SDS in PAGE: The Definitive Guide to Denaturation, Separation, and Analysis for Life Science Researchers
This article provides a comprehensive examination of the role of Sodium Dodecyl Sulfate (SDS) in Polyacrylamide Gel Electrophoresis (PAGE), a cornerstone technique in biochemistry and molecular biology.
SDS in PAGE: The Definitive Guide to Denaturation, Separation, and Analysis for Life Science Researchers
Abstract
This article provides a comprehensive examination of the role of Sodium Dodecyl Sulfate (SDS) in Polyacrylamide Gel Electrophoresis (PAGE), a cornerstone technique in biochemistry and molecular biology. Tailored for researchers, scientists, and drug development professionals, the content spans from foundational principles to advanced applications. It covers the mechanism by which SDS denatures proteins and confers uniform charge, the standard SDS-PAGE protocol and its critical steps, common troubleshooting scenarios for poor band resolution, and a comparative analysis with alternative electrophoretic methods like Native PAGE and BN-PAGE. The article also explores innovative adaptations, such as NSDS-PAGE, that preserve protein function, highlighting the technique's evolving role in proteomics and diagnostic research.
SDS Unmasked: How a Simple Detergent Enables Pure Size-Based Protein Separation
Sodium dodecyl sulfate (SDS) is an anionic detergent that serves a critical function in molecular biology by denaturing proteins and conferring upon them a uniform negative charge. This foundational process enables the separation of complex protein mixtures by molecular weight via polyacrylamide gel electrophoresis (SDS-PAGE). This technical guide explores the chemical properties and mechanisms of SDS, detailing its indispensable role in protein analysis. Framed within the context of proteomic research and drug development, we provide detailed methodologies, quantitative data on SDS-protein interactions, and essential resource guidance for research implementation, underscoring how SDS-PAGE remains a cornerstone technique for protein characterization, purity assessment, and diagnostic applications.
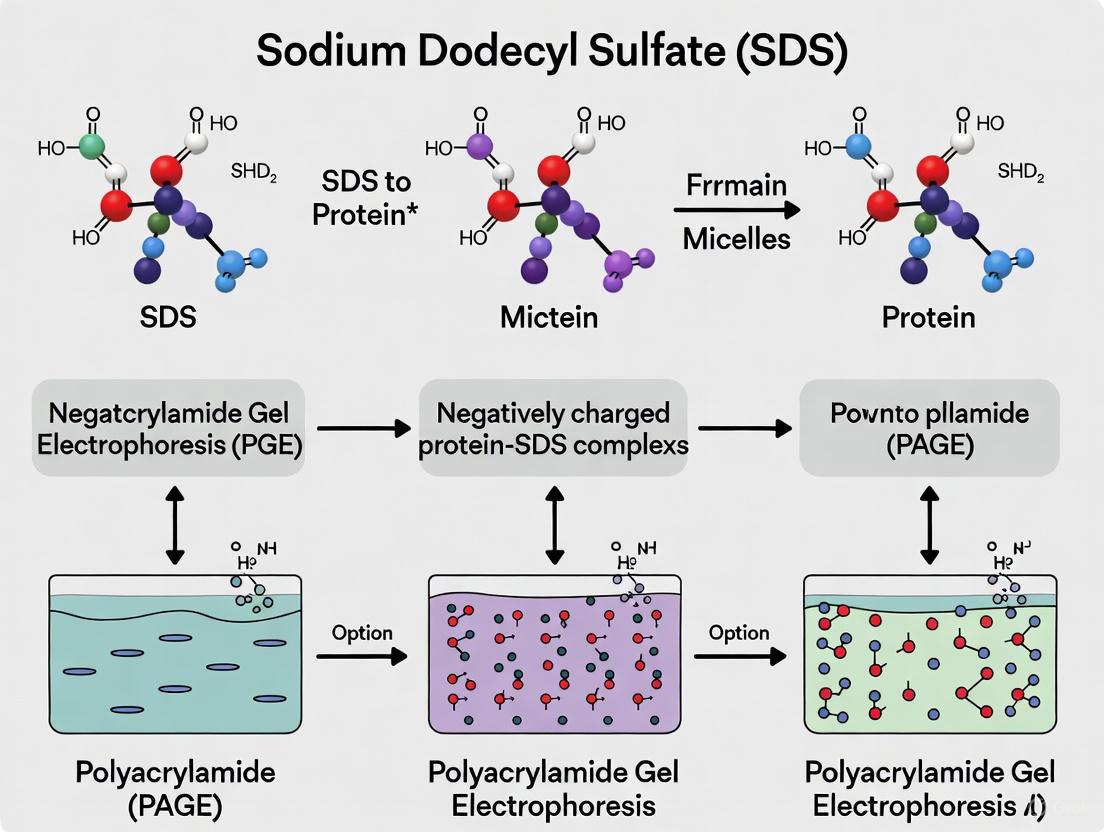
In the realm of protein biochemistry, the ability to separate, visualize, and analyze proteins based on molecular weight is a fundamental requirement. SDS-PAGE fulfills this need, and its efficacy hinges almost entirely on the properties of sodium dodecyl sulfate (SDS). This anionic surfactant is integral to the technique's name and function [1]. The primary objective of SDS-PAGE is to separate proteins solely on the basis of their molecular weight, eliminating confounding variables such as innate protein charge or three-dimensional shape [2]. SDS achieves this through a dual mechanism: it efficiently denatures protein structures, unfolding them into linear polypeptides, and simultaneously coats them with a uniform negative charge [3] [4]. This process ensures that when an electric field is applied, all proteins migrate through the polyacrylamide gel matrix toward the anode at a rate inversely proportional to their size [5]. The critical role of SDS extends across basic research, clinical diagnostics, and biopharmaceutical development, making a thorough understanding of its action essential for research scientists and drug development professionals.
Chemical Identity and Core Properties of SDS
SDS is a key member of the alkyl sulfate family of anionic detergents. Its amphipathic nature is defined by a distinct hydrophobic "tail", a 12-carbon alkyl chain (dodecyl), and a hydrophilic "head", the sulfate group [1] [4]. This structure is the source of its protein-denaturing power and its effectiveness as a surfactant.
In aqueous solutions, SDS molecules exhibit critical aggregation behavior. Below the critical micelle concentration (CMC) of 7 to 10 millimolar, SDS exists predominantly as monomers. However, above the CMC, SDS monomers self-assemble into spherical micelles, with each micelle consisting of approximately 62 SDS molecules [6]. It is crucial to note that only the SDS monomers are responsible for binding to and denaturing proteins, while the micelles remain in solution and do not adsorb proteins [6].
Table 1: Fundamental Physicochemical Properties of SDS
| Property | Description | Significance in Protein Analysis |
|---|---|---|
| Chemical Name | Sodium Dodecyl Sulfate | Anionic detergent used for protein denaturation [6]. |
| Molecular Structure | Amphipathic: 12-carbon hydrophobic tail, anionic sulfate head group [1]. | Tail disrupts hydrophobic protein core; head confers charge [1]. |
| Critical Micelle Concentration (CMC) | 7-10 mM [6] | Monomers below CMC denature proteins; micelles form above this concentration [6]. |
| Micelle Structure | ~62 molecules per spherical micelle [6] | Micelles do not bind protein backbones but are part of the electrophoretic system [6]. |
The hydrophobic tail readily interacts with nonpolar regions of proteins, while the ionic sulfate group disrupts electrostatic interactions and provides a strong negative charge. This combination is the foundation for SDS's potent denaturing capability and its ability to mask a protein's intrinsic charge [1].
The Dual Mechanism of SDS Action
SDS exerts its effect on proteins through two synergistic mechanisms that are essential for successful electrophoretic separation.
Protein Denaturation and Unfolding
SDS fundamentally disrupts the native structure of proteins. Its hydrophobic region interacts with and embeds into the hydrophobic core of the protein, while its ionic part disrupts salt bridges and other non-covalent interactions that stabilize secondary and tertiary structures [1]. This results in the loss of a protein's higher-order structures—unfolding it into a random coil or rigid rod-like conformation [4]. For complete denaturation, SDS treatment is typically combined with heat (95°C for several minutes) to break hydrogen bonds, and reducing agents like β-mercaptoethanol (BME) or dithiothreitol (DTT) to cleave disulfide bonds, thereby linearizing the polypeptide into its primary structure [1].
Charge Conferment and Masking
Following denaturation, SDS binds to the unfolded protein backbone at a nearly constant weight ratio of 1.4 grams of SDS per 1 gram of protein [6] [3]. This corresponds to approximately one SDS molecule for every two amino acid residues [6]. This saturation binding coats the entire polypeptide in a uniform layer of negative charge. The intrinsic charge of the amino acids becomes negligible compared to the overwhelming negative charge provided by the bound SDS [6] [2]. Consequently, all SDS-protein complexes possess a similar net negative charge and a nearly identical charge-to-mass ratio, ensuring that during electrophoresis, separation is based solely on molecular size and not on the protein's original charge or shape [3] [1].
Diagram 1: SDS-Mediated Protein Denaturation and Linearization Pathway
SDS-PAGE: Experimental Protocol and Workflow
The following section outlines a standard protocol for SDS-PAGE, highlighting the critical role of SDS at each stage. This procedure is adaptable for both mini-gel (8 x 8 cm) and larger formats [2].
Sample Preparation
- Lysate Preparation: Solubilize proteins from whole tissue or cell culture using mechanical homogenization (e.g., blender, sonicator) in a lysis buffer containing detergents and protease inhibitors [7].
- Protein Quantification: Determine the protein concentration of the lysate using a standard assay (e.g., BCA, Bradford).
- Denaturation: Mix the protein sample with an SDS-based sample buffer (Laemmli buffer). A typical buffer contains:
- Heat Denaturation: Heat the sample-protein buffer mixture at 95°C for 3-5 minutes (or 70°C for 10 minutes) to complete the denaturation process [5] [7]. Cool briefly and centrifuge before loading.
Gel Casting and the Discontinuous System
SDS-PAGE employs a discontinuous buffer system using two distinct gel layers to achieve high-resolution separation [4].
- Resolving Gel (Separating Gel): This lower gel is poured first. It typically has a higher acrylamide concentration (8-15%, depending on target protein size) and a pH of 8.8. This gel is responsible for the size-based separation of proteins [5] [1].
- Stacking Gel: This upper gel is poured after the resolving gel has polymerized. It has a lower acrylamide concentration (4-5%) and a pH of 6.8. Its function is to concentrate all protein samples into a sharp, unified band before they enter the resolving gel, resulting in tighter, clearer bands [1] [4].
The polymerization of both gels is catalyzed by ammonium persulfate (APS) and TEMED, which generate free radicals to initiate the cross-linking of acrylamide and bisacrylamide monomers [1].
Electrophoresis and the Role of Buffers
- Assembly: Place the cast gel cassette into the electrophoresis chamber and fill the upper and lower chambers with running buffer.
- Running Buffer Composition: The standard running buffer contains Tris, glycine, and SDS at pH 8.3 [4]. The SDS in the running buffer helps maintain the denatured state of the proteins during their migration.
- Loading and Run: Load prepared samples and a molecular weight marker into the wells. Apply a constant voltage (100-200V). The stacking effect occurs as the glycine in the running buffer (pH 8.3) enters the stacking gel (pH 6.8), becoming a zwitterion with low mobility, creating a voltage gradient that "stacks" proteins into a sharp line [4]. Once the stacked proteins enter the resolving gel (pH 8.8), the glycine becomes fully negatively charged and migrates faster, leaving the proteins to be separated by size in the resolving gel matrix.
Table 2: Quantitative Guide to Polyacrylamide Gel Concentration
| Target Protein Size (kDa) | Recommended Acrylamide Concentration (%) | Purpose and Notes |
|---|---|---|
| < 25 | 15% | High percentage for optimal resolution of small proteins/peptides [7]. |
| 25 - 50 | 12% | Standard concentration for medium-sized proteins [7]. |
| 50 - 100 | 10% | Standard concentration for many common proteins [7]. |
| > 100 | 8% | Low percentage for large proteins to facilitate migration [7]. |
| Broad Range | 4-20% (Gradient) | A single gel that can resolve a wide spectrum of protein sizes [3]. |
Advanced Research Context: NSDS-PAGE and Functional Retention
While traditional SDS-PAGE is a powerful denaturing tool, a modified method known as Native SDS-PAGE (NSDS-PAGE) has been developed for applications where retaining protein function or non-covalently bound cofactors (like metal ions) is desirable [8]. This technique addresses a key shortcoming of standard SDS-PAGE, which destroys functional properties [8].
The NSDS-PAGE protocol involves significant modifications:
- Sample Buffer: Removal of SDS and EDTA from the sample buffer and omission of the heating step [8].
- Running Buffer: Reduction of SDS concentration in the running buffer from the standard 0.1% to 0.0375%, along with the deletion of EDTA [8].
These milder conditions have been shown to dramatically increase the retention of bound metal ions in proteomic samples from 26% to 98% and preserve the enzymatic activity of most model enzymes tested, while still maintaining high-resolution separation [8]. This demonstrates that the role of SDS in electrophoresis can be precisely modulated to serve broader research goals, particularly in metallomics and functional proteomics.
The Scientist's Toolkit: Essential Reagents for SDS-PAGE
Table 3: Key Research Reagent Solutions for SDS-PAGE
| Reagent / Solution | Core Function | Technical Specification / Rationale |
|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | Denatures proteins and confers uniform negative charge [1] [4]. | Typically used at 1-2% in sample buffer; binds 1.4g per 1g protein [6] [3]. |
| Reducing Agents (DTT, BME) | Cleaves disulfide bonds to fully linearize proteins [1]. | DTT (10-100 mM) or BME (5% v/v) are common; DTT is less odorous [6] [1]. |
| Acrylamide / Bis-Acrylamide | Forms the porous gel matrix for molecular sieving [3]. | Total concentration (e.g., 8-15%) and crosslinker ratio determine pore size [3] [1]. |
| APS & TEMED | Catalyzes the polymerization of the polyacrylamide gel [1]. | APS provides free radicals; TEMED accelerates polymerization [1]. |
| Tris-Based Buffers | Provides controlled pH environment for gel polymerization and electrophoresis [4]. | Discontinuous system: Stacking gel (pH ~6.8), Resolving gel (pH ~8.8), Running buffer (pH ~8.3) [4]. |
| Glycine | Key ion in discontinuous buffer system for protein stacking [4]. | In running buffer (pH 8.3); charge state changes at different gel pHs to enable stacking [4]. |
| Molecular Weight Markers | Allows estimation of protein size from migration distance [2]. | Pre-stained or unstained protein ladders with known molecular weights [2]. |
| N-(4-bromophenyl)-4-nitroaniline | N-(4-Bromophenyl)-4-nitroaniline CAS 40932-71-6 | |
| 1-(5-methyl-1H-pyrazol-3-yl)propan-2-amine | 1-(5-methyl-1H-pyrazol-3-yl)propan-2-amine, CAS:1025087-55-1, MF:C7H13N3, MW:139.2 g/mol | Chemical Reagent |
Sodium dodecyl sulfate is far more than a simple detergent; it is the fundamental component that enables robust, reproducible protein separation by molecular weight. Its dual action of denaturing proteins and masking their intrinsic charge is a masterpiece of biochemical application, simplifying complex protein mixtures into a parameter that can be easily analyzed. From its foundational role in the standard SDS-PAGE protocol to more nuanced applications like NSDS-PAGE, SDS continues to be an indispensable tool. For researchers and drug development professionals, a deep and mechanistic understanding of SDS is not merely academic—it is a practical necessity for designing experiments, interpreting electrophoretograms, and advancing our knowledge of the proteome in health and disease. As protein-based therapeutics and diagnostics continue to grow, the role of SDS-PAGE, and by extension SDS, remains secure as a cornerstone of modern biochemical analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stands as a cornerstone technique in molecular biology and biochemistry, enabling researchers to separate proteins based primarily on their molecular weight [6]. The revolutionary development of this method by Ulrich Laemmli in 1970 incorporated SDS to largely eliminate the influences of protein structure and inherent charge, allowing separation based predominantly on polypeptide chain length [5] [9]. This technique has become indispensable in modern laboratories, with applications spanning from basic protein characterization to quality control in biopharmaceutical development [10] [11].
The fundamental breakthrough of SDS-PAGE lies in its use of SDS to execute a dual-action mechanism on proteins. This mechanism ensures that proteins unfold into linear chains and acquire a uniform negative charge distribution, effectively standardizing their behavior during electrophoresis [5] [6]. By masking intrinsic charge differences and eliminating the effects of complex three-dimensional structures, SDS allows researchers to determine molecular weight with reasonable accuracy, assess sample purity, and prepare samples for downstream applications like western blotting [9]. Understanding this dual-action mechanism is crucial for researchers and drug development professionals who rely on this technique for accurate protein analysis.
The Molecular Mechanism of SDS Action
Protein Linearization and Denaturation
The first critical action of SDS involves the systematic denaturation of proteins into their linear form. SDS is a potent anionic detergent with strong protein-denaturing properties [5]. When proteins are treated with SDS, particularly at concentrations above 1 mM, the detergent disrupts nearly all non-covalent bonds that maintain the protein's secondary and tertiary structure, including hydrogen bonds and hydrophobic interactions [6] [9]. This disruption occurs as the hydrophobic tail of SDS inserts into the protein core, while the hydrophilic sulfate head group remains exposed to the aqueous environment.
This denaturation process unfolds the native three-dimensional structure of proteins, converting them into random coil conformations [12]. The resulting SDS-protein complexes adopt a rod-like shape with a consistent charge-to-mass ratio, effectively eliminating differences in molecular shape as a factor in electrophoretic separation [6]. For complete denaturation, samples are typically heated to 95°C for several minutes in the presence of SDS and reducing agents like β-mercaptoethanol or dithiothreitol (DTT), which cleave disulfide bonds that covalently stabilize protein structures [6] [13]. This comprehensive linearization ensures that protein migration depends solely on molecular dimensions rather than structural complexities.
Negative Charge Shielding and Charge Masking
The second crucial action of SDS involves imparting a uniform negative charge to all proteins. SDS binds to the protein backbone at an approximately constant ratio of 1.4 grams of SDS per gram of protein [6] [13]. This binding ratio corresponds to approximately one SDS molecule for every two amino acid residues, creating a nearly continuous "shield" of negative charges along the entire polypeptide chain [6].
This extensive binding masks the proteins' intrinsic charges, whether positive or negative, effectively overwhelming them with the negative charges from SDS [9] [12]. Consequently, all proteins acquire a similar net negative charge density, standardizing their charge-to-mass ratios [6]. During electrophoresis, this charge uniformity ensures that all proteins migrate toward the anode (positive electrode) at rates determined primarily by their size rather than their inherent charge characteristics [5]. This charge masking represents a fundamental aspect of the SDS-PAGE technique, enabling molecular weight estimation with an error margin typically around ±10% [6].
Figure 1: Dual-Action Mechanism of SDS in Protein Denaturation and Charge Shielding
Quantitative Aspects of SDS-Protein Interactions
The effectiveness of SDS-PAGE relies on precise quantitative relationships between SDS and proteins. Understanding these parameters is essential for optimizing experimental conditions and interpreting results accurately.
Table 1: Key Quantitative Parameters in SDS-PAGE
| Parameter | Value/Range | Functional Significance | Experimental Impact |
|---|---|---|---|
| SDS Binding Ratio | 1.4 g SDS / 1 g protein [6] [13] | Ensures complete charge masking | Critical for accurate molecular weight determination |
| SDS Monomer Concentration | > 1 mM for protein denaturation [6] | Maintains denaturing conditions | Prevents protein refolding during electrophoresis |
| Critical Micelle Concentration (CMC) | 7-10 mM in aqueous solutions [6] | Determines SDS monomer availability | Ensures sufficient SDS for protein binding |
| Typical SDS in Running Buffer | 0.1% (standard) [6] to 0.0375% (native SDS-PAGE) [8] | Maintains protein linearity during separation | Affects resolution and protein stability |
| Optimal Sample Heating | 95°C for 3-5 minutes [5] [6] | Ensures complete denaturation | Incomplete heating causes smearing |
The binding interaction between SDS and proteins exhibits some variability depending on protein characteristics. Hydrophobic proteins may bind more SDS, while proteins with post-translational modifications such as phosphorylation and glycosylation may bind less SDS [12]. These variations, though generally minimal, can occasionally cause anomalous migration and should be considered when proteins run at unexpected molecular weights [12]. Additionally, the presence of SDS micelles in solutions above the critical micellar concentration provides a reservoir of SDS monomers for sustained protein binding throughout the electrophoresis process [6].
Experimental Protocols for SDS-PAGE
Sample Preparation Methodology
Proper sample preparation is crucial for successful SDS-PAGE separation. The following protocol ensures complete protein denaturation and linearization:
Sample Buffer Preparation: Prepare 2× Laemmli buffer containing 4% SDS, 20% glycerol, 0.004% bromophenol blue, 100 mM Tris-HCl (pH 6.8), and 10% β-mercaptoethanol (added fresh) or 10-100 mM DTT as reducing agent [13] [12].
Sample Denaturation: Mix protein sample with an equal volume of 2× sample buffer. Heat the mixture at 95°C for 3-5 minutes in a heat block or boiling water bath [5] [6]. For heat-sensitive proteins, alternative denaturation at 70°C for 10 minutes may be used [6].
Centrifugation: Briefly centrifuge the denatured samples at 15,000 rpm for 1 minute to collect condensation and ensure the entire sample is at the bottom of the tube [5].
Loading: Load 20-50 μg of protein per well for Coomassie staining or 1-10 μg for silver staining [13]. Include appropriate molecular weight markers in a separate well.
This protocol ensures complete protein denaturation, reduction of disulfide bonds, and proper charge masking. The glycerol in the buffer adds density to facilitate loading, while bromophenol blue serves as a tracking dye to monitor electrophoresis progress [12].
Gel Composition and Electrophoresis Conditions
The discontinuous gel system fundamental to SDS-PAGE consists of two distinct layers with different pore sizes and pH values:
Table 2: Standard Gel Compositions for SDS-PAGE
| Component | Stacking Gel (pH 6.8) | Separating Gel (pH 8.8) | Function |
|---|---|---|---|
| Acrylamide | 4-5% [6] [13] | 6-15% (depending on target protein size) [5] [9] | Creates porous matrix for separation |
| Tris-HCl Buffer | 0.5-1.0 M, pH 6.8 [13] | 1.5 M, pH 8.8 [13] | Maintains appropriate pH for stacking and separation |
| SDS | 0.1% [13] | 0.1% [13] | Maintains protein denaturation |
| Ammonium Persulfate (APS) | 0.05% [13] | 0.05% [13] | Polymerization initiator |
| TEMED | 0.1% [13] | 0.1% [13] | Polymerization catalyst |
| Glycerol | - | - | Adds density for loading |
Electrophoresis Protocol:
Gel Casting: Assemble glass plates with spacers. Prepare separating gel solution, pour between plates, and overlay with water-saturated isopropanol or water to prevent oxygen inhibition of polymerization. Allow to polymerize for 20-30 minutes. Pour stacking gel solution over polymerized separating gel and insert combs. Polymerize for 15-20 minutes [5] [13].
Electrophoresis Setup: Mount gel in electrophoresis apparatus filled with running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3) [13]. Load prepared samples and molecular weight markers into wells.
Electrophoresis Run: Apply constant voltage of 80V until dye front enters separating gel, then increase to 100-150V until dye front reaches bottom of gel [13] [9]. Cooling the apparatus with an ice bath or circulating water cooler is recommended for high-voltage runs to prevent heat-induced artifacts [13].
The discontinuous buffer system creates a stacking effect at the interface between the stacking and separating gels, concentrating protein samples into sharp bands before separation, thereby significantly enhancing resolution [6] [12].
Figure 2: SDS-PAGE Experimental Workflow from Sample Preparation to Analysis
Advanced Applications and Modifications
Native SDS-PAGE for Functional Analysis
A significant modification of the standard technique, termed Native SDS-PAGE (NSDS-PAGE), has been developed to address the limitation of complete protein denaturation [8]. This method reduces SDS concentration in the running buffer from 0.1% to 0.0375% and eliminates both EDTA from sample buffers and the heating step [8]. These modifications result in retention of Zn²⺠bound in proteomic samples increasing from 26% to 98% compared to standard SDS-PAGE, with seven of nine model enzymes maintaining activity after separation [8].
This approach bridges the gap between the high resolution of traditional SDS-PAGE and the functional preservation of native electrophoresis methods like Blue-Native PAGE [8]. NSDS-PAGE is particularly valuable for metalloprotein analysis, enzyme activity studies, and investigations of protein complexes that maintain stability in mild detergent conditions [8].
Capillary Electrophoresis as an Advanced Alternative
Capillary electrophoresis SDS (CE-SDS) represents a technological evolution from traditional slab gel SDS-PAGE [11]. This automated approach provides several advantages, including higher resolution, superior reproducibility, quantitative precision, reduced analysis time, and elimination of manual gel casting [11]. The method uses narrow-bore capillaries filled with separation matrix, with detection via UV absorption or fluorescence, enabling accurate quantification without staining procedures [11].
CE-SDS has been widely adopted in biopharmaceutical industries for characterization of therapeutic proteins, including monoclonal antibodies, antibody-drug conjugates, and fusion proteins, where quantitative analysis and regulatory compliance are essential [11]. The method maintains the fundamental SDS-mediated separation principles while offering enhanced precision and automation.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Essential Research Reagents for SDS-PAGE Experiments
| Reagent/Material | Function | Technical Specifications | Considerations |
|---|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | Protein denaturation and charge masking | >99% purity; 10-20% stock solution in water | Critical for consistent results; filter stock solutions |
| Acrylamide/Bis-acrylamide | Gel matrix formation | Typically 29:1 or 37.5:1 ratio of acrylamide to bis-acrylamide | Neurotoxin - handle with gloves in fume hood |
| TEMED | Gel polymerization catalyst | >99% purity; store at 4°C | Accelerates polymerization; add just before casting |
| Ammonium Persulfate (APS) | Gel polymerization initiator | 10% solution in water; prepare fresh weekly | Degrades with time; affects polymerization efficiency |
| Tris Buffer | pH maintenance | 1.0 M, pH 6.8 (stacking gel); 1.5 M, pH 8.8 (separating gel) | Essential for discontinuous buffer system |
| Glycine | Running buffer component | Electrophoresis grade; running buffer: 25 mM Tris, 192 mM glycine, 0.1% SDS | Zwitterionic properties enable stacking effect |
| DTT or β-mercaptoethanol | Disulfide bond reduction | DTT: 10-100 mM; β-mercaptoethanol: 5% by volume | Essential for complete unfolding of proteins with disulfide bonds |
| Molecular Weight Markers | Size calibration | Pre-stained or unstained; cover expected size range | Include in every gel for accurate molecular weight determination |
| Coomassie Brilliant Blue | Protein staining | 0.1% in 40% ethanol, 10% acetic acid | Standard sensitivity; compatible with mass spectrometry |
| 4-(4-Chlorophenyl)-2,5-dimethylthiazole | 4-(4-Chlorophenyl)-2,5-dimethylthiazole|High Purity | Get 4-(4-Chlorophenyl)-2,5-dimethylthiazole for research. This thiazole derivative is used in medicinal chemistry and material science. For Research Use Only. Not for human or veterinary use. | Bench Chemicals |
| H-Leu-Ala-Pro-OH | H-Leu-Ala-Pro-OH Tripeptide | H-Leu-Ala-Pro-OH is a synthetic tripeptide for research use. This product is For Research Use Only and not intended for diagnostic or therapeutic procedures. | Bench Chemicals |
The selection and quality of reagents directly impact the success and reproducibility of SDS-PAGE experiments. High-purity SDS is particularly critical as impurities can affect binding consistency and migration patterns. Similarly, fresh preparation of reducing agents ensures complete disruption of disulfide bonds. Commercial pre-cast gel systems provide convenience and consistency, particularly for standardized applications, while hand-cast gels offer flexibility in acrylamide concentrations and formulations for specialized separations [10].
The dual-action mechanism of SDS - protein linearization and negative charge shielding - remains fundamental to the widespread utility of SDS-PAGE in protein research. By systematically unfolding complex three-dimensional structures and masking intrinsic charge variations, SDS enables separation based primarily on molecular weight, providing researchers with a robust analytical tool. While modifications like Native SDS-PAGE and technological advancements like CE-SDS have expanded the applications and precision of SDS-based separations, the core mechanism established decades ago continues to underpin this essential methodology.
For drug development professionals and research scientists, understanding these mechanistic principles allows for proper experimental design, accurate interpretation of results, and troubleshooting when anomalies occur. As protein therapeutics and proteomics continue to advance, the principles of SDS-mediated separation maintain their relevance, ensuring this technique remains a cornerstone of biochemical analysis for the foreseeable future.
In the realm of proteomics and drug development, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stands as a cornerstone technique for protein analysis. The fundamental breakthrough of this method lies in its ability to separate proteins almost exclusively based on their molecular weight, a feat achieved by manipulating the inherent properties of proteins through a simple yet powerful detergent [2]. For researchers and pharmaceutical professionals tasked with characterizing complex protein mixtures, from enzyme therapeutics to monoclonal antibodies, this technique provides the reproducible, high-resolution data essential for quality control and diagnostic applications [14] [15]. This technical guide explores the core principle of achieving a uniform charge-to-mass ratio, a concept that has made SDS-PAGE an indispensable tool in life science research for over half a century [10].
The Fundamental Principle: From Native Structure to Linearized Chains
In their native state, proteins exhibit complex three-dimensional structures with charges determined by their amino acid composition, leading to variations in both net charge and molecular shape [16]. When an electric field is applied to these native proteins, their migration rate depends on a combination of charge, size, and shape, preventing separation based solely on molecular weight [2].
