26]. This application note provides detailed protocols for implementing DD platforms specifically tailored for discovering STAT SH2 domain inhibitors, enabling researchers to efficiently navigate ultralarge chemical spaces while maintaining high accuracy in hit identification.
Table 1: Performance Metrics of Deep Docking Against STAT SH2 Domains
| Target Protein | Library Size | Compounds Docked | Hit Rate | Fold Enrichment | Data Reduction |
|---|---|---|---|---|---|
| STAT3-SH2 | 5.51 billion (Enamine REAL) | ~120,000 | 50.0% | ~6,000x | ~100-fold |
| STAT5b-SH2 | 5.59 million (Mcule-in-stock) | ~120,000 | 42.9% | N/A | N/A |
| Typical DD Performance (Multiple Targets) | 1.36 billion (ZINC15) | 1 million per iteration | Varies by target | Up to 6,000x | Up to 100-fold |
Deep Docking Platform Fundamentals
Core Architecture and Mechanism
The Deep Docking platform operates on an iterative active learning principle that combines traditional docking with deep neural networks (DNNs) to predict docking outcomes for the vast majority of compounds without actually docking them [26]. The fundamental innovation lies in using QSAR models trained on docking scores of small, representative subsets of a chemical library to approximate docking results for remaining entries, thereby enabling the systematic prioritization of likely hits for actual docking while excluding unlikely candidates [26]. This approach effectively breaks the computational bottleneck that has traditionally limited virtual screening to libraries of only a few million compounds.
The platform's efficiency stems from its ability to learn and progressively refine its predictions through multiple cycles. Initially, the system docks a randomly selected subset of compounds to establish baseline structure-activity relationships. As iterations progress, the model becomes increasingly accurate at identifying regions of chemical space that contain high-scoring compounds, focusing computational resources exclusively on these promising areas [26]. This iterative enrichment process typically achieves up to 100-fold data reduction while retaining the majority of true hits, making billion-compound screening feasible on standard high-performance computing infrastructure [26].
Key Advantages for SH2 Domain Targets
SH2 domains present particular challenges for inhibitor discovery due to their shallow, solvent-exposed phosphotyrosine-binding sites, which complicate traditional structure-based drug design approaches [1] [19]. Deep Docking offers specific advantages for these difficult targets by enabling the comprehensive exploration of diverse chemotypes that might be missed in smaller, traditionally screened libraries. Recent studies have demonstrated that AI-based ultralarge virtual screening can achieve exceptional hit rates of 50.0% for STAT3-SH2 and 42.9% for STAT5b-SH2 domains, far exceeding typical screening outcomes [1].
The platform's ability to process ultralarge libraries is particularly valuable for SH2 domains because these protein-interaction domains require compounds that can effectively compete with native phosphopeptide ligands. The extensive chemical diversity available in billion-compound libraries increases the probability of identifying novel scaffolds with sufficient affinity and specificity to effectively inhibit these challenging targets [1]. Furthermore, the Deep Docking approach has proven effective even for more difficult protein-protein interaction-type targets like STAT proteins, where the reliability of underlying docking models is traditionally harder to assess [1].
Experimental Protocols and Implementation
The Deep Docking workflow consists of seven key stages that are repeated iteratively until convergence criteria are met. Before beginning, ensure all necessary computational resources and software dependencies are installed and configured, including a docking program (such as FRED, AutoDock Vina, or RosettaVS), deep learning frameworks (such as TensorFlow or PyTorch), and cheminformatics toolkits (such as RDKit) for descriptor calculation [26].
Initialization Phase: Prepare the target protein structure by removing water molecules, adding hydrogen atoms, and defining the binding site coordinates. For STAT SH2 domains, the binding site should encompass the phosphotyrosine pocket and adjacent specificity determinants [19]. Compute standard sets of ligand-based QSAR descriptors (such as molecular fingerprints) for every entry in the ultralarge docking database. This one-time preprocessing step enables rapid similarity searching and model training throughout the DD process [26].
Critical Setup Parameters:
- Training set size: 1 million compounds for initial sampling
- Docking protocol: Standardized for consistency across iterations
- Fingerprint type: Extended-connectivity fingerprints (ECFP4) recommended
- Deep learning architecture: Fully connected deep neural networks
- Convergence criterion: Stable recall values (90% of virtual hits retrieved)
Iterative Deep Docking Protocol
Step 1: Initial Random Sampling and Docking Randomly select 1 million compounds from the preprocessed chemical library as the initial training subset. This sample size has been empirically determined to provide sufficient chemical diversity while remaining computationally manageable [26]. Perform conventional docking of this subset against the STAT SH2 domain target using standardized parameters. Record docking scores and binding poses for all successfully docked compounds.
Step 2: Deep Neural Network Training Train a deep neural network model to relate the 2D molecular descriptors of the training compounds to their empirical docking scores. Divide the training compounds into virtual hits (scoring below a predetermined cutoff) and non-hits (scoring above the cutoff) based on their docking scores. The model learns to recognize complex patterns in chemical structures that correlate with favorable binding to the SH2 domain [26].
Step 3: Prediction and Selection Use the trained DNN model to predict docking outcomes for all undocked compounds in the library. Randomly select a predetermined number of compounds predicted to be virtual hits (typically 1 million) to augment the training set in the next iteration. This selection strategy balances exploration of chemical space with exploitation of predicted high-scoring regions [26].
Step 4: Iteration and Convergence Repeat Steps 1-3 using the augmented training set. Monitor convergence by tracking the recall value (percentage of actual virtual hits retrieved) across iterations. The process typically requires 5-10 iterations to stabilize, with the final output being a significantly enriched subset representing 1-2% of the original library that contains the majority of true hits [26] [1].
Table 2: Deep Docking Protocol Parameters for STAT SH2 Domains
| Parameter | Recommended Setting | Alternative Options | Notes |
|---|---|---|---|
| Training Set Size | 1,000,000 compounds | 250,000 - 2,000,000 compounds | Larger sizes improve model accuracy |
| Molecular Descriptors | ECFP4 Fingerprints | MACCS keys, other 2D fingerprints | Fast computation essential |
| DNN Architecture | Fully connected (3-5 hidden layers) | Varies by implementation | Sufficient complexity for QSAR |
| Iterations | Until convergence (5-10 cycles) | Fixed number (e.g., 8) | Monitor recall stability |
| Selection per Iteration | 1,000,000 predicted hits | 500,000 - 2,000,000 | Balance exploration/exploitation |
| Docking Program | FRED | AutoDock Vina, RosettaVS, Glide | Consistency critical |
Validation and Hit Confirmation
Following the completion of the Deep Docking protocol, validate the final enriched subset by docking all retained compounds using a more rigorous docking protocol or multiple docking programs to minimize scoring function bias [27]. For STAT SH2 domains specifically, prioritize compounds that form key interactions with the conserved arginine residue in the βB5 position of the phosphotyrosine binding pocket and demonstrate complementary interactions with specificity-determining regions [19].
Select top-ranking compounds for experimental validation using biochemical assays such as fluorescence polarization, surface plasmon resonance, or enzymatic activity assays. For STAT proteins, cellular assays measuring phosphorylation status or downstream transcriptional activity provide functional validation of SH2 domain inhibition [1].
Table 3: Research Reagent Solutions for Deep Docking Implementation
| Resource Category | Specific Tools & Resources | Function in Deep Docking Workflow | Implementation Notes |
|---|---|---|---|
| Chemical Libraries | ZINC15, Enamine REAL, Mcule-in-stock | Source of compounds for virtual screening | Enamine REAL offers >5 billion make-on-demand compounds |
| Docking Software | FRED, AutoDock Vina, RosettaVS | Generate training data through conventional docking | FRED used in original DD publication [26] |
| Deep Learning Frameworks | TensorFlow, PyTorch, Keras | Build and train QSAR models for score prediction | Pre-built DD scripts available on GitHub [26] |
| Cheminformatics | RDKit, Open Babel | Compute molecular descriptors and fingerprints | Essential for pre-processing entire chemical library |
| Computing Infrastructure | HPC clusters, Cloud computing | Execute docking and training computations | 3000 CPUs can screen billion compounds in days [27] |
| SH2 Domain Resources | PDB structures, Crystallography | Provide accurate target structures for docking | STAT3/5b SH2 domains available (1BG1, 1Y1U) |
| Validation Assays | Fluorescence polarization, SPR | Confirm binding of computational hits | Critical for establishing experimental correlation |
Technical Considerations for STAT SH2 Domain Targets
SH2 Domain Structural Features
STAT-type SH2 domains exhibit distinctive structural characteristics that must be considered when implementing Deep Docking protocols. Unlike SRC-type SH2 domains, STAT SH2 domains lack the βE and βF strands and have a split αB helix, adaptations that facilitate the dimerization required for STAT-mediated transcriptional regulation [19]. The phosphotyrosine binding pocket contains a highly conserved arginine residue (βB5) that forms a critical salt bridge with the phosphate moiety of phosphotyrosine-containing ligands [19].
Successful inhibitors must compete with native phosphopeptide ligands that typically bind with moderate affinity (Kd 0.1-10 μM) [19]. When preparing the STAT SH2 domain structure for docking, ensure the binding site definition includes not only the phosphotyrosine pocket but also adjacent specificity determinants that interact with residues C-terminal to the phosphotyrosine in native peptides. These secondary interactions contribute significantly to binding affinity and specificity [19].
Performance Optimization Strategies
To maximize Deep Docking efficiency for STAT SH2 domains, implement several optimization strategies. First, ensure the initial random sampling adequately represents the chemical diversity of the full library, as this foundation critically impacts all subsequent iterations [26]. Second, adjust the docking score cutoff used to define virtual hits based on target characteristics; for challenging PPI targets like STAT SH2 domains, a less stringent cutoff may be appropriate in early iterations [1].
Leverage the fact that Deep Docking performs effectively even with smaller training set sizes for focused libraries. Studies screening millions (rather than billions) of compounds against STAT5b-SH2 achieved 42.9% hit rates while docking only approximately 120,000 compounds, representing an extremely economic workflow [1]. This suggests that for initial exploratory campaigns, smaller diverse libraries may provide sufficient chemical space coverage while significantly reducing computational demands.
Deep Docking represents a transformative approach to virtual screening that effectively bridges the gap between traditional docking limitations and the opportunities presented by ultralarge chemical libraries. For challenging targets like STAT SH2 domains, this AI-powered workflow enables the efficient identification of novel inhibitors with exceptional hit rates, dramatically accelerating the early drug discovery process. The protocols outlined in this application note provide researchers with a comprehensive framework for implementing Deep Docking in their STAT inhibitor programs, offering specific guidance tailored to the unique characteristics of SH2 domain targets. As the field continues to evolve, the integration of advanced deep learning approaches with structure-based drug design promises to further enhance our ability to target these clinically important but challenging protein-interaction domains.
The field of computer-aided drug discovery is undergoing a transformative shift with the emergence of ultra-large make-on-demand compound libraries, such as the Enamine REAL space, which now contain billions of readily available compounds [28] [29]. This expansion presents both a golden opportunity and a significant computational challenge for virtual screening, particularly when accounting for receptor flexibility during docking procedures [28]. The RosettaEvolutionaryLigand (REvoLd) algorithm represents a novel approach to this problem, utilizing an evolutionary algorithm to efficiently search combinatorial make-on-demand chemical space without enumerating all possible molecules [28] [29]. This methodology is particularly relevant for targeting challenging drug targets such as the STAT3 SH2 domain, a key therapeutic target in multiple cancers including gastric cancer, where conventional screening approaches have yielded inhibitors with weak binding affinities due to domain flexibility [30] [31].
REvoLd exploits the fundamental architecture of make-on-demand compound libraries, which are constructed from defined lists of substrates and chemical reactions [29]. Unlike exhaustive screening methods that require substantial computational resources, REvoLd implements an evolutionary optimization process that progressively refines potential ligands through generations of selection, mutation, and crossover operations [28] [29]. Benchmark studies conducted on five drug targets have demonstrated improvements in hit rates by factors between 869 and 1,622 compared to random selections, highlighting the algorithm's robust enrichment capabilities [28] [32]. The first prospective validation of REvoLd occurred during the CACHE challenge #1, where it successfully identified novel binders for the WDR40 domain of LRRK2, a target associated with Parkinson's disease [33].
REvoLd Algorithm Implementation and Workflow
Core Algorithmic Framework
REvoLd implements an evolutionary algorithm that mimics Darwinian evolution through selective pressure based on docking scores [29]. The algorithm begins with a population of randomly generated ligands constructed by selecting a random reaction and suitable synthons from the combinatorial library [34]. Each individual molecule in the population is then docked against the target protein using the RosettaLigand protocol, which incorporates full ligand and receptor flexibility [28] [29]. The resulting interface energies between ligand and protein are used as fitness scores to drive the evolutionary process [34].
The evolutionary optimization cycle consists of multiple generations where fit individuals are selected for reproduction through mutation and crossover operations [29]. Mutation operations alter small parts of promising molecules by switching single fragments to low-similarity alternatives or changing the reaction scheme, while crossover recombines fragments from two parent molecules to create novel offspring [28] [29]. This approach maintains strict adherence to the synthetically accessible chemical space defined by the make-on-demand library, ensuring that all proposed compounds can be readily synthesized [29]. The algorithm incorporates multiple selection strategies, including TournamentSelector and RouletteSelector, which introduce non-deterministic elements to help escape local minima and explore broader chemical space [29].
Computational Workflow
The following diagram illustrates the complete REvoLd workflow, from initial population generation to final hit selection:
REvoLd Evolutionary Optimization Workflow
STAT3 SH2 Domain Targeting Considerations
For STAT3 SH2 domain inhibition, particular considerations must be incorporated into the REvoLd workflow. The high flexibility of the STAT3 SH2 domain necessitates special treatment, as conventional rigid docking may miss potential binders [31]. Molecular dynamics simulations can generate an ensemble of receptor conformations for docking, creating "induced-active site" receptor models that account for domain flexibility [31] [35]. Additionally, the scoring function can be optimized to prioritize compounds that interact with key residues in the pY+0 binding pocket, particularly R609 and S613, which are critical for STAT3 function [31]. This targeted approach has previously led to the identification of uncharged STAT3 inhibitors with improved cell penetration capabilities compared to previously identified compounds containing negatively charged moieties [35].
Research Reagent Solutions and Experimental Setup
Table 1: Essential Research Reagents and Computational Resources for REvoLd Implementation
| Resource Type | Specific Solution | Function in Workflow |
|---|---|---|
| Combinatorial Library | Enamine REAL Space | Provides synthetically accessible chemical space; 20-30+ billion compounds defined through fragment combinations [28] [33] |
| Software Suite | Rosetta Software Suite | Core platform for REvoLd implementation and RosettaLigand flexible docking [34] |
| Reaction Definition | SMARTS-formatted Reactions | Defines chemical rules for fragment coupling and compound generation [34] [33] |
| Fragment Library | SMILES-formatted Reagents | Building blocks for combinatorial library construction; includes synton identifiers [34] |
| Target Preparation | Molecular Dynamics Software (AMBER) | Generates receptor conformational ensembles for flexible docking [31] [33] |
| Computational Resources | MPI-enabled High Performance Computing | Enables parallel execution; recommended: 50-60 CPUs per run, 200-300GB RAM [34] |
REvoLd Application Protocol for STAT3 Inhibitor Discovery
Target Preparation and Binding Site Definition
The first critical step in implementing REvoLd for STAT3 SH2 domain inhibitor discovery involves comprehensive target preparation. The crystal structure of STAT3 complexed with a small-molecule inhibitor (PDB ID: 6NJS) should be obtained from the Protein Data Bank, with particular focus on the SH2 domain where most small-molecule inhibitors bind [30]. To account for domain flexibility, molecular dynamics simulations should be performed using the AMBER force field, with the system minimized, heated to 303K, and production runs conducted for 1.5 μs in replicates [33]. The resulting trajectories should be clustered based on Cα-root-mean square deviation using DBSCAN with an ε-value of 1.4 Å to generate representative receptor conformations for docking [33]. The active pocket should be defined as the ligand-binding region located in the SH2 domain, with explicit consideration of the pY+0 binding pocket residues R609 and S613 [31].
REvoLd Configuration and Execution
REvoLd requires specific configuration parameters to optimize performance for STAT3 SH2 domain screening. The algorithm should be compiled with MPI support to enable parallel execution, with recommendations of 20-60 CPUs per run and 200-300GB of RAM [34]. Key command line options must include the protein structure file, RosettaScript for docking, centroid position for initial ligand placement, and paths to the reagent and reaction files [34]. The evolutionary parameters should be set with a population size of 200 individuals, reduced to 50 through selective pressure each generation, with optimization conducted over 30 generations [28]. Multiple independent runs (10-20) with different random seeds are recommended to sample diverse regions of the chemical space [28] [34].
