Ancient Guardians of Signaling: Evolutionary Conservation and Clinical Targeting of STAT-type SH2 Domains
This article provides a comprehensive analysis of the STAT-type Src Homology 2 (SH2) domain, tracing its evolutionary origins and exploring its profound structural and functional conservation.
Ancient Guardians of Signaling: Evolutionary Conservation and Clinical Targeting of STAT-type SH2 Domains
Abstract
This article provides a comprehensive analysis of the STAT-type Src Homology 2 (SH2) domain, tracing its evolutionary origins and exploring its profound structural and functional conservation. We detail how this ancient protein module, essential for phosphotyrosine signal transduction, evolved prior to the divergence of plants and animals and served as a template for subsequent SH2 domain diversification. For an audience of researchers and drug development professionals, the review synthesizes foundational knowledge with modern methodological approaches for studying these domains. It further addresses key challenges in the field, offers comparative analyses with other SH2 domain types, and validates their significance through the lens of human genetic constraint and a burgeoning pipeline of clinical inhibitors, ultimately framing STAT-type SH2 domains as high-value therapeutic targets.
Tracing the Deep Evolutionary Roots of STAT-type SH2 Domains
Src homology 2 (SH2) domains represent a cornerstone of phosphotyrosine signaling in eukaryotic organisms. This review examines the evolutionary provenance of SH2 domains, tracing their origin to the early Unikonta and their subsequent expansion alongside protein tyrosine kinases and phosphatases. Genomic analyses across diverse eukaryotic species reveal that SH2 domains first emerged in unicellular organisms at the pre-metazoan boundary, with the transcription factor STAT's linker-SH2 domain identified as one of the most ancient functional versions. The rapid elaboration of SH2 domain-containing proteins alongside developing multicellularity underscores their crucial role in the evolution of complex cell communication networks. This whitepaper synthesizes current understanding of SH2 domain origins, structural diversification, and experimental approaches for their study, providing researchers with essential insights into the evolution of tyrosine kinase signaling networks with implications for therapeutic development.
The Src homology 2 (SH2) domain is a structurally conserved protein domain of approximately 100 amino acids that functions as a phosphotyrosine-specific binding module, facilitating regulated protein-protein interactions in intracellular signaling pathways [1] [2]. SH2 domains recognize and bind to phosphorylated tyrosine residues on target proteins, thereby enabling the transmission of signals controlling diverse cellular functions including proliferation, differentiation, and migration [1] [3]. As the prototypical modular protein-protein interaction domain in tyrosine kinase signaling, SH2 domains play indispensable roles in metazoan cell communication [1] [4].
Understanding the evolutionary origins of SH2 domains provides crucial insights into the development of complex signaling systems in multicellular organisms. The emergence and expansion of SH2 domain-containing proteins coincided with the development of tyrosine kinase-based signaling, representing a key adaptation in the transition from unicellular to multicellular life [5] [4]. This review examines the phylogenetic distribution, structural diversification, and experimental characterization of SH2 domains from their first appearance in early eukaryotes, with particular emphasis on the evolutionary conservation of STAT-type SH2 domains and their implications for modern drug discovery.
Evolutionary Emergence of SH2 Domains
Phylogenetic Distribution Across Eukaryota
Comprehensive genomic analyses of 21 eukaryotic species have revealed that SH2 domains first appeared in the early Unikonta, one of the two major divisions of eukaryotes (including opisthokonts and amoebozoans) [5]. The examination of SH2 domain-containing proteins across Bikonta and Unikonta lineages demonstrates that:
- Earliest Origins: SH2 domains are absent in prokaryotes with rare exceptions in bacterial pathogens like Legionella, which likely acquired them through horizontal gene transfer [6]. The domains first emerged in unicellular eukaryotes approximately 900-1,000 million years ago [5] [4].
- Limited Presence in Bikonta: The Bikonta division, including plants like Arabidopsis thaliana, generally contains very few SH2 domains, with Arabidopsis possessing only two novel genes carrying STAT-type linker-SH2 domains [7].
- Expansion in Unikonta: SH2 domains expanded considerably in the choanoflagellate and metazoan lineages alongside the development of tyrosine kinases, leading to rapid elaboration of phosphotyrosine signaling in early multicellular animals [5].