The key innovation of SDS-PAGE is the use of the anionic detergent sodium dodecyl sulfate (SDS) to eliminate these variables. The sample preparation process involves three critical steps that transform native proteins into a uniform state [16]:
- Denaturation: SDS disrupts hydrophobic interactions and hydrogen bonds, destroying the protein's secondary and tertiary structure.
- Reduction: Adding a reducing agent like DTT or β-mercaptoethanol cleaves disulfide bonds, separating protein subunits.
- Binding: SDS binds to the denatured protein backbone at a nearly constant ratio of 1.4 g SDS per 1 g of protein [17].
This process results in the formation of SDS-polypeptide complexes that adopt a rod-like shape with a length proportional to the protein's molecular weight [16] [17]. Most importantly, the intrinsic charge of the polypeptide becomes insignificant compared to the overwhelming negative charge provided by the bound SDS molecules, resulting in complexes that all possess a uniform negative charge density [2] [16].
Table 1: Key Steps in Protein Denaturation for SDS-PAGE
| Step | Reagents | Primary Function | Resulting Protein State |
|---|---|---|---|
| Denaturation | SDS, Heat (70-100°C) | Disrupts non-covalent interactions | Unfolded polypeptide chain |
| Reduction | DTT or β-mercaptoethanol | Cleaves disulfide bridges | Separate polypeptide subunits |
| Charge Masking | Excess SDS | Coats polypeptide backbone | Linear complex with uniform negative charge |
The Gel Matrix: A Molecular Sieve for Separation
The polyacrylamide gel serves as a molecular sieve that imposes a frictional force on the migrating proteins [5] [16]. This matrix consists of cross-linked acrylamide polymers whose pore size can be precisely controlled by varying the concentrations of acrylamide and bisacrylamide [2].
The separation occurs because smaller proteins navigate the porous network more easily than larger proteins, causing them to migrate faster through the gel [5]. This differential migration rate, combined with the uniform charge-to-mass ratio of all proteins, enables separation based primarily on polypeptide chain length [5] [16].
Table 2: Polyacrylamide Gel Concentrations and Optimal Separation Ranges
| Acrylamide Concentration (%) | Effective Separation Range (kDa) | Primary Application |
|---|---|---|
| 7% | 50 - 500 | Large proteins |
| 10% | 20 - 300 | Standard protein mixture |
| 12% | 10 - 200 | Standard protein mixture |
| 15% | 3 - 100 | Small proteins and peptides |
For separating proteins of vastly different sizes or those with similar molecular weights, gradient gels with increasing acrylamide concentration (e.g., 4-20%) provide enhanced resolution across a broader molecular weight range [2].
The Discontinuous Buffer System: Focusing the Sample
A crucial innovation in standard SDS-PAGE is the use of a discontinuous buffer system (often called the Laemmli system), which incorporates both a stacking gel and a resolving gel [16]. This system ensures that all proteins enter the resolving gel simultaneously as sharp, focused bands, significantly improving resolution.
The process relies on controlling the charge states of ions in the buffer system, particularly glycine, which exists in different charge states depending on pH [16]. The diagram below illustrates this focusing mechanism and the subsequent separation.
Diagram 1: Protein stacking and separation in SDS-PAGE.
The separation of Clâ» ions from the Tris counter-ion creates a narrow zone with a steep voltage gradient that pulls the glycine ions along behind it, resulting in two narrowly separated fronts of migrating ions [16]. All proteins in the sample have an electrophoretic mobility intermediate between the extreme mobility of the glycine and Clâ», so when these fronts sweep through the sample well, the proteins are concentrated into a narrow zone between them [16]. This procession continues until it hits the running gel, where the pH switches to 8.8, causing glycine molecules to become mostly negatively charged and migrate faster than the proteins, leaving them to separate based on size in the resolving gel [16].
Experimental Protocol: Standard SDS-PAGE Methodology
Sample Preparation and Gel Casting
The following protocol provides a detailed methodology for performing standard SDS-PAGE, adapted from multiple technical sources [5] [2]:
Materials Needed:
- Protein samples
- SDS-PAGE sample buffer (containing SDS, reducing agent, glycerol, and tracking dye)
- Precast or self-cast polyacrylamide gels
- Electrophoresis apparatus and power supply
- Running buffer (e.g., Tris-Glycine-SDS buffer)
Procedure:
Sample Preparation:
Gel Preparation:
- Use precast gels or prepare resolving gel by mixing acrylamide/bisacrylamide solution, Tris-HCl buffer (pH 8.8), SDS, ammonium persulfate (APS), and TEMED [2].
- Pour resolving gel and overlay with water or alcohol to prevent oxygen inhibition of polymerization.
- After polymerization (20-30 minutes), prepare and pour stacking gel (lower acrylamide concentration, Tris-HCl pH 6.8) and insert combs [5].
Electrophoresis:
- Assemble gel cassette in electrophoresis chamber filled with running buffer.
- Load prepared samples and molecular weight markers into wells.
- Apply constant voltage (typically 150-200V) for approximately 45 minutes or until dye front reaches bottom [5].
Post-Electrophoresis Analysis:
- Dismantle apparatus and carefully remove gel from plates.
- Proceed with protein detection methods such as Coomassie Blue staining, silver staining, or western blotting [2].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for SDS-PAGE
| Reagent/Material | Composition/Type | Function in SDS-PAGE |
|---|---|---|
| SDS Sample Buffer | Tris-HCl, SDS, Glycerol, Bromophenol Blue, Reducing Agent | Denatures proteins, provides density for loading, and tracking dye |
| Running Buffer | Tris-Glycine with 0.1% SDS | Conducts current and maintains SDS coating during electrophoresis |
| Polyacrylamide Gel | Acrylamide-Bisacrylamide matrix polymerized with APS/TEMED | Forms molecular sieve for protein separation based on size |
| Molecular Weight Markers | Pre-stained or unstained protein standards of known mass | Provides reference for estimating sample protein molecular weights |
| Stacking Gel | Low-concentration acrylamide (4-5%) at pH 6.8 | Concentrates protein samples into sharp bands before separation |
| Methanesulfonamide, N-(trimethylsilyl)- | Methanesulfonamide, N-(trimethylsilyl)-, CAS:999-96-2, MF:C4H13NO2SSi, MW:167.3 g/mol | Chemical Reagent |
| 3-Chloro-4-fluoro-3'-iodobenzophenone | 3-Chloro-4-fluoro-3'-iodobenzophenone, CAS:951890-19-0, MF:C13H7ClFIO, MW:360.55 g/mol | Chemical Reagent |
Advanced Applications and Recent Innovations
Native SDS-PAGE: Preserving Functional Properties
A significant innovation in electrophoretic techniques is Native SDS-PAGE (NSDS-PAGE), which modifies standard conditions to preserve certain functional properties of proteins while maintaining high resolution [8]. This method eliminates SDS and EDTA from the sample buffer, omits the heating step, and reduces SDS concentration in the running buffer (e.g., to 0.0375%) [8]. These modifications dramatically increase the retention of bound metal ions in metalloproteins from 26% in standard SDS-PAGE to 98% in NSDS-PAGE, with seven of nine model enzymes retaining activity after separation [8]. This advancement bridges the gap between the high resolution of denaturing SDS-PAGE and the functional preservation of native-PAGE, opening new possibilities for metalloprotein analysis [8].
Methodological Enhancements and Market Evolution
Recent technical improvements include the use of colored stacking gels containing acidic dyes (tartrazine, brilliant blue FCF, or new coccine) to facilitate well visualization and sample loading without affecting separation performance [18]. The electrophoresis market continues to evolve with trends toward automation, miniaturization, and integration of digital technologies [14] [10]. Capillary electrophoresis systems and microfluidic platforms are gaining traction for their ability to provide faster run times, reduced sample volumes, and automated data analysis, particularly in pharmaceutical quality control settings [10] [15]. Artificial intelligence is increasingly being applied to automate image analysis, band quantification, and pattern evaluation, reducing human error and enhancing reproducibility [15].
The principle of achieving a uniform charge-to-mass ratio through SDS binding remains the foundational concept that enables reliable protein separation by molecular weight. This technique continues to be indispensable in biotechnology and pharmaceutical industries, particularly for the characterization of therapeutic proteins and monoclonal antibodies [15]. While sophisticated alternatives like mass spectrometry have emerged, SDS-PAGE maintains its relevance due to its simplicity, cost-effectiveness, and visual clarity [19]. Ongoing innovations in electrophoretic methodology ensure that this decades-old technique will continue to evolve, maintaining its critical role in proteomic research and drug development [8] [10].
Sodium dodecyl sulfate (SDS) is a foundational reagent in protein biochemistry, most notably for its role in denaturing gel electrophoresis. Its functionality is governed by a critical physical property: the critical micelle concentration (CMC). This technical guide elucidates the molecular mechanism whereby SDS monomers, but not micelles, bind to and denature protein substrates. We detail the hydrophobic and electrostatic forces driving this selective interaction, summarize key quantitative data on SDS-protein binding, and provide validated experimental methodologies for investigating these interactions. Within the broader context of SDS-polyacrylamide gel electrophoresis (SDS-PAGE) research, understanding this monomer-centric mechanism is paramount, as it is the fundamental process that confers a uniform negative charge to polypeptides, enabling their separation by molecular mass rather than intrinsic charge.
Sodium dodecyl sulfate (SDS) is an anionic surfactant with a 12-carbon alkyl tail attached to a sulfate head group [20]. In aqueous solutions, its behavior is concentration-dependent. Below a specific threshold known as the critical micelle concentration (CMC), SDS exists as individual molecules, or monomers. Above the CMC, these monomers self-associate into spherical aggregates called micelles, wherein the hydrophobic tails are sequestered inward, and the negatively charged sulfate groups are exposed to the aqueous environment [21]. The CMC for SDS is typically in the range of 6–8 mM (approximately 0.17–0.23% w/v) [6] [21]. It is this monomeric form of SDS that is responsible for the initial binding and denaturation of proteins, a cornerstone of the SDS-PAGE technique [22].
The principle of SDS-PAGE relies on overcoming the inherent variations in protein charge and shape to separate them based solely on polypeptide chain length. This is achieved because SDS binds to proteins in a constant weight ratio, masking their intrinsic charge and imparting a similar charge-to-mass ratio [5] [6]. The ensuing separation through a polyacrylamide gel matrix, which acts as a molecular sieve, allows for the determination of protein molecular weight with an error of approximately ±10% [6].
The Molecular Mechanism of SDS-Protein Binding
The Primacy of Monomeric SDS
The fundamental tenet of SDS-protein interaction is that only the monomeric form of the amphiphile binds to proteins, not the micellar form [22]. This specificity arises from the structural and thermodynamic properties of the micelle. The SDS micelle is anionic on its surface and does not adsorb protein [6]. The hydrophobic core of the micelle is energetically stable, and incorporating a protein chain would be highly unfavorable. Instead, the cooperative binding process is driven by individual monomer units.
The process begins at very low SDS concentrations. At concentrations above 0.1 mM, the unfolding of proteins commences, and above 1 mM, most proteins are denatured [6]. The binding is cooperative, meaning the binding of one SDS molecule increases the probability that another will bind to the same protein chain [21]. This cooperative process saturates the protein backbone, with approximately 1.4 grams of SDS binding per gram of protein [6]. This ratio corresponds to roughly one SDS molecule per two amino acid residues, effectively coating the polypeptide chain [6].
Forces Driving Monomer Binding
The binding of SDS monomers to proteins is primarily hydrophobic in nature but is stabilized by electrostatic interactions [22].
- Hydrophobic Interactions: The aliphatic dodecyl chain of the SDS monomer interacts with non-polar regions and hydrophobic amino acid side chains on the protein. This interaction disrupts the hydrophobic core of the protein, leading to unfolding.
- Electrostatic Interactions: The negatively charged sulfate head group of SDS interacts with positively charged amino acid residues (e.g., lysine, arginine) on the protein. This interaction further destabilizes the protein's native structure by neutralizing positive charges.
Molecular dynamics simulation studies on human ubiquitin have shown that at high temperatures, SDS monomers disrupt the first hydration shell and expand the hydrophobic core, leading to complete protein unfolding [23]. The simulations also suggest that SDS can induce or stabilize α-helical structures in certain contexts, demonstrating the complex nature of the interaction [23].
Structural Evidence for Specific SDS Binding
While SDS binding is often considered non-specific, high-resolution structural studies have revealed that SDS can bind to pre-formed cavities in certain proteins. For instance, the X-ray crystal structure of the SDS complex with horse-spleen apoferritin showed that a single SDS molecule binds specifically in an internal cavity, with the alkyl tail bent into a horseshoe shape and the charged head group positioned at the cavity opening [24]. Isothermal titration calorimetry determined the dissociation constant for this specific interaction to be 24 ± 9 µM at 293 K, which is well below the CMC, confirming monomeric binding [24]. This demonstrates that beyond generalized coating, SDS can exhibit specific, high-affinity binding at discrete sites on some proteins.
Quantitative Data on SDS-Protein Interactions
The following tables summarize key quantitative data essential for understanding and experimenting with SDS-protein interactions.
Table 1: Key Properties of Sodium Dodecyl Sulfate (SDS)
| Property | Value | Conditions / Notes | Reference |
|---|---|---|---|
| Critical Micelle Concentration (CMC) | 6–8 mM (0.17–0.23% w/v) | In aqueous solution | [21] |
| Molecular Weight (Monomer) | 288 Da | [21] | |
| Aggregation Number | 62 | Molecules per micelle | [6] [21] |
| Molecular Weight (Micelle) | ~18 kDa | [21] | |
| Typical SDS-PAGE Running Buffer Concentration | 0.1% (w/v) | ~3.5 mM, which is below the CMC | [6] |
| Typical SDS-PAGE Sample Buffer Concentration | 1-2% (w/v) | ~35-70 mM, well above the CMC | [20] |
| Average SDS Binding Ratio | 1.4 g SDS / 1 g protein | Corresponds to ~1 SDS molecule per 2 amino acids | [6] |
Table 2: Experimental Techniques for Studying SDS-Protein Interactions
| Technique | Application | Key Measurable Parameters | Reference |
|---|---|---|---|
| Isothermal Titration Calorimetry (ITC) | Direct measurement of binding affinity and thermodynamics. | Dissociation constant (Kd), enthalpy change (ΔH), stoichiometry (n). | [24] |
| X-ray Crystallography | High-resolution structural determination of SDS-protein complexes. | Atomic-level coordinates of SDS binding sites and protein conformational changes. | [24] |
| Molecular Dynamics (MD) Simulation | Theoretical study of binding pathways, kinetics, and unfolding mechanisms. | Root-mean-square deviation (RMSD), solvent-accessible surface area (SASA), residue-specific interactions. | [23] |
| Circular Dichroism (CD) Spectroscopy | Monitoring changes in protein secondary structure upon SDS binding. | α-helical and β-sheet content, unfolding transitions. | [24] [23] |
Experimental Protocols
Protocol 1: Isothermal Titration Calorimetry (ITC) for SDS Binding
This protocol is adapted from studies investigating SDS binding to apoferritin [24].
Objective: To determine the binding affinity (Kd), stoichiometry (n), and thermodynamic parameters (ΔH, ΔS) of SDS monomer binding to a target protein.
Materials:
- Purified target protein (e.g., apoferritin)
- High-purity SDS
- ITC instrument (e.g., MicroCal VP-ITC)
- Dialysis buffer (e.g., 130 mM NaCl, 20 mM sodium phosphate, pH 7.0)
- Dialysis tubing
Method:
- Sample Preparation:
- Dialyze the target protein extensively against the chosen buffer to ensure precise buffer matching.
- Dissolve SDS in the same dialysate buffer used for the protein. The SDS concentration should be substantially higher than the protein concentration in the cell.
- Instrument Setup:
- Degas all solutions to prevent bubble formation during the titration.
- Load the sample cell (typically 1.4 mL) with the protein solution (e.g., 0.01 mM apoferritin).
- Load the syringe with the SDS solution (e.g., 3.47 mM).
- Titration Procedure:
- Set the reference power and stirring speed as per instrument guidelines.
- Program the titration parameters: initial delay, number of injections, injection volume (e.g., 1 µL first injection, followed by 15 µL injections), and duration between injections (e.g., 5 minutes).
- Begin the titration at a constant temperature (e.g., 293 K).
- Control Experiment:
- Perform an identical titration of SDS solution into buffer alone to measure the heat of dilution.
- Data Analysis:
- Subtract the control data from the protein titration data.
- Fit the corrected isotherm to an appropriate binding model (e.g., "single set of identical sites") using the instrument's software to extract Kd, n, and ΔH.
Protocol 2: Native SDS-PAGE (NSDS-PAGE) for Functional Analysis
This protocol demonstrates how modifying SDS concentration can preserve protein function, underscoring the role of controlled monomer binding [8].
Objective: To separate proteins with high resolution while retaining native enzymatic activity and/or bound metal cofactors.
Materials:
- Standard SDS-PAGE equipment and precast Bis-Tris gels
- Protein sample (e.g., LLC-PK1 cell proteome fraction)
- Standard SDS-PAGE reagents (MOPS, Tris, etc.)
- NSDS-PAGE Sample Buffer: 100 mM Tris HCl, 150 mM Tris base, 10% (v/v) glycerol, 0.01875% (w/v) Coomassie G-250, 0.00625% (w/v) Phenol Red, pH 8.5. No SDS or EDTA.
- NSDS-PAGE Running Buffer: 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7. No EDTA.
Method:
- Sample Preparation:
- Mix 7.5 µL of protein sample with 2.5 µL of 4X NSDS sample buffer. Do not heat the sample.
- Gel Pre-run:
- Mount the precast gel in the electrophoresis apparatus.
- Run the gel at 200V for 30 minutes in double-distilled H2O to remove storage buffer and unpolymerized acrylamide.
- Electrophoresis:
- Replace the water in the buffer chambers with NSDS-PAGE running buffer.
- Load the prepared samples and molecular weight markers.
- Run electrophoresis at a constant voltage of 200V for approximately 45 minutes until the dye front reaches the gel bottom.
- Analysis:
- Proteins can be analyzed for activity using in-gel zymography or for metal content using techniques like laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS). This method has been shown to increase Zn²⺠retention in proteomic samples from 26% (standard SDS-PAGE) to 98% [8].
Visualizing the Mechanism and Workflow
The following diagrams illustrate the core concepts and experimental workflows described in this guide.
Mechanism of SDS Monomer Binding vs. Micelle Formation
Diagram 1: SDS Monomer Binding vs. Micelle Formation. This flowchart illustrates the concentration-dependent fate of SDS in solution and its consequence for protein binding. Below the CMC, monomers are available to cooperatively bind and unfold proteins. Above the CMC, stable micelles form which do not bind protein substrates.
Experimental Workflow for ITC Binding Studies
Diagram 2: ITC Workflow for SDS Binding. This workflow outlines the key steps for a successful Isothermal Titration Calorimetry experiment to quantify SDS-protein interactions, highlighting the critical need for buffer matching and control measurements.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Studying SDS-Protein Interactions
| Reagent / Material | Function / Description | Example Application |
|---|---|---|
| High-Purity SDS | Anionic detergent; core ligand for binding studies. Minimizes impurities that can interfere with assays. | All binding and electrophoresis studies. |
| Apoferritin | Model four-helix bundle protein with a defined internal cavity for specific SDS binding. | Structural and thermodynamic binding studies [24]. |
| Ubiquitin | Small, heat-stable model protein with mixed α/β structure. | Molecular dynamics and unfolding studies [23]. |
| ITC Instrument | Measures heat released or absorbed during molecular binding events. | Direct measurement of binding constants and thermodynamics [24]. |
| Precast Bis-Tris Gels | Polyacrylamide gels with near-neutral pH; stable and reduce protein modification. | Standard and Native SDS-PAGE [8] [6]. |
| MOPS Buffer | Buffer for SDS-PAGE running buffer (pH ~7.7). | Maintaining stable pH during electrophoresis [8]. |
| Tris-Glycine Buffer | Discontinuous buffer system for standard SDS-PAGE. | Stacking and separating proteins based on size [6] [25]. |
| Dithiothreitol (DTT) | Reducing agent; cleaves disulfide bonds to ensure complete unfolding. | Standard SDS-PAGE sample preparation [6]. |
| CHAPS Detergent | Zwitterionic, non-denaturing detergent. Used as a milder alternative for comparison. | Membrane protein solubilization without denaturation [21]. |
| 1-(3-Chloro-4-methylphenyl)urea | 1-(3-Chloro-4-methylphenyl)urea|CAS 13142-64-8 | 1-(3-Chloro-4-methylphenyl)urea is a chemical for research use only (RUO). It is a phenylurea compound studied in environmental analysis and medicinal chemistry. Not for human or veterinary use. |
| 2-t-Butyl-4-quinoline carboxylic acid | 2-t-Butyl-4-quinoline carboxylic acid, MF:C14H15NO2, MW:229.27 g/mol | Chemical Reagent |
In the field of protein research, Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) stands as a fundamental analytical technique for separating proteins based on their molecular weight. The core principle of SDS-PAGE relies on the complete denaturation of proteins into their linear polypeptide forms, and this is where the essential partnership between SDS and reducing agents comes into play. While SDS is responsible for disrupting non-covalent bonds and imparting a uniform negative charge, it is incapable of breaking the strong covalent disulfide bonds that stabilize tertiary and quaternary protein structures. These disulfide bridges, formed between cysteine residues, can maintain structural domains even in the presence of detergents, potentially leading to inaccurate molecular weight determination and poor separation efficiency. The introduction of reducing agents such as Dithiothreitol (DTT) and β-Mercaptoethanol (BME) is therefore critical to achieve complete protein denaturation by specifically targeting and reducing these disulfide bonds, enabling proteins to be separated solely based on polypeptide chain length.
This whitepaper provides an in-depth technical examination of the synergistic relationship between SDS and reducing agents in protein biochemistry. Designed for researchers, scientists, and drug development professionals, it details the mechanisms, applications, and practical protocols essential for effective protein analysis, with a specific focus on the comparative advantages of DTT and BME in experimental workflows.
The Fundamental Principles of SDS-PAGE
The Indispensable Role of SDS
SDS (Sodium Dodecyl Sulfate) is a powerful anionic detergent that serves two primary functions in protein denaturation for electrophoresis. First, it effectively disrupts nearly all non-covalent interactions—including hydrogen bonds, hydrophobic forces, and ionic bonds—that maintain a protein's secondary and tertiary structure [26]. This action "unfolds" the protein, destroying its higher-order organization. Second, SDS binds to the denatured protein backbone at a relatively constant ratio of approximately 1.4 g of SDS per gram of polypeptide [27]. This uniform binding masks the protein's intrinsic charge and imparts a large, negative net charge that is roughly proportional to the protein's molecular mass [5] [28]. The result is the formation of SDS-polypeptide complexes that share a similar charge-to-mass ratio, ensuring that separation during electrophoresis is based primarily on molecular size rather than native charge or shape [26].
The Limitation of SDS and the Need for Reducing Agents
Despite its effectiveness against non-covalent bonds, SDS has a critical limitation: it is incapable of breaking covalent disulfide bonds (-S-S-). These bonds, which form between the sulfur atoms of cysteine residues, are a key feature of the three-dimensional structure of many proteins and are essential for stabilizing the quaternary structure of multimetric proteins [29]. If left intact, disulfide bonds can prevent complete protein unfolding, leading to aberrant migration during electrophoresis and inaccurate molecular weight estimates. This creates an imperative for reducing agents, which are specifically designed to reduce these disulfide bonds into free sulfhydryl groups (-SH), thereby completing the denaturation process initiated by SDS [29].
The following diagram illustrates the synergistic denaturation process involving both heat, SDS, and a reducing agent (like DTT or BME) to fully unfold a protein for SDS-PAGE.
Synergistic Protein Denaturation for SDS-PAGE
The Science of Reducing Agents
Mechanism of Disulfide Bond Reduction
Reducing agents function by participating in a thiol-disulfide exchange reaction, wherein their own free thiol (-SH) groups nucleophilically attack the sulfur-sulfur bond in a protein's disulfide bridge. This reaction reduces the protein's disulfide bond, converting it into two free thiol groups, while the reducing agent itself becomes oxidized [30]. For the reduction to be effective in a typical biochemical context, the reducing agent must possess a lower redox potential than the protein's disulfide bond, making the reaction thermodynamically favorable. The efficiency of this process is further enhanced by the application of heat (95°C), which increases molecular motion and accelerates both the denaturation by SDS and the reduction of disulfide bonds [31]. This combination of chemical reduction and thermal energy ensures that proteins are fully unfolded into linear polypeptides, ready for accurate electrophoretic separation.
Dithiothreitol (DTT) - The Reagent of Choice
Dithiothreitol (DTT), also known as Cleland's reagent, is a potent reducing agent that has become the standard in many protein biochemistry applications. Its mechanism involves two sequential thiol-disulfide exchange reactions. First, a mixed disulfide intermediate is formed between one of DTT's thiol groups and the protein's disulfide bond. Subsequently, an intramolecular cyclization of DTT occurs, resulting in a stable six-membered ring (a cyclic disulfide) and the release of the fully reduced protein with its free thiol groups [30]. This cyclic reaction is highly favorable, driving the reduction to completion.
DTT is particularly valued for its strong reducing power, lower volatility, and significantly less unpleasant odor compared to BME [30]. A typical working concentration for DTT in sample buffer is between 40-160 mM [29]. However, DTT has a key limitation: its reducing power diminishes in acidic conditions (pH < 7) due to the protonation of its thiol groups, which are necessary for the nucleophilic attack [30]. Furthermore, DTT solutions are prone to oxidation by air and must be prepared fresh or stored frozen in aliquots to maintain efficacy.
β-Mercaptoethanol (BME) - A Traditional Agent
β-Mercaptoethanol (BME) is a traditional reducing agent that has been widely used for decades, famously featured in Laemmli buffer. It operates through a mechanism similar to DTT, using its single thiol group to reduce protein disulfide bonds, resulting in the formation of oxidized BME dimers. However, BME is generally considered less effective than DTT due to its weaker reducing power. It is also highly volatile, which contributes to its characteristically strong, unpleasant odor that can permeate laboratory environments [30] [29]. This volatility can also lead to a gradual loss of reducing capacity from an opened container. Despite these drawbacks, BME remains in use due to its lower cost and established history in certain protocols.
Comparative Analysis of DTT and BME
The choice between DTT and BME can significantly impact experimental outcomes, cost, and laboratory working conditions. The following table provides a detailed, quantitative comparison to guide researchers in selecting the appropriate agent.
Table 1: Quantitative Comparison of DTT and β-Mercaptoethanol
| Parameter | Dithiothreitol (DTT) | β-Mercaptoethanol (BME) |
|---|---|---|
| Chemical Structure | HOOC-CH(NHâ‚‚)-CHâ‚‚-SH | HO-CHâ‚‚-CHâ‚‚-SH |
| Mechanism | Two-step reaction forming a stable cyclic disulfide [30] | Simple thiol-disulfide exchange, forming oxidized dimers |
| Typical Working Concentration | 40-160 mM [29] | Often used at ~1% (v/v) or ~140 mM in sample buffer [31] |
| Reducing Power | Stronger reducing agent [30] | Weaker reducing agent [30] |
| Odor & Volatility | Lower volatility, less unpleasant odor [30] | High volatility, very strong and unpleasant odor [30] [29] |
| Stability in Solution | Prone to oxidation; prepare fresh or store at -20°C [30] | Solutions lose potency over time due to volatility and oxidation |
| Cost (Example) | $56.25 for 10 g [30] | Generally less expensive |
| Effective pH Range | Most effective at pH > 7 [30] | Effective over a broader pH range |
Advanced Applications and Methodological Considerations
Detailed Experimental Protocol for SDS-PAGE with Reducing Agents
A robust, reproducible protocol is essential for high-quality protein separation. The following detailed methodology incorporates the critical steps for effective protein denaturation using reducing agents.
Table 2: Reagent Solutions for SDS-PAGE Sample Preparation
| Reagent | Composition / Purpose | Typical Concentration / Note |
|---|---|---|
| 4X Sample Loading Buffer (Laemmli Buffer) | Tris-HCl (pH 6.8), SDS, Glycerol, Bromophenol Blue, Reducing Agent [28] | Contains 2% SDS, 20% Glycerol, 160 mM DTT (or 1-5% BME) [29] |
| SDS | Anionic detergent; denatures proteins and imparts charge [26] | Final conc. 1-2% in sample [29] |
| DTT | Reducing agent; breaks disulfide bonds [30] | Final conc. 40-160 mM; preferred over BME [29] |
| BME | Alternative reducing agent [28] | Final conc. ~1-5% (v/v); strong odor [31] |
| Glycerol | Increases sample density for easy well loading [29] | 10-20% final concentration |
| Bromophenol Blue | Tracking dye for monitoring electrophoresis progress [29] | ~0.05 mg/ml final concentration |
Step-by-Step Procedure:
- Sample Preparation: Dilute your protein sample to a predetermined concentration in an appropriate buffer. A final concentration of 1-2 mg/mL after mixing with sample buffer is often suitable for complex mixtures, though this may require optimization [29].
- Denaturation Mix Preparation: Combine the protein sample with an equal volume of the 4X Sample Loading Buffer containing the chosen reducing agent (DTT or BME). For instance, mix 10 µL of protein sample with 2.5 µL of 4X buffer and 7.5 µL of water, ensuring the final concentration of SDS is ~1% and DTT is ~40-160 mM [29] [31].
