Activating vs. Inactivating STAT SH2 Domain Mutations: Mechanisms, Methods, and Clinical Implications
This article provides a comprehensive analysis of gain-of-function (GOF) and loss-of-function (LOF) mutations within the STAT protein SH2 domain, a critical hotspot in oncology and immunology.
Activating vs. Inactivating STAT SH2 Domain Mutations: Mechanisms, Methods, and Clinical Implications
Abstract
This article provides a comprehensive analysis of gain-of-function (GOF) and loss-of-function (LOF) mutations within the STAT protein SH2 domain, a critical hotspot in oncology and immunology. Aimed at researchers and drug development professionals, it synthesizes foundational knowledge of STAT SH2 structure with advanced methodological approaches for characterizing mutations. The content explores the divergent pathological consequences of activating versus inactivating mutations, using specific variants like STAT5B-Y665F and STAT5B-Y665H as paradigmatic examples. It further examines emerging therapeutic strategies, including small-molecule inhibitors, and discusses the integration of computational and functional validation techniques to bridge molecular understanding with clinical application in precision medicine.
The STAT SH2 Domain: Structure, Function, and Mutation Hotspots
Canonical Structure of the STAT SH2 Domain and Its Role in JAK-STAT Signaling
The Signal Transducer and Activator of Transcription (STAT) proteins represent a critical component of the JAK-STAT signaling pathway, an evolutionarily conserved system that transmits information from extracellular cytokine signals directly to the nucleus to regulate gene transcription [1] [2]. Among the various domains comprising STAT proteins, the Src homology 2 (SH2) domain serves an indispensable role, functioning as the central module that governs activation, dimerization, and nuclear translocation of STATs [3] [2]. This domain's ability to recognize and bind specific phosphotyrosine motifs establishes the binary "on-off" switch of the pathway, making it a focal point for both physiological regulation and pathogenic mutations [2] [4]. Within the broader context of STAT SH2 domain mutation research, understanding the precise structural mechanisms that differentiate activating from inactivating mutations provides crucial insights for therapeutic development. This guide systematically compares the canonical structure of the STAT SH2 domain against disease-associated mutations, supported by experimental data that highlights the domain's function as a molecular switch in health and disease.
Canonical Structure of the STAT SH2 Domain
Architectural Features and Classification
The STAT SH2 domain belongs to a distinct subclass of SH2 domains characterized by a unique αβββα structural motif [3] [5]. This core architecture consists of a central anti-parallel β-sheet (composed of βB, βC, and βD strands) flanked by two α-helices (αA and αB) [3]. What distinguishes the STAT-type SH2 domain from the more common Src-type is the presence of a C-terminal αB' helix rather than the additional β-sheets (βE and βF) found in Src-type domains [3] [5] [6]. This structural variation is not merely incidental; it represents an ancient evolutionary template from which other SH2 domains may have diversified [5] [6].
The STAT SH2 domain contains two functionally critical sub-pockets:
- Phosphotyrosine-binding pocket (pY pocket): Formed by the αA helix, BC loop, and one face of the central β-sheet, this pocket recognizes and binds phosphotyrosine residues [3].
- Specificity pocket (pY+3 pocket): Created by the opposite face of the β-sheet along with residues from the αB helix and CD and BC* loops, this pocket determines sequence specificity by accommodating residues C-terminal to the phosphotyrosine, particularly the +3 residue [3].
A defining feature of STAT SH2 domains is their hydrophobic system - a cluster of non-polar residues at the base of the pY+3 pocket that stabilizes the β-sheet conformation and maintains overall domain integrity [3]. Additionally, the αB, αB', and BC* loop participate in critical cross-domain interactions that facilitate STAT dimerization [3].
Structural Determinants of SH2 Domain Function
The STAT SH2 domain mediates two essential functions in JAK-STAT signaling: phosphopeptide recognition and STAT dimerization. In conventional phosphopeptide binding, the target peptide aligns perpendicular to the β-sheet, with the phosphotyrosine inserting into the pY pocket and C-terminal residues extending across the SH2 domain into the pY+3 pocket [3]. This binding mode is conserved across SH2 domains, but STAT-type domains exhibit unique flexibility, with the accessible volume of the pY pocket varying dramatically even on sub-microsecond timescales [3].
Table 1: Key Structural Motifs of the Canonical STAT SH2 Domain
| Structural Motif | Location | Functional Role | Conservation |
|---|---|---|---|
| Central β-sheet (βB-βD) | Core domain | Forms binding surface for phosphopeptides | High across STAT family |
| αA helix | N-terminal region | Contributes to pY pocket formation | High across STAT family |
| αB helix | C-terminal region | Forms part of pY+3 pocket and dimerization interface | High across STAT family |
| αB' helix | C-terminal extension | STAT-type SH2 domain signature; mediates dimerization | Unique to STAT-type SH2 domains |
| BC loop | Between βB-βC | Forms part of pY pocket; hotspot for mutations | Variable; mutation prone |
| Hydrophobic system | Base of pY+3 pocket | Stabilizes β-sheet conformation | High across STAT family |
For STAT dimerization, the SH2 domain facilitates reciprocal phosphotyrosine-SH2 interactions between two STAT monomers, forming either parallel homo- or heterodimers [3] [4]. This "phosphotyrosine switch" mechanism represents the fundamental activation step that enables nuclear accumulation and DNA binding of STAT transcription factors [1] [4].
Methodologies for Investigating STAT SH2 Domain Structure and Function
Structural Biology Approaches
X-ray crystallography has been instrumental in elucidating the atomic-level structure of STAT SH2 domains. The methodology typically involves:
- Protein Expression and Purification: Cloning, expressing, and purifying recombinant STAT SH2 domains, often as fusion proteins to enhance solubility [3].
- Crystallization: Optimizing conditions for crystal formation using vapor diffusion or microfluidic approaches [3].
- Data Collection and Structure Determination: Collecting diffraction data at synchrotron facilities and solving structures through molecular replacement or experimental phasing [3].
A significant challenge in crystallizing STAT SH2 domains is their inherent flexibility, which can result in crystals that capture different conformational states [3]. This dynamic behavior underscores the importance of complementing crystallographic data with other biophysical techniques.
Functional Characterization of Mutations
Site-directed mutagenesis coupled with functional assays represents the cornerstone for validating the impact of SH2 domain mutations. A standard experimental workflow includes:
- Mutation Introduction: Using PCR-based methods or CRISPR/Cas9 gene editing to introduce specific point mutations into STAT genes [7].
- Functional Assays:
- Phosphorylation Analysis: Western blotting with phospho-specific STAT antibodies to assess activation status [7].
- Transcriptional Reporter Assays: Luciferase-based reporters under control of STAT-responsive promoters to measure transcriptional activity [7] [8].
- Electrophoretic Mobility Shift Assays (EMSAs): Evaluating STAT-DNA binding capability [7].
- Subcellular Localization Studies: Immunofluorescence microscopy to track nuclear translocation [7] [8].
Table 2: Key Experimental Assays for Characterizing STAT SH2 Domain Mutations
| Assay Type | Measured Parameters | Applications in SH2 Domain Research |
|---|---|---|
| Tyrosine Phosphorylation Assays | STAT phosphorylation kinetics and magnitude | Determine impact on activation threshold |
| Transcriptional Reporter Assays | Luciferase activity driven by STAT-responsive elements | Quantify functional consequences on gene regulation |
| Co-Immunoprecipitation | Protein-protein interaction strength | Assess dimerization capability and receptor binding |
| Chromatin Immunoprecipitation (ChIP) | Genomic binding profiles | Evaluate DNA binding specificity and efficiency |
| Cellular Proliferation/Differentiation | Growth curves, marker expression | Determine phenotypic consequences in relevant cell types |
For in vivo validation, researchers have employed knock-in mouse models where human disease-associated mutations are introduced into the endogenous mouse STAT genes [7]. These models allow assessment of mutation impacts on mammalian development, immune function, and tissue homeostasis under physiological conditions [7].
Comparative Analysis of STAT SH2 Domain Mutations
Mutation Hotspots and Functional Consequences
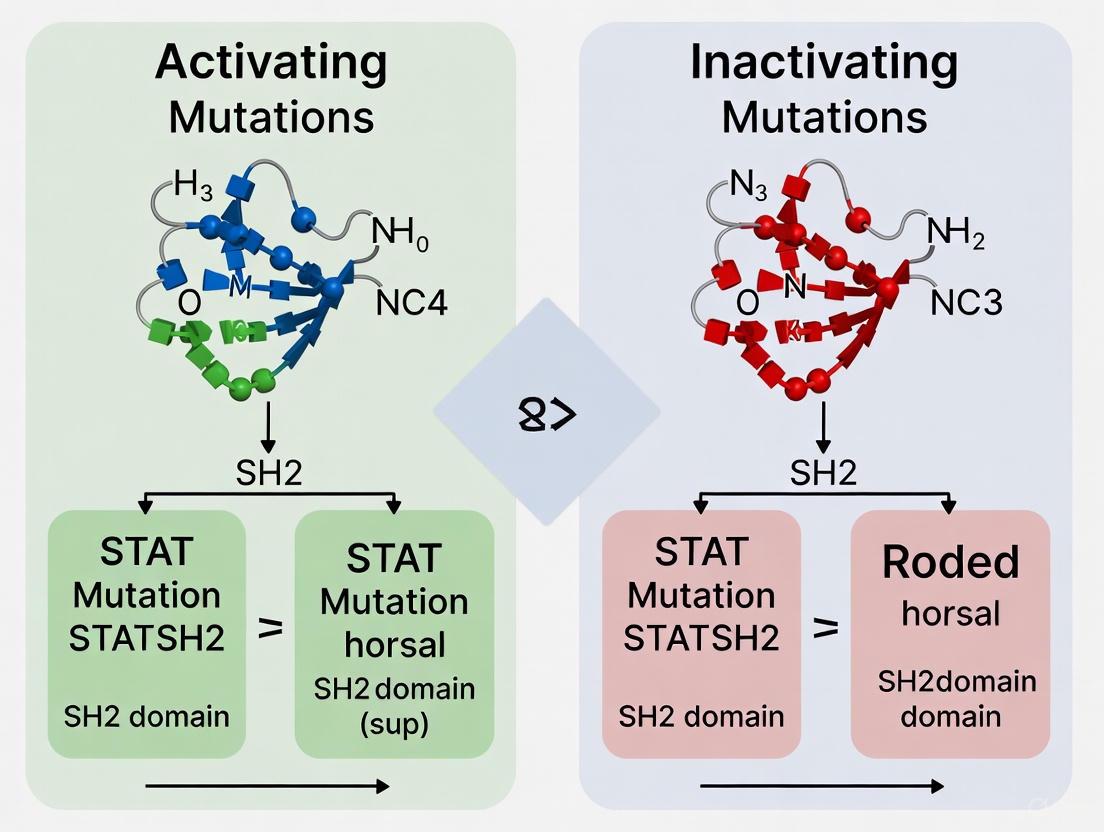
Sequencing analyses of patient samples have identified the SH2 domain as a hotspot for mutations in both STAT3 and STAT5B, with distinct clusters occurring in structurally and functionally critical regions [3]. The majority of disease-associated mutations localize to the pY pocket, pY+3 pocket, and the BC loop that connects βB and βC strands [3]. These mutations can have either gain-of-function (GOF) or loss-of-function (LOF) consequences, sometimes with different substitutions at the same residue producing opposite effects [3].
Table 3: Comparative Analysis of Disease-Associated STAT SH2 Domain Mutations
| Mutation | STAT Protein | Location in SH2 | Functional Consequence | Associated Disease(s) |
|---|---|---|---|---|
| Y665F | STAT5B | pY pocket | Gain-of-Function | T-cell leukemias [7] |
| Y665H | STAT5B | pY pocket | Loss-of-Function | Lactation failure, impaired mammary development [7] |
| S614R | STAT3 | BC loop | Gain-of-Function | T-cell large granular lymphocytic leukemia, NK-cell LGLL [3] |
| K591E/M | STAT3 | αA helix | Loss-of-Function | Autosomal-dominant Hyper IgE Syndrome [3] |
| R609G | STAT3 | βB strand | Loss-of-Function | Autosomal-dominant Hyper IgE Syndrome [3] |
| S611N/I | STAT3 | βB strand | Loss-of-F-function | Autosomal-dominant Hyper IgE Syndrome [3] |
Molecular Mechanisms of Pathogenic Mutations
The biochemical and structural mechanisms through which SH2 domain mutations disrupt normal STAT function include:
Loss-of-Function Mechanisms:
- Disrupted phosphotyrosine binding: Mutations in the pY pocket (e.g., STAT5B Y665H) impair recognition of phosphotyrosine motifs, preventing STAT activation and nuclear translocation [7].
- Impaired dimer stability: Mutations affecting the dimerization interface (e.g., in the αB or αB' helices) compromise stable dimer formation even when phosphorylation occurs [3].
- Structural destabilization: Mutations in the hydrophobic core (e.g., STAT3 V637L) can destabilize the entire SH2 domain fold, leading to protein misfolding or accelerated degradation [3].
Gain-of-Function Mechanisms:
- Enhanced phosphopeptide affinity: Certain mutations (e.g., STAT5B Y665F) may increase binding affinity for phosphotyrosine motifs, lowering the activation threshold [7].
- Constitutive dimerization: Mutations that mimic the phosphorylated state (e.g., STAT3 S614R) can promote dimer formation independent of activation signals [3].
- Altered specificity: Mutations in the pY+3 pocket can broaden binding specificity, enabling activation by non-cognate cytokines [3].
Visualizing STAT SH2 Domain Structure and Signaling
The following diagrams illustrate key structural and functional aspects of the STAT SH2 domain using Graphviz (DOT language).
STAT SH2 Domain Architecture
SH2 Domain Role in JAK-STAT Signaling
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for STAT SH2 Domain Investigations
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| Phospho-Specific Antibodies | Anti-STAT1 (pY701), Anti-STAT3 (pY705), Anti-STAT5 (pY694) | Detection of activated STATs in Western blot, flow cytometry, and immunofluorescence |
| Recombinant STAT Proteins | Wild-type and mutant SH2 domains expressed in E. coli or insect cells | Structural studies (crystallography), in vitro binding assays, biophysical characterization |
| JAK/STAT Reporter Cell Lines | Luciferase reporters under STAT-responsive promoters (e.g., M67/SIE, IRF1 GAS) | Functional assessment of STAT transcriptional activity in high-throughput screens |
| Cytokine Stimuli | IFN-γ, IL-6, IL-2, Prolactin, G-CSF, and other STAT-activating cytokines | Pathway activation under controlled conditions to study mutation impacts |
| CRISPR/Cas9 Components | sgRNAs targeting STAT genes, Cas9 nucleases, homology-directed repair templates | Generation of isogenic cell lines with specific SH2 domain mutations |
| Kinase Inhibitors | JAK inhibitors (Ruxolitinib, Tofacitinib), Src family kinase inhibitors | Pathway modulation to dissect specific versus redundant activation mechanisms |
| Structural Biology Reagents | Crystallization screens, size-exclusion chromatography matrices, cryo-protectants | Protein purification and structure determination of SH2 domains |
The canonical structure of the STAT SH2 domain represents a precisely evolved molecular module whose functional integrity is essential for proper cytokine signaling. Systematic comparison of disease-associated mutations reveals that the SH2 domain embodies a structural compromise - maintaining conserved motifs necessary for phosphotyrosine recognition while accommodating specific variations that enable STAT family functional diversity [3]. The observation that both activating and inactivating mutations cluster in similar regions, particularly the pY pocket and BC loop, highlights the delicate evolutionary balance required for proper STAT function [3].
From a therapeutic perspective, the STAT SH2 domain presents both challenges and opportunities. While the shallow, flexible nature of the pY and pY+3 pockets complicates small-molecule inhibitor development [3], the increasing understanding of allosteric networks within the SH2 domain may reveal novel targeting strategies [3] [4]. Furthermore, the systematic categorization of SH2 domain mutations enhances our ability to interpret variants of unknown significance emerging from clinical sequencing efforts [9].
Future research directions should focus on elucidating the structural dynamics of SH2 domain function in full-length STAT proteins, developing more sophisticated mouse models that recapitulate human disease mutations [7], and exploiting emerging structural insights to design next-generation therapeutics that can selectively target pathological STAT signaling in cancer and autoimmune disorders [3] [4].
The Src Homology 2 (SH2) domain is a critical modular unit that arose within metazoan signaling pathways approximately 600 million years ago, making it fundamentally tied to complex cellular communication in multicellular organisms [10]. In humans, 121 SH2 domains are encoded within 111 different proteins, including kinases, phosphatases, adaptors, and other signaling molecules [11] [12]. These domains function as readers of phosphotyrosine (pTyr) signaling information, directing myriad cellular processes by mediating specific protein-protein interactions [11]. In STAT (Signal Transducers and Activators of Transcription) proteins, the SH2 domain is particularly indispensable for canonical activation, nuclear translocation, and transcriptional functions [10]. This guide provides a comprehensive comparison of three essential functional interfaces within STAT SH2 domains: the phosphotyrosine-binding pocket, the dimerization surface, and the recently characterized lipid-binding regions, with particular emphasis on how mutations at these interfaces create a spectrum of activating and inactivating phenotypes with significant pathological consequences.
Structural Architecture of STAT-Type SH2 Domains
The SH2 domain maintains a conserved structural architecture consisting of a central anti-parallel β-sheet (composed of βB, βC, and βD strands) flanked by two α-helices (αA and αB), forming an αβββα motif [10]. This core structure partitions the domain into two primary functional subpockets:
- pY Pocket (Phosphate-Binding Pocket): Formed by the αA helix, BC loop, and one face of the central β-sheet, this pocket contains conserved residues that directly interact with the phosphotyrosine moiety [10].
- pY+3 Pocket (Specificity Pocket): Created by the opposite face of the β-sheet along with residues from the αB helix and CD and BC* loops, this pocket determines binding specificity by accommodating residues C-terminal to the phosphotyrosine [10].
STAT-type SH2 domains contain unique features that distinguish them from Src-type SH2 domains, most notably an α-helix (αB') at the C-terminus instead of a β-sheet [10]. This region, known as the evolutionary active region (EAR), contains additional potential drug-targeting clefts. Furthermore, a cluster of non-polar residues forms a hydrophobic system at the base of the pY+3 pocket that stabilizes the β-sheet conformation and maintains overall SH2 domain integrity [10].
Table 1: Core Structural Elements of STAT SH2 Domains
| Structural Element | Description | Functional Role |
|---|---|---|
| Central β-sheet | Anti-parallel βB, βC, βD strands | Structural scaffold that partitions the domain |
| αA helix | Flanks one side of β-sheet | Forms part of pY pocket |
| αB helix | Flanks opposite side of β-sheet | Forms part of pY+3 pocket and dimerization interface |
| BC loop | Connects βB and βC strands | Contributes to pY pocket formation |
| pY pocket | Binding cleft formed by αA, BC loop, and β-sheet | Binds phosphotyrosine moiety |
| pY+3 pocket | Binding cleft formed by αB, CD/BC* loops, and β-sheet | Determines binding specificity |
| Hydrophobic system | Cluster of non-polar residues at base of pY+3 pocket | Stabilizes β-sheet and domain integrity |
The Phosphotyrosine (pY)-Binding Pocket
Structure and Function
The pY-binding pocket is characterized by a highly conserved cationic surface that specifically recognizes and binds phosphotyrosine residues. This pocket employs arginine residues from the conserved FLVRES motif to form critical hydrogen bonds and electrostatic interactions with the phosphate group of the phosphotyrosine [10] [12]. The precise geometry and chemical environment of this pocket ensure both phosphorylation dependence and sequence specificity for proper target recognition.
Mutational Analysis and Pathological Implications
Mutations within the pY pocket frequently disrupt phosphopeptide binding and have been linked to both activating and inactivating phenotypes depending on the specific residue altered and the consequent structural impact.
Table 2: Disease-Associated Mutations in the STAT SH2 pY-Binding Pocket
| Mutation | Location | Pathology | Type | Functional Impact |
|---|---|---|---|---|
| STAT3 K591E/M | αA2 helix | AD-HIES | Germline | Loss-of-function; disrupts conserved pY binding residue |
| STAT3 R609G | βB5 strand | AD-HIES | Germline | Loss-of-function; affects Sheinerman & Signature motif |
| STAT3 S611N/G/I | βB7 strand | AD-HIES | Germline | Loss-of-function; key pY pocket residue |
| STAT3 S614R | BC loop | T-LGLL, NK-LGLL, ALCL | Somatic | Gain-of-function; enhances dimerization stability |
| STAT3 E616K/G | BC loop | DLBCL, NKTL | Somatic | Gain-of-function; alters binding specificity/affinity |
The dual nature of mutations at the same structural location highlights the delicate evolutionary balance maintained in wild-type STAT proteins. For instance, while most mutations in the βB7 strand (S611) cause loss-of-function leading to AD-HIES, mutations in the adjacent BC loop (S614, E616) can create activating phenotypes associated with lymphomas and leukemias [10]. This demonstrates how subtle alterations in the pY pocket can either destabilize functional binding or create constitutively active configurations.
Experimental Assessment of pY-Binding Function
Protocol: Surface Plasmon Resonance (SPR) for Binding Affinity Measurement
- Immobilization: Covalently immobilify purified SH2 domains or mutant variants onto a CMS sensor chip using standard amine coupling chemistry.
- Analyte Preparation: Synthesize phosphopeptides corresponding to known STAT binding motifs (e.g., pYXXQ for STAT1) and serially dilute in HBS-EP buffer.
- Binding Measurements: Inject peptide analytes at varying concentrations over immobilized SH2 domains at a flow rate of 30 μL/min.
- Regeneration: Remove bound analyte using a quick pulse of 10 mM glycine-HCl (pH 2.0).
- Data Analysis: Determine kinetic parameters (ka, kd) by fitting sensorgrams to a 1:1 Langmuir binding model and calculate equilibrium dissociation constants (KD) from the ratio kd/ka.
This approach enables quantitative comparison of binding affinities for wild-type versus mutant SH2 domains, directly assessing the functional impact of pY pocket mutations [13].
The Dimerization Interface
Structural Basis of STAT Dimerization
The SH2 domain mediates one of the most critical interactions in STAT signaling: reciprocal phosphotyrosine-SH2 domain engagement between two STAT monomers to form active dimers. The crystal structure of tyrosine-phosphorylated STAT-1 dimer bound to DNA reveals that the dimer forms a contiguous C-shaped clamp around DNA, stabilized by specific interactions between the SH2 domain of one monomer and the phosphotyrosine-containing C-terminal segment of the other monomer [14]. This phosphotyrosine-binding site is coupled structurally to the DNA-binding domain, suggesting the SH2-phosphotyrosine interaction helps stabilize DNA interacting elements [14].
Beyond STAT proteins, SH2 domain-mediated dimerization serves as an activation mechanism for other signaling proteins. For SH2-B and APS adapter proteins, an N-terminal domain mediates homodimerization, creating heterotetrameric JAK2-(SH2-B)2-JAK2 complexes that facilitate JAK2 transactivation [15]. This demonstrates the broader paradigm of SH2 domain involvement in higher-order complex formation.
Dimerization Dynamics and Regulation
SH2 domains can undergo dimerization themselves, which may represent a regulatory mechanism. The Fyn SH2 domain forms an intertwined dimer in solution that dissociates upon phosphopeptide binding [16]. This dimerization utilizes an extended βE-EF-βF region that creates an altered configuration compared to the canonical SH2 fold [16]. Analytical gel filtration and circular dichroism experiments confirm the presence of both monomeric and dimeric states, with the dimer showing increased β-sheet content [16]. The biological significance of such SH2 dimerization may include regulation of accessibility for partner binding or controlled sequestration of signaling elements.
Figure 1: STAT Activation Pathway and SH2 Domain-Mediated Dimerization
Experimental Analysis of Dimerization
Protocol: Size Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS)
- Sample Preparation: Purify recombinant wild-type or mutant STAT SH2 domains and concentrate to 5-10 mg/mL in phosphate-buffered saline.
- Chromatography System: Equip HPLC system with size exclusion column (e.g., Superdex 75 Increase 10/300) and connect to MALS detector and refractive index detector.
- Calibration: Perform system calibration using bovine serum albumin as molecular weight standard.
- Sample Analysis: Inject 100 μL of protein sample at 0.5 mL/min flow rate with continuous monitoring of light scattering and refractive index.
- Data Interpretation: Calculate absolute molecular weight from light scattering data independent of column calibration. Shifts in oligomeric state between wild-type and mutant proteins indicate dimerization defects.
This methodology provides unambiguous determination of dimerization capability and stoichiometry for STAT SH2 domain variants [16].
Lipid-Binding Regions
Discovery and Prevalence of SH2 Lipid Binding
A systematic genomic-scale analysis of human SH2 domains revealed that approximately 90% of SH2 domains bind plasma membrane lipids, with many exhibiting specific phosphoinositide preferences [11]. This lipid binding occurs through surface cationic patches distinct from the pY-binding pocket, enabling simultaneous or competitive binding to both lipids and pY motifs [11]. The lipid-binding sites typically form grooves for specific lipid headgroup recognition or flat surfaces for non-specific membrane interactions [11].
Table 3: Lipid-Binding Properties of Selected SH2 Domains
| SH2 Domain | Kd for PM Vesicles (nM) | Lipid Specificity | Biological Role of Lipid Binding |
|---|---|---|---|
| STAT6-SH2 | 20 ± 10 | Not specified | Not characterized |
| ZAP70-cSH2 | 340 ± 35 | PIP3 > PI45P2 > others | Sustained T-cell activation |
| p85αN-cSH2 | 220 ± 20 | Not specified | PI3K pathway regulation |
| Abl-SH2 | Not determined | PIP2 | Mutually exclusive with pY binding |
| C1-Ten/Tensin2 | Not determined | PIP3 | Activation and targeting to IRS-1 |
Molecular Mechanisms and Functional Consequences
Lipid binding can either promote or inhibit SH2 domain function depending on the cellular context and specific domain. For the Abl SH2 domain, phosphatidylinositol-4,5-bisphosphate (PIP2) interacts via an electrostatic mechanism at a site overlapping with the phosphotyrosine-binding pocket, creating a potentially mutually exclusive binding scenario [12]. In ZAP70, the C-terminal SH2 domain binds PIP3 and other anionic lipids, contributing to sustained activation during T lymphocyte signaling [11] [12]. These interactions provide a mechanism for membrane recruitment and spatial control of SH2 domain-containing proteins within cellular compartments.
Experimental Characterization of Lipid Interactions
Protocol: Lipid Protein Overlay Assay
- Membrane Strip Preparation: Spot various biologically relevant lipids (PIP, PIP2, PIP3, PC, PS, etc.) onto nitrocellulose membranes in a dilution series.
- Blocking: Incubate membranes in blocking buffer (3% fatty acid-free BSA in TBST) for 1 hour.
- Protein Probing: Incubate membranes with purified SH2 domains (0.5-1 μg/mL) in blocking buffer for 2 hours.
- Detection: Incubate with domain-specific primary antibody followed by HRP-conjugated secondary antibody.
- Visualization: Develop using enhanced chemiluminescence and quantify spot intensity.
This approach provides a rapid assessment of lipid binding specificity and relative affinity, guiding more quantitative biophysical analyses [11].
Cross-Interface Functional Integration in Disease Mutations
The functional interfaces of SH2 domains do not operate in isolation; rather, they form an integrated network where perturbation at one interface can affect others. This is particularly evident in disease-associated mutations where single amino acid substitutions can have cascading effects across multiple functional surfaces.
Interdependence in STAT Activation
The coiled-coil domain of STAT proteins, while distinct from the SH2 domain, plays an essential role in SH2 domain-mediated receptor binding and subsequent activation. Systematic deletion analysis of Stat3 revealed that the coiled-coil domain is essential for Stat3 recruitment to the receptor and subsequent tyrosine phosphorylation [17]. Single mutation of Asp170 in α-helix 1 diminishes both receptor binding and tyrosine phosphorylation, despite the SH2 domain remaining functionally intact for DNA binding when phosphorylated [17]. This demonstrates the allosteric integration between distal domains and the SH2 interface.
Mutational Hotspots and Therapeutic Targeting
The SH2 domain represents a hotspot in the mutational landscape of STAT proteins [10]. The genetic volatility of specific regions can result in either activating or inactivating mutations at the same site, underscoring the delicate evolutionary balance of wild-type STAT structural motifs. Understanding these mutational patterns is driving therapeutic development, with the relatively shallow binding surfaces of SH2 domains presenting both challenges and opportunities for small molecule inhibitor design [10].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for SH2 Domain Interface Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Recombinant SH2 Domains | Purified wild-type and mutant STAT3-SH2, STAT5B-SH2 | Biophysical analysis, structural studies, in vitro binding assays |
| Phosphopeptide Libraries | pYXXQ motifs, Cantley peptide library | Specificity profiling, binding affinity measurements |
| Lipid Vesicles | PM-mimetic vesicles, PIP2/PIP3-containing liposomes | Lipid binding assays, membrane recruitment studies |
| Antibody Tools | Anti-phospho-Tyr705-Stat3, FLAG-tag antibodies | Immunoprecipitation, Western blotting, cellular localization |
| Cell-Based Reporter Systems | STAT-responsive luciferase constructs, GFP-tagged SH2 domains | Functional assessment of mutants, pathway activity monitoring |
| Structural Biology Resources | Crystallization screens, NMR isotope-labeled proteins | High-resolution structure determination of interfaces |
The functional interfaces of STAT SH2 domains—the pY-binding pocket, dimerization surface, and lipid-binding regions—represent interconnected modules whose precise coordination enables specific cellular signaling outcomes. Mutations at these interfaces disrupt this delicate balance, leading to either constitutive activation or loss-of-function across various disease states. The comprehensive characterization of these interfaces through structural, biophysical, and cellular approaches provides the foundation for targeted therapeutic intervention in STAT-driven pathologies. Future research will undoubtedly continue to elucidate the dynamic interplay between these interfaces and their regulation in both normal physiology and disease, potentially revealing new opportunities for precision medicine in oncology and immunology.