Table 1: SH2 Domain Distribution Across Representative Eukaryotic Species
| Organism | Group | SH2 Proteins | Key Findings |
|---|---|---|---|
| Homo sapiens (Human) | Metazoa | 111-121 | Maximum expansion; complex signaling networks |
| Monosiga brevicollis (Choanoflagellate) | Choanozoa | ~30 | Intermediate expansion; pre-metazoan lineage |
| Dictyostelium discoideum (Social amoeba) | Amoebozoa | 10+ | Early Unikont with functional pTyr signaling |
| Arabidopsis thaliana (Thale cress) | Viridiplantae | 2 | STAT-type only; limited pTyr signaling |
| Saccharomyces cerevisiae (Yeast) | Fungus | 1 | Minimal SH2 presence |
Coevolution with Tyrosine Kinases and Phosphatases
The evolutionary expansion of SH2 domains occurred in tight coordination with the development of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), forming the essential triad of phosphotyrosine signaling [5] [4]. Analysis across unicellular and multicellular Unikonts reveals a striking correlation (r = 0.95) between the percentage of PTKs and SH2 domains in their respective genomes [5]. This coevolution suggests coordinated emergence and increasing sophistication of phosphotyrosine signaling during eukaryotic evolution.
The essential triad consists of:
- Protein Tyrosine Kinases (PTKs): "Writers" that phosphorylate tyrosine residues
- Protein Tyrosine Phosphatases (PTPs): "Erasers" that dephosphorylate tyrosine residues
- SH2 Domains: "Readers" that recognize and bind phosphorylated tyrosines [3]
This coordinated system enabled the development of complex, dynamic signaling networks essential for metazoan multicellularity and tissue specialization [4].
Figure 1: Evolutionary Origin and Expansion of SH2 Domains in Eukaryotes
Structural Phylogeny and the Ancient STAT-Type SH2 Domain
Structural Classification of SH2 Domains
SH2 domains share a conserved structural fold characterized by a central antiparallel β-sheet flanked by two α-helices, creating binding pockets for phosphotyrosine recognition [2] [3]. Despite this conserved architecture, SH2 domains can be divided into two major structural subgroups:
- Src-Type SH2 Domains: Characterized by a basic "αβββα" structure with an extra β-strand (βE or βE-βF motif). These represent the most prevalent SH2 structure in metazoans [7] [2].
- STAT-Type SH2 Domains: Distinct in that they lack the βE and βF strands and contain a split αB helix with an additional αB' motif. This structural adaptation facilitates dimerization, a critical step in STAT-mediated transcriptional regulation [7] [2].
The STAT-Type SH2 Domain as an Ancient Template
Secondary structural alignment and phylogenetic analysis reveal that the linker-SH2 domain of the transcription factor STAT represents one of the most ancient and fully developed functional SH2 domains [7]. Key evidence supporting this conclusion includes:
- Broad Phylogenetic Distribution: STAT-type SH2 domains are found in diverse eukaryotic lineages, including plants, amoebozoans, and metazoans, suggesting an origin prior to the divergence of plants and animals [7].
- Ancestral Structural Features: The STAT-type SH2 domain lacks the more complex C-terminal structural elements of Src-type domains, representing a more primordial architecture that may have served as a template for SH2 domain evolution [7] [2].
- Conserved Functional Role: STAT proteins function in transcriptional regulation, a role that has been conserved from social amoebae like Dictyostelium to humans, indicating the early integration of SH2 domains into nuclear signaling pathways [2].
Table 2: Comparative Features of Src-Type vs. STAT-Type SH2 Domains
| Structural Feature | Src-Type SH2 Domains | STAT-Type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB with additional β-strands | αA-βB-βC-βD-αB with split αB helix |
| βE and βF Strands | Present | Absent |
| C-terminal Adjoining Loop | Present | Absent |
| Dimerization Capability | Limited | Enhanced; critical for STAT function |
| Evolutionary Appearance | Later diversification | Early emergence; ancestral form |
| Representative Examples | Src, Fyn, Grb2 | STAT transcription factors |
Experimental Approaches for Studying SH2 Domain Evolution and Function
Genomic Identification and Classification
The identification and classification of SH2 domains across diverse organisms relies on bioinformatic approaches using predictive algorithms such as:
- Protein Families Database (Pfam): Uses hidden Markov models to identify domain signatures [5] [4]
- Simple Modular Architecture Research Tool (SMART): Identifies protein domains and analyzes domain architectures [5]
- Conserved Domains Database (CDD): Provides functional annotations of conserved domains [4]
These tools enable researchers to systematically identify SH2 domain-containing proteins by scanning genomic sequences, followed by multiple sequence alignment and phylogenetic analysis to classify SH2 domains into distinct families and trace their evolutionary trajectories [5] [4].