- Heat Denaturation: Cap the tubes securely and heat the mixture at 90-100°C for 3-10 minutes in a heat block or boiling water bath [5] [31]. This critical step accelerates the denaturation by SDS and the reduction of disulfide bonds by DTT/BME. Caution: Tube caps may pop open due to pressure build-up; use tube clamps if available.
- Brief Centrifugation: Pulse-centrifuge the heated samples (e.g., 15,000 rpm for 1 minute) to collect any condensation and ensure the entire sample is at the bottom of the tube [31].
- Gel Loading and Electrophoresis: Load the denatured, reduced samples into the wells of a pre-cast polyacrylamide gel. Run the gel at a constant voltage (e.g., 120-150 V for the separating gel) until the bromophenol blue dye front reaches the bottom of the gel [26].
The workflow below summarizes the key steps in preparing and running a reducing SDS-PAGE experiment.
SDS-PAGE Sample Prep Workflow
Specialized Applications in Research and Industry
The synergy of SDS and reducing agents extends far beyond basic protein analysis, playing a vital role in advanced research and industrial quality control.
- Protein Purity and Expression Analysis: SDS-PAGE is a cornerstone for assessing the purity of recombinant protein preparations and analyzing protein expression levels in cell lysates. The complete denaturation ensured by SDS and DTT/BME allows researchers to visualize individual polypeptide chains and identify contaminants [8].
- Western Blotting: The technique is a prerequisite for western blotting, where proteins separated by SDS-PAGE are transferred to a membrane for immunodetection. Consistent and complete reduction is critical for antibody recognition of linear epitopes [8].
- Food Science and Quality Assurance: In the food industry, SDS-PAGE is used to characterize protein ingredients, verify authenticity, detect adulteration, and assess the impact of processing (e.g., heat or enzymatic hydrolysis) on protein molecular weight profiles [32].
- Metalloprotein Studies and Alternative Methods: While standard SDS-PAGE destroys native protein function, modified methods like Native SDS-PAGE (NSDS-PAGE) have been developed. This technique uses minimal SDS and omits reducing agents and heat, allowing for the separation of proteins while retaining bound metal ions and, for some enzymes, catalytic activity [8]. This highlights a specific use case where the omission of DTT/BME is deliberate to preserve a functional characteristic.
The powerful synergy between SDS and reducing agents like DTT and BME is a cornerstone of modern protein science. While SDS unfolds protein structures and standardizes charge, it is the specific action of DTT and BME in breaking resilient disulfide bonds that ensures complete denaturation into linear polypeptides. This partnership is fundamental to the success of SDS-PAGE, enabling the high-resolution separation of proteins based on molecular weight that underpins countless applications in research, diagnostics, and product development. The choice between reducing agents, particularly the more potent and less odorous DTT versus the traditional BME, requires careful consideration of the specific experimental needs and conditions. As protein analysis continues to evolve, the precise control of reduction states—whether for full denaturation or for the preservation of native complexes in techniques like NSDS-PAGE—will remain an essential skill for scientists driving innovation in biotechnology and drug development.
Mastering the SDS-PAGE Protocol: From Sample Prep to Precise Molecular Weight Determination
In the realm of protein research, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stands as a cornerstone technique for separating proteins based on their molecular weight. [5] [33] This method's unparalleled effectiveness hinges on a critical preliminary step: the complete denaturation of protein samples using an SDS-based buffer and controlled heat. [34] [35] Within the broader context of understanding SDS's role in polyacrylamide gel electrophoresis research, sample preparation emerges not merely as a routine procedure but as the foundational process that determines the entire experiment's validity and resolution. The intentional and complete unfolding of proteins is what allows SDS-PAGE to separate molecules primarily by size, effectively neutralizing the influence of innate protein charge and complex three-dimensional structure. [33] [6] For researchers and drug development professionals, mastering this denaturation process is therefore not a mere technicality but a prerequisite for obtaining accurate, reproducible, and interpretable data in applications ranging from western blotting and mass spectrometry to protein purity assessment and molecular weight estimation. [34] [35]
The Chemical Basis of Denaturation
The Action of Sodium Dodecyl Sulfate (SDS)
Sodium dodecyl sulfate (SDS) is a powerful anionic detergent that serves as the primary denaturing agent in the sample buffer. [36] Its mechanism of action is twofold. First, the hydrophobic hydrocarbon tail of SDS interacts with and dissolves the hydrophobic regions of the protein, while the ionic sulfate group disrupts non-covalent ionic bonds that maintain secondary and tertiary structure. [34] [36] This concerted action causes the protein to lose its higher-order structures and unfold into a linear polypeptide chain. [36]
Second, SDS binds to the unfolded protein backbone at a remarkably constant weight ratio of approximately 1.4 grams of SDS per 1 gram of protein. [6] [35] This uniform coating imparts a strong negative charge to the polypeptide that is directly proportional to its chain length. Consequently, all proteins in the sample achieve a similar charge-to-mass ratio, ensuring that their electrophoretic mobility through the gel becomes a function of molecular size alone, rather than a combination of size, shape, and intrinsic charge. [5] [33] [35] It is this fundamental principle, established during sample preparation, that underpins the entire SDS-PAGE technique.
The Supporting Role of Reducing Agents and Heat
While SDS is the principal denaturant, its effect is significantly potentiated by reducing agents and heat, which target the remaining structural elements holding the protein in a native conformation.
- Reducing Agents: Compounds such as β-mercaptoethanol (β-ME) or dithiothreitol (DTT) are added to the sample buffer to cleave disulfide bonds, which are covalent linkages that stabilize tertiary and quaternary structures. [34] [35] By breaking these sulfur bridges, reducing agents ensure that multimeric proteins dissociate into their individual subunits and that all proteins are fully linearized, further promoting the spaghettification of the polypeptide chain. [34]
- Heat: The application of heat, typically 95°C for 3-5 minutes or 70°C for 10 minutes, provides the kinetic energy needed to overcome hydrogen bonding and other stabilizing interactions that SDS alone may not disrupt. [5] [6] Boiling also serves a practical purpose by homogenizing the sample, particularly for cell lysates that may contain viscous DNA. The heat melts the DNA, reducing gumminess and making the sample easier to pipette into the gel wells. [34]
Table 1: Key Components of SDS Sample Denaturation Buffer and Their Functions
| Component | Typical Concentration | Primary Function | Mechanism of Action |
|---|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | 1-2% [36] | Denaturant & Charge Provider | Disrupts hydrophobic/ionic bonds; coats proteins with uniform negative charge. [34] [36] |
| Reducing Agent (e.g., β-mercaptoethanol or DTT) | β-ME: 1-5% [6]DTT: 10-100 mM [6] | Disulfide Bond Reduction | Cleaves covalent -S-S- bridges, ensuring full dissociation and linearization. [6] [35] |
| Tris-HCl Buffer | 50-200 mM, pH ~6.8 [36] | pH Stabilization | Maintains stable pH environment for the denaturation process. [36] |
| Glycerol | 10-20% [36] | Density Agent | Adds density to sample, allowing it to sink to bottom of loading well. [36] [35] |
| Bromophenol Blue | Trace | Tracking Dye | Visualizes sample migration during electrophoresis. [36] [35] |
Detailed Experimental Protocol for Optimal Denaturation
The following section provides a detailed, step-by-step methodology for the denaturation of protein samples prior to SDS-PAGE. Adherence to this protocol is critical for achieving consistent and reliable results.
Reagent Preparation
Laemmli Sample Buffer (2X Concentrate) A standard, widely used formulation is the Laemmli buffer. [36] To prepare 10 mL of a 2X stock solution:
- 4.0 mL of 1.0 M Tris-HCl, pH 6.8
- 2.0 mL of 20% (w/v) SDS (Final concentration ~4%)
- 2.0 mL of Glycerol (Final concentration 20%)
- 0.5 mL of β-Mercaptoethanol (Final concentration 5%) or 0.77 g of DTT (Final concentration ~0.5 M)
- 1.5 mL of Deionized Water
- A few grains of Bromophenol Blue (approx. 0.002%)
Mix the components thoroughly. The buffer can be aliquoted and stored at -20°C for several months. Avoid repeated freeze-thaw cycles for aliquots containing reducing agents.
Step-by-Step Denaturation Procedure
Sample and Buffer Mixing:
Heat Denaturation:
- Secure the cap of the microcentrifuge tube to prevent popping.
- Place the tube in a pre-heated heat block or water bath set to 95°C for 3-5 minutes. [5] [6]
- Critical Note: The optimal heating time can be protein-dependent. Large or complex proteins may require extended heating for complete denaturation, while prolonged boiling can degrade smaller proteins. Empirical testing is recommended for new protein systems. [34]
Brief Centrifugation:
- After heating, centrifuge the samples at high speed (e.g., 15,000 rpm) for 1 minute at room temperature or 4°C. [5]
- This step collects any condensation from the tube walls and sediments any insoluble material.
Sample Loading:
- The sample is now ready for loading onto the polyacrylamide gel. Use the supernatant for electrophoresis, being careful not to disturb any pellet. [5]
The workflow below summarizes the sample preparation process.
Troubleshooting and Optimization
Even with a standardized protocol, researchers may encounter issues stemming from suboptimal denaturation. The table below outlines common problems, their potential causes, and recommended solutions.
Table 2: Troubleshooting Guide for Sample Denaturation in SDS-PAGE
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Smearing Bands | Incomplete denaturation [34]; Insufficient reducing agent; Protein degradation. | Ensure fresh reducing agent is used; Increase heating time or temperature; Perform all steps on ice with protease inhibitors. |
| Atypical Band Migration | Over-heating leading to protein degradation [34]; Incomplete disaggregation. | Optimize heating time; Ensure sample is fully mixed and dissolved in buffer. |
| Poor Resolution of Similar Sized Proteins | Inefficient stacking due to improper buffer pH or ionic content. [36] | Verify pH of sample buffer and gel buffers; Use fresh running buffer. |
| No or Weak Bands | Over-heating of small, labile proteins [34]; Insufficient protein loaded. | Reduce heating time for small proteins; Concentrate protein sample prior to loading. |
The Researcher's Toolkit: Essential Reagents for SDS-PAGE Sample Preparation
Successful and reproducible sample denaturation requires precise formulation of reagents. The following table details the essential materials for this critical step.
Table 3: Essential Research Reagent Solutions for SDS-PAGE Sample Preparation
| Item | Specifications & Function | Technical Notes |
|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | >99% purity; Anionic detergent for protein denaturation and charge conferment. [36] | Prepare as 10-20% (w/v) stock solution in water. Filter through a 0.22 µm filter. |
| Dithiothreitol (DTT) | High-purity; Reducing agent for cleaving disulfide bonds. [6] | Preferred over β-ME for lower odor. Prepare as 1M stock, aliquot, and store at -20°C. |
| Tris-HCl Buffer | 1.0 M, pH 6.8; Provides optimal pH environment for denaturation and stacking. [36] | Confirm pH at room temperature. Sterile filter for long-term storage. |
| Glycerol | Molecular biology grade; Adds density to sample for easy gel loading. [35] | |
| Bromophenol Blue | Tracking dye for monitoring electrophoresis progress. [36] [35] | Typically added in trace amounts to the sample buffer. |
| Laemmli Buffer (2X) | Ready-to-use denaturing buffer containing all above components. [36] | Available commercially for convenience and consistency. |
| 2-(2-Chlorophenyl)acetohydrazide | 2-(2-Chlorophenyl)acetohydrazide, CAS:22631-60-3, MF:C8H9ClN2O, MW:184.62 g/mol | Chemical Reagent |
| 3-(4-(Chlorosulfonyl)phenyl)propanoic acid | 3-(4-(Chlorosulfonyl)phenyl)propanoic acid, CAS:63545-54-0, MF:C9H9ClO4S, MW:248.68 g/mol | Chemical Reagent |
The denaturation of proteins with SDS buffer and heat is a deceptively simple yet profoundly critical step that dictates the success of subsequent SDS-PAGE analysis. This process, which intentionally dismantles native protein structures to create uniformly charged linear polypeptides, is the very foundation upon which the technique's principle of size-based separation is built. [5] [33] [35] A thorough understanding of the biochemical roles of SDS, reducing agents, and heat—coupled with meticulous execution of the preparation protocol—empowers researchers to generate high-quality, interpretable data. As SDS-PAGE continues to be an indispensable tool in proteomics, biomarker discovery, and biopharmaceutical development, the precision applied in these initial steps remains a fundamental determinant of experimental rigor and reliability.
Within the framework of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), a technique foundational to modern biochemistry and drug development, the stacking gel performs a critical, yet often overlooked, function. This in-depth technical guide elucidates the science behind this essential first step. The stacking gel leverages a discontinuous buffer system to concentrate disparate protein samples into ultrasharp bands before they enter the resolving gel, thereby ensuring the high-resolution separation that SDS-PAGE is renowned for. This article will deconstruct the underlying principles of this stacking phenomenon, provide detailed methodologies, and present quantitative data, firmly framing the discussion within the broader context of SDS's role in revolutionizing protein analysis by conferring a uniform charge-to-mass ratio and denaturing proteins to allow separation primarily by molecular weight [2] [37].
SDS-PAGE is the workhorse method for protein separation, and its efficacy hinges on the action of sodium dodecyl sulfate (SDS). This anionic detergent binds to proteins in a constant weight ratio (approximately 1.4 g SDS per 1 g of protein), performing two critical functions: it denatures proteins, disrupting their secondary, tertiary, and quaternary structures, and it imparts a uniform negative charge to the resulting polypeptide chains [2] [3] [37]. This process masks the intrinsic charges of proteins, creating SDS-polypeptide complexes that have similar charge-to-mass ratios and shapes, thus ensuring their migration through the gel is determined almost solely by molecular weight [2].
However, a challenge remains. Protein samples are loaded into wells that can be a centimeter deep, and if these samples entered the resolving gel in such a diffuse state, the result would be smeared, poorly resolved bands [38]. The stacking gel resolves this issue. It is a distinct gel layer, cast on top of the resolving gel, with a specific composition designed to concentrate all protein molecules into a single, sharp band. This process is fundamental to achieving the clarity and resolution required for applications ranging from purity assessment to western blotting [2] [3].
The Principle of the Discontinuous Buffer System
The stacking effect is not achieved by magic but by sophisticated electrochemistry. The system employs three points of discontinuity between the stacking and resolving gels: pH, gel pore size, and ionic composition [2] [38]. The core of the mechanism involves the manipulation of the mobility of three key ionic species: the chloride ion (Clâ»), the glycine ion, and the SDS-protein complex.
The table below outlines the standard composition of the key components in a Tris-Glycine SDS-PAGE system, highlighting their roles and differences.
Table 1: Composition and Roles of SDS-PAGE Buffer System Components
| Component | Stacking Gel | Resolving Gel | Running Buffer | Primary Function |
|---|---|---|---|---|
| pH | 6.8 [38] | 8.8 [38] | 8.3 [38] | Controls the charge state of glycine. |
| Acrylamide | Low concentration (~4%) [2] | Higher concentration (e.g., 8-15%) [3] | Not Applicable | Creates a porous matrix; stacking gel has larger pores for free movement. |
| Buffer Ions | Tris-HCl [38] | Tris-HCl [2] | Tris-Glycine, SDS [38] | Tris provides buffering capacity; Clâ» and glycine are the leading and trailing ions, respectively. |
The process can be broken down into two main phases, as illustrated in the following workflow diagram:
Diagram 1: The Stacking Gel Mechanism.
The Stacking Phase: When the electric current is applied in the running buffer (pH 8.3), glycine exists primarily as a glycinate anion. Upon entering the low-pH (6.8) environment of the stacking gel, glycine's charge state shifts dramatically. Its carboxyl group is protonated, resulting in a molecule that is predominantly a zwitterion with a net charge close to zero [38] [39]. This neutral state drastically reduces glycine's electrophoretic mobility, making it the "trailing ion." In contrast, the Clâ» ions from the Tris-HCl in the gel are small and fully negatively charged, giving them high mobility as the "leading ion." The SDS-protein complexes, with their uniform negative charge, have an intermediate mobility. This setup creates a steep voltage gradient between the fast Clâ» front and the slow glycine front. All SDS-protein complexes, regardless of size, are compressed or "stacked" into this narrow, moving boundary, entering the resolving gel as a single, sharp band [2] [38].
The Transition to Separation: As this stacked band reaches the resolving gel, it encounters a higher pH (8.8). At this pH, glycine loses a proton and is converted back into the highly mobile glycinate anion [38]. It rapidly accelerates, overtaking the protein stack and dissipating the steep voltage gradient. The proteins, now released from the stacking boundary and entering the gel with smaller pores, begin to be separated based on their molecular weight [2].
Experimental Protocols and Methodologies
Standard SDS-PAGE Gel Casting and Sample Preparation
The following step-by-step protocol, adapted from common laboratory practice, details the process of preparing and running a gel with a stacking layer [5].
Gel Casting:
- Assemble the Gel Cassette: Thoroughly clean the glass plates with ethanol and assemble them with spacers to form a leak-proof cassette [5].
- Prepare and Pour the Resolving Gel: Mix the components for the resolving gel—including acrylamide/bis-acrylamide at the desired percentage, Tris-HCl (pH 8.8), and SDS. Add the polymerization catalysts, ammonium persulfate (APS) and TEMED, and immediately pipette the solution into the gel cassette. Carefully overlay the gel solution with water or isopropanol to create a flat, level interface and prevent inhibition of polymerization by oxygen [2] [5]. Allow the gel to polymerize completely (typically 20-30 minutes).
- Prepare and Pour the Stacking Gel: After removing the overlay liquid, prepare the stacking gel solution with a lower percentage of acrylamide, Tris-HCl (pH 6.8), and SDS. Add APS and TEMED, pour the solution on top of the polymerized resolving gel, and immediately insert a clean comb. Allow it to polymerize [5].
Sample Preparation:
- Mix Sample with Laemmli Buffer: Combine the protein sample with an equal volume of 2X Laemmli buffer [39]. This buffer contains:
- SDS: To denature proteins and confer negative charge.
- A reducing agent (e.g., DTT or β-mercaptoethanol): To break disulfide bonds [3] [39].
- Glycerol: To add density for easy loading into wells [38] [39].
- Tris-HCl (pH 6.8): To provide the correct buffering environment.
- A tracking dye (Bromophenol Blue): To visualize sample migration [38] [39].
- Denature Samples: Heat the mixture at 95-100°C for 3-5 minutes to ensure complete denaturation [5].
- Centrifuge: Briefly centrifuge the samples to collect all liquid at the bottom of the tube before loading [5].
Methodological Variations and Recent Innovations
Colored Stacking Gels: A recent innovation addresses the challenge of visualizing the transparent wells of a standard stacking gel. Adding an acidic dye (e.g., tartrazine, brilliant blue FCF, or new coccine) to the stacking gel solution allows for easy visualization of wells, facilitating smoother sample loading without affecting the performance of the gel in subsequent protein separation or western blotting analyses [18].
Native SDS-PAGE (NSDS-PAGE): While standard SDS-PAGE is denaturing, a modified protocol known as Native SDS-PAGE (NSDS-PAGE) has been developed. This method omits the heating step and reduces the SDS concentration in the sample and running buffers. The goal is to achieve high-resolution separation while retaining native protein function and bound metal ions, which is impossible under full denaturation. In one study, this method increased Zn²⺠retention in proteomic samples from 26% to 98% and preserved the activity of most model enzymes tested [8].
The Scientist's Toolkit: Essential Reagents
The following table catalogs the key reagents required for the stacking gel process and their specific functions.
Table 2: Essential Research Reagents for Stacking Gel Electrophoresis
| Reagent | Function in Stacking/Process |
|---|---|
| Acrylamide/Bis-acrylamide | Forms the cross-linked polyacrylamide gel matrix; lower concentration in stacking gel creates larger pores [2] [3]. |
| Tris-HCl (pH 6.8) | Provides the specific acidic pH environment in the stacking gel necessary for glycine to become a zwitterion [38] [39]. |
| Ammonium Persulfate (APS) | Initiates the free-radical polymerization of acrylamide and bisacrylamide [2]. |
| TEMED | Catalyzes the polymerization reaction by accelerating the production of free radicals from APS [2]. |
| Glycine | Key trailing ion; its charge-state change between the stacking (zwitterion) and resolving (anion) gels is central to the discontinuous buffer system [38]. |
| SDS (Sodium Dodecyl Sulfate) | Denatures proteins and provides a uniform negative charge, creating SDS-polypeptide complexes with similar charge-to-mass ratios [2] [37]. |
| Laemmli Buffer | Sample buffer containing SDS, reducing agent, glycerol, Tris, and tracking dye to prepare samples for loading [39]. |
| Bromophenol Blue | Anionic tracking dye that migrates ahead of the proteins, allowing visualization of the electrophoresis progress [38] [39]. |
| 2-[3-(Trifluoromethyl)phenyl]propanedial | 2-[3-(Trifluoromethyl)phenyl]propanedial Supplier |
| 1-Boc-5-Cyano-3-hydroxymethylindole | 1-Boc-5-Cyano-3-hydroxymethylindole, CAS:914349-11-4, MF:C15H16N2O3, MW:272.3 g/mol |
The stacking gel is a masterpiece of practical electrochemistry, a critical enabler of the high-resolution protein separations that SDS-PAGE provides. By understanding and leveraging the principles of the discontinuous buffer system—the careful manipulation of pH, pore size, and ion mobility—researchers can ensure their samples are perfectly concentrated at the start of the separation journey. This foundational technique, born from Laemmli's work in 1970, continues to be indispensable in laboratories worldwide, forming the bedrock of protein analysis in fundamental research and drug development [40]. As innovations like colored stacking gels and native modifications emerge, the core science of the stacking gel remains as relevant as ever, ensuring that the first step in SDS-PAGE is a sharp one.
In sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the resolving gel serves as the critical molecular sieve that enables high-resolution separation of protein mixtures based on molecular weight. This sieving function is directly governed by the polyacrylamide matrix formation, created through the polymerization of acrylamide monomers cross-linked by bisacrylamide [2]. The precise control over this matrix density through varying acrylamide percentages represents a fundamental parameter that laboratory researchers must optimize to achieve effective protein separation. Within the broader context of SDS-PAGE methodology, SDS plays the crucial role of masking proteins' intrinsic charges and conferring a uniform negative charge density, thereby eliminating separation based on charge or conformation [6] [41]. This denaturation and charge normalization allows the polyacrylamide matrix to perform its singular function: separating proteins based on polypeptide chain length through molecular sieving [5]. The resolving gel, with its carefully calibrated pore structure, therefore constitutes the physical manifestation of the molecular sieve principle that underpins this ubiquitous laboratory technique.
The pore size of the polyacrylamide gel is inversely related to its total acrylamide concentration, with higher percentages creating smaller pores and a denser sieving matrix [2] [42]. This relationship directly controls the size range of proteins that can be effectively resolved, making the selection of appropriate gel percentage a critical experimental decision. Understanding how polyacrylamide concentration affects separation efficiency and range is essential for researchers across biological disciplines, from fundamental proteomic studies to applied drug development workflows where protein characterization is paramount.
The Biochemical Principle of Molecular Sieving
Polyacrylamide Gel Structure and Pore Formation
The molecular sieving properties of polyacrylamide gels originate from their precise chemical structure and polymerization mechanics. Polyacrylamide gels are formed through the polymerization of acrylamide monomers cross-linked by N,N'-methylenebisacrylamide (bisacrylamide) [2]. This reaction, initiated by ammonium persulfate (APS) and catalyzed by tetramethylethylenediamine (TEMED), creates a three-dimensional mesh-like network with predictable pore sizes [43] [41]. The pore dimensions are determined primarily by two factors: the total concentration of acrylamide and bisacrylamide (%T), and the concentration of the cross-linker relative to the total acrylamide (%C) [42]. Standard SDS-PAGE typically employs a bisacrylamide-to-acrylamide ratio of approximately 1:35, though this can be varied for special purposes [42].
The average pore diameter in polyacrylamide gels ranges between 20 and 150 nm, significantly smaller than the pores in agarose gels used for nucleic acid separation [43] [41]. These pores act as a molecular sieve, retarding the migration of proteins based on their hydrodynamic radius in SDS-denatured conditions. The higher the polyacrylamide percentage, the smaller the pore size, creating a denser matrix that provides greater resistance to protein migration [2] [42]. This sieving effect allows smaller proteins to navigate the pores more readily than their larger counterparts, establishing the foundation for size-based separation.
The Role of SDS in Normalizing Protein Charge
For the molecular sieving mechanism to separate proteins strictly by molecular weight, SDS plays the indispensable role of eliminating the influence of protein charge and structure. SDS is a strong anionic detergent that binds to protein backbones at a constant ratio of approximately 1.4 g SDS per 1 g of polypeptide, corresponding to roughly one SDS molecule per two amino acids [6] [42]. This uniform SDS coating, combined with the disruption of hydrogen bonds through heating and cleavage of disulfide bonds by reducing agents like β-mercaptoethanol or dithiothreitol (DTT), unfolds proteins into linear chains [5] [41]. The result is that all SDS-coated polypeptides assume a similar rod-like shape with consistent negative charge proportional to their length [6] [2].
This charge and conformation normalization is crucial because it ensures that during electrophoresis, all proteins experience identical electrostatic driving forces toward the anode, with migration differences arising solely from the differential frictional resistance encountered when passing through the gel matrix [5] [42]. Without SDS, proteins would separate according to their inherent charge, shape, and size, creating complex migration patterns that would preclude straightforward molecular weight determination. The combination of SDS treatment and polyacrylamide sieving thus creates a system where protein migration distance correlates inversely with the logarithm of molecular weight, enabling both analytical separations and molecular weight estimations [42].
Table 1: Key Reagents in SDS-PAGE and Their Functions
| Reagent | Function | Technical Specification |
|---|---|---|
| Acrylamide | Polymerizable monomer forming gel matrix backbone | Neurotoxic in monomer form; typically used at 5-25% total concentration |
| Bisacrylamide | Cross-linking agent creating porous network | Creates three-dimensional mesh; standard ratio ~1:35 (bis:acrylamide) |
| Ammonium Persulfate (APS) | Free radical initiator for polymerization | Typically used at 0.1% concentration; generates free radicals for chain reaction |
| TEMED | Polymerization catalyst | Accelerates free radical production from APS; final concentration ~0.1% |
| SDS (Sodium Dodecyl Sulfate) | Protein denaturant and charge normalizer | Binds ~1.4g per 1g protein; masks intrinsic charge; concentration 0.1-0.2% in buffers |
| Tris-HCl Buffer | pH maintenance during electrophoresis | Stacking gel: pH 6.8; Resolving gel: pH 8.8; maintains optimal charge states |
Optimization of Polyacrylamide Percentage for Target Protein Separation
Gel Percentage Selection Based on Protein Size Range
The selection of appropriate polyacrylamide concentration is perhaps the most critical factor in achieving optimal protein separation. The relationship between gel percentage and effective separation range follows a predictable pattern where lower percentage gels (with larger pore sizes) resolve high molecular weight proteins better, while higher percentage gels (with smaller pore sizes) provide superior resolution for low molecular weight proteins [2]. This principle enables researchers to select gel percentages tailored to their specific protein targets, as detailed in Table 2.
Table 2: Polyacrylamide Gel Percentage Recommendations for Protein Separation
| Gel Percentage (%) | Optimal Separation Range (kDa) | Application Notes |
|---|---|---|
| 6-8% | 50-200 | Ideal for high molecular weight proteins; fragile and difficult to handle |
| 10% | 15-100 | Standard workhorse gel for general protein separation |
| 12% | 10-70 | Common choice for moderate molecular weight range |
| 15% | 12-45 | Suitable for smaller proteins; may appear turbid |
| Up to 20% | 4-40 | Essential for resolving very small proteins and peptides |
The migration of proteins through these different percentage gels follows a predictable pattern where the distance traveled is inversely proportional to the logarithm of molecular weight [42]. This relationship enables the creation of standard curves using molecular weight markers, allowing estimation of unknown protein sizes. However, certain proteins exhibit anomalous migration patterns due to factors such as extensive glycosylation, high proline content, or membrane-associated hydrophobic domains that affect SDS binding efficiency [42]. For such problematic proteins, gradient gels often provide superior resolution.
Gradient Gels: Expanding the Separation Range
Gradient gels represent a sophisticated solution to the limitation of fixed-concentration gels, which optimally separate only a limited range of protein sizes. These gels are formulated with a continuous gradient of polyacrylamide concentration, typically from low to high percentage, creating a corresponding pore size gradient [44]. As proteins migrate through such gels, they encounter progressively smaller pores, creating a stacking effect that results in sharper bands and improved resolution across a broader molecular weight range [44].
The advantages of gradient gels include their ability to resolve a wider spectrum of protein sizes on a single gel, produce sharper bands due to the continuous decrease in pore size, and better separate similarly-sized proteins through extended separation distances [44]. When selecting gradient ranges, researchers should choose gradients that bracket their proteins of interest, with popular configurations including 4-20% for discovery work (separating proteins from 4-250 kDa), 8-15% for more targeted approaches, and 10-12.5% for resolving similarly-sized proteins [44]. Although gradient gels require more sophisticated preparation using gradient mixers or specialized pouring techniques, they often provide superior results, particularly for complex protein mixtures with diverse molecular weights.
Experimental Protocol: Resolving Gel Preparation and Electrophoresis
Standard Protocol for Discontinuous SDS-PAGE Gel Casting
The following detailed protocol outlines the preparation of resolving gels for discontinuous SDS-PAGE, the most widely used variant for protein separation [6] [41]. This procedure assumes the use of a standard mini-gel format (approximately 8 × 8 cm) with 1.0 mm spacers.