In the study of disease genetics, mutations are fundamentally categorized by their functional consequences on the resulting protein. Loss-of-function (LOF) mutations disrupt normal protein activity, typically through reduced stability, impaired binding, or complete absence of the protein. In contrast, gain-of-function (GOF) mutations confer novel, often pathogenic activities that can include enhanced signaling, new interaction partners, or resistance to normal regulatory mechanisms [18]. The distinction between these mutation types is critical for understanding disease mechanisms and developing targeted therapies, particularly in cancer and developmental disorders where specific pathways are dysregulated.
This compendium focuses on mutation hotspots within key signaling proteins, with a specialized analysis of the STAT family's SH2 domains where a delicate balance exists between activating and inactivating mutations. The structural and functional consequences of these mutations reveal intricate mechanisms of pathogenicity that inform both biological understanding and therapeutic development. Through systematic comparison of GOF and LOF variants, we provide a landscape view of how specific amino acid changes can drive divergent disease phenotypes through opposing effects on protein function and pathway signaling.
Fundamental Mechanisms of GOF and LOF Mutations
Structural and Functional Consequences
GOF and LOF mutations operate through distinct structural mechanisms that perturb normal protein function in predictable ways. LOF mutations typically occur in structured protein domains and often affect folding, stability, or catalytic activity [18]. These mutations follow a predictable pattern where the loss of a specific function leads to impaired signaling or regulatory capacity. In tumor suppressor genes like TP53, LOF mutations eliminate critical cell cycle control and DNA damage response functions, allowing uncontrolled proliferation [19].
GOF mutations demonstrate more diverse mechanisms, including acquisition of novel structural domains that enable new protein-protein interactions, formation of novel intrinsically disordered regions (IDRs) that alter interaction networks, creation of short linear motifs (SLiMs) that mediate new binding events, and generation of novel transcription factor binding sites in noncoding regions [18]. For example, in the multi-domain phosphatase SHP2, GOF mutations at the N-SH2/PTP interface disrupt autoinhibition, leading to constitutive phosphatase activity that promotes Ras/Erk and JAK-STAT signaling in cancers and developmental disorders [20].
Table 1: Mechanisms of Gain-of-Function Mutations in Cancer
| Mechanism | Functional Consequence | Example |
|---|---|---|
| Gain of Structural Domains | Enables novel protein-protein interactions | PIK3CA E545K gains ability to associate with IRS1 [18] |
| Gain of Novel IDRs | Perturbs disorder-mediated processes and signaling networks | c-Myc uses gained IDRs to perform diverse interactions in cancer [18] |
| Gain of SLiMs | Creates new protein-binding modules | β-catenin mutations perturb DEGSCFTRCP1_1 SLiM [18] |
| Disruption of Auto-inhibitory Interfaces | Causes constitutive activation | SHP2 E76K disrupts N-SH2/PTP interface [20] |
Signaling Pathway Dysregulation
The JAK-STAT pathway exemplifies how GOF and LOF mutations in the same protein domains can cause divergent diseases. This pathway communicates information from chemical signals outside the cell to the nucleus, activating genes through transcription [21]. JAKs (JAK1, JAK2, JAK3, TYK2) phosphorylate STAT transcription factors (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, STAT6), which dimerize via SH2 domain interactions and translocate to the nucleus [1] [21]. The SH2 domain is particularly mutation-prone in STAT proteins, with specific variants causing either hyperactivation or refractoriness to normal activation signals [3].
The delicate evolutionary balance in STAT SH2 domains means that mutations at identical positions can have opposing functional effects. This structural vulnerability creates mutation hotspots where different amino acid substitutions produce divergent phenotypes. For instance, in the STAT3 SH2 domain, specific mutations cause autosomal-dominant hyper IgE syndrome (AD-HIES) through LOF mechanisms, while other mutations in the same domain drive leukemias and lymphomas through GOF mechanisms [3].
Diagram 1: JAK-STAT signaling pathway and mutation impacts. GOF mutations (red) enhance signaling while LOF mutations (blue) disrupt it.
Experimental Approaches for Characterizing Mutations
Deep Mutational Scanning Technologies
Deep mutational scanning represents a powerful high-throughput method for characterizing mutation effects across a protein. This approach combines selection assays on pooled mutant libraries with deep sequencing to profile mutational effects with comprehensive coverage [20]. The experimental workflow involves creating saturation mutagenesis libraries covering the target protein, introducing these libraries into a model system (such as yeast), applying functional selection pressure, and sequencing pre- and post-selection populations to calculate enrichment scores for each variant.
In a landmark study of SHP2, researchers divided the protein into 15 sub-libraries (tiles) and conducted selection assays in yeast where cell growth was dependent on SHP2 catalytic activity [20]. This system allowed differentiation between GOF and LOF mutants based on their ability to rescue growth from tyrosine kinase toxicity. The resulting datasets provided activity profiles for over 11,000 SHP2 mutants, revealing unexpected mutational hotspots including activating mutations in the N-SH2 domain core and inactivating mutations at the C-SH2/PTP interface [20].
Table 2: Key Research Reagents for Mutation Characterization
| Reagent/Technique | Application | Functional Role |
|---|---|---|
| Deep Mutational Scanning | Comprehensive mutation profiling | High-throughput functional characterization of thousands of variants [20] |
| Saturation Mutagenesis Libraries | Mutant library generation | Creates comprehensive collections of point mutants for scanning studies [20] |
| Yeast Growth Rescue Assay | Functional selection | Links cell survival to protein activity, enabling selection-based enrichment [20] |
| Co-transformed Src Kinase | Selection pressure | Provides toxic tyrosine kinase activity that must be counterbalanced by phosphatase function [20] |
Diagram 2: Deep mutational scanning workflow for functional characterization of mutations.
Structural and Biophysical Methods
Structural biology approaches provide mechanistic insights into how mutations alter protein function at the atomic level. X-ray crystallography and molecular dynamics simulations reveal how GOF mutations disrupt autoinhibitory interfaces in multi-domain proteins like SHP2 [20]. For STAT proteins, structural analysis shows how SH2 domain mutations affect phospho-tyrosine binding specificity and dimerization stability [3].
Biophysical characterization of mutant proteins includes measuring catalytic efficiency (kcat/KM), protein stability, and binding affinity. For SHP2 mutants, purification and enzymatic assays validated deep mutational scanning results, showing strong correlation between catalytic efficiency and enrichment scores in selection assays [20]. These approaches confirm that basal catalytic activity is the major determinant of functional effects for many pathogenic mutations.
STAT SH2 Domain Mutations: A Case Study in GOF/LOF Hotspots
Molecular Anatomy of STAT SH2 Domains
The SH2 domain represents a critical mutational hotspot in STAT proteins, with sequencing analyses of patient samples identifying numerous point mutations associated with diverse diseases [3]. STAT-type SH2 domains possess a conserved structure consisting of a central anti-parallel β-sheet (βB-βD strands) flanked by two α-helices (αA and αB) in an αβββα motif [3]. This structure forms two functionally critical subpockets: the phospho-tyrosine (pY) binding pocket and the pY+3 specificity pocket that determines peptide binding selectivity.
The structural flexibility of STAT SH2 domains makes them particularly susceptible to mutational disruption. Molecular dynamics simulations reveal that these domains exhibit substantial flexibility even on sub-microsecond timescales, with the accessible volume of the pY pocket varying dramatically [3]. This inherent flexibility creates an evolutionary compromise where critical structural motifs are preserved while maintaining peptide recognition capacity, making specific residues vulnerable to both activating and inactivating mutations.
Disease-Associated STAT SH2 Mutations
STAT3 and STAT5B SH2 domain mutations demonstrate how different amino acid substitutions at the same positions can cause either GOF or LOF phenotypes. In STAT3, specific SH2 domain mutations (e.g., K591E, K591M, R609G) cause autosomal-dominant hyper IgE syndrome (AD-HIES) through LOF mechanisms that impair STAT3-mediated Th17 T-cell responses [3]. These mutations typically reduce phospho-tyrosine binding affinity or disrupt dimerization stability.
Conversely, other STAT3 SH2 domain mutations (e.g., S614R, E616K, E616G) drive lymphoid malignancies through GOF mechanisms that enhance STAT3 transcriptional activity [3]. The S614R mutation appears in T-cell large granular lymphocytic leukemia (T-LGLL), natural killer cell LGLL (NK-LGLL), anaplastic large cell lymphoma (ALK-ALCL), and hepatosplenic T-cell lymphoma (HSTL) [3]. These mutations often enhance dimer stability or enable cytokine-independent activation.
Table 3: Disease-Associated Mutations in STAT3 SH2 Domain
| Mutation | Location | Domain Position | Disease Association | Mutation Type |
|---|---|---|---|---|
| K591E/M | αA2 helix | pY pocket | AD-HIES | LOF [3] |
| R609G | βB5 strand | pY pocket | AD-HIES | LOF [3] |
| S611N/I/G | βB7 strand | pY pocket | AD-HIES | LOF [3] |
| S614R | BC loop | pY pocket | T-LGLL, NK-LGLL, ALK-ALCL, HSTL | GOF [3] |
| E616K/G | BC loop | pY pocket | DLBCL, NKTL | GOF [3] |
STAT5B SH2 domain mutations show similar divergence between GOF and LOF variants. The N642H hotspot mutation is a well-characterized GOF variant found in hematopoietic malignancies, particularly T-cell prolymphocytic leukemia [3]. This mutation enhances STAT5B dimerization and transcriptional activity through mechanisms that stabilize the active conformation. In contrast, other STAT5B SH2 mutations cause growth hormone insensitivity through LOF mechanisms that impair STAT5B activation and nuclear translocation [3].
Therapeutic Targeting of GOF and LOF Mutations
Drug Development Strategies
Targeting pathogenic mutations therapeutically requires distinct approaches for GOF versus LOF variants. For GOF mutations, strategies include allosteric inhibitors that stabilize autoinhibited states, competitive inhibitors that block protein-protein interactions, and degraders that target mutant proteins for destruction. The JAK-STAT pathway has been successfully targeted by small molecule inhibitors like tofacitinib (JAK inhibitor for rheumatoid arthritis) and ruxolitinib (JAK1/JAK2 inhibitor for primary myelofibrosis) [22] [1]. These compounds typically target the ATP-binding pocket of hyperactive kinases resulting from GOF mutations.
For LOF mutations, therapeutic approaches are more challenging and include gene therapy, read-through compounds for nonsense mutations, and chaperones that stabilize misfolded proteins. In the case of STAT3 LOF mutations causing AD-HIES, strategies to enhance residual STAT3 function or modulate upstream activators may provide therapeutic benefit, though no targeted therapies are yet approved [3].
Mutation-Specific Precision Medicine
The comprehensive characterization of mutation hotspots enables mutation-specific therapeutic strategies. For example, in SHP2-related diseases, GOF mutations at the N-SH2/PTP interface (e.g., E76K) are susceptible to allosteric inhibitors that stabilize the autoinhibited state, while other mutations may require alternative targeting strategies [20]. Similarly, in TP53 GOF mutants, compounds like APR-246 and COTI-2 that reactivate wild-type conformation or destabilize mutant p53 have entered clinical trials [19].
Deep mutational scanning data increasingly informs therapeutic development by predicting mutation-specific drug sensitivity. The functional characterization of thousands of variants across proteins like SHP2 provides resources for interpreting clinical variants and predicting their pathogenicity and drug response [20]. This approach enables stratification of mutations by functional consequence and therapeutic vulnerability, moving beyond simple location-based classification to mechanism-based targeting.
The landscape of disease-associated mutations reveals complex relationships between genetic variation, protein function, and disease phenotype. The compendium of GOF and LOF hotspots presented here highlights the importance of functional characterization beyond mere mutation identification. The STAT SH2 domain exemplifies how the same protein region can harbor both activating and inactivating mutations with divergent clinical consequences.
Future mutation classification will increasingly integrate structural data, deep mutational scanning profiles, and clinical annotations to predict functional consequences and therapeutic vulnerabilities. As functional datasets expand across human signaling proteins, precision medicine approaches will leverage mutation-specific mechanisms to develop targeted therapies matched to individual variants. The systematic comparison of GOF and LOF mutations provides both a biological framework for understanding disease pathogenesis and a clinical roadmap for developing mutation-informed therapeutics.
The Src Homology 2 (SH2) domain of STAT5B is a critical hotspot for mutations, with tyrosine 665 (Y665) representing a key residue where single nucleotide substitutions can drive opposing functional consequences. This guide provides a structured comparison of the Y665F and Y665H mutations, detailing their divergent impacts on STAT5B structure, activity, and physiological outcomes. We summarize quantitative biochemical and cellular data, present detailed experimental methodologies for assessing these mutations, and catalog essential research tools. This resource is designed to inform drug development efforts targeting pathogenic STAT5B signaling.
The STAT5B SH2 domain is indispensable for cytokine-induced activation, mediating JAK-dependent tyrosine phosphorylation, STAT dimerization, nuclear translocation, and the establishment of functional transcriptional enhancers [23]. Disease-associated mutations within this domain are frequently identified in hematologic malignancies, particularly in T-cell large granular lymphocytic leukemia (T-LGLL) and T-cell prolymphocytic leukemia (T-PLL) [23] [3] [24]. Among these, mutations at tyrosine 665 serve as a paradigm for how subtle genetic changes can profoundly alter protein function. The Y665F substitution is a well-recognized, recurrent somatic mutation in leukemia, whereas the Y665H substitution is far less common and exhibits distinct functional properties [23]. Understanding the precise mechanisms underlying their divergent behaviors is crucial for developing targeted therapeutic interventions.
Comparative Analysis of Y665F and Y665H Mutations
Structural and In Silico Predictions
In silico modeling and structural analyses reveal how the Y665F and Y665H mutations exert opposing effects on STAT5B stability and dimerization.
- Location and Role of Y665: Tyrosine 665 is located at a critical interface involved in STAT5B homodimerization and is highly conserved across vertebrate species [23]. It plays a key role in intramolecular interactions that support the active dimer conformation.
- Divergent Energetic Impacts: Computational tools like COORDinator predict that the Y665F substitution stabilizes the protein structure, potentially by promoting favorable aromatic stacking interactions with phenylalanine 711 (F711). In contrast, the Y665H substitution is destabilizing, likely due to the introduction of an imidazole ring that disrupts these same interactions [23] [25].
Table 1: In Silico Pathogenicity Predictions for STAT5B Y665 Mutations
| Mutation | AlphaMissense Score (Prediction) | CADD PHRED Score | REVEL Score | PolyPhen-2 Score (Prediction) |
|---|---|---|---|---|
| Y665F | 0.173 (Benign) | 24.3 | 0.535 | 0.93 (Probably Damaging) |
| Y665H | 0.383 (Benign) | 23.1 | 0.304 | 0.084 (Benign) |
The table above summarizes predictions from multiple state-of-the-art computational tools, which collectively suggest a higher probability of pathogenicity for the Y665F variant compared to Y665H [23] [25].
Functional Outcomes in Cellular and Animal Models
Experimental data from in vitro and in vivo models clearly delineate the gain-of-function (GOF) versus loss-of-function (LOF) nature of these mutations.
- Biochemical and Transcriptional Activity: In primary T cells and following cytokine activation, the STAT5B-Y665F variant displays enhanced STAT5 phosphorylation, increased DNA binding, and greater transcriptional activity compared to wild-type STAT5B. Conversely, the STAT5B-Y665H variant resembles a null allele, with diminished phosphorylation and transcriptional output [23] [25].
- Immune Phenotypes in Murine Models: Knock-in mouse models recapitulate these functional divergences. The Stat5b-Y665F GOF mutation leads to an accumulation of CD8+ effector and memory T cells and CD4+ regulatory T cells, altering CD8+/CD4+ ratios. In contrast, Stat5b-Y665H LOF mice show diminished populations of these T-cell subsets [23] [25].
- Mammary Gland Development: The mutations also exert opposing effects in other STAT5B-dependent tissues. Stat5b-Y665F mice exhibit accelerated mammary development during pregnancy, while Stat5b-Y665H mice initially fail to develop functional mammary tissue, resulting in lactation failure [26].
- Unexpected Protective Role: Recent research uncovered that the Stat5b-Y665F variant protects against acute kidney injury in a mouse model, inducing transcriptomic shifts that modulate inflammation and amino acid transport in renal epithelium [27]. This highlights that the effects of a single nucleotide polymorphism can extend beyond its primary disease association.
Table 2: Experimental Functional Outcomes of STAT5B Y665 Mutations
| Parameter | STAT5B-Y665F | STAT5B-Y665H |
|---|---|---|
| Functional Classification | Gain-of-Function (GOF) [23] [26] [25] | Loss-of-Function (LOF) [23] [26] [25] |
| Phosphorylation Status | Increased [23] [25] [27] | Diminished (resembles null) [23] [25] |
| DNA Binding & Transcription | Enhanced [23] [25] [27] | Impaired [23] [25] |
| T Cell Phenotype (in vivo) | Accumulation of CD8+ effector/memory and CD4+ T-reg cells [23] [25] | Diminished CD8+ effector/memory and CD4+ T-reg cells [23] [25] |
| Mammary Gland Phenotype | Accelerated development [26] | Failure of functional development (initial pregnancy) [26] |
| Leukemic Potential | Does not directly induce malignancy [23] [25] [27] | Not associated with cancer in major databases [23] |
Experimental Protocols for Functional Characterization
This section outlines key methodologies used to generate the comparative data cited in this guide.
Generation of Mutant Mouse Models
The CRISPR/Cas9 and base editing techniques were used to introduce the Y665F and Y665H mutations into the mouse genome, creating knock-in models that faithfully replicate the human genetic variants [26].
- For the Y665H mutation: Adenine base editor (ABE 7.10) mRNA and a specific sgRNA were co-microinjected into the cytoplasm of fertilized C57BL/6 N mouse eggs. This base editing approach directly converts the target adenosine, creating the desired histidine codon.
- For the Y665F mutation: A Cas9 protein-sgRNA ribonucleoprotein (RNP) complex was co-electroporated with a single-strand oligonucleotide donor template into zygotes. The donor template contained the tyrosine (TAC) to phenylalanine (TTT) change and a silent mutation to disrupt the sgRNA protospacer adjacent motif (PAM) site, preventing repeated Cas9 cleavage after successful homology-directed repair.
Embryos were implanted into foster mothers, and founders were genotyped using PCR, Sanger sequencing, and/or TaqMan-based assays [26].
Assessment of Immune Phenotypes
Comprehensive flow cytometric analysis of immune cell populations in primary lymphoid organs and peripheral blood of mutant mice and their wild-type littermates is essential.
- Protocol: Single-cell suspensions are prepared from spleen, thymus, and lymph nodes. Red blood cells are lysed. Cells are stained with fluorescently labeled antibodies against surface markers, including CD3, CD4, CD8, CD44, CD62L, and CD25, and the transcription factor FoxP3 (for regulatory T cells). Data acquisition is performed on a flow cytometer, and populations are analyzed to quantify naive, effector, and memory T cell subsets, as well as regulatory T cells [23] [25].
Transcriptomic and Epigenomic Profiling
RNA-seq and ChIP-seq are used to determine the global transcriptional and enhancer landscape changes driven by the mutations.
- RNA-seq: Total RNA is extracted from tissues of interest (e.g., mammary gland, kidney, T cells). Ribosomal RNA is removed, and cDNA libraries are prepared and sequenced on a platform such as Illumina's NovaSeq 6000. Reads are aligned to the reference genome (e.g., mm10), and differential gene expression analysis is performed using tools like DESeq2 [26] [27].
- ChIP-seq: Tissues or cells are cross-linked, and chromatin is sheared. STAT5B is immunoprecipitated using a specific antibody (e.g., recognizing total STAT5B or phosphorylated STAT5B). After reversing cross-links and purifying DNA, libraries are constructed and sequenced. Aligned reads are used to identify STAT5B-binding peaks and define enhancer and super-enhancer regions [26] [27].
Signaling Pathway and Experimental Workflow
The following diagram illustrates the structural and functional divergence stemming from the Y665 mutations, and the key experimental workflows used to characterize them.
The Scientist's Toolkit: Key Research Reagents
The table below catalogues essential materials and reagents used in the featured studies for investigating STAT5B Y665 mutations.
Table 3: Essential Research Reagents and Resources
| Reagent / Resource | Function and Application | Example Source / Citation |
|---|---|---|
| CRISPR/Cas9 & Base Editing Systems | Precise genome editing to introduce point mutations in mouse models or cell lines. | ABE 7.10; Cas9 protein RNP [26] |
| Phospho-STAT5 Specific Antibody | Detection of activated, tyrosine-phosphorylated STAT5 by Western blot or flow cytometry. | Used in functional validation [23] [27] |
| STAT5B ChIP-grade Antibody | Immunoprecipitation of STAT5B-bound chromatin for genome-wide binding site mapping (ChIP-seq). | Used for epigenomic profiling [26] [27] |
| Flow Cytometry Antibodies (CD3, CD4, CD8, CD44, CD62L, FoxP3) | Immunophenotyping of T-cell populations in primary tissues from mutant mice. | Used for immune profiling [23] [25] |
| TruSeq Stranded Total RNA Library Prep Kit | Preparation of sequencing libraries from total RNA for transcriptomic analysis (RNA-seq). | Illumina [26] [27] |
| C57BL/6 N Mice | Genetic background for generating and maintaining knock-in mouse models. | Charles River Laboratories [26] |
Evolutionary Conservation and Structural Determinants of Pathogenicity
This guide provides a comparative analysis of activating and inactivating mutations within STAT SH2 domains, focusing on their structural mechanisms, functional consequences, and implications for drug development. We objectively evaluate mutational impacts through integrated structural biology, deep mutational scanning, and in vivo models, presenting quantitative data on how evolutionary conservation patterns correlate with pathogenicity mechanisms. The analysis reveals how specific residues dictate functional outcomes through precise structural determinants, enabling researchers to interpret mutation effects and prioritize therapeutic targets.
Src Homology 2 (SH2) domains are approximately 100 amino acid protein modules that specifically recognize phosphotyrosine (pY) motifs, serving as crucial mediators in metazoan signal transduction [28] [29]. These domains first emerged in unicellular eukaryotes and expanded alongside tyrosine kinases throughout metazoan evolution, with humans encoding approximately 110 SH2 domain-containing proteins [29]. The STAT (Signal Transducer and Activator of Transcription) family of transcription factors contains specialized SH2 domains that are essential for cytokine-mediated signaling, dimerization, and nuclear translocation [3]. Mutations within STAT SH2 domains, particularly in STAT3 and STAT5B, represent hotspots in disease pathogenesis, with specific alterations driving either gain-of-function (GOF) or loss-of-function (LOF) outcomes through distinct structural mechanisms [3] [30]. Understanding the evolutionary conservation and structural determinants governing these mutational outcomes provides critical insights for targeted therapeutic development.
Structural Framework of STAT SH2 Domains
Conserved Architecture and Functional Motifs
All SH2 domains share a conserved αβββα structural fold centered on a three-stranded antiparallel β-sheet flanked by two α-helices [28]. The STAT-type SH2 domains contain distinctive features including an additional α-helix (αB') at the C-terminal region of the pY+3 binding pocket, known as the evolutionary active region (EAR) [3]. This domain is partitioned into two functionally specialized subpockets:
- pY (phosphate-binding) pocket: Formed by the αA helix, BC loop, and one face of the central β-sheet, this pocket contains an invariant arginine residue (βB5) that directly engages the phosphotyrosine moiety through a salt bridge [28] [31]
- pY+3 (specificity) pocket: Created by the opposite face of the β-sheet along with residues from the αB helix and CD/BC* loops, this region determines binding specificity for downstream signaling partners [3]
Table 1: Key Structural Elements and Their Functional Roles in STAT SH2 Domains
| Structural Element | Location | Functional Role | Conservation |
|---|---|---|---|
| βB5 arginine | pY pocket | Direct phosphotyrosine binding | Invariant across 118/121 human SH2 domains |
| FLVR motif | pY pocket | Phosphate recognition | Highly conserved |
| BC loop | pY pocket | Domain flexibility and communication | Variable length |
| αB' helix (EAR) | pY+3 pocket | STAT-specific dimerization interface | Unique to STAT-type SH2 domains |
| Hydrophobic system | pY+3 pocket base | Stabilizes β-sheet architecture | Conserved |
Evolutionary Conservation Patterns
Analysis of evolutionary and population constraint reveals that missense-depleted sites (under strong constraint) are significantly enriched in buried residues and binding interfaces, while missense-enriched sites typically reside on protein surfaces [32]. This constraint pattern correlates strongly with deep evolutionary conservation measured across species, indicating that structural and functional necessities shape both long-term evolutionary patterns and contemporary human population variation [32]. The development of Missense Enrichment Score (MES) has enabled residue-level quantification of population constraint, demonstrating that combining evolutionary and population metrics provides enhanced prediction of structurally and functionally critical residues [32].
Comparative Analysis of STAT SH2 Domain Mutations
Mutation Hotspots and Functional Classification
Sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in STAT proteins [3]. The functional impact of these mutations can be objectively categorized through biochemical, cellular, and organismal phenotypes:
Table 2: Functional Classification of STAT SH2 Domain Mutations
| Mutation Type | Structural Impact | Biochemical Consequence | Cellular Phenotype | Disease Association |
|---|---|---|---|---|
| GOF Mutations | Disrupt autoinhibition; Enhance dimerization | Increased phosphorylation and DNA binding | Enhanced proliferation and survival | T-cell leukemias (T-LGLL, T-PLL) |
| LOF Mutations | Impair phosphopeptide binding or dimerization | Reduced phosphorylation and nuclear translocation | Immunodeficiency, growth defects | AD-HIES, growth hormone insensitivity |
| Dual-Potential Mutations | Context-dependent structural effects | Variable signaling output | Tissue-specific phenotypes | Complex immune dysregulation |
Structural Mechanisms of Pathogenicity: STAT5B Y665 Case Study
The STAT5B tyrosine 665 residue represents an instructive model for understanding how subtle structural alterations dictate divergent pathogenic outcomes. Comparative analysis of Y665F and Y665H mutations reveals opposing functional impacts through distinct mechanisms:
STAT5B Y665F Gain-of-Function Mechanism
The Y665F substitution replaces tyrosine with phenylalanine, removing the hydroxyl group while maintaining aromatic character. Computational modeling using COORDinator predicts this mutation stabilizes intramolecular aromatic stacking interactions with F711, facilitating constitutive activation [23]. Experimental validation demonstrates:
- Enhanced dimerization: Increased STAT5 phosphorylation and DNA binding after cytokine activation [30]
- Transcriptional amplification: Elevated enhancer formation and target gene expression [7]
- Immunophenotypic consequences: Accumulation of CD8+ effector/memory and CD4+ regulatory T cells, altering CD8+/CD4+ ratios [30]
STAT5B Y665H Loss-of-Function Mechanism
In contrast, the Y665H substitution introduces an imidazole group that disrupts critical hydrophobic packing interactions. COORDinator predictions indicate this mutation destabilizes binding of the C-terminal tail, impairing dimerization [23]. Experimental observations confirm:
- Dimerization deficiency: Reduced STAT5 phosphorylation and nuclear translocation [30]
- Transcriptional impairment: Compromised enhancer establishment and alveolar differentiation [7]
- Immunophenotypic consequences: Diminished CD8+ effector/memory and CD4+ regulatory T cells [30]
- Developmental impact: Failure in functional mammary tissue development and lactation [7]
Experimental Methodologies for Mutation Analysis
Deep Mutational Scanning Approaches
Deep mutational scanning enables high-throughput functional characterization of comprehensive mutation libraries. The application to SHP2 (containing two SH2 domains) illustrates methodology transferable to STAT analysis:
- Library construction: Saturation mutagenesis with SHP2 divided into 15 sub-libraries using mutagenesis by integrated tiles (MITE) [20]
- Selection system: Yeast viability rescue from tyrosine kinase toxicity through SHP2 catalytic activity [20]
- Quantitative readouts: Enrichment scores calculated from deep sequencing before/after selection [20]
- Validation: Correlation with catalytic efficiency (kcat/KM) measurements of purified mutants [20]
This approach successfully identified mutational hotspots beyond characterized autoinhibitory interfaces, including activating mutations in the N-SH2 core and around the catalytic WPD loop [20].
In Vivo Modeling Using CRISPR/Cas9 and Base Editing
Precise mouse models incorporating human disease mutations enable physiological assessment of mutational impact:
- CRISPR/Cas9-mediated homology-directed repair: For Y665F mutation introduction using single-strand oligonucleotide donors [7]
- Adenine base editing (ABE): For Y665H mutation without double-strand breaks using ABE mRNA and sgRNA co-microinjection [7]
- Phenotypic characterization: Multi-system analysis of immune function, mammary development, and transcriptomic/epigenomic alterations [7] [30]
Computational Prediction and Energetic Profiling
Integrated computational approaches provide mechanistic insights into mutational impacts:
- Structure prediction: AlphaFold3 generates homodimer structures for interface analysis [23]
- Energetic profiling: COORDinator neural network predicts stability effects of substitutions using backbone structure [23]
- Pathogenicity prediction: Combined annotation from AlphaMissense, CADD, and REVEL algorithms [23]
Signaling Pathways and Experimental Workflows
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Materials and Their Applications
| Reagent/Resource | Function | Application Example | Experimental Context |
|---|---|---|---|
| AlphaFold3 | Protein structure prediction | STAT5B SH2 dimer modeling | Computational structural analysis [23] |
| COORDinator | Energetic effect prediction | Y665F/H mutation impact quantification | Computational pathogenicity assessment [23] |
| CRISPR/Cas9 with ABE | Precise genome editing | Y665F and Y665H mouse model generation | In vivo physiological studies [7] |
| Deep mutational scanning libraries | Saturation mutagenesis | Comprehensive SHP2 mutant activity profiling | High-throughput functional characterization [20] |
| Yeast viability assay | Selection based on phosphatase activity | SHP2 mutant functional screening | Controlled genetic system [20] |
| gnomAD database | Population variant frequency | Missense Enrichment Score calculation | Constraint and conservation analysis [32] |
| ClinVar database | Pathogenic variant annotations | Clinical correlation of mutations | Disease association studies [32] |
The comparative analysis of STAT SH2 domain mutations reveals that evolutionary conservation patterns provide powerful predictors of structural determinants governing pathogenicity. The precise structural alteration—not merely mutation location—dictates functional outcome, as demonstrated by the opposing impacts of Y665F and Y665H mutations. Integrated computational, high-throughput screening, and physiological approaches enable comprehensive mutation characterization, providing the foundational knowledge required for targeted therapeutic development. Future research should focus on expanding deep mutational scanning to STAT family members and developing small molecules that specifically counter pathogenic mechanisms at the SH2 domain interface.