Table 2: Key REvoLd Configuration Parameters and Recommended Settings
| Parameter Category | Specific Parameter | Recommended Setting | Rationale |
|---|---|---|---|
| Population Settings | Initial Population Size | 200 individuals | Balances diversity with computational cost [28] |
| Generations | 30 | Provides balance between convergence and exploration [28] | |
| Selective Pressure | Reduce to 50 individuals | Maintains fittest solutions while controlling population growth [28] | |
| Scoring Parameters | Docking Runs per Ligand | 150 | Provides sufficient sampling of binding poses [34] |
| Fitness Function | lid_root2 | Interface energy normalized by cube root of heavy atoms [34] | |
| Execution Parameters | Independent Runs | 10-20 | Samples diverse chemical space regions [28] [34] |
| Computational Resources | 50-60 CPUs, 200-300GB RAM | Enables efficient parallel execution [34] |
Hit Validation and Expansion
Following REvoLd screening, top-ranking compounds should undergo comprehensive validation. Initial filtering should prioritize molecules that form distinct hydrogen bonds with the SH2 domain of STAT3, particularly those interacting with key residues R609 and S613 [30]. Molecular dynamics simulations of 50 ns duration should be performed using GROMACS with the GAFF force field to assess complex stability [30]. Validated hits should then serve as starting points for a second round of REvoLd screening to explore analogous regions of the chemical space, leveraging the fragment-based nature of the approach to identify derivatives with improved binding properties [33]. This iterative optimization process mirrors the successful strategy employed in the CACHE challenge, where an initial binder was identified and subsequently optimized through derivative screening [33].
Performance Metrics and Benchmarking Results
REvoLd has demonstrated exceptional performance in both retrospective benchmarks and prospective applications. In benchmark studies across five drug targets, REvoLd improved hit rates by factors between 869 and 1,622 compared to random selection, highlighting its robust enrichment capabilities [28] [32]. The algorithm typically samples between 1,000 and 4,000 unique ligands per run, representing an extremely efficient exploration of the billion-compound chemical space [34]. In the CACHE challenge #1, REvoLd successfully identified novel binders for the WDR40 domain of LRRK2, with three of the five selected molecules showing measurable dissociation constants (KD) better than 150 μM [33]. This prospective validation confirmed the algorithm's ability to identify genuine binders for challenging protein targets.
The STAT3 signaling pathway and REvoLd's mechanism of interference can be visualized as follows:
STAT3 Signaling Pathway and REvoLd Intervention
REvoLd represents a significant advancement in virtual screening methodology, particularly for challenging targets like the STAT3 SH2 domain. Its evolutionary algorithm approach enables efficient exploration of ultra-large combinatorial libraries while maintaining synthetic accessibility and accounting for receptor flexibility. The successful application of REvoLd in both benchmark studies and prospective challenges demonstrates its potential to accelerate the discovery of novel inhibitors for difficult drug targets. Future developments will likely focus on integrating advanced machine learning approaches with the evolutionary framework and expanding the application to even more complex target classes, further solidifying its role in the modern computational drug discovery pipeline.
Structure-based molecular docking is a cornerstone of modern computational drug discovery, enabling the prediction of how small molecule ligands interact with biological targets. However, a significant limitation of traditional docking methods is their treatment of the receptor as a rigid body, which fails to capture the dynamic nature of protein binding sites. This is particularly problematic for challenging targets like STAT SH2 domains, where conformational flexibility plays a crucial role in ligand binding and inhibitor design [19]. The Src Homology 2 (SH2) domain is a approximately 100-amino acid module that specifically recognizes phosphotyrosine (pY) motifs, facilitating protein-protein interactions in critical signaling pathways [19]. In STAT proteins, which are transcription factors with roles in oncogenesis, the SH2 domain mediates dimerization through reciprocal phosphotyrosine-SH2 interactions, making it a prime target for therapeutic intervention [1] [19].
The rigid receptor approximation becomes especially limiting when targeting protein-protein interactions (PPIs), such as those mediated by SH2 domains. These interfaces tend to be large, shallow, and flexible, making them notoriously difficult to target with small molecules [1]. Recent advances in addressing these challenges have led to the development of sophisticated protocols that incorporate both receptor and ligand flexibility, dramatically improving docking accuracy and the success of virtual screening campaigns for STAT inhibitors [36] [1].
Key Methodological Advances in Flexible Docking
Accounting for Receptor Flexibility
Multiple computational strategies have emerged to incorporate receptor flexibility into docking workflows. These approaches vary in their complexity, computational requirements, and applicability to different stages of drug discovery.
Table 1: Strategies for Incorporating Receptor Flexibility in Docking
| Methodology | Key Principle | Advantages | Limitations |
|---|---|---|---|
| Ensemble Docking [36] [37] | Docking against multiple receptor conformations from experimental structures or simulations | Comprehensive sampling of conformational space; straightforward implementation | Requires pre-generated structures; weighting conformations can be challenging |
| FlexCovDock for KRASG12C [36] | Modified covalent docking protocol allowing protein conformational mobility | Specifically designed for covalent inhibitors; improved success rates from 55% to 89% | Specialized for covalent binding scenarios |
| Normal Mode-Based Approaches [38] | Perturbation of receptor structure along relevant low-frequency normal modes | Physically meaningful representation of backbone flexibility; no need for multiple structures | Computationally intensive; limited to small-scale loop rearrangements |
| Homology Modeling Integrated Docking [37] | @TOME server interfacing protein structure modeling with flexible ligand docking | Allows virtual screening against multiple modeled conformations; accessible web server | Dependent on template availability and model quality |
Advanced Sampling and Affinity Prediction
Accurately predicting binding affinities for flexible systems presents additional challenges beyond pose prediction. Free energy perturbation (FEP) methods provide more rigorous binding energy calculations but struggle with large conformational changes. For the flexible switch-II pocket in KRASG12C, researchers developed an innovative solution using targeted protein mutations to accelerate conformational transitions, reducing the mean unsigned error in binding affinity prediction from 1.44 to 0.89 kcal/mol [36].
For ultra-large libraries, AI-assisted workflows like Deep Docking have emerged, where a deep learning model is trained on a subset of docking results to predict binding scores for the remaining compounds, dramatically reducing computational requirements [1]. This approach has proven particularly valuable for challenging PPI targets like STAT3, achieving exceptional hit rates of up to 50.0% in virtual screening campaigns [1].
Application to STAT SH2 Domain Inhibitor Discovery
STAT SH2 Domain Structure and Flexibility
STAT SH2 domains belong to a distinct structural subclass characterized by the absence of βE and βF strands and a split αB helix, adaptations that facilitate STAT dimerization [19]. The phosphotyrosine (pY) binding pocket is divided into three sub-pockets: pY+X (hydrophobic side), pY+0 (binds pY705), and pY+1 (binds L706) [17]. This architecture creates a flexible binding interface that accommodates specific peptide motifs while maintaining moderate binding affinity (Kd 0.1–10 μM) to allow for reversible signaling interactions [19].
The flexibility of these domains necessitates specialized docking approaches. Recent successful virtual screening campaigns against STAT3 and STAT5b have employed everything from traditional brute-force docking to AI-accelerated workflows, demonstrating the importance of method selection based on available resources and project goals [1].
Quantitative Performance of Flexible Docking Methods
Table 2: Performance Metrics of Flexible Docking Methods for STAT Inhibitor Discovery
| Method/Workflow | Target | Library Size | Performance Metric | Result |
|---|---|---|---|---|
| Deep Docking [1] | STAT3-SH2 | Billion-scale | Hit Rate | 50.0% |
| Deep Docking (Economic) [1] | STAT5b-SH2 | Million-scale | Hit Rate | 42.9% |
| FlexCovDock [36] | KRASG12C (flexible pocket) | Cross-docking test set | Pose Prediction Success | 89% (vs. 55% baseline) |
| FEP with Loop Mutations [36] | KRASG12C (flexible pocket) | 14 compounds | Binding Affinity MUE | 0.89 kcal/mol (vs. 1.44 kcal/mol baseline) |
| Standard Docking [17] | STAT3-SH2 | 182,455 natural compounds | Compounds Advanced to XP Docking | 55,872 |
Detailed Experimental Protocols
Flexible Receptor Docking Protocol for SH2 Domains
This protocol outlines the steps for performing flexible receptor docking against STAT SH2 domains, adaptable for both covalent and non-covalent inhibitors.
Step 1: Receptor Preparation and Conformational Sampling
- Retrieve STAT SH2 domain structures from PDB (e.g., 6NJS for STAT3 at 2.70 Å resolution) [17] [39]
- Prepare protein structure using Protein Preparation Wizard (Schrödinger):
- Add hydrogen atoms
- Fill missing side chains and loops using Prime
- Optimize hydrogen bonding networks
- Minimize structure using OPLS3e or OPLS4 force field
- Generate receptor conformational ensemble:
- Collect existing experimental structures (holo and apo forms)
- Use molecular dynamics simulations to sample flexibility
- Alternatively, employ normal mode analysis for backbone flexibility [38]
Step 2: Binding Site Definition and Grid Generation
- Define binding site around known functional regions:
- For multiple conformation docking, generate grid files for each receptor structure with consistent binding site definition
- Validate grid quality by redocking native ligand and calculating RMSD (<2.0 Å acceptable)
Step 3: Ligand Preparation
- Prepare ligand library using LigPrep (Schrödinger) or similar tools:
- Generate possible tautomers and protonation states at physiological pH (7.4 ± 0.5)
- Determine chiralities and generate stereoisomers
- Apply energy minimization using appropriate force field
Step 4: Docking Execution and Analysis
- Perform docking against receptor ensemble using flexible docking protocol:
- Initial screening with High-Throughput Virtual Screening (HTVS) mode
- Follow with Standard Precision (SP) docking for top compounds
- Final refinement with Extra Precision (XP) docking [17]
- For covalent docking scenarios (e.g., cysteine-targeting inhibitors), use specialized protocols like FlexCovDock [36]
- Analyze results based on docking scores, binding modes, and interaction patterns with key residues
AI-Accelerated Ultra-High-Throughput Virtual Screening Protocol
For screening billion-compound libraries, the following Deep Docking protocol has demonstrated success against STAT SH2 domains [1].
Step 1: Library Curation and Preparation
- Select appropriate compound library (e.g., Enamine REAL, ZINC, or vendor-specific collections)
- Apply property-based filtering (Lipinski's Rule of Five, Veber criteria)
- Remove pan-assay interference compounds (PAINS) and reactive compounds
- For large libraries, prepare distributed database for efficient access
Step 2: Initial Docking and Model Training
- Randomly select representative subset (1-5% of total library)
- Perform standard docking on subset to generate training data
- Train deep neural network to predict docking scores from molecular fingerprints/descriptors
- Validate model performance on held-out test set
Step 3: Iterative Screening and Model Refinement
- Use trained model to predict docking scores for entire library
- Select top-ranking compounds for next iteration of docking
- Re-train model with expanded training set
- Repeat for 5-10 iterations or until convergence
Step 4: Final Selection and Validation
- Perform detailed docking (XP mode) on final candidate set
- Apply additional filters (ADMET properties, structural diversity)
- Select compounds for experimental validation
Table 3: Key Research Reagent Solutions for Flexible Docking Studies
| Resource Category | Specific Tools/Sources | Function/Application | Key Features |
|---|---|---|---|
| Protein Structures | RCSB PDB (6NJS, 6NUQ for STAT3) [17] [39] | Source of experimental receptor structures | High-resolution SH2 domain structures with bound ligands |
| Compound Libraries | ZINC15, Enamine REAL, Life Chemicals [1] [17] [39] | Source of screening compounds | Billions of synthetically accessible compounds; filtered for drug-likeness |
| Specialized Libraries | OTAVAchemicals SH2 Domain Targeted Library [1] | Knowledge-based screening | Pre-filtered compounds targeting SH2 domain pharmacophores |
| Docking Software | Schrödinger Suite (Glide), AutoDock, DOCK3.7 [36] [40] [17] | Flexible ligand and receptor docking | Multiple precision modes; covalent docking capabilities |
| Web Servers | @TOME 3.0 [37] | Integrated modeling and docking | Combines homology modeling with flexible docking |
| MD Simulation | GROMACS, Desmond [17] [39] | Conformational sampling and validation | Refines docking poses; calculates binding free energies |
The integration of receptor and ligand flexibility has transformed structure-based docking from a rigid modeling exercise into a dynamic simulation of molecular recognition. For challenging targets like STAT SH2 domains, these advanced protocols have demonstrated remarkable success, with hit rates exceeding 50% in some virtual screening campaigns [1]. The continued development of methods like FlexCovDock for flexible covalent docking [36] and AI-accelerated workflows for billion-compound screening [1] represents the cutting edge of this evolution.
As structural biology and computational power continue to advance, we anticipate further refinement of these protocols, particularly in more accurate treatment of entropy contributions and solvation effects. The integration of machine learning across the virtual screening pipeline, from protein structure prediction to binding affinity estimation, promises to further enhance the efficiency and success of docking-based drug discovery for STAT inhibitors and other therapeutically relevant targets.
Virtual screening represents a cornerstone of modern computer-aided drug discovery, enabling researchers to efficiently identify potential therapeutic compounds from vast chemical libraries. Within this domain, ligand-based and hybrid approaches provide powerful strategies when structural information about the target protein is limited or incomplete. These methods primarily utilize the known biological and structural information of active compounds to discover new chemical entities with similar or improved properties. This application note details the implementation of these methodologies within research focused on inhibiting STAT SH2 domains, crucial therapeutic targets in oncology and inflammatory diseases. The approaches outlined here leverage pharmacophore modeling and similarity searching to identify novel inhibitors through a structured computational protocol.
Theoretical Background and Significance
The Pharmacophore Concept in Drug Discovery
A pharmacophore is defined by the International Union of Pure and Applied Chemistry (IUPAC) as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response" [41]. In practical terms, a pharmacophore is an abstract model that represents the key molecular interaction capabilities of a compound or series of compounds, rather than specific chemical structures or functional groups. This abstraction makes pharmacophore models particularly valuable for identifying novel chemotypes through a process known as "scaffold hopping" [42].
Modern pharmacophore modeling encompasses several distinct approaches:
- Ligand-based modeling: Derives common chemical features from a set of known active molecules
- Structure-based modeling: Extracts interaction points from protein-ligand complexes
- Hybrid approaches: Combines elements of both strategies for enhanced accuracy
STAT SH2 Domains as Therapeutic Targets
The Src Homology 2 (SH2) domain is a protein module of approximately 100 amino acids that recognizes and binds to phosphorylated tyrosine residues in specific sequence contexts. In STAT (Signal Transducer and Activator of Transcription) proteins, the SH2 domain plays a critical role in facilitating dimerization and subsequent nuclear translocation, which is essential for their function as transcription factors [17]. Dysregulated STAT signaling, particularly through STAT3 and STAT5, is implicated in various cancers, autoimmune disorders, and inflammatory conditions, making the STAT SH2 domain an attractive target for therapeutic intervention.
The structural organization of STAT3's SH2 domain features a central anti-parallel β-sheet flanked by two α-helices (αA and αB), forming an αβββα motif. The phosphotyrosine (pY) binding pocket is divided into three sub-pockets: pY+X (hydrophobic side), pY+0 (binds to pY705), and pY+1 (binds to L706) [17]. Disrupting the interaction at this site prevents STAT dimerization and subsequent transcriptional activity, providing a viable strategy for therapeutic development.
Table 1: Key Sub-Pockets in the STAT3 SH2 Domain
| Sub-Pocket | Function | Key Residues |
|---|---|---|
| pY+0 | Binds phosphotyrosine705 (pY705) to stabilize dimerization | Arg609, Glu594, Lys591 |
| pY+1 | Binds leucine706 (L706) | Ser611, Ser636, Thr640 |
| pY+X | Provides hydrophobic interaction surface | Tyr657, Trp623, Gln644 |
Computational Methodologies
Ligand-Based Pharmacophore Modeling
Ligand-based pharmacophore modeling begins with the collection and curation of known active compounds. This approach is particularly valuable when the three-dimensional structure of the target protein is unavailable. The methodology involves several key steps:
Training Set Compilation and Preparation
- Active compound selection: Assemble a structurally diverse set of confirmed active molecules. For STAT SH2 domain inhibitors, this might include known inhibitors such as Stattic, SD-36, and related compounds [17].
- Conformational analysis: Generate representative conformational ensembles for each molecule. Typically, 50 conformers per compound are generated using tools like LigPrep in Schrödinger Suite [43].
- Molecular alignment: Align training set compounds based on common structural elements or pharmacophoric features using alignment algorithms.
Pharmacophore Hypothesis Generation
- Feature identification: The aligned molecules are analyzed to identify common chemical features. Standard pharmacophore features include:
- Model generation: Using software such as Phase in Schrödinger [43] or Discovery Studio [24], common pharmacophore hypotheses are generated from the aligned training set.
- Model validation: The resulting pharmacophore models are validated using the Güner-Henry (GH) method and enrichment factor (EF) calculations [24]. A GH score above 0.6 is generally considered acceptable.