Gel Casting Materials:
- Clean glass plates, spacers, and comb
- Acrylamide/bisacrylamide solution (typically 30-40% stock, 37.5:1 ratio)
- Separating gel buffer: 1.5 M Tris-HCl, pH 8.8
- Stacking gel buffer: 0.5 M Tris-HCl, pH 6.8
- 10% sodium dodecyl sulfate (SDS)
- 10% ammonium persulfate (freshly prepared)
- TEMED
- Water-saturated isobutanol or isopropanol
- Gel casting cassette and stand
Resolving Gel Preparation:
- Assemble Gel Cassette: Thoroughly clean and dry glass plates before assembly with spacers to prevent leaks [5].
- Prepare Resolving Gel Solution: For a standard 12% resolving gel, mix 4.0 mL of 30% acrylamide/bis solution, 2.5 mL of 1.5 M Tris-HCl (pH 8.8), 3.4 mL deionized water, 0.1 mL of 10% SDS, 0.1 mL of 10% APS, and 0.004 mL TEMED [2]. Add TEMED last to initiate polymerization.
- Cast Resolving Gel: Pipette the solution between glass plates, leaving space for stacking gel (approximately 2 cm from top).
- Overlay with Alcohol: Carefully add water-saturated isobutanol or isopropanol to cover gel surface, eliminating oxygen which inhibits polymerization and creating a crisp, flat interface [5] [42].
- Polymerize: Allow gel to set for 20-30 minutes until polymerization is complete; a distinct schlieren line will be visible at the alcohol-gel interface.
- Remove Overlay: Pour off overlay liquid and rinse gel surface thoroughly with deionized water.
Stacking Gel Preparation:
- Prepare Stacking Gel Solution: Mix 0.67 mL of 30% acrylamide/bis, 1.25 mL of 0.5 M Tris-HCl (pH 6.8), 3.0 mL deionized water, 0.05 mL of 10% SDS, 0.05 mL of 10% APS, and 0.005 mL TEMED [2].
- Cast Stacking Gel: Pour solution onto polymerized resolving gel, immediately insert clean comb without trapping air bubbles.
- Complete Polymerization: Allow stacking gel to polymerize for 20-30 minutes before carefully removing comb.
Sample Preparation and Electrophoresis Conditions
Protein Sample Preparation:
- Mix with Sample Buffer: Combine protein sample with 4X SDS-PAGE sample buffer (final 1X concentration: 62.5 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue) with or without 5% β-mercaptoethanol or 100 mM DTT as reducing agent [6] [41].
- Denature Proteins: Heat samples at 95-100°C for 3-5 minutes or 70°C for 10 minutes to linearize proteins [5] [6].
- Centrifuge: Briefly spin samples at 15,000 × g for 1 minute to pellet insoluble debris [5].
Electrophoresis Execution:
- Assemble Apparatus: Mount gel in electrophoresis tank, fill upper and lower chambers with running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3-8.5) [6] [41].
- Load Samples: Carefully pipette prepared samples and molecular weight markers into wells; typical mini-gel loading volume is 10-20 μL.
- Apply Current: Run gel at constant voltage (100-200 V for mini-gels) until bromophenol blue tracking dye reaches bottom of gel (approximately 45-60 minutes) [5] [6].
- Terminate Run: Turn off power supply, disassemble apparatus, and carefully remove gel from plates for subsequent staining or transfer.
Diagram 1: SDS-PAGE Workflow from Sample Preparation to Separation
Technical Variations and Advanced Applications
Gradient Gel Preparation Techniques
For researchers requiring enhanced separation capabilities, gradient gels offer significant advantages. Two primary methods exist for creating gradient gels:
Gradient Mixer Method:
- Setup: Use a two-chamber gradient mixer connected to gel cassette via tubing.
- Prepare Solutions: Place low-percentage acrylamide solution in "reservoir" chamber and high-percentage solution in "mixing" chamber; both contain APS and TEMED.
- Initiate Flow: Open connection between chambers and start flow to cassette, creating continuous gradient.
- Overlay and Polymerize: Carefully overlay with solvent as with standard gels [44].
Pipette Mixing Method (Simplified Alternative):
- Prepare Solutions: Have low and high percentage acrylamide solutions with APS/TEMED in separate tubes.
- Layer in Pipette: Aspirate half total volume needed from low percentage, then half from high percentage into serological pipette.
- Create Air Bubble: Gently aspirate ~0.5 mL air, allowing bubble to travel up pipette to mix solutions during dispensing.
- Pour Gradient: Slowly pipette mixed solution into gel cassette [44].
Alternative Buffer Systems and Native Electrophoresis
While the Tris-glycine-SDS buffer system is most common, alternative buffers offer specialized advantages. Tris-acetate-SDS buffers provide better resolution for high molecular weight proteins (up to 400 kDa), while Tris-tricine-SDS systems optimize separation of small proteins and peptides (1-100 kDa) [6] [41]. For native PAGE, where protein structure and function are preserved, SDS is omitted from all buffers, no reducing agents are added, and samples are not heated before loading [2] [41]. This approach separates proteins based on both charge and size, maintaining enzymatic activity and protein complexes [2].
A modified approach called native SDS-PAGE (NSDS-PAGE) reduces SDS concentration in running buffer to 0.0375% and eliminates EDTA and heating steps, resulting in high resolution separation while retaining enzymatic activity and metal cofactors in many proteins [8]. This innovation demonstrates how understanding the fundamental principles of molecular sieving enables methodological refinements for specialized applications.
The resolving gel in SDS-PAGE functions as a precision molecular sieve whose separation characteristics are directly controlled by polyacrylamide percentage. Through the charge-normalizing action of SDS, proteins migrate according to size as they navigate the porous matrix, with smaller proteins migrating more rapidly than larger counterparts. The strategic selection of gel percentage—whether a single concentration optimized for a specific molecular weight range or a gradient gel for broader separation—represents a critical experimental decision that directly impacts resolution quality. This technical understanding enables researchers to tailor electrophoretic conditions to their specific protein separation needs, ensuring optimal results across diverse applications from routine protein analysis to advanced proteomic research. The precise control over polyacrylamide concentration remains a fundamental aspect of experimental design in protein biochemistry, making the resolving gel not merely a support matrix but an active molecular sieve whose properties determine separation success.
Within the framework of Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE), a technique foundational to biochemical research and drug development, the running buffer is not merely a supportive reagent but a critical component that ensures the success and reproducibility of the separation. The broader thesis on the role of SDS in polyacrylamide gel electrophoresis research establishes that SDS acts as a powerful denaturant and charge-masking agent, conferring a uniform negative charge to all proteins and thereby allowing separation based almost exclusively on molecular weight [45] [6] [46]. However, this fundamental role of SDS can only be effectively realized within the precise electrochemical environment maintained by the running buffer. This technical guide delves into the composition and function of the running buffer, elucidating how it works in concert with SDS to facilitate high-resolution protein separation by rigorously managing pH and conductivity throughout the electrophoretic process.
Chemical Composition of Standard SDS-PAGE Running Buffer
The most prevalent running buffer system, based on the discontinuous Laemmli method, is a Tris-Glycine-SDS buffer [6] [47]. It is typically prepared as a concentrated stock solution (e.g., 10X) for convenience and diluted to its working concentration (1X) before use. The table below details the standard components and their final concentrations in the working buffer.
Table 1: Chemical Composition of a Standard 1X Tris-Glycine-SDS Running Buffer
| Component | Molecular Weight (g/mol) | Final Concentration | Role in Electrophoresis |
|---|---|---|---|
| Tris Base | 121.14 | 25 mM | Maintains a stable basic pH (8.3) for the system; primary buffer agent [48] [47]. |
| Glycine | 75.07 | 192 mM (250 mM in some protocols) | A trailing ion that works with Tris to create the discontinuous buffer system essential for protein stacking [6] [46]. |
| SDS (Sodium Dodecyl Sulfate) | 288.38 | 0.1% (w/v) | Reinforces the denaturation of proteins and maintains a uniform negative charge density during electrophoresis [48] [6]. |
| Deionized Water | - | To volume | Solvent for all components. |
The preparation of the running buffer is a straightforward process. For a 1-liter volume of 10X stock solution, 30.3 g of Tris base and 144.0 g of glycine are dissolved in approximately 800 mL of distilled water. Then, 10 g of SDS is added. The solution is gently stirred and heated if necessary to solubilize the SDS completely, after which the volume is adjusted to 1 L with distilled water. The pH of the 10X stock is typically around 8.3 and does not require adjustment. For use, this stock is diluted ten-fold with deionized water to achieve the 1X working concentration [49] [47].
The Functional Role of Running Buffer in Maintaining pH and Conductivity
The running buffer is the central ionic medium that governs the electrophoretic process. Its functions are multifaceted and critical for achieving sharp, well-resolved protein bands.
Primary Functions and Mechanisms
Establishing Electrical Conductivity: The running buffer contains ions (Tris+, Cl-, glycinate-) that are essential for conducting electric current through the gel apparatus. Without these ions, the applied voltage would not create a current, and protein migration would not occur [46]. The conductivity of the solution is directly proportional to the ion concentration and mobility.
Maintaining a Stable pH Environment: The Tris-Glycine buffer system is designed to maintain a stable pH of 8.3 in the electrode chambers [48] [46]. This alkaline environment is crucial for the function of the discontinuous buffer system, particularly for controlling the charge state of glycine, as detailed below.
Replenishing SDS for Protein Stability: The SDS in the running buffer (0.1%) helps ensure that proteins remain denatured and uniformly coated with negative charge as they migrate through the gel, preventing re-folding and aggregation that could lead to smeared or distorted bands [48] [6].
The Discontinuous Buffer System and the Stacking Effect
A key innovation in modern SDS-PAGE is the discontinuous buffer system, which relies critically on the running buffer's composition and pH. This system involves a stacking gel (pH ~6.8) and a separating gel (pH ~8.8) in addition to the running buffer (pH ~8.3) [6] [47]. The mechanism hinges on the changing ionization state of glycine.
In the running buffer at pH 8.3, glycine exists predominantly as a glycinate anion, which is highly mobile. However, when this ion enters the low-pH environment of the stacking gel, its carboxyl group becomes protonated, converting it into a neutral zwitterion with a much lower electrophoretic mobility. This creates an ion gradient where chloride ions (from Tris-HCl in the gels) are the fast "leading" ions, and the glycine zwitterions are the slow "trailing" ions [6] [46].
The proteins, with a mobility intermediate to the leading and trailing ions, are compressed into a very narrow zone between these two fronts. This "stacking" effect concentrates all protein samples into sharp layers before they enter the separating gel, dramatically improving resolution [47]. When the stacked proteins reach the separating gel with its higher pH (~8.8), the glycine zwitterions regain their negative charge, become mobile again, and overtake the proteins. The proteins then separate based on their size as they migrate through the sieving matrix of the separating gel.
Diagram: Glycine's charge transition drives protein stacking and separation.
Experimental Protocol for Buffer Preparation and Electrophoresis
A reliable protocol is essential for generating consistent and high-quality results. The following section outlines the standard methodology for preparing and using Tris-Glycine running buffer.
Reagent Preparation
- 10X Running Buffer Stock: Dissolve 30.3 g of Tris base, 144.0 g of glycine, and 10.0 g of SDS in approximately 800 mL of deionized water. Once fully dissolved, bring the final volume to 1 L with deionized water. The pH should be approximately 8.3 and does not require adjustment. Store at room temperature [49] [47].
- 1X Working Solution: Dilute the 10X stock solution 1:10 with deionized water. For example, to prepare 1 L of 1X running buffer, mix 100 mL of 10X stock with 900 mL of deionized water [48].
Electrophoresis Procedure
- Gel Assembly: After polymerizing the stacking and separating gels, assemble the gel cassette into the electrophoresis tank according to the manufacturer's instructions.
- Buffer Introduction: Fill the inner (upper) and outer (lower) chambers of the electrophoresis apparatus with the freshly prepared 1X running buffer. Ensure that the gel wells in the upper chamber are completely submerged to prevent arcing and sample evaporation [47].
- Sample Loading: Mix protein samples with an SDS-containing sample buffer, denature by heating (95°C for 5 minutes), and load into the wells. Include a molecular weight marker in one well.
- Electrophoresis Run: Place the lid on the tank, connect the electrodes to the power supply (cathode at the top, anode at the bottom), and apply a constant voltage. For a standard mini-gel, 100-150 V is typical. Run until the dye front (bromophenol blue) migrates to the bottom of the gel [6].
- Post-Run Analysis: Turn off the power supply, disassemble the apparatus, and carefully remove the gel for subsequent staining (e.g., Coomassie, silver stain) or transfer to a membrane for Western blotting [45] [48].
Table 2: Troubleshooting Common Running Buffer and Electrophoresis Issues
| Problem | Potential Cause | Solution |
|---|---|---|
| Smiling Bands | Excessive heat during run. | Use a lower voltage, run in a cold room, or use a cooling apparatus [50]. |
| Smeared Bands | Old or degraded running buffer; incomplete protein denaturation. | Prepare fresh running buffer and ensure samples are properly heated in SDS sample buffer [50] [48]. |
| Slow Migration | Buffer concentration or pH is incorrect; low voltage. | Check buffer dilution and pH; ensure power supply is functioning correctly. |
| Poor Resolution | Insufficient buffering capacity; incorrect gel percentage. | Use fresh, correctly prepared buffer. Choose a gel percentage appropriate for the target protein size range [50]. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful SDS-PAGE relies on a suite of carefully formulated reagents. The table below details the key components beyond the running buffer.
Table 3: Essential Reagents for SDS-PAGE Experimentation
| Reagent | Composition | Function |
|---|---|---|
| SDS Sample Buffer | Tris-HCl, SDS, glycerol, bromophenol blue, often with a reducing agent (β-mercaptoethanol or DTT) [6] [46]. | Denatures proteins, provides negative charge, adds density for loading, and includes a tracking dye. Reducing agents break disulfide bonds. |
| Separating Gel | Acrylamide/bis-acrylamide, Tris-HCl (pH 8.8), SDS, APS, TEMED [6] [47]. | Forms the sieving matrix that resolves proteins by molecular weight. The concentration (%T) determines the separation range. |
| Stacking Gel | Acrylamide/bis-acrylamide, Tris-HCl (pH 6.8), SDS, APS, TEMED [6] [47]. | A low-concentration gel that focuses all proteins into a sharp starting zone before they enter the separating gel. |
| Transfer Buffer | Tris, Glycine, Methanol (for wet transfer) [48]. | A conductive medium used in Western blotting to electrophoretically transfer separated proteins from the gel onto a membrane. |
| 2-Chloro-5-cyanobenzenesulfonamide | 2-Chloro-5-cyanobenzenesulfonamide, CAS:1939-76-0, MF:C7H5ClN2O2S, MW:216.65 g/mol | Chemical Reagent |
| 3-Bromo-5-(3-chlorophenoxy)pyridine | 3-Bromo-5-(3-chlorophenoxy)pyridine, CAS:28232-65-7, MF:C11H7BrClNO, MW:284.53 g/mol | Chemical Reagent |
The running buffer is an indispensable component of SDS-PAGE, whose role extends far beyond being a simple conductive liquid. By working synergistically with the overarching function of SDS—to impart a uniform charge-to-mass ratio—the Tris-Glycine-SDS running buffer establishes the precise electrochemical conditions necessary for the discontinuous separation. Its ability to maintain a stable pH and controlled conductivity is what enables the critical stacking phenomenon and subsequent high-resolution separation of proteins by size. As SDS-PAGE continues to be a cornerstone technique in scientific research and biopharmaceutical development, from analyzing therapeutic proteins to detecting allergens in food, a deep and practical understanding of the running buffer's composition and role remains fundamental to experimental success [45] [51].
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) represents a foundational methodology in biochemical research, providing a robust platform for protein analysis. This technical guide examines three core applications of SDS-PAGE: molecular weight estimation, purity assessment, and preparation for western blotting. The central role of SDS in denaturing protein complexes and imparting a uniform negative charge enables precise protein separation based solely on molecular dimensions. Through detailed protocols, troubleshooting guidelines, and analytical frameworks, this whitepaper serves as a comprehensive resource for researchers and drug development professionals seeking to implement these critical techniques in their experimental workflows.
SDS-PAGE operates on the principle that proteins denatured by sodium dodecyl sulfate (SDS) and reducing agents can be separated based primarily on polypeptide chain length rather than native structure or intrinsic charge [5]. SDS, an anionic detergent with strong protein-denaturing capability, binds to protein backbones at an approximately constant ratio of 1.4 grams SDS per gram of protein [6]. This binding confers a uniform negative charge density, effectively masking proteins' intrinsic charges and creating similar charge-to-mass ratios across different protein species [6].
The denaturing process unfolds proteins into linear chains, with SDS disrupting hydrogen bonds and hydrophobic interactions while reducing agents cleave disulfide bonds critical for proper folding [5] [52]. When subjected to an electric field within a polyacrylamide gel matrix, these SDS-coated proteins migrate toward the anode at rates inversely proportional to the logarithm of their molecular mass [53]. The polyacrylamide gel serves as a molecular sieve, with smaller proteins experiencing less resistance and migrating faster through the gel matrix [5]. This system enables researchers to separate proteins with molecular masses typically between 5 and 250 kDa [6], though separation ranges can be optimized by adjusting gel composition.
Estimating Molecular Weight
Principle and Methodology
Molecular weight estimation via SDS-PAGE relies on the logarithmic relationship between protein mobility and molecular mass. Proteins are separated based on polypeptide chain length, as SDS binding confers a uniform negative charge proportional to protein size [5]. The polyacrylamide gel matrix acts as a molecular sieve, with smaller proteins migrating faster than larger ones [53].
The procedure involves several critical steps. First, protein samples are prepared with Laemmli buffer (containing SDS and reducing agents like β-mercaptoethanol or DTT) and heated to 95°C for 5 minutes to ensure complete denaturation [6]. The denatured samples are then loaded into wells of a polyacrylamide gel alongside molecular weight markers containing proteins of known sizes [53]. Electrophoresis is performed at constant voltage (typically 100-200V) until the dye front (bromophenol blue) reaches the gel bottom [5] [8].
To estimate molecular weight, the relative distance of migration (Rf) is calculated for both standards and unknown proteins [53]. A standard curve is generated by plotting the logarithm of molecular weights of standard proteins against their migration distances, enabling interpolation of unknown protein masses from this calibration curve [6].
Optimization Strategies for Accurate Molecular Weight Determination
Table 1: Gel Percentage Selection Based on Target Protein Size
| Acrylamide Percentage | Optimal Separation Range | Application Notes |
|---|---|---|
| 15% | 10-50 kDa | Ideal for small proteins and peptides |
| 12% | 40-100 kDa | Standard range for most applications |
| 10% | 70 kDa and above | Suitable for large proteins |
| 4-20% Gradient | 5-200 kDa | Broad range separation |
Several factors influence the accuracy of molecular weight estimation. Gel concentration must be appropriate for the target protein size, with higher acrylamide percentages providing better resolution for smaller proteins [53]. Gradient gels (e.g., 4-20% acrylamide) offer extended separation ranges ideal for complex samples [6]. Sample preparation must ensure complete denaturation and reduction, as incomplete unfolding can alter migration patterns [52]. Additionally, post-translational modifications such as glycosylation or phosphorylation may reduce SDS binding, causing anomalous migration [54]. For proteins with such modifications, molecular weight estimates may deviate from actual values by approximately ±10% [6].
Figure 1: Workflow for Molecular Weight Estimation Using SDS-PAGE
Assessing Protein Purity
Analytical Framework for Purity Assessment
SDS-PAGE provides a rapid, sensitive method for evaluating protein purity by visualizing contaminants through differential staining patterns. In a pure preparation, a single protein should yield a single band at the expected molecular weight, while impurities appear as additional bands [6]. The high resolution of polyacrylamide gels allows detection of contaminants as small as 5-10% of the total protein content when using sensitive staining methods [53].
The assessment process begins with resolving the protein sample alongside appropriate controls on an SDS-PAGE gel optimized for the target protein size. Following electrophoresis, the gel is stained using Coomassie Brilliant Blue for standard sensitivity (detection limit ~50-100 ng) or silver staining for enhanced sensitivity (detection limit ~0.1-1 ng) [6]. Stained gels are then analyzed for band number, intensity, and position relative to molecular weight standards.
Troubleshooting Common Purity Issues
Table 2: Troubleshooting Common Purity Assessment Problems
| Issue Observed | Potential Causes | Recommended Solutions |
|---|---|---|
| Multiple bands | Protein degradation, contaminating proteins, incomplete denaturation | Use protease inhibitors, improve purification, ensure complete denaturation with fresh reducing agents |
| Smeared bands | Insufficient reduction/denaturation, overloading, high salt concentration | Add fresh reducing agent, boil samples for 5+ minutes at 100°C, reduce salt concentration below 500 mM |
| Unexpected band positions | Post-translational modifications, anomalous SDS binding | Consider glycosylation/phosphorylation, use deglycosylation enzymes |
| High background | Incomplete destaining, membrane blocking issues | Optimize destaining times, test alternative blocking agents |
Unexpected banding patterns often indicate specific purity issues. Multiple bands may suggest protein degradation, which can be mitigated by adding protease inhibitors to samples [53]. Smearing often results from incomplete denaturation, remedied by adding fresh reducing agents and ensuring adequate heating [52]. High salt concentrations (>500 mM) can cause smearing and should be reduced before loading [53]. For membrane-associated proteins, detergent choice in extraction buffers significantly impacts solubility and apparent purity [55].
Negative controls are essential for proper interpretation, including samples from cells or tissues known not to express the target protein [53]. Loading controls such as housekeeping proteins (e.g., β-actin) confirm consistent sample loading across wells, crucial for quantitative purity assessments [53].
Preparing for Western Blotting
SDS-PAGE as a Critical Precursor to Immunoblotting
SDS-PAGE serves as the essential first separation step in western blotting, enabling subsequent protein identification and characterization through antibody-based detection [56]. The denaturing conditions of SDS-PAGE are particularly suited for western blotting as they linearize proteins, expose internal epitopes, and inactivate proteases that might otherwise degrade samples during the extended blotting procedure [52].
The process involves separating proteins via SDS-PAGE as described in previous sections, followed by electrophoretic transfer of the separated proteins from the gel to a membrane support [56]. This transfer must maintain the spatial separation achieved during electrophoresis while rendering proteins accessible for antibody binding.
Optimizing SDS-PAGE Conditions for Western Blotting
Several parameters require optimization to ensure successful western blotting. Gel composition should be chosen based on target protein size, with lower percentage gels (8-10%) preferred for large proteins (>100 kDa) and higher percentages (12-15%) for smaller proteins [53]. The inclusion of SDS in both sample preparation and running buffers is critical for maintaining protein denaturation throughout separation [54].
Sample preparation must balance complete denaturation with antigen preservation. Standard conditions (heating to 95°C for 5 minutes in Laemmli buffer) work for most targets, but some sensitive epitopes may require milder heating (70°C for 10-15 minutes) [6] [8]. Additionally, the transfer buffer typically contains a small amount of SDS (0.01-0.025%) to maintain protein solubility during transfer, though excessive SDS can interfere with protein-membrane binding [56].
Figure 2: Western Blotting Workflow Following SDS-PAGE Separation
Essential Reagents and Methodologies
Research Reagent Solutions
Table 3: Essential Reagents for SDS-PAGE Applications
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Denaturing Agents | Sodium dodecyl sulfate (SDS) | Unfolds proteins, imparts uniform charge; use at 0.1-1% concentration |
| Reducing Agents | β-mercaptoethanol, DTT, DTE | Breaks disulfide bonds; DTT preferred for stronger reduction |
| Gel Components | Acrylamide, bis-acrylamide | Forms porous matrix; concentration determines separation range |
| Polymerization Catalysts | Ammonium persulfate (APS), TEMED | Initiates and accelerates gel polymerization |
| Buffers | Tris-glycine, Tris-HCl, Bis-Tris | Maintains pH; discontinuous systems enhance band resolution |
| Tracking Dyes | Bromophenol blue | Visualizes migration front during electrophoresis |
| Molecular Weight Markers | Prestained, unstained standards | Provides size calibration; prestained markers monitor transfer |
| Transfer Reagents | PVDF/nitrocellulose membranes, transfer buffer | Immobilizes proteins for western blotting |
Detailed Experimental Protocol: SDS-PAGE for Western Blotting
Gel Preparation:
- Assemble clean glass plates with spacers (typically 0.75-1.5 mm thickness) [6].
- Prepare separating gel solution (appropriate percentage for target protein size), add TEMED and APS to initiate polymerization, and pour between plates [5].
- Overlay with water-saturated butanol or isopropanol to exclude oxygen and create a flat interface [6].
- After polymerization (20-30 minutes), remove overlay and prepare stacking gel (4-6% acrylamide) [5].
- Pour stacking gel, insert comb without creating bubbles, and allow to polymerize [5].
Sample Preparation:
- Mix protein samples with Laemmli buffer (2X concentration) containing SDS and reducing agent [52].
- Heat at 95°C for 5 minutes or 70°C for 10 minutes to denature proteins [6].
- Centrifuge at 15,000 rpm for 1 minute to collect condensed sample [5].
Electrophoresis:
- Mount gel in electrophoresis apparatus and fill chambers with running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3) [54].
- Remove comb and load samples (10-50 μg total protein per lane) alongside molecular weight markers [5].
- Run at constant voltage (100-150V for mini-gels) until dye front reaches bottom (approximately 45-90 minutes) [8].
Post-Electrophoresis Processing:
- For western blotting, proceed to transfer step [56].
- For direct analysis, stain with Coomassie Blue (30-60 minutes) or silver stain (higher sensitivity) [6].
- Destain and image gel for molecular weight estimation or purity assessment [5].
SDS-PAGE remains an indispensable technique in protein research, with core applications spanning molecular weight estimation, purity assessment, and preparation for western blotting. The fundamental role of SDS in creating uniform charge densities across diverse protein species enables separation based primarily on molecular dimensions. Through optimized protocols and appropriate troubleshooting approaches, researchers can leverage these applications for diverse protein characterization needs. As methodologies continue to evolve, including the development of precast gels and specialized buffer systems, the utility of SDS-PAGE in basic research and drug development remains firmly established.
Beyond the Basics: Troubleshooting Common SDS-PAGE Issues for Publication-Quality Gels
Diagnosing and Fixing Smeared or Distorted Protein Bands
The Fundamental Role of SDS in Electrophoresis
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) remains a cornerstone technique in biochemical research, particularly in protein characterization for drug development. The essential role of SDS, an anionic detergent, is to mask the protein's intrinsic charge and confer a relatively uniform charge-to-mass ratio by binding to the protein backbone with high affinity. [6] This binding, approximately 1.4 grams of SDS per gram of protein, unfolds higher-order structures, transforming proteins into linear chains. [6] This process is crucial because it eliminates the influence of native protein charge and three-dimensional shape, ensuring that separation occurs primarily based on molecular mass as proteins migrate through the polyacrylamide gel matrix toward the anode. [57] [6] Understanding this fundamental mechanism is the first step in diagnosing separation anomalies, as most issues with band smearing and distortion relate to the failure of this denaturation process or subsequent experimental parameters.
Systematic Diagnosis of Band Anomalies
Smeared or distorted bands indicate a failure in the system to properly separate proteins by size. The following workflow outlines a systematic approach to diagnose the root cause of these common issues. This process begins with the most frequent culprits in sample preparation before moving to gel and electrophoresis parameters.
Comprehensive Troubleshooting Guide and Solutions
Once the diagnostic workflow identifies potential causes, the following table provides detailed solutions and methodological adjustments to resolve specific issues. This guide consolidates quantitative data and protocols for easy reference.
Table 1: Troubleshooting Smeared and Distorted Protein Bands
| Problem Category | Specific Issue | Recommended Solution | Experimental Protocol Adjustment |
|---|---|---|---|
| Sample Preparation | Insufficient Denaturation | Ensure complete unfolding by optimizing SDS, reductant, and heat. | Increase boiling time to 5 minutes at 98°C; include fresh β-mercaptoethanol (5% v/v) or DTT (10-100 mM); place samples on ice immediately after heating to prevent renaturation. [58] [6] |
| Protein Overloading | Reduce amount of protein loaded per well. | Load a maximum of 10-20 µg of protein per well for a standard mini-gel; perform a protein concentration assay prior to loading. [59] [58] | |
| High Salt Concentration | Desalt samples to prevent current disruption and band skewing. | Dialyze the sample, precipitate protein with TCA, or use a desalting column. [60] | |
| Protein Aggregation | Solubilize hydrophobic or precipitating proteins. | Add 4-8 M urea to the sample buffer; for heat-sensitive samples, incubate at 60°C instead of 95°C. [59] [60] | |
| Gel System | Incorrect Acrylamide % | Match gel pore size to target protein size. | Use low-percentage gels (e.g., 4-12%) for high MW proteins (>100 kDa); use high-percentage gels (e.g., 12-20%) for low MW proteins (<30 kDa); use a 4-20% gradient gel for a broad separation range. [58] [60] |
| Incomplete Polymerization | Ensure gel is fully formed and uniform. | Confirm fresh TEMED and ammonium persulfate (APS) are used; allow sufficient time (30+ minutes) for complete polymerization before removing comb. [58] [60] | |
| Electrophoresis | Voltage Too High | Prevent "smile effects" and overheating. | Decrease voltage by 25-50%; run gels at a constant lower voltage (e.g., 80-100V) for a longer time; use a cooled apparatus or run in a cold room. [58] [60] |
| Old or Depleted Buffers | Maintain proper pH and ionic strength for conduction. | Prepare fresh running buffer before each run; ensure SDS is present in the running buffer (e.g., 0.1% in standard protocols). [58] |
Advanced Methodological Considerations
The Role of Buffer Systems and Additives
The discontinuous buffer system, typically involving Tris-glycine, is fundamental to achieving sharp band stacking. In the stacking gel (pH ~6.8), glycine exists as a zwitterion with low mobility, creating a voltage gradient that concentrates proteins into a tight band before they enter the resolving gel (pH ~8.8). [57] [6] Any disruption to this pH balance or ionic composition can cause band broadening. For difficult samples, especially those containing hydrophobic proteins, the addition of urea (4-8 M) to the sample buffer can disrupt hydrophobic interactions and prevent aggregation that leads to smearing. [59] [60]
Native SDS-PAGE as a Comparative Tool
A modified technique known as Native SDS-PAGE (NSDS-PAGE) can be employed to retain protein function while still achieving good separation. This method involves omitting SDS and EDTA from the sample buffer, removing the heating step, and reducing the SDS concentration in the running buffer to 0.0375%. [8] This protocol results in 98% retention of bound metal ions like Zn²⺠and preserves the activity of many enzymes, a stark contrast to the complete denaturation of standard SDS-PAGE. [8] While not a solution for smearing in denaturing gels, understanding this alternative highlights the critical role of SDS concentration and sample treatment in determining electrophoretic outcomes.