Advanced Techniques for Characterizing STAT SH2 Mutations
The accurate classification of genetic variants is a cornerstone of precision medicine, directly influencing diagnosis, treatment strategies, and therapeutic development. For researchers investigating specific mutational patterns, such as those in the STAT SH2 domain, selecting the most appropriate computational tools is critical. Among the plethora of available in silico predictors, AlphaMissense, CADD (Combined Annotation Dependent Depletion), and REVEL (Rare Exome Variant Ensemble Learner) have emerged as widely used and powerful methods. This guide provides an objective, data-driven comparison of these three tools, framing their performance within the context of activating versus inactivating mutations, with a specific focus on STAT SH2 domain mutations to illustrate key practical considerations for researchers and drug development professionals.
Understanding the underlying algorithms and data types used by each tool is essential for interpreting their predictions correctly. The following table summarizes the core methodologies of AlphaMissense, CADD, and REVEL.
Table 1: Fundamental Characteristics of AlphaMissense, CADD, and REVEL
| Tool | Primary Methodology | Input Data & Features | Output Score & Range | Key Distinction |
|---|---|---|---|---|
| AlphaMissense | Deep learning model (based on AlphaFold) | Evolutionary conservation from multiple sequence alignments, protein structure (AlphaFold2) [33] [34] | Pathogenicity probability (0-1); classified as Benign, Ambiguous, or Pathogenic [35] | Unsupervised; does not rely on clinical labels, reducing human annotation bias [33]. |
| CADD | Supervised machine learning (Support Vector Machine) | 63+ diverse genomic annotations, including conservation, epigenomic marks, and transcriptomic features [36] | Phred-scaled score (1-99+); higher scores indicate more deleteriousness [36] | Models the difference between derived alleles and simulated variants that have become fixed in evolution [36]. |
| REVEL | Ensemble method (meta-predictor) | Combits the scores of 13 individual missense pathogenicity predictors, including SIFT, PolyPhen-2, and MutPred [36] | Pathogenicity probability (0-1); higher scores indicate higher probability of pathogenicity [36] | Trained on known pathogenic and benign missense variants from HumVar [36]. |
The workflow for utilizing these tools, from variant identification to final classification, involves several key stages that integrate computational predictions with biological evidence.
Performance Benchmarking and Comparative Analysis
Independent benchmarking studies across various diseases and variant types provide critical insights into the real-world performance of these tools.
Quantitative Performance Metrics
Recent evaluations on carefully curated datasets allow for a direct comparison of the predictive accuracy of each tool.
Table 2: Performance Comparison on Epilepsy-Associated Genes and Somatic Variants
| Tool | AUROC (Epilepsy Genes) [33] | Performance Tier (Somatic Variants) [37] | Notes on Clinical Utility |
|---|---|---|---|
| AlphaMissense | 0.93, 0.88, 0.95 (across 3 datasets) | Not specifically ranked in [37] | Top performer in epilepsy genes; also excels in identifying known cancer drivers (AUROC 0.98) [34]. |
| REVEL | 0.93, 0.88, 0.93 (across 3 datasets) | Top Tier | Robust and consistent high performer across both germline and somatic contexts; useful for VUS reclassification. |
| CADD | Not among top performers in [33] | Top Tier | Widely used but may have limited value for VUS in specific diseases like ALS; general deleteriousness score [36]. |
A study on epilepsy-associated genes, which used blind test sets not part of the tools' training data, found that AlphaMissense and REVEL showed the best classification performance, also outperforming other tools in the number of classified variants [33]. In the somatic variant context, a benchmark of 4,319 somatic single-nucleotide variants classified both REVEL and CADD as top-performing predictors [37].
Performance in Cancer Genomics
The utility of these tools extends to somatic mutations in cancer, where distinguishing driver from passenger mutations is crucial. A 2025 pan-cancer study found that methods incorporating protein structure or functional genomic data, like AlphaMissense, outperformed methods trained only on evolutionary data [34]. In this analysis, AlphaMissense significantly outperformed other deep learning-based methods as well as other best-in-class methods in predicting oncogenic mutations, achieving an AUROC of 0.98 for both oncogenes and tumor suppressor genes at the population level [34].
Case Study: Application to STAT SH2 Domain Mutations
The practical application of these tools can be illustrated by their performance on specific STAT SH2 domain mutations, which are critical in leukemogenesis. Research on STAT5B tyrosine 665 (Y665) mutations provides a compelling case study for comparing tool predictions against experimental validation.
Table 3: Divergent Predictions for STAT5B Y665 Mutations
| STAT5B Mutation | AlphaMissense [25] | CADD (PHRED) [25] | REVEL [25] | PolyPhen-2 [25] | Experimental Validation [25] |
|---|---|---|---|---|---|
| Y665F | 0.173 (Benign) | 24.3 (Deleterious) | 0.535 (Pathogenic) | 0.93 (Probably Damaging) | Gain-of-Function (Increased phosphorylation, DNA binding) |
| Y665H | 0.383 (Benign) | 23.1 (Deleterious) | 0.304 (Uncertain) | 0.084 (Benign) | Loss-of-Function (Resembles null phenotype) |
This case highlights critical insights for researchers:
- Tool Discordance is Informative: The starkly different predictions for Y665F and Y665H across tools underscore that these mutations likely have divergent biological mechanisms, a finding confirmed by functional experiments [25].
- No Single Tool is Infallible: AlphaMissense classified both Y665F and Y665H as "benign" despite their clear functional impacts demonstrated in vivo [25].
- Combined Analysis is Powerful: Using REVEL and PolyPhen-2 together would have correctly flagged Y665F as likely pathogenic, highlighting the value of a multi-tool approach.
The STAT signaling pathway and the critical location of the Y665 mutation within the SH2 domain can be visualized as follows:
Best Practices and Experimental Protocols
Recommended Workflow for Pathogenicity Assessment
Based on the comparative data, researchers should adopt the following workflow for robust pathogenicity assessment:
- Multi-Tool Approach: Never rely on a single tool. Start with a combination of AlphaMissense, REVEL, and a structure-based analysis where possible.
- Context-Specific Thresholds: Use established, context-specific thresholds when available. For example, in ALS research, a predetermined REVEL threshold showed clinical value, while CADD did not [36].
- Interpret Discordant Results: Discordant predictions between tools, as seen with STAT5B Y665F/H, can reveal biologically significant differences between variants that warrant further investigation.
- Integration with Experimental Data: Computational predictions should be combined with functional data. For STAT5B mutations, in silico modeling with tools like COORDinator to predict energetic effects on homodimerization provided crucial insights that complemented the pathogenicity predictions [25].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents and Resources for In Silico Validation
| Resource Category | Specific Tools / Databases | Primary Function in Analysis |
|---|---|---|
| Pathogenicity Predictors | AlphaMissense, REVEL, CADD, PolyPhen-2, SIFT | Provide computational evidence for variant impact on protein function [37] [25] [33]. |
| Variant Databases | ClinVar, gnomAD, OncoKB, COSMIC | Curated repositories of variant classifications and population frequencies for benchmarking [33] [36] [34]. |
| Structural Modeling | AlphaFold3, COORDinator, PyMOL | Predict and visualize protein structures; model mutational impact on stability and interactions [25]. |
| Variant Annotation | Ensembl VEP (Variant Effect Predictor) | Critical pipeline component for annotating variants with functional consequences and predictor scores [36] [38]. |
| Functional Assay Resources | Primary T cells, Genetically engineered mouse models (e.g., STAT5B knock-in) | Experimental validation of computational predictions through in vitro and in vivo functional characterization [25]. |
AlphaMissense, CADD, and REVEL each offer distinct strengths for pathogenicity prediction. AlphaMissense demonstrates leading performance in multiple independent benchmarks, leveraging structural and deep learning approaches. REVEL remains a robust, high-performing ensemble method particularly valuable for missense variant interpretation. CADD, while widely used as a general deleteriousness metric, shows more variable performance in disease-specific contexts. For researchers studying STAT SH2 domain mutations or similar pathogenic mechanisms, a consensus approach using multiple tools, with careful attention to discordant predictions and integration with structural modeling, provides the most robust strategy for accurate variant classification and functional insight.
The Src Homology 2 (SH2) domain is a critical modular unit in metazoan signal transduction, particularly within the STAT (Signal Transducer and Activator of Transcription) family of proteins. STAT proteins are central to cytokine and growth-factor signaling, and their conventional activation is initiated by SH2 domain-mediated recruitment to phosphorylated cytoplasmic domains of activated receptors [3]. Subsequent phosphorylation, dimerization via reciprocal SH2 domain-phosphotyrosine interactions, and nuclear translocation enable transcription of genes governing proliferation and survival [3]. The SH2 domain is a documented hotspot for mutations in diseases like cancer and autoimmune disorders, where single amino acid changes can lead to either constitutive activation or loss of function, fundamentally altering cellular transcriptional programs [3] [39].
Computational structural biology has become indispensable for elucidating the molecular mechanisms of such mutations. The recent release of AlphaFold 3 (AF3) represents a transformative advancement, enabling high-accuracy prediction of complexes containing proteins, nucleic acids, and small molecules within a unified deep-learning framework [40]. This guide provides an objective comparison of AlphaFold3's performance against its predecessors and specialized alternatives, with a focused analysis on its application in modeling STAT SH2 domain mutations and predicting their structural and energetic impacts.
AlphaFold3: Architectural Advances and Performance Comparison
AlphaFold3 introduces a substantially updated architecture compared to AlphaFold 2 (AF2), moving away from a structure module that operated on amino-acid-specific frames and side-chain torsion angles. Instead, AF3 employs a diffusion-based model that predicts raw atom coordinates directly [40] [41]. This approach uses a generative process where random noise is iteratively denoised to produce a final structure. The multiscale nature of diffusion allows the model to learn both local stereochemistry and large-scale structural organization without requiring complex stereochemical violation penalties during training [40]. Furthermore, AF3 de-emphasizes multiple sequence alignment (MSA) processing by replacing the evoformer with a simpler "pairformer" module, enhancing its efficiency and capability to handle diverse biomolecules [40].
Quantitative Performance Comparison Across Biomolecular Complexes
Extensive benchmarking reveals that AlphaFold3 achieves state-of-the-art accuracy across a wide range of interaction types, often surpassing specialized prediction tools [40].
Table 1: Performance Comparison of AlphaFold3 Against Specialized Tools
| Complex Type | Benchmark Set | AlphaFold3 Performance | Comparative Tool Performance | Key Metric |
|---|---|---|---|---|
| Protein-Ligand | PoseBusters (428 complexes) | Far greater accuracy [40] | Vina, RoseTTAFold All-Atom | % with pocket-aligned ligand RMSD < 2 Å |
| Protein-Protein | Recent protein-protein benchmarks | Substantially higher accuracy [40] [41] | AlphaFold-Multimer v2.3 | Interface TM-score / DockQ |
| Protein-Nucleic Acid | Nucleic-acid-specific benchmarks | Much higher accuracy [40] | Specialized nucleic-acid predictors | Nucleotide-level RMSD |
| Antibody-Antigen | Antibody-antigen benchmarks | Substantially improved accuracy [40] | AlphaFold-Multimer v2.3 | Interface LDDT |
As shown in Table 1, AF3's unified framework outperforms even traditional docking tools like Vina, which benefit from using solved protein structures as input—information that is not available in a true blind prediction scenario [40]. For protein-protein interactions, AF3 shows marked improvement over its predecessor, AlphaFold-Multimer v2.3 [40] [41].
Limitations and Considerations for Structure-Based Analysis
Despite its high structural accuracy, independent evaluations urge caution when applying AF3 predictions for downstream thermodynamic and functional analyses. A key study found that while AF3's initial prediction accuracy for protein-protein complexes is high, major inconsistencies from experimental structures can exist in:
- The compactness of the complex.
- Intermolecular polar interactions, with more than two hydrogen bonds often incorrectly predicted.
- Apolar-apolar packing at the interface [42].
Furthermore, when AF3-predicted structures are subjected to molecular dynamics (MD) simulation relaxation, the quality of the structural ensemble can deteriorate significantly, indicating potential instability in the predicted intermolecular packing [42]. This has direct consequences for energy calculations. Alanine scanning simulations to identify "hot-spot" residues for binding affinity, conducted using AF3-predicted structures as starting points, consistently underperform compared to those using experimental structures. The correlation between structural deviation metrics (like RMSD) and the quality of affinity calculations is poor, meaning a high-quality static structure prediction does not guarantee reliable thermodynamic profiling [42].
Table 2: AlphaFold3 Workflow Considerations for Energetic Studies
| Stage | AlphaFold3 Strength | Consideration for Energetic Analysis |
|---|---|---|
| Initial Structure Prediction | High-accuracy static models of complexes. | May contain subtle errors in interfacial packing and polar networks. |
| Conformational Sampling | Generates a single, low-energy conformation. | Cannot natively capture protein dynamics, flexibility, or alternative folds. |
| Binding Affinity Prediction | Not a direct function of the model. | Structures may be unstable in MD simulations, leading to unreliable free energy estimates. |
| Hot-Spot Identification | Provides a high-resolution structural context. | Alanine scanning results are less accurate than when using experimental structures. |
Application to STAT SH2 Domain Mutations: Activating vs. Inactivating Profiles
The SH2 domain structure consists of a central antiparallel β-sheet (βB-βD) flanked by two α-helices (αA and αB), forming an αβββα motif [3] [31]. It features two primary subpockets: the phospho-tyrosine (pY) pocket, which binds the phosphorylated tyrosine, and the pY+3 specificity pocket, which confers binding selectivity [3] [31]. STAT-type SH2 domains are distinct, lacking the βE and βF strands found in Src-type domains and instead featuring a split αB helix, an adaptation that facilitates STAT dimerization [3] [31].
Mutations within this domain can have divergent functional consequences. A compelling example is found in STAT5B, where mutations at a single tyrosine residue (Y665) are associated with leukemia but have opposite effects:
- The STAT5BY665F mutation is activating. It enhances STAT5B activity, establishes transcriptional enhancers, and expands CD8+ and regulatory CD4+ T-cell populations in mouse models, leading to progressive dermatitis [39].
- The STAT5BY665H mutation is largely inactivating. It fails to induce the interleukin-regulated enhancer landscape and gene expression, and does not drive the same T-cell expansion, though it still modifies immune cell profiles [39].
This dichotomy underscores the exquisitely tuned evolutionary balance of the SH2 domain, where subtle structural perturbations can fundamentally redirect transcriptional programs and immune responses [3] [39]. A systematic analysis of mutation prevalence shows that the SH2 and transactivation domains (TAD) of STAT genes are among the most heavily mutated in the general population, highlighting their genetic volatility [9].
Workflow for Computational Analysis of STAT SH2 Mutations
The following diagram illustrates a robust experimental-computational workflow for characterizing STAT SH2 domain mutations, integrating AlphaFold3 modeling with validation and functional analysis.
Successfully executing the workflow above requires a combination of computational tools, datasets, and experimental reagents.
Table 3: Key Research Reagent Solutions for STAT SH2 Domain Investigation
| Category | Item / Resource | Function and Application | Example / Source |
|---|---|---|---|
| Computational Modeling | AlphaFold3 Server / Model | Predicts 3D structures of STAT complexes with proteins, nucleic acids, or ligands. | Isomorphic Labs/DeepMind [40] |
| AutoDockFR | Specialized docking software for flexible ligands and receptors; useful for probing pY pocket binding. | CCSB, Scripps Research [43] | |
| Molecular Dynamics Software | Simulates dynamic behavior and refines predicted structures (e.g., GROMACS, AMBER). | [42] | |
| Data & Databases | Protein Data Bank (PDB) | Repository of experimentally solved protein structures for validation and template-based studies. | RCSB PDB [40] |
| COSMIC Database | Catalogs somatic mutations in cancer; identifies disease-associated STAT SH2 mutations. | Catalogue of Somatic Mutations in Cancer [9] | |
| All of Us Database | Provides population-level genetic variation data; contrasts mutation prevalence in healthy vs. diseased cohorts. | NIH "All of Us" Research Program [9] | |
| GEO Accession Viewer | Archives functional genomics datasets (e.g., scRNA-seq) to link mutations to transcriptional programs. | GSE276312 [39] | |
| Experimental Reagents | Mutant STAT Constructs | Plasmid DNA for expressing wild-type and mutant STAT proteins (e.g., Y665F, Y665H). | Custom gene synthesis [39] |
| Phospho-specific Antibodies | Antibodies detecting phosphorylated STATs to assay activation status. | Commercial suppliers (e.g., Cell Signaling Tech) | |
| Cell-based Reporter Assays | Systems to measure STAT transcriptional activity downstream of cytokine stimulation. | Luciferase-based kits |
AlphaFold3 represents a monumental leap in computational structural biology, providing researchers with an unparalleled tool for generating accurate models of STAT proteins and their complexes. Its ability to predict the structural consequences of SH2 domain mutations is a powerful asset for generating hypotheses about molecular dysfunction. However, the tool is not a panacea. For investigations into the energetic impact of mutations—crucial for understanding the precise mechanism of activation or inactivation and for rational drug design—AF3-predicted structures should be viewed as a starting point. They require robust validation and refinement through molecular dynamics simulations and, ultimately, correlation with experimental data on protein function and transcriptional output. The path forward lies in the intelligent integration of AF3's formidable predictive power with advanced simulation techniques and rigorous experimental biology to fully unravel the complexities of STAT signaling in health and disease.
Transcription factors (TFs) are pivotal regulators of gene expression, and their dysfunction is a common driver of disease pathogenesis. Signal Transducers and Activators of Transcription (STAT) proteins, particularly STAT5B, represent a critical TF family whose activity is modulated by phosphorylation and protein-protein interactions mediated through their Src Homology 2 (SH2) domains. Disease-associated mutations frequently cluster within the STAT SH2 domain, altering phosphorylation status, DNA binding capacity, and transcriptional output. This guide provides a comparative analysis of experimental approaches for quantifying these functional parameters, with a specific focus on distinguishing between activating and inactivating STAT SH2 domain mutations to support basic research and drug discovery efforts.
Comparative Analysis of STAT SH2 Domain Mutations
The SH2 domain is essential for STAT activation, mediating recruitment to activated cytokine receptors and facilitating STAT dimerization through phospho-tyrosine-SH2 domain interactions. Different mutations within this domain can produce strikingly opposite functional consequences, as demonstrated by recent investigations into STAT5B mutations identified in human diseases.
Table 1: Functional Characteristics of STAT5B SH2 Domain Mutations
| Mutation | Location | Pathology | Type | DNA Binding | Transcriptional Output | Molecular Consequence |
|---|---|---|---|---|---|---|
| Y665F | αB' Helix (EAR) | T-cell Leukemia | Gain-of-Function (GOF) | Enhanced | Elevated enhancer formation | Disrupts hydrophobic system, promotes constitutive activation [3] [7] |
| Y665H | αB' Helix (EAR) | T-cell Leukemia | Loss-of-Function (LOF) | Impaired | Defective enhancer establishment | Compromises structural integrity of pY+3 pocket, reducing dimerization capacity [3] [7] |
| S614R | BC Loop (pY pocket) | T-LGLL, NK-LGLL | GOF | Enhanced/Altered | Increased | Alters phospho-peptide binding specificity [3] |
| K665E/M | αA Helix (pY pocket) | AD-HIES | LOF | Diminished | Reduced | Disrupts conserved phosphate-binding residues [3] |
The evolutionary active region (EAR) of the STAT SH2 domain, containing an additional αB' helix, serves as a particular hotspot for mutations with significant functional impact. The Y665F and Y665H mutations exemplify how single amino acid substitutions at the same residue can drive opposing pathological states through distinct biophysical mechanisms [3] [7].
Experimental Protocols for Functional Characterization
Assessing Phosphorylation Status
A. Simple Western Capillary-Based Immunoassay This automated, high-sensitivity approach represents a significant advancement over traditional Western blotting for detecting phosphorylation events.
Protocol Summary:
- Sample Preparation: Lyse cells under appropriate conditions to preserve phosphorylation status, including phosphatase inhibitors.
- Protein Separation: Utilize charge-based (isoelectric focusing) or size-based separation in capillaries. Charge-based separation effectively resolves different phosphorylation states of the same protein.
- Immunodetection: Incubate with phospho-specific primary antibodies followed by HRP-conjugated or fluorescent secondary antibodies.
- Quantification: Automated detection and quantification of signal intensity, with normalization to total protein levels.
Advantages: Capillary-based systems like Jess offer up to 100x greater sensitivity than traditional Western blotting, enable precise quantification of phosphorylation stoichiometry, and require smaller sample volumes while providing superior reproducibility [44].
B. Cell-Based ELISA This microplate-based format allows high-throughput quantification of protein phosphorylation in intact cells.
Protocol Summary:
- Cell Culture and Treatment: Plate cells in 96-well plates, grow to appropriate density, and apply experimental treatments.
- Fixation and Blocking: Fix cells directly in culture wells, then block non-specific binding sites.
- Antibody Incubation: Simultaneously detect phospho-protein and total protein using specific antibodies with different detection channels (e.g., colorimetric or fluorometric).
- Signal Detection and Normalization: Measure absorbance or fluorescence, normalizing phospho-signal to total protein signal to correct for well-to-well variation [44].
Advantages: Enables high-throughput screening of multiple conditions, preserves cellular context, and provides internal normalization for improved accuracy [44].
Evaluating DNA Binding Capacity
A. Electrophoretic Mobility Shift Assay (EMSA) EMSA remains a foundational technique for detecting sequence-specific DNA-protein interactions through differential migration in non-denaturing gels.
Protocol Summary:
- Probe Preparation: Generate double-stranded DNA probes containing the transcription factor binding sequence (e.g., GAS motif for STATs: TTCnnnGAA), labeled with biotin, fluorophores, or radioisotopes.
- Binding Reaction: Incubate nuclear extracts or purified protein with labeled DNA probe in appropriate binding buffer with non-specific competitors (e.g., poly-dIdC).
- Electrophoresis and Detection: Separate protein-DNA complexes from free DNA via non-denaturing PAGE, then transfer to membrane and detect using appropriate methods (chemiluminescence for biotinylated probes) [45] [46].
Advantages: Directly visualizes specific DNA-protein complexes, allows assessment of binding stoichiometry, and can be adapted for competition experiments to determine binding specificity [45] [46].
B. DNA Binding Scintillation Proximity Assay (SPA) This solution-based homogenous assay provides a quantitative, higher-throughput alternative to EMSA.
Protocol Summary:
- Probe Preparation: Create DNA probes containing the transcription factor binding site, labeled with radioisotopes or other detectable tags.
- Binding Reaction: Incubate protein extracts with labeled DNA in microplate wells coated with scintillant-containing beads that capture DNA-protein complexes.
- Signal Detection: Measure bound complexes through scintillation counting, where energy transfer occurs only when the radioactive label is in close proximity to the scintillant bead [46].
Advantages: Amenable to higher-throughput formats, provides quantitative binding data, and eliminates the need for separation/wash steps [46].
Quantifying Transcriptional Activity
A. Transcription Factor Enrichment Analysis (TFEA) This computational method leverages high-throughput genomic data to infer transcription factor activity by detecting positional motif enrichment associated with transcriptional changes.
Protocol Summary:
- Data Input Preparation: Generate genome-wide data informing on transcriptional regulation, such as nascent transcription (PRO-Seq), chromatin accessibility (ATAC-Seq), or histone modifications (H3K27ac ChIP-Seq).
- Region of Interest Definition: Use tools like muMerge to combine data from multiple replicates into a consensus set of transcriptionally active regions with high positional accuracy.
- Motif Enrichment Analysis: Apply TFEA to calculate enrichment scores that incorporate both the magnitude of transcriptional changes and the precise positioning of TF motifs relative to regions of interest.
- Statistical Validation: Compare observed enrichment against empirically derived null distributions to assign statistical significance [47].
Advantages: Circumvents limitations of steady-state RNA-seq by focusing on direct transcriptional outputs, enables temporal resolution of regulatory networks, and identifies master regulator TFs from genomic data alone [47].
B. Priori Transcription Factor Activity Inference This method predicts TF activity from RNA-seq data by leveraging prior biological knowledge of TF-target relationships.
Protocol Summary:
- Network Construction: Extract literature-supported TF-target gene relationships from curated databases (e.g., Pathway Commons).
- Model Fitting: Apply linear models to determine the direction and magnitude of TF regulation on its known target genes.
- Activity Score Calculation: Generate composite activity scores that reflect the aggregate expression changes of target genes weighted by their regulatory relationship to the TF [48].
Advantages: Grounds predictions in established biology rather than covariance alone, demonstrates superior sensitivity and specificity in detecting perturbed TFs, and identifies significant determinants of clinical outcomes in patient datasets [48].
Signaling Pathway and Experimental Workflows
STAT Signaling and Functional Assays Workflow. This diagram illustrates the STAT protein activation pathway from cytokine stimulus to target gene transcription, with corresponding functional assays mapped to specific stages where they provide quantitative measurements. SH2 domain mutations disrupt phospho-tyrosine-mediated dimerization, affecting downstream functions [3] [7].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for STAT Functional Analysis
| Reagent / Method | Function / Application | Key Features |
|---|---|---|
| Phospho-specific Antibodies | Detection of phosphorylated STAT isoforms | Essential for Western blot, ELISA; must be validated for specificity [44] |
| Simple Western Systems | Automated capillary-based immunoassay | High-sensitivity phospho-isoform resolution; 100x more sensitive than traditional Western [44] |
| Biotinylated DNA Probes | EMSA and DNA binding assays | Contains STAT binding motifs (GAS sequences); enables detection of specific complexes [46] |
| Pathway Commons Database | Source of prior biological knowledge | Curated TF-target relationships for activity inference methods like Priori [48] |
| muMerge Software | ROI consolidation from genomic data | Statistically principled combination of regions across replicates for TFEA [47] |
| Nuclear Extraction Kits | Preparation of protein extracts for DNA binding assays | Isolates nuclear proteins including transcription factors [46] |
The comprehensive functional characterization of STAT SH2 domain mutations requires an integrated experimental approach assessing phosphorylation status, DNA binding capacity, and transcriptional output. The complementary methodologies presented here enable researchers to distinguish between activating and inactivating mutations, elucidate their molecular mechanisms, and identify potential therapeutic targets. The selection of appropriate assays should be guided by research objectives, available resources, and required throughput, with particular attention to the distinct advantages each method offers for quantifying specific aspects of transcription factor function in health and disease.
The Src Homology 2 (SH2) domain is a critical modular unit found in numerous signaling proteins, enabling specific recognition of phosphotyrosine (pY) motifs and facilitating the assembly of complex signaling networks [31]. In Signal Transducers and Activators of Transcription (STAT) proteins, the SH2 domain is indispensable for canonical activation: it mediates recruitment to activated cytokine receptors, facilitates JAK-mediated tyrosine phosphorylation, and drives STAT dimerization through reciprocal phosphotyrosine-SH2 interactions [3]. This dimerization is a prerequisite for nuclear translocation and the transcription of target genes governing proliferation, survival, and differentiation [3] [49]. The structural integrity of the STAT SH2 domain is therefore paramount for precise signal transduction. Consequently, this domain is a mutational hotspot in human diseases, with single amino acid substitutions capable of fundamentally altering signaling output, leading to either gain-of-function (GOF) hyperactivation or loss-of-function (LOF) deficiencies [3] [7]. This guide compares the in vivo phenotypic outcomes of activating versus inactivating mutations within the STAT SH2 domain, using knock-in mouse models to delineate the profound physiological consequences of these dysregulated signaling states.
Comparative Analysis of SH2 Domain Mutations: In Vivo Phenotypes
Knock-in mouse models provide the most physiologically relevant platform for dissecting the impact of human disease-associated mutations. The table below summarizes the contrasting phenotypes driven by two specific mutations at tyrosine 665 (Y665) in the STAT5B SH2 domain.
Table 1: Phenotypic Comparison of STAT5B SH2 Domain Mutations in Knock-in Mouse Models
| Feature | STAT5BY665F (GOF Mutation) | STAT5BY665H (LOF Mutation) |
|---|---|---|
| Molecular & Cellular Phenotype | ||
| STAT5 Phosphorylation | Enhanced and/or sustained phosphorylation after cytokine stimulation [30] | Greatly diminished phosphorylation, resembling a null state [30] |
| Transcriptional Activity & Enhancer Formation | Elevated transcriptional activity and increased enhancer establishment [7] | Impaired enhancer establishment and gene regulation [7] |
| DNA Binding | Increased DNA binding capacity [30] | Impaired DNA binding [30] |
| Immune Cell Populations | Accumulation of CD8+ effector/memory and CD4+ regulatory T cells; altered CD8+/CD4+ ratios [30] | Diminished CD8+ effector/memory and CD4+ regulatory T cells [30] |
| Organ & Systemic Phenotype | ||
| Mammary Gland Development | Accelerated mammary gland development during pregnancy [7] | Failure to develop functional mammary tissue; lactation failure [7] |
| Lactation | Successful lactation [7] | Lactation failure (unless rescued by persistent hormonal stimulation over multiple pregnancies) [7] |
| Associated Human Diseases | Somatic mutations found in T-cell leukemias (e.g., T-LGLL, T-PLL) [30] | Identified in a case of T-PLL; model reflects growth hormone insensitivity and immune pathology [30] |
Experimental Protocols for Modeling and Analysis
Generation of Knock-in Mouse Models
The detailed methodology for creating these precise genetic models is foundational to their phenotypic analysis.
Protocol 1: CRISPR/Cas9 and Base Editing for Knock-in Model Generation
- Genetic Engineering Strategy: The STAT5BY665F and STAT5BY665H mutations are introduced into the mouse genome using CRISPR/Cas9 technology combined with homology-directed repair or adenine base editing (ABE) [7].
- Microinjection/Electroporation: For the Y665H mutation, ABE mRNA and a single-guide RNA (sgRNA) are co-microinjected into the cytoplasm of fertilized C57BL/6 N mouse eggs. The base editor directly converts the target adenine to guanine, creating the His (H) codon [7]. For the Y665F mutation, a more complex strategy is employed: Cas9 protein is pre-complexed with the Y665F sgRNA to form a ribonucleoprotein (RNP) complex. This RNP is co-electroporated into zygotes along with a single-stranded oligonucleotide donor template containing the desired Tyr (TAC) to Phe (TTT) mutation [7].