Table 2: Common Pharmacophore Features and Their Characteristics
| Feature Type | Description | Geometric Representation |
|---|---|---|
| Hydrogen Bond Donor (HBD) | Atom that can donate a hydrogen bond | Vector with target interaction point |
| Hydrogen Bond Acceptor (HBA) | Atom that can accept a hydrogen bond | Vector with target interaction point |
| Hydrophobic (H) | Non-polar region that engages in van der Waals interactions | Sphere |
| Positive Ionizable (PI) | Group that can carry a positive charge | Sphere |
| Negative Ionizable (NI) | Group that can carry a negative charge | Sphere |
| Aromatic Ring (AR) | Pi-electron system for cation-pi or stacking interactions | Ring plane with normal vector |
Similarity Searching Methods
Similarity searching provides a complementary approach to pharmacophore modeling for identifying potential inhibitors. This methodology relies on the "similarity property principle," which states that structurally similar molecules tend to have similar properties.
Molecular Descriptor Calculation
- Structural fingerprints: Generate binary bit strings representing the presence or absence of specific structural patterns (e.g., ECFP, FCFP fingerprints)
- Physicochemical descriptors: Calculate properties such as molecular weight, logP, polar surface area, hydrogen bond donors/acceptors
- Pharmacophore fingerprints: Encode the presence of specific pharmacophore patterns within molecules
Similarity Metrics and Screening
- Tanimoto coefficient: The most widely used similarity metric for chemical structures
- Cosine similarity: Alternative metric particularly effective for high-dimensional data
- Database searching: Screen large chemical databases (e.g., ZINC, ChEMBL) to identify compounds with similarity above a defined threshold to known active molecules
Hybrid Virtual Screening Approaches
Hybrid approaches combine the strengths of multiple computational methods to enhance screening efficiency and hit rates. A typical hybrid workflow for STAT SH2 domain inhibitor discovery might include:
Pharmacophore-Based Pre-screening
- Apply a validated pharmacophore model as an initial filter to reduce chemical space
- Focus on compounds that match essential interaction features required for STAT SH2 domain binding
Similarity-Based Enrichment
- Apply 2D and 3D similarity searching to the pre-screened compound set
- Prioritize compounds that are structurally similar to known active inhibitors but possess novel scaffolds
Structure-Based Verification
- Perform molecular docking studies on the enriched compound subset
- Use ensemble docking against multiple STAT SH2 domain structures to account for protein flexibility [43]
- Apply binding free energy calculations (MM-GBSA/PBSA) to refine the selection of candidate compounds
Diagram 1: Hybrid Virtual Screening Workflow for STAT SH2 Domain Inhibitors (VS: 76 characters)
Application to STAT SH2 Domain Inhibitor Discovery
Case Study: Identification of Novel STAT3 SH2 Domain Inhibitors
A recent study demonstrated the application of these methodologies to identify natural compounds targeting the SH2 domain of STAT3 [17]. The research employed a comprehensive virtual screening approach:
Database Preparation
- 182,455 natural compounds were retrieved from the ZINC15 database
- Compounds were prepared using LigPrep to generate 3D structures with optimized ionization states at physiological pH (7.4 ± 0.5)
- Conformational ensembles were generated for each compound
Multi-Step Virtual Screening
- Initial high-throughput virtual screening (HTVS) using molecular docking
- Followed by standard precision (SP) docking of top-ranked compounds
- Final extra precision (XP) docking with binding affinity cut-off of -6.5 kcal/mol
Hit Identification and Validation
- Four compounds (ZINC255200449, ZINC299817570, ZINC31167114, and ZINC67910988) were identified as potential STAT3 inhibitors
- ZINC67910988 demonstrated superior stability in molecular dynamics simulations
- Network pharmacology analysis revealed multi-target potential of the identified hits
Case Study: p56lck SH2 Domain Inhibitor Discovery
Another study focused on identifying inhibitors of the p56lck SH2 domain, employing ligand-based e-pharmacophore modeling combined with ensemble docking [43]:
Pharmacophore Model Development
- 26 known active compounds were used as a training set
- A five-point pharmacophore model (DHRRR_1) was generated containing:
- One hydrogen bond donor (D)
- One hydrophobic group (H)
- Three aromatic rings (R)
- The model was validated and used for virtual screening of 782,000 compounds from ZINC15
Ensemble Docking Strategy
- Seven different protein structures of the SH2 domain were used for docking
- Multi-step docking protocol: HTVS → SP → XP with retention of top 10% at each step
- This approach accounted for protein flexibility and improved docking accuracy
Hit Optimization
- Core hopping was performed on top hits to enhance protein-ligand interactions
- ADMET property prediction was conducted to ensure drug-like properties
- Six novel top hits were identified for further experimental validation
Table 3: Key Research Reagent Solutions for STAT SH2 Domain Inhibitor Screening
| Resource/Software | Type | Primary Function | Application in STAT SH2 Research |
|---|---|---|---|
| ZINC15 Database | Compound Library | Provides commercially available compounds for screening | Source of natural products & synthetic compounds for STAT SH2 inhibition [17] |
| Schrödinger Suite | Software Platform | Integrated computational drug discovery platform | Pharmacophore modeling, molecular docking, & ADMET prediction [43] |
| Protein Data Bank (PDB) | Structural Database | Repository of 3D protein structures | Source of STAT SH2 domain structures (e.g., 6NJS for STAT3) [17] |
| ChEMBL Database | Bioactivity Database | Curated database of bioactive molecules | Source of known active compounds for training set creation [44] |
| Discovery Studio | Software Platform | Modeling and simulation suite | Structure-based pharmacophore generation & validation [24] |
Experimental Protocols
Protocol 1: Ligand-Based Pharmacophore Model Generation for STAT SH2 Domain Inhibitors
Objective: To generate a validated ligand-based pharmacophore model for virtual screening of STAT SH2 domain inhibitors.
Materials and Software
- Workstation with Schrödinger Suite (Phase module) or Discovery Studio
- Set of known STAT SH2 domain inhibitors (minimum 10-15 structurally diverse compounds)
- Chemical databases for screening (e.g., ZINC15, in-house compound libraries)
Procedure
- Training Set Compilation
- Curate a set of known STAT SH2 domain inhibitors with confirmed activity (IC50 or Ki values)
- Ensure structural diversity to avoid bias toward specific chemotypes
- Prepare compounds using LigPrep or similar tools: generate 3D structures, optimize geometry, and generate possible ionization states at pH 7.0 ± 2.0
Conformational Analysis
- Generate 50 conformers per compound using a mixed torsional/low-mode sampling approach
- Apply an energy window of 10 kcal/mol to exclude high-energy conformations
- Retain duplicate conformers based on RMSD threshold of 1.0 Å
Molecular Alignment and Pharmacophore Generation
- Align training set compounds using common structural features or pharmacophore points
- Use the "common features pharmacophore" generation algorithm in Phase or Discovery Studio
- Generate multiple pharmacophore hypotheses with varying numbers of features
Model Validation
- Prepare a decoy set containing known active compounds and presumed inactives (ratio ~1:50)
- Calculate Güner-Henry (GH) score and enrichment factor (EF)
- Select the model with GH score > 0.6 and highest EF for virtual screening
Troubleshooting Tips
- If model yields too many false positives: Increase feature stringency or add exclusion volumes
- If model is too restrictive and misses known actives: Define one or more features as optional
- If model shows poor enrichment: Re-evaluate training set composition and ensure adequate diversity
Protocol 2: Hybrid Virtual Screening for STAT SH2 Domain Inhibitors
Objective: To implement a hybrid virtual screening workflow combining pharmacophore modeling, similarity searching, and molecular docking for identifying novel STAT SH2 domain inhibitors.
Materials and Software
- Validated pharmacophore model (from Protocol 1)
- STAT SH2 domain protein structure (PDB: 6NJS for STAT3)
- Chemical database for screening (e.g., ZINC15 natural compounds subset)
- Schrödinger Suite or equivalent software platform
Procedure
- Pharmacophore-Based Pre-screening
- Screen the entire database using the validated pharmacophore model
- Use "fast" screening mode for initial filtering
- Apply "best flexible" search method for compounds passing initial filter
- Retain compounds that map all essential pharmacophore features
Similarity-Based Enrichment
- Calculate molecular fingerprints (ECFP4) for known active compounds and pharmacophore hits
- Compute Tanimoto similarity between active compounds and pharmacophore hits
- Retain compounds with similarity score > 0.7 to any known active
- Apply property-based filtering (Lipinski's Rule of Five, molecular weight 250-500 Da)
Structure-Based Verification
- Prepare protein structure: remove water molecules, add hydrogens, optimize H-bond network
- Generate receptor grid centered on the SH2 domain pY705 binding pocket
- Perform hierarchical docking: HTVS → SP → XP with retention of top 10% at each step
- Calculate binding free energies using MM-GBSA for top-ranked compounds
ADMET Profiling
- Predict key ADMET properties for final hits: solubility, hepatotoxicity, plasma protein binding
- Apply filters for desirable drug-like properties
- Select 10-20 top-ranked compounds for experimental validation
Expected Outcomes
- Identification of 5-20 potential STAT SH2 domain inhibitors with predicted binding affinity <-8.0 kcal/mol
- Hit rates of 5-40% in subsequent experimental validation, significantly higher than random screening
Diagram 2: Molecular Docking Protocol for STAT SH2 Domain (VS: 76 characters)
Ligand-based and hybrid virtual screening approaches represent powerful strategies for identifying novel STAT SH2 domain inhibitors. By leveraging pharmacophore models and similarity searching, researchers can efficiently explore vast chemical spaces while focusing resources on compounds with the highest probability of activity. The protocols outlined in this application note provide a structured framework for implementing these methodologies in drug discovery campaigns targeting STAT proteins and other challenging therapeutic targets. When properly validated and applied, these computational approaches can significantly accelerate the identification of novel chemical starting points for drug development, with reported hit rates typically ranging from 5% to 40% in prospective screening campaigns [45]. As computational methods continue to advance, integrating these approaches with experimental validation will remain essential for successful STAT-targeted therapeutic development.
Overcoming Virtual Screening Pitfalls: Scoring, Flexibility, and Selectivity
Virtual screening, particularly for challenging protein-protein interaction (PPI) targets like the STAT3 and STAT5b SH2 domains, has become an indispensable tool in modern drug discovery. The SH2 domain is a well-established pharmaceutical target due to its critical role in phosphotyrosine recognition and subsequent STAT dimerization and activation [19]. However, the initial molecular docking phase of virtual screening is notoriously approximate, leading to high false-positive and false-negative rates. This limitation has prompted widespread adoption of rescoring strategies—applying more sophisticated, physics-based methods to refine docking results and improve hit rates. In practice, however, rescoring often fails to deliver the anticipated improvements, creating a significant bottleneck in inhibitor development pipelines. This Application Note examines the fundamental causes of rescoring failure within STAT SH2 domain research and provides validated protocols to mitigate these risks, enabling more reliable identification of true bioactive compounds.
The Quantitative Evidence: Documented Limitations of Rescoring
Multiple independent studies have systematically evaluated rescoring performance across various target classes, revealing consistent patterns of limitation. The data demonstrate that while rescoring can provide marginal improvements, it rarely delivers transformative discrimination between true and false positives.
Table 1: Documented Performance Limitations of Rescoring Methods
| Evaluation Context | Rescoring Method(s) | Key Performance Finding | Reference |
|---|---|---|---|
| Ultra-large library screening against STAT SH2 domains | Deep Learning, Quantum Mechanics, Force Fields | "True positive and false positive ligands remain hard to discriminate, whatever the complexity of the chosen scoring function." [46] | [46] |
| Model cavity sites (L99A, L99A/M102Q, W191G) | MM-GBSA | Rescoring rescued 23 docking false negatives but introduced 10 new false positives. | [47] |
| Plasmodium falciparum dihydrofolate reductase | BEAR (MM-PB(GB)SA) | Significantly improved enrichment factors over docking alone, but performance remains target-dependent. | [48] |
| Diverse protein targets | Semiempirical QM, Force Fields with implicit solvation | "Neither method performed significantly better than empirical machine-learning scoring functions." [46] | [46] |
Root Causes of Rescoring Failure
Fundamental Methodological Limitations
Rescoring failure stems from several interconnected methodological constraints. First, inadequate pose sampling often persists through rescoring workflows; if the initial docking pose is incorrect, even perfect scoring cannot recover the true binding mode [46] [47]. Second, implicit solvent models struggle with accurately capturing desolvation penalties, particularly in buried binding pockets like those found in SH2 domains [47]. Third, most rescoring approaches, including MM-PB(GB)SA, typically ignore configurational entropy contributions due to computational expense, creating systematic errors in binding affinity predictions [47]. Finally, the handling of key water molecules, ions, and cofactors is often oversimplified or ignored, despite their critical roles in mediating ligand interactions [46].
Target-Specific Challenges with STAT SH2 Domains
The STAT SH2 domains present particular challenges for rescoring methodologies. These domains feature large, solvent-exposed PPI interfaces rather than deep, well-defined binding pockets, complicating pose prediction and affinity estimation [1]. Additionally, SH2 domains exhibit structural flexibility, particularly in their CD, EF, and BG loops, which control access to ligand specificity pockets and can undergo significant conformational changes upon ligand binding [19]. The phosphotyrosine (pY) binding pocket contains a highly conserved arginine residue that forms a strong salt bridge with the phosphate moiety, creating electrostatic interactions that can be challenging for scoring functions to properly evaluate [19]. Recent research also indicates that many SH2 domains, including those in STAT proteins, can bind lipid molecules at sites adjacent to the pY-binding pocket, potentially creating allosteric effects that conventional rescoring misses [19].
Mitigation Strategies and Protocols
Integrated Workflow for Rescoring Validation
The following diagram outlines a comprehensive workflow that integrates multiple mitigation strategies to address common rescoring failure points in STAT SH2 domain inhibitor identification:
Multi-Method Rescoring Protocol
This protocol implements a consensus approach to rescoring for STAT SH2 domain virtual screening hits, specifically designed to mitigate individual method failures.
Protocol 1: Multi-Method Consensus Rescoring
- Objective: To improve discrimination between true and false positives from initial docking screens against STAT3/STAT5b SH2 domains through consensus rescoring.
Experimental Context: Follows initial high-throughput docking of ultra-large libraries (e.g., Enamine REAL, Mcule-in-stock) or focused libraries (e.g., OTAVAchemicals SH2 Domain Targeted Library) [1].
Materials:
- Software Requirements: Molecular dynamics simulation package (AMBER, GROMACS), Python/R for data analysis, structure visualization software (PyMOL, Chimera).
- Computational Resources: High-performance computing cluster with CPU and GPU nodes.
- Structural Data: High-resolution crystal structure of STAT SH2 domain (e.g., PDB IDs for STAT3/STAT5b SH2 domains).
Procedure:
- Input Preparation: Extract top 1,000-10,000 ranked compounds from initial docking screen. Prepare protein-ligand complexes using standardized structure preparation protocols (add hydrogens, assign charges, optimize hydrogen bonding).
- Multi-Method Execution:
- MM/GBSA Rescoring: Perform molecular mechanics optimization with generalized Born solvation using the BEAR (Binding Estimation After Refinement) protocol [48]:
- System setup: Apply AMBER ff03 force field to protein, GAFF to ligands, AM1-BCC charges.
- Minimization: 2,000 steps without restraints, distance-dependent dielectric ε = 4r, 12 Å cutoff.
- MD simulation: 100 ps at 300 K with SHAKE on, 2.0 fs timestep (ligand unrestrained).
- Final minimization: 2,000 steps on entire complex.
- Energy calculation: MM-PBSA and MM-GBSA binding free energy estimation.
- Deep Learning Rescoring: Apply deep neural network-based scoring functions (e.g., DeepDock) trained on known SH2 domain binders [1].
- Binding Pose Refinement: Execute short (5-10 ns) molecular dynamics simulations in explicit solvent for top-ranked diverse compounds to assess pose stability.
- MM/GBSA Rescoring: Perform molecular mechanics optimization with generalized Born solvation using the BEAR (Binding Estimation After Refinement) protocol [48]:
- Consensus Ranking: Normalize scores from each method (Z-score or percentile ranking). Generate weighted consensus rank based on retrospective validation performance of each method for SH2 domains.
- Experimental Triaging: Select compounds for experimental validation based on consensus ranking, chemical diversity, and favorable drug-like properties.
Pose Validation and Filtering Protocol
This critical pre-rescoring protocol addresses the fundamental issue of incorrect starting poses, which is a major contributor to rescoring failure.
Protocol 2: Pre-Rescoring Pose Validation
- Objective: To identify and eliminate geometrically unrealistic docking poses before committing to computationally expensive rescoring.
Experimental Context: Applied to all docking hits before proceeding to rescoring in Protocol 1.
Materials:
- Software Requirements: Structure visualization software (PyMOL, Chimera), molecular interaction analysis tools (PLIP, LigPlot+), scripting environment (Python/R).
- Reference Data: Known crystal structures of ligand-SH2 domain complexes for comparison.
Procedure:
- Strain Analysis: Calculate ligand strain energy using molecular mechanics; filter compounds with strain energy > 10-15 kcal/mol above their global minimum.