Essential Research Reagent Solutions
The following table details key reagents and materials critical for successful SDS-PAGE execution, along with their specific functions in ensuring clear, sharp protein bands.
Table 2: Essential Reagents for SDS-PAGE
| Reagent/Material | Function & Importance in Band Resolution |
|---|---|
| SDS (Sodium Dodecyl Sulfate) | Linearizes proteins by breaking hydrogen bonds and masks intrinsic charge, allowing separation by size. Use a final concentration well above 1 mM for full denaturation. [61] [6] |
| DTT (Dithiothreitol) or β-Mercaptoethanol | Reducing agents that break disulfide bonds, ensuring complete protein unfolding. Critical for analyzing oligomeric proteins. [6] |
| Acrylamide/Bis-acrylamide | Forms the porous gel matrix. The ratio and total percentage (%T) determine pore size and must be matched to the target protein's molecular weight. [58] [6] |
| TEMED & Ammonium Persulfate (APS) | Catalyzes the free-radical polymerization of acrylamide. Fresh reagents are essential for forming a uniform gel with consistent pore size. [58] [6] |
| Tris-Glycine Buffers | Creates the discontinuous pH system for stacking and resolving. Fresh buffers are necessary for maintaining correct pH and ionic strength. [58] [57] |
| Glycerol | Adds density to the sample buffer, allowing the sample to sink to the bottom of the well and preventing leakage and diffusion. [59] |
| Urea | A chaotropic agent added to sample buffer (4-8 M) to solubilize hydrophobic proteins and prevent aggregation. [59] [60] |
Effective diagnosis and resolution of smeared or distorted protein bands in SDS-PAGE require a methodical approach grounded in the core principle of the technique: the uniform negative charge conferred by SDS enables separation by molecular size. By systematically addressing sample preparation, gel composition, and electrophoresis conditions as detailed in this guide, researchers can achieve the high-resolution separation necessary for accurate protein analysis, a non-negotiable standard in rigorous scientific and drug development workflows.
Addressing Poor Band Resolution and Unusual Migration Patterns
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is a foundational technique in biochemical research, essential for separating proteins based on molecular weight. The method, initially developed by Laemmli in 1970, relies critically on the properties of SDS to denature proteins and confer a uniform negative charge, thereby allowing separation primarily by size [9] [6]. This denaturation is crucial for reliable results, as proteins naturally exhibit diverse charges and complex three-dimensional structures that would otherwise impede straightforward separation [37].
Despite its widespread use, researchers often encounter analytical challenges, including poor band resolution and unusual migration patterns (gel shifting). These issues can compromise data integrity, leading to inaccurate molecular weight estimations and misinterpretation of protein composition. Such problems are particularly prevalent in the analysis of membrane proteins and complex biological mixtures [62]. This guide addresses the core principles of SDS-PAGE, identifies common pitfalls, and provides detailed protocols to troubleshoot and optimize protein separation, ensuring reliable data for drug development and basic research.
Core Principles: How SDS Enables Separation by Molecular Weight
SDS is an anionic detergent that plays two indispensable roles in protein electrophoresis [9] [37]:
- Protein Denaturation: SDS disrupts hydrogen bonds and van der Waals forces, effectively unfolding proteins and masking their intrinsic three-dimensional structures.
- Uniform Charge Conferral: SDS binds to the protein backbone at a consistent ratio of approximately 1.4 g SDS per gram of protein, imparting a uniform negative charge density [6]. This creates a near-identical charge-to-mass ratio for most polypeptides.
When subjected to an electric field within a polyacrylamide gel matrix, these denatured, SDS-coated proteins migrate toward the anode. The gel acts as a molecular sieve, where smaller proteins navigate the pores more easily and migrate faster, while larger proteins are hindered and migrate more slowly [9]. This process results in separation primarily by molecular size. The polyacrylamide gel concentration can be optimized to resolve proteins within specific size ranges, as detailed in Table 1.
Table 1: Recommended Gel Percentages for Optimal Resolution of Different Protein Sizes
| Protein Size Range (kDa) | Recommended Gel Percentage (%) | Remarks |
|---|---|---|
| < 30 | 12-20 | Use high-percentage gels or Tricine-SDS-PAGE for small proteins/peptides [9] [45] |
| 15 - 100 | 10 | Standard range for many applications [9] |
| 25 - 200 | 8 | Optimal for larger proteins [9] |
| Broad/Complex Mixture | 4-12 or 8-16 | Gradient gels provide the best resolution across a wide mass range [9] [63] |
Troubleshooting Poor Band Resolution
Poor band separation manifests as smeared, diffuse, or poorly defined bands, complicating analysis and quantification. The causes are often rooted in sample preparation, gel polymerization, or electrophoresis conditions. A systematic workflow for diagnosing these issues is presented below.
Diagram 1: Troubleshooting Band Resolution
Key Methodologies for Improving Resolution
The following protocols provide detailed steps to address the most common causes of poor resolution.
Protocol 1: Optimized Sample Preparation for Complete Denaturation Incomplete denaturation is a primary cause of smearing, as folded proteins migrate anomalously [58].
- Sample Buffer Composition: Use Laemmli buffer containing SDS (e.g., 2% LDS) and a reducing agent (e.g., 50-100 mM DTT or 5% β-mercaptoethanol) to break disulfide bonds [6].
- Denaturation: Heat samples at 95-98°C for 5 minutes [58] [6]. Alternatively, heating at 70°C for 10 minutes can be used for heat-sensitive proteins.
- Cooling: Immediately after heating, place samples on ice to prevent gradual cooling and protein renaturation [58].
- Centrifugation: Briefly spin cooled samples in a microcentrifuge to collect condensation before loading.
Protocol 2: Vertical PAGE with Optimized Electrophoresis Parameters Suboptimal running conditions can cause band broadening and "smiling" or "frowning" bands [9].
- Buffer: Always use freshly prepared running buffer (e.g., Tris-Glycine-SDS) to ensure correct ionic strength and pH [58].
- Voltage/Temperature: Run gels at a constant voltage of 100-150 V for standard mini-gels. If bands show distortion (e.g., smiling), reduce the voltage or employ an active cooling system to prevent excessive Joule heating [58] [63].
- Run Time: Continue electrophoresis until the dye front (e.g., bromophenol blue) has migrated to the bottom of the gel, typically 40-60 minutes [9].
Protocol 3: Native SDS-PAGE (NSDS-PAGE) for Functional Analysis A modified SDS-PAGE protocol can be employed to separate proteins while retaining native enzymatic activity or bound metal cofactors, which is valuable in metalloprotein research and drug discovery [8].
- Sample Buffer (4X NSDS): 100 mM Tris HCl, 150 mM Tris base, 10% (v/v) glycerol, 0.0185% (w/v) Coomassie G-250, 0.00625% (w/v) Phenol Red, pH 8.5. Omit SDS and reducing agents. Do not heat the sample [8].
- Running Buffer: 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7. Note the significantly reduced SDS concentration compared to standard denaturing PAGE [8].
- Electrophoresis: Pre-run the gel in ddH₂O for 30 minutes. Load samples and run at 200V for approximately 45 minutes using the specified running buffer. Under these conditions, proteins remain native, and Zn²+ retention can increase from 26% to over 98% compared to standard SDS-PAGE [8].
Investigating Unusual Migration Patterns (Gel Shifting)
Anomalous migration, where proteins run at sizes inconsistent with their known molecular weight, is a frequent challenge. This "gel shifting" is often systematic and can provide insights into protein properties.
Principal Causes and Underlying Mechanisms
The primary factors causing unusual migration are summarized in the table below.
Table 2: Common Causes of Unusual Protein Migration in SDS-PAGE
| Cause of Anomaly | Impact on Migration | Mechanism | Example Proteins |
|---|---|---|---|
| High Hydrophobicity (e.g., Membrane Proteins) | Faster migration (Apparent MW < Formula MW) | Altered SDS binding: hydrophobic domains embed in micelle interiors, leading to higher SDS binding (up to 10g SDS/g protein) and a more compact protein-detergent complex [62]. | Glycophorin (-33%), KcsA tetramer (-21%) [62] |
| Post-Translational Modifications (e.g., Glycosylation) | Slower migration (Apparent MW > Formula MW) | PTMs like added carbohydrate chains alter the protein's hydrodynamic radius and can interfere with SDS binding, reducing mobility through the gel matrix [64] [9]. | Glycoproteins, Phosphoproteins |
| Robust Tertiary/Quaternary Structure | Variable (Often Faster) | Compact, stable structures (e.g., disulfide-bonded or SDS-resistant complexes) resist complete denaturation, resulting in a more compact shape that migrates faster [62] [6]. | OmpA (folded vs. denatured) [62] |
| Protein-Protein Interactions | Slower migration (High MW complexes) | Non-covalent interactions that persist despite SDS treatment can cause proteins to run as oligomers or higher-order complexes [6]. | SDS-resistant complexes |
The relationship between protein structure, SDS binding, and migration is a key area of study. Research on helix-loop-helix membrane proteins has demonstrated a strong correlation (R² = 0.8) between the amount of bound SDS and the resulting gel shift, confirming that altered detergent binding is a fundamental explanation for anomalous migration [62].
Reference Database and Diagnostic Protocols
Protocol 4: Utilizing a Reference Migration Database To distinguish between expected and truly anomalous migration, researchers can consult publicly available empirical data.
- Resource: Access the PUUMBA database (https://pumba.dcsr.unil.ch/), which contains accurate electrophoretic migration patterns for approximately 10,000 human proteins, obtained by SDS-PAGE coupled with mass spectrometry [64].
- Application: Query the database with your protein of interest to obtain its empirically determined migration weight. Compare this to the migration of your experimental sample.
- Interpretation: A match supports proper identification. A discrepancy may indicate specific PTMs, differential splicing, or issues with your sample or gel conditions [64].
Protocol 5: Diagnostic SDS-PAGE for Membrane Proteins Membrane proteins are notorious for anomalous migration. This diagnostic approach helps characterize the behavior.
- Vary Denaturation Conditions: Prepare identical samples under: a) Standard denaturing conditions (SDS + DTT, heated to 95°C), b) Non-reducing conditions (SDS, no DTT, heated), and c) Mild denaturation (SDS, no DTT, no heat) [62] [6].
- Electrophoresis: Run all samples on the same gel alongside a standard molecular weight marker.
- Analysis: Compare migration differences. Faster migration in non-reducing or non-heated conditions suggests the presence of a compact, SDS-resistant structure stabilized by disulfide bonds or strong non-covalent interactions [62].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table catalogs key reagents and materials critical for successful and reliable SDS-PAGE experiments.
Table 3: Essential Reagents and Materials for SDS-PAGE Research
| Reagent/Material | Function & Role in Experimentation |
|---|---|
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent responsible for denaturing proteins and conferring a uniform negative charge; the core agent enabling separation by mass [9] [37]. |
| DTT (Dithiothreitol) or β-Mercaptoethanol | Reducing agents that cleave disulfide bonds, ensuring complete unfolding of proteins and dissociation of subunits [6]. |
| Acrylamide/Bis-acrylamide | Monomer and cross-linker that polymerize to form the porous gel matrix, which acts as a molecular sieve [6]. |
| TEMED & Ammonium Persulfate (APS) | Catalyst (TEMED) and initiator (APS) for the free-radical polymerization of polyacrylamide gels [6]. |
| Molecular Weight Markers | Pre-stained or unstained protein standards of known molecular weight, essential for estimating the size of unknown proteins and monitoring run progress [6]. |
| Tris-Glycine Buffer System | The discontinuous buffer system (stacking gel pH ~6.8, resolving gel pH ~8.8) that stacks proteins into sharp bands before separation in the resolving gel [6]. |
| Coomassie Brilliant Blue / Silver Stain | Dyes used for post-electrophoretic visualization of protein bands in the gel, with silver stain offering higher sensitivity [9]. |
| Colored Acidic Dyes (e.g., Tartrazine) | Added to the stacking gel to visualize wells, enabling smoother and more accurate sample loading [18]. |
Advanced Techniques and Future Directions
Innovations in electrophoresis continue to enhance resolution and address longstanding limitations.
Horizontal PAGE with Double-Deck Electrodes: Traditional horizontal systems have electrodes only on top of the gel, creating an uneven electric field that broadens bands. A new design employing double-deck flat electrodes applies the electric field simultaneously from the top and bottom. This innovation produces perpendicular protein bands and enhances resolution. When combined with Field Inversion Gel Electrophoresis (FIGE)—which periodically reverses the electric field polarity—band focusing is further improved, achieving the highest level of separation clarity [63]. The workflow for this advanced setup is illustrated below.
Diagram 2: Advanced Horizontal PAGE Workflow
Two-Dimensional Electrophoresis (2-DE): For the analysis of complex protein mixtures, 2-DE separates proteins first by their isoelectric point (pI) using isoelectric focusing, and subsequently by molecular weight using SDS-PAGE in the second dimension. This technique allows for the resolution of thousands of proteins in a single gel, proving invaluable for proteomic studies and the analysis of post-translational modifications [9].
SDS-PAGE remains an indispensable tool in the molecular biologist's arsenal, with SDS itself being the cornerstone that enables predictable separation by molecular weight. Effectively troubleshooting poor band resolution and anomalous migration requires a systematic approach, addressing factors from sample preparation to gel electrophoresis parameters. Furthermore, understanding that migration anomalies can provide valuable biological insights—such as the presence of hydrophobic domains, stable protein structures, or specific post-translational modifications—transforms a potential pitfall into an analytical opportunity. By applying the detailed protocols and diagnostic strategies outlined in this guide, researchers and drug development professionals can significantly enhance the reliability and interpretability of their protein analysis data.
Optimizing Gel Percentage and Voltage for Specific Protein Sizes
The Critical Role of SDS in Electrophoretic Research
In polyacrylamide gel electrophoresis (PAGE) research, sodium dodecyl sulfate (SDS) plays a fundamental role by standardizing protein charge and structure, thereby enabling separation primarily based on molecular weight. This anionic detergent binds to proteins in a constant ratio, approximately 1.4 grams of SDS per gram of protein, effectively masking the proteins' intrinsic charges and conferring a uniform negative charge density [6]. Simultaneously, SDS acts as a strong denaturing agent, unfolding proteins into linear polypeptide chains by disrupting hydrogen bonds and hydrophobic interactions [6] [8]. This dual action of charge standardization and structural denaturation eliminates the influence of protein shape and native charge, ensuring that migration through the polyacrylamide gel matrix depends almost exclusively on polypeptide chain length [5] [6]. Consequently, SDS-PAGE provides researchers with a robust, inexpensive, and relatively accurate method for analyzing protein samples based on molecular mass, making it an indispensable technique in biochemical and biomedical research [5].
Optimizing Gel Percentage for Target Protein Sizes
The polyacrylamide gel acts as a molecular sieve, with its pore size determined by the acrylamide concentration. Selecting the appropriate gel percentage is paramount for achieving optimal resolution of target proteins.
Gel Percentage Selection Guide
The table below summarizes recommended gel percentages based on the molecular weight of your target proteins [65] [66]:
| Acrylamide % | Optimal Molecular Weight Range | Example Proteins |
|---|---|---|
| 6% | > 200 kDa | Spectrin, Titin, large IgG complexes [65] |
| 8% | 50 - 200 kDa [66] / 100 - 200 kDa [65] | Fibrinogen, β-galactosidase [65] |
| 10% | 15 - 100 kDa [66] / 60 - 150 kDa [65] | BSA, GAPDH, actin, HSP70 [65] |
| 12% | 10 - 70 kDa [66] / 20 - 100 kDa [65] | Histones, caspases, transcription factors [65] |
| 15% | 12 - 45 kDa [66] / < 30 kDa [65] | Small peptides, cytokines, ubiquitin [65] |
Advanced Gel Formulations
For complex samples, consider these advanced approaches:
- Gradient Gels: Gels with a gradient of acrylamide (e.g., 4-20%) offer a broad separation range and are excellent for analyzing proteins of unknown molecular weight or samples containing proteins with vastly different sizes [6] [65]. The increasing pore size creates a pore barrier effect, sharpening bands as they migrate.
- High-Resolution Applications: When separating closely sized proteins (e.g., 40 kDa and 45 kDa), a higher percentage gel (12-15%) will provide superior band separation compared to a standard 10% gel [65].
Optimizing Electrophoresis Voltage and Running Conditions
The application of an electric field drives protein migration. Proper management of electrical parameters is crucial to prevent artifacts and ensure clear separation.
Electrical Parameter Settings
The table below outlines the key considerations for different power supply settings [67]:
| Setting Type | Mechanism | Advantages | Disadvantages |
|---|---|---|---|
| Constant Voltage | Voltage (V) is fixed; current decreases as resistance increases during the run. | Limits heat production; simple to use [67]. | Protein migration slows down later in the run [67]. |
| Constant Current | Current (I) is fixed; voltage increases to maintain current as resistance rises. | Consistent run timing across multiple gels [67]. | Voltage (and heat) increases, potentially causing "smiling bands" or warped gels [67]. |
| Constant Power | Power (P = I x V) is fixed; both V and I can fluctuate. | May limit heat while maintaining a more consistent speed [67]. | "Constant" conditions are hard to define due to two fluctuating variables [67]. |
Standard Running Protocol
A common and effective strategy for mini-gels is a two-stage voltage approach [67] [68]:
- Stacking Phase: Begin electrophoresis at a low voltage of 50-80 V for approximately 20-30 minutes. This allows the proteins to migrate slowly and concentrate into a sharp band as they move through the stacking gel and enter the separating gel.
- Separation Phase: Once the samples have entered the separating gel, increase the voltage to 100-120 V. A general rule of thumb is 5-15 V per centimeter of gel length [67]. Continue running until the bromophenol blue dye front reaches the bottom of the gel. Total run time is typically between 45 minutes to 2 hours, depending on gel concentration and voltage [66] [67]. For higher percentage gels (e.g., 15%), the run time may need to be slightly extended [68].
Managing Heat Production
Heat is a critical factor in SDS-PAGE. While it can aid in protein denaturation, excessive heat causes gel expansion, leading to distorted "smiling" bands and making gels difficult to handle [67]. To mitigate heat buildup, especially when using constant current, consider running the gel in a cold room or submerging the electrophoresis apparatus in an ice bath [67].
Detailed Experimental Protocol for SDS-PAGE
- Assemble Gel Cassette: Thoroughly clean glass plates and spacers with ethanol, then assemble the casting mold sealed at the bottom and sides.
- Prepare and Pour Separating Gel: Mix components for the desired acrylamide percentage (see formulation table below), adding ammonium persulfate (APS) and TEMED last to initiate polymerization. Pour the solution immediately into the gel cassette.
- Overlay with Solvent: Carefully layer water-saturated butanol or isopropanol over the gel to exclude oxygen and create a flat meniscus. Allow polymerization for 20-30 minutes.
- Prepare and Pour Stacking Gel: After polymerization, pour off the overlay, rinse the gel surface, and remove excess liquid. Pour the stacking gel mixture (see table) on top of the polymerized separating gel.
- Insert Comb: Insert a clean sample comb without introducing bubbles. Allow the stacking gel to polymerize for 20-30 minutes.
- Denature Proteins: Mix protein sample with SDS-PAGE sample buffer (containing SDS and a reducing agent like β-mercaptoethanol or DTT).
- Heat Samples: Heat the samples at 95-100 °C for 5 minutes (or 70 °C for 10 minutes) to fully denature the proteins [6].
- Brief Centrifugation: Centrifuge heated samples briefly (e.g., 15,000 rpm for 1 minute) to collect condensation.
- Load Samples: Load 10-50 µg of protein from cell lysate or 10-100 ng of purified protein into the wells [66]. Always include a molecular weight marker in one lane. Ensure consistent total volume across wells; if necessary, fill empty wells with 1x loading buffer to prevent sample diffusion [68].
Gel Formulations for Hand-Casting
The table below provides a standard recipe for casting SDS-PAGE gels [69]:
| Component | 10% Resolving Gel (for 15 mL) | Stacking Gel (for 5 mL) |
|---|---|---|
| Water | 5.9 mL | 3.4 mL |
| 30% Acrylamide Mix | 5.0 mL | 830 µL |
| 1.5 M Tris-HCl (pH 8.8) | 3.8 mL | - |
| 0.5 M Tris-HCl (pH 6.8) | - | 630 µL |
| 10% SDS | 150 µL | 50 µL |
| 10% Ammonium Persulfate (APS) | 150 µL | 50 µL |
| TEMED | 12 µL | 8 µL |
Essential Reagents and Materials
The following table details key reagents and their critical functions in the SDS-PAGE workflow:
| Reagent/Material | Function |
|---|---|
| Sodium Dodecyl Sulfate (SDS) | Denatures proteins and confers a uniform negative charge, enabling separation by size [5] [6]. |
| Acrylamide/Bis-acrylamide | Forms the cross-linked porous gel matrix that acts as a molecular sieve [5] [69]. |
| Tris-HCl Buffers | Maintains the appropriate pH for the stacking (pH ~6.8) and separating (pH ~8.8) gels, crucial for the discontinuous system [6] [69]. |
| TEMED & Ammonium Persulfate (APS) | Catalyzes the polymerization of acrylamide to form the polyacrylamide gel [6] [69]. |
| Reducing Agents (DTT, β-ME) | Cleaves disulfide bonds to ensure complete protein unfolding [6]. |
| Glycine | A trailing ion in the Tris-Glycine buffer system that is critical for the stacking effect at the gel interface [6]. |
| Bromophenol Blue | Tracking dye that allows visualization of protein migration during electrophoresis [6]. |
Experimental Workflow and SDS Mechanism
The following diagram illustrates the complete SDS-PAGE workflow and the fundamental role of SDS.
Advanced Technique: Native SDS-PAGE
A modification of the standard protocol, known as Native SDS-PAGE (NSDS-PAGE), can be employed when retaining protein function or non-covalently bound cofactors (e.g., metal ions) is desirable. This method involves omitting the heating step and reducing or eliminating SDS and EDTA from the sample and running buffers [8]. Research has shown that this approach can preserve the enzymatic activity of many proteins and significantly increase the retention of bound metal ions (e.g., Zn²⺠retention increased from 26% to 98% in one study) while still providing high-resolution separation, though it may not be suitable for all applications [8].
Ensuring Complete Gel Polymerization and Fresh Buffer Preparation
Within the framework of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) research, the reliability of experimental data is profoundly dependent on two foundational technical elements: the complete polymerization of polyacrylamide gels and the consistent preparation of fresh electrophoresis buffers. SDS-PAGE separates proteins primarily based on their molecular weight by using the anionic detergent SDS to denature proteins and confer a uniform negative charge, and a polyacrylamide gel matrix to act as a molecular sieve [5] [6] [35]. This technique is a cornerstone in biochemistry and molecular biology for analyzing protein mixtures, determining protein size, and assessing sample purity [9] [35]. Any imperfection in the gel matrix or degradation of the running buffer directly compromises the sieving effect and charge uniformity provided by SDS, leading to poor resolution, distorted band patterns, and erroneous molecular weight estimations. This guide provides a detailed examination of standardized protocols and troubleshooting methodologies to ensure optimal gel polymerization and buffer integrity, thereby safeguarding the accuracy and reproducibility of protein analysis.
The development of SDS-PAGE, notably refined by Ulrich Laemmli in 1970, revolutionized protein biochemistry by enabling high-resolution separation of polypeptides based on size [6] [9]. The principle hinges on the ability of SDS to bind to proteins at a nearly constant ratio (approximately 1.4 g SDS per gram of protein), masking their intrinsic charge and unfolding them into linear chains [6] [35]. This creates a uniform charge-to-mass ratio, ensuring that during electrophoresis, proteins migrate through the polyacrylamide gel based solely on polypeptide chain length [5] [70]. The polyacrylamide gel itself, formed through the polymerization of acrylamide and a cross-linker like bis-acrylamide, creates a porous mesh that acts as a molecular sieve [71] [35].
The integrity of this molecular sieve is paramount. An incompletely polymerized gel has an inconsistent pore structure, which distorts protein migration and hampers accurate size determination [9]. Similarly, the running buffer, typically a Tris-glycine-SDS system, serves multiple critical functions: it conducts current, maintains a stable pH for the separation process, and provides SDS to keep proteins denatured [6] [72]. Old or improperly prepared buffer can lead to insufficient buffering capacity, altered ionic strength, and inadequate SDS levels, resulting in poor stacking, smiling/frowning bands, and incomplete protein separation [9] [72]. Therefore, rigorous attention to gel polymerization and buffer preparation is not merely a procedural step but a fundamental requirement for achieving the high-resolution separation that defines reliable SDS-PAGE analysis.
The Science of Gel Polymerization
Chemistry of Polyacrylamide Gel Formation
Polyacrylamide gel formation is a process of free radical polymerization. The monomer, acrylamide, and the cross-linker, most commonly N,N'-methylenebisacrylamide (bis-acrylamide), copolymerize to form a three-dimensional network. The polymerization reaction is initiated by the generation of free radicals from ammonium persulfate (APS), which is catalyzed by the base N,N,N',N'-Tetramethylethylenediamine (TEMED) [73] [71]. TEMED accelerates the decomposition of APS into sulfate free radicals, which then attack the vinyl groups of acrylamide monomers, initiating a chain reaction that propagates until terminated [71].
The concentration of acrylamide and bis-acrylamide determines the gel's properties. The total acrylamide concentration (%T) defines the average pore size, with higher percentages creating smaller pores for better separation of lower molecular weight proteins [5] [35]. The cross-linking ratio (%C) influences the rigidity and porosity of the gel. A standard ratio is 29:1 or 37.5:1 (acrylamide to bis-acrylamide) [73]. The polymerization process is highly sensitive to environmental factors, particularly oxygen, which acts as a free radical scavenger and can inhibit polymerization, leading to soft, uneven gels [5].
Protocol for Reliable Gel Polymerization
Materials:
- Acrylamide/Bis-acrylamide solution (e.g., 30% stock, 29:1 ratio)
- Separating Gel Buffer (e.g., 1.5 M Tris-HCl, pH 8.8)
- Stacking Gel Buffer (e.g., 0.5 M Tris-HCl, pH 6.8)
- Ammonium Persulfate (APS): 10% (w/v) solution in water, prepared fresh
- TEMED
- Water-saturated isopropanol or n-butanol
- Gel casting system (glass plates, spacers, combs)
Methodology for Casting a Discontinuous SDS-PAGE Gel:
- Assemble the Gel Cassette: Thoroughly clean and dry the glass plates and spacers. Assemble the cassette and ensure it is leak-proof [5] [73].
- Prepare the Separating Gel Solution: In a clean beaker or flask, mix the components for the separating gel in the order listed in the table below. Avoid introducing bubbles while mixing.
Table 1: Example Formulations for Separating Gels (for ~20 mL volume)
| Component | 7.5% Gel | 10% Gel | 12% Gel |
|---|---|---|---|
| diHâ‚‚O | 9.70 mL | 8.02 mL | 6.70 mL |
| 30% Acrylamide/Bis | 5.00 mL | 6.66 mL | 8.00 mL |
| 1.5 M Tris-HCl (pH 8.8) | 5.00 mL | 5.00 mL | 5.00 mL |
| 10% SDS | 200 µL | 200 µL | 200 µL |
| 10% APS | 100 µL | 100 µL | 100 µL |
| TEMED | 20 µL | 20 µL | 20 µL |
Adapted from [73]
- Catalyze and Pour: Add APS and TEMED last, and mix gently by swirling. Immediately pour the solution into the gel cassette, leaving space for the stacking gel.
- Overlay and Polymerize: Carefully overlay the gel solution with a thin layer of water-saturated isopropanol or n-butanol to exclude oxygen and create a flat interface. Allow the gel to polymerize completely for 20-30 minutes at room temperature [5]. A distinct schlieren line will appear at the gel-alcohol interface upon polymerization.
- Prepare the Stacking Gel: After polymerization, pour off the overlay liquid and rinse the gel surface with diHâ‚‚O. Dry the area with filter paper. Prepare the stacking gel solution as shown below.
Table 2: Example Formulation for a Stacking Gel (for ~10 mL volume)
| Component | 4% Stacking Gel |
|---|---|
| diHâ‚‚O | 6.00 mL |
| 30% Acrylamide/Bis | 1.32 mL |
| 0.5 M Tris-HCl (pH 6.8) | 2.52 mL |
| 10% SDS | 100 µL |
| 10% APS | 50 µL |
| TEMED | 20 µL |
Adapted from [73]
- Complete the Gel Assembly: Add APS and TEMED to the stacking gel solution, pour it onto the polymerized separating gel, and immediately insert a clean comb. Allow the stacking gel to polymerize for 20-30 minutes. The gel can be used immediately or stored wrapped in moist paper towels and plastic film at 4°C for a short period [73].