- Embryo Transfer and Genotyping: The successfully injected or electroporated embryos are cultured to the 2-cell stage and then implanted into the oviducts of pseudopregnant surrogate mothers. Founder mice are genotyped using PCR amplification of tail DNA followed by Sanger sequencing or TaqMan-based assays to confirm the presence of the intended mutation [7].
Phenotypic Analysis of Mutant Mice
Comprehensive characterization of the knock-in models involves a multi-faceted approach to capture molecular, cellular, and organismal phenotypes.
Protocol 2: Multi-level Phenotypic Characterization
- Immune Phenotyping by Flow Cytometry: Lymphocytes are isolated from spleen, lymph nodes, or blood. Single-cell suspensions are stained with fluorescently labeled antibodies against cell surface markers (e.g., CD3, CD4, CD8, CD44, CD62L) to analyze immune cell populations, activation status, and memory differentiation using flow cytometry. This protocol revealed the altered CD8+/CD4+ T-cell ratios in the mutant mice [30].
- Transcriptomic Analysis (RNA-seq): Total RNA is extracted from tissues of interest (e.g., mammary gland, liver) using homogenization and commercial kits. RNA quality is assessed (RIN > 8.0), and ribosomal RNA is removed. Sequencing libraries are prepared and sequenced on platforms like Illumina NovaSeq. Bioinformatic analysis of the data identifies differentially expressed genes and dysregulated pathways, highlighting the failure to induce milk protein genes in STAT5BY665H mutants [7].
- Molecular Phenotyping:
- Western Blotting: Protein lysates from tissues or stimulated cells are analyzed by Western blot to assess STAT5 phosphorylation status (using phospho- specific antibodies) and total protein levels [30].
- Electrophoretic Mobility Shift Assay (EMSA): Nuclear extracts are incubated with a labeled DNA probe containing a STAT5 binding consensus sequence. The DNA-protein complexes are resolved on a native gel to evaluate STAT5 DNA-binding activity, which is heightened in GOF and diminished in LOF mutants [30].
Visualizing STAT Signaling and Mutational Impact
The following diagram illustrates the canonical STAT5 activation pathway and the points where SH2 domain mutations exert their effect.
Figure 1: Canonical JAK-STAT5 Signaling Pathway and SH2 Domain Function. The pathway depicts cytokine-induced STAT5 activation. SH2 domain mutations (e.g., Y665) disrupt critical steps: phosphotyrosine (pY) recognition during receptor recruitment and reciprocal SH2-pY interaction during dimerization [3] [50].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for STAT Knock-in Research
| Reagent / Solution | Function in Research | Example Application in STAT Models |
|---|---|---|
| CRISPR/Cas9 System | Enables precise genome editing to introduce point mutations. | Generation of STAT5BY665F and Y665H knock-in alleles in mouse embryos [7]. |
| Adenine Base Editor (ABE) | Directly converts A•T to G•C base pairs without double-strand DNA breaks. | Used to create the STAT5BY665H mutation efficiently [7]. |
| Phospho-STAT5 Antibodies | Detect activated, tyrosine-phosphorylated STAT5 via Western blot or flow cytometry. | Confirmation of hyperphosphorylation (GOF) or lack of phosphorylation (LOF) in mutant cells [30]. |
| Flow Cytometry Antibody Panels | Identify and quantify specific immune cell populations. | Analysis of CD8+/CD4+ T-cell imbalances in primary lymphocytes from mutant mice [30]. |
| STAT SH2 Domain Inhibitors | Small molecules that disrupt SH2 domain function, used for mechanistic and therapeutic studies. | Compounds like S3I-201.1066 bind the STAT3 SH2 domain, block dimerization, and have shown antitumor effects in models [51]. |
The Src Homology 2 (SH2) domain is a critical regulatory module within metazoan signaling pathways, particularly in Signal Transducers and Activators of Transcription (STAT) proteins [3]. In STAT-mediated signaling, the SH2 domain facilitates phosphotyrosine-dependent recruitment, dimerization, and nuclear translocation of activated STATs, ultimately driving the transcription of genes involved in proliferation, survival, and differentiation [3]. Mutations within this domain can profoundly alter STAT function, leading to either constitutive activation or loss of function, which are implicated in various diseases, including immunodeficiencies and cancers [3] [30].
Multi-omics approaches, which integrate data from transcriptomic and epigenomic platforms, are vital for understanding the hierarchical complexity of biological systems [52] [53]. These methods allow researchers to collectively analyze molecular data from different biological layers, providing a systems-level view of how genetic variations disrupt normal cellular function [52]. In the context of STAT SH2 domain mutations, integrating RNA sequencing (RNA-seq) with epigenomic techniques like Reduced-Representation Bisulfite Sequencing (RRBS) can uncover how single amino acid substitutions reshape enhancer landscapes and gene expression programs, revealing mechanisms of pathogenicity [52] [7].
This guide focuses on the application of multi-omics integration to compare the functional consequences of activating (gain-of-function) and inactivating (loss-of-function) mutations within the STAT SH2 domain. We will objectively compare the molecular and phenotypic outputs driven by different mutant STAT alleles, provide detailed experimental protocols for profiling these effects, and visualize the underlying signaling pathways and workflows.
STAT SH2 Domain Structure and Mutational Landscape
The STAT SH2 domain consists of a central anti-parallel β-sheet (strands βB-βD) flanked by two α-helices (αA and αB), forming a characteristic αβββα motif [3]. This structure creates two primary functional pockets: the phospho-tyrosine (pY) binding pocket and the pY+3 specificity pocket [3]. The pY pocket, formed by the αA helix, BC loop, and one face of the central β-sheet, binds phosphorylated tyrosine residues. The pY+3 pocket, created by the opposite face of the β-sheet, the αB helix, and CD and BC* loops, determines peptide binding specificity [3]. A unique feature of STAT-type SH2 domains is the presence of an additional α-helix (αB') in the C-terminal region of the pY+3 pocket, known as the evolutionary active region (EAR) [3].
Mutations within the SH2 domain represent a hotspot in the STAT mutational landscape identified through patient sequencing [3]. These mutations can have divergent functional impacts, even when occurring at the same residue. For instance, in STAT5B, mutations at tyrosine 665 (Y665) to phenylalanine (Y665F) or histidine (Y665H) lead to gain-of-function (GOF) and loss-of-function (LOF) phenotypes, respectively [7] [30]. The structural locations and clinical associations of key STAT3 and STAT5B SH2 domain mutations are summarized in Table 1.
Table 1: Disease-Associated Mutations in STAT3 and STAT5B SH2 Domains
| Protein | Mutation | Domain Location | Functional Type | Associated Pathology |
|---|---|---|---|---|
| STAT3 | S614R | BC loop (pY pocket) | Activating (GOF) | T-LGLL, NK-LGLL, ALK-ALCL [3] |
| STAT3 | K591E/M | αA helix (pY pocket) | Inactivating (LOF) | AD-HIES (Germline) [3] |
| STAT3 | S611N/G/I | βB strand (pY pocket) | Inactivating (LOF) | AD-HIES (Germline) [3] |
| STAT5B | Y665F | SH2 Domain | Activating (GOF) | T-LGLL, T-PLL [30] |
| STAT5B | Y665H | SH2 Domain | Inactivating (LOF) | T-PLL (Single Case) [30] |
| STAT5B | N642H | SH2 Domain | Activating (GOF) | T-LGLL [30] |
Figure 1: STAT protein domain architecture and SH2 domain functional pockets. Mutations at critical residues like Y665 can alter phosphopeptide binding and dimerization.
Comparative Multi-Omics Profiling of STAT5B Y665F vs. Y665H Mutations
The contrasting phenotypes of STAT5B Y665F (GOF) and Y665H (LOF) mutations provide an ideal model for comparing how single amino acid changes can differentially rewire transcriptomic and epigenomic programs. Recent studies using genetically engineered mouse models have delineated their distinct impacts on mammary gland development and immune function [7] [30].
Phenotypic and Molecular Consequences
Table 2: Comparative Analysis of STAT5B Y665F and Y665H Mutations
| Parameter | STAT5B Y665F (GOF) | STAT5B Y665H (LOF) | Wild-Type STAT5B |
|---|---|---|---|
| Mammary Gland Development | Accelerated alveolar development during pregnancy [7] | Failure of functional mammary tissue development, lactation failure [7] | Normal, pregnancy-induced development [7] |
| T Cell Populations (Mouse Model) | Accumulation of CD8+ effector/memory and CD4+ regulatory T cells; altered CD8+/CD4+ ratio [30] | Diminished CD8+ effector/memory and CD4+ regulatory T cells [30] | Normal T cell homeostasis [30] |
| STAT5 Phosphorylation & DNA Binding | Enhanced and sustained after cytokine activation [30] | Greatly diminished, resembling a null state [30] | Transient and cytokine-dependent [30] |
| Enhancer & Super-Enhancer Formation | Elevated enhancer formation and activity [7] | Impaired establishment of enhancers and alveolar differentiation [7] | Hormonally and cytokine-induced establishment [7] |
| Transcriptomic Profile | Hyper-activation of STAT5 target genes (e.g., milk proteins) [7] | Failure to activate STAT5-dependent genetic programs [7] | Context-dependent activation of target genes [7] |
Multi-Omics Integration for Mechanistic Insights
Integrative analysis of transcriptomics (RNA-seq) and epigenomics (ATAC-seq, ChIP-seq) data is crucial for understanding how these mutations alter gene regulatory networks.
- Transcriptomics (RNA-seq): In STAT5BY665H mice, RNA-seq from mammary tissue revealed a profound failure to induce the expression of key milk protein genes (e.g., Csn1s1, Csn2, Wap) during pregnancy, consistent with a LOF phenotype. In contrast, STAT5BY665F mice showed precocious and elevated expression of these genes [7].
- Epigenomics (ChIP-seq): Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) and chromatin immunoprecipitation followed by sequencing (ChIP-seq) for H3K27ac and STAT5B itself demonstrated that the GOF mutant increased the number and strength of STAT5-bound enhancers and super-enhancers. The LOF mutant, however, was defective in hormone-driven enhancer establishment [7].
- Integrated Analysis: Correlation of open chromatin regions or active enhancer marks with differentially expressed genes allows for the direct linking of epigenetic alterations to transcriptional outcomes. For example, a candidate quadripartite super-enhancer regulating the Olah gene was identified as being under the control of STAT5B [7].
Figure 2: Signaling cascade and functional outcomes of wild-type and mutant STAT5B.
Experimental Protocols for Multi-Omics Profiling
This section outlines detailed methodologies for generating and integrating transcriptomic and epigenomic data to profile the effects of STAT SH2 domain mutations, drawing from established protocols [52] [7].
Transcriptomic Profiling with RNA Sequencing
Objective: To identify genome-wide differences in gene expression between GOF and LOF STAT mutants and wild-type controls.
Protocol:
- Sample Collection and RNA Extraction: Isolate total RNA from target tissues or cell lines (e.g., mammary gland, T cells) using a commercial kit (e.g., PureLink RNA Mini Kit). Assess RNA integrity and purity using an Agilent Bioanalyzer, accepting only samples with an RNA Integrity Number (RIN) > 8.0 [7].
- Library Preparation: Deplete ribosomal RNA from 1 μg of total RNA. Synthesize cDNA using reverse transcriptase (e.g., SuperScript III). Prepare sequencing libraries with a stranded total RNA library preparation kit (e.g., TruSeq Stranded Total RNA Library Prep Kit) [7].
- Sequencing and Data Analysis: Sequence the libraries on an Illumina platform (e.g., NovaSeq 6000) to generate paired-end reads. Align the raw sequencing reads to the reference genome (e.g., mm10 for mouse) using a splice-aware aligner like STAR. Quantify gene expression counts and perform differential expression analysis using software packages such as DESeq2 or edgeR in R/Bioconductor [52] [7].
Epigenomic Profiling with ATAC-Seq and ChIP-Seq
Objective: To map changes in chromatin accessibility (ATAC-seq) and histone modifications or transcription factor binding (ChIP-seq).
Protocol for ATAC-Seq:
- Tagmentation: Treat nuclei from fresh cells with the hyperactive Tn5 transposase. This enzyme simultaneously fragments the genome and inserts sequencing adapters into open chromatin regions.
- Library Amplification and Sequencing: Purify the tagmented DNA and amplify it by PCR using primers compatible with the Illumina sequencing platform. Sequence the resulting library to identify genome-wide regions of accessible chromatin [52].
Protocol for ChIP-Seq (e.g., for H3K27ac or STAT5):
- Cross-linking and Sonication: Cross-link proteins to DNA in cells using formaldehyde. Lyse cells and shear the chromatin by sonication to fragment sizes of 200–500 bp.
- Immunoprecipitation: Incubate the sheared chromatin with a specific antibody targeting the protein or histone mark of interest (e.g., anti-H3K27ac, anti-STAT5). Capture the antibody-chromatin complexes.
- Library Preparation and Sequencing: Reverse the cross-linking, purify the DNA, and construct sequencing libraries from the immunoprecipitated DNA. Sequence the libraries to map the binding sites or histone modifications genome-wide [7].
Data Integration Workflow
Objective: To correlate epigenomic changes with transcriptomic outputs for a unified biological interpretation.
Protocol:
- Quality Control: Perform rigorous quality control on all datasets. For RNA-seq, check for 3' bias and GC content. For epigenomic data, use tools like FastQC and ChIPQC.
- Peak Calling and Annotation: For ATAC-seq and ChIP-seq data, use peak-calling software (e.g., MACS2) to identify significant regions of signal. Annotate these peaks to genomic features (e.g., promoters, enhancers) using tools like ChIPseeker.
- Correlative Analysis: Integrate the datasets within a computational environment like an R/Bioconductor Jupyter notebook. Overlap differentially expressed genes from RNA-seq with nearby or linked differentially accessible chromatin regions or active enhancer marks from ATAC/ChIP-seq. Functional enrichment analysis (e.g., GO, KEGG) of the correlated gene sets can reveal impacted biological pathways [52].
- Visualization: Use genome browsers (e.g., IGV) and plotting packages in R (e.g., ggplot2) to visualize integrated data, such as plotting ATAC-seq signal tracks alongside RNA-seq read counts at specific genomic loci [52].
Figure 3: Integrated multi-omics workflow for profiling mutant effects, from sample preparation to mechanistic insight.
Successful multi-omics research requires a suite of reliable reagents, computational tools, and data resources. The table below lists key solutions for studying STAT SH2 domain mutations.
Table 3: Essential Research Reagents and Resources for Multi-Omics Profiling
| Category | Item | Specific Example / Catalog Number | Function in Protocol |
|---|---|---|---|
| Animal Models | Genetically Engineered Mice | STAT5BY665F and STAT5BY665H knock-in [7] [30] | Provide in vivo context to study physiological impact of mutations. |
| RNA Analysis | RNA Extraction Kit | PureLink RNA Mini Kit (Thermo Fisher Scientific) [7] | Isolves high-quality, intact total RNA from tissues/cells. |
| RNA Analysis | rRNA Depletion & Library Prep | TruSeq Stranded Total RNA Library Prep Kit (Illumina) [7] | Constructs sequencing libraries from total RNA. |
| Epigenomics | Chromatin Shearing | Covaris S220 or Bioruptor | Sonication for shearing cross-linked chromatin for ChIP-seq. |
| Epigenomics | Specific Antibodies | Anti-STAT5, Anti-H3K27ac [7] | Immunoprecipitation of target proteins or histone marks for ChIP-seq. |
| Epigenomics | Transposase | Illumina Tagment DNA TDE1 Enzyme | Fragments DNA and adds adapters for ATAC-seq library prep. |
| Computational | Cloud Computing Platform | Google Cloud Platform (Vertex AI, Cloud Storage) [52] | Provides scalable resources for data storage and analysis. |
| Computational | Analysis Environment | Jupyter Notebook with R/Bioconductor kernel [52] | Interactive environment for executing analysis workflows. |
| Computational | Public Data Repository | Gene Expression Omnibus (GEO) [52] | Source for procuring and sharing public datasets. |
| Bioinformatics | Alignment Software | STAR (RNA-seq), BWA (ChIP-seq/ATAC-seq) [7] | Aligns sequencing reads to a reference genome. |
| Bioinformatics | Differential Analysis | DESeq2 (RNA-seq), MACS2 (Peak calling) [52] | Identifies statistically significant differences between samples. |
The integration of transcriptomic and epigenomic profiling provides a powerful, systems-level framework for deciphering the mechanistic consequences of disease-associated mutations. The comparative analysis of STAT5B SH2 domain mutations Y665F and Y665H demonstrates how single amino acid substitutions can cause divergent phenotypes through opposing alterations in enhancer function and transcriptional programs. The detailed experimental protocols and resource toolkit outlined here offer a roadmap for researchers to objectively characterize the functional impact of genetic variants, accelerating the discovery of novel therapeutic targets and personalized treatment strategies in oncology and immunology.
Challenges in Mutation Analysis and Emerging Therapeutic Targeting
In the field of molecular biology and drug development, interpreting the functional impact of genetic variants, particularly within critical signaling domains like the STAT SH2 domain, presents a significant challenge. A persistent issue faced by researchers is the frequent discrepancy between in silico computational predictions and subsequent experimental functional data. These conflicts can stall variant classification, hinder mechanistic studies, and delay therapeutic development. This guide objectively compares the performance of various computational and experimental methods used to characterize activating versus inactivating mutations in the STAT SH2 domain, providing a structured framework for resolving such discrepancies. We focus on the specific case of STAT SH2 domain mutations, a hotspot in cancer and immunodeficiencies, to illustrate a systematic approach for data reconciliation [3].
Understanding the STAT SH2 Domain and Its Mutational Landscape
The Src Homology 2 (SH2) domain is a modular protein unit approximately 100 amino acids long that specifically binds to phosphorylated tyrosine (pY) motifs, thereby facilitating critical protein-protein interactions in signal transduction networks [31]. In STAT (Signal Transducer and Activator of Transcription) proteins, the SH2 domain is indispensable for canonical activation. It mediates recruitment to activated cytokine receptors, subsequent tyrosine phosphorylation by JAK kinases, and ultimately, the dimerization of two STAT monomers via reciprocal phospho-tyrosine-SH2 domain interactions. This dimerization is a prerequisite for nuclear translocation and the transcription of target genes [3] [23].
STAT-type SH2 domains possess unique structural features that distinguish them from Src-type SH2 domains, most notably a C-terminal α-helix instead of a β-sheet [3] [31]. Structurally, the core SH2 domain consists of a central anti-parallel β-sheet flanked by two α-helices. This architecture forms two key sub-pockets:
- The pY (phosphate-binding) pocket: Binds the phosphorylated tyrosine residue and contains highly conserved amino acids.
- The pY+3 (specificity) pocket: Determines binding specificity by interacting with residues C-terminal to the phosphotyrosine [3].
The mutational landscape of the STAT SH2 domain is a hotspot in diseases like cancer and autosomal-dominant Hyper IgE Syndrome (AD-HIES). Mutations can have opposing effects: deactivating (loss-of-function, LOF) mutations in STAT3, for example, are linked to AD-HIES, while activating (gain-of-function, GOF) mutations in both STAT3 and STAT5B are drivers of leukemias such as T-cell large granular lymphocytic leukemia (T-LGLL) [3] [23]. The precise molecular effect of a mutation—whether it hyperactivates or deactivates the protein—depends on its location within the SH2 domain and how it alters the delicate structural balance governing dimerization and phospho-peptide binding [3].
A Framework for Resolving Discrepant Data
The process of reconciling conflicting data is a multi-stage investigative workflow. The following diagram and subsequent sections detail this logical pathway.
Re-evaluating the Basis ofIn SilicoPredictions
When conflict arises, the first step is a critical re-examination of the computational predictions. Different algorithms are trained on distinct data and principles, leading to varied outputs. For instance, a mutation might be predicted as stabilizing by a biophysics-based model but damaging by an evolution-based model. Key considerations include:
- Prediction Algorithm and Training Data: Understand the fundamental principles of the tools used. AlphaMissense, for example, is trained on evolutionary data and protein structure, while COORDinator predicts the energetic effect of substitutions on protein stability [23]. METL is a newer protein language model pretrained on biophysical simulation data to capture sequence-structure-energy relationships [54].
- Score Interpretation: Note that scores have different scales and interpretations. A CADD PHRED score above 20 suggests a potentially deleterious variant, while AlphaMissense scores below 0.5 are often classified as benign. However, these are probabilistic estimates, not functional measurements [23].
- Structural Context: Always map the mutation onto a high-quality protein structure. A mutation predicted to be benign might occur at a critical residue for stabilizing the hydrophobic core of the SH2 domain or for forming the dimer interface, which would only be apparent through structural analysis [3] [23].
Interrogating the Functional Assay Context
The biological readout from a functional assay is not infallible. Its design and execution must be scrutinized.
- Physiological Relevance: An in vitro kinase assay might confirm a STAT mutant can be phosphorylated, but it cannot reveal defects in nuclear translocation or DNA binding that would be apparent in a transcriptional reporter assay. The choice of cell line is also critical, as signaling pathways differ between cell types [55].
- Assay Dynamic Range and Sensitivity: The assay must be robust enough to distinguish between different levels of function. Multiplexed assays of variant effect (MAVEs) must demonstrate sufficient dynamic range to separate LOF and GOF variants from wild-type activity clearly. Inadequate range can lead to misclassification [55].
- Cellular Expression and Folding: A mutation causing protein misfiling and degradation may appear as LOF in a cellular assay, even if the mutant protein itself, when stabilized, is functional. Controls measuring protein expression and stability are essential to rule out these trivial explanations [20].
Investigating the Molecular Mechanism
Reconciliation often requires deeper mechanistic studies to understand how the mutation exerts its effect.
- Dimerization and Binding Studies: Use techniques like co-immunoprecipitation, surface plasmon resonance (SPR), or analytical ultracentrifugation to directly measure the impact of the mutation on STAT dimerization affinity or SH2 domain binding to phospho-peptides [3] [23].
- In Vivo Functional Profiling: Move beyond cell lines to model organisms. Introducing the STAT5B Y665F and Y665H mutations into the mouse genome provided the definitive evidence that Y665F is a GOF mutation in vivo, while Y665H is a LOF mutation, despite initial in vitro data suggesting both were GOF [23] [7]. This in vivo context captures the full complexity of tissue-specific signaling and physiological feedback.
Case Study: STAT5B Tyrosine 665 Mutations
The mutations at tyrosine 665 in STAT5B provide a quintessential example of conflicting data and its resolution.
Table 1: Conflicting Predictions and Outcomes for STAT5B Y665 Mutations
| Mutation | In Silico Predictions (Conflicting) | Initial In Vitro Data | Definitive In Vivo Functional Data | Resolved Classification |
|---|---|---|---|---|
| STAT5BY665F | CADD: 24.3 (Deleterious)AlphaMissense: 0.173 (Benign)REVEL: 0.535 (Pathogenic) [23] | Reported as gain-of-function in some cellular studies [23] | Accumulation of CD8+ effector/memory T cells; enhanced phospho-STAT5, DNA binding, and transcription; accelerated mammary development [23] [7] | GOF |
| STAT5BY665H | CADD: 23.1 (Deleterious)AlphaMissense: 0.383 (Benign)REVEL: 0.304 (Uncertain) [23] | Reported as gain-of-function in some cellular studies [23] | Diminished CD8+ effector/memory T cells; reduced STAT5 phosphorylation and transcription; failure in mammary gland development and lactation [23] [7] | LOF |
Resolution: The discrepancy was resolved by moving to more physiologically relevant models. In silico modeling with COORDinator suggested Y665F would stabilize the SH2 domain's interaction with the C-terminal tail, while Y665H would destabilize it [23]. This prediction was confirmed in vivo using knock-in mouse models, which revealed the starkly opposite phenotypes. This case underscores that some mutations require the full in vivo context—including appropriate cell types, developmental stages, and hormonal signals—for their true functional impact to be manifested [7].
Experimental Protocols for Key Assays
To generate reliable data, robust and well-controlled experimental protocols are essential. Below are detailed methodologies for two key approaches used in characterizing STAT SH2 domain mutations.
Deep Mutational Scanning (DMS) Protocol
DMS is a high-throughput method for characterizing thousands of protein variants simultaneously [55] [20].
- Library Construction: Create a saturation mutagenesis library covering the entire STAT SH2 domain. This can be achieved via oligonucleotide-based synthesis or error-prone PCR, generating a plasmid library where each molecule contains a single amino acid substitution.
- Functional Selection in a Model System:
- Platform: Use a yeast-based viability assay [20] or a mammalian cell culture system with a fluorescent or selectable reporter gene under the control of a STAT-responsive promoter.
- Selection: Introduce the variant library into the chosen host system and apply selective pressure. For example, in a yeast growth assay, express a toxic tyrosine kinase and rely on functional STAT phosphatase activity (e.g., SHP2) to rescue growth [20]. For STATs directly, use cytokine stimulation to activate signaling, linking STAT function to cell survival or fluorescence.
- Sequencing and Enrichment Scoring: Isolate genomic DNA from the population before and after selection. Amplify the STAT SH2 domain coding region by PCR and subject it to high-throughput sequencing. Calculate an enrichment score for each variant by comparing its frequency before and after selection. Variants with scores >1 are enriched (potential GOF), while those with scores <1 are depleted (potential LOF) [20].
- Validation: Purify a subset of variant proteins (both predicted GOF and LOF) and measure their activity using low-throughput, gold-standard assays like in vitro phosphorylation dimerization assays or catalytic efficiency measurements to validate the DMS scores [20].
In Vivo Mouse Model Characterization Protocol
This protocol is for validating the physiological impact of a STAT SH2 mutation identified in prior screens [23] [7].
- Generation of Knock-in Mice:
- Method: Use CRISPR/Cas9 and a single-strand oligonucleotide donor template or base editing (e.g., ABE system) to introduce the specific point mutation (e.g., Y665F or Y665H) into the endogenous Stat5b locus of C57BL/6 mouse embryos [7].
- Controls: Always use wild-type littermates as controls to eliminate effects of genetic background.
- Immunophenotyping:
- Flow Cytometry: Isolate immune cells from spleen, lymph nodes, and blood. Stain cells with fluorescently labeled antibodies against T-cell markers (e.g., CD4, CD8, CD44, CD62L) and analyze by flow cytometry to assess changes in T-cell subsets, effector, and memory populations [23].
- Assessment of Molecular and Functional Phenotypes:
- Phospho-STAT5 Analysis: Stimulate splenocytes with IL-2 or other relevant cytokines. Fix and permeabilize cells, then stain intracellularly with an antibody against phosphorylated STAT5 (pY694) for analysis by flow cytometry to measure signaling capacity [23].
- Mammary Gland Development: For STAT5B, analyze mammary tissue from pregnant mice. Dissect mammary glands, perform whole-mount staining, and examine histologically for alveolar bud development and differentiation. Assess lactation capability by monitoring pup growth [7].
- Transcriptomic Analysis (RNA-seq): Extract total RNA from target tissues (e.g., mammary gland, T-cells). Prepare libraries and sequence. Analyze differential gene expression of known STAT5 target genes (e.g., Cish) and perform gene set enrichment analysis [7].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Resources for STAT SH2 Domain Research
| Reagent / Resource | Function and Application | Key Considerations |
|---|---|---|
| Saturation Mutagenesis Library | A plasmid pool containing all possible single amino acid substitutions within the STAT SH2 domain, used for DMS. | Coverage should be as complete as possible. Quality control via sequencing is critical to ensure even representation [55] [20]. |
| CRISPR/Cas9 with Base Editor | A genome editing system for introducing precise point mutations into the mouse genome to create knock-in models. | Allows for modeling of specific human mutations without introducing selectable markers or large sequence changes [7]. |
| Phospho-Specific STAT5 Antibody | An antibody that recognizes STAT5 phosphorylated at tyrosine 694 (for STAT5A/B), used in flow cytometry and Western blotting. | Essential for directly measuring the activation status of STAT5 in cells upon cytokine stimulation [23]. |
| Recombinant Cytokines (e.g., IL-2) | Ligands that activate the JAK-STAT pathway, used to stimulate cells in functional assays. | Must be of high purity and activity. Titration is required to determine optimal stimulating concentrations. |
| Multiplexed Assay of Variant Effect (MAVE) | A comprehensive framework for generating, analyzing, and clinically interpreting high-throughput functional data. | Following standardized guidelines ensures data quality, reproducibility, and clinical utility [55]. |
| Biophysics-Aware Protein Language Models (e.g., METL) | Deep learning models pretrained on molecular simulation data to predict variant effects from sequence. | Particularly powerful for generalizing from small training sets and for position extrapolation tasks in protein engineering [54]. |
Visualization of Key Signaling and Experimental Pathways
Understanding the pathway context and experimental workflow is crucial. The diagram below illustrates the canonical JAK-STAT signaling pathway and the points where SH2 domain mutations exert their influence.
Addressing Protein Flexibility and Dynamics in SH2 Domain Drug Discovery
The Src Homology 2 (SH2) domain is a critical protein-protein interaction module that specifically recognizes sequences containing a phosphorylated tyrosine, serving as a fundamental component in eukaryotic cell signaling [56]. These approximately 100-amino-acid domains function as crucial "readers" in phosphotyrosine (pTyr) signaling networks, inducing proximity between protein tyrosine kinases (PTKs) and their substrates to propagate cellular signals [31]. Despite considerable research efforts, the inherent flexibility and structural dynamics of SH2 domains present formidable challenges for drug discovery initiatives. This adaptability is particularly pronounced in STAT-type SH2 domains, which exhibit substantial conformational flexibility even on sub-microsecond timescales, with the accessible volume of their phosphate-binding (pY) pockets varying dramatically [3]. This review comprehensively compares experimental methodologies and strategic approaches designed to address these challenges, with a specific focus on how activating versus inactivating mutations in STAT SH2 domains inform therapeutic targeting strategies.