- Intermolecular Geometry Check: Identify poses with unsatisfied hydrogen bonds, buried charged groups without solvation, or polar groups in apolar subpockets.
- Conserved Interaction Validation: For STAT SH2 domains, verify poses maintain critical interaction with conserved arginine in the pY-binding pocket (βB5 position) [19].
- Cluster Analysis: Perform clustering of docking poses; prioritize consensus binding modes over singleton poses.
- Visual Inspection: Mandatory expert visual inspection of top 200-500 poses to identify geometric anomalies missed by automated filters.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Research Reagents for STAT SH2 Domain Virtual Screening
| Reagent / Resource | Function / Application | Example Sources / Specifications |
|---|---|---|
| STAT SH2 Domain Targeted Library | Focused library with compounds designed using SH2 domain pharmacophore models; improves initial hit rates. | OTAVAchemicals (1,807 compounds) [1] |
| Natural Product Library | Source of complex, 3D-shaped molecules with potential for PPI inhibition. | Compiled from LifeChemicals, ChemBridge, Asinex, ChemDiv (193,757 compounds) [1] |
| Enamine REAL Library | Ultra-large synthetically accessible virtual library for extensive chemical space sampling. | 5.51 billion compounds complying with Lipinski's rule of five and Veber criteria [1] |
| Mcule-in-stock Library | Commercially available compounds for rapid experimental follow-up. | 5.59 million purchasable compounds [1] |
| BEAR (Binding Estimation After Refinement) | Software tool for post-docking refinement with MD and MM-PB(GB)SA rescoring. | Implements AMBER modules for pose refinement and binding free energy estimation [48] |
| Deep Docking Workflow | AI-based method to reduce computational cost of screening ultra-large libraries. | Uses deep learning to prioritize compounds for docking [1] |
Rescoring docking hit lists remains a valuable but imperfect strategy in virtual screening campaigns against STAT SH2 domains. The documented failures stem from fundamental methodological limitations coupled with target-specific challenges presented by the SH2 domain structure and chemistry. By implementing the mitigation protocols outlined here—particularly multi-method consensus scoring, rigorous pre-rescoring pose validation, and expert-informed triaging—research teams can significantly reduce false positives and rescue valuable true positives that might otherwise be missed. As virtual screening continues to evolve toward ultra-large libraries, the integration of these careful rescoring validation strategies with emerging AI-based approaches will be essential for advancing STAT inhibitor discovery.
Integrating Machine Learning Rescoring with CNN-Score and RF-Score-VS
Virtual screening is a cornerstone of modern drug discovery, providing a cost-effective method for identifying potential hit compounds. The integration of machine learning (ML) scoring functions, particularly Convolutional Neural Network (CNN)-Score and RF-Score-VS, into structure-based virtual screening (SBVS) pipelines has demonstrated significant improvements in identifying active compounds, especially for challenging targets like the STAT3 and STAT5b SH2 domains [49] [1]. These domains are critical mediators of oncogenic signaling in various cancers, yet their relatively flat, solvent-exposed protein-protein interaction interfaces make them difficult to target with small molecules [1] [19]. Traditional docking scoring functions often struggle to achieve sufficient enrichment in such cases. ML rescoring functions address this limitation by learning complex features of protein-ligand interactions from structural data, enabling them to better distinguish true binders from decoys [50] [49]. This application note details the protocols and quantitative benefits of integrating CNN-Score and RF-Score-VS into virtual screening workflows focused on STAT SH2 domain inhibitors.
Quantitative Performance Benchmarking
The performance of ML rescoring functions has been rigorously evaluated in benchmark studies, demonstrating their ability to substantially enhance early enrichment in virtual screening campaigns.
Table 1: Virtual Screening Enrichment Performance of ML Rescoring Functions
| Target Protein | Docking Method | Rescoring Method | Performance Metric | Result | Citation |
|---|---|---|---|---|---|
| Wild-Type PfDHFR | PLANTS | CNN-Score | EF₁% | 28.0 | [49] |
| Quadruple-Mutant PfDHFR | FRED | CNN-Score | EF₁% | 31.0 | [49] |
| STAT3 SH2 Domain | AutoDock Vina (Baseline) | Deep Docking (CNN-based) | Hit Rate | 50.0% | [1] |
| STAT5b SH2 Domain | AutoDock Vina (Baseline) | Deep Docking (CNN-based) | Hit Rate | 42.9% | [1] |
| Multiple DUD-E Targets | AutoDock Vina (Baseline) | RF-Score-VS | Avg. Hit Rate (Top 1%) | >3x DOCK3.7 | [49] |
Key Performance Insights:
- CNN-Score has shown exceptional performance, achieving an Enrichment Factor at 1% (EF₁%) of 31 for a resistant variant of the Plasmodium falciparum enzyme DHFR, indicating a powerful ability to prioritize active compounds early in the ranking list [49].
- In prospective screens against the challenging STAT3 SH2 domain, an AI-driven workflow using a deep learning model achieved a remarkable 50% experimental hit rate, far exceeding typical virtual screening outcomes [1].
- RF-Score-VS also demonstrates a substantial advantage, with benchmark studies reporting its average hit rate at the top 1% of ranked molecules is more than three times higher than that of the classical scoring function DOCK3.7 [49].
- Importantly, re-scoring with CNN and RF-Score-VS has been shown to rescue the performance of docking programs that initially perform poorly, turning worse-than-random screening results into better-than-random enrichments [49].
Experimental Protocols
Core Workflow for ML Rescoring in Virtual Screening
The standard pipeline involves an initial docking step followed by a separate rescoring phase using the ML models.
Protocol 1: Structure Preparation
Objective: Generate high-quality, ready-to-dock protein structures.
- Source Structures: Obtain crystal structures of the target protein from the Protein Data Bank (PDB). For STAT SH2 domains, relevant PDB codes include those for STAT3 and STAT5b.
- Preparation Steps:
- Remove Non-Essential Components: Strip away water molecules, crystallization agents, and redundant protein chains not involved in binding [49].
- Add Hydrogen Atoms: Use tools like PDBFixer [13] or the protein preparation utilities in molecular modeling suites to add and optimize hydrogen atoms, correcting for proper protonation states at physiological pH [13] [49].
- Define the Binding Site: Identify the canonical phosphotyrosine (pY) binding pocket of the SH2 domain. This site is characterized by a conserved arginine residue (e.g., ArgβB5) in the FLVR motif that forms a salt bridge with the phosphate group [13] [19].
Protocol 2: Ligand Library Preparation
Objective: Prepare a library of small molecules for docking and screening.
- Compound Sourcing: Curate compound libraries from commercial or public databases (e.g., ZINC15, Broad Repurposing Hub, Enamine REAL) [13] [1].
- Preparation Steps:
- Format Conversion and Tautomer Generation: Use tools like OpenBabel or RDKit to ensure proper file formats and generate likely tautomeric states [13] [49].
- Generate 3D Conformers: For docking methods that require pre-generated conformers, use software such as Omega to sample low-energy 3D structures [49].
- Filtering: Apply filters to remove compounds with undesirable properties, such as Pan-Assay Interference Compounds (PAINS) [1].
Protocol 3: Molecular Docking for Pose Generation
Objective: Generate a diverse set of plausible binding poses for each compound in the library.
- Software Selection: Commonly used programs include AutoDock Vina, FRED, and PLANTS [49].
- Procedure:
- Set Up the Docking Grid: Define a grid box centered on the binding site of interest. For the STAT3 SH2 domain, a box with dimensions approximately 21.33Å × 25.00Å × 19.00Å has been used successfully [49].
- Run Docking: Execute the docking calculation with an appropriate level of exhaustiveness. For instance, using smina (a variant of Vina) with parameters
--seed 0 --exhaustiveness 50provides a good balance of thoroughness and speed [50]. - Output Multiple Poses: Retain multiple top-scoring poses per ligand (e.g., 5-20) to provide a conformational ensemble for the subsequent ML rescoring step [50].
Protocol 4: Machine Learning Rescoring
Objective: Re-rank the docked poses using pre-trained ML scoring functions to improve the prioritization of true binders.
- Model Selection: Employ pre-trained models of CNN-Score and RF-Score-VS v2 [49].
- Procedure:
- Input Preparation: Convert the docked protein-ligand complexes (poses) into the required input format for the ML model. For CNN-Score, this typically involves creating a 3D grid representation of the complex, analogous to an image, capturing atom types and positions [50] [49].
- Rescoring Execution: Run the ML models on the prepared inputs to generate new binding scores or probabilities for each pose.
- Ranking: Re-rank all compounds based on their best ML score obtained from any of their poses. The final output is a prioritized list for experimental testing.
The Scientist's Toolkit: Essential Research Reagents & Software
A successful ML-rescoring virtual screening campaign relies on a suite of specialized software tools and databases.
Table 2: Key Resources for ML-Enhanced Virtual Screening
| Category | Tool/Resource | Primary Function | Relevance to STAT SH2 Screening |
|---|---|---|---|
| Docking Software | AutoDock Vina / smina | Flexible ligand docking and initial pose scoring | Robust, widely-used baseline method for generating input poses for ML rescoring [50] [49]. |
| ML Scoring Functions | CNN-Score | Predict binding affinity using 3D convolutional neural networks | Excels in early enrichment; proven on challenging PPI targets like PfDHFR [50] [49]. |
| RF-Score-VS v2 | Predict binding affinity using random forest algorithm | Provides significant enrichment over classical functions; effective for virtual screening [49]. | |
| Chemical Libraries | ZINC15 / Broad Repurposing Hub | Source of commercially available or repurposable compounds | Provides large, diverse, and synthetically accessible small molecules for screening [13] [1]. |
| Structure Preparation | PDBFixer / OpenBabel | Add missing atoms, correct residues, and optimize hydrogen bonding | Crucial for preparing STAT SH2 domain structures (e.g., PDB: 2SHP) for accurate docking [13]. |
| Benchmarking Sets | DEKOIS 2.0 | Public benchmark sets for evaluating virtual screening methods | Used for rigorous performance validation of docking/rescoring protocols [49]. |
Biological Context: Targeting STAT SH2 Domains in Cancer
The strategic rationale for applying advanced virtual screening techniques to STAT SH2 domains is rooted in their critical role in oncogenesis.
Basis for Targeting:
- The SH2 domain of STAT proteins, including STAT3 and STAT5b, is essential for their activation. It recognizes and binds to phosphorylated tyrosine (pY) residues on cytokine receptors, facilitating the recruitment and subsequent phosphorylation of the STAT protein itself [1] [19].
- Once phosphorylated, STATs form homo- or heterodimers via reciprocal SH2-pY interactions, leading to their translocation to the nucleus where they drive the transcription of genes promoting cell survival, proliferation, and differentiation [1] [19].
- STAT3 and STAT5b are established oncoproteins. Their constitutive activation is a common feature in many leukemias, lymphomas, and solid tumors, making them high-value therapeutic targets [1].
- The Targeting Challenge: The SH2 domain's binding interface is relatively shallow and involved in protein-protein interactions, which are traditionally difficult to disrupt with small-molecule inhibitors. This makes them an ideal test case for advanced ML-based screening methods, which can learn complex patterns in binding data that elude traditional scoring functions [1].
The integration of machine learning rescoring functions, specifically CNN-Score and RF-Score-VS, into structure-based virtual screening pipelines represents a significant advancement in computational drug discovery. The quantitative benchmarking data and detailed protocols provided herein demonstrate that these methods can dramatically improve early enrichment and hit rates, even for challenging targets like the STAT3 and STAT5b SH2 domains. By leveraging these advanced computational strategies, researchers can accelerate the identification of novel, potent inhibitors for oncology and other therapeutic areas.
Addressing Protein Flexibility and Solvation Effects in PPI Interfaces
The Src Homology 2 (SH2) domain of Signal Transducer and Activator of Transcription (STAT) proteins represents a classic yet challenging protein-protein interaction (PPI) interface for therapeutic targeting. STAT proteins, particularly STAT3 and STAT5b, play pivotal roles in cancer progression and immune evasion through their SH2 domain-mediated dimerization, which is essential for activation and subsequent nuclear translocation [51] [1]. Unlike traditional drug targets with well-defined deep cavities, the STAT SH2 domain presents a relatively flat, solvent-exposed binding interface that requires sophisticated virtual screening approaches to identify effective inhibitors [52] [1].
The intrinsic flexibility of STAT proteins and critical solvation effects at the PPI interface complicate inhibitor discovery. Molecular dynamics simulations reveal that constrained flexibility within protein subunits can promote a defined range of architectures rather than nonspecific aggregation [53]. Furthermore, the release of energetically "unhappy" waters from hydrophobic pockets upon binding contributes significantly to the entropy of interaction [52]. These characteristics necessitate specialized computational protocols that move beyond traditional rigid docking approaches to address the dynamic nature of these interfaces and their intricate solvent interactions.