The following workflow diagram summarizes the key stages of the gel polymerization process:
Troubleshooting Gel Polymerization
Common issues and their solutions are outlined in the table below.
Table 3: Troubleshooting Gel Polymerization Problems
| Problem | Possible Cause | Solution |
|---|---|---|
| Slow or No Polymerization | Old or degraded APS/TEMED; Oxygen inhibition; Incorrect pH | Use fresh APS (10% solution); Ensure no leaks in cassette; Check buffer pH [73]. |
| Gel is Too Soft or Sticky | Incomplete polymerization; Incorrect acrylamide concentration | Ensure proper APS/TEMED levels and mixing; Verify recipe calculations. |
| Non-parallel Gel Front | Uneven polymerization; Improper casting | Ensure cassette is level during polymerization; Mix catalysts thoroughly [9]. |
| Bubbles in Gel | Improper pouring | Pour solutions slowly down the corner of the cassette to avoid bubbles. |
The Criticality of Fresh Buffer Preparation
Composition and Function of SDS-PAGE Buffers
The discontinuous buffer system in SDS-PAGE consists of three key components, each with a specific role in achieving high-resolution separation [6] [72].
- Sample Buffer (Laemmli Buffer): This buffer contains SDS to denature and charge proteins, a reducing agent (e.g., β-mercaptoethanol or DTT) to break disulfide bonds, glycerol to add density for loading, and a tracking dye (bromophenol blue) to monitor migration [9] [74] [72].
- Running Buffer (Tris-Glycine-SDS): This buffer provides the ions necessary for conducting current and maintaining the discontinuous system. Tris acts as the buffering agent. Glycine is the key "trailing ion" whose charge state changes with pH, enabling the stacking effect. SDS in the running buffer helps maintain protein denaturation during electrophoresis [6] [72].
- Gel Buffers (Tris-HCl): The stacking gel (pH ~6.8) and separating gel (pH ~8.8) buffers create the pH discontinuity essential for the stacking process, where proteins are concentrated into a sharp zone before entering the separating gel [72] [35].
Protocol for Preparing and Using Fresh Buffer
Materials:
- Tris base
- Glycine
- SDS
- Deionized water
Methodology for Running Buffer Preparation:
- 10X Running Buffer Stock (1 L): Dissolve 30.3 g of Tris base, 144.1 g of glycine, and 10.0 g of SDS in approximately 800 mL of deionized water. Once completely dissolved, bring the final volume to 1 L with deionized water. The pH of this 10X stock should be approximately 8.3 and does not require adjustment [73].
- 1X Working Solution: Dilute the 10X stock to a 1X concentration with deionized water before use (e.g., 100 mL of 10X stock + 900 mL deionized water) [74].
- Storage and Stability: The 10X stock solution can be stored at room temperature for several weeks. However, the 1X working solution should ideally be prepared fresh for each electrophoresis run. If reused, it should not be used more than 2-3 times, as buffer depletion (the "buffer depletion effect") can occur, leading to increased resistance, slower run times, and distorted bands [9].
Consequences of Using Compromised Buffers
Using old, improperly prepared, or excessively reused running buffer leads to several artifacts:
- Smiling or Frowning Bands: Caused by uneven heating and current distribution across the gel, often linked to buffer ion depletion or incorrect ionic strength [9].
- Poor Stacking and Band Resolution: Results from incorrect pH or degraded SDS, preventing the proper formation of the stacking zone between chloride and glycine ions [72].
- Slow Migration or Protein Precipitation: Can occur if SDS precipitates or is omitted from the buffer, causing proteins to lose their uniform charge and potentially aggregate [9].
The Scientist's Toolkit: Essential Reagents for SDS-PAGE
The following table details key reagents and their critical functions in SDS-PAGE protocols.
Table 4: Essential Research Reagents for SDS-PAGE
| Reagent | Function / Role in the Experiment |
|---|---|
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent that denatures proteins and confers a uniform negative charge, enabling separation by size rather than native charge or shape [6] [70]. |
| Acrylamide / Bis-acrylamide | Monomer and cross-linker that polymerize to form the porous gel matrix, which acts as a molecular sieve [71] [35]. |
| Ammonium Persulfate (APS) | Initiator that provides free radicals to start the polymerization reaction of acrylamide and bis-acrylamide [73] [71]. |
| TEMED | Catalyst that accelerates the decomposition of APS into free radicals, thus controlling the rate of gel polymerization [73] [71]. |
| Tris Buffers | Maintain the required pH in the stacking gel (pH ~6.8) and separating gel (pH ~8.8) to enable the discontinuous buffer system for sharp band stacking [72] [35]. |
| Glycine | "Trailing ion" in the running buffer; its charge state changes with pH, which is critical for the stacking effect in the discontinuous buffer system [6] [72]. |
| β-Mercaptoethanol / DTT | Reducing agents that break disulfide bonds in proteins, ensuring complete denaturation into polypeptide subunits [9] [74]. |
| Coomassie Brilliant Blue | A common protein stain used to visualize separated protein bands on the gel post-electrophoresis [9] [73]. |
Concluding Remarks
The fidelity of data generated through SDS-PAGE is inextricably linked to the foundational quality of its core components: a perfectly polymerized polyacrylamide gel and a freshly prepared running buffer. As detailed in this guide, the meticulous preparation of these elements is not a mere prelude to the experiment but is as critical as the electrophoretic run itself. Adherence to standardized protocols for gel casting, with attention to reagent freshness and polymerization conditions, ensures a consistent molecular sieve. Similarly, the preparation of fresh buffer guarantees the proper ionic environment and charge distribution necessary for sharp band resolution. For researchers in drug development and basic science, where conclusions are drawn from band patterns, intensities, and molecular weight estimates, mastering these fundamentals is paramount. By rigorously applying these protocols, scientists can minimize experimental variability, enhance reproducibility, and fortify the reliability of their protein analytical data.
Solving Sample Leakage and Aggregation in Wells
In polyacrylamide gel electrophoresis (PAGE) research, Sodium Dodecyl Sulfate (SDS) serves a critical function by ensuring that protein separation occurs solely on the basis of molecular weight, independent of the protein's inherent charge or three-dimensional structure [75]. This anionic detergent binds to proteins in a uniform ratio, approximately 1.4 grams of SDS per gram of protein, conferring a ubiquitous negative charge and linearizing the polypeptides by disrupting hydrophobic interactions and hydrogen bonds [45] [75]. The success of this process, however, is contingent upon proper sample preparation. When this preparatory stage is compromised, two frequent technical challenges arise: sample leakage from wells and sample aggregation within wells. These issues not only impede the electrophoretic separation but also fundamentally undermine the core principle of SDS-PAGE—that migration distance is inversely proportional to the log of molecular weight. This guide details the mechanistic causes and procedural solutions for these problems, providing researchers and drug development professionals with robust protocols to ensure data accuracy and reliability.
Core Principles and the Mechanism of SDS
A deep understanding of how SDS prepares proteins for electrophoresis is essential for diagnosing and resolving issues that occur at the sample loading stage.
The Chemistry and Function of SDS
SDS possesses a unique amphipathic structure, featuring a polar sulfate head group (ionic part) and a non-polar hydrocarbon tail (hydrophobic region) [75]. Its action is twofold:
- Uniform Charge Masking: The ionic part of SDS interacts with the aqueous environment and confers a strong negative charge to the protein, overwhelming the molecule's intrinsic charge. This creates a uniform charge-to-mass ratio, directing all proteins to migrate towards the anode during electrophoresis [75].
- Protein Denaturation: The hydrophobic tails of SDS molecules embed themselves into the hydrophobic regions of the protein, effectively disrupting the protein's secondary and tertiary structure. This interaction, coupled with the electrostatic repulsion from the ionic groups, forces the protein into a rod-like shape [75].
Table 1: Key Reagents for Effective SDS-PAGE Sample Preparation
| Reagent | Primary Function | Mechanism of Action |
|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | Denaturant & Charge Provider | Binds protein backbone, masks intrinsic charge, linearizes structure [75]. |
| DTT (Dithiothreitol) / BME (Beta-Mercaptoethanol) | Reducing Agent | Breaks disulfide bridges between cysteine residues, aiding full denaturation [76] [75]. |
| Glycerol | Density Agent | Increases sample density, ensuring it sinks to the bottom of the well during loading [76]. |
| Urea | Solubilizing Agent | Disrupts hydrogen bonds and hydrophobic interactions, preventing aggregation of hydrophobic proteins [76]. |
The following diagram illustrates the core workflow of SDS-PAGE, integrating the role of SDS and highlighting the stages where leakage and aggregation typically occur, linking them to the underlying causes discussed in this article.
Solving Sample Leakage
Sample leakage manifests as distorted, smeared bands that appear to spread outwards from the well. This occurs when the protein sample fails to remain confined within the well during or immediately after loading [76].
Root Causes and Corrective Methodologies
Table 2: Troubleshooting Guide for Sample Leakage
| Root Cause | Underlying Principle | Corrective Protocol |
|---|---|---|
| Insufficient Glycerol | The density of the sample is lower than the running buffer in the tank. | Ensure the loading buffer contains 5-10% glycerol. The sample should be sufficiently dense to sink and remain in the well [76]. |
| Air Bubbes in Wells | Air displaces sample and creates an uneven surface for loading. | Prior to loading, use a pipette tip to gently flush each well with a small amount of running buffer to displace air bubbles [76]. |
| Overfilled Wells | The physical capacity of the well is exceeded. | Do not load the well beyond 3/4 of its total capacity. Use a calibrated pipette and load samples carefully [76]. |
| Damaged Well Integrity | The gel's physical structure is compromised, allowing sample to escape. | When casting the gel, avoid pushing the comb all the way to the bottom of the cassette. Remove the comb carefully and steadily after polymerization to prevent tearing [77]. |
Solving Sample Aggregation
Sample aggregation is observed as protein clumping within the wells, leading to poor or no migration into the resolving gel. This is often a consequence of incomplete denaturation or protein precipitation [76].
Root Causes and Corrective Methodologies
Aggregation occurs when proteins are not fully linearized and solubilized, causing them to form large complexes that cannot enter the gel matrix.
Table 3: Troubleshooting Guide for Sample Aggregation
| Root Cause | Underlying Principle | Corrective Protocol |
|---|---|---|
| Incomplete Denaturation | Secondary/tertiary structures and disulfide bonds are not fully broken. | 1. Heat Denaturation: Heat samples at 95°C for 5-10 minutes to disrupt hydrogen bonds [75]. 2. Reducing Agents: Include 1-5% DTT or β-mercaptoethanol in the loading buffer to break disulfide bridges [76] [75]. |
| Protein Overload | The well capacity for soluble protein is exceeded. | Do not exceed 10-20 µg of total protein per well for a standard mini-gel. Determine protein concentration accurately before loading [76]. |
| High Salt Concentration | Salt ions interfere with SDS binding and protein solubility. | Dilute the sample in nuclease-free water or dialyze it into a low-salt buffer before adding the SDS-PAGE loading buffer [77]. |
| Hydrophobic Proteins | Hydrophobic interactions cause proteins to precipitate. | Add a chaotrope like 4-8 M urea to the lysis and loading buffers to disrupt hydrophobic interactions and maintain solubility [76]. |
| Presence of Contaminants | Non-protein cellular components (e.g., lipids, nucleic acids) can trap proteins. | Clarify the protein lysate by high-speed centrifugation (e.g., 12,000-16,000 x g for 15 min) to remove insoluble debris before loading the supernatant [76]. |
Experimental Workflow for Preventing Aggregation
The following diagram outlines a detailed, step-by-step protocol designed to prevent sample aggregation, from initial preparation to gel loading.
Advanced Considerations for Drug Development
In biopharmaceutical development, protein aggregation is not merely a technical obstacle but a critical quality attribute with direct implications for drug efficacy and safety [78]. Aggregates can alter bioactivity, increase immunogenicity, and compromise product stability. While SDS-PAGE is a fundamental tool for identifying aggregates that survive denaturation, it has limitations. It may not detect non-covalent aggregates that dissociate under denaturing conditions [78]. Therefore, for comprehensive aggregation profiling in a drug development context, SDS-PAGE should be complemented by orthogonal, in-solution biophysical techniques. These advanced methods, such as those employed by FIDA (Flow-Induced Dispersion Analysis), can quantitatively measure the number and size of aggregates in their native formulation without requiring purification, providing a more complete picture of product quality during screening and stability studies [78].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table catalogs the key reagents and materials required for effective SDS-PAGE analysis, with an emphasis on their role in preventing leakage and aggregation.
Table 4: Essential Reagents for Robust SDS-PAGE
| Reagent/Material | Function in Workflow | Specific Role in Preventing Leakage/Aggregation |
|---|---|---|
| SDS Loading Buffer | Sample preparation matrix. | Provides SDS for denaturation/charging, glycerol for density, and a tracking dye [76] [75]. |
| DTT or Dithiothreitol | Reducing agent in loading buffer. | Breaks inter- and intra-molecular disulfide bonds more stably and with less odor than BME, preventing aggregation [76] [75]. |
| Ultrapure Urea | Chaotrope in lysis/buffer. | Solubilizes hydrophobic and aggregated proteins at 4-8 M concentrations [76]. |
| Pre-cast Gels | Standardized separation matrix. | Ensure consistent well integrity and polyacrylamide composition, reducing variability and well damage [79]. |
| TEMED & APS (Ammonium Persulfate) | Catalysts for gel polymerization. | Initiate the free-radical reaction that forms the polyacrylamide mesh; fresh preparation is critical for proper gel structure [75]. |
| Protein Ladder/Marker | Molecular weight standard. | Contains pre-stained, denatured proteins of known weight, serving as a control for the electrophoresis process itself [79]. |
Sample leakage and aggregation in SDS-PAGE wells are not isolated technical failures but are instead symptoms of a deviation from the core principles of the technique, primarily centered on the effective role of SDS. By understanding that SDS must uniformly charge and linearize every protein molecule, researchers can systematically diagnose and resolve these issues through meticulous sample preparation. The protocols and guidelines provided herein—from optimizing reagent concentrations to adhering to precise thermal denaturation cycles—empower scientists to achieve the high-resolution, reproducible separations that are the hallmark of reliable protein analysis. For the drug development professional, this rigor is indispensable, forming the foundation upon which accurate characterization, quality control, and ultimately, patient safety are built.
SDS-PAGE in Context: Validation, Limitations, and Comparison with Native Techniques
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is a foundational technique in biochemistry, enabling protein separation based primarily on molecular weight. This method relies on SDS to denature proteins and confer a uniform negative charge, effectively masking intrinsic charge differences and structural variations. However, certain post-translational modifications, notably glycosylation and phosphorylation, can significantly alter protein electrophoretic mobility, leading to anomalous migration and potential misinterpretation of results. This technical guide examines the mechanisms by which these modifications disrupt standard SDS-PAGE separation principles, provides experimental evidence of their effects, and offers methodological approaches to identify and address these artifacts. Understanding these limitations is crucial for researchers employing SDS-PAGE in protein characterization, disease biomarker discovery, and drug development.
SDS-PAGE revolutionized protein analysis by providing a simple, reproducible method for separating complex protein mixtures. The technique's effectiveness hinges on the properties of sodium dodecyl sulfate (SDS), an anionic detergent that binds to protein backbones at a relatively constant ratio of approximately 1.4 grams of SDS per 1 gram of protein [6]. This binding achieves two critical functions:
- Protein Denaturation: SDS disrupts nearly all non-covalent interactions—including hydrogen bonds, hydrophobic interactions, and ionic bonds—unfolding proteins into linear polypeptide chains [3].
- Charge Masking: The uniform SDS coating imparts a consistent negative charge density along the protein backbone, rendering the intrinsic charge of amino acids negligible in comparison [6].
When subjected to an electric field within a polyacrylamide gel matrix, which acts as a molecular sieve, SDS-bound proteins migrate toward the anode at rates inversely proportional to their molecular weights [5]. This relationship enables molecular weight estimation with typical accuracy of ±10% when compared to standardized protein ladders [6].
The discontinuous buffer system further enhances resolution. Proteins are first concentrated in a stacking gel (pH ~6.8) with large pores before entering the separating gel (pH ~8.8) with smaller pores, where actual size-based separation occurs [3]. This elegant system creates the foundational assumption that migration distance correlates directly with molecular weight—an assumption compromised by specific post-translational modifications.
Glycosylation: Mechanisms and SDS-PAGE Anomalies
Glycosylation Mechanisms and Structural Impact
Glycosylation, one of the most abundant and complex post-translational modifications, involves the enzymatic attachment of carbohydrate chains (glycans) to specific amino acid residues [80]. The major glycosylation types include:
- N-linked Glycosylation: Attachment of oligosaccharides to asparagine residues within the consensus sequence Asn-X-Ser/Thr, where X is any amino acid except proline. This process occurs in the endoplasmic reticulum and Golgi apparatus, producing complex, hybrid, or high-mannose glycan structures [80].
- O-linked Glycosylation: Primarily involves the attachment of N-acetylgalactosamine (GalNAc) to serine or threonine residues (mucin-type), or β-N-acetylglucosamine (O-GlcNAc) to serine/threonine residues in nuclear and cytoplasmic proteins [80].
- Other Types: Including C-linked glycosylation (tryptophan linkage) and glycosylphosphatidylinositol (GPI) anchor attachments [80].
Recent research has highlighted the prevalence of O-glycosylation in intrinsically disordered regions (IDRs) of proteins. A 2025 study demonstrated that extracytoplasmic serine/threonine-rich IDRs in streptococcal membrane-associated proteins are extensively glycosylated, with Streptococcus mutans utilizing GalNAc modifications via a Pgf-dependent mechanism [81].
Impact on SDS-PAGE Mobility
Glycosylation significantly alters protein migration in SDS-PAGE through several interconnected mechanisms:
- Altered SDS Binding: The bulky, hydrophilic glycan structures can sterically hinder uniform SDS binding to the protein backbone, resulting in less negative charge per unit mass than predicted [80].
- Increased Hydrodynamic Volume: Extensive glycosylation increases the apparent size of proteins without contributing proportionally to SDS binding, creating a larger molecular entity that migrates more slowly than non-glycosylated proteins of equivalent mass [80].
- Molecular Sieving Effects: The branched, hydrated structure of glycans interacts differently with the polyacrylamide matrix than polypeptide chains, further retarding migration.
These effects can cause glycoproteins to appear 10-50% larger than their actual molecular weights on SDS-PAGE, with the discrepancy proportional to the glycan's size and complexity [80].
Table 1: Documented SDS-PAGE Migration Anomalies of Glycosylated Proteins
| Protein Example | Actual MW (kDa) | Apparent MW (kDa) | Modification Type | Biological Context |
|---|---|---|---|---|
| Sec24C [82] | ~110 | Variable shifts | O-GlcNAcylation | COPII vesicle protein; cell cycle regulation |
| Streptococcal PrsA [81] | ~45 | ~55-60 | O-GalNAc (Pgf-dependent) | Post-translocation chaperone |
| Cnm adhesin [81] | ~120 | ~150+ | O-N-acetylhexosamines | Streptococcus mutans virulence factor |
| PD-L1 [80] | ~33 | ~45-55 | N-glycosylation | Immune checkpoint protein in triple-negative breast cancer |
Phosphorylation: Electrophoretic Consequences
Phosphorylation Mechanisms
Protein phosphorylation involves the reversible addition of phosphate groups to specific amino acid residues—primarily serine, threonine, and tyrosine—catalyzed by protein kinases [82]. This modification is a fundamental regulatory mechanism controlling nearly all cellular processes, including signal transduction, cell cycle progression, and metabolic pathways. Phosphatases mediate phosphate removal, creating dynamic regulation.
Impact on SDS-PAGE Mobility
While phosphorylation adds only ~80 Da per modification, its effects on SDS-PAGE migration can be substantial due to:
- Altered Protein Conformation: Phosphorylation can induce conformational changes that affect SDS binding efficiency and protein compactness [82].
- Charge Introduction: Each phosphate group adds two negative charges at neutral pH, increasing the protein's net negative charge beyond what SDS alone provides [82].
- Gel Interaction Variability: The additional negative charges can alter interactions with the gel matrix, particularly in the stacking gel where charge-based concentration occurs.
The 2025 study on Sec24C demonstrated that phosphorylation, in concert with O-GlcNAcylation, creates dynamic migration patterns throughout the cell cycle, with phosphorylated forms often migrating faster than their non-phosphorylated counterparts due to increased charge density [82].
Table 2: Phosphorylation-Induced Mobility Shifts in SDS-PAGE
| Protein | Migration Change | Biological Context | Functional Impact |
|---|---|---|---|
| Sec24C [82] | Cell cycle-dependent mobility shifts | Mitotic regulation of ER export sites | Coordinates secretion pausing during division |
| Casein | Detectable shift with multiple phosphorylation | Model phosphoprotein | Standard for assessing phosphorylation effects |
| Tau protein | Multiple band patterns | Neurodegenerative disease | Hyperphosphorylation associated with pathology |
Experimental Evidence and Case Studies
Sec24C: Dynamic PTM Regulation During Cell Cycle
A compelling example of combined glycosylation and phosphorylation effects comes from the study of Sec24C, a core component of the COPII vesicle coat complex. Research published in 2025 revealed that Sec24C undergoes O-GlcNAcylation during interphase, which is rapidly removed upon mitotic entry coinciding with increased phosphorylation [82].
Experimental Methodology:
- Cell Synchronization: Double thymidine block (DTB) to arrest cells at G1/S boundary, followed by release into cell cycle progression.
- Immunoprecipitation: Anti-Sec24C antibodies used to isolate the protein from synchronized cell populations.
- LC-MS/MS Analysis: Liquid chromatography tandem mass spectrometry to identify and quantify phosphorylation sites and O-GlcNAcylation.
- Electron-Transfer Dissection (ETD): Specialized fragmentation technique to preserve labile post-translational modifications during mass spectrometry.
- Electrophoretic Analysis: Parallel SDS-PAGE runs of interphase and mitotic Sec24C, showing clear mobility differences corresponding to PTM status [82].
This study demonstrated that the coordinated action of phosphorylation and O-GlcNAcylation regulates Sec24C localization during mitosis, with clear electrophoretic mobility shifts reflecting the changing modification status [82].
Streptococcal Glycosylation Systems
The 2025 Nature Communications article provided systematic analysis of glycosylation in streptococcal membrane proteins, revealing that glycosylation of intrinsically disordered regions protects these proteins from proteolytic degradation and is critical for biological function [81].
Experimental Approach:
- Genetic Manipulation: Construction of glycosyltransferase-deficient mutants (ΔgtrB in S. pyogenes) and complemented strains.
- Lectin Affinity Chromatography: Concanavalin A (ConA) sepharose to isolate glycoproteins from membrane fractions.
- Metabolic Labeling: UDP-[3H]Glc incorporation assays to track glycosylation activity.
- Biofilm Assays: Functional assessment of glycosylation-deficient mutants under ethanol-stressed conditions.
- SDS-PAGE Analysis: Demonstrated absence of ConA-reactive glycoproteins only in glycosylation-deficient strains [81].
This research established that O-linked glycosylation of serine/threonine-rich IDRs in streptococcal proteins like PrsA causes noticeable upward shifts in SDS-PAGE migration, with the non-glycosylated forms migrating faster than their glycosylated counterparts [81].
Methodological Approaches for Accurate Analysis
Detecting and Characterizing Glycosylation Artifacts
When glycosylation is suspected to cause anomalous SDS-PAGE migration, several experimental approaches can confirm and characterize the modification:
- Enzymatic Deglycosylation: Treatment with specific glycosidases (PNGase F for N-linked glycans, O-glycosidases for O-linked glycans) followed by comparative SDS-PAGE.
- Lectins and Chemical Detection: Staining with periodic acid-Schiff (PAS) or lectin-based blotting to identify carbohydrate components.
- Mass Spectrometry Analysis: Intact mass measurement or glycoproteomic approaches to identify glycosylation sites and compositions.
Addressing Phosphorylation Effects
For phosphorylation-related mobility shifts:
- Phosphatase Treatment: Alkaline phosphatase or specific protein phosphatases to remove phosphate groups, followed by mobility comparison.
- Phos-tag Gels: Incorporation of phosphate-binding molecules into acrylamide gels to retard phosphorylated proteins.
- Two-Dimensional Electrophoresis: Combining isoelectric focusing (separating by charge) with SDS-PAGE (separating by size) to resolve phosphorylation isoforms.
Alternative Electrophoretic Techniques
When conventional SDS-PAGE proves inadequate due to extensive modifications:
- Native SDS-PAGE (NSDS-PAGE): A modified approach using reduced SDS concentration (0.0375% in running buffer) and no heating step preserves some native protein features while maintaining reasonable separation resolution [8]. This method retains metal cofactors and enzymatic activity in 7 of 9 tested model enzymes compared to complete denaturation in standard SDS-PAGE [8].
- Blue Native PAGE (BN-PAGE): Uses Coomassie G-250 to impart charge while preserving protein complexes and modifications, though with reduced resolution for complex mixtures [8].
- CTAB-PAGE: Utilizes the cationic detergent cetyltrimethylammonium bromide, which can provide different separation characteristics for highly acidic or modified proteins [6].
Diagram 1: SDS-PAGE Workflow Showing PTM Interference Points and Solutions
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Research Reagent Solutions for Addressing PTM-Related Mobility Issues
| Reagent/Method | Function/Application | Key Considerations |
|---|---|---|
| PNGase F [80] | Removes N-linked glycans by cleaving between GlcNAc and asparagine | Requires denatured substrate; effective on most N-glycans |
| O-Glycosidase [80] | Removes core 1 and core 3 O-linked disaccharides | Limited to specific O-glycan structures; often used in enzyme cocktails |
| Alkaline Phosphatase [82] | Removes phosphate groups from serine, threonine, tyrosine residues | Broad specificity; requires appropriate buffer conditions |
| Concanavalin A (ConA) [81] | Lectin affinity chromatography for α-mannose/α-glucose glycoproteins | Useful for glycoprotein enrichment prior to analysis |
| Phos-tag Acrylamide | Gel additive that retards phosphorylated proteins | Creates discrete bands for different phosphorylation states |
| NSDS-PAGE Reagents [8] | Modified electrophoresis preserving metal binding and activity | Reduced SDS (0.0375%), no heating, no EDTA in buffers |
| Lectin Blotting | Detection of specific glycan structures after transfer | Requires appropriate lectin-horseradish peroxidase conjugates |
| Anti-phospho Antibodies [82] | Immunodetection of specific phosphorylation sites | Site-specific antibodies required for meaningful interpretation |
SDS-PAGE remains an indispensable tool in protein research, but its limitations in analyzing post-translationally modified proteins must be recognized. Glycosylation and phosphorylation can significantly alter electrophoretic mobility, leading to inaccurate molecular weight estimates and potential misinterpretation of protein composition. Understanding these effects is particularly crucial in disease contexts where aberrant glycosylation patterns are linked to cancer progression [80] and phosphorylation dynamics regulate fundamental cellular processes [82].
Researchers must employ confirmatory techniques—including enzymatic treatments, alternative electrophoretic methods, and mass spectrometric analysis—to validate findings when PTMs are suspected. The continuing development of modified electrophoretic approaches, such as NSDS-PAGE that preserves certain native protein features [8], provides valuable alternatives for challenging samples. As protein-based therapeutics and biomarkers continue to gain importance in drug development, recognizing and accounting for these SDS-PAGE limitations becomes increasingly critical for accurate protein characterization.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) represents a cornerstone technique in biochemical research, whose utility is fundamentally anchored in the denaturing action of the SDS detergent. The role of SDS is to bind to protein backbones at a constant weight ratio, typically 1.4 g SDS per 1 g of polypeptide, effectively masking intrinsic protein charges and conferring a uniform negative charge density [5] [2]. This binding, coupled with heating at 70-100°C in the presence of reducing agents like β-mercaptoethanol or DTT, unfolds proteins into linear chains, transforming complex separation parameters into a single variable: molecular weight [83] [2]. This revolutionary simplification enabled researchers to separate proteins based primarily on polypeptide chain length with remarkable resolution, making SDS-PAGE an indispensable tool for molecular weight determination, purity assessment, and expression analysis [2].
However, this denaturing strength constitutes the technique's primary limitation—the deliberate destruction of native protein structure and function. This fundamental trade-off between resolution and biological relevance sparked the development and refinement of native electrophoresis techniques, particularly Blue Native PAGE (BN-PAGE), which preserves protein complexes in their functional state while offering respectable separation power [8]. Within this evolutionary context, recent innovations like native SDS-PAGE (NSDS-PAGE) have emerged, attempting to balance the high resolution of denaturing systems with the functional preservation of native approaches by significantly modifying standard SDS-PAGE conditions [8]. This technical progression reflects an ongoing effort to expand the analytical capabilities of polyacrylamide gel electrophoresis to meet increasingly complex research demands in proteomics and drug development.
Principles of Separation: A Comparative Framework
Fundamental Separation Mechanisms
The core distinction between these electrophoretic techniques lies in their treatment of protein structure during separation, which directly determines the type of biological information they can reveal.
SDS-PAGE operates under denaturing conditions where proteins are separated primarily by molecular mass. The anionic detergent SDS binds extensively to polypeptide chains, unfolding them and imparting a uniform negative charge. This SDS-protein complex migrates through the polyacrylamide gel matrix toward the anode, with smaller proteins moving faster due to less resistance from the gel mesh [83] [2]. The discontinuous buffer system, incorporating a stacking gel with larger pores and different pH, concentrates protein samples into sharp bands before they enter the separating gel, significantly enhancing resolution [5].