Structural Dynamics of SH2 Domains: Implications for Drug Design
Fundamental SH2 Architecture and Plasticity
All SH2 domains share a conserved structural fold featuring a central anti-parallel β-sheet (strands βB-βD) flanked by two α-helices (αA and αB), forming an αβββα motif [3]. This core structure partitions the domain into two functionally critical subpockets: the pY pocket that binds the phosphotyrosine moiety, and the pY+3 pocket that determines ligand specificity [3]. Despite this conserved architecture, SH2 domains exhibit significant structural diversity, particularly in their C-terminal regions. STAT-type SH2 domains are distinguished from Src-type domains by the presence of a C-terminal α-helix (αB') instead of β-sheets, an adaptation that facilitates the dimerization required for STAT-mediated transcriptional regulation [31]. This structural divergence is evolutionarily significant, reflecting ancestral functions that predate animal multicellularity [31].
The flexibility of loop regions within SH2 domains substantially contributes to their dynamic behavior. The length and conformation of the CD-loop, for instance, varies considerably between different protein families, with enzymatic proteins typically possessing longer loops compared to non-enzymatic proteins like STATs [31]. These structural variations directly influence phosphopeptide binding accessibility and specificity. Particularly relevant for drug discovery is the observation that crystal structures do not necessarily preserve targetable pockets in accessible states, underscoring the critical importance of accounting for protein dynamics in structure-based drug design [3].
Comparative Structural Features of STAT-type versus Src-type SH2 Domains
Table 1: Structural and Functional Comparison of SH2 Domain Types
| Feature | STAT-type SH2 Domains | Src-type SH2 Domains |
|---|---|---|
| C-terminal Structure | α-helix (αB') | β-sheets (βE and βF) |
| Dimerization Role | Critical for STAT dimerization and nuclear translocation | Less central to dimerization |
| Loop Characteristics | Generally shorter CD loops | Often longer, more variable loops |
| Evolutionary Origin | More ancient, predates animal multicellularity | More recent adaptation |
| Domain Cooperation | SH2 domain essential for activation and DNA binding | Often involved in autoinhibitory functions |
| Drug Targeting Challenges | High flexibility in pY pocket | More stable, defined pockets |
Experimental Approaches for Probing SH2 Dynamics and Binding
Quantitative Affinity Profiling Using Display Technologies
Recent methodological advances have enabled researchers to transition from qualitative classification to quantitative affinity modeling for SH2 domain interactions. An integrated experimental-computational framework combining bacterial peptide display, enzymatic phosphorylation, affinity selection, and next-generation sequencing (NGS) has proven particularly powerful for profiling SH2 domain binding across highly diverse ligand libraries [57]. This approach employs ProBound, a statistical learning method that generates quantitative sequence-to-affinity models capable of predicting binding free energy across the full theoretical ligand sequence space [57].
The experimental workflow involves several critical steps: First, random peptide libraries are displayed on bacterial surfaces and phosphorylated enzymatically. Subsequent affinity selection with purified SH2 domains enriches for high-affinity binders across multiple selection rounds. NGS of the selected pools provides deep sequencing data suitable for training additive models that accurately predict binding free energy. For the resulting models, relative binding affinity is defined as ∆∆G, with the optimal sequence set to one and all other sequences taking values between zero and one [57]. This methodology represents a significant advancement over traditional position-specific scoring matrices (PSSMs) by providing biophysically interpretable affinity predictions rather than simple binary classifications.
Structural Biology and Biophysical Methods
X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy have provided fundamental insights into SH2 domain structure and dynamics. To date, the structures of approximately 70 SH2 domains have been experimentally determined with varying resolution [31]. These structural studies reveal that while SH2 domains maintain a conserved fold, they display considerable diversity in loop conformations and binding pocket architectures. Crystallographic analyses have been particularly valuable for identifying unique features of STAT-type SH2 domains, including their distinctive C-terminal αB' helix and the organization of their hydrophobic systems that stabilize β-sheet conformation [3].
NMR spectroscopy offers complementary advantages for characterizing SH2 domain flexibility, particularly in mapping dynamic regions and quantifying conformational exchange processes. This approach is exceptionally valuable for detecting structural fluctuations that occur on microsecond to millisecond timescales—precisely the motions relevant for molecular recognition and drug binding. For STAT SH2 domains, NMR has revealed substantial backbone flexibility in the pY and pY+3 pockets, explaining the challenges in targeting these regions with small molecules [3].
STAT SH2 Domain Mutations: A Comparative Analysis of Activation Mechanisms
Functional Impact of SH2 Domain Mutations in STAT Proteins
The SH2 domain serves as a critical functional hotspot in STAT proteins, mediating both receptor recruitment through phosphopeptide binding and STAT dimerization through reciprocal SH2-pTyr interactions [3]. Sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in the STAT protein landscape, with mutations producing diverse and sometimes opposing functional consequences [3]. These clinical observations provide natural experiments that reveal structure-function relationships and validate potential therapeutic targets.
Loss-of-function (LOF) mutations frequently disrupt conserved residues essential for phosphotyrosine binding or SH2 domain structural integrity. In STAT3, mutations such as K591E, R609G, and S611N—located in critical phosphate-binding regions—are associated with autosomal-dominant Hyper IgE Syndrome (AD-HIES) due to impaired Th17 T-cell responses [3]. Similarly, the STAT5B Y665H mutation, identified in T-cell leukemias, acts as a LOF variant that impairs enhancer establishment and alveolar differentiation in mammary gland development, resulting in lactation failure in mouse models [7].
Conversely, gain-of-function (GOF) mutations typically enhance STAT dimerization stability or prolong phosphorylation status. The STAT5B Y665F mutation, also found in T-cell leukemias, functions as a GOF mutation that accelerates mammary development during pregnancy and elevates enhancer formation [7]. In STAT2, the Y631F mutation confers sustained signaling and induction of interferon-stimulated genes by resisting dephosphorylation by nuclear tyrosine phosphatase TcPTP, ultimately promoting IFN-α-induced apoptosis [58].
Table 2: Functional Classification of Disease-Associated STAT SH2 Domain Mutations
| STAT Protein | Mutation | Location/Type | Functional Effect | Associated Pathology |
|---|---|---|---|---|
| STAT3 | K591E/M | αA2 helix, pY pocket | Loss-of-function | AD-HIES |
| STAT3 | R609G | βB5 strand, pY pocket | Loss-of-function | AD-HIES |
| STAT3 | S611N | βB7 strand, pY pocket | Loss-of-function | AD-HIES |
| STAT3 | S614R | BC loop, pY pocket | Gain-of-function | T-LGLL, NK-LGLL |
| STAT5B | Y665H | SH2 domain | Loss-of-function | Lactation failure, T-cell leukemia |
| STAT5B | Y665F | SH2 domain | Gain-of-function | Enhanced mammary development, T-cell leukemia |
| STAT2 | Y631F | PYTK motif | Gain-of-function | Prolonged interferon signaling |
Structural Mechanisms of Mutation Effects
The structural basis for how mutations effect functional changes illuminates key aspects of SH2 domain dynamics. Loss-of-function mutations typically disrupt essential binding interactions or destabilize the SH2 fold. For example, mutations affecting the invariant arginine at position βB5 (part of the FLVR motif found in most SH2 domains) directly impair phosphotyrosine binding through loss of critical salt bridge formation [31]. Other LOF mutations may destabilize the hydrophobic core that maintains the integrity of the β-sheet and overall SH2 domain structure [3].
Gain-of-function mutations operate through more diverse mechanisms. Some GOF mutations, like STAT3 S614R, may enhance binding affinity for phosphopeptide ligands or stabilize active dimer conformations [3]. Others, including STAT2 Y631F, prolong signaling by impairing dephosphorylation kinetics without affecting initial activation [58]. This mutation in the conserved PYTK motif of STAT2 confers resistance to nuclear tyrosine phosphatase TcPTP, leading to sustained STAT1 and STAT2 tyrosine phosphorylation, prolonged nuclear retention, and enhanced apoptotic responses to IFN-α stimulation [58].
Emerging Targeting Strategies for Flexible SH2 Domains
Allosteric and Protein-Protein Interaction Inhibitors
Traditional drug discovery efforts for SH2 domains have focused primarily on developing competitive inhibitors that target the phosphotyrosine-binding pocket. However, the highly conserved and polar nature of this site, combined with its conformational flexibility, has presented significant challenges for drug development [3]. Consequently, researchers are increasingly exploring allosteric inhibition strategies that target alternative sites on the SH2 domain. The evolutionary active region (EAR) at the C-terminal region of the pY+3 pocket represents one promising allosteric target, particularly as it contains the distinctive αB' helix in STAT-type SH2 domains [3]. Similarly, the hydrophobic system that stabilizes the β-sheet conformation may offer opportunities for allosteric modulation of SH2 domain structure and function.
Emerging research also indicates that many SH2 domains interact with membrane lipids—nearly 75% according to recent studies—with cationic regions near the pY-binding pocket serving as lipid-binding sites [31]. These interactions modulate cellular signaling of SH2-containing proteins, as demonstrated by PIP3 binding to the SYK SH2 domain, which is required for noncatalytic activation of STAT3/5 [31]. Disease-causing mutations frequently localize within these lipid-binding pockets, suggesting they represent functionally critical regions amenable to therapeutic targeting [31]. The successful development of nonlipidic inhibitors of Syk kinase that target its lipid-protein interaction demonstrates the feasibility of this approach [31].
Exploiting Liquid-Liquid Phase Separation in Signaling Modulation
Recent studies have revealed that proteins with SH2 domains contribute to the formation of intracellular condensates through liquid-liquid phase separation (LLPS), a process driven by multivalent interactions involving modules like SH2 and SH3 domains [31]. In T-cells, interactions among GRB2, Gads, and the LAT receptor promote LLPS formation that enhances T-cell receptor signaling [31]. Similarly, in kidney podocyte cells, LLPS increases the membrane dwell time of N-WASP and Arp2/3 complexes, promoting actin polymerization [31]. These findings suggest that modulators of phase separation behavior may offer novel therapeutic opportunities for targeting SH2 domain-mediated signaling pathways, potentially by altering the spatiotemporal organization of signaling complexes rather than directly inhibiting binding interactions.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Key Experimental Resources for SH2 Domain Dynamics Research
| Resource/Methodology | Primary Application | Key Advantages | Technical Considerations |
|---|---|---|---|
| Bacterial Peptide Display | Affinity profiling of SH2 binding | Compatible with highly diverse random libraries (>10^7 sequences) | Requires enzymatic phosphorylation of displayed peptides |
| ProBound Computational Platform | Sequence-to-affinity modeling | Quantitative ∆∆G predictions across full sequence space | Requires multi-round selection NGS data |
| STAT SH2 Domain Mutants (Y665F/H) | Functional studies of STAT activation | Well-characterized GOF/LOF variants available | Context-dependent effects require physiological models |
| SH2 Domain Lipid Binding Assays | Profiling lipid-protein interactions | Reveals non-canonical SH2 functions | Membrane mimic systems required for relevance |
| Phase Separation Assays | Study LLPS in signaling | Examines higher-order signaling organization | Requires careful control of component concentrations |
| NMR Dynamics Measurements | Characterizing flexibility | Resolves atomic-level motions on multiple timescales | Technical expertise and specialized equipment needed |
The dynamic nature of SH2 domains presents both challenges and opportunities for drug discovery. The comparative analysis of activating versus inactivating STAT SH2 domain mutations reveals the exquisite sensitivity of these domains to structural perturbations and provides valuable insights for targeted therapeutic development. Emerging methodologies—from quantitative affinity profiling using display technologies to the investigation of non-canonical SH2 functions in lipid binding and phase separation—are expanding the toolkit available for probing SH2 domain flexibility and function. As our understanding of SH2 domain dynamics continues to evolve, so too will our ability to develop innovative strategies for targeting these critical signaling modules in human disease.
Signal Transducer and Activator of Transcription (STAT) 5A and STAT5B are highly homologous transcription factors that have long been considered functionally redundant. However, emerging evidence reveals critical distinctions in their roles in hematopoiesis, immune regulation, and cancer pathogenesis. This comparison guide objectively analyzes the differential functions of STAT5A and STAT5B through the lens of recent structural, genetic, and functional studies. We provide a comprehensive framework for researchers to dissect their non-redundant roles, with particular focus on SH2 domain mutations that demonstrate opposing functional consequences. The experimental data and methodologies presented herein offer valuable insights for drug development professionals targeting the JAK-STAT pathway with greater precision.
The STAT5 proteins, STAT5A and STAT5B, are paralogs encoded by separate genes on chromosome 17q11.2 that share over 90% amino acid sequence identity [59] [60]. For decades, their extreme homology led to the presumption of functional redundancy, with early murine studies suggesting largely overlapping roles in cytokine responses [61]. However, clinical observations from human deficiencies and cancer genomics have fundamentally challenged this paradigm, revealing that STAT5A and STAT5B fulfill both complementary and unique biological functions [59] [62].
The context of SH2 domain mutations provides a particularly illuminating model for understanding how subtle structural differences translate to significant functional divergence. The SH2 domain is essential for STAT activation, mediating phosphotyrosine-dependent dimerization and nuclear translocation [3]. Recent investigations into leukemia-associated mutations within this domain have revealed that STAT5A and STAT5B not only exhibit different mutation frequencies in human disease but may also respond differently to equivalent mutations [23] [30]. This guide systematically compares STAT5A and STAT5B through integrated analysis of their structural biology, expression patterns, physiological functions, and pathological roles, providing researchers with experimental frameworks to overcome the challenge of functional redundancy.
Structural and Molecular Distinctions
Protein Architecture and Domain Specificity
STAT5A and STAT5B proteins contain six conserved domains: N-terminal domain, coiled-coil domain, DNA-binding domain, linker region, Src homology 2 (SH2) domain, and transactivation domain [60]. Despite their high overall similarity, critical differences localize to specific regions that dictate their functional specialization.
Table 1: Key Structural Differences Between STAT5A and STAT5B
| Structural Feature | STAT5A | STAT5B | Functional Implication |
|---|---|---|---|
| C-terminal length | 12 additional amino acids | Shorter C-terminus | Differential protein-protein interactions [59] |
| Phosphotyrosyl tail | Shortened by 5 residues | Standard length | Alters phosphopeptide binding properties [59] |
| DNA-binding domain | Unique 5 amino acids | Distinct 5 amino acids | Different DNA binding affinities and specificity [59] [60] |
| Tyrosine phosphorylation site | Y694 | Y699 | Conservation of activation mechanism [59] |
| Additional phosphorylation sites | S127/S128, T682/T683 | S193, Y725, Y740, Y743 | Different regulatory inputs [59] |
The C-terminal variations are particularly significant for transcriptional activity and protein interactions. STAT5A contains 12 additional amino acids at its C-terminus compared to STAT5B, while STAT5B has a complete phosphotyrosyl tail segment that STAT5A lacks [59] [60]. These differences, though subtle, create distinct interaction surfaces that enable unique protein partnerships and potentially different transcriptional outcomes.
DNA Binding and Transcriptional Specificity
The DNA-binding domains of STAT5A and STAT5B differ by just five amino acids, yet these differences significantly impact their DNA binding preferences [59]. STAT5B homodimers demonstrate more efficient DNA binding compared to STAT5A homodimers and can recognize a broader range of GAS (gamma-interferon activation site) motifs, specifically TTCT/CnnnGAA sequences with 4-base pair spacers [63]. STAT5A, in contrast, preferentially forms tetramers when two weak STAT5 affinity sites are in close proximity, a property not prominently observed with STAT5B [63].
These biochemical differences translate to distinct genomic binding patterns. Chromatin immunoprecipitation sequencing (ChIP-seq) in human CD4+ T cells has revealed both shared and unique target genes: STAT5A specifically binds genes involved in neural development and function (NDRG1, DNAJC6, SSH2), while STAT5B uniquely regulates genes critical for T cell function (DOCK8, SNX9, FOXP3, IL2RA) [64]. Both proteins redundantly regulate genes involved in fundamental cellular processes like proliferation and apoptosis, exemplified by their shared regulation of SGK1 [64].
Figure 1: Canonical JAK-STAT5 Signaling Pathway. STAT5A and STAT5B undergo parallel activation processes but form both homodimers and heterodimers with potentially different DNA binding properties and transcriptional outcomes.
Expression Patterns and Physiological Functions
Tissue-Specific Expression and Hematopoietic Roles
STAT5B demonstrates higher expression than STAT5A across most hematopoietic cell types, including erythrocytes, megakaryocytes, natural killer (NK) cells, CD4+ and CD8+ T cells, and B cells [60]. STAT5A expression predominates only in CD34+ hematopoietic stem cells [60]. This differential expression pattern provides the first clue to their non-redundant functions, with STAT5B playing a more prominent role in differentiated immune cells.
In the hematopoietic system, both STAT5A and STAT5B are activated by cytokines including IL-2, IL-3, IL-5, IL-7, IL-9, IL-15, and IL-21 [60]. However, they exhibit distinct roles within specific lineages:
B cell development: STAT5 activation regulates B cell lymphopoiesis via IL-7R signaling, promoting cell survival and immunoglobulin gene rearrangement in pro-B cells [60]. Complete Stat5a/b null mice show a developmental block between pro- and pre-B cell stages [60].
T cell biology: STAT5B plays a more critical role in regulatory T (Treg) cell maintenance and function [62]. Human STAT5B deficiency results in reduced FOXP3 expression and impaired Treg function, which cannot be compensated by STAT5A [62].
NK cells: Both proteins contribute to NK cell development, but STAT5B appears dominant, with STAT5B-deficient patients showing reduced NK cell numbers [60] [62].
Non-Hematopoietic Functions and Mouse Models
Genetic studies in mice have revealed distinctive non-redundant functions outside the immune system. STAT5A is essential for prolactin-dependent mammary gland development and lactation, while STAT5B plays a more critical role in growth hormone signaling and body growth regulation [59] [61]. The sexual dimorphism observed in Stat5b-deficient mice (affecting males more significantly) contrasts with human STAT5B deficiency, which impacts growth in both sexes, highlighting important species-specific differences [64].
Table 2: Functional Specialization of STAT5A and STAT5B in Physiology and Disease
| Biological Context | STAT5A Role | STAT5B Role | Experimental Evidence |
|---|---|---|---|
| Mammary gland development | Essential: mediates prolactin signaling, alveolar differentiation [59] | Supporting role | Stat5a-/- mice: failed lactation; Stat5b-/- mice: less severe defects [59] |
| Body growth regulation | Moderate effect | Critical: mediates growth hormone signaling [59] | Stat5b-/- mice: growth impairment; Humans: STAT5B mutations cause growth failure [59] [62] |
| Treg cell function | Secondary role | Primary role: maintains FOXP3 expression and suppressive function [62] | STAT5B-deficient patients: reduced Treg numbers/function despite normal STAT5A [62] |
| Leukemogenesis | Less frequently mutated | Hotspot for gain-of-function mutations (e.g., N642H, Y665F) [23] | T-LGLL and T-PLL harbor STAT5B not STAT5A mutations [23] [30] |
| B cell development | Redundant with STAT5B | Redundant with STAT5A | Stat5a/b-/- mice: block at pro-B cell stage [60] |
SH2 Domain Mutations: A Paradigm for Functional Dissection
Mutation Hotspots and Pathogenic Mechanisms
The SH2 domain represents a critical mutational hotspot in STAT5B, with the N642H and Y665F substitutions being most frequently identified in T-cell leukemias including T-cell large granular lymphocytic leukemia (T-LGLL) and T-cell prolymphocytic leukemia (T-PLL) [23] [3]. Notably, equivalent mutations are far less common in STAT5A, suggesting intrinsic structural or functional constraints [59].
Recent structural analyses reveal that tyrosine 665 (Y665) occupies a critical position at the STAT5B homodimerization interface [23] [30]. In silico modeling predicts that Y665F and Y665H substitutions have opposing effects on protein stability and function: Y665F stabilizes the SH2 domain through enhanced intramolecular aromatic stacking with F711, while Y665H introduces an imidazole group that destabilizes C-terminal tail binding [23]. These predictions are confirmed by experimental data showing that STAT5BY665F exhibits gain-of-function (GOF) properties with enhanced phosphorylation, DNA binding, and transcriptional activity, whereas STAT5BY665H behaves as a loss-of-function (LOF) mutant resembling STAT5B null alleles [23] [30].
Functional Consequences of SH2 Domain Mutations
The physiological impacts of these mutations have been elucidated through knock-in mouse models:
STAT5BY665F (GOF): Leads to accumulation of CD8+ effector and memory T cells, expanded CD4+ regulatory T cells, and altered CD8+/CD4+ ratios, but does not directly initiate malignant transformation [23] [30].
STAT5BY665H (LOF): Results in diminished CD8+ effector and memory T cells, reduced CD4+ regulatory T cells, and impaired immune function [23].
In mammary gland biology, these mutations produce equally divergent phenotypes: STAT5BY665H mice fail to develop functional mammary tissue and experience lactation failure, while STAT5BY665F mice exhibit accelerated mammary development during pregnancy [7]. Transcriptomic and epigenomic analyses reveal that the Y665H mutation impairs enhancer establishment and alveolar differentiation, while the Y665F mutation enhances enhancer formation [7].
Figure 2: Opposing Functional Impacts of STAT5B SH2 Domain Mutations. Single amino acid substitutions at tyrosine 665 produce structurally and functionally divergent proteins with distinct physiological consequences.
Experimental Approaches for Functional Dissection
Methodologies for Differentiating STAT5A and STAT5B Roles
Gene Targeting and Knockdown Approaches:
- siRNA-mediated knockdown: Selective silencing of STAT5A or STAT5B in human primary T cells followed by transcriptomic analysis reveals differentially regulated genes. STAT5B knockdown significantly reduces FOXP3 and IL-2R expression, while STAT5A knockdown preferentially affects Bcl-X expression [62].
- CRISPR/Cas9 mutagenesis: Introduction of patient-derived point mutations (e.g., Y665F, Y665H) into the mouse genome enables physiological assessment of mutation impacts in specific tissue contexts [23] [7].
- Conditional knockout mice: Cell-type specific deletion of Stat5a versus Stat5b elucidates lineage-specific requirements that may be masked in complete knockouts [59].
Genome-Wide Binding and Expression Profiling:
- Chromatin Immunoprecipitation Sequencing (ChIP-seq): Using isoform-specific antibodies to map STAT5A versus STAT5B binding sites genome-wide in human CD4+ T cells [64]. Critical parameters include cross-linking conditions (1% formaldehyde for 10 min), sonication (200-500 bp fragments), and specific antibodies (anti-STAT5A sc-1081, anti-STAT5B 135300) [64].
- RNA Sequencing: Transcriptomic profiling following isoform-specific perturbation identifies unique transcriptional networks regulated by each paralog [7] [64].
- Epigenomic Analysis: ATAC-seq or histone modification profiling reveals how STAT5A versus STAT5B binding influences chromatin accessibility and enhancer establishment [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for STAT5A/STAT5B Investigation
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Isoform-Specific Antibodies | STAT5A (sc-1081), STAT5B (135300) | Immunoblotting, immunofluorescence, ChIP-seq validation | Verify specificity using knockout/knockdown controls [64] |
| Cell Line Models | Ba/F3, HEK293T, primary T cells | Functional assays, transformation studies | Primary cells best reflect physiological signaling [23] |
| Cytokine Stimulation | IL-2, IL-3, IL-7, GM-CSF, GH, Prolactin | Pathway activation, phosphorylation studies | Concentration and timing critical for specific activation [60] [64] |
| Genomic Tools | ChIP-seq, siRNA, CRISPR/Cas9 | Binding site mapping, functional dissection | Multiple sgRNAs recommended for genetic perturbation [23] [64] |
| Mouse Models | Stat5a-/-, Stat5b-/-, conditional alleles, knock-in mutations | Physiological context, tissue-specific functions | Consider genetic background effects [59] [23] |
The experimental evidence unequivocally demonstrates that STAT5A and STAT5B have evolved distinct biological functions despite their extensive homology. Their non-redundancy manifests at multiple levels: expression patterns, DNA binding preferences, transcriptional programs, and pathological mutations. The SH2 domain mutation paradigm illustrates how single amino acid changes can dictate opposing functional outcomes, providing a powerful experimental framework for dissecting structure-function relationships.
For drug development professionals, these insights carry important implications. First, therapeutic strategies targeting STAT5 must account for isoform-specific functions, particularly in immune regulation where STAT5B dominates Treg biology. Second, the mutational landscape suggests STAT5B represents a more promising direct therapeutic target in hematologic malignancies. Finally, the structural characterization of SH2 domain mutations reveals potential allosteric mechanisms that could be exploited for selective inhibition.
Overcoming functional redundancy between STAT5A and STAT5B requires integrated approaches combining structural biology, genomic techniques, and physiological models. The methodologies and reagents outlined herein provide a roadmap for researchers to precisely dissect their unique contributions in health and disease, ultimately enabling more targeted therapeutic interventions in cancer and immune disorders.
Src homology 2 (SH2) domains are protein interaction modules approximately 100 amino acids in length that specifically recognize and bind to phosphorylated tyrosine (pTyr) residues on target proteins [31] [65]. These domains form a crucial component of the phosphotyrosine signaling network, functioning as primary "readers" of tyrosine phosphorylation events immediately downstream of protein tyrosine kinases [66]. The human genome encodes approximately 110 SH2 domain-containing proteins that participate in diverse cellular processes, including development, homeostasis, immune responses, and transcriptional regulation [31] [66]. SH2 domains achieve signaling specificity by recognizing both the phosphotyrosine residue and the sequence context of surrounding amino acids in their ligand proteins [66] [67]. This sophisticated recognition system allows SH2 domains to direct the formation of specific protein complexes in response to phosphorylation events, thereby ensuring fidelity in signal transduction pathways. The critical role of SH2 domains in coordinating cellular communication, coupled with their involvement in numerous diseases when dysregulated, has positioned them as attractive targets for therapeutic intervention.
Structural Basis of SH2 Domain Function and Specificity
Conserved Architecture and Ligand Recognition
All SH2 domains share a conserved structural fold despite significant sequence variation among family members [31] [65]. The core structure consists of a central anti-parallel β-sheet flanked by two α-helices, forming an αββα sandwich configuration [3] [68]. This structural arrangement creates two primary binding clefts separated by the central β-sheet: a phosphotyrosine (pY) pocket that coordinates the phosphate moiety and a specificity (pY+3) pocket that engages residues C-terminal to the phosphotyrosine [3] [65].
The pY pocket contains highly conserved residues, notably an invariant arginine at position βB5 (from the FLVR motif) that forms critical bidentate hydrogen bonds with the phosphate group of phosphotyrosine [31] [68]. This interaction provides approximately half of the total binding energy for SH2 domain-phosphopeptide interactions [65]. The specificity pocket, formed by the opposite face of the β-sheet along with residues from the αB helix and various loops, determines ligand selectivity by accommodating specific amino acids at positions C-terminal to the phosphotyrosine [3] [65]. The structural diversity in the EF and BG loops that regulate access to these specificity pockets contributes significantly to the distinct recognition properties of different SH2 domains [31] [65].
STAT-Type Versus Src-Type SH2 Domains
SH2 domains are broadly classified into two major subgroups: STAT-type and Src-type, which differ in their C-terminal structural elements [31] [3]. STAT-type SH2 domains lack the βE and βF strands present in Src-type domains and feature a split αB helix, which is believed to be an adaptation that facilitates STAT dimerization—a critical step in STAT-mediated transcriptional regulation [31] [3]. This structural distinction reflects the specialized function of STAT SH2 domains in mediating both receptor recruitment and dimerization of phosphorylated STAT monomers [3] [69].
The unique architecture of STAT SH2 domains creates particular challenges for drug discovery. These domains exhibit significant flexibility even on sub-microsecond timescales, with the accessible volume of the pY pocket varying dramatically [3]. Additionally, crystal structures do not always preserve targetable pockets in accessible states, emphasizing the importance of accounting for protein dynamics in STAT-directed drug discovery efforts [3].
Disease-Associated Mutations in STAT SH2 Domains
Spectrum and Impact of STAT SH2 Domain Mutations
Sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in STAT proteins, particularly STAT3 and STAT5 [3]. These mutations can have either activating or inactivating effects on STAT function, sometimes with mutations at the same residue producing opposite phenotypic consequences depending on the specific amino acid substitution [3]. This genetic volatility underscores the delicate evolutionary balance maintained in wild-type STAT structural motifs to ensure precise levels of cellular activity.
The table below summarizes representative disease-associated mutations in the STAT3 SH2 domain and their pathological consequences:
Table 1: Disease-Associated Mutations in the STAT3 SH2 Domain
| Mutation | Location | Pathology | Mutation Type | Functional Impact |
|---|---|---|---|---|
| K591E/M | αA2 helix, pY pocket | AD-HIES | Germline | Loss-of-function [3] |
| R609G | βB5 strand, pY pocket | AD-HIES | Germline | Loss-of-function [3] |
| S611N | βB7 strand, pY pocket | AD-HIES | Germline | Loss-of-function [3] |
| S614R | BC loop, pY pocket | T-LGLL, NK-LGLL, ALCL | Somatic | Gain-of-function [3] |
| E616K | BC loop, pY pocket | NKTL | Somatic | Gain-of-function [3] |
| G617R | BC loop, pY pocket | AD-HIES | Germline | Loss-of-function [3] |
| D661Y | βD4 strand | ALK-ALCL | Somatic | Gain-of-function [3] |
Functional Consequences of STAT SH2 Domain Mutations
Mutations in the STAT SH2 domain can disrupt normal function through multiple mechanisms. Loss-of-function mutations, typically associated with autosomal-dominant hyper IgE syndrome (AD-HIES), often impair phosphopeptide binding or SH2 domain stability, leading to diminished STAT3-mediated Th17 T-cell responses and consequent immunological deficiencies [3]. In contrast, gain-of-function mutations, frequently identified in various leukemias and lymphomas, enhance STAT dimerization or DNA binding affinity, resulting in constitutive transcriptional activation of proliferation and survival genes such as BCL-XL, MCL-1, and C-MYC [3].
The bidirectional nature of these mutations highlights the critical importance of precise SH2 domain function in maintaining cellular homeostasis. Understanding the molecular mechanisms by which different mutations alter STAT function provides valuable insights for developing targeted therapeutic strategies that can either restore or inhibit SH2 domain function depending on the pathological context.