Key Challenges in STAT SH2 Domain Targeting
Structural Flexibility and Conformational Diversity
STAT proteins exhibit significant structural flexibility that impacts inhibitor binding:
- Domain Dynamics: The SH2 domain itself maintains a conserved αβββα motif with three sub-pockets (pY+0, pY+1, and pY+X) that exhibit subtle conformational variations [51]
- Oligomorphic Assemblies: Computational designs have revealed that local structural flexibility in protein subunits can drive unexpected oligomorphic outcomes, with assemblies adopting multiple distinct architectures [53]
- Conserved Binding Sites: The high conservation of the phosphotyrosine (pY+0) binding pocket across STAT family members (particularly between STAT1 and STAT3) creates significant challenges for achieving inhibitor specificity [54]
Solvation Effects and Hydrophobic Pockets
Solvation effects play a critical role in SH2 domain interactions:
- Water-Mediated Binding: Deep "anchor" pockets within the SH2 domain often contain structured water molecules whose displacement upon binding contributes favorably to binding entropy [52]
- Hydrophobic Interactions: The pY+1 and pY+X sub-pockets provide hydrophobic interactions that enhance binding affinity through water release to bulk solvent [52] [51]
- Solvent-Exposed Interfaces: The large, solvent-exposed nature of the PPI interface requires careful treatment of solvation energy in binding affinity calculations [1] [55]
Table 1: Key Challenges in Targeting STAT SH2 Domains
| Challenge Category | Specific Manifestation | Impact on Drug Discovery |
|---|---|---|
| Structural Flexibility | Concerted folding and binding of partner proteins | Difficulty in predicting binding modes |
| Multiple conformational states of SH2 domain | Reduced docking accuracy | |
| Interdomain flexibility in full-length STATs | Challenges in structural modeling | |
| Solvation Effects | Energetically unfavorable interface waters | Entropic contributions to binding |
| Hydrophobic sub-pockets (pY+1, pY+X) | Selectivity optimization opportunities | |
| Solvent-exposed binding surfaces | Imprecise binding affinity predictions |
Computational Methodologies
Molecular Dynamics for Flexibility Analysis
Molecular dynamics (MD) simulations provide powerful approaches for addressing protein flexibility:
Explicit Solvent MD Protocol:
- System Preparation: Solvate the STAT SH2 domain in a TIP3P water box with 10Å padding and add physiological ion concentration [56] [55]
- Energy Minimization: Perform 5,000 steps of steepest descent minimization to relieve steric clashes
- Equilibration: Conduct 100ps equilibration with positional restraints on protein heavy atoms, followed by 100ps without restraints
- Production Run: Execute 100ns-1μs production simulation using a 2fs timestep at 300K and 1atm pressure [57]
- Analysis: Calculate root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and residue correlation matrices to identify flexible regions
Accelerated Sampling Techniques:
- Gaussian Accelerated MD (GaMD): Adds a harmonic boost potential to reduce energy barriers and enhance conformational sampling [57]
- Replica Exchange MD (REMD): Parallel simulations at different temperatures to overcome energy barriers
- Metadynamics: Uses history-dependent bias potential to explore free energy landscapes
Solvation Modeling Approaches
Implicit Solvent Models:
- Generalized Born (GB) Models: Efficient approximation to Poisson-Boltzmann equation suitable for molecular dynamics [55]
- Solvent-Accessible Surface Area (SASA): Models nonpolar solvation contributions based on exposed surface area [55]
- Poisson-Boltzmann (PB) Methods: Numerical solution of dielectric continuum equations for electrostatic contributions [56] [55]
Explicit Solvent Models:
- TIP3P/TIP4P Water Models: Three-site and four-site water models with fixed point charges [56]
- Polarizable Force Fields: Models with interactive atomic dipoles (AMOEBA) for enhanced electrostatic accuracy [56]
- WaterMap Analysis: Identifies and characterizes hydration sites and their energetic properties [51]
Hybrid Solvent Methods:
- QM/MM with Implicit Solvent: Quantum mechanical treatment of binding site with molecular mechanics for protein and implicit solvent for bulk water [56]
- Reference Interaction Site Model (RISM): Statistical mechanical theory of molecular liquids that captures local solvent density fluctuations [56]
Table 2: Solvation Models for PPI Interface Characterization
| Solvent Model | Methodology | Advantages | Limitations | Typical Applications |
|---|---|---|---|---|
| Generalized Born (GB) | Continuum dielectric approximation | Computational efficiency; Suitable for MD | Less accurate for nonpolar solvation | High-throughput docking; MD simulations |
| Poisson-Boltzmann (PB) | Numerical solution of dielectric equation | Accurate electrostatic treatment | Computationally intensive; Single conformation | Binding affinity calculations |
| Explicit Water | Atomic representation of water molecules | Specific water interactions; Realistic dynamics | Extreme computational cost; Sampling challenges | Detailed binding mechanism studies |
| WaterMap | Identification of hydration sites | Energetic characterization of water networks | Requires prior MD simulation | Binding hotspot identification |
Advanced Docking and Virtual Screening
Ensemble Docking Protocol:
- Structure Selection: Compile multiple STAT SH2 domain structures from MD simulations or experimental sources [16]
- Receptor Grid Generation: Create docking grids for each structure with centroid at conserved binding residues [16]
- Multi-Conformation Docking: Dock compound libraries against all ensemble members
- Consensus Scoring: Rank compounds based on average binding affinity across ensemble
Ultra-High-Throughput Virtual Screening (uHTVS):
- AI-Assisted Workflows: Deep Docking approaches use deep learning to prioritize compounds for docking [1]
- Library Preparation: Filter billion-compound libraries (e.g., Enamine REAL) using drug-like properties [1]
- Multi-Step Docking: HTVS → SP → XP docking with 10% retention at each step [51] [16]
- MM-GBSA Refinement: Calculate binding free energies for top hits using molecular mechanics with generalized Born and surface area solvation [51]
Integrated Application Notes & Protocols
Comprehensive Protocol for STAT SH2 Inhibitor Screening
Phase 1: System Preparation (1-2 days)
- Target Selection: Obtain STAT SH2 domain structure (PDB: 6NJS recommended for STAT3) [51]
- Structure Preparation:
- Add missing side chains and loops using Prime [51]
- Optimize hydrogen bonding network and assign protonation states at pH 7.4
- Perform restrained minimization using OPLS3e or OPLS4 force field
- Molecular Dynamics Simulation:
- Solvate system in orthorhombic water box with 10Å buffer
- Neutralize with NaCl to 0.15M concentration
- Equilibrate using protocol in Section 3.1
Phase 2: Flexibility Analysis (3-5 days)
- Conformational Sampling:
- Perform 500ns GaMD simulation
- Cluster trajectories using RMSD-based clustering to identify representative conformations
- Select 5-10 diverse structures for ensemble docking
- Binding Site Analysis:
- Calculate pocket volumes for each conformation using POVME or MDpocket
- Map conserved water sites from simulations
- Identify sub-pocket flexibility in pY+0, pY+1, and pY+X sites
Phase 3: Virtual Screening (2-7 days, depending on library size)
- Library Preparation:
- Download natural compound libraries (e.g., ZINC15, 182,455 compounds) [51]
- Prepare ligands using LigPrep at pH 7.4±0.5 with OPLS3e force field
- Filter using Lipinski's Rule of Five and PAINS patterns
- Ensemble Docking:
- Generate receptor grids for each ensemble member centered on pY+0 binding pocket
- Perform HTVS docking → SP docking → XP docking workflow
- Retain top 1% at each stage for subsequent analysis
- Binding Affinity Refinement:
- Calculate MM-GBSA binding energies for top 1000 compounds
- Perform WaterMap analysis on top 100 compounds
- Select 20-50 compounds for experimental validation
Phase 4: Specificity Assessment (1-2 days)
- Comparative Modeling:
- ADMET Prediction:
- Calculate pharmacokinetic properties using QikProp
- Predict toxicity and metabolic stability
Case Study: Successful Application to STAT3-SH2 Domain
A recent study demonstrated the effectiveness of this integrated approach for STAT3-SH2 inhibitor identification [51]. The researchers screened 182,455 natural compounds from the ZINC15 database using multi-step docking (HTVS → SP → XP) followed by MM-GBSA calculations and molecular dynamics simulations. Through this approach, they identified four potential STAT3 inhibitors (ZINC255200449, ZINC299817570, ZINC31167114, and ZINC67910988) with ZINC67910988 showing superior stability in 100ns MD simulations [51].
Key success factors included:
- Ensemble Docking: Using multiple STAT3 conformations to account for binding site flexibility
- WaterMap Analysis: Identifying and targeting unfavorable hydration sites for displacement
- Specificity Validation: Cross-docking against other STAT SH2 domains to ensure selectivity
- Stability Assessment: Confirming binding mode stability through MD simulations
The protocol achieved an exceptional hit rate of 50.0% in experimental validation, significantly higher than traditional virtual screening approaches [1].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for STAT SH2 Domain Studies
| Reagent/Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| STAT SH2 Domain Structures | PDB: 6NJS (STAT3), 1BF5 (STAT1), 1Y1U (STAT5A) | Experimental templates for modeling | High-resolution structures with complete SH2 domains |
| Chemical Libraries | ZINC15 Natural Products, Enamine REAL, OTAVA SH2-Targeted Library | Source of potential inhibitors | Drug-like compounds with SH2 domain targeting potential |
| Computational Software | Schrödinger Suite, AutoDock Vina, GROMACS, AMBER | Molecular modeling and simulation | Compatibility with force fields and solvation models |
| Force Fields | OPLS3e/OPLS4, CHARMM36, AMBER ff19SB | Molecular mechanics parameterization | Accurate protein and small molecule representation |
| Solvation Models | GBSA, PBSA, TIP3P, TIP4P | Solvent effects treatment | Balance of accuracy and computational efficiency |
| Known Inhibitors | Stattic, SD-36, Fludarabine | Positive controls and validation | Established activity against STAT SH2 domains |
Troubleshooting and Optimization Guidelines
Common Challenges and Solutions
Low Hit Rates in Experimental Validation:
- Potential Cause: Overly rigid receptor structures in docking
- Solution: Increase conformational diversity in ensemble docking (7-10 structures)
- Solution: Incorporate protein side-chain flexibility during docking
Poor Compound Specificity:
- Potential Cause: Targeting overly conserved pY+0 pocket
- Solution: Focus on pY+1 and pY+X sub-pockets with greater sequence variation
- Solution: Implement comparative docking against all STAT family members
Inaccurate Binding Affinity Predictions:
- Potential Cause: Inadequate treatment of solvation effects
- Solution: Combine MM-GBSA with explicit solvent water mapping
- Solution: Include entropy estimates from MD simulations
Protocol Adaptation for Different STAT Family Members
The general protocol requires specific adaptations for different STAT targets:
- STAT1: Higher specificity achievable through less conserved pY+1 sub-pocket [54]
- STAT5B: Recent success targeting N-terminal domain in addition to SH2 domain [1]
- STAT2: Distinct binding characteristics requiring customized grid placement
Addressing protein flexibility and solvation effects is essential for successful inhibitor discovery against challenging PPI targets like the STAT SH2 domain. The integrated computational workflow presented here—combining ensemble docking from molecular dynamics simulations, sophisticated solvation modeling, and comprehensive specificity assessment—provides a robust framework for identifying potent and selective STAT inhibitors. As demonstrated in recent successful applications, this approach can achieve exceptional hit rates and identify novel chemical matter worthy of further development. Continuing advances in molecular dynamics methods, solvation models, and machine learning-assisted screening promise to further enhance our ability to target these therapeutically important but challenging PPI interfaces.
Optimizing Protocols for Drug-Resistant Mutants and Multi-Parameter Optimization
The Signal Transducer and Activator of Transcription (STAT) protein family comprises seven structurally related transcription factors (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) that are crucial for cellular signaling in response to cytokines, growth factors, and pathogens [59] [54]. Among their conserved structural domains, the Src Homology 2 (SH2) domain is particularly critical for STAT function, mediating phosphotyrosine-dependent protein-protein interactions that facilitate STAT dimerization and subsequent nuclear translocation for gene transcription [59] [19]. This domain, approximately 100 amino acids in length, contains a highly conserved phosphotyrosine (pY) binding pocket with an invariant arginine residue (βB5) that forms a salt bridge with phosphorylated tyrosine motifs [19]. The critical role of STAT proteins in fundamental cellular processes, combined with their abnormal activation in numerous diseases—including various cancers, inflammatory conditions, autoimmune disorders, and viral infections—has established them as highly attractive therapeutic targets [59].
STAT3 and STAT5B have received particular attention as oncological targets because their inhibition can cause cancer-derived cells to undergo growth arrest or apoptosis while leaving healthy cells largely unaffected [1]. However, targeting these transcription factors presents significant challenges due to their large, solvent-exposed protein-protein interaction interfaces and the high conservation of their SH2 domains across STAT family members [1] [54]. This conservation often leads to cross-binding specificity of inhibitors, complicating the development of selective therapeutic compounds [54]. The emergence of drug-resistant mutants further underscores the need for optimized screening protocols and multi-parameter optimization strategies in STAT inhibitor development.
Virtual Screening Strategies for STAT SH2 Domain Inhibitors
Virtual screening has emerged as a powerful, cost-effective methodology for identifying potent small-molecule STAT inhibitors, offering significantly higher hit rates compared to experimental high-throughput screening [1]. Two primary computational strategies have been developed for this purpose: knowledge-based approaches that leverage specialized compound libraries with known affinity for SH2 domains, and AI-based ultrahigh-throughput virtual screening (uHTVS) that utilizes deep learning models to efficiently screen ultralarge chemical libraries [1].
Table 1: Comparison of Virtual Screening Strategies for STAT SH2 Domains
| Screening Strategy | Compound Libraries | Key Features | Reported Hit Rates |
|---|---|---|---|
| Knowledge-Based | OtavaSH2 Library (1,807 compounds) | Pharmacophore models targeting SH2 domains; pre-filtered for PAINS | Benchmark for comparison |
| Natural Product Library (193,757 compounds) | Natural products and derivatives; complex 3D structures | Benchmark for comparison | |
| AI-Based uHTVS | Enamine REAL (5.51 billion compounds) | Synthetically accessible compounds complying with Lipinski's rule and Veber criteria | Up to 50.0% for STAT3-SH2 |
| Mcule-in-stock (5.59 million compounds) | Purchasable compounds; economic workflow | 42.9% for STAT5b-SH2 |
Experimental Protocol: Deep Docking Workflow
The following protocol outlines the Deep Docking methodology for identifying STAT SH2 domain inhibitors:
Target Preparation: Select appropriate X-ray crystal structures of the STAT SH2 domain (STAT3 or STAT5b). Validate structure quality and prepare for docking by adding hydrogen atoms, assigning partial charges, and defining flexible residues in the binding pocket.
Library Curation: Obtain synthetically accessible compound libraries (e.g., Enamine REAL, Mcule-in-stock). Apply pre-filtering to remove pan-assay interference compounds (PAINS) and ensure compliance with drug-like criteria such as Lipinski's Rule of Five and Veber criteria [1].
Benchmark Set Creation: Select a chemically diverse subset (e.g., 117,500 compounds) from the main library using diversity picking algorithms (e.g., RDKit Diversity Picker in KNIME) to create a representative benchmark dataset [1].
Initial Docking Phase: Perform molecular docking of the benchmark dataset against the prepared STAT SH2 domain structure using validated docking software and parameters. Record docking scores and poses for all compounds.
Model Training: Use the docking results (scores and compound structures) from the benchmark set to train a deep neural network model. This model learns to predict docking scores based on chemical features, reducing the need for exhaustive docking.
Iterative Screening: Apply the trained model to score the entire large library. Select the top-ranked compounds (e.g., highest predicted affinity) for actual docking in the next iteration.
Model Refinement: Retrain the deep learning model with new docking results, improving its predictive accuracy with each iteration.
Hit Identification: After multiple iterations (typically 5-10), select the highest-ranking compounds from the final model for experimental validation.
This protocol enables the efficient screening of billion-compound libraries while docking only a small fraction (e.g., 120,000 compounds) of the total chemical space, making it computationally feasible without supercomputer resources [1].
Addressing Cross-Binding Specificity
A significant challenge in STAT inhibitor development is the cross-binding specificity resulting from high conservation in SH2 domains across STAT family members [54]. Research has demonstrated that inhibitors like stattic (originally reported as a STAT3 inhibitor) and fludarabine (initially characterized as a STAT1 inhibitor) show substantial cross-reactivity with multiple STAT proteins [54]. Comparative in silico docking studies reveal that this lack of specificity occurs because these compounds primarily target the highly conserved pY+0 binding pocket within the SH2 domain [54]. To address this challenge, screening protocols should incorporate selectivity profiling across multiple STAT SH2 domains early in the discovery process. This can be achieved through:
- Comparative docking studies against SH2 domains of STAT1, STAT3, and other relevant STAT family members
- Focus on less conserved sub-pockets beyond the pY+0 site, such as the pY+1 and pY-X hydrophobic pockets that show greater sequence variation
- Experimental validation of computational predictions using cell-based assays measuring phosphorylation of different STAT proteins
Multi-Parameter Optimization for STAT Inhibitors
Principles of Multi-Parameter Optimization
Multi-parameter optimization (MPO) represents a critical process in drug discovery aimed at identifying lead compounds that achieve an optimal balance of multiple properties, including target potency, appropriate absorption, distribution, metabolism, and elimination (ADME) characteristics, and an acceptable safety profile [60] [61]. For STAT SH2 domain inhibitors, this involves optimizing often conflicting requirements between biochemical potency, selectivity, and drug-like properties. MPO approaches range from simple "rules of thumb" like Lipinski's Rule of Five to more sophisticated methods including desirability functions, Pareto optimization, and probabilistic approaches that account for predictive error and experimental variability [60].
Key Compound Properties for STAT Inhibitors
Table 2: Multi-Parameter Optimization Framework for STAT SH2 Inhibitors
| Parameter Category | Specific Properties | Target Values/Ranges | Optimization Strategy |
|---|---|---|---|
| Potency & Efficacy | IC50 against target STAT | < 1 µM | Structure-based design focusing on SH2 domain interactions |
| Selectivity over other STATs | >10-fold | Exploit differences in pY+1 and pY-X subpockets | |
| Physicochemical Properties | Molecular weight | ≤500 Da | Fragment-based approaches, pruning non-essential moieties |
| LogP | 1-3 | Introduce polar groups, adjust hydrophobic character | |
| Hydrogen bond donors/acceptors | ≤5/≤10 | Balance polarity for membrane permeability and solubility | |
| Pharmacokinetics | Metabolic stability | High (low hepatic clearance) | Introduce metabolic blockers, reduce labile functional groups |
| Plasma protein binding | Moderate to low | Optimize lipophilicity and ionization | |
| Safety & Toxicity | hERG inhibition | Low risk | Reduce basic pKa, decrease lipophilicity |
| PAINS filters | Clean | Remove problematic structural motifs early |
Experimental Protocol: Lead Optimization Workflow
The following structured protocol outlines the multi-parameter optimization process for STAT SH2 domain inhibitors:
Compound Profiling:
- Determine IC50 values against target STAT protein using standardized biochemical assays (e.g., fluorescence polarization, TR-FRET)
- Assess selectivity against other STAT family members (minimum STAT1, STAT3, STAT5B)
- Evaluate cellular activity in relevant disease models (e.g., cancer cell lines)
Physicochemical Characterization:
- Measure lipophilicity (LogD at pH 7.4) using chromatographic methods (e.g., UPLC-derived LogD)
- Assess solubility in biologically relevant media (PBS, simulated intestinal fluid)
- Determine permeability using PAMPA or cell-based models (Caco-2, MDCK)
In Vitro ADME Screening:
- Evaluate metabolic stability in liver microsomes (human and relevant species)
- Assess cytochrome P450 inhibition (key isoforms: 3A4, 2D6, 2C9)
- Determine plasma protein binding using equilibrium dialysis
Early Safety Assessment:
- Screen for hERG channel inhibition using patch clamp or binding assays
- Evaluate cytotoxicity in relevant cell lines
- Assess genotoxicity potential (Ames test, micronucleus)
Data Integration and Compound Selection:
- Apply desirability functions to normalize and weight key parameters
- Use probabilistic scoring to account for experimental variability
- Prioritize compounds based on balanced profile rather than single-parameter excellence
Structural Design Cycle:
- Employ matched molecular pair analysis (MMPA) to identify structural transformations that improve multiple parameters simultaneously
- Utilize structure-activity relationships (SAR) and structure-property relationships (SPR) to guide chemical design
- Iterate chemical design based on integrated data
This protocol emphasizes the parallel assessment of multiple parameters rather than sequential optimization, enabling more efficient identification of high-quality STAT inhibitors with balanced properties [61].