Affinity Profiling and Specificity Determination
Understanding SH2 domain function requires characterization of their binding specificities and affinities. Recent methodological advances include:
- Peptide Library Display: Bacterial or phage display of random phosphopeptide libraries coupled with next-generation sequencing enables high-throughput profiling of SH2 domain binding specificities [8].
- Quantitative Affinity Models: Computational approaches like ProBound can analyze selection data to generate quantitative sequence-to-affinity models that predict binding free energies across theoretical ligand sequence space [8].
- Position-Specific Scoring Matrices (PSSMs): Traditional approach for distinguishing binding specificities using score thresholds [8].
These methods have revealed that SH2 domains typically exhibit moderate binding affinities (Kd = 0.1-10 μM), which is crucial for allowing transient interactions required for dynamic signaling responses [8] [3].
Figure 2: Experimental Workflow for SH2 Domain Specificity Profiling
Structural Characterization Techniques
Elucidation of SH2 domain structures and their binding mechanisms employs:
- X-ray Crystallography: To date, the structures of approximately 70 unique SH2 domains have been experimentally solved, providing atomic-level insights into phosphopeptide recognition [2] [3].
- Comparative Structural Analysis: Systematic comparison of SH2 domain structures reveals conserved features and variations that underlie functional diversification [2].
- Evolutionary Trace Analysis: Mapping of conserved residues and functional sites across SH2 domain families to identify critical structural determinants [9].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for SH2 Domain Investigation
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Bioinformatics Tools | Pfam, SMART, CDD | Identification and classification of SH2 domains in genomic sequences |
| Peptide Display Systems | Bacterial display, Phage display | High-throughput profiling of SH2 domain binding specificities |
| Quantitative Modeling Software | ProBound | Generation of sequence-to-affinity models from selection data |
| Structural Biology Resources | X-ray crystallography, NMR | Determination of SH2 domain structures and binding mechanisms |
| SH2 Domain Constructs | Wild-type and mutant SH2 domains | Functional characterization of binding specificity and affinity |
| Peptide Libraries | Random phosphopeptide libraries, Proteome-derived peptides | Comprehensive profiling of SH2 domain binding landscapes |
| NDM-1 inhibitor-5 | NDM-1 inhibitor-5, MF:C24H23NO4, MW:389.4 g/mol | Chemical Reagent |
| Mcl-1 inhibitor 16 | Mcl-1 inhibitor 16, MF:C25H29Cl2N3Pt, MW:637.5 g/mol | Chemical Reagent |
The evolutionary history of SH2 domains reveals their crucial role in the development of complex cell signaling systems in eukaryotes. From their first appearance in early Unikonta to their expansion and diversification in metazoans, SH2 domains have coevolved with tyrosine kinases and phosphatases to enable sophisticated phosphotyrosine-based communication networks. The STAT-type SH2 domain stands out as an ancient template that predates the plant-animal divergence and has been conserved in its role in transcriptional regulation.
Understanding the evolutionary origins and structural diversification of SH2 domains has significant implications for biomedical research and drug development. Many human diseases, including cancer, immunodeficiencies, and metabolic disorders, involve mutations in SH2 domain-containing proteins or dysregulation of phosphotyrosine signaling pathways [5] [2]. The insights gained from evolutionary studies of SH2 domains can inform the development of targeted therapeutics that exploit natural structural variations and specificity determinants. Furthermore, the identification of bacterial SH2 domains in pathogens like Legionella reveals how microbes have hijacked eukaryotic signaling components, opening new avenues for antimicrobial development [6].
As research continues to unravel the complexities of SH2 domain evolution and function, integrating phylogenetic, structural, and biochemical approaches will be essential for comprehending their roles in health and disease and for harnessing this knowledge for therapeutic innovation.
The Src Homology 2 (SH2) domain represents a crucial protein interaction module that recognizes phosphotyrosine motifs in eukaryotic signal transduction pathways. While SH2 domains are prevalent in metazoans, their presence in simpler organisms provides critical insights into the evolutionary origins of phosphotyrosine signaling. This whitepaper synthesizes evidence from genomic studies of plants and amoebae to demonstrate that the STAT-type SH2 domain represents an ancient structural template that predates the divergence of plants and animals. We present comparative structural analysis, experimental data from diverse eukaryotic models, and quantitative genomic findings that establish the early emergence and functional conservation of STAT-type SH2 domains across evolutionary boundaries. The conservation of these domains in organisms lacking sophisticated tyrosine kinase networks suggests their fundamental role in the early development of eukaryotic signaling systems.