Emerging Therapeutic Strategies Targeting SH2 Domains
Conventional and Novel Targeting Approaches
Traditional approaches to targeting SH2 domains have focused on developing phosphotyrosine mimetics that compete with native phosphopeptides for binding to the pY pocket [31]. However, these strategies have faced challenges due to the charged nature of phosphate-mimicking groups, which often result in poor cellular permeability and bioavailability [31]. More recent strategies have expanded to target alternative sites and mechanisms, including:
- Specificity pocket inhibitors: Compounds that target the more variable specificity pocket to achieve greater selectivity [31] [3]
- Dimerization disruptors: Molecules that interfere with STAT dimerization by targeting the SH2 domain interface [3]
- Allosteric inhibitors: Compounds that bind outside the canonical pY pocket to modulate SH2 domain function [31]
- Lipid-binding pocket targeting: Emerging approach focusing on cationic lipid-binding regions near the pY pocket [31]
The latter approach is particularly promising based on recent research showing that nearly 75% of SH2 domains interact with lipid molecules, primarily phosphatidylinositol-4,5-bisphosphate (PIP2) or phosphatidylinositol-3,4,5-trisphosphate (PIP3), with disease-causing mutations often localized within these lipid-binding pockets [31]. The successful development of nonlipidic inhibitors of Syk kinase that target its lipid-protein interaction interface demonstrates the potential of this strategy [31].
SH2 Domain Targeting in Neurodegenerative Diseases
Emerging research has revealed the therapeutic potential of targeting SH2 domain-containing proteins in neurodegenerative diseases, particularly through modulation of SHP2 (Src homology 2-containing protein tyrosine phosphatase 2) [70]. SHP2 contains two SH2 domains that regulate its phosphatase activity and mediate protein-protein interactions [70] [68]. In its basal state, SHP2 exists in an autoinhibited conformation where the N-SH2 domain blocks the catalytic cleft; binding of phosphopeptides to the SH2 domains releases this inhibition, activating the phosphatase [70].
SHP2 has demonstrated bidirectional effects in neurodegenerative contexts, functioning as both a neuroprotective "checkpoint" and a promoter of degenerative lesions depending on cellular context and disease state [70]. In Alzheimer's disease models, SHP2 inhibition has been shown to enhance phosphorylation of amyloid-β protein precursor (AβPP), reducing Aβ accumulation in neuronal cells [70]. The complex role of SHP2 in multiple neurodegenerative pathways, including those regulating oxidative stress, mitochondrial dysfunction, neuroinflammation, and apoptosis, positions it as a compelling target for therapeutic intervention [70].
Experimental Approaches for Studying SH2 Domain Interactions
High-Throughput Screening Technologies
Advanced experimental platforms have been developed to comprehensively profile SH2 domain binding specificities. One particularly powerful approach is high-density peptide chip technology, which enables probing the affinity of most SH2 domains against a large fraction of the entire complement of tyrosine phosphopeptides in the human proteome [67] [71]. This technology involves:
- SPOT synthesis of thousands of phosphopeptides on cellulose membranes
- Punch-pressing peptide spots into microtiter plates
- Peptide release from cellulose discs
- Printing onto aldehyde-modified glass surfaces to create high-density chips
This approach has been used to experimentally identify thousands of putative SH2-peptide interactions for more than 70 different SH2 domains, revealing 17 distinct specificity classes based on recognition preferences [67]. Interestingly, the correlation between SH2 domain sequence homology and peptide recognition specificity is relatively poor (Pearson correlation coefficient = 0.30), indicating that subtle sequence variations can significantly alter binding preferences [67].
Binding Affinity and Specificity Assays
Quantitative assessment of SH2 domain interactions employs various biochemical and biophysical techniques:
- Fluorescence polarization: Measures interactions between SH2 domains and soluble fluorescently labeled peptides to determine binding affinities (typical Kd values range from 0.1-10 μM) [66] [65]
- Surface plasmon resonance: Provides real-time kinetic data for SH2 domain-phosphopeptide interactions
- Isothermal titration calorimetry: Determines thermodynamic parameters of binding, including enthalpy and entropy changes
- Peptide array profiling: Semiquantitative analysis of domain-peptide interactions using membrane-bound peptides [66]
These approaches have revealed that SH2 domains achieve remarkable selectivity through complex linguistics that involves recognition of both permissive residues that enhance binding and non-permissive residues that oppose binding [66]. This contextual dependence substantially increases the information content accessible to SH2 domains for discriminating between similar peptide ligands.
Research Reagent Solutions for SH2 Domain Studies
Table 2: Essential Research Reagents for SH2 Domain Investigations
| Reagent/Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Recombinant SH2 Domains | GST-tagged SH2 domains; 70+ human SH2 domains available [67] | Binding assays, structural studies, inhibitor screening | Soluble expression; tag facilitation purification and detection [67] |
| Peptide Libraries | Oriented peptide libraries; Physiological peptide arrays (6,202 phosphopeptides) [67] | Specificity profiling, motif identification | Comprehensive coverage of human phosphoproteome; high-density format [67] |
| Binding Assay Systems | Fluorescence polarization; Surface plasmon resonance; Peptide chips [66] [67] | Affinity measurements; kinetic analysis | Quantitative data generation; high-throughput capability [67] |
| Computational Tools | Artificial neural networks (NetSH2) [67] | Prediction of SH2 binding; in silico screening | Trained on experimental peptide chip data; average PCC = 0.4 [67] |
| Structural Biology Resources | SH2 domain-ligand complex structures (70+ unique structures in PDB) [65] | Structure-based drug design; mutational analysis | Guides rational inhibitor design; elucidates specificity determinants [31] [65] |
The strategic disruption of SH2 domain-mediated interactions represents a promising therapeutic approach for numerous diseases driven by aberrant tyrosine kinase signaling. The comprehensive characterization of SH2 domain structure, function, and specificity has enabled the development of increasingly sophisticated targeting strategies that move beyond traditional phosphotyrosine mimetics. Future directions in this field will likely focus on several key areas:
First, the exploitation of alternative binding surfaces, such as lipid-binding pockets and dimerization interfaces, may yield inhibitors with improved selectivity and drug-like properties [31] [3]. Second, the integration of structural biology with molecular dynamics simulations will enhance our understanding of SH2 domain flexibility and its implications for drug design [3]. Finally, the application of emerging screening technologies, including high-density peptide arrays and artificial intelligence-driven prediction tools, will accelerate the identification and optimization of novel SH2 domain-targeted therapeutics [67].
As our understanding of SH2 domain biology continues to evolve, so too will our ability to precisely modulate these critical signaling modules for therapeutic benefit across a spectrum of human diseases, from cancer to neurodegenerative disorders. The continued refinement of targeting strategies that disrupt specific SH2 domain-mediated interactions while sparing others will be essential for realizing the full potential of this approach in clinical practice.
Src homology 2 (SH2) domains, approximately 100 amino acids in length, are modular protein domains that canonically recognize and bind phosphorylated tyrosine (pY) residues, thereby facilitating phospho-dependent protein-protein interactions in signal transduction networks [28]. Emerging research now reveals that SH2 domains participate in non-canonical functions that extend beyond simple pY-recognition, including specific lipid binding and active participation in liquid-liquid phase separation (LLPS) [28] [72]. These findings necessitate a re-evaluation of SH2 domain functionality, particularly for STAT family transcription factors whose SH2 domains are mutation hotspots in human disease [3]. This review compares these non-canonical roles, detailing the experimental evidence, biophysical principles, and biological implications, with a specific focus on how activating versus inactivating mutations in STAT SH2 domains influence these processes.
The canonical role of the SH2 domain involves a conserved structure—a central β-sheet flanked by two α-helices—that forms a pY-binding pocket and a specificity pocket that recognizes residues C-terminal to the pY [3] [28]. In STAT proteins, this domain is essential for receptor recruitment, JAK-mediated phosphorylation, and subsequent STAT dimerization via reciprocal SH2-pY interactions [3]. However, the discovery of non-canonical roles indicates a more complex functional landscape. Nearly 75% of SH2 domains are now predicted to interact with lipid molecules, particularly phosphoinositides like PIP₂ and PIP₃, and a growing number are implicated in driving or regulating the formation of biomolecular condensates via LLPS [28]. Understanding these mechanisms is crucial for dissecting the full spectrum of SH2 domain pathophysiology.
Lipid Binding: SH2 Domains as Membrane Interaction Hubs
Mechanisms and Functional Consequences
The non-canonical lipid-binding function of SH2 domains challenges the traditional view that these domains are solely protein-interaction modules. Lipid association is often mediated by cationic regions near the pY-binding pocket, which are typically flanked by aromatic or hydrophobic side chains that facilitate interaction with the lipid head groups [28]. This allows many SH2-containing proteins to be recruited to specific membrane compartments, thereby positioning them to respond to localized signaling events.
Table 1: Key Examples of SH2 Domain Lipid Interactions and Functional Outcomes
| Protein | Lipid Moiety | Function of Lipid Association |
|---|---|---|
| SYK | PIP₃ | PIP₃-dependent membrane binding is required for the non-catalytic activation of STAT3/5 scaffolding function [28]. |
| ZAP70 | PIP₃ | Essential for facilitating and sustaining ZAP70 interactions with the TCR-ζ chain [28]. |
| LCK | PIP₂, PIP₃ | Modulates the interaction of LCK with its binding partners in the TCR signaling complex [28]. |
| ABL | PIP₂ | Mediates membrane recruitment and modulation of Abl kinase activity [28]. |
| VAV2 | PIP₂, PIP₃ | Modulates the interaction of VAV2 with membrane receptors, e.g., EphA2 [28]. |
| C1-Ten/Tensin2 | PIP₃ | Regulates Abl activity and the phosphorylation of IRS-1 in insulin signaling pathways [28]. |
These interactions have profound effects on protein function. For instance, the lipid-binding activity of the TNS2 SH2 domain is critical for regulating insulin receptor substrate-1 (IRS-1) phosphorylation, directly linking this non-canonical function to metabolic signaling [28]. Furthermore, disease-causing mutations are frequently localized within these lipid-binding pockets, underscoring the physiological importance of this feature [28].
Experimental Protocols for Investigating SH2-Lipid Interactions
A primary method for studying membrane-associated protein behavior is the Supported Lipid Bilayer (SLB) assay coupled with Total Internal Reflection Fluorescence (TIRF) Microscopy [72]. This protocol allows for real-time observation of protein condensation on a two-dimensional membrane surface.
Detailed Protocol:
- SLB Preparation: Create model membranes typically from phosphatidyl choline (e.g., DOPC or POPC) incorporating a small percentage (1-5%) of a modified lipid such as DGS-NTA(Ni) to enable the specific attachment of His-tagged proteins to the bilayer [72].
- Protein Purification: Express and purify the SH2 domain or full-length protein of interest, often with a fluorescent tag (e.g., GFP) for visualization.
- Imaging and Analysis: Incubate the purified protein with the SLB. Use TIRFM to excite fluorophores only within a very thin evanescent field (~100 nm) above the glass slide, providing high-contrast imaging of membrane-bound events without interference from the solution above [72]. Analyze the resulting images for protein clustering, diffusion, and condensate formation.
A key insight from these experiments is that the critical concentration required for LLPS is an order of magnitude lower when constrained to a 2D membrane surface compared to in 3D solution. For example, components of T cell signaling clusters like Grb2 and Sos1 undergo phase separation in the nM range on membranes, whereas μM concentrations are required in solution [72].
SH2 Domains in Liquid-Liquid Phase Separation (LLPS)
Driving Biomolecular Condensate Formation in Signaling
LLPS is a process whereby biomolecules demix from the surrounding nucleoplasm or cytoplasm to form concentrated, dynamic, membraneless organelles, also known as biomolecular condensates [73]. SH2 domains contribute to LLPS by engaging in multivalent, weak, and transient interactions—a key driver of phase separation. These interactions often involve simultaneous engagement with other modular domains (e.g., SH3 domains) and their binding partners, creating a dense interaction network that separates from the dilute phase [28] [72].
Table 2: Signaling Complexes Driven by SH2 Domain-Mediated Phase Separation
| Condensate Complex | Key SH2-Containing Proteins | Biological Role |
|---|---|---|
| LAT-GRB2-SOS1 | GRB2, PLCγ1 | Enhances T-cell receptor (TCR) signaling and activation by concentrating signaling components [28] [72]. |
| FGFR2:SHP2:PLCγ1 | SHP2, PLCγ1 | Increases the activity and efficiency of RTK signaling [28]. |
| N-WASP–NCK | NCK | Promotes actin polymerization via the Arp2/3 complex in T-cell signaling [28]. |
| SLP65, CIN85 | SLP65 | Facilitates efficient B-cell receptor signaling [28]. |
In the context of T cell activation, the adapter protein LAT, when phosphorylated, nucleates condensates by recruiting the SH2 domain of GRB2, which in turn binds GADS and SOS1, forming a dense network that phase separates [72]. This condensate serves to concentrate signaling components, enhancing kinase activity and signal amplification. Similarly, in kidney podocytes, phase separation of the adapter protein NCK, which contains an SH2 domain, increases the membrane dwell time of N-WASP and Arp2/3 complexes, promoting actin polymerization [28].
Experimental Protocols for Analyzing SH2-Dependent LLPS
A common methodology for studying phase separation involves in vitro reconstitution followed by fluorescence microscopy and Fluorescence Recovery After Photobleaching (FRAP) to assess material properties [73] [74].
Detailed Protocol:
- In Vitro Reconstitution: Express and purify the protein(s) of interest (e.g., a specific SH2 domain or a multi-domain protein) from bacterial or insect cells. The solution conditions (pH, salt, temperature, RNA) are carefully controlled, as LLPS is highly sensitive to these factors [73].
- Droplet Visualization: Incubate the purified protein under conditions that drive phase separation (e.g., by adding a crowding agent like PEG). Use bright-field Differential Interference Contrast (DIC) or fluorescence microscopy (if the protein is fluorescently labeled) to visualize the formation of spherical liquid droplets [74].
- FRAP Assay: Photobleach a region of the condensate with a high-intensity laser and monitor the recovery of fluorescence into the bleached area over time. Rapid and complete recovery indicates liquid-like properties and dynamic exchange of molecules, whereas slow or incomplete recovery suggests a transition to a more gel-like or solid state [73] [74]. For example, liquid-like FUS protein droplets show rapid FRAP recovery, which is lost as they age into fibrous aggregates [74].
For intracellular studies, optogenetics-based systems like optoDroplet are employed. This involves fusing the protein of interest's intrinsically disordered regions (IDRs) to the Cry2 protein domain, which oligomerizes upon blue light activation, allowing spatiotemporal control over condensate formation in live cells [73].
The Pathophysiological Nexus: STAT SH2 Domain Mutations
The SH2 domain is a recognized mutational hotspot in STAT proteins, particularly STAT3 and STAT5B, with single amino acid substitutions leading to either gain-of-function (GOF) or loss-of-function (LOF) phenotypes in diseases ranging from immunodeficiencies to cancer [3] [7]. These mutations can disrupt both canonical and non-canonical functions.
- Loss-of-Function Mutations: In STAT3, germline LOF mutations (e.g., K591E, S611I, G617R) are associated with Autosomal-Dominant Hyper IgE Syndrome (AD-HIES), characterized by reduced Th17 T-cell responses and recurrent infections [3]. These mutations often impair phospho-tyrosine binding or STAT dimerization. A striking example in STAT5B is the Y665H mutation, identified in T-cell leukemias. When introduced into mice, this mutation acts as a LOF allele, impairing enhancer establishment and alveolar differentiation in mammary glands, leading to lactation failure [7].
- Gain-of-Function Mutations: Somatic GOF mutations in STAT3 (e.g., S614R, E616K) and STAT5B are frequently found in leukemias and lymphomas [3]. They often lead to constitutive, cytokine-independent activation. The STAT5B Y665F mutation is a prime example of a GOF mutation. In mouse models, this mutation accelerates mammary gland development during pregnancy and elevates enhancer formation [7].
The precise impact of these mutations on non-canonical functions like lipid binding and LLPS is an area of active investigation. It is plausible that mutations altering the charge or conformation of the SH2 domain could disrupt its interaction with membrane lipids or its valency in multivalent interaction networks, thereby altering the formation or function of signaling condensates.
Integrated Signaling and Research Toolkit
The non-canonical roles of SH2 domains are not isolated; they integrate with canonical functions to regulate complex signaling pathways. The following diagram synthesizes how SH2 domains in STAT proteins coordinate membrane recruitment, phase separation, and transcriptional activation, and how disease-associated mutations perturb this system.
Diagram Title: Integration of SH2 Domain Functions in STAT Signaling
The Scientist's Toolkit: Key Research Reagents and Methods
Table 3: Essential Reagents and Methods for Studying Non-Canonical SH2 Functions
| Tool / Reagent | Function / Application | Key Utility |
|---|---|---|
| Supported Lipid Bilayers (SLBs) | A model membrane system for reconstituting 2D protein-lipid and protein-protein interactions. | Enables study of membrane-associated phase separation using TIRF microscopy [72]. |
| OptoDroplet System | An optogenetics tool using Cry2 oligomerization controlled by blue light to induce IDR-mediated condensate formation in live cells. | Allows spatiotemporal, controlled induction of LLPS to study its functional consequences in vivo [73]. |
| Fluorescence Recovery After Photobleaching (FRAP) | A microscopy technique that measures the mobility of molecules within condensates. | Used to determine the material properties (liquid-like vs. solid-like) of biomolecular condensates in vitro and in cells [73] [74]. |
| ProBound Computational Tool | A statistical learning method for analyzing data from peptide display and NGS. | Builds quantitative sequence-to-affinity models, predicting how mutations affect SH2 binding specificity and affinity [57]. |
| D2P2 Database | A database that curates predictions on protein disorder, binding sites, and post-translational modifications. | Helps predict the intrinsic disorder in proteins, a key feature of proteins that undergo LLPS [73]. |
The traditional view of SH2 domains as simple phospho-tyrosine binding modules is no longer sufficient. It is now evident that these domains are multifunctional hubs that also engage in specific lipid binding and drive the formation of biomolecular condensates via LLPS. These non-canonical functions work in concert with canonical signaling to ensure precise spatiotemporal control of cellular processes. The high prevalence of disease-associated mutations in the STAT SH2 domain underscores its functional importance. Future research must systematically evaluate how these mutations impact all SH2 domain functions—canonical and non-canonical—to fully understand disease mechanisms. The integration of biophysical methods (e.g., in vitro reconstitution, FRAP), cell biological tools (e.g., optogenetics), and computational models will be essential to dissect this complexity and pave the way for novel therapeutic strategies that target these multifaceted domains.
Comparative Pathophysiology and Validation of Mutation Impacts
The Signal Transducer and Activator of Transcription 5B (STAT5B) protein plays a defining role in cytokine signaling within the hematopoietic system, regulating genetic programs essential for immune function, growth, and metabolism [23] [60]. As a signal-dependent transcription factor, its activation is exquisitely controlled by cytokines and growth factors via the JAK-STAT pathway. The Src Homology 2 (SH2) domain of STAT5B is particularly critical for its function, mediating phosphotyrosine-dependent recruitment, homo-dimerization, and nuclear translocation [23] [3]. Sequencing of patient samples has identified the SH2 domain as a hotspot for mutations in various pathologies, with tyrosine 665 (Y665) representing a key mutational target [23] [75] [76]. This comparison guide provides a comprehensive functional analysis of two specific mutations at this residue—Y665F and Y665H—which exhibit strikingly opposing biological impacts despite their proximity within the protein structure [23] [30]. Understanding their distinct mechanisms and functional consequences is essential for researchers investigating STAT5B biology, immune regulation, and targeted therapeutic development.
Structural and Computational Predictions
In silico modeling and structural analysis reveal how minimal amino acid substitutions at position 665 lead to substantial functional divergence.
Location and Conservation
Tyrosine 665 is located at a critical interface within the STAT5B SH2 domain that is directly involved in homodimerization [23]. This residue is highly conserved across vertebrate species, underscoring its structural and functional importance [23]. Structural predictions generated by AlphaFold3 show that Y665 participates in key interactions that stabilize the STAT5B homodimer configuration [23].
Divergent Energetic and Pathogenicity Profiles
Computational analyses using COORDinator predict divergent energetic impacts for the two mutations:
- STAT5B-Y665F: The substitution of tyrosine with phenylalanine, which lacks the tyrosine hydroxyl group but maintains the aromatic ring, is predicted to stabilize the protein structure, potentially through enhanced intramolecular aromatic stacking interactions with phenylalanine 711 (F711) [23].
- STAT5B-Y665H: The introduction of histidine, with its imidazole side chain, is predicted to destabilize binding of the C-terminal tail, disrupting critical intramolecular interactions [23].
Pathogenicity assessment tools further highlight their functional differences:
Table 1: Computational Pathogenicity Predictions for STAT5B Mutations
| Mutation | AlphaMissense Score | CADD PHRED Score | REVEL Score | Predicted Functional Impact |
|---|---|---|---|---|
| Y665F | 0.173 (Benign) | 24.3 | 0.535 | Higher probability of pathogenicity |
| Y665H | 0.383 (Benign) | 23.1 | 0.304 | Lower probability of pathogenicity |
Molecular and Functional Characterization
Experimental data from multiple systems demonstrate the opposing functional consequences of these mutations at the molecular level.
Signaling Capacity and Transcriptional Activity
- STAT5B-Y665F: Displays enhanced STAT5 phosphorylation, increased DNA binding, and elevated transcriptional activity following cytokine activation [23] [30] [77]. This gain-of-function (GOF) profile is characterized by prolonged activation and resistance to deactivation mechanisms.
- STAT5B-Y665H: Resembles a null variant with diminished phosphorylation, DNA binding, and transcriptional response to cytokine stimulation, consistent with a loss-of-function (LOF) profile [23] [30].
Dimerization and Stability
Biophysical studies suggest that the Y665F mutation promotes sustained interchain cross-domain interactions, conferring kinetic stability to the mutant anti-parallel dimer [78]. In contrast, the Y665H mutation impairs proper dimerization, disrupting the structural integrity required for STAT5B activation and nuclear function [23].
Figure 1: STAT5B Signaling Pathway and Mutation Impacts. The diagram illustrates the canonical JAK-STAT signaling pathway and highlights the divergent effects of Y665F (enhancing) and Y665H (impairing) mutations on key activation steps.
Cellular and Immune Phenotypes
The opposing molecular functions of these mutations translate to distinct phenotypic outcomes in immune cells and animal models.
T Cell Populations and Homeostasis
Table 2: Immune Phenotypes in STAT5B Mutation Knock-in Mice
| Immune Parameter | STAT5B-Y665F (GOF) | STAT5B-Y665H (LOF) |
|---|---|---|
| CD8+ T cells | Accumulation of effector and memory populations | Diminished effector and memory populations |
| CD4+ T cells | Increased regulatory T cells (Tregs) | Reduced regulatory T cells (Tregs) |
| CD8+/CD4+ Ratio | Altered (increased) | Not specifically reported |
| Overall Impact | Enhanced lymphocyte accumulation | Reduced lymphocyte populations |
Mammary Gland Development
Beyond immune functions, these mutations exert opposing effects on mammary gland development during pregnancy:
- STAT5B-Y665F: Accelerated mammary development during pregnancy with elevated enhancer formation [7].
- STAT5B-Y665H: Failed development of functional mammary tissue resulting in lactation failure, impaired enhancer establishment, and compromised alveolar differentiation [7].
Disease Associations and Clinical Relevance
The distinct functional properties of these mutations correlate with different clinical manifestations and disease associations.
- STAT5B-Y665F: Frequently identified in patients with T-cell large granular lymphocytic leukemia (T-LGLL) and T-cell prolymphocytic leukemia (T-PLL) [23] [75] [76]. This mutation is reproducibly associated with blood cancers in multiple databases, with 53 cases documented in the Munich Leukemia Laboratory database and 12 cases in the COSMIC database [23].
- STAT5B-Y665H: Only a single case reported in T-PLL, with no association with cancer in major databases [23]. This aligns with its LOF characterization and suggests it may not directly drive malignant transformation.
Notably, neither mutation alone directly induces hematopoietic malignancy in mouse models, indicating that additional cooperating factors are likely required for full leukemogenesis [23] [30].
Experimental Approaches and Methodologies
Key Experimental Protocols
Generation of Mutant Mouse Models
The STAT5B-Y665F and Y665H mutations were introduced into the mouse genome using CRISPR/Cas9 and base editing techniques [7]:
- Y665H Mutant Mice: ABE mRNA (50 ng/μl) and Y665H sgRNA (20 ng/μl) were co-microinjected into the cytoplasm of fertilized eggs collected from superovulated C57BL/6 N females.
- Y665F Mutant Mice: A single-strand oligonucleotide donor containing the Y (TAC) to F (TTT) change plus a silent C to G change (to destroy the sgRNA PAM site) was electroporated with Cas9 protein and Y665F sgRNA into zygotes.
- Embryos that reached the 2-cell stage were implanted into pseudopregnant surrogate mothers, and resulting mice were genotyped by PCR amplification and Sanger sequencing.
Transcriptomic and Epigenomic Analyses
- RNA Sequencing: Total RNA was extracted from frozen tissues with quality assessment (RIN >8.0). Ribosomal RNA was removed, and cDNA was synthesized using SuperScript III. Libraries were prepared with TruSeq Stranded Total RNA Library Prep Kit and sequenced to assess gene expression profiles [7].
- Epigenomic Analyses: Chromatin immunoprecipitation followed by sequencing (ChIP-seq) was employed to map STAT5B binding sites and enhancer landscapes, revealing differential enhancer establishment between the mutants [7].
In Vitro Functional Assays
- STAT5 Luciferase Reporter Assay: HeLa cells or primary T cells were transfected with STAT5B constructs and a STAT5-responsive luciferase reporter. Following cytokine stimulation (e.g., IL-2), luciferase activity was measured to quantify transcriptional activity [23] [76].
- Phosphorylation Analysis: Western blotting with phospho-STAT5 specific antibodies was used to assess tyrosine phosphorylation status under various cytokine stimulation conditions [23] [76].
Figure 2: Experimental Workflow for STAT5B Mutation Studies. The diagram outlines the multi-disciplinary approach combining in silico predictions, animal model generation, cellular assays, molecular analyses, and multi-omics integration.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for STAT5B Mutation Studies
| Reagent / Tool | Application | Function in Research |
|---|---|---|
| CRISPR/Cas9 with ABE | Generation of knock-in mouse models (Y665H) | Precise adenine base editing without double-strand breaks |
| Single-strand Oligo Donors | Generation of knock-in mouse models (Y665F) | Template for homologous recombination with desired mutation |
| STAT5-Responsive Luciferase Reporter | In vitro transcriptional activity assessment | Quantification of STAT5-dependent transcriptional activation |
| Phospho-STAT5 Specific Antibodies | Western blot, flow cytometry | Detection of activated/phosphorylated STAT5 |
| TruSeq Stranded Total RNA Kit | RNA sequencing library preparation | Generation of strand-specific RNA-seq libraries for transcriptomic analysis |
| IL-2 and other γc cytokines | Cell culture stimulation | Activation of JAK-STAT signaling pathway in lymphocytes |
| Magnetic cell separation beads | Immune cell isolation (CD4+, CD8+, CD57+) | Purification of specific lymphocyte populations from mixed samples |
The head-to-head comparison of STAT5B-Y665F and Y665H mutations reveals how minimal amino acid changes at a critical structural position can drive opposing functional outcomes. The Y665F mutation demonstrates characteristic GOF properties including enhanced phosphorylation, dimerization stability, and transcriptional activity, leading to altered immune homeostasis and association with lymphoproliferative disorders. In contrast, the Y665H mutation exhibits LOF features across all assay systems, with impaired signaling and developmental defects. These mutations represent valuable natural experiments for understanding STAT5B structure-function relationships and their pathophysiological consequences. Future research should focus on identifying cooperating factors that enable full malignant transformation in the context of these mutations, and developing targeted therapeutic strategies that can selectively modulate these aberrant signaling states. The experimental approaches outlined here provide a framework for systematic characterization of disease-associated STAT5B variants and their functional impacts across biological systems.
The Src Homology 2 (SH2) domain present in STAT (Signal Transducer and Activator of Transcription) proteins serves as a critical regulatory module for immune cell signaling and fate determination. These domains facilitate phosphotyrosine-dependent protein-protein interactions that are essential for JAK-STAT pathway activation, which governs fundamental processes in T-cell development, differentiation, and function [79]. Research has revealed that single amino acid substitutions within the SH2 domain can dramatically alter STAT protein function, with profound consequences for T-cell populations and their role in leukemogenesis [23] [7]. This review systematically compares the immunological impacts of activating versus inactivating STAT SH2 domain mutations, with particular emphasis on the STAT5B Y665F (gain-of-function) and Y665H (loss-of-function) variants identified in T-cell leukemias [23]. Understanding these contrasting immune phenotypes provides crucial insights for developing targeted therapies that can either augment or suppress STAT signaling in specific disease contexts.
Structural and Functional Basis of STAT SH2 Domain Mutations
Molecular Architecture of STAT SH2 Domains
The SH2 domain of STAT proteins is a conserved structural module of approximately 100 amino acids that facilitates phosphotyrosine-dependent dimerization and subsequent nuclear translocation [79]. This domain recognizes and binds to specific phosphotyrosine motifs on cytokine receptors and, critically, engages in reciprocal phosphotyrosine-SH2 interactions between STAT monomers to form active parallel dimers [23]. Tyrosine 665 (Y665) in STAT5B is located at a crucial interface involved in STAT5B homodimerization, where it contributes to stabilizing the intramolecular interactions that support the dimer conformation [23]. In silico modeling predicts that substitutions at this position exert divergent energetic effects on homodimerization with varying pathogenicity [23].