Research Reagent Solutions
Table 3: Essential Research Reagents for STAT SH2 Domain Studies
| Reagent Category | Specific Examples | Function/Application | Commercial Sources |
|---|---|---|---|
| STAT Inhibitors | Stattic, Fludarabine, STX-0119, OPB-31121 | Tool compounds for validation; reference standards for screening | Sigma-Aldrich, MedChemExpress |
| Specialized Screening Libraries | Otava SH2 Domain Targeted Library | Knowledge-based screening; focused library with predicted SH2 domain affinity | Otava Chemicals |
| Natural Product Libraries | Identification of complex, 3D-shaped inhibitors against PPI interfaces | LifeChemicals, ChemBridge, Asinex, ChemDiv | |
| Synthetically Accessible Libraries | Enamine REAL, Mcule-in-stock | Ultrahigh-throughput virtual screening; billions of synthesizable compounds | Enamine, Mcule |
| Antibodies | Phospho-STAT1 (Tyr701), Phospho-STAT3 (Tyr705), Total STAT1/3 | Cellular validation of inhibition; Western blot, immunofluorescence | Santa Cruz Biotechnology, Cell Signaling Technology |
| Cell Lines | Cancer cell lines with STAT dependency (e.g., MDA-MB-231, DU145) | Cellular efficacy assessment; mechanism of action studies | ATCC, DSMZ |
| Recombinant Proteins | STAT1, STAT3, STAT5B SH2 domains | Biochemical assays; structural studies; crystallography | R&D Systems, Abcam |
The development of effective STAT SH2 domain inhibitors requires integrated protocols that address both the challenges of targeting protein-protein interactions and the optimization of multiple drug-like properties. Virtual screening strategies, particularly AI-enhanced uHTVS approaches, have demonstrated remarkable efficiency in identifying novel chemotypes with hit rates exceeding 40% in prospective applications [1]. These computational methods must be coupled with rigorous experimental validation to address the critical issue of cross-binding specificity among STAT family members [54]. Furthermore, the implementation of systematic multi-parameter optimization frameworks ensures that identified hits can be successfully advanced to leads with balanced potency, selectivity, and developability profiles [60] [61]. As STAT inhibitors continue to show promise for therapeutic applications in oncology, inflammation, and viral infections, these optimized protocols provide a roadmap for navigating the complex landscape of targeting transcription factors with small molecules.
Benchmarking VS Performance: Hit Rates, Enrichment, and Experimental Validation
The Src Homology 2 (SH2) domain of Signal Transducer and Activator of Transcription 3 (STAT3) is a critical therapeutic target due to its essential role in STAT3 dimerization and activation, a process implicated in numerous cancers [62] [19]. Targeting this domain offers a strategic approach to inhibit the oncogenic signaling of STAT3. However, the traditional virtual screening method of "brute-force" docking faces immense computational challenges when applied to ultralarge chemical libraries containing billions of "make-on-demand" compounds [26] [1].
Deep Docking (DD) has emerged as a powerful artificial intelligence (AI) accelerated platform that addresses this bottleneck. By training deep learning models on the docking scores of a small, iteratively selected subset of a chemical library, DD can rapidly approximate docking outcomes for the vast majority of unprocessed compounds [26]. This approach achieves dramatic data reduction and enrichment of high-scoring molecules, enabling the efficient screening of gigascale chemical spaces that were previously inaccessible [26] [27]. This application note details a benchmark case study where the Deep Docking workflow was deployed against the STAT3-SH2 domain, resulting in an exceptional experimental hit rate of 50.0% [1].
Deep Docking Methodology and Workflow
The Deep Docking protocol is an iterative workflow that combines fast molecular descriptor calculation, conventional docking, and deep learning to efficiently prioritize potential hits from an ultralarge library.
Core DD Pipeline
The pipeline, as introduced in its foundational form, relies on the following consecutive steps [26]:
- Descriptor Calculation: For each molecule in the ultralarge docking database (e.g., ZINC15, Enamine REAL), standard ligand-based QSAR descriptors (e.g., 2D molecular fingerprints) are computed.
- Initial Training Set Sampling: A reasonably sized training subset is randomly sampled from the database and docked against the target protein using a conventional docking program.
- Deep Model Training: The generated docking scores are related to the 2D molecular descriptors through a Deep Neural Network (DNN). A docking score cutoff is used to classify training compounds as virtual hits or non-hits.
- Prediction and Augmentation: The trained QSAR model predicts the docking outcomes for all unprocessed database entries. A predefined number of molecules predicted to be virtual hits are then randomly sampled to augment the training set.
- Iteration: Steps 2 through 4 are repeated iteratively. With each iteration, the model improves, and the hit prediction cutoff typically becomes more stringent. The process continues until a predefined number of iterations is reached, yielding a final, highly enriched subset of molecules for conventional docking.
Workflow Implementation for STAT3-SH2
The following diagram illustrates the integrated workflow of the Deep Docking process, from library preparation to final hit selection.
Benchmarking Case Study: STAT3-SH2 Domain
Experimental Setup and Performance
A study benchmarked the Deep Docking workflow against the STAT3-SH2 domain, a challenging protein-protein interaction (PPI) target [1]. The performance of the AI-accelerated uHTVS was compared to a "brute-force" docking of a smaller, diversity-picked subset and traditional knowledge-based approaches using specialized libraries.
Table 1: Virtual Screening Performance Against STAT3-SH2 Domain
| Screening Approach | Library Screened | Library Size | Compounds Actually Docked | Experimental Hit Rate |
|---|---|---|---|---|
| Deep Docking (AI-based) | Enamine REAL | 5.51 billion | ~120,000 (economic workflow) | 50.0% [1] |
| Brute-Force Docking | Mcule Benchmark Set | 117,500 | 117,500 | Not specified (lower than DD) [1] |
| Knowledge-Based (Traditional) | Otava SH2-Targeted Library | 1,807 | 1,807 | Not specified (lower than DD) [1] |
| Knowledge-Based (Traditional) | Natural Product Library | 193,757 | 193,757 | Not specified (lower than DD) [1] |
The Deep Docking protocol demonstrated exceptional efficiency and effectiveness. By applying an "economic workflow," it achieved its remarkable hit rate while docking only about 120,000 compounds—a minute fraction (0.002%) of the 5.51-billion-compound Enamine REAL library [1]. This represents a computational data reduction of nearly 50,000-fold. Furthermore, the study confirmed that Deep Docking is also highly effective with smaller, million-compound libraries, achieving a 42.9% hit rate against the related STAT5b-SH2 domain using the Mcule-in-stock library [1].
STAT3 Signaling and SH2 Domain Inhibition
To understand the therapeutic significance of this work, it is essential to contextualize the role of the STAT3 SH2 domain. STAT3 is a transcription factor that is constitutively activated in many cancers [30] [63]. Its activation is dependent on phosphorylation at Tyr705, which is facilitated by the SH2 domain.
Diagram: STAT3 Activation Pathway and SH2 Domain Inhibition
As shown in the pathway, the SH2 domain binds to the phosphorylated Tyr705 (pY705) of another STAT3 monomer, forming an active dimer that translocates to the nucleus to drive the expression of oncogenes [62] [19]. Small-molecule inhibitors identified by Deep Docking bind directly to the STAT3 SH2 domain, disrupting this critical protein-protein interaction and subsequent dimerization [1] [62].
Detailed Experimental Protocols
This section provides a detailed methodology for replicating the Deep Docking workflow for a novel target, based on the established protocol [26] [1].
Protocol 1: Deep Docking Setup and Execution
Objective: To reduce an ultralarge chemical library to a manageable subset enriched with potential binders for a specific protein target.
Materials:
- Hardware: High-Performance Computing (HPC) cluster.
- Software: Publicly available DD scripts from GitHub (https://github.com/vibudh2209/D2) [26].
- Chemical Library: Database of synthesizable compounds (e.g., Enamine REAL, ZINC15). Precompute 2D molecular fingerprints for all entries.
- Target Preparation: A prepared protein structure file of the target (e.g., STAT3-SH2 domain, PDB: 6NJS).
Procedure:
- Initialization: Configure the DD platform on your HPC cluster. Input the database of precomputed molecular descriptors and the prepared protein target structure.
- Iteration 1: The DD platform will randomly sample an initial training set (e.g., 50,000-100,000 compounds). Dock this subset using your chosen docking program (e.g., FRED, AutoDock Vina).
- Model Training 1: Train the first deep learning model on the docking scores from the initial set. Set a lenient score cutoff to classify a broad set of virtual hits.
- Prediction & Selection 1: Use the model to predict the docking scores of all undocked compounds. Randomly select a new set of compounds from the top-ranked predictions to augment the training set.
- Iteration 2-n: Repeat the docking, model training, and prediction/selection steps. With each iteration, the model becomes more accurate, and the hit prediction cutoff can be made more stringent to further enrich for the best binders.
- Final Output: After 5-10 iterations, the DD platform will output a final list of 100,000-500,000 prioritized compounds. This list represents the highly enriched subset.
Protocol 2: Validation of Top Hits
Objective: To experimentally validate the top-ranking compounds from the final DD output for STAT3-SH2 inhibitory activity.
Materials:
- Compounds: Purchased samples of the top 50-100 virtual hits.
- Cell Lines: STAT3-dependent cancer cell lines (e.g., prostate cancer LNCaP cells, gastric cancer MGC803 cells) [1] [30] [62].
- Reagents: Antibodies for pSTAT3 (Tyr705), total STAT3; IL-6 cytokine; cell viability assay kits (e.g., alamarBlue); luciferase-based STAT3 reporter assay.
Procedure:
- In Vitro Binding Validation:
- Perform a fluorescence polarization (FP) assay to confirm direct binding to the STAT3 SH2 domain by measuring the ability of hits to disrupt the binding of a fluorescently labeled phosphopeptide (e.g., GpYLPQTV) [62].
- Cellular Activity Assessment:
- Treat IL-6-stimulated cells with the hit compounds and analyze lysates via western blot to measure inhibition of STAT3 phosphorylation at Tyr705 [30] [62].
- Transfert cells with a STAT3-responsive luciferase reporter construct. Treat with compounds and measure luciferase activity to quantify inhibition of STAT3-mediated transcription [62].
- Functional Phenotypic Assays:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for STAT3-SH2 Targeted Discovery
| Category | Item / Resource | Function / Description | Example Source / Citation |
|---|---|---|---|
| Chemical Libraries | Enamine REAL Library | Ultralarge library of synthetically accessible compounds for uHTVS. | [1] |
| ZINC15 Database | Publicly available database of commercial compounds for virtual screening. | [26] | |
| Software & Tools | Deep Docking (DD) Platform | AI-accelerated workflow for screening ultralarge libraries. | [26] |
| RosettaVS / OpenVS | Open-source, physics-based VS platform with high accuracy. | [27] | |
| Docking Programs (FRED, AutoDock Vina) | Conventional docking software used within the DD iterative steps. | [26] [27] | |
| STAT3 Reagents | STAT3-SH2 Domain Protein | Recombinant protein for in vitro binding assays (FP, SPR). | [62] |
| Phospho-STAT3 (Tyr705) Antibody | Detects activated STAT3 in cellular assays (Western Blot). | [30] [62] | |
| STAT3 Reporter Assay | Luciferase-based construct to measure STAT3 transcriptional activity. | [62] | |
| Reference Inhibitors | S3I-201 | A well-characterized STAT3-SH2 domain inhibitor for benchmark comparisons. | [62] |
| Stattic | A non-peptidic small molecule inhibitor of STAT3 activation. | [17] |
This benchmarking case study demonstrates that the Deep Docking platform is a transformative tool for drug discovery, particularly against challenging targets like the STAT3-SH2 domain. The ability to achieve a 50.0% experimental hit rate from a 5.51-billion-compound library establishes a new benchmark for the efficiency and success of virtual screening campaigns [1]. The detailed protocols and toolkit provided here offer a roadmap for researchers to implement this powerful AI-accelerated strategy. Applying Deep Docking to other therapeutically relevant SH2 domains and difficult-to-drug targets promises to significantly accelerate the identification of novel, potent lead compounds in oncology and beyond.
Comparative Analysis of Docking Tools and Screening Strategies
The Signal Transducer and Activator of Transcription (STAT) family of proteins represents a critical node in cellular signaling, regulating processes such as proliferation, survival, and differentiation. Under physiological conditions, STAT activity is tightly regulated, but constitutive activation occurs in a broad range of human cancers, inflammation, and autoimmune diseases [64]. The Src Homology 2 (SH2) domains of STAT proteins are particularly attractive therapeutic targets as they facilitate critical protein-protein interactions required for STAT dimerization and activation [58]. Despite more than a decade of research, no STAT-targeting drug has gained FDA approval, highlighting the challenges in developing effective inhibitors [58].
Virtual screening has emerged as a powerful computational approach to identify novel STAT inhibitors, but the success of these campaigns depends critically on the selection of appropriate docking tools and screening strategies. This application note provides a comparative analysis of current molecular docking programs and integrated screening methodologies, with specific application to STAT SH2 domain inhibitor discovery. We present quantitative performance data, detailed experimental protocols, and practical recommendations to guide researchers in designing effective virtual screening pipelines for STAT-targeted drug discovery.
Performance Benchmarking of Docking Software
Key Performance Metrics for Docking Tools
The virtual screening accuracy of molecular docking software is typically evaluated using several key metrics. The enrichment factor (EF) measures the ability of a docking program to identify true binders early in the screening process, calculated as the ratio of true positives in the top X% of ranked compounds compared to random selection [27]. Root-mean-square deviation (RMSD) quantifies the accuracy of predicted ligand binding poses by measuring the deviation from experimentally determined crystal structures [65]. Area under the curve (AUC) of the receiver operating characteristic (ROC) curve provides an overall measure of screening performance across all ranking thresholds [27].
Comparative Performance Analysis
Table 1: Performance Comparison of Molecular Docking Software in Virtual Screening Benchmarks
| Docking Software | Sampling Algorithm | Scoring Function | RMSD Performance | Screening Power (EF1%) | Best Use Cases |
|---|---|---|---|---|---|
| RosettaVS (VSH mode) | Genetic Algorithm | RosettaGenFF-VS (Physics-based) | ~1.5-2.0 Å | 16.72 [27] | High-accuracy screening with receptor flexibility |
| DOCK 6 | Anchor-and-grow | Force field-based | 1.5-2.5 Å [65] | N/A | RNA targets, general virtual screening |
| AutoDock Vina | Monte Carlo | Empirical & Knowledge-based | ~2.0 Å | Lower than RosettaVS [27] | Standard protein-ligand docking, balance of speed/accuracy |
| GOLD | Genetic Algorithm | Empirical (ChemScore, GoldScore) | N/A | N/A | High-accuracy pose prediction |
| Glide | Hierarchical filters | Empirical (GlideScore) | N/A | ~11.9 [27] | Commercial high-throughput screening |
| rDock | Stochastic algorithm | Empirical | >2.5 Å [65] | N/A | Nucleic acid targets |
| RLDOCK | Monte Carlo | Force field-based | Least accurate [65] | N/A | Nucleic acid targets (with limitations) |
Recent benchmarking studies reveal significant differences in performance across docking programs. In comprehensive evaluations, RosettaVS demonstrated superior performance in both docking accuracy and virtual screening enrichment, achieving an enrichment factor of 16.72 at the 1% cutoff on the CASF-2016 benchmark—significantly outperforming other methods [27]. DOCK 6 showed strong performance for ribosomal targets, accurately replicating native ligand binding poses in 4 out of 11 tested structures [65]. However, the performance of docking programs can be target-dependent, with some methods struggling with specific target classes like RNA pockets due to high flexibility [65].
Integrated Screening Strategies for STAT Inhibitors
Cell-Based Functional Screening
Cell-based transcriptional reporter assays provide a powerful functional screening approach for STAT inhibitors. This strategy involves stably transfecting cells with a luciferase reporter gene under the control of a STAT-responsive promoter [64]. When stimulated with cytokines such as IL-6, STAT activation leads to luciferase expression quantifiable by luminometry. To ensure STAT-specific effects, researchers have utilized STAT1-deficient human fibrosarcoma cells, eliminating confounding activation of other STAT family members [64].
A critical component of this approach involves implementing counter-screens to exclude compounds acting through non-specific mechanisms. Parallel screening using NFκB-dependent reporter systems effectively identifies and filters out non-specific inhibitors, such as DNA damaging agents and protein synthesis inhibitors [64]. This functional screening strategy offers the advantage of identifying STAT inhibitors working through novel mechanisms beyond direct SH2 domain binding, including effects on nuclear import or upstream pathway components.
Structure-Based Virtual Screening Pipeline
For STAT SH2 domain targets, we propose a consensus virtual screening protocol integrating multiple computational approaches:
Comparative Homology Modeling: Develop high-quality 3D structure models for all human STAT SH2 domains using homology modeling based on existing crystal structures (STAT1: 1BF5, STAT3: 1BG1, STAT5A: 1Y1U) [58].
Multi-Tool Docking Campaign: Employ multiple docking programs (RosettaVS, DOCK 6, AutoDock Vina) in parallel to screen compound libraries against STAT SH2 domains.
Pharmacophore Filtering: Apply structure-based pharmacophore models derived from known active compounds to prioritize candidates with key interaction features.