Src Homology 2 (SH2) domains are structurally conserved protein modules of approximately 100 amino acids that specifically bind to phosphorylated tyrosine residues, thereby facilitating regulated protein-protein interactions in intracellular signaling pathways [1] [2]. These domains constitute essential components of the phosphotyrosine signaling triad, working in concert with protein tyrosine kinases (PTKs) as "writers" and protein tyrosine phosphatases (PTPs) as "erasers" of phosphorylation marks [4]. The human genome encodes approximately 110-120 SH2 domains contained within 115 proteins, representing one of the largest families of phosphotyrosine recognition modules [1] [5].
Evolutionary analyses reveal that SH2 domains first emerged in the early Unikonta, with subsequent expansion coinciding with the development of multicellularity in metazoans [4] [5]. The number of SH2 domains correlates strongly with organismal complexity, ranging from a single SH2 domain in Saccharomyces cerevisiae to over 100 in humans [5]. This expansion occurred alongside the diversification of tyrosine kinases, suggesting coordinated evolution of phosphotyrosine signaling components [4]. SH2 domains are structurally classified into two major subgroups: Src-type and STAT-type, distinguished by characteristic structural features in their C-terminal regions [10] [7] [2].
STAT-type SH2 Domains: Structural Hallmarks and Classification
STAT-type SH2 domains exhibit distinctive structural characteristics that differentiate them from Src-type SH2 domains. While both share the conserved central antiparallel β-sheet flanked by two α-helices (the αβββα motif), STAT-type domains are characterized by unique C-terminal structural elements [10] [7].
Defining Structural Features
The STAT-type SH2 domain contains a split αB helix and lacks the βE and βF strands present in Src-type SH2 domains [2]. Instead, STAT-type domains feature an additional α-helix (αB') in the evolutionary active region (EAR) at the C-terminus [10]. This structural configuration creates a continuous binding surface that facilitates both phosphopeptide binding and STAT dimerization through reciprocal SH2-phosphotyrosine interactions [10] [2]. The N-terminal region of STAT-type SH2 domains is highly conserved and contains the deep phosphate-binding pocket with the invariant arginine residue at position βB5 that forms critical salt bridges with the phosphotyrosine moiety [2].
Functional Implications of STAT-type Architecture
The unique structural organization of STAT-type SH2 domains supports their dual functionality in both phosphopeptide recognition and STAT dimerization [10] [2]. This integrated architecture enables STAT proteins to function as both signal transducers and transcription factors, with SH2-mediated dimerization representing a critical regulatory step in canonical STAT activation pathways [10]. The structural flexibility observed in STAT-type SH2 domains, particularly in the phosphate-binding pocket, may facilitate allosteric regulation and contribute to the dynamic range of STAT signaling responses [10].
Table 1: Structural Comparison of STAT-type versus Src-type SH2 Domains
| Structural Feature | STAT-type SH2 Domain | Src-type SH2 Domain |
|---|---|---|
| Core Structure | αβββα motif | αβββα motif |
| C-terminal Elements | αB' helix | βE and βF strands |
| Dimerization Capability | Direct participation in STAT dimerization | Primarily phosphopeptide binding |
| Binding Specificity | Moderate (Kd 0.1-10 μM) | Moderate to high (Kd 0.1-10 μM) |
| Evolutionary Appearance | Early eukaryotes | Later in metazoans |
| Representative Proteins | STATs, plant STATL proteins | Src, Grb2, ZAP70 |
Genomic Evidence from Amoebozoan Organisms
Dictyostelium discoideum as a Model System
The social amoeba Dictyostelium discoideum represents a pivotal model for understanding the early evolution of SH2 domain function. Genomic analyses reveal that Dictyostelium possesses 13 SH2 domain-containing proteins, a notable expansion compared to unicellular eukaryotes like yeast but considerably fewer than metazoans [11] [5]. This intermediate number positions Dictyostelium as a crucial evolutionary link in the development of phosphotyrosine signaling systems.