Native PAGE maintains proteins in their folded, functional state throughout separation. Without denaturing agents, migration depends on the protein's intrinsic charge, size, and three-dimensional shape [2]. Proteins with higher negative charge density migrate faster toward the anode, while the gel matrix creates a sieving effect that retards larger molecules [83] [2]. This technique preserves protein function, enzymatic activity, and non-covalent interactions, including subunit associations in multimeric complexes [84].
Blue Native PAGE (BN-PAGE), a specialized native technique, employs Coomassie Brilliant Blue G-250 dye, which binds to protein surfaces without denaturation. The dye confers additional negative charges, ensuring even basic proteins migrate toward the anode while improving solubility, particularly for membrane proteins [8] [84]. This method excels at resolving native protein complexes within the 100 kDa to 10 MDa range, making it ideal for studying protein-protein interactions and macromolecular assemblies [84].
Quantitative Comparison of Electrophoretic Techniques
Table 1: Comprehensive comparison of SDS-PAGE, Native PAGE, and BN-PAGE characteristics
| Parameter | SDS-PAGE | Native PAGE | BN-PAGE |
|---|---|---|---|
| Separation Basis | Molecular weight | Size, intrinsic charge, & shape | Size, charge, & native mass |
| Gel Conditions | Denaturing | Non-denaturing | Non-denaturing |
| SDS Presence | Present (0.1-0.2%) | Absent | Absent |
| Reducing Agents | DTT or β-mercaptoethanol present | Absent | Absent |
| Sample Preparation | Heating at 70-100°C | No heating | No heating |
| Protein State | Denatured, linearized | Native, folded | Native, folded |
| Protein Function | Lost | Retained | Retained |
| Protein Recovery | Not recoverable | Recoverable | Recoverable |
| Molecular Weight Determination | Accurate | Approximate | Approximate for complexes |
| Key Additives | SDS, reducing agents | None | Coomassie G-250 dye |
| Typical Running Temperature | Room temperature | 4°C [83] | Room temperature [8] |
| Primary Applications | MW determination, purity assessment, expression analysis | Enzyme activity studies, oligomerization state | Membrane protein complexes, protein-protein interactions |
Advanced Technical Variations
Beyond these core techniques, several specialized variants address specific research needs:
Clear Native PAGE (CN-PAGE) represents a milder alternative to BN-PAGE that eliminates Coomassie dye, relying solely on proteins' intrinsic charges for separation [84]. While offering superior preservation of delicate protein assemblies and enzymatic activities, it provides lower resolution, particularly for proteins with isoelectric points above 5.4 [84].
Quantitative Preparative Native Continuous PAGE (QPNC-PAGE) employs continuous buffer systems at specific pH values (typically 8.00 or 10.00) to separate metaloproteins while preserving metal cofactor binding and tertiary structure [84]. This technique enables quantitative recovery of biologically active proteins for functional studies.
Native SDS-PAGE (NSDS-PAGE) represents a hybrid approach that modifies standard SDS-PAGE conditions by reducing SDS concentration in running buffers to 0.0375%, eliminating EDTA from sample buffers, and omitting the heating step [8]. This protocol preserves enzymatic activity in seven of nine model enzymes tested and dramatically increases zinc ion retention in metalloproteins from 26% to 98% compared to standard SDS-PAGE, while maintaining high resolution separation [8].
Experimental Protocols and Methodologies
Standard SDS-PAGE Protocol
The following procedure outlines the widely adopted SDS-PAGE method for protein separation based on molecular weight [5]:
Gel Preparation: Assemble glass plates with spacers. Prepare separating gel solution (typically 8-15% acrylamide depending on target protein size) containing 375 mM Tris-HCl (pH 8.8), 0.1% SDS, ammonium persulfate (APS), and TEMED catalyst. Pour between plates, overlay with water or alcohol to ensure even polymerization, and allow to set for 20-30 minutes.
Stacking Gel Formation: After separating gel polymerization, pour stacking gel solution (4-5% acrylamide) containing 125 mM Tris-HCl (pH 6.8), 0.1% SDS, APS, and TEMED. Insert comb immediately and allow to polymerize for 20-30 minutes.
Sample Preparation: Mix protein samples with SDS-PAGE sample buffer (typically containing 50-100 mM Tris-HCl, 2% SDS, 10% glycerol, 0.01% bromophenol blue, and 50-100 mM DTT or β-mercaptoethanol). Heat at 70-100°C for 3-10 minutes to denature proteins [5].
Electrophoresis: Mount gel in electrophoresis apparatus filled with running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3). Load samples and molecular weight markers into wells. Apply constant voltage (120-200 V depending on gel size) until dye front reaches bottom of gel (typically 45-90 minutes).
Post-Electrophoresis Analysis: Disassemble gel apparatus, remove gel, and proceed with staining (Coomassie Blue, silver stain, etc.), western blotting, or other analytical techniques.
Blue Native PAGE (BN-PAGE) Protocol
BN-PAGE follows a distinct procedure optimized for preserving protein complexes [8]:
Gel Preparation: Cast gradient gels (4-16% acrylamide) using specific BN-PAGE formulations. The cathode buffer contains 50 mM BisTris, 50 mM Tricine, and 0.02% Coomassie G-250 (pH 6.8), while anode buffer contains 50 mM BisTris and 50 mM Tricine (pH 6.8).
Sample Preparation: Mix protein samples with BN-PAGE sample buffer (50 mM BisTris, 50 mM NaCl, 10% glycerol, 0.001% Ponceau S, pH 7.2). Do not heat samples.
Electrophoresis: Load samples onto gel and run at constant voltage (150 V) for approximately 90-95 minutes at room temperature until dye front reaches gel bottom.
Post-Electrophoresis Analysis: Proteins can be visualized with Coomassie staining or other native-compatible detection methods. For two-dimensional analysis, excised BN-PAGE lanes can be applied to SDS-PAGE gels for second-dimension separation under denaturing conditions.
Native SDS-PAGE (NSDS-PAGE) Protocol
This modified SDS-PAGE method balances resolution with functional preservation [8]:
Gel Preparation: Use standard Bis-Tris gels (e.g., 12%) but pre-run in double-distilled water for 30 minutes at 200V to remove storage buffers and unpolymerized acrylamide.
Sample Preparation: Mix protein samples with NSDS sample buffer (100 mM Tris-HCl, 150 mM Tris base, 10% glycerol, 0.0185% Coomassie G-250, 0.00625% Phenol Red, pH 8.5). Omit heating step.
Electrophoresis: Run gels in modified running buffer (50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7) at 200V for standard separation times.
Diagram 1: PAGE technique selection workflow based on research objectives
Technical Considerations and Research Applications
Buffer Compositions and Electrophoretic Conditions
Table 2: Detailed buffer compositions for different PAGE techniques
| Component | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Sample Buffer | 106 mM Tris HCl, 141 mM Tris Base, 0.51 mM EDTA, 0.22 mM SERVA Blue G-250, 0.175 mM Phenol Red, 2% LDS, 10% Glycerol, pH 8.5 [8] | 50 mM BisTris, 50 mM NaCl, 16 mM HCl, 10% Glycerol, 0.001% Ponceau S, pH 7.2 [8] | 100 mM Tris HCl, 150 mM Tris Base, 0.01875% Coomassie G-250, 0.00625% Phenol Red, 10% Glycerol, pH 8.5 [8] |
| Running Buffer | 50 mM MOPS, 50 mM Tris Base, 1 mM EDTA, 0.1% SDS, pH 7.7 [8] | Cathode: 50 mM BisTris, 50 mM Tricine, 0.02% Coomassie G-250, pH 6.8Anode: 50 mM BisTris, 50 mM Tricine, pH 6.8 [8] | 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 [8] |
| Critical Additives | SDS (denaturant), DTT/β-mercaptoethanol (reducing agent) | Coomassie G-250 (charge conferring dye) | Reduced SDS (0.0375%), Coomassie G-250 |
| Key Modifications | Heating samples (70-100°C) | No heating, dye in cathode buffer | No heating, reduced SDS concentration |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents for PAGE experiments and their functions
| Reagent | Function | Technique |
|---|---|---|
| SDS (Sodium Dodecyl Sulfate) | Denatures proteins, confers negative charge | SDS-PAGE |
| DTT/β-mercaptoethanol | Reduces disulfide bonds | SDS-PAGE |
| Coomassie G-250 | Binds proteins, confers negative charge without denaturation | BN-PAGE |
| Coomassie R-250/G-250 | Stains proteins post-electrophoresis | All techniques |
| APS (Ammonium Persulfate) | Initiates acrylamide polymerization | All techniques |
| TEMED | Catalyzes acrylamide polymerization | All techniques |
| Acrylamide/Bis-acrylamide | Forms cross-linked gel matrix | All techniques |
| Tris-based buffers | Maintain pH during electrophoresis | All techniques |
| Glycine/MOPS/BisTris | Conducting ions in running buffers | All techniques |
| Glycerol | Increases sample density for well loading | All techniques |
| Bromophenol Blue | Tracking dye for migration monitoring | SDS-PAGE, NSDS-PAGE |
Research Applications and Technique Selection
Each electrophoretic method addresses distinct research questions based on its inherent strengths and limitations:
SDS-PAGE Applications:
- Molecular weight determination using protein standards [2]
- Assessing protein purity and homogeneity
- Analyzing protein expression levels
- Quality control in protein purification protocols
- Sample preparation for western blotting and mass spectrometry [2]
Native PAGE Applications:
- Studying oligomerization states and quaternary structure [2]
- Enzyme activity assays after separation (zymography) [2]
- Protein folding studies under non-denaturing conditions
- Analysis of charge variants and isoforms
- Purification of functional proteins for downstream applications [83]
BN-PAGE Applications:
- Resolution of native membrane protein complexes [84]
- Analysis of protein-protein interactions [84]
- Mitochondrial respiratory chain studies
- First-dimension separation for 2D BN/SDS-PAGE [8]
- Molecular weight estimation of native complexes
Diagram 2: Evolutionary relationships between PAGE techniques showing methodological developments
The comparative analysis of SDS-PAGE, Native PAGE, and BN-PAGE reveals a sophisticated toolkit for protein research, with each technique offering distinct advantages tailored to specific experimental goals. SDS-PAGE remains the gold standard for molecular weight determination and analytical separation when protein function preservation is not required. Native PAGE techniques, particularly BN-PAGE, provide critical insights into protein complexes and functional interactions that would be destroyed by denaturing conditions. The ongoing innovation in this field, exemplified by hybrid approaches like NSDS-PAGE, continues to expand the analytical capabilities available to researchers.
The role of SDS in this technological ecosystem is foundational—its introduction revolutionized protein electrophoresis by simplifying separation parameters to primarily molecular weight. While this denaturing action comes at the cost of biological context, understanding this trade-off enables researchers to strategically select the most appropriate technique. For drug development professionals and research scientists, this comparative framework supports informed methodological choices based on whether the research question prioritizes analytical resolution or biological relevance, ultimately accelerating discovery in proteomics and biomedical research.
The Foundational Role of SDS in Polyacrylamide Gel Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is a cornerstone technique in biochemistry and molecular biology for separating proteins based on their molecular weight. The critical innovation that enables this precise separation is the use of SDS, an anionic detergent [5] [6] [9].
SDS serves two essential functions in protein analysis. First, it acts as a powerful denaturing agent that binds to the protein backbone at a constant ratio of approximately 1.4 grams of SDS per gram of protein, effectively disrupting hydrogen bonds and van der Waals forces that maintain secondary and tertiary structures [6] [9]. This binding unfolds proteins into linear polypeptide chains, masking their intrinsic charges and conferring a uniform negative charge proportional to their molecular weight [5] [6] [9]. When subjected to an electric field, these SDS-coated proteins migrate through the polyacrylamide gel matrix solely based on size, with smaller proteins moving faster through the gel's pores [5] [53] [9].
This denaturation process is typically enhanced by heating samples to 95°C for several minutes and including reducing agents like dithiothreitol (DTT) or β-mercaptoethanol to break disulfide bonds [5] [6] [85]. The result is a system that eliminates the influence of native protein structure and charge, allowing researchers to separate complex protein mixtures with excellent resolution based primarily on polypeptide chain length [5] [9].
Experimental Protocols for SDS-PAGE
Standard SDS-PAGE Protocol:
- Gel Preparation: Assemble glass plates with spacers. Prepare separating gel solution (typically 10-12% acrylamide), pour between plates, and overlay with water-saturated butanol to prevent oxygen inhibition. After polymerization (20-30 minutes), remove overlay and prepare stacking gel (4-6% acrylamide) with comb inserted [5] [6].
- Sample Preparation: Mix protein samples with SDS-containing sample buffer. Include fresh reducing agent (DTT or β-mercaptoethanol). Heat denature at 95°C for 3-5 minutes, then cool on ice to prevent renaturation [5] [85].
- Electrophoresis: Mount gel in apparatus, fill chambers with running buffer (typically Tris-glycine with 0.1% SDS). Load samples and molecular weight markers. Run at constant voltage (100-150V) until dye front reaches bottom [5] [6] [9].
- Post-Electrophoresis Analysis: Proteins can be visualized by staining with Coomassie Blue, silver stain, or other detection methods, or transferred to membrane for western blotting [6] [9].
Modified Native SDS-PAGE Protocol for Functional Studies: For experiments requiring retention of protein function or metal cofactors, a modified approach reduces denaturing conditions [8]:
- Prepare sample buffer without SDS or EDTA
- Omit heating step
- Reduce SDS concentration in running buffer to 0.0375%
- Include Coomassie G-250 and phenol red in sample buffer
- Run pre-cast Bis-Tris gels at 200V for 30 minutes [8]
Protein Ladders: Essential Tools for Molecular Weight Determination and Quality Control
Protein ladders, also known as molecular weight markers, are mixtures of highly purified proteins of known molecular weights that serve as critical reference standards in SDS-PAGE [86] [53]. These tools enable researchers to estimate the size of unknown proteins, monitor electrophoresis progress, and verify transfer efficiency during western blotting [86] [53].
Types of Protein Ladders and Their Applications
Table 1: Comparison of Prestained Protein Ladders
| Product Name | Molecular Weight Range | Number of Bands | Primary Applications | Visualization Methods |
|---|---|---|---|---|
| PageRuler Plus Prestained Protein Ladder | 10-250 kDa | 9 | Routine applications | Colorimetric, NIR fluorescence |
| Spectra Multicolor Broad Range Protein Ladder | 10-260 kDa | 10 | Improved visualization during separation and transfer | Colorimetric, NIR fluorescence, RGB fluorescence |
| HiMark Prestained Protein Standard | 31-460 kDa | 9 | Analysis of high molecular weight proteins | Colorimetric |
| SeeBlue Prestained Standard | 3-200 kDa | 9 | Monitoring gel separation and transfer efficiency | Colorimetric |
| iBright Prestained Protein Ladder | 11-250 kDa | 12 | Visible, IgG or fluorescent western blot detection | Colorimetric, NIR fluorescence, IgG binding |
Table 2: Comparison of Unstained Protein Ladders and Specialty Markers
| Product Name/Type | Molecular Weight Range | Number of Bands | Key Features |
|---|---|---|---|
| PageRuler Unstained Protein Ladder | 10-200 kDa | 14 | Superior accuracy with Strep-tag II for immunodetection |
| PageRuler Unstained Broad Range Protein Ladder | 5-250 kDa | 11 | Accurate estimation across broader range |
| HiMark Unstained Protein Standard | 40-500 kDa | 9 | Analysis of high molecular weight proteins |
| NativeMark Unstained Protein Standard | ~20-1236 kDa | 8 | Native PAGE applications |
| BenchMark His-tagged Protein Standard | 10-160 kDa | 10 | Detection with His-tag stain |
| IEF Marker | 3.5-10.7 pI | 13 | pI calibration for isoelectric focusing |
Selection Guidelines for Protein Ladders
Choosing the appropriate protein ladder requires consideration of several factors. Prestained markers allow real-time monitoring of electrophoresis progression and transfer efficiency but may exhibit slightly altered migration compared to unstained proteins due to the attached dye molecules [86]. Unstained ladders provide more accurate molecular weight determination and are ideal for precise size estimation when visualized by protein stains like Coomassie or silver [86]. Specialty markers including those for native PAGE, isoelectric focusing, or detecting specific tags (His-tag, phosphoproteins, glycoproteins) address specialized research needs [86].
For optimal results, match the ladder's molecular weight range to your protein of interest and use the recommended gel type (e.g., Tris-acetate gels for high molecular weight proteins) [86]. Follow manufacturer recommendations for loading volumes, typically 5-10 μL per well for 1.0 mm gels [86].
Critical Controls for Experimental Validation
Beyond molecular weight markers, appropriate experimental controls are essential for validating SDS-PAGE and western blotting results, ensuring specificity, and enabling accurate interpretation.
Types of Essential Controls
Loading Controls assess consistency in sample loading and protein transfer [53]. Housekeeping proteins (e.g., β-actin, GAPDH) constitutively expressed in cells and tissues are commonly used, confirming equal protein loading across wells and allowing normalization for quantitative analysis [53]. Alternatively, spiked protein controls can be added to each sample to verify consistent transfer from gel to membrane during electroblotting [53].
Negative Controls identify non-specific detection by primary or secondary antibodies [53]. These include:
- Tissue or cell samples known not to express the target protein
- No-primary antibody controls where the primary antibody is omitted from the detection step
- Isotype controls using non-specific immunoglobulins of the same species and isotype as the primary antibody
Positive signals in negative controls indicate non-specific binding that must be addressed through protocol optimization [53].
Experimental and Technical Controls address specific methodological variables. For reduction-sensitive analyses, include both reduced and non-reduced samples to examine disulfide bond effects [9]. Process control samples identical to experimental samples throughout preparation identify degradation or modification artifacts [53]. Buffer-only controls detect contamination in sample preparation or reagent systems [53].
Troubleshooting Common SDS-PAGE Issues
Even with proper controls, technical issues can arise. The table below outlines common problems and their solutions.
Table 3: Troubleshooting Common SDS-PAGE Issues
| Issue | Potential Causes | Solutions |
|---|---|---|
| Smiling or frowning bands | Uneven heating, improper buffer composition, irregular current distribution | Ensure even sample loading, monitor voltage, confirm proper buffer preparation [53] [9] |
| Smeared bands | Incomplete denaturation, insufficient reduction, high salt concentration | Add fresh reducing agent, boil samples 5 minutes at 100°C, reduce salt concentration below 500 mM [53] |
| Weak/faint bands | Too little protein loaded | Calculate protein concentration using Bradford, Lowry, or BCA assay before loading [53] |
| Multiple/unexpected bands | Protein degradation, modification, or aggregation | Use protease inhibitors, include phosphatase inhibitors, check for truncation with bioinformatics tools [53] |
| Sample doesn't migrate but ladder does | Improper sample preparation, missing SDS or reducing agents | Ensure sample buffer contains SDS, use fresh reducing agents, include heating step [85] |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Essential Reagents for SDS-PAGE Research
| Reagent/Category | Function/Purpose | Examples/Specifications |
|---|---|---|
| Protein Ladders | Molecular weight calibration, process monitoring | Prestained, unstained, and specialty markers covering relevant size ranges [86] |
| Acrylamide/Bis-acrylamide | Gel matrix formation | Varying percentages (6-15%) for different separation ranges; 29:1 or 37.5:1 acrylamide:bis-acrylamide ratios [5] [6] |
| SDS (Sodium Dodecyl Sulfate) | Protein denaturation and charge conferral | 10% solution in sample buffer; critical micelle concentration ~7-10 mM [6] [9] |
| Reducing Agents | Disruption of disulfide bonds | DTT (10-100 mM), β-mercaptoethanol (5%), TCEP [6] [85] |
| Buffers | Maintain pH and conductivity | Tris-glycine (pH 8.3-8.8), MOPS, Bis-Tris; stacking gel (pH 6.8) and resolving gel (pH 8.8) buffers [6] [53] |
| Catalysts | Polyacrylamide polymerization | Ammonium persulfate (APS) and TEMED [6] |
| Protein Stains | Visualizing separated proteins | Coomassie Blue, silver stain, fluorescent dyes (varying sensitivity) [6] [9] |
| Protease/Phosphatase Inhibitors | Preventing sample degradation | PMSF, complete protease inhibitor cocktails, phosphatase inhibitors [53] |
Optimizing SDS-PAGE Conditions for Reliable Results
Gel Percentage Selection
The acrylamide concentration significantly impacts resolution. Lower percentages (8-10%) separate higher molecular weight proteins more effectively, while higher percentages (12-15%) provide better resolution for smaller proteins [53] [9]. Gradient gels (e.g., 4-12% or 4-20%) offer broad separation ranges in a single gel, ideal for complex samples with proteins of diverse sizes [6] [9].
Electrophoresis Conditions
Standard conditions for mini-gels typically use constant voltage between 100-150V for 40-60 minutes, or until the dye front reaches the bottom [9]. Higher voltages reduce run time but may generate excessive heat, leading to band distortion [53] [9]. For large format gels, lower voltages (100-120V) with longer run times provide better resolution [9].
Sample Preparation Considerations
Proper sample preparation is critical for success. Ensure adequate protein concentration (typically 10-50 μg per lane for Coomassie staining, less for western blotting) using quantification assays like BCA or Bradford [53]. Fresh reducing agents are essential for complete denaturation of disulfide-rich proteins [85]. For difficult samples, consider alternative detergents or chaotropic agents to improve solubility and denaturation [9].
Advanced Applications and Future Directions
The fundamental principles of SDS-PAGE continue to support evolving applications in proteomics and drug development. Two-dimensional electrophoresis combines isoelectric focusing with SDS-PAGE to separate thousands of proteins simultaneously [9]. Advanced western blotting techniques depend on high-quality SDS-PAGE separation for sensitive protein detection [53]. The development of modified approaches like NSDS-PAGE demonstrates ongoing innovation, enabling researchers to balance the need for high resolution with preservation of protein function for specialized applications [8].
SDS-PAGE Experimental Workflow with Critical Validation Points
Protein ladders and controls are not mere accessories but fundamental components that transform SDS-PAGE from a simple separation technique to a robust, quantitative analytical method. When employed strategically within the denaturing framework established by SDS, these validation tools provide the critical reference points needed for accurate molecular weight determination, specificity confirmation, and experimental reproducibility. As SDS-PAGE continues to evolve through techniques like NSDS-PAGE and specialized applications, the thoughtful implementation of appropriate standards and controls remains essential for generating reliable, interpretable data that advances scientific discovery and therapeutic development.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is a cornerstone technique in molecular biology, primarily used to separate protein mixtures based on molecular weight [2]. The role of SDS, an anionic detergent, in this process is fundamental: it denatures proteins by binding to the polypeptide backbone at a constant ratio, masking intrinsic charges and imparting a uniform negative charge density [2] [6]. This allows separation based almost exclusively on polypeptide chain length rather than native charge or structure [5] [2]. However, this very denaturing capability that makes SDS-PAGE so effective for molecular weight determination also constitutes its primary limitation for functional studies, as it destroys native protein structure, quaternary interactions, enzymatic activity, and non-covalently bound cofactors such as metal ions [8].
The field has sought alternatives to overcome these limitations. Blue Native (BN)-PAGE was introduced to retain functional properties, but it comes at the cost of the high resolution that characterizes SDS-PAGE [8]. This trade-off created a persistent gap in the researcher's toolkit—a need for a method offering both high resolution and the retention of native functionality. This whitepaper explores Native SDS-PAGE (NSDS-PAGE) as a novel adaptation designed to bridge this gap. By strategically modifying standard SDS-PAGE conditions, NSDS-PAGE aims to preserve the excellent resolution of denaturing electrophoresis while maintaining proteins in a native, functional state, thereby expanding the role of SDS from a mere denaturant to a potential facilitator of native-state analysis [8].
NSDS-PAGE: Core Principles and Methodological Innovations
The innovation of NSDS-PAGE lies not in discarding SDS, but in precisely controlling its concentration and omitting other denaturing conditions to preserve protein function without sacrificing separation quality. The core hypothesis is that while SDS is necessary for imparting charge and enabling electrophoretic mobility, its denaturing effect can be mitigated below a critical threshold, allowing proteins to refold into their native conformations after electrophoresis [8].
Key Methodological Modifications
The NSDS-PAGE protocol makes several critical modifications to the standard denaturing SDS-PAGE method, each targeting a different denaturing factor [8]:
- Elimination of Denaturing Steps in Sample Preparation: The sample buffer is reconfigured to remove SDS and EDTA (a chelating agent that strips essential metal cofactors). Furthermore, the heating step traditionally used to denature samples is omitted entirely. This alone prevents the initial and deliberate unfolding of proteins before they even enter the gel [8].
- Critical Reduction of SDS in the Running Buffer: The SDS concentration in the running buffer is drastically reduced from the standard 0.1% to 0.0375% [8]. This sub-micellar concentration is crucial. Research indicates that SDS denatures proteins most effectively at concentrations above its critical micelle concentration (CMC: 7-10 mM, equivalent to ~0.22-0.31% w/v) [6]. While unfolding begins above 0.1 mM SDS, using a concentration of 0.0375% (~1.3 mM) likely falls below the threshold required for sustained denaturation during the brief electrophoresis period, allowing for potential refolding post-separation.
- Buffer Reformation: The running buffer also has EDTA removed to preserve metal-protein interactions [8]. The sample buffer is reformulated with Tris, glycerol, and minimal Coomassie G-250 and Phenol Red as tracking dyes, creating a non-denaturing environment for the sample [8].
Table 1: Quantitative Comparison of Key Buffer Components Across PAGE Methods
| Component | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Sample Buffer SDS | Present (in LDS) | Absent | Absent |
| Sample Heating | 70°C for 10 min | Not specified | Omitted |
| Running Buffer [SDS] | 0.1% | Absent | 0.0375% |
| EDTA | 1 mM (Running Buffer) | Not specified | Absent |
| Key Additive | SDS | Coomassie G-250 | Minimal Coomassie G-250 |
Theoretical Workflow and Logical Rationale
The logical flow and key decision points for implementing NSDS-PAGE are summarized in the following workflow. This diagram outlines the parallel processes of standard and native methods, highlighting the critical modifications that enable functional preservation.
Experimental Validation and Key Findings
The development of NSDS-PAGE was validated through rigorous experiments comparing its performance against standard SDS-PAGE and BN-PAGE, with a focus on metal retention and enzymatic activity.
Quantitative Assessment of Metal Retention and Enzymatic Activity
Research quantitatively demonstrates the superior performance of NSDS-PAGE in preserving functional protein characteristics. The data below summarizes key findings from these validation experiments.
Table 2: Quantitative Functional Outcomes of NSDS-PAGE vs. Other Methods
| Assessment Parameter | SDS-PAGE | BN-PAGE | NSDS-PAGE |
|---|---|---|---|
| Zn²⺠Retention in Proteome | 26% | Not Specified | 98% [8] |
| Active Enzymes (from 9 models) | 0 of 9 | 9 of 9 | 7 of 9 [8] |
| Example Active Zn²⺠Enzymes | None | Zn-ADH, Zn-AP, Cu,Zn-SOD, Zn-CA | Zn-ADH, Zn-AP, Cu,Zn-SOD, Zn-CA [8] |
| Key Functional Advantage | Molecular Weight Determination | Full Activity Retention | High Resolution + Mostly Intact Function |
The 98% retention of Zn²⺠in a complex proteome sample, compared to only 26% in standard SDS-PAGE, provides direct evidence that the modified conditions successfully prevent the stripping of metal cofactors, which is critical for the function of many metalloproteins [8]. Furthermore, the fact that seven out of nine model enzymes, including four zinc-binding proteins (alcohol dehydrogenase, alkaline phosphatase, superoxide dismutase, and carbonic anhydrase), retained activity after NSDS-PAGE confirms that the proteins not only hold their metals but also refold into their functional three-dimensional structures post-electrophoresis [8]. While BN-PAGE was able to preserve activity in all nine enzymes, NSDS-PAGE achieves this functional retention while offering a significant improvement in resolution [8].
Detailed Experimental Protocol for NSDS-PAGE
The following section provides a step-by-step methodology for conducting an NSDS-PAGE experiment, as derived from the primary literature [8]. This protocol is designed for researchers to replicate and implement the technique.
I. Sample Preparation
- Prepare Sample Buffer (4X): 100 mM Tris HCl, 150 mM Tris Base, 10% (v/v) glycerol, 0.0185% (w/v) Coomassie G-250, 0.00625% (w/v) Phenol Red, pH 8.5 [8]. Note the absence of SDS and EDTA.
- Mix Sample: Combine 7.5 μL of protein sample with 2.5 μL of the 4X NSDS sample buffer. Do not heat the mixture. Mix gently by pipetting or flicking the tube [8].
II. Gel Preparation and Pre-electrophoresis
- Use Standard Pre-cast Gels: Commercially available precast NuPAGE Novex 12% Bis-Tris 1.0 mm mini-gels are suitable [8].
- Pre-run the Gel: Mount the gel in the electrophoresis apparatus and run it at 200V for 30 minutes in double-distilled Hâ‚‚O. This critical step removes the storage buffer and any unpolymerized acrylamide, which could interfere with the native conditions [8].
III. Electrophoresis
- Prepare Running Buffer (1X): 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7. Ensure the SDS is thoroughly dissolved [8].
- Load and Run: Replace the water in the apparatus with the running buffer. Load the prepared samples into the wells. Include an appropriate molecular weight marker. Run the gel at a constant voltage of 200V for approximately 45 minutes (or until the dye front reaches the bottom) at room temperature [8].