ADMET Profiling: Predict absorption, distribution, metabolism, excretion, and toxicity properties using tools like SwissADME to filter compounds with undesirable properties [66].
Consensus Scoring: Rank compounds based on integrated scores from multiple docking programs and screening approaches.
This consensus approach has demonstrated improved performance over single-method screening, particularly for challenging targets like tubulin-microtubule system inhibitors [66].
AI-Accelerated Screening Platforms
Recent advances in artificial intelligence have enabled the development of accelerated virtual screening platforms capable of screening billion-compound libraries in practical timeframes. The OpenVS platform incorporates active learning techniques that simultaneously train target-specific neural networks during docking computations to triage and select promising compounds for more expensive physics-based docking calculations [27]. This approach has reduced screening times for multi-billion compound libraries to under seven days using a 3000-CPU cluster, while maintaining high accuracy through methods like RosettaVS with its virtual screening express (VSX) and virtual screening high-precision (VSH) modes [27].
Experimental Protocols
Protocol 1: STAT-Specific Virtual Screening Workflow
Objective: Identify specific STAT SH2 domain inhibitors using structure-based virtual screening.
Materials:
- STAT SH2 domain 3D structures (from homology modeling or crystal structures)
- Compound library (ZINC, Enamine, in-house collections)
- Computational resources (HPC cluster recommended)
- Docking software (RosettaVS, DOCK 6, AutoDock Vina)
Procedure:
- Target Preparation:
- Generate homology models for all STAT SH2 domains using MODELLER or SWISS-MODEL
- Optimize structures using molecular mechanics force fields (AMBER, CHARMM)
- Define binding site around phosphotyrosine pocket with 4.5Å radius
Library Preparation:
- Download compound library in SMILES format
- Generate 3D conformations using OMEGA or CORINA
- Add hydrogens and assign partial charges using AM1-BCC
- Filter for drug-like properties (Lipinski's Rule of Five)
Multi-Stage Docking:
- Stage 1: High-throughput screening with RosettaVS VSX mode
- Stage 2: High-precision docking with RosettaVS VSH mode for top 1% compounds
- Stage 3: Consensus docking with DOCK 6 and AutoDock Vina
Post-Docking Analysis:
- Cluster compounds by structural similarity
- Visualize top poses for key interactions with SH2 domain
- Select diverse compounds for experimental validation
Validation: Include known STAT inhibitors as positive controls and assess enrichment during screening.
Protocol 2: Cell-Based STAT Transcriptional Activity Assay
Objective: Functionally validate STAT inhibitors identified through virtual screening.
Materials:
- STAT-responsive luciferase reporter construct
- STAT-deficient cell lines (e.g., STAT1-/- fibrosarcoma)
- Cytokines for STAT activation (IL-6 for STAT3)
- Luciferase assay kit
- Test compounds dissolved in DMSO
Procedure:
- Cell Line Development:
- Stably transfect STAT-deficient cells with STAT-responsive luciferase reporter
- Clone selection using antibiotic resistance (e.g., G418)
- Validate responsiveness to STAT-activating cytokines
Compound Screening:
- Seed cells in 96-well plates (10,000 cells/well)
- Pre-treat with test compounds (1-100 µM) for 1 hour
- Stimulate with STAT-activating cytokine (e.g., IL-6 at 10 ng/mL) for 6 hours
- Measure luciferase activity using luminometer
Counter-Screening:
- Parallel screening with NFκB-responsive reporter cells
- Cytotoxicity assessment using MTT or resazurin assays
Data Analysis:
- Normalize luminescence to vehicle control (0% inhibition) and unstimulated control (100% inhibition)
- Calculate IC50 values using non-linear regression
- Prioritize compounds with STAT-specific inhibition and minimal cytotoxicity
Validation: Include known STAT inhibitors (e.g., Stattic) as positive controls and assess assay robustness using Z-factor calculations.
Visualization of Screening Workflows and Signaling Pathways
STAT Inhibitor Screening Workflow
STAT Activation and Inhibition Pathway
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagents for STAT Inhibitor Screening
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| STAT SH2 Domain Structures | Structure-based screening | Crystal structures: STAT1 (1BF5), STAT3 (1BG1), STAT5A (1Y1U); Homology models for other STATs |
| Compound Libraries | Source of potential inhibitors | ZINC, Enamine, BIOFACQUIM, Prestwick library (~1,200 compounds) |
| Docking Software | Pose prediction and scoring | RosettaVS, DOCK 6, AutoDock Vina, GOLD, Glide |
| STAT-Responsive Reporter Constructs | Functional validation | Luciferase under STAT-responsive promoter (e.g., M67 SIE mutant) |
| STAT-Deficient Cell Lines | Specificity controls | STAT1-/- human fibrosarcoma cells, other STAT-isogenic pairs |
| Cytokines/Growth Factors | STAT pathway activation | IL-6 (STAT3), IFN-γ (STAT1), EGF, PDGF |
| Luciferase Assay Systems | Reporter gene quantification | Commercial kits (Promega, Thermo Fisher) with luminometer detection |
| ADMET Prediction Tools | Compound prioritization | SwissADME, pkCSM, ProTox-II |
| High-Performance Computing | Computational screening | CPU/GPU clusters (3000+ CPUs for billion-compound screening) |
The field of STAT inhibitor discovery is advancing rapidly with the development of more sophisticated docking tools and integrated screening strategies. The emergence of AI-accelerated platforms like OpenVS and improved physics-based methods such as RosettaVS are addressing critical challenges in screening accuracy and efficiency [27]. However, important limitations remain, including the accurate prediction of binding affinities and the need for better models of receptor flexibility [67].
Future directions in STAT inhibitor screening will likely involve increased integration of machine learning approaches, more sophisticated treatment of protein flexibility through ensemble docking, and the development of target-specific scoring functions optimized for STAT SH2 domains. The implementation of comprehensive multi-parameter optimization balancing potency, specificity, and drug-like properties early in the screening process will be essential for translating computational hits into clinically viable STAT inhibitors [67].
As these technologies mature, virtual screening campaigns targeting STAT proteins and other challenging transcription factor targets will become increasingly successful, potentially unlocking new therapeutic opportunities in oncology, inflammation, and autoimmune diseases where STAT pathways play a central pathogenic role.
The Src Homology 2 (SH2) domain is a critical protein module approximately 100 amino acids long that specifically recognizes phosphorylated tyrosine (pTyr) residues to mediate intracellular signaling cascades [19]. In the context of Signal Transducer and Activator of Transcription (STAT) proteins, particularly the oncogenic STAT3 and STAT5b, the SH2 domain facilitates dimerization through reciprocal phosphotyrosine-SH2 interactions, which is essential for their activation and subsequent nuclear translocation [1] [17]. This dimerization mechanism makes the STAT SH2 domain a prominent target for therapeutic intervention in cancers and other diseases [1] [19].
Virtual screening has emerged as a powerful, cost-effective computational approach for identifying potential inhibitors, especially for challenging protein-protein interaction (PPI) interfaces like the STAT SH2 domain [1] [17]. The screening process involves evaluating ultra-large chemical libraries, often containing millions to billions of compounds, to select promising candidates for experimental validation [1]. The success of these virtual screening campaigns depends on robust evaluation metrics that can accurately discriminate true inhibitors from inactive compounds and ensure the identification of chemically diverse leads. This application note details the critical success metrics—Enrichment Factors (EF), Area Under the Curve (AUC), and Chemotype Diversity—within the specific context of STAT SH2 domain inhibitor discovery.
Core Success Metrics in Virtual Screening
Area Under the Curve (AUC) of the ROC Curve
The Area Under the Receiver Operating Characteristic (ROC) Curve (AUC) serves as a fundamental metric for evaluating the overall performance of a virtual screening workflow in distinguishing active compounds from inactive ones [1]. The ROC curve itself plots the true positive rate (TPR) against the false positive rate (FPR) across all possible classification thresholds.
In practice for STAT SH2 domain screening, a retrospective virtual screening is performed using a benchmark dataset containing known active compounds and decoy molecules. The resulting AUC value quantifies the probability that a randomly selected active compound will be ranked higher than a randomly selected decoy by the screening protocol [1]. An AUC of 0.5 indicates performance equivalent to random selection, while an AUC of 1.0 represents perfect separation of actives from inactives. For challenging PPI targets like the STAT3-SH2 domain, AI-enhanced screening workflows have demonstrated exceptional performance, achieving hit rates as high as 50.0%, which implies a high AUC value in the underlying docking model [1].
Enrichment Factors (EF)
Enrichment Factors (EF) measure the concentration of active compounds in the top fraction of a screened library compared to their random distribution throughout the entire library. This metric is particularly valuable for assessing the practical utility of a virtual screening method in a real-world drug discovery context, where researchers are primarily interested in the top-ranked compounds.
The EF is calculated as follows: ( \text{EF} = \frac{\text{(Number of actives in top } \%) / (\text{Total number of actives})}{\text{(Total compounds in top } \%) / (\text{Total compounds in library})} )
For STAT SH2 domain screens, EF is typically evaluated at the top 1%, 2%, and 5% of the ranked database [1]. The following table summarizes expected EF and AUC values for successful STAT SH2 domain screens based on benchmark studies:
Table 1: Interpretation of Enrichment Factor and AUC Values in STAT SH2 Domain Screening
| Performance Tier | Top 1% EF | Top 2% EF | Top 5% EF | AUC Value | Practical Significance |
|---|---|---|---|---|---|
| Excellent | >10 | >15 | >20 | >0.8 | High probability of finding multiple actives in a small subset |
| Good | 5-10 | 8-15 | 10-20 | 0.7-0.8 | Useful for hit identification with manageable experimental follow-up |
| Moderate | 3-5 | 5-8 | 7-10 | 0.6-0.7 | May require screening of larger top-ranked fractions |
| Random | ~1 | ~1 | ~1 | ~0.5 | No enrichment; method fails to distinguish actives |
Chemotype Diversity
Chemotype Diversity refers to the structural and chemical variety present within a set of hit compounds. It is a crucial metric for ensuring that virtual screening does not simply identify multiple analogs of the same chemical scaffold, but rather provides a foundation for a robust drug discovery campaign with multiple lead series [1]. Assessing chemotype diversity helps to:
- Mitigate the risk of compound attrition due to shared off-target effects or toxicity profiles
- Provide backup compounds should a primary lead series fail during optimization
- Enable exploration of different binding modes within the STAT SH2 domain pocket
In successful screens against the STAT5b SH2 domain, researchers have identified novel, diverse chemotypes with high hit rates (42.9%), demonstrating that effective virtual screening can yield chemically varied starting points for optimization [1]. Diversity analysis typically involves calculating molecular descriptors (e.g., molecular weight, logP, topological polar surface area) and employing clustering methods based on molecular fingerprints to group compounds with similar structural features.
Experimental Protocols for Metric Evaluation
Protocol 1: Retrospective Screening for AUC and EF Calculation
Objective: To validate a virtual screening workflow for STAT SH2 domains by calculating AUC and Enrichment Factors using a benchmark dataset.
Materials:
- STAT SH2 domain crystal structure (e.g., PDB ID: 6NJS for STAT3)
- Known active compounds for the target (e.g., 69 known STAT3 actives from ChEMBL)
- Decoy molecules (e.g., generated using the DUD-E database)
- Molecular docking software (e.g., GLIDE, AutoDock Vina, GROMACS)
- Computing infrastructure
Procedure:
- Dataset Preparation: Compile a benchmark dataset containing known active compounds and decoy molecules. Filter out pan-assay interference compounds (PAINS) [1].
- Structure Preparation: Prepare the STAT SH2 domain protein structure by adding hydrogen atoms, filling missing side chains, and optimizing hydrogen bonding networks using tools like the Protein Preparation Wizard (Schrödinger) or PDBFixer [13] [17].
- Grid Generation: Define the binding site for docking. For STAT SH2 domains, this typically encompasses the phosphotyrosine-binding pocket. Generate a receptor grid file centered on the co-crystallized ligand or known binding site [17].
- Docking Execution: Dock all compounds in the benchmark dataset against the prepared STAT SH2 domain structure using standardized parameters [1] [17].
- Result Analysis: Rank all compounds based on their docking scores. Calculate the AUC value and Enrichment Factors at the top 1%, 2%, and 5% of the ranked list using statistical analysis tools [1].
Protocol 2: Chemotype Diversity Analysis of Screening Hits
Objective: To assess the structural diversity of compounds identified as hits in a virtual screen against STAT SH2 domains.
Materials:
- List of potential hit compounds with structures
- Cheminformatics software (e.g., RDKit, Schrödinger Canvas)
- Clustering algorithms and visualization tools
Procedure:
- Descriptor Calculation: For all hit compounds, calculate molecular descriptors including molecular weight, logP, hydrogen bond donors/acceptors, topological polar surface area, and number of rotatable bonds [43] [17].
- Fingerprint Generation: Generate molecular fingerprints (e.g., ECFP4, FCFP4) for each compound to encode their structural features [1].
- Clustering Analysis: Perform clustering using appropriate algorithms (e.g., Butina clustering, k-means) based on the fingerprint similarity to group compounds with related structures [1].
- Diversity Assessment: Evaluate the number of distinct clusters, cluster sizes, and inter-cluster distances. A diverse hit list will contain multiple clusters with significant structural differences between them [1].
- Structural Representation: Select representative compounds from each major cluster for further experimental validation to maximize structural diversity in the initial testing phase.
Research Reagent Solutions for STAT SH2 Domain Screening
Table 2: Essential Research Reagents and Resources for STAT SH2 Domain Virtual Screening
| Category | Specific Resource | Function in Screening | Example Sources |
|---|---|---|---|
| Protein Structures | STAT SH2 domain crystal structures | Provides structural basis for docking experiments | PDB IDs: 6NJS (STAT3), other STAT structures |
| Compound Libraries | ZINC15 Database [13] [17] | Source of commercially available compounds for screening | University of California, San Francisco |
| Broad Repurposing Hub [13] | Collection of FDA-approved, clinical, and preclinical compounds | Broad Institute | |
| Enamine REAL Database [1] | Ultra-large library of synthetically accessible compounds | Enamine | |
| SH2 Domain Targeted Library | Curated library of compounds with predicted SH2 domain affinity | OTAVAchemicals [1] | |
| Software Tools | Molecular Docking Software | Predicts binding poses and scores of ligands | GLIDE [43] [17], AutoDock Vina [13], GROMACS [13] |
| Molecular Dynamics Software | Simulates protein-ligand dynamics and stability | GROMACS [13], Desmond [17] | |
| Cheminformatics Platforms | Analyzes compound properties and diversity | RDKit [13] [1], Schrödinger Suite [43] [17] | |
| Benchmark Datasets | Known STAT Actives | For retrospective validation of screening methods | ChEMBL [1] |
| Decoy Molecules | Inactive compounds for control and validation | DUD-E database [1] |
Workflow Visualization for Metric Evaluation
Virtual Screening Evaluation Workflow: This diagram illustrates the integrated process for evaluating virtual screening success metrics, from initial preparation through experimental validation of diverse hit compounds.
The rigorous evaluation of virtual screening campaigns for STAT SH2 domain inhibitors requires the integrated assessment of Enrichment Factors, AUC, and Chemotype Diversity. These metrics provide complementary insights: AUC offers an overall assessment of the screening method's ability to distinguish actives from inactives; EF quantifies the practical enrichment in the top-ranked compounds most likely to be tested experimentally; and chemotype diversity ensures that the resulting hit list provides multiple, structurally distinct starting points for lead optimization.
For the challenging target class of STAT SH2 domains, contemporary virtual screening approaches, including AI-enhanced methods like Deep Docking, have demonstrated the capability to achieve high performance across all these metrics, with hit rates exceeding 40% in prospective studies [1]. By implementing the standardized protocols and metrics outlined in this application note, researchers can more effectively prioritize computational resources and identify high-quality, diverse chemical matter for one of the most promising target classes in oncology and immunology drug discovery.
The discovery of inhibitors targeting the Src Homology 2 (SH2) domains of STAT (Signal Transducers and Activators of Transcription) proteins represents a significant challenge and opportunity in drug development, particularly in oncology and inflammatory diseases. The STAT family, comprising STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6, shares a highly conserved SH2 domain that facilitates phosphotyrosine-dependent protein-protein interactions essential for STAT dimerization, nuclear translocation, and transcriptional activation [68] [69]. Abnormal activation of STAT signaling pathways, especially STAT3 and STAT1, is implicated in numerous malignancies, including breast cancer, melanoma, prostate cancer, and multiple myeloma, making these proteins attractive therapeutic targets [69].
Virtual screening (VS) has emerged as a powerful computational approach to identify initial hit compounds that potentially disrupt the phosphotyrosine-SH2 interaction, thereby inhibiting STAT dimerization [70] [69]. However, the high conservation among STAT-SH2 domains presents a major challenge for achieving specificity, and many previously reported STAT3 inhibitors demonstrate significant cross-binding with other STAT family members [69]. This reality underscores the critical importance of robust experimental validation strategies to confirm both the potency and specificity of computationally identified hits before investing resources in lead optimization. This application note outlines established best practices for this validation process within the context of STAT SH2 domain inhibitor research.