Table 1: Structural and Functional Characteristics of STAT5B SH2 Domain Mutations
| Parameter | STAT5BY665F (GOF) | STAT5BY665H (LOF) |
|---|---|---|
| Structural Prediction | Stabilizes intramolecular aromatic stacking with F711 | Introduces imidazole group that destabilizes C-terminal tail binding |
| Pathogenicity Scores | CADD: 24.3; REVEL: 0.535 | CADD: 23.1; REVEL: 0.304 |
| Phosphorylation Status | Enhanced STAT5 phosphorylation after cytokine activation | Diminished phosphorylation resembling null phenotype |
| DNA Binding | Increased binding affinity and transcriptional activity | Impaired DNA binding capacity |
| Dimerization | Stabilized active dimer conformation | Disrupted dimer formation |
Mechanism of Pathogenic Mutations
The contrasting effects of Y665F and Y665H mutations exemplify how minimal genetic alterations can divergently reprogram immune cell fate. The Y665F substitution replaces tyrosine with phenylalanine, promoting intramolecular aromatic stacking interactions with F711 that stabilize the active conformation [23]. This results in prolonged STAT5 phosphorylation, enhanced DNA binding, and increased transcriptional activity after cytokine stimulation [23]. Conversely, the Y665H mutation introduces a histidine residue with an imidazole side chain that sterically and electrostatically disrupts binding of the C-terminal tail, destabilizing the functional dimeric structure and impairing transcriptional activity [23]. These structural perturbations manifest as fundamentally opposed immunological phenotypes, with Y665F driving proliferative expansion and Y665H resulting in lymphoid deficiency.
Contrasting Immune Phenotypes in Experimental Models
T-cell Population Alterations in Murine Models
In vivo studies using genetically engineered mouse models reveal striking contrasts in how these mutations reshape the immune landscape. Mice harboring the gain-of-function Stat5bY665F mutation demonstrate marked accumulation of CD8+ effector and memory T cells alongside expanded CD4+ regulatory T cell (Treg) populations, substantially altering CD8+/CD4+ T cell ratios [23]. This phenotype reflects sustained STAT5B signaling that promotes T-cell survival, proliferation, and effector differentiation. The immunological landscape directly contrasts with STAT5BY665H "knock-in" mice, which show diminished CD8+ effector and memory T cells and reduced CD4+ regulatory T cell compartments [23]. These differential effects underscore STAT5B's critical role in maintaining T-cell homeostasis, with hyperactivation driving expansion and hypoactivation resulting in deficiency.
Association with Leukemogenic Transformation
The gain-of-function STAT5B Y665F mutation is frequently identified in T-cell large granular lymphocytic leukemia (T-LGLL) and T-cell prolymphocytic leukemia (T-PLL), where it promotes clonal expansion of cytotoxic T cells [23] [80]. Single-cell transcriptomic analyses of T-LGLL patients reveal that these leukemic clonotypes exhibit heightened cytotoxicity markers (GZMB, PRF1, NKG7) alongside exhaustion signatures (LAG3, TIGIT), representing a dysfunctional hyperactivated state [80]. Interestingly, despite its strong association with human leukemia, the STAT5BY665F mutation alone does not directly induce malignant transformation in mouse models, suggesting requirement for cooperating factors in full leukemogenesis [23]. In contrast, the loss-of-function Y665H variant has only been reported in a single T-PLL case and demonstrates no leukemogenic potential in experimental systems [23].
Table 2: Immune Phenotypes and Leukemogenic Potential of STAT5B Mutations
| Immune Parameter | STAT5BY665F (GOF) | STAT5BY665H (LOF) |
|---|---|---|
| CD8+ T-cell Compartment | Expanded effector and memory populations | Diminished effector and memory populations |
| CD4+ Treg Cells | Increased frequency and number | Decreased frequency and number |
| CD8+/CD4+ Ratio | Significantly altered | Reduced |
| Cytotoxic Potential | Enhanced GZMB, PRF1, NKG7 expression | Diminished cytotoxic function |
| Exhaustion Markers | Elevated LAG3, TIGIT | Not characterized |
| Leukemia Association | T-LGLL, T-PLL | Rare in T-PLL only |
| Transforming Potential | Requires cooperating factors | Non-transforming |
Experimental Approaches for Characterizing STAT Mutations
In Silico Structural Prediction and Pathogenicity Assessment
Computational approaches provide the first layer of mutation characterization through structural prediction and pathogenicity assessment. AlphaFold3-generated structures of STAT5A and STAT5B SH2 domain homodimers reveal that Y665 is located at a critical interface involved in STAT5B homodimerization [23]. The COORDinator algorithm predicts energetic contributions of residue substitutions, highlighting that Y665F stabilizes while Y665H destabilizes intramolecular interactions [23]. Pathogenicity prediction tools including AlphaMissense, Combined Annotation Dependent Depletion (CADD), and Rare Exome Variant Ensemble Learner (REVEL) provide complementary assessments of mutation impact, with Y665F consistently scoring higher for potential deleterious effects [23]. These computational approaches enable prioritization of mutations for functional validation and offer mechanistic hypotheses regarding their structural consequences.
Signaling and Functional Assays
Comprehensive functional characterization employs diverse signaling and transcriptional assays to quantify mutation impacts. Phospho-specific flow cytometry enables tracking of STAT phosphorylation kinetics following cytokine stimulation, revealing that Y665F exhibits enhanced and prolonged phosphorylation compared to wild-type STAT5B [23] [79]. Electrophoretic mobility shift assays (EMSAs) demonstrate increased DNA binding activity for Y665F mutants, while Y665H shows impaired binding capacity [23]. Transcriptional reporter assays using GAS (gamma-activated sequence) elements further confirm heightened transactivation potential for Y665F and diminished activity for Y665H [23]. These assays collectively establish the functional consequences of SH2 domain mutations on STAT5B signaling output.
Advanced Single-Cell Technologies
Single-cell RNA sequencing coupled with T-cell receptor profiling (scRNA+TCRαβ-seq) enables unprecedented resolution of leukemic and non-leukemic T-cell populations in STAT-mutant leukemias [80]. This approach has revealed that T-LGLL clonotypes exhibit elevated cytotoxicity-associated transcripts (GZMB, PRF1, NKG7) and exhaustion markers (LAG3, TIGIT) compared to healthy reactive clonotypes [80]. Additionally, these technologies uncover aberrant cell-cell communication networks between leukemic clones and non-leukemic immune cells via costimulatory interactions and cytokine signaling [80]. Mass cytometry (CyTOF) using metal isotope-tagged antibodies further enables high-dimensional immunophenotyping of STAT-mutant samples, quantifying changes in both lymphoid and myeloid compartments [79].
The Scientist's Toolkit: Essential Research Reagents and Methods
Table 3: Essential Research Reagents and Experimental Solutions
| Reagent/Method | Specific Example | Research Application |
|---|---|---|
| Genetically Engineered Mouse Models | STAT5B Y665F and Y665H knock-in mice | In vivo assessment of immune phenotypes and leukemogenic potential |
| Single-cell Multi-omics | scRNA+TCRαβ-seq (10X Genomics) | Unbiased characterization of leukemic and non-leukemic T-cell repertoires |
| High-dimensional Phenotyping | Mass cytometry (CyTOF) with metal-tagged antibodies | Deep immunoprofiling of >40 parameters simultaneously |
| Phospho-specific Flow Cytometry | Phospho-STAT5 (Tyr694/699) antibodies | Signaling dynamics in response to cytokine stimulation |
| Structural Prediction Tools | AlphaFold3, COORDinator | In silico modeling of mutation effects on protein structure |
| Pathogenicity Prediction | CADD, REVEL, AlphaMissense | Computational assessment of mutation deleteriousness |
| Transcriptional Reporter Assays | GAS element luciferase constructs | Quantification of STAT transcriptional activity |
| Cell Culture Models | STAT-deficient U3A cells + reconstitution | Controlled assessment of STAT mutation function |
Discussion and Clinical Implications
Therapeutic Targeting of STAT Signaling Pathways
The contrasting immune phenotypes resulting from STAT5B SH2 domain mutations highlight the delicate balance in STAT signaling that governs T-cell homeostasis and leukemogenesis. Gain-of-function mutations like Y665F create a hyperactive signaling state that promotes clonal expansion while simultaneously driving T-cell exhaustion - a paradoxical combination that represents both the driver of pathology and a potential therapeutic vulnerability [80]. Several strategic approaches have emerged for targeting dysregulated STAT signaling, including direct SH2 domain inhibitors that disrupt phosphotyrosine binding, JAK inhibitors that attenuate upstream activation, and combinatorial therapies that target both STAT signaling and complementary pathways [81] [20]. The differential responses of mutant versus wild-type STAT proteins to these interventions remain an active area of investigation.
Diagnostic and Prognostic Applications
Understanding the specific immune phenotypes associated with STAT SH2 domain mutations enables more precise stratification of hematologic malignancies. The detection of STAT5B Y665F in T-LGLL correlates with distinct clinical features including severe neutropenia and autoimmune manifestations [23] [80]. Single-cell technologies further reveal that the non-leukemic T-cell repertoire in T-LGLL patients is also abnormally mature, cytotoxic, and clonally restricted compared to healthy individuals or those with other immune disorders [80]. These findings suggest that STAT mutations create a permissive immune environment that extends beyond the leukemic clone itself, with implications for monitoring minimal residual disease and assessing treatment efficacy.
The comparative analysis of STAT SH2 domain mutations reveals how minimal genetic alterations at critical structural interfaces can generate dramatically opposed immune phenotypes. The STAT5B Y665F gain-of-function mutation promotes expansion of cytotoxic T-cell populations with exhausted features, ultimately predisposing to leukemogenesis, while the Y665H loss-of-function mutation results in lymphoid deficiency. These contrasting outcomes underscore the precision required for therapeutic targeting of STAT signaling pathways and highlight the importance of comprehensive immunophenotyping in characterizing mutation-specific effects. Future research should focus on elucidating the cooperative genetic events that complete malignant transformation and developing mutation-specific therapeutic strategies that can either augment or suppress STAT signaling based on disease context.
The Signal Transducer and Activator of Transcription 5B (STAT5B) protein serves as a crucial transcription factor that regulates genetic programs essential for mammary gland development and function. While extensively studied in hematopoietic contexts, STAT5B's role in non-hematopoietic tissues, particularly the mammary gland, represents a critical area of investigation with significant implications for understanding mammary development, lactation biology, and reproductive medicine. The Src homology 2 (SH2) domain of STAT5B enables its recruitment to phosphorylated tyrosine residues on cytokine receptors, facilitating STAT5B's own phosphorylation, dimerization, nuclear translocation, and DNA binding activity [31] [30]. Naturally occurring missense mutations within this domain, initially identified in patients with T-cell leukemias, exhibit divergent functional impacts when introduced into physiological systems [30].
This comparison guide objectively analyzes two specific STAT5B SH2 domain mutations—tyrosine 665 to phenylalanine (Y665F) and tyrosine 665 to histidine (Y665H)—and their opposing effects on mammary gland development and lactation capacity. By examining direct experimental evidence from murine models, we delineate how single amino acid substitutions at codon 665 generate contrasting phenotypic outcomes through fundamental alterations in STAT5B transcriptional regulation. This analysis provides researchers and drug development professionals with a structured comparison of how gain-of-function versus loss-of-function STAT5B mutations manifest in mammary physiology, offering insights for therapeutic targeting and diagnostic approaches.
Comparative Analysis of Mutant Phenotypes
Mammary Gland Development and Lactation Performance
Table 1: Phenotypic Comparison of STAT5B Mutations in Mammary Gland Development and Function
| Parameter | STAT5BY665F (GOF) | STAT5BY665H (LOF) | Wild-Type STAT5B |
|---|---|---|---|
| Mammary development during pregnancy | Accelerated alveolar development and expansion | Severely impaired functional tissue development; failure to form lobuloalveolar structures | Normal, hormonally-regulated development |
| Lactation capability | Successful milk production | Complete lactation failure in initial pregnancy | Normal lactation onset post-parturition |
| Lactation rescue potential | Not applicable | Possible after persistent hormonal stimulation through multiple pregnancies | Not applicable |
| Enhancer landscape establishment | Elevated formation of STAT5-dependent enhancers and super-enhancers | Impaired enhancer establishment; failure to activate lactogenic program | Hormonally-induced enhancer formation |
| Milk protein gene expression | Enhanced expression of Wap, Csn1s1, Csn2, Csn1s2a, Csn1s2b, Csn3 | Severely reduced expression in initial pregnancy | Appropriate temporal expression during pregnancy and lactation |
| Transcriptional programs | Hyperactivation of STAT5B-driven genetic networks | Failure to induce interleukin-regulated genetic programs | Balanced activation of pregnancy and lactation programs |
Molecular and Functional Characterization
Table 2: Molecular and Biochemical Properties of STAT5B SH2 Domain Mutations
| Characteristic | STAT5BY665F | STAT5BY665H | Experimental Evidence |
|---|---|---|---|
| Classification | Gain-of-Function (GOF) | Loss-of-Function (LOF) | In vitro and in vivo functional assays [7] [30] |
| STAT5 phosphorylation | Enhanced and sustained after cytokine activation | Diminished; resembles STAT5B-null state | Phospho-STAT5 western blotting [30] |
| DNA binding capacity | Increased binding to GAS motifs (TTCnnnGAA) | Severely impaired DNA binding | ChIP-seq against STAT5B [7] |
| Transcriptional activity | Elevated reporter gene activation | Minimal activation above background | Luciferase reporter assays [30] |
| Dimerization potential | Enhanced homodimerization stability | Impaired dimerization capability | In silico modeling and biochemical assays [30] |
| Enhancer function | Increased H3K27ac marks at target enhancers | Failed establishment of active enhancer landscape | Chromatin immunoprecipitation [7] |
Experimental Models and Methodologies
Murine Model Generation
The comparative analysis of STAT5B SH2 domain mutations relies on precisely engineered murine models that recapitulate human mutations. Researchers employed distinct genome editing approaches to introduce the specific amino acid substitutions at tyrosine 665:
STAT5BY665H Model: Generated using adenine base editing (ABE) technology, with ABE mRNA (50 ng/μL) and Y665H sgRNA (20 ng/μL) co-microinjected into the cytoplasm of fertilized C57BL/6N eggs [7]. This approach directly converts the tyrosine (TAC) codon to histidine (CAC) without creating double-strand DNA breaks.
STAT5BY665F Model: Created using CRISPR-Cas9-mediated homology-directed repair, with Cas9 protein complexed with Y665F sgRNA (forming a ribonucleoprotein complex) co-electroporated with a single-stranded oligonucleotide donor template into zygotes [7]. The donor template contained the Y665F (TAC→TTT) mutation plus a silent C→G change to disrupt the protospacer adjacent motif and prevent continued Cas9 cleavage.
Both models were backcrossed to C57BL/6 backgrounds, and wild-type littermates served as controls in all experiments to ensure genetically matched comparisons [7]. This rigorous approach eliminates confounding variables from mixed genetic backgrounds.
Analytical Methodologies
Comprehensive phenotyping employed multimodal approaches to assess molecular, cellular, and physiological outcomes:
Mammary Gland Whole Mount Analysis: Intact mammary glands were harvested at defined developmental timepoints (virgin, pregnancy days 1, 6, 12, 18, lactation day 1), fixed, and stained with carmine alum to visualize ductal branching, alveolar bud formation, and lobuloalveolar development [7].
Transcriptomic Profiling: Total RNA sequencing was performed on mammary tissues during pregnancy and lactation phases following ribosomal RNA depletion, cDNA synthesis using SuperScript III, and library preparation with TruSeq Stranded Total RNA Library Prep Kit [7]. Differential gene expression analysis identified STAT5B-dependent genetic programs.
Epigenomic Mapping: Chromatin immunoprecipitation followed by sequencing (ChIP-seq) for STAT5B, H3K27ac (active enhancer mark), and RNA polymerase II was conducted to define enhancer landscapes and transcriptional regulatory mechanisms [7].
Quantitative Phenotypic Assessment: Milk protein gene expression (Csn1s1, Csn2, Csn1s2a, Csn1s2b, Csn3, Wap) was quantified by RT-qPCR using TaqMan probes, normalized to Gapdh, and analyzed via comparative CT method [7].
Figure 1: STAT5B Signaling Pathway and Mutation Impacts. The diagram illustrates how Y665F (GOF) and Y665H (LOF) mutations divergently alter STAT5B activation, dimerization, and transcriptional outcomes in mammary gland development.
Research Reagent Solutions
Table 3: Essential Research Reagents for STAT5B Mammary Gland Studies
| Reagent/Category | Specific Examples | Research Application | Functional Role |
|---|---|---|---|
| Genetically Engineered Mouse Models | STAT5BY665F knock-in, STAT5BY665H knock-in | In vivo functional studies | Model human STAT5B mutations in physiological context |
| Cell Line Models | HC11 mouse mammary epithelial cells | In vitro differentiation studies | Assess STAT5B-dependent gene regulation in mammary epithelium |
| Antibodies for Detection | Anti-STAT5B, anti-pY-STAT5, H3K27ac, RNA Pol II | Protein detection, ChIP-seq | Identify STAT5B expression, activation, and genomic localization |
| RNA Analysis Tools | TaqMan probes (Csn1s1, Csn2, Wap, etc.), TruSeq Stranded Total RNA Kit | Gene expression quantification | Measure milk protein gene expression and transcriptional programs |
| Genome Editing Tools | ABE mRNA, sgRNAs, Cas9 protein, single-strand oligonucleotide donors | Model generation | Introduce specific point mutations into endogenous STAT5B locus |
| Histological Reagents | Carmine alum stain, E-cadherin antibodies, α-SMA antibodies | Tissue morphology analysis | Visualize mammary gland structure and cellular organization |
Discussion: Implications for Research and Therapeutics
The comparative analysis of STAT5B SH2 domain mutations reveals how single amino acid substitutions at tyrosine 665 generate profoundly divergent developmental outcomes in mammary gland physiology. The STAT5BY665F gain-of-function mutation enhances STAT5B transcriptional activity through stabilized dimerization and increased DNA binding, resulting in accelerated mammary development and elevated enhancer formation [7] [30]. In stark contrast, the STAT5BY665H loss-of-function mutation impairs STAT5B activation, disrupting the establishment of lactogenic enhancer landscapes and causing complete lactation failure during initial pregnancy [7].
Notably, the mammalian system exhibits remarkable plasticity in compensating for STAT5B deficiency. STAT5BY665H homozygous mutants eventually achieve functional lactation after persistent hormonal stimulation through multiple pregnancies, indicating that sustained endocrine signals can partially overcome the molecular deficit through compensatory mechanisms potentially involving STAT5A or related signaling pathways [7]. This adaptive capacity highlights the robustness of reproductive systems and suggests potential therapeutic avenues for lactation disorders.
These findings extend beyond mammary gland biology to inform drug development strategies targeting STAT5B. The opposing molecular phenotypes arising from mutations at the same residue demonstrate the structural precision of SH2 domain function and illustrate how minor alterations can dramatically rewire transcriptional programs. For researchers investigating STAT5B-associated pathologies, these models provide validated systems for testing therapeutic interventions that either enhance STAT5B function in deficiency states or suppress hyperactive STAT5B in neoplastic contexts. Furthermore, the molecular insights gained from these contrasting mutations illuminate fundamental principles of how somatic mutations fine-tune transcription factor activity to modulate tissue homeostasis and physiological adaptation.
The Signal Transducer and Activator of Transcription (STAT) family of proteins represents critical signaling molecules that mediate cellular responses to cytokines and growth factors. Among these, STAT3 and STAT5B play particularly vital roles in immunity, cellular growth, and survival, with their dysregulation frequently driving oncogenic transformation [3]. The Src Homology 2 (SH2) domain, which arose approximately 600 million years ago within metazoan signaling pathways, serves as a crucial structural and functional module in STAT proteins [3]. This domain facilitates STAT activation through phosphotyrosine-mediated recruitment to cytokine receptors, subsequent tyrosine phosphorylation, and STAT dimerization—events essential for nuclear translocation and transcriptional activity [3] [82]. Recent sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in both STAT3 and STAT5B, with these mutations exhibiting diverse functional consequences across various diseases [3] [23] [82]. This review systematically compares the commonalities and differences between STAT3 and STAT5B SH2 domain mutations, providing a structured analysis of their molecular mechanisms, functional impacts, and clinical implications to inform targeted therapeutic development.
Structural Organization of STAT-Type SH2 Domains
STAT-type SH2 domains share a conserved structural architecture centered on a central anti-parallel β-sheet (comprising βB-βD strands) flanked by two α-helices (αA and αB), forming an αβββα motif [3]. This core structure partitions the domain into two functionally critical subpockets: the phospho-tyrosine (pY) binding pocket and the pY+3 specificity pocket [3]. The pY pocket, formed by the αA helix, BC loop, and one face of the central β-sheet, accommodates the phosphorylated tyrosine residue, while the pY+3 pocket, created by the opposite face of the β-sheet along with residues from the αB helix and CD/BC* loops, determines peptide binding specificity [3].
STAT-type SH2 domains are distinguished from Src-type domains by the presence of a C-terminal α-helix (αB') rather than a β-sheet in what is termed the evolutionary active region (EAR) [3]. This region, along with a hydrophobic system of non-polar residues at the base of the pY+3 pocket, stabilizes the β-sheet and maintains overall SH2 domain integrity [3]. Notably, the αB, αB', and BC* loop also participate in SH2-mediated STAT dimerization through critical cross-domain interactions, giving residues in the pY+3 pocket dual influence over both STAT dimerization capacity and phospho-peptide binding [3].
Table 1: Key Structural Elements of STAT SH2 Domains
| Structural Element | Location | Functional Role | Conservation in STAT3/STAT5B |
|---|---|---|---|
| Central β-sheet (βB-βD) | Core domain | Forms backbone; partitions pY and pY+3 pockets | Highly conserved |
| αA helix | N-terminal to β-sheet | Contributes to pY pocket formation | Highly conserved |
| αB helix | C-terminal to β-sheet | Forms part of pY+3 pocket; dimerization interface | Highly conserved |
| BC loop | Connects βB-βC strands | Forms part of pY pocket; mutational hotspot | Highly conserved |
| pY pocket | Between αA helix and β-sheet | Binds phosphotyrosine moiety | Critical residues conserved |
| pY+3 pocket | Between β-sheet and αB helix | Determines binding specificity | Critical residues conserved |
| EAR (αB' helix) | C-terminal extension | STAT-type specific feature; functional modulation | Conserved with variations |
Despite these shared structural features, STAT SH2 domains exhibit remarkable flexibility, particularly in the accessible volume of the pY pocket, even on sub-microsecond timescales [3]. This inherent dynamics complicates drug discovery efforts, as crystal structures may not preserve targetable pockets in accessible states, underscoring the importance of accounting for protein flexibility in therapeutic development [3].
Mutation Spectra and Hotspots
STAT3 SH2 Domain Mutations
The SH2 domain of STAT3 represents a well-established mutational hotspot in numerous hematologic and immunologic disorders. Patient sequencing has identified multiple point mutations, predominantly clustered in specific regions critical for phosphopeptide binding and dimerization [3] [83]. Key mutational hotspots include residues K591 and R593 in the αA helix; R609, S611, and S614 in the βB strand and BC loop; and E616, G617, and G618 in the BC loop [3]. These mutations manifest in diverse pathologies, with germline mutations typically causing autosomal-dominant Hyper IgE Syndrome (AD-HIES), while somatic mutations drive various malignancies, including T-cell large granular lymphocytic leukemia (T-LGLL), natural killer (NK) cell LGL leukemia, and diffuse large B-cell lymphoma [3].
Notably, the S614R mutation appears in multiple malignancies, including T-LGLL, NK-LGLL, ALK-negative anaplastic large cell lymphoma, and hepatosplenic T-cell lymphoma [3]. Similarly, E616K and E616G mutations have been identified in NK/T-cell lymphoma and diffuse large B-cell lymphoma, respectively [3]. The distribution of these mutations across structural elements highlights the functional importance of specific regions within the SH2 domain, with particular concentration in the pY pocket and adjacent loops that mediate critical protein interactions.
STAT5B SH2 Domain Mutations
STAT5B SH2 domain mutations similarly cluster in specific hotspots, with N642H and Y665 representing the most frequently mutated residues [23] [82]. The N642H mutation is particularly prevalent in γδ-T-cell lymphomas, hepatosplenic T-cell lymphomas, and enteropathy-associated T-cell lymphoma type II [82]. This mutation demonstrates robust oncogenic potential, promoting increased STAT5 phosphorylation, enhanced DNA binding, and upregulation of target genes including IL2Rα, BCL-XL, BCL2, MIR155HG, and HIF2α [82].
The Y665 residue exhibits divergent mutational patterns, with substitution to phenylalanine (Y665F) representing a well-validated gain-of-function mutation identified in T-LGLL and T-cell prolymphocytic leukemia [23] [30]. In contrast, the Y665H substitution demonstrates loss-of-function characteristics despite its initial identification in a T-PLL case [23] [30]. This mutation paradox highlights how different amino acid substitutions at identical residues can produce opposing functional consequences, reflecting the precise structural requirements for STAT activation.
Table 2: Comparative Mutation Profiles of STAT3 and STAT5B SH2 Domains
| Feature | STAT3 | STAT5B |
|---|---|---|
| Key Hotspot Residues | Y640, S614, D661, G618 | N642, Y665 |
| Most Common Mutation | Y640F [82] | N642H [82] |
| Germline Mutation Diseases | AD-HIES [3] | Growth hormone insensitivity (Laron syndrome) [23] [7] |
| Somatic Mutation Diseases | T-LGLL, NK-LGLL, lymphomas [3] [83] | T-LGLL, T-PLL, γδ-T-cell lymphomas [23] [82] |
| GOF/LOF Potential | Both GOF and LOF at same site [3] | Both GOF and LOF possible (e.g., Y665F vs Y665H) [23] |
| Mutation Distribution | Concentrated in pY pocket and BC loop [3] | pY pocket and dimerization interface [23] |
Cross-STAT Mutation Patterns
Comparative analysis reveals both shared and distinct mutational patterns between STAT3 and STAT5B. Both proteins experience mutations that cluster in the pY pocket and adjacent regions, reflecting the critical nature of these areas for STAT function [3] [82]. Additionally, both STATs demonstrate the potential for either gain-of-function (GOF) or loss-of-function (LOF) mutations at identical residues, underscoring the delicate evolutionary balance in wild-type STAT structural motifs [3]. However, disease associations differ, with STAT3 mutations more prevalent in AD-HIES and CD8+ T-LGLL, while STAT5B mutations strongly associate with γδ-T-cell malignancies and growth pathway disorders [3] [23] [82].
Functional Consequences of SH2 Domain Mutations
Molecular Mechanisms of Pathogenicity
SH2 domain mutations in STAT3 and STAT5B exert their functional effects through distinct molecular mechanisms that alter protein dynamics, stability, and interaction networks. For STAT3, SH2 domain GOF mutants exhibit increased homodimer stability, which enhances DNA binding and transcriptional activity [84]. In contrast, SH2 domain LOF mutants demonstrate reduced conformational stability as both monomers and homodimers, leading to impaired phosphopeptide recruitment, tyrosine phosphorylation, dimerization, nuclear localization, and DNA binding [84].
For STAT5B, the molecular mechanisms have been particularly well-characterized for the Y665F and Y665H mutations. Structural modeling indicates that Y665 is located at a critical homodimerization interface [23]. The Y665F substitution promotes intramolecular aromatic stacking interactions with F711, stabilizing the SH2 domain structure and enhancing function [23]. Conversely, the Y665H substitution introduces an imidazole group that destabilizes C-terminal tail binding, resulting in LOF characteristics [23]. Similarly, the prevalent N642H mutation increases binding affinity between the phosphotyrosine (Y699) and the mutant histidine residue, prolonging phospho-STAT5B persistence and enhancing binding to target genomic sites [82].
Signaling and Transcriptional Alterations
Mutational impacts extend to downstream signaling and transcriptional programs, with distinct patterns emerging for STAT3 versus STAT5B. STAT3 GOF mutants drive overexpression of anti-apoptotic (BCL-2, BCL-XL, MCL-1), proliferative (C-MYC, D-type cyclins), and metabolic (HIF) genes [3] [84]. In the immune context, STAT3 LOF mutations impair Th17 differentiation through reduced RORγt expression, diminishing IL-17 and IL-22 production and compromising antimicrobial immunity [3].
STAT5B GOF mutants similarly upregulate proliferative and anti-apoptotic pathways but demonstrate particular potency in modulating enhancer function [7]. The Y665F mutation elevates cytokine-driven enhancer formation in mammary tissue, accelerating development during pregnancy [7]. In contrast, the Y665H mutation impairs enhancer establishment and alveolar differentiation, though persistent hormonal stimulation through multiple pregnancies can partially compensate for this deficit [7].
Cellular and Physiological Impacts
The physiological manifestations of SH2 domain mutations reflect the distinct tissue-specific functions of STAT3 versus STAT5B. STAT3 mutations profoundly impact immune homeostasis, with LOF mutations causing AD-HIES characterized by recurrent infections, eczema, and eosinophilia due to disrupted Th17 development [3]. GOF mutations drive malignant transformation in lymphoid lineages, particularly in T and NK cells [83] [82].
STAT5B mutations exert broad effects on growth, metabolism, and tissue development beyond their oncogenic roles [23] [7]. LOF mutations cause growth hormone insensitivity and immune deficiencies, while GOF mutations promote accumulation of CD8+ effector/memory T cells and CD4+ regulatory T cells, altering CD8+/CD4+ ratios [23]. In mammary tissue, STAT5B GOF mutations accelerate development during pregnancy, while LOF mutations cause lactation failure due to impaired alveolar differentiation [7].
Figure 1: Functional consequences of STAT3 and STAT5B SH2 domain mutations
Diagnostic and Clinical Implications
Disease Associations and Diagnostic Signatures
STAT3 and STAT5B SH2 domain mutations demonstrate distinctive disease associations that inform diagnostic approaches. STAT3 mutations are highly prevalent in T-LGLL (40-73% of cases) and are also found in NK/T-cell lymphomas, γδ-T-cell lymphomas, and inflammatory hepatocellular adenomas [83] [82]. The distribution of specific mutations varies by disease subtype, with D661 and Y640F variants more prevalent in lymphoid neoplasms, while S614R and G618R variants occur in both lymphoid and myeloid neoplasms [85].
STAT5B mutations show strong association with γδ-T-cell malignancies, particularly hepatosplenic T-cell lymphoma and enteropathy-associated T-cell lymphoma type II, where the N642H mutation appears especially frequent [82]. In T-LGLL, STAT5B mutations are relatively rare compared to STAT3 mutations (approximately 4% versus 92% of STAT-mutant LGLLs) and associate with the CD4+ T-LGLL subtype [23] [85].