IV. Post-Electrophoresis Analysis
- Functional Assays: For activity staining, incubate the gel in an appropriate substrate solution specific to the enzyme of interest (e.g., a substrate that produces a colored precipitate upon cleavage) [8] [87].
- Metal Detection: Use laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) for direct metal quantification or in-gel fluorescence staining with a metal-specific fluorophore like TSQ for zinc [8].
- Standard Staining: Proteins can also be visualized using standard Coomassie or silver staining protocols [88].
The Scientist's Toolkit: Essential Reagents for NSDS-PAGE
Successful implementation of NSDS-PAGE relies on specific reagents. The following table details key solutions and their functions, forming a core toolkit for researchers.
Table 3: Essential Research Reagent Solutions for NSDS-PAGE
| Reagent / Solution | Composition / Key Feature | Function in NSDS-PAGE |
|---|---|---|
| NSDS Sample Buffer (4X) | 100 mM Tris HCl, 150 mM Tris Base, 10% Glycerol, pH 8.5 (No SDS/EDTA) [8] | Maintains sample pH and density without initiating denaturation; glycerol aids loading. |
| NSDS Running Buffer (1X) | 50 mM MOPS, 50 mM Tris Base, 0.0375% SDS, pH 7.7 [8] | Provides conducting medium and controlled, sub-denaturing SDS to impart charge for migration. |
| Bis-Tris Polyacrylamide Gels | Precast 12% Bis-Tris gels, continuous buffer system, near-neutral pH [8] [6] | Separation matrix; neutral pH minimizes protein damage and acrylamide hydrolysis. |
| Model Zn-Metalloproteins | e.g., Alcohol Dehydrogenase (ADH), Alkaline Phosphatase (AP), Carbonic Anhydrase (CA) [8] | Positive controls for validating metal retention and enzymatic activity post-electrophoresis. |
| In-Gel Activity Assay Reagents | Specific enzyme substrates (e.g., for phosphatases, dehydrogenases) [8] [87] | To directly visualize and confirm retention of native enzymatic function after separation. |
| Metal Detection Probes | Fluorophore TSQ for Zn²âº; LA-ICP-MS for broad metal analysis [8] | To quantitatively measure the success of metal cofactor retention in the separated proteins. |
NSDS-PAGE represents a significant innovative adaptation in electrophoretic methodology. It redefines the role of SDS from a mere denaturant for mass-based separation to a finely tuned reagent that can be leveraged for high-resolution separation of functional, native proteins. By systematically modifying the concentration of SDS and eliminating auxiliary denaturing conditions, this technique successfully bridges the critical gap between the high resolution of SDS-PAGE and the functional preservation of BN-PAGE [8].
The implications for research, particularly in drug development and metallomics, are substantial. The ability to separate complex protein mixtures with high resolution while retaining enzymatic activity and metal-binding properties enables direct functional screening and analysis from a gel. This positions NSDS-PAGE not just as an alternative protocol, but as a powerful enabling technology for functional proteomics, facilitating the discovery and characterization of proteins based on their biological activity rather than size alone.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) stands as a cornerstone analytical technique in the biopharmaceutical industry for characterizing protein-based therapeutics. Within the framework of pharmacopoeial methods, SDS-PAGE provides a reliable, reproducible, and cost-effective means to assess critical quality attributes such as molecular weight, purity, and subunit composition of biological products. The technique's importance is underscored by its recognition in compendial standards and its alignment with Quality by Design (QbD) principles outlined in ICH guidelines, which emphasize a systematic, scientific, and risk-based approach to analytical method development and validation [89]. This technical guide explores the regulatory and scientific context of SDS-PAGE, detailing its fundamental principles, standardized methodologies, and applications essential for ensuring the safety, efficacy, and quality of biopharmaceuticals.
Fundamental Principles: The Critical Role of SDS
Mechanism of Protein Denaturation and Charge Uniformity
The resolving power of SDS-PAGE hinges on the action of sodium dodecyl sulfate (SDS), an anionic detergent that fundamentally transforms protein structure. SDS binds to the protein backbone at an approximate ratio of 1.4g SDS per 1g of protein, effectively disrupting non-covalent bonds—including hydrogen bonds, hydrophobic interactions, and ionic bonds—that maintain secondary and tertiary structures [3]. This binding denatures proteins into linear polypeptides and confers a uniform negative charge density along the entire polypeptide chain. This process masks the intrinsic charge differences among proteins, ensuring that migration during electrophoresis depends solely on molecular weight rather than shape or native charge [3] [5].
Molecular Sieving in the Polyacrylamide Gel Matrix
The polyacrylamide gel serves as a molecular sieve, with its pore size determined by the concentrations of acrylamide and the crosslinker N,N'-methylenebisacrylamide (Bis) [3]. Under an electric field, smaller protein molecules navigate through the gel's porous matrix more rapidly than larger molecules, resulting in separation based primarily on polypeptide chain length [5]. The polyacrylamide gel's strength allows for easy handling, making it suitable for routine laboratory analysis [5].
The Discontinuous Buffer System
SDS-PAGE employs a discontinuous buffer system comprising two distinct gel layers with different pore sizes and pH values to enhance resolution:
- Stacking Gel (pH 6.8): Features low acrylamide concentration with large pores that concentrate protein samples into narrow bands before they enter the separating gel, utilizing glycine's zwitterionic state at this pH [3].
- Separating Gel (pH 8.8): Contains higher acrylamide concentration with smaller pores where actual size-based separation occurs as glycine becomes fully negatively charged, accelerating migration [3].
This discontinuous system ensures proteins enter the separating gel simultaneously as sharp, concentrated bands, significantly improving resolution compared to a continuous system.
Regulatory Framework and Pharmacopoeial Standards
Quality by Design (QbD) and ICH Guidelines
The application of SDS-PAGE in biopharmaceutical analysis aligns with the Quality by Design (QbD) framework, a systematic approach emphasized in ICH guidelines to enhance product quality through thorough understanding and control of both product and process variables [89]. Within this framework, SDS-PAGE serves as a critical analytical procedure for:
- Assessing identity through molecular weight confirmation
- Determining purity and detecting impurities
- Monitoring product consistency across manufacturing batches
- Supporting comparability studies following process changes
Adherence to QbD principles for analytical methods ensures robust, reproducible SDS-PAGE protocols that can detect variability and contribute to continuous improvement throughout the product lifecycle [89].
SDS-PAGE in the Context of Complementary Techniques
SDS-PAGE represents one essential component in a comprehensive analytical toolbox for biopharmaceutical characterization, which typically includes techniques such as:
- Size Exclusion Chromatography (SEC): For assessing aggregation and molecular size distribution
- High-Performance Liquid Chromatography (HPLC): For precise purity quantification and variant analysis
- Western Blotting: For specific antigen detection following electrophoretic separation
- Enzyme-Linked Immunosorbent Assays (ELISA): For quantitative analysis of specific proteins or impurities [89]
The orthogonal data generated by these complementary techniques provide a holistic understanding of protein therapeutic attributes, with SDS-PAGE offering particular strengths in visualizing protein integrity, detecting fragments, and assessing general purity profile.
Experimental Protocols and Methodologies
Standard SDS-PAGE Protocol: Step-by-Step
The following comprehensive protocol ensures reproducible results compliant with regulatory standards:
Gel Preparation
- Assemble Gel Casting Mold: Thoroughly clean glass plates with ethanol and assemble with spacers [5].
- Prepare Separating Gel: Mix acrylamide, bis-acrylamide, SDS, buffer (pH 8.8), and polymerize using ammonium persulfate (APS) and TEMED [3]. Pour between glass plates and overlay with water to prevent oxygen inhibition [5].
- Prepare Stacking Gel: After separating gel polymerization (20-30 minutes), remove water and pour stacking gel solution (pH 6.8) with comb inserted [5].
Sample Preparation
- Dilute Protein Samples: Mix with equal volume of 2X Laemmli sample buffer containing SDS and reducing agent (DTT or β-mercaptoethanol) [90].
- Denature Proteins: Heat samples at 95°C for 5 minutes [90] or 100°C for 3 minutes [5] to ensure complete denaturation.
- Centrifuge: Spin samples at 15,000 rpm for 1-3 minutes to pellet debris [90] [5].
Electrophoresis
- Assemble Apparatus: Place gel in electrophoresis chamber, fill with running buffer, and remove air bubbles from wells [5].
- Load Samples: Load prepared samples and molecular weight markers (5-35 μL per lane) [90].
- Run Electrophoresis: Apply constant voltage (150V) until dye front reaches bottom (45-90 minutes) [90].
Post-Electrophoresis Analysis
- Disassemble Apparatus: Carefully remove gel from plates using a spatula [5].
- Proceed with Detection: Use appropriate staining, Western blotting, or other analytical methods based on experimental objectives.
Critical Method Parameters and Optimization
Gel Concentration Selection Table 1: Gel Concentration Guidelines for Optimal Separation
| Acrylamide Concentration | Separation Range (kDa) | Typical Applications |
|---|---|---|
| 8-8% | 100-500 | Large proteins, complexes |
| 10% | 20-200 | Standard protein separation |
| 12% | 10-100 | Most routine applications |
| 15% | 5-50 | Small peptides, fragments |
| 4-20% Gradient | 10-200 | Broad range separation |
Sample Preparation Variables
- Reducing vs. Non-Reducing Conditions: Reducing agents (DTT, β-mercaptoethanol) break disulfide bonds to analyze subunit composition; omission preserves native quaternary structure [3].
- Protein Load Optimization: 0.5-1.0 μg sufficient for purified proteins; 10 μg typically needed for complex lysates in Coomassie-stained gels [90].
- Buffer Composition: Ensuring appropriate ionic strength and pH for specific protein types.
Essential Reagents and Materials
Research Reagent Solutions
Table 2: Essential SDS-PAGE Reagents and Their Functions
| Reagent/Material | Function | Critical Quality Attributes |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Denatures proteins, confers uniform charge | High purity, minimal aldehydes and alcohols |
| Acrylamide/Bis-acrylamide | Forms porous gel matrix for molecular sieving | Electrophoresis grade, neurotoxin handling precautions |
| Ammonium Persulfate (APS) | Initiates polymerization reaction | Fresh preparation recommended |
| TEMED | Catalyzes polymerization reaction | Stored properly to prevent degradation |
| DTT or β-mercaptoethanol | Reduces disulfide bonds | Fresh reducing agents for consistent results |
| Tris Buffers | Maintains pH during electrophoresis | Ultrapure, correct pH adjustment |
| Glycine | Mobile ion in discontinuous buffer system | Electrophoresis grade |
| Protein Molecular Weight Markers | Reference for size determination | Pre-stained or unstained based on application |
| Coomassie Blue/Silver Stain | Visualizes separated proteins | Sensitivity appropriate to detection needs |
Applications in Biopharmaceutical Development
Critical Quality Attribute Assessment
SDS-PAGE serves as a fundamental tool for evaluating multiple critical quality attributes of biopharmaceutical products:
Purity and Impurity Analysis SDS-PAGE enables direct visualization of product-related impurities including:
- Protein fragments resulting from cleavage or degradation
- Aggregates under non-reducing conditions
- Host cell protein contaminants in early purification stages A single, sharp band indicates a pure sample, while multiple or smeared bands suggest impurities, degradation, or heterogeneity [3].
Molecular Weight Determination and Identity Testing By comparing protein migration distances to calibrated molecular weight markers, researchers confirm protein identity and estimate size with an accuracy typically within 5-10% [3] [90]. This application is particularly valuable for:
- Confirmatory testing of recombinant protein therapeutics
- Analysis of biosimilar similarity to reference products
- Detection of unexpected post-translational modifications that alter mobility
Subunit Composition Analysis Under reducing conditions, multi-subunit proteins (e.g., antibodies, enzyme complexes) dissociate into individual subunits, allowing characterization of:
- Monoclonal antibody heavy and light chains
- Hemoglobin subunit patterns for diagnostic applications [3]
- Oligomeric state confirmation for complex biologics
Process-Related Applications
Purification Process Monitoring SDS-PAGE provides a rapid, cost-effective method for monitoring protein purification efficiency across different chromatography steps, enabling:
- Fraction analysis during process development
- Contaminant clearance validation
- Column performance assessment
Stability and Comparability Studies The technique supports regulatory filings through:
- Forced degradation studies to identify vulnerable cleavage sites
- Real-time stability testing for shelf-life determination
- Comparability assessments following manufacturing process changes
Method Validation and Regulatory Considerations
Analytical Performance Characteristics
For inclusion in regulatory submissions, SDS-PAGE methods should demonstrate adequate performance through validation of these key parameters:
Specificity
- Ability to distinguish target protein from degradants and impurities
- Demonstration through stressed samples or spiked impurities
Precision
- Repeatability (same analyst, same conditions)
- Intermediate precision (different days, different analysts)
- Typically expressed as %RSD of migration distance or band intensity
Range and Linearity
- Demonstration of adequate separation across relevant molecular weight range
- Linear relationship between log(MW) and migration distance
Robustness
- Deliberate variations in key parameters (gel concentration, running time, voltage)
- Identification of critical method parameters for control
Emerging Trends and Technological Advancements
The SDS-PAGE landscape continues to evolve with several notable trends impacting biopharmaceutical applications:
Automation and High-Throughput Systems Market analysis indicates increasing demand for automated electrophoresis platforms that enhance:
- Reproducibility through standardized protocols
- Throughput for increased sample numbers in development
- Data integrity through digital capture and analysis [10]
Digital Imaging and Advanced Analysis Software innovations now provide:
- Automated band detection and molecular weight calculation
- Quantitative densitometry for impurity quantification
- Normalization against internal standards to reduce user bias [10]
Precast Gel Systems The market has seen significant growth in precast gel adoption due to:
- Enhanced convenience and reduced preparation variability
- Consistent performance with quality-controlled manufacturing
- Specialized formulations for specific applications [10]
Capillary Electrophoresis Systems Emerging capillary electrophoresis technologies offer:
- Automated operation with reduced manual intervention
- Improved quantitative capabilities
- Direct compatibility with quality control environments [10]
SDS-PAGE remains an indispensable analytical technique within the biopharmaceutical quality control arsenal, providing critical data on molecular weight, purity, and structural attributes of protein therapeutics. Its alignment with pharmacopoeial standards and QbD principles ensures continued relevance in regulatory frameworks, while technological advancements address evolving needs for precision, throughput, and data integrity. As biopharmaceuticals grow increasingly complex, SDS-PAGE will maintain its foundational role in confirming product quality, supporting process understanding, and ultimately ensuring patient safety through rigorous analytical characterization.
Conclusion
Sodium Dodecyl Sulfate remains the pivotal reagent that makes high-resolution protein separation by mass a routine and reliable procedure. Its fundamental role in denaturing proteins and standardizing their charge is irreplaceable for standard analytical workflows, from basic research to quality control in drug development. However, an understanding of its limitations is equally important. The emergence of modified techniques like NSDS-PAGE, which can preserve metal cofactors and enzymatic activity, points to a future where electrophoretic methods are increasingly tailored for functional proteomics. For the modern researcher, mastering the principles, applications, and troubleshooting of SDS-PAGE is not merely a technical skill but a foundational competency. This knowledge enables critical evaluation of data, informed selection of the most appropriate separation technique, and contributes to advancements in characterizing complex biological systems and developing novel biotherapeutics.
References
- 1. Protein Electrophoresis Using SDS-PAGE: A Detailed ... [https://www.goldbio.com/blogs/articles/protein-electrophoresis-sds-page-overview?srsltid=AfmBOoqwDRHw_BWHTYxNSwT8ZH3RihU4xkSmUvayiYw6KEORYUOYhMJx]
- 2. Overview of Protein Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 3. SDS-PAGE: A Cornerstone of Protein Analysis in Modern ... [https://www.metwarebio.com/sds-page-protein-analysis/]
- 4. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOooFr3BOFjt07nj0TxsBK4wU6nikwxGdZe_dRxaauQPe9XBSOnSH]
- 5. The principle and method of polyacrylamide gel ... [https://www.mblbio.com/bio/g/support/method/sds-page.html]
- 6. SDS-PAGE [https://en.wikipedia.org/wiki/SDS-PAGE]
- 7. Sodium-Dodecyl-Sulfate-polyacrylamid gel electrophoresis [https://www.hycultbiotech.com/sds-page/]
- 8. Native SDS-PAGE: High Resolution Electrophoretic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4517606/]
- 9. SDS-PAGE guide: Sample preparation to analysis [https://www.abcam.com/en-us/knowledge-center/western-blot/sds-page-guide]
- 10. SDS-PAGE Electrophoresis Market Size & Share 2025-2032 [https://www.360iresearch.com/library/intelligence/sds-page-electrophoresis]
- 11. The Evolution of SDS-Based Protein Separation [https://www.bio-techne.com/resources/blogs/advantages-of-ce-sds]
- 12. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOorj3r-cL9_2BAnz9wk6ctCtKuAg8mahwYcjEC38JrIKWWsl2swo]
- 13. How Does SDS-PAGE Work? A Comprehensive Guide [https://www.metwarebio.com/how-sds-page-works-guide/]
- 14. With an anticipated CAGR of 6.2%, the SDS-PAGE ... [https://www.linkedin.com/pulse/anticipated-cagr-62-sds-page-electrophoresis-market-xpopf]
- 15. Electrophoresis Market Trends, Share and Forecast, 2025- ... [https://www.coherentmarketinsights.com/market-insight/electrophoresis-market-2666]
- 16. How SDS-PAGE Works [https://bitesizebio.com/580/how-sds-page-works/]
- 17. SDS Polyacrylamide Gel Electrophoresis of Proteins [https://pubmed.ncbi.nlm.nih.gov/20512673/]
- 18. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis ... [https://www.sciencedirect.com/science/article/pii/S000326972200207X]
- 19. Application of SDS-PAGE in Protein Characterization [https://www.mtoz-biolabs.com/application-of-sds-page-in-protein-characterization.html]
- 20. SDS protein interactions [https://www.sciencedirect.com/science/article/abs/pii/S0301462225001462]
- 21. Detergents for Cell Lysis and Protein Extraction [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/detergents-cell-lysis-protein-extraction.html]
- 22. Binding of Dodecyl Sulfate to Proteins at High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC283150/]
- 23. The Molecular Basis of the Sodium Dodecyl Sulfate Effect ... [https://www.nature.com/articles/s41598-018-20669-7]
- 24. a binding site for sodium dodecyl sulfate (SDS) in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3335284/]
- 25. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOopLvV_YZvyT7fVmoPFvZMkHqBJDf2qpNAViJ9nEget3dsjFCrHa]
- 26. SDS-PAGE Electrophoresis [https://www.bocsci.com/blog/sds-page-separation-of-proteins-based-on-size/?srsltid=AfmBOoqZ9QDUDMw2Uu2GcPYdQ8aF32M7rZVaKCdUTWonXDUa7KXVsY-2]
- 27. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis ... [https://pubmed.ncbi.nlm.nih.gov/34853120/]
- 28. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoqyQ3nK-950h8v9Q9XdcmPDsAfiCrM8X6YdtcRqB9cHPm7nMfn6]
- 29. Preparing protein samples for sds-page [https://www.ruf.rice.edu/~bioslabs/studies/sds-page/denature.html]
- 30. Dithiothreitol (DTT) Common Applications [https://agscientific.com/blog/dithiothreitol-dtt-applications.html?srsltid=AfmBOoq2Xl0I6PpaQ6RDLlh4y_YzA599igg9kDIxzHQFDqTw6qhXoHWA]
- 31. SDS PAGE [https://cafgroup.lbl.gov/protocols/protein-expression-and-purification/sds-page]
- 32. SDS-PAGE in Food Science: Elevating Protein Insight and ... [https://www.eurofinsus.com/food-testing/resources/sds-page-in-food-science-elevating-protein-insight-and-product-integrity/]
- 33. Overview of Protein Electrophoresis [https://www.thermofisher.com/br/pt/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-electrophoresis.html]
- 34. The Nature of Denaturing (Protein Gels, that is!) [https://bitesizebio.com/20794/the-nature-of-denaturing-protein-gels-that-is/]
- 35. Overview of SDS-PAGE [https://www.creative-proteomics.com/resource/overview-of-sds-page.htm]
- 36. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOordJ9PHL_UmUx7a1Ibw7-EpAjPHcM4hjg92wn4oapbAFMrK4Jhr]
- 37. What Is the Role of SDS in Protein Gel Electrophoresis? [https://synapse.patsnap.com/article/what-is-the-role-of-sds-in-protein-gel-electrophoresis]
- 38. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOopZbFPNMJAkwqKkNyGw2bBZIg2h6sy6aswpxrpRXUa7KwtKlNiG]
- 39. Laemmli Buffer: The 5 Critical Components [https://bitesizebio.com/44540/laemmli-buffer-what-is-it-for-anyway/]
- 40. Ulrich Laemmli's Development of SDS Polyacrylamide Gel ... [https://fnl.mit.edu/ulrich-laemmlis-development-of-sds-polyacrylamide-gel-electrophoresis/]
- 41. Polyacrylamide Gel Electrophoresis, How It Works, ... [https://www.technologynetworks.com/analysis/articles/polyacrylamide-gel-electrophoresis-how-it-works-technique-variants-and-its-applications-359100]
- 42. Polyacrylamide gel electrophoresis [https://en.wikipedia.org/wiki/Polyacrylamide_gel_electrophoresis]
- 43. Steps in Nucleic Acid Gel Electrophoresis [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/na-electrophoresis-education/na-electrophoresis-workflow.html]
- 44. A Guide to Gradient Gels: The Why's and How's [https://bitesizebio.com/47184/gradient-gels/]
- 45. Deciphering food proteins: The applications of SDS-PAGE ... [https://www.sciencedirect.com/science/article/abs/pii/S2212429225003025]
- 46. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoqr1QNo5X3k15gfju86E7ZM32aFRC7uaJdiBY64kYfKtpWo_gBv]
- 47. Principle and Protocol of Sodium Dodecyl Sulphate- ... [https://www.creativebiomart.net/blog/principle-and-protocol-of-sodium-dodecyl-sulphate-polyacrylamide-gel-electrophoresis-sds-page]
- 48. What's the Difference between Running and Transfer Buffer? [https://azurebiosystems.com/blog/electrophoresis-101-the-difference-between-running-and-transfer-buffer/]
- 49. SDS-PAGE SDS Running Buffer (10x) Preparation and ... [https://www.aatbio.com/resources/buffer-preparations-and-recipes/sds-page-10x-sds-running-buffer]
- 50. SDS PAGE [https://www.bostonbioproducts.com/research-and-discovery/sds-page/]
- 51. Revolutionary Advances in SDS-PAGE Technology Drive [https://www.openpr.com/news/4047935/revolutionary-advances-in-sds-page-technology-drive]
- 52. How to Master SDS-PAGE and Western Blot in 5 Easy Steps [https://www.neobiotechnologies.com/resources/sds-page-western-blot/?srsltid=AfmBOooDOq-r_Pc7GlRgGyp_JWCXvIvPsyqVHHzu-viu7R3Utpb--tIe]
- 53. Western blotting guide: Part 2, Protein separation by SDS- ... [https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blotting-guide-part-2/]
- 54. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoq_Sab35UBFU00L6rWt8BIrrvaWNKKRdCX9ASI25KEaq_YBs4PW]
- 55. SDS -PAGE and Western Blotting Techniques [https://pubmed.ncbi.nlm.nih.gov/21340897/]
- 56. The principle and method of Western blotting (WB) [https://www.mblbio.com/bio/g/support/method/westernblotting.html]
- 57. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOooxthuLoLfUVuJZ3Ncf35Ihae2c8d6ZnXI3eXeVDcwAU4sbGOUm]
- 58. 6 Best Tips for Troubleshooting Poor Band Separation on ... [https://azurebiosystems.com/blog/6-tips-for-troubleshooting-poor-band-separation-on-sds-page-gels/]
- 59. Troubleshooting SDS-PAGE Sample Preparation Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-sample-preparation-issues?srsltid=AfmBOorteCPOp7LmuHAiuw1axc-NBcbUoRO85rdAMZXwwF-GdXDNODMa]
- 60. Troubleshooting Sodium Dodecyl Sulfate- Polyacrylamide ... [https://www.hycultbiotech.com/sds-page/troubleshooting/]
- 61. SDS protein interactions [https://pubmed.ncbi.nlm.nih.gov/41086667/]
- 62. Detergent binding explains anomalous SDS-PAGE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2644111/]
- 63. Improved Separation in Horizontal Protein SDS-PAGE with ... [https://www.mdpi.com/2409-9279/6/6/106]
- 64. A Database of Accurate Electrophoretic Migration Patterns ... [https://www.sciencedirect.com/science/article/pii/S0022283622005605]
- 65. How to Choose the Right Gel % for Your Protein Sample [https://nusep.us/blogs/precast-gel-education-center/how-to-choose-the-right-gel-for-your-protein-sample?srsltid=AfmBOoqBfS-Z4EDTeLFcUm4tgyuunt86RGIWNFJ_SQhLqbzBEOQsk_2W]
- 66. Western Blot SDS-PAGE [https://www.novusbio.com/support/support-by-application/western-blot-sds-page?srsltid=AfmBOooJH41qQj59MjY-0VWuZsmLswQNW843RaaJgrjhPXbLqN5x6OhD]
- 67. How current, voltage, and power settings affect SDS-PAGE [https://www.biossusa.com/blogs/news/how-current-voltage-and-power-settings-affect-sds-page?srsltid=AfmBOoqR5hgEx8bDs4n65JVYn6Y3YoMcTy1bt63RzHFUyyvLZtWSiOI6]
- 68. Optimizing Gel Electrophoresis: Best Practices for Sample ... [https://www.gelepchina.com/news/optimizing-gel-electrophoresis-best-practices-for-sample-volume-voltage-and-timing/]
- 69. An Easy SDS-PAGE Gel Recipe & 10-Step Protocol [https://bitesizebio.com/59429/sds-page-gel-recipe/]
- 70. What is the role/function of SDS in SDS-PAGE? [https://www.aatbio.com/resources/faq-frequently-asked-questions/What-is-the-role-function-of-SDS-in-SDS-PAGE]
- 71. 13.11: Lab Technique - SDS-PAGE [https://bio.libretexts.org/Courses/West_Los_Angeles_College/Biotechnology/13%3A_Biotechnology_Lab_Protocols/13.11%3A_Lab_Technique_-_SDS-PAGE]
- 72. SDS-PAGE: The science behind all those bubbles [https://www.antibodiesinc.com/pages/sds-page-demystified?srsltid=AfmBOoprOcfR4_EvWBDTy9ndFhj_TWpnrrbJar3KLLLdsN1b-u4_T9kP]
- 73. Polyacrylamide Gel Electrophoresis (SDS-PAGE) [https://www.protocols.io/view/polyacrylamide-gel-electrophoresis-sds-page-9b7h2rn.html]
- 74. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOoqfCgxTR7fMzrWp0VVY6nLDai0iS69o0Pe5PQN_bNe0gNJY2oeL]
- 75. Protein Electrophoresis Using SDS-PAGE: A Detailed ... [https://www.goldbio.com/blogs/articles/protein-electrophoresis-sds-page-overview?srsltid=AfmBOoqVn_rnDfbbSenjpnGBCpS09cDniC4UR64duMGiThSuU3OcVccq]
- 76. Troubleshooting SDS-PAGE Sample Preparation Issues [https://www.goldbio.com/blogs/articles/troubleshooting-sds-page-sample-preparation-issues?srsltid=AfmBOooQr3rm-HYqRfX5Y1U4m4l07IPj3AJUnF_rEhfpeWkn8KeGyZVt]
- 77. Nucleic Acid Gel Electrophoresis Troubleshooting [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/na-electrophoresis-education/na-electrophoresis-troubleshooting.html]
- 78. Aggregation in drug development: a challenge that can be ... [https://www.fidabio.com/blogs/aggregation-in-drug-development-a-challenge-that-can-be-avoided]
- 79. What is SDS-PAGE? Uses, How It Works & Top Companies ... [https://www.linkedin.com/pulse/what-sds-page-uses-how-works-top-companies-2025-clearsedge-research-kztwf]
- 80. Glycosylation: mechanisms, biological functions and clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11298558/]
- 81. Glycosylation of serine/threonine-rich intrinsically ... [https://www.nature.com/articles/s41467-025-58692-8]
- 82. Dynamic regulation of Sec24C by phosphorylation and O ... [https://www.sciencedirect.com/science/article/pii/S0021925825023063]
- 83. Differences between SDS PAGE and Native PAGE [https://www.geeksforgeeks.org/biology/differences-between-sds-page-and-native-page/]
- 84. éžå˜æ€§èšä¸™çƒ¯é…°èƒºå‡èƒ¶ç”µæ³³(Native-PAGE)的分类 [https://www.biomart.cn/lab-web/news/article/31m31tcgo4t13.html]
- 85. Foolproof Guide to SDS-Page Ladder Migration [https://azurebiosystems.com/blog/sds-page-troubleshooting-protein-separation-but-my-ladder-is-fine/]
- 86. Protein Standards & Ladders [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-gel-electrophoresis/protein-standards-ladders.html]
- 87. High Resolution Clear Native Electrophoresis for In-gel ... [https://www.sciencedirect.com/science/article/pii/S153594762031690X]
- 88. gel band contrast - proteins [https://biology.stackexchange.com/questions/72786/gel-band-contrast]
- 89. Vol 7 No 1 (2025) | Journal of Pharmaceutical and ... [https://www.syncsci.com/journal/JPBR/issue/current]
- 90. SDS-PAGE Protocol [https://www.rockland.com/resources/sds-page-protocol/?srsltid=AfmBOoq49ubyvMyocWEZyn5Rk4NT_B1SC5WhU8iVkJVWPF7S_iLwWY07]