Hit Quality Assessment and Triage
Following a virtual screen, the first step is a thorough computational assessment of hit quality to prioritize compounds for experimental testing. This process involves evaluating multiple chemical and physicochemical parameters to identify compounds with the highest potential for success in subsequent experimental assays and development.
Table 1: Key Criteria for Hit Quality Assessment and Triage
| Assessment Category | Specific Criteria/Parameters | Target Values/Rationale |
|---|---|---|
| Potency & Efficiency | IC₅₀, Ki, % Inhibition, Ligand Efficiency (LE) | Low micromolar activity (e.g., 1-25 µM); LE ≥ 0.3 kcal/mol/heavy atom for fragment-like hits [70]. |
| Chemical Tractability | Synthetic feasibility, presence of reactive or toxicophores | Avoid Pan-Assay Interference Compounds (PAINS); assess potential for chemical optimization [71]. |
| Drug-Likeness | Molecular Weight, cLogP, Topological Polar Surface Area (TPSA) | Adherence to established rules (e.g., Lipinski's Rule of Five) to improve likelihood of favorable ADMET properties [71]. |
| Selectivity Potential | STAT-Comparative Binding Affinity Value (STAT-CBAV), Ligand Binding Pose Variation (LBPV) | Computational metrics to prioritize compounds with predicted specificity for a single STAT member over others [69]. |
The transition from in-silico hit to confirmed active compound requires clear hit-calling criteria. While a minority of VS studies predefine such criteria, establishing them is essential. For STAT inhibitors, hit identification often relies on an activity cutoff in the low to mid-micromolar range (e.g., 1-50 µM), as the primary goal is to identify a novel scaffold for further optimization rather than a final drug candidate [70]. The use of ligand efficiency metrics, which normalize biological activity by molecular size, is highly recommended as it helps identify hits whose potency is not merely a function of large molecular weight [70].
Experimental Validation Workflow: From Binding to Cellular Activity
A rigorous, multi-stage experimental validation funnel is crucial for confirming the activity and specificity of virtual screening hits targeting the STAT SH2 domain. The following workflow diagram outlines this sequential process, from initial binding confirmation to ultimate mechanistic validation in disease-relevant models.
Diagram 1: The experimental validation workflow for virtual screening hits, progressing from binding confirmation to mechanistic studies.
Orthogonal Biophysical Binding Assays
The initial validation step involves confirming direct physical binding between the hit compound and the STAT SH2 domain using biophysical techniques. These assays provide label-free, direct evidence of interaction that is less prone to the false positives common in primary screening assays [71].
- Surface Plasmon Resonance (SPR): SPR is a powerful technique for quantifying binding affinity (KD), kinetics (kon, koff), and stoichiometry in real-time without requiring labels [71]. It is highly sensitive and can be used to study the interaction of small molecules with immobilized STAT SH2 domains.
- Isothermal Titration Calorimetry (ITC): ITC measures the heat change associated with binding, providing a direct readout of the binding affinity (KD), enthalpy (ΔH), entropy (ΔS), and stoichiometry (n) in a single experiment [71]. It is considered a gold standard for confirming binding as it is a label-free, solution-based technique.
- Nuclear Magnetic Resonance (NMR): NMR-based methods, such as chemical shift perturbation or line broadening, can confirm binding and even map the ligand-binding site on the STAT SH2 domain [71]. This is particularly valuable for confirming that the compound binds to the intended pTyr-binding pocket.
- Thermal Shift Assay (TSA): Also known as differential scanning fluorimetry, TSA monitors the thermal stabilization of the STAT protein upon ligand binding. A shift in the protein's melting temperature (ΔTm) indicates compound binding and can be used as a medium-throughput secondary confirmation method [71].
Functional Activity and Counter-Screens
After confirming direct binding, the next step is to demonstrate that the binding event translates into the desired functional outcome—inhibition of STAT phosphorylation or dimerization.
- STAT Phosphorylation Assays: Using cell lysates or in vitro systems, these assays measure the compound's ability to inhibit cytokine-induced phosphorylation of the target STAT (e.g., STAT3 Tyr705) via Western blot or ELISA. This confirms the compound can achieve its primary functional goal in a biologically relevant context [69].
- Electrophoretic Mobility Shift Assay (EMSA): EMSA assesses the compound's ability to prevent STAT dimerization and subsequent DNA binding. A reduction in the gel-shift band indicates successful disruption of the STAT-DNA complex formation [69] [68].
- Counter-Screens for Specificity: To address the critical issue of STAT cross-binding, hits must be screened against other STAT family members and unrelated targets. This can be done using the same functional or biophysical assays configured for different STAT proteins (e.g., STAT1, STAT5) [68] [69]. This step is vital for identifying truly specific inhibitors.
- Assay Interference Counter-Screens: These assays rule out false positives caused by compound aggregation, oxidation/reduction, or fluorescence. Examples include testing compounds in the presence of detergents like Triton X-100 to disrupt aggregates, or using assay formats that are insensitive to such interferences [71].
Cellular Phenotypic and Mechanism-of-Action Studies
The final validation stage demonstrates activity in a live-cell, physiologically relevant environment.
- Cell Viability and Proliferation Assays: For STAT3 inhibitors in oncology, assays like WST-1 or MTT are used to measure the reduction in viability of STAT3-dependent cancer cell lines [69]. This provides a direct link between target inhibition and a desired phenotypic outcome.
- Gene Expression Reporter Assays: These assays utilize constructs with a STAT-responsive promoter driving a luciferase or GFP reporter. A decrease in reporter signal indicates functional inhibition of STAT-mediated transcription within the cell [69] [68].
- Downstream Target Analysis: Validation includes measuring the reduction in mRNA or protein levels of known STAT target genes (e.g., Bcl-2, c-Myc for STAT3) using qPCR or Western blot, confirming downstream biological consequences [69].
The Scientist's Toolkit: Research Reagent Solutions
Successful experimental validation relies on a suite of specialized reagents and tools. The following table details key materials essential for studying STAT SH2 domain inhibitors.
Table 2: Essential Research Reagents for STAT Inhibitor Validation
| Reagent / Material | Function and Application | Specific Examples / Notes |
|---|---|---|
| Recombinant STAT SH2 Domains | Purified protein for biophysical assays (SPR, ITC) and in vitro functional assays. | Essential for direct binding studies; can be wild-type or mutant forms to probe binding site specificity [69]. |
| Phospho-STAT Specific Antibodies | Detect inhibited phosphorylation of target STAT in cellular assays via Western blot. | Antibodies specific for pY-STAT3, pY-STAT1; critical for functional validation in cell lysates [69]. |
| STAT-Dependent Cell Lines | Cellular models for phenotypic and mechanistic studies. | Human cancer cell lines with constitutive STAT3 signaling (e.g., breast, melanoma) [69]. |
| Reporter Gene Constructs | Measure STAT transcriptional activity in live cells. | Plasmids with STAT-responsive elements (e.g., M67 SIE) driving luciferase expression [68]. |
| Known STAT Inhibitors | Serve as positive controls in validation assays to benchmark new hits. | Compounds like Stattic (for STAT3) or previously reported inhibitors with known activity profiles [69]. |
A Practical Case Study: STAT1/STAT3 Specific Inhibitor Screening
A comprehensive study by Szeląg et al. provides a exemplary model for the application of these best practices [69]. The researchers developed a novel pipeline to identify STAT-specific inhibitors, directly addressing the problem of cross-binding. Their approach involved:
- Comparative In-Silico Docking: They generated 3D structure models for all human STATs and performed virtual screening of multi-million compound libraries against each STAT-SH2 domain.
- Novel Selection Criteria: Instead of relying solely on binding affinity, they introduced the "STAT-comparative binding affinity value" (STAT-CBAV) and "ligand binding pose variation" (LBPV) as metrics to prioritize compounds with predicted specificity for either STAT1 or STAT3.
- Experimental Validation: The top-ranked, specificity-predicted compounds were then subjected to experimental validation to confirm both their activity and, crucially, their STAT1 or STAT3 specificity, as predicted by the computational model.
This case study highlights the power of integrating advanced computational filtering with a rigorous, multi-tiered experimental validation protocol to solve a central challenge in the field.
The journey from a computational prediction to a biologically active, specific STAT SH2 domain inhibitor is fraught with potential for false positives and promiscuous binders. A systematic and stringent validation strategy, incorporating orthogonal biophysical binding assays, functional and counter-screens for specificity, and culminating in cellular mechanistic studies, is non-negotiable for success. By adhering to these best practices and leveraging the appropriate toolkit of reagents and assays, researchers can confidently triage virtual screening hits, thereby laying a solid foundation for the development of high-quality lead compounds with genuine therapeutic potential.
Conclusion
The virtual screening landscape for STAT SH2 domain inhibitors has been revolutionized by approaches capable of navigating ultra-large chemical spaces, with AI-driven and evolutionary algorithms demonstrating remarkable efficiency and hit rates exceeding 50% in benchmark studies. The integration of robust benchmarking, machine learning rescoring, and hybrid strategies that combine ligand- and structure-based methods is crucial for success against these challenging PPI targets. Future directions must focus on improving scoring function accuracy, better accounting for full system flexibility, and streamlining the transition from computational hits to validated leads. As these methodologies mature, they hold significant promise for delivering novel, potent, and selective STAT inhibitors, ultimately impacting the treatment paradigms for STAT-driven cancers and other diseases.
References
- 1. Ultrahigh-Throughput Virtual Screening Strategies against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12308805/]
- 2. Review Article STAT3 axis in cancer and cancer stem cells [https://www.sciencedirect.com/science/article/abs/pii/S0304419X25002033]
- 3. Structural Implications of STAT3 and STAT5 SH2 Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6895964/]
- 4. A High-Throughput Fluorescence Polarization-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9781101/]
- 5. High-throughput profiling of sequence recognition by ... [https://elifesciences.org/articles/82345]
- 6. The Prognostic Role of STAT5B Across Cancer Types and ... [https://www.mdpi.com/2218-273X/15/11/1503]
- 7. SH2 domains: role, structure and implications for molecular ... [https://pubmed.ncbi.nlm.nih.gov/14987415/]
- 8. SH2 domain [https://en.wikipedia.org/wiki/SH2_domain]
- 9. Phosphotyrosine recognition domains: the typical, the atypical ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-10-32]
- 10. Specificity and regulation of phosphotyrosine signaling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7337045/]
- 11. SH2db, an information system for the SH2 domain - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10320075/]
- 12. SH2 Domain - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/sh2-domain]
- 13. Determination of promising inhibitors for N-SH2 domain of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11612003/]
- 14. Selective Targeting of SH2 Domain–Phosphotyrosine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5417323/]
- 15. High-Throughput Phosphotyrosine Profiling Using SH2 ... [https://www.sciencedirect.com/science/article/pii/S1097276507003346]
- 16. Identifying p56 lck SH2 Domain Inhibitors Using Molecular ... [https://www.mdpi.com/2076-3417/14/10/4277]
- 17. Computational screening and molecular dynamics of ... [https://link.springer.com/article/10.1007/s11030-024-11075-5]
- 18. Update on Structure and Function of SH2 Domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC12470777/]
- 19. Update on Structure and Function of SH2 Domains [https://www.mdpi.com/1422-0067/26/18/9060]
- 20. Cytosolic delivery of peptidic STAT3 SH2 domain inhibitors [https://pmc.ncbi.nlm.nih.gov/articles/PMC7294595/]
- 21. Identification of the Linker-SH2 Domain of STAT as ... [https://www.sciencedirect.com/science/article/pii/S1535947620338639]
- 22. Review The SH2 domain: versatile signaling module and ... [https://www.sciencedirect.com/science/article/abs/pii/S1570963904002882]
- 23. SH2 Domain Binding: Diverse FLVRs of Partnership [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2020.575220/full]
- 24. Discovery of Novel Allosteric SHP2 Inhibitor Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11280062/]
- 25. Enamine REAL Space [https://enamine.net/compound-collections/real-compounds/real-space-navigator]
- 26. Deep Docking: A Deep Learning Platform for Augmentation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7318080/]
- 27. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 28. Ultra-large library screening with an evolutionary algorithm ... [https://www.nature.com/articles/s42004-025-01758-x]
- 29. REvoLd: Ultra-Large Library Screening with an ... [https://arxiv.org/html/2404.17329v1]
- 30. Discovery of potent STAT3 inhibitors using structure-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10652556/]
- 31. Novel STAT3 small-molecule inhibitors identified by ... [https://scholars.houstonmethodist.org/en/publications/novel-stat3-small-molecule-inhibitors-identified-by-structure-bas/]
- 32. Ultra-large library screening with an evolutionary algorithm in ... [https://pubmed.ncbi.nlm.nih.gov/41203805/]
- 33. Cache: Utilizing ultra-large library screening in Rosetta to ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01084-3]
- 34. RosettaEvolutionaryLigand (REvoLd) [https://docs.rosettacommons.org/docs/latest/revold]
- 35. Abstract 17: Novel STAT3 inhibitors identified by Structure- ... [https://www.scilit.com/publications/8e96cd658a93e70daa74aa031a22251d]
- 36. Modeling receptor flexibility in the structure-based design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9512760/]
- 37. Interfacing Protein Structure Modeling and Ligand Docking [https://www.sciencedirect.com/science/article/pii/S0022283624003139]
- 38. Representing receptor flexibility in ligand docking through ... [https://pubmed.ncbi.nlm.nih.gov/15984891/]
- 39. Discovery of potent STAT3 inhibitors using structure-based ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2023.1287797/full]
- 40. A practical guide to large-scale docking [https://www.nature.com/articles/s41596-021-00597-z]
- 41. Drug Design by Pharmacophore and Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145410/]
- 42. Strategies for 3D pharmacophore-based virtual screening [https://www.sciencedirect.com/science/article/abs/pii/S1740674910000375]
- 43. Identifying p56lck SH2 Domain Inhibitors Using Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11906178/]
- 44. Machine learning accelerates pharmacophore-based ... [https://www.nature.com/articles/s41598-024-58122-7]
- 45. Concepts and Applications Exemplified on Hydroxysteroid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6332202/]
- 46. On the failure of rescoring in virtual screening [https://medchemash.substack.com/p/on-the-failure-of-rescoring-in-virtual]
- 47. Rescoring Docking Hit Lists for Model Cavity Sites [https://www.sciencedirect.com/science/article/abs/pii/S002228360800096X]
- 48. Refinement and Rescoring of Virtual Screening Results [https://pmc.ncbi.nlm.nih.gov/articles/PMC6637856/]
- 49. Benchmarking the Structure-Based Virtual Screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12363558/]
- 50. Protein-Ligand Scoring with Convolutional Neural Networks [https://pmc.ncbi.nlm.nih.gov/articles/PMC5479431/]
- 51. Computational screening and molecular dynamics of natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12638358/]
- 52. Flexibility and small pockets at protein–protein interfaces [https://pmc.ncbi.nlm.nih.gov/articles/PMC4726663/]
- 53. Local structural flexibility drives oligomorphism in ... [https://www.nature.com/articles/s41594-025-01490-z]
- 54. In silico simulations of STAT1 and STAT3 inhibitors predict ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299913008224]
- 55. Design and application of implicit solvent models in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4045398/]
- 56. Solvent model [https://en.wikipedia.org/wiki/Solvent_model]
- 57. The key role of the dynamics and flexibility of proteins in ... [https://pubmed.ncbi.nlm.nih.gov/41101376/]
- 58. Comparative screening and validation as a novel tool ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299914004099]
- 59. SINBAD, structural, experimental and clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8971479/]
- 60. Multi-parameter optimization: identifying high quality ... [https://pubmed.ncbi.nlm.nih.gov/22316157/]
- 61. An overview of compound properties, multiparameter ... [https://www.sciencedirect.com/science/article/pii/S0223523423002660]
- 62. Novel STAT3 Inhibitors Targeting STAT3 Dimerization by ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.836724/full]
- 63. Novel STAT3 Small-Molecule Inhibitors Identified by Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8217378/]
- 64. Screening approaches to generating STAT inhibitors [https://pmc.ncbi.nlm.nih.gov/articles/PMC3670287/]
- 65. Comparative Assessment of Docking Programs for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10044086/]
- 66. Consensus Virtual Screening Protocol Towards the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10078098/]
- 67. Where Docking and Virtual Screening Can Drop the Ball in ... [https://deeporigin.com/blog/where-docking-and-virtual-screening-drop-the-ball-in-drug-discovery]
- 68. Comparative screening and validation as a novel... [https://www.ovid.com/journals/eujoph/fulltext/10.1016/j.ejphar.2014.05.047~comparative-screening-and-validation-as-a-novel-tool-to]
- 69. Identification of STAT1 and STAT3 Specific Inhibitors Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4339377/]
- 70. Hit Identification and Optimization in Virtual Screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC3772997/]
- 71. Hit-to-Lead: Hit Validation and Assessment [https://www.sciencedirect.com/science/article/abs/pii/S0076687918303859]