Discriminatory features in diagnostic sequencing include variant allele frequency (VAF) patterns, with STAT3/STAT5B mutations in LGLLs typically showing VAFs between 5-18%, while myeloid neoplasms demonstrate broader VAF distributions including subclonal populations [85]. Furthermore, LGLLs with STAT3/STAT5B mutations typically show fewer concomitant mutations (1.7 variants per patient versus 4.2 in myeloid neoplasms) and STAT3/STAT5B variants typically represent the founding clone [85].
Table 3: Diagnostic Differentiation of STAT3/STAT5B-Mutant Neoplasms
| Diagnostic Feature | Lymphoid Neoplasms (LGLL) | Myeloid Neoplasms |
|---|---|---|
| STAT3 vs STAT5B Prevalence | STAT3: 92%, STAT5B: 4% [85] | STAT3: 65%, STAT5B: 34% [85] |
| Median VAF of STAT3/STAT5B | 8.8% (range 1.4-48.6%) [85] | 12.0% (range 1.1-65.2%) [85] |
| Concomitant Mutations | 35% of cases [85] | 92% of cases [85] |
| Mutation Burden | 1.7 variants per patient [85] | 4.2 variants per patient [85] |
| Clonal Hierarchy | STAT3/STAT5B as founder clone (100%) [85] | STAT3/STAT5B as founder clone (52%) [85] |
| Karyotype | Normal/low-risk (64%) [85] | Complex karyotypes more frequent (64%) [85] |
Therapeutic Implications and Targeting Strategies
The therapeutic implications of STAT3 versus STAT5B SH2 domain mutations are increasingly informing targeted intervention strategies. For STAT3-driven malignancies, small molecule inhibitors targeting the phosphopeptide-binding pocket show promise, with TTI-101 demonstrating potent inhibition of pY-peptide binding and cell growth driven by STAT3 SH2 domain GOF mutants [84]. Additionally, the STAT3 Y640F mutation has been shown to predict therapeutic response to methotrexate in LGL leukemia, with all patients harboring this mutation responding after at least four treatment cycles [83].
For STAT5B-driven pathologies, JAK1/2 inhibitors partially suppress growth-promoting activity of STAT5B mutants, suggesting potential utility in managing hyperactive STAT5B signaling [82]. However, the differential responses of specific mutations highlight the need for mutation-specific therapeutic approaches. Notably, neither the STAT5B Y665F nor Y665H mutation directly induces malignant transformation in mouse models, despite their clear effects on lymphocyte homeostasis, suggesting that additional cooperating events are necessary for full leukemogenesis [23].
Experimental Approaches and Methodologies
Key Experimental Protocols
Research characterizing STAT3 and STAT5B SH2 domain mutations employs sophisticated experimental approaches spanning structural biology, molecular profiling, and functional validation. Key methodologies include:
Structural Prediction and Energetic Profiling: Computational approaches using AlphaFold3 and COORDinator neural networks predict structural impacts of mutations and calculate energetic contributions of residues to dimerization and domain stability [23]. These in silico methods enable pathogenicity prediction and mechanistic hypothesis generation.
Site-Directed Mutagenesis and Functional Characterization: Introduction of specific mutations into STAT genes via CRISPR/Cas9 and base editing technologies, followed by comprehensive functional assessment [23] [7]. This includes measurement of phosphorylation kinetics, DNA binding capacity (EMSA), nuclear translocation (imaging), and transcriptional activity (reporter assays).
Transcriptomic and Epigenomic Profiling: RNA sequencing and chromatin immunoprecipitation with sequencing (ChIP-seq) identify altered gene expression programs and enhancer landscapes driven by STAT mutants [7] [82]. These approaches reveal how GOF versus LOF mutations reshape the regulatory genome.
Primary Cell and Animal Modeling: Introduction of human STAT mutations into mouse genomes via CRISPR/Cas9 and base editing in C57BL/6 N mice [7]. These models enable physiological assessment of mutation impacts on immune function, mammary development, and overall organismal homeostasis.
In Vitro Functional Assays: Lentiviral transduction of STAT mutants into cell lines (e.g., KAI3 NK cells) and primary human NK cells with growth monitoring under limiting cytokine conditions [82]. Western blotting assesses phosphorylation status, while ChIP-qPCR quantifies DNA binding at specific target loci.
The Scientist's Toolkit
Table 4: Essential Research Reagents and Experimental Tools
| Reagent/Technology | Specific Application | Function and Utility |
|---|---|---|
| AlphaFold3 | Structural prediction | Models SH2 domain structures and dimer interfaces [23] |
| COORDinator | Energetic calculation | Predicts stability effects of amino acid substitutions [23] |
| CRISPR/Cas9 with Base Editing | Mouse model generation | Introduces precise human mutations into mouse genome [7] |
| Adenine Base Editor (ABE 7.10) | Y665H mutation modeling | Converts A•T to G•C base pairs for specific mutation introduction [7] |
| ChIP-seq | Enhancer mapping | Identifies STAT5B-bound genomic regions and enhancer landscapes [7] |
| RNA-seq | Transcriptome profiling | Reveals global gene expression changes in mutant tissues [7] |
| JAK1/2 Inhibitors | Pathway inhibition | Tests dependency on JAK-STAT signaling in mutant cells [82] |
| TTI-101 | STAT3-specific inhibition | Targets SH2 domain to block pY-peptide binding [84] |
| Phospho-STAT Antibodies | Activation assessment | Measures phosphorylation status via Western blot [82] |
Figure 2: Integrated experimental workflow for characterizing STAT SH2 domain mutations
The comparative analysis of STAT3 and STAT5B SH2 domain mutations reveals a complex landscape of shared and distinct pathogenic mechanisms. Both STATs experience mutational clustering in the pY pocket and critical dimerization interfaces, with single residue substitutions capable of producing either GOF or LOF consequences depending on the specific amino acid change [3] [23]. However, the disease associations and physiological impacts diverge, reflecting the unique biological functions of each STAT family member. STAT3 mutations predominantly affect immune homeostasis and drive lymphoid malignancies [3] [83], while STAT5B mutations additionally disrupt growth pathways, metabolism, and mammary development [23] [7].
Structurally, both STATs rely on SH2 domain integrity for phosphopeptide binding, dimerization, and nuclear function, yet the precise molecular mechanisms differ. STAT3 pathogenesis is closely linked to altered stability of monomers and homodimers [84], while STAT5B mutations particularly impact enhancer establishment and chromatin remodeling [7]. These distinctions inform therapeutic strategies, with STAT3 showing susceptibility to SH2 domain-targeted inhibitors like TTI-101 [84], while STAT5B-driven signaling remains partially dependent on JAK kinase activity [82].
Future research directions should include comprehensive structural studies of mutant STAT complexes, development of mutation-specific therapeutic agents, and exploration of combinatorial treatment approaches targeting both STAT proteins and cooperating signaling pathways. The continued refinement of experimental models, particularly those incorporating physiological cytokine signaling and tissue microenvironmental factors, will be essential for translating mechanistic insights into targeted clinical interventions for STAT-driven diseases.
The Src Homology 2 (SH2) domain is a critical regulatory module found in numerous signaling proteins, including STAT (Signal Transducer and Activator of Transcription) family transcription factors. It specifically recognizes and binds to phosphorylated tyrosine residues, facilitating the assembly of multiprotein signaling complexes and controlling pivotal cellular processes such as proliferation, differentiation, and immune responses [31]. Research into activating versus inactivating STAT SH2 domain mutations provides a powerful framework for understanding how discrete molecular alterations drive divergent clinical phenotypes in hematologic malignancies, particularly T-cell large granular lymphocyte leukemia (T-LGLL). This domain, approximately 100 amino acids in length, maintains a highly conserved structure—a sandwich of antiparallel beta-sheets flanked by alpha-helices—with an invariant arginine residue in the βB strand that is essential for phosphotyrosine binding [31]. The functional integrity of the SH2 domain is paramount for STAT protein dimerization, nuclear translocation, and the transcriptional regulation of target genes. Mutations disrupting this domain can therefore fundamentally rewire cellular signaling networks, creating a direct link between molecular lesion and disease pathology that serves as an exemplary model for bench-to-bedside correlation.
Molecular Mechanisms: Structural and Functional Impact of STAT Mutations
SH2 Domain Dynamics and Mutational Hotspots
The SH2 domain of STAT proteins, particularly STAT3 and STAT5B, serves as a critical hub for regulating transcriptional activity through its role in cytokine-induced phosphorylation, dimerization, and nuclear translocation. Structural analyses reveal that the SH2 domain forms a highly conserved protein-interaction module characterized by a three-stranded antiparallel beta-sheet flanked by two alpha-helices [31]. This architecture creates a deep binding pocket that recognizes phosphotyrosine motifs through a conserved arginine residue (βB5) within the FLVR signature motif. In STAT proteins, this domain mediates both receptor interaction and the reciprocal phosphotyrosine-SH2 engagement that stabilizes active transcription factor dimers. The tyrosine 665 (Y665) residue in STAT5B represents a critical mutational hotspot located at the dimerization interface, where structural alterations can profoundly influence protein function [23]. Computational modeling and experimental data indicate that substitutions at this position can either stabilize or destabilize the homodimer interface and intramolecular interactions with phenylalanine 711 (F711), leading to either constitutive activation or functional impairment [23].
Functional Consequences of Specific Mutations
Research has elucidated how specific amino acid substitutions at critical positions generate divergent functional outcomes. The STAT5B Y665F mutation (tyrosine to phenylalanine) demonstrates gain-of-function (GOF) properties through enhanced STAT5 phosphorylation, increased DNA binding capacity, and elevated transcriptional activity following cytokine stimulation [23] [7]. In silico analyses predict that this substitution promotes intramolecular aromatic stacking interactions with F711, thereby stabilizing the active conformation. Conversely, the STAT5B Y665H mutation (tyrosine to histidine) exhibits loss-of-function (LOF) characteristics, with the introduced imidazole group destabilizing binding of the C-terminal tail and impairing dimerization capability [23] [7]. This mutation results in diminished CD8+ effector and memory T cells and reduced CD4+ regulatory T cells in mouse models, reflecting its impaired transcriptional activity. Similarly, in STAT3, the N646H mutation within the SH2 domain represents a frequent GOF alteration that promotes constitutive dimerization and signaling, driving oncogenic programs in T-LGLL [86] [87].
Table 1: Functional Characteristics of STAT SH2 Domain Mutations in T-LGLL
| Mutation | Type | Structural Impact | Functional Consequence | Transcriptional Activity |
|---|---|---|---|---|
| STAT5B Y665F | Gain-of-Function | Stabilizes dimer interface; enhanced intramolecular stacking | Increased phospho-STAT5, DNA binding, and transcriptional activation | Enhanced STAT5-responsive gene expression |
| STAT5B Y665H | Loss-of-Function | Disrupts C-terminal tail binding; impairs dimerization | Reduced phospho-STAT5, diminished DNA binding capacity | Impaired STAT5-responsive gene expression |
| STAT3 N646H | Gain-of-Function | Promotes constitutive dimerization | Enhanced STAT3 phosphorylation and nuclear translocation | Upregulation of proliferation and survival genes |
Signaling Pathway Dysregulation
The following diagram illustrates how STAT SH2 domain mutations disrupt normal JAK-STAT signaling and contribute to T-LGLL pathogenesis:
Diagram 1: JAK-STAT signaling pathway disruption by SH2 domain mutations. GOF mutations cause constitutive activation promoting survival and dysregulation, while LOF mutations impair dimerization.
Experimental Approaches: Methodologies for Characterizing STAT Mutations
Computational and Structural Analysis Protocols
In silico modeling approaches provide the initial framework for hypothesizing functional impacts of STAT mutations. The experimental workflow typically begins with structural prediction using AlphaFold3 to generate high-confidence models of SH2 domain homodimers, identifying critical interfacial residues like Y665 in STAT5B [23]. Researchers then employ computational tools such as COORDinator to predict energetic contributions of specific residues to dimer stability, comparing configurations with and without homodimeric counterparts to distinguish dimerization-specific effects from general domain stability [23]. Pathogenicity assessment utilizes multiple prediction algorithms including AlphaMissense for functional impact scores, CADD (Combined Annotation Dependent Depletion) for deleteriousness prediction (with scores above 20 considered potentially impactful), and REVEL (Rare Exome Variant Ensemble Learner) for pathogenicity probability [23]. These computational approaches guide subsequent experimental design by generating testable hypotheses about mutation effects.
Functional Validation in Cellular and Animal Models
Primary T-cell assays represent a critical methodology for validating computational predictions. The standard protocol involves introducing STAT mutations into primary human T-cells via viral transduction, followed by stimulation with relevant cytokines (e.g., IL-2). Functional readouts include quantification of STAT phosphorylation by flow cytometry or Western blot, electrophoretic mobility shift assays (EMSAs) to assess DNA binding capacity, and luciferase reporter assays measuring transcriptional activity of STAT-responsive promoters [23]. For in vivo validation, researchers employ knock-in mouse models generated through CRISPR/Cas9 and base editing techniques to introduce precise human mutations (e.g., STAT5B Y665F or Y665H) into the mouse genome [23] [7]. Phenotypic characterization includes comprehensive immunophenotyping of T-cell subsets (CD4+, CD8+, regulatory T-cells), assessment of effector and memory cell populations, and evaluation of pathological consequences such as clonal expansions resembling human T-LGLL.
Transcriptomic and Epigenomic Profiling
RNA-sequencing of purified T-LGLs from patient subgroups stratified by STAT mutation status provides insights into pathway dysregulation. The standard methodology involves magnetic bead-based purification of T-LGLs from peripheral blood mononuclear cells (achieving >98% purity), followed by RNA extraction with quality assessment (RIN >8.0), library preparation with ribosomal RNA depletion, and high-throughput sequencing on platforms such as Illumina HiSeq3000 [86]. Bioinformatics pipelines typically include alignment with BWA or STAR, transcript quantification with StringTie, differential expression analysis with DESeq2, and gene set enrichment analysis (GSEA) to identify dysregulated pathways [86]. For epigenomic assessment, ChIP-sequencing for histone modifications (H3K27ac) and STAT binding identifies enhancer and super-enhancer alterations, revealing how mutations rewire the regulatory landscape in T-LGLL [7].
Table 2: Key Experimental Methods for STAT Mutation Analysis
| Method Category | Specific Techniques | Primary Readouts | Utility in STAT Research |
|---|---|---|---|
| Computational Analysis | AlphaFold3, COORDinator, AlphaMissense, CADD, REVEL | Structural models, stability predictions, pathogenicity scores | Initial mutation characterization and hypothesis generation |
| In Vitro Cellular Assays | Viral transduction, phospho-flow cytometry, EMSA, luciferase reporter | Phosphorylation status, DNA binding, transcriptional activity | Functional validation of mutation effects in relevant cell types |
| Animal Models | CRISPR/Cas9 knock-in, base editing, immunophenotyping | T-cell subsets, clonal expansions, pathological manifestations | In vivo validation of physiological and pathological impacts |
| Omics Profiling | RNA-seq, ChIP-seq, TCR-seq | Gene expression, enhancer activity, clonality | Systems-level understanding of downstream effects |
Clinical Correlations: From Molecular Lesions to Patient Presentation
Genotype-Clinical Feature Relationships
The distinct molecular properties of STAT SH2 domain mutations translate into specific clinical manifestations in T-LGLL patients. STAT3 mutations, predominantly found in CD8+ T-LGLL, associate with symptomatic disease characterized by severe neutropenia and autoimmune manifestations, particularly rheumatoid arthritis [86] [88] [87]. Transcriptomic profiling reveals that CD8+ STAT3-mutated cases display extensive gene expression dysregulation with upregulation of oncogenic pathways including EZH2 and MDM2, and de-repression of proliferation and cell cycle pathways [86]. This molecular signature correlates with more aggressive clinical behavior and increased treatment requirements. In contrast, STAT5B mutations occur more frequently in CD4+ T-LGLL and typically follow an indolent clinical course [86] [87]. The transcriptional impact of STAT5B mutations is more limited, with PIM1 serine/threonine kinase overexpression identified as a relevant feature in STAT5B-mutated CD4+ T-LGLL [86]. This genotypic-clinical correlation underscores how different STAT family members, despite structural similarities, drive distinct disease entities with unique presentation and management considerations.
Immunological and Hematological Manifestations
The functional impact of STAT mutations directly shapes the immunological landscape and hematological manifestations in T-LGLL. GOF mutations promote clonal expansion of cytotoxic T lymphocytes through enhanced survival signaling and resistance to apoptosis [88] [87]. These expanded clones exhibit sustained JAK-STAT pathway activation regardless of STAT mutation status, suggesting both mutation-dependent and microenvironment-driven mechanisms [89] [87]. The resulting clinical picture often includes cytopenias—particularly neutropenia and anemia—through multiple proposed mechanisms including Fas/Fas-ligand mediated neutrophil apoptosis, humoral factor secretion, and direct bone marrow suppression [87]. Notably, a paradoxical combination of clonal expansion alongside broader immunodeficiency features is frequently observed, with approximately 77% of T-LGLL patients exhibiting lymphocytopenia and/or hypogammaglobulinemia [89]. This suggests that maladaptive CTL expansions in T-LGLL may stem from underlying immunodeficiency traits, with recent research identifying inborn errors of immunity (IEI) variants in 36% of patients [89].
Therapeutic Implications and Research Applications
Targeting JAK-STAT Signaling in T-LGLL
The central role of JAK-STAT signaling in T-LGLL pathogenesis makes this pathway an attractive therapeutic target. Current approaches include JAK inhibitors that target upstream kinase activity, though their efficacy varies based on mutation status. For STAT3-mutated cases, direct STAT3 inhibitors represent a more targeted strategy, with compounds like Stattic demonstrating ability to induce apoptosis in leukemic LGLs in experimental models [86]. The recognition that epigenetic vulnerabilities coexist with JAK-STAT dysregulation has prompted investigation of combination therapies targeting multiple pathways simultaneously [90]. Notably, T-cell malignancies demonstrate marked sensitivity to epigenetically targeted drugs including histone deacetylase (HDAC) inhibitors and EZH2 inhibitors, with emerging data suggesting combinations of epigenetic agents may potentially replace historical chemotherapy regimens [90]. This therapeutic approach aligns with the understanding that T-cell neoplasms represent prototypical epigenetic diseases enriched for mutations in genes governing epigenetic biology, including TET2, DNMT3A, and IDH2 [90].
Research Toolkit for STAT SH2 Domain Investigation
The following table outlines essential research reagents and methodologies for investigating STAT SH2 domain mutations:
Table 3: Research Reagent Solutions for STAT SH2 Domain Studies
| Research Tool Category | Specific Examples | Application/Function | Experimental Context |
|---|---|---|---|
| Computational Prediction Tools | AlphaFold3, COORDinator, AlphaMissense, CADD, REVEL | Structural modeling, stability prediction, pathogenicity assessment | Initial characterization of novel STAT variants |
| Cell-based Assay Systems | Primary T-cell transduction, Luciferase reporter constructs, EMSA | Functional validation of phosphorylation, dimerization, DNA binding | In vitro assessment of mutation impact on STAT function |
| Animal Models | STAT5B Y665F/Y665H knock-in mice, Immunodeficient mouse reconstitution | In vivo pathophysiological validation, preclinical therapeutic testing | Physiological context for mutation effects on immune function and transformation |
| Signaling Inhibitors | Stattic (STAT3 inhibitor), JAK inhibitors (e.g., tofacitinib), HDAC inhibitors | Pathway modulation, functional rescue experiments, therapeutic targeting | Mechanistic studies and preclinical therapeutic development |
| Omics Technologies | RNA-seq, ChIP-seq, Whole exome sequencing, TCR-seq | Comprehensive molecular profiling, pathway analysis, clonality assessment | Systems-level understanding of mutation impacts across biological layers |
The investigation of STAT SH2 domain mutations in T-LGLL provides a compelling paradigm for translational research, demonstrating how precise molecular alterations dictate clinical disease phenotypes. The structural-functional continuum from atomic-level interactions in the SH2 domain dimerization interface to systemic clinical manifestations offers a complete bench-to-bedside correlation model. Research in this area continues to evolve, with emerging evidence suggesting maladaptive CTL expansions in T-LGLL may originate from cryptic immunodeficiency traits, opening new horizons connecting inborn errors of immunity to clonal hematopoiesis and bone marrow failure [89]. Future research directions include developing more selective STAT inhibitors, exploring rational combination therapies targeting parallel survival pathways, and leveraging multi-omics profiling to identify patient subgroups most likely to benefit from specific interventions. The continued dissection of how specific SH2 domain mutations rewire cellular signaling networks will undoubtedly yield further insights with broad implications for precision oncology across hematologic malignancies.
Conclusion
The characterization of STAT SH2 domain mutations reveals a delicate structural balance where single amino acid substitutions can push signaling into opposing pathological states, exemplified by the Y665F (GOF) and Y665H (LOF) variants. A multidisciplinary approach, integrating cutting-edge computational predictions with robust in vitro and in vivo validation, is paramount for accurate functional annotation. These findings underscore the SH2 domain as a critical therapeutic node. Future research must focus on elucidating the full spectrum of mutations in diverse physiological contexts, understanding their role in condensate formation via phase separation, and accelerating the development of targeted inhibitors that can selectively correct pathological signaling driven by these mutational events, thereby enabling new strategies for precision oncology and immunology.
References
- 1. The JAK/STAT signaling pathway: from bench to clinic [https://www.nature.com/articles/s41392-021-00791-1]
- 2. Evolution of the JAK-STAT pathway - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3670263/]
- 3. Structural Implications of STAT3 and STAT5 SH2 Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6895964/]
- 4. SnapShot: Jak-STAT signaling II - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11533724/]
- 5. Identification of the linker-SH2 domain of STAT as ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/15073273/]
- 6. Identification of the Linker-SH2 Domain of STAT as ... [https://www.sciencedirect.com/science/article/pii/S1535947620338639]
- 7. Disease-Associated Mutations of the STAT5B SH2 Domain ... [https://link.springer.com/article/10.1007/s10911-025-09582-8]
- 8. The role of JAK-STAT signaling pathway and its regulators in ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-017-0177-y]
- 9. Spotlight on amino acid changing mutations in the JAK ... [https://www.nature.com/articles/s41598-025-90788-5]
- 10. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6895964/]
- 11. SH2 domains serve as lipid binding modules for pTyr ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4826312/]
- 12. Novel Roles of SH2 and SH3 Domains in Lipid Binding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8152464/]
- 13. Binding Specificity of SH2 Domains: Insight from Free Energy Simulations - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2635922/]
- 14. Crystal Structure of a Tyrosine Phosphorylated STAT-1 ... [https://www.sciencedirect.com/science/article/pii/S0092867400814439]
- 15. Kinase Activation through Dimerization by Human SH2-B - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1061652/]
- 16. Structural insights into the intertwined dimer of fyn SH2 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4815226/]
- 17. The Coiled-Coil Domain of Stat3 Is Essential for Its SH2 Domain-Mediated Receptor Binding and Subsequent Activation Induced by Epidermal Growth Factor and Interleukin-6 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC86266/]
- 18. Gain-of-Function Mutations: An Emerging Advantage for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6944532/]
- 19. Gain of Function (GOF) Mutant p53 in Cancer—Current ... [https://www.mdpi.com/1422-0067/23/21/13287]
- 20. Deep mutational scanning of the multi-domain ... [https://www.nature.com/articles/s41467-025-60641-4]
- 21. JAK-STAT signaling pathway [https://en.wikipedia.org/wiki/JAK-STAT_signaling_pathway]
- 22. Small molecule drug discovery targeting the JAK-STAT ... [https://pubmed.ncbi.nlm.nih.gov/38777110/]
- 23. STAT5B leukemic mutations, altering SH2 tyrosine 665, have ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11999048/]
- 24. Structural Implications of STAT3 and STAT5 SH2 Domain ... [https://www.mdpi.com/2072-6694/11/11/1757]
- 25. STAT5B leukemic mutations, altering SH2 tyrosine 665, have ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11722272/]
- 26. Disease-Associated Mutations of the STAT5B SH2 Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11958444/]
- 27. Y665F variant of mouse Stat5b protects against acute ... [https://www.nature.com/articles/s41598-025-15812-0]
- 28. Update on Structure and Function of SH2 Domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC12470777/]
- 29. The SH2 Domain–Containing Proteins in 21 Species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4255630/]
- 30. STAT5B leukemic mutations, altering SH2 tyrosine 665 ... [https://www.life-science-alliance.org/content/8/7/e202503222]
- 31. Update on Structure and Function of SH2 Domains [https://www.mdpi.com/1422-0067/26/18/9060]
- 32. A unified analysis of evolutionary and population constraint ... [https://www.nature.com/articles/s42003-024-06117-5]
- 33. Evaluating novel in silico tools for accurate pathogenicity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11647429/]
- 34. AI cancer driver mutation predictions are valid in real-world ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474978/]
- 35. AlphaMissense [https://alphamissense.hegelab.org/]
- 36. Evaluating the Utility of REVEL and CADD for Interpreting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11919158/]
- 37. Comparison of Pathogenicity Prediction Tools on Somatic ... [https://www.sciencedirect.com/science/article/pii/S1525157820304839]
- 38. Evaluation of in silico pathogenicity prediction tools for the ... [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-023-01454-6]
- 39. GSE276312 - GEO Accession viewer - NIH [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE276312]
- 40. Accurate structure prediction of biomolecular interactions ... [https://www.nature.com/articles/s41586-024-07487-w]
- 41. AlphaFold3: An Overview of Applications and Performance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12027460/]
- 42. AlphaFold3 prediction of protein-protein complex: Is it ... [https://www.sciencedirect.com/science/article/pii/S2001037025004180]
- 43. Center for Computational Structural Biology [https://ccsb.scripps.edu/]
- 44. Methods for Detecting Protein Phosphorylation [https://www.bio-techne.com/resources/scientific-articles/methods-for-detecting-protein-phosphorylation]
- 45. DNA–protein interaction studies: a historical and comparative ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-021-00780-z]
- 46. Three assays show differences in binding of wild-type ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2917581/]
- 47. Transcription factor enrichment analysis (TFEA) quantifies ... [https://www.nature.com/articles/s42003-021-02153-7]
- 48. Predicting transcription factor activity using prior biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10918219/]
- 49. Mutational analysis of the STAT6 SH2 domain [https://pubmed.ncbi.nlm.nih.gov/9651359/]
- 50. In Vivo Analysis of Growth Hormone Receptor Signaling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC538772/]
- 51. A novel small-molecule disrupts Stat3 SH2 domain– ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295210000043]
- 52. Transcriptomics and epigenetic data integration learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11299028/]
- 53. Transcriptomics and epigenetic data integration learning ... [https://pubmed.ncbi.nlm.nih.gov/39101486/]
- 54. Biophysics-based protein language models for ... [https://www.nature.com/articles/s41592-025-02776-2]
- 55. Recommendations for the collection and use of multiplexed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6925490/]
- 56. SH2 Domains: Folding, Binding and Therapeutical Approaches [https://pmc.ncbi.nlm.nih.gov/articles/PMC9783222/]
- 57. Accurate affinity models for SH2 domains from peptide binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12521615/]
- 58. A Mutation in the SH2 Domain of STAT2 Prolongs Tyrosine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1924825/]
- 59. STAT5A and STAT5B—Twins with Different Personalities ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6895831/]
- 60. STAT5b: A master regulator of key biological pathways - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9899847/]
- 61. Article Stat5a and Stat5b Proteins Have Essential ... [https://www.sciencedirect.com/science/article/pii/S0092867400814440]
- 62. Differentiating the roles of STAT5B and STAT5A in human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4169138/]
- 63. Implications of STAT3 and STAT5 signaling on gene ... [https://www.nature.com/articles/s41375-018-0117-x]
- 64. Identification of STAT5A and STAT5B Target Genes in Human ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0086790]
- 65. Phosphotyrosine recognition domains: the typical, the atypical ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-10-32]
- 66. SH2 Domains Recognize Contextual Peptide Sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2984226/]
- 67. The SH2 domain interaction landscape - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4110347/]
- 68. SH2 Domain - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/sh2-domain]
- 69. Mutational Analysis of the STAT6 SH2 Domain [https://www.sciencedirect.com/science/article/pii/S002192581880535X]
- 70. Targeting Shp2 as a therapeutic strategy for ... [https://www.nature.com/articles/s41398-024-03222-1]
- 71. The SH2 Domain Interaction Landscape [https://www.sciencedirect.com/science/article/pii/S2211124713001083]
- 72. Membrane-associated phase separation: organization and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8339363/]
- 73. Liquid–liquid phase separation in human health and ... [https://www.nature.com/articles/s41392-021-00678-1]
- 74. Visualizing liquid-liquid phase separation and protein ... [https://www.nature.com/articles/s42004-025-01756-z]
- 75. Identification of novel STAT5B mutations and ... [https://www.nature.com/articles/s41408-022-00630-8]
- 76. Discovery of somatic STAT5b mutations in large granular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3668487/]
- 77. STAT5B leukemic mutations, altering SH2 tyrosine 665, have ... [https://pubmed.ncbi.nlm.nih.gov/39803507/]
- 78. Structural and functional consequences of the STAT5B ... [https://www.nature.com/articles/s41467-019-10422-7]
- 79. Systems Immunology Analyses of STAT1 Gain-of-Function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9623573/]
- 80. Single-cell characterization of leukemic and non ... [https://www.nature.com/articles/s41467-022-29173-z]
- 81. Alterations of T Cell Mediated Immunity in Acute Myeloid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7234277/]
- 82. Activating mutations of STAT5B and STAT3 in lymphomas ... [https://www.nature.com/articles/ncomms7025]
- 83. Activating somatic mutations outside the SH2-domain of ... [https://www.nature.com/articles/leu2015263]
- 84. Aberrant function of pathogenic STAT3 mutant proteins is ... [https://www.sciencedirect.com/science/article/pii/S0006497122077667]
- 85. Distinguishing STAT3/STAT5B-mutated large granular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9813527/]
- 86. Transcriptomic landscape of CD8+ and CD4 + T-LGL ... [https://www.nature.com/articles/s41375-025-02708-1]
- 87. Large Granular Lymphocytic Leukemia: Clinical Features, ... [https://www.mdpi.com/2072-6694/16/7/1307]
- 88. Intersection Between Large Granular Lymphocyte ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.869205/full]
- 89. Inborn errors of immunity underlie clonal T cell expansions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12043085/]
- 90. Management of T‐cell malignancies - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12223354/]