The Dictyostelium genome encodes STAT-type SH2 proteins that function in transcriptional regulation during the multicellular stage of its life cycle [11] [2]. Specifically, the CudA protein contains a STAT-like DNA-binding domain upstream of an SH2 domain and regulates prespore gene expression, including the cotC spore coat protein gene [11]. Chromatin immunoprecipitation analyses demonstrate direct binding of CudA to the cotC promoter, establishing its function as a transcription factor [11]. This configuration parallels metazoan STAT proteins, suggesting an ancient evolutionary origin for this architectural motif.
Expansion of Amoebozoan STAT Proteins
Beyond Dictyostelium, genomic studies have identified STAT proteins in other Amoebozoan lineages, including Acanthamoeba castellanii and various slime molds [12]. Acanthamoeba castellanii STAT protein contains domains similar to Dictyostelium STAT proteins: a coiled coil region, STAT DNA-binding domain, and SH2 domain [12]. Phylogenetic analyses reveal four distinct clades of STAT proteins within slime molds, with Acanthamoeba STAT branching alongside Mycetozoa STATc proteins [12]. This phylogenetic distribution demonstrates that STAT proteins form a monophyletic lineage within Amoebozoa, separate from other eukaryotic groups [12].
Table 2: SH2 Domain Distribution in Selected Eukaryotic Organisms
| Organism | Classification | Total SH2 Proteins | STAT-type SH2 Proteins | Reference |
|---|---|---|---|---|
| Homo sapiens | Metazoa | 115 | 5 (STAT1-5, plus others) | [1] [10] |
| Dictyostelium discoideum | Amoebozoa | 13 | Multiple (including CudA, STATa) | [11] [5] |
| Acanthamoeba castellanii | Amoebozoa | Not quantified | 1 (STAT protein) | [12] |
| Arabidopsis thaliana | Plantae | 2 | 2 (AtSHA, AtSHB) | [11] [7] |
| Oryza sativa | Plantae | 1 | 1 (OsSHA) | [11] |
| Saccharomyces cerevisiae | Fungi | 1 | 0 | [5] |
Plant Genomes Encode STAT-like SH2 Domain Proteins
Identification of Plant STAT-type SH2 Domains
Genomic analyses of plant species have revealed the presence of STAT-type SH2 domains in both vascular and non-vascular plants [11] [7]. Arabidopsis thaliana encodes two proteins containing SH2 domains (AtSHA and AtSHB), while Oryza sativa (rice) encodes a single such protein (OsSHA) [11]. These plant SH2 domain-containing proteins were initially enigmatic, as they lacked readily identifiable DNA-binding domains in initial annotations [11].
Secondary structure prediction and comparative sequence analysis demonstrated that these plant proteins contain STAT-type SH2 domains with an associated linker region but lack the characteristic N-terminal domains of metazoan STAT proteins [7]. These plant STAT-type proteins have been designated STATL (STAT-type linker-SH2 domain) factors [7]. The conservation of the linker-SH2 domain architecture across plants and animals suggests this structural motif represents an ancient evolutionary template that predates the divergence of these kingdoms [7].
DNA Binding Specificity Conservation
Remarkably, the CudA protein from Dictyostelium recognizes DNA sequences with half-sites (GAA) identical to metazoan STAT binding sites, though with reversed orientation of the dyad symmetry [11]. This conservation of DNA recognition specificity across evolutionary boundaries provides compelling evidence for the deep evolutionary origin of STAT-type DNA binding and its functional association with SH2 domains. The CudA protein forms homodimers via its SH2 domain, mirroring the dimerization mechanism of metazoan STAT proteins [11].
Experimental Methodologies for STAT-type SH2 Domain Characterization
Genomic Identification and Bioinformatics Approaches
The identification of STAT-type SH2 domains in diverse organisms employs sophisticated bioinformatic pipelines combining sequence analysis with secondary structure prediction [7]. Primary sequence alignment alone often fails to identify divergent SH2 domains due to sequence degeneration, necessitating complementary structural approaches.
Protocol 1: Secondary Structure-Based SH2 Domain Identification
- Sequence Retrieval: Compile protein sequences from target genomes using standardized annotation pipelines [13].
- Hidden Markov Model Screening: Scan proteomes with established SH2 domain profiles from Pfam, SMART, and Conserved Domains Database [4] [5].
- Secondary Structure Prediction: Apply algorithms such as PSIPRED or JPRED to predict secondary structural elements [7].
- Structural Alignment: Map predicted secondary structures against known SH2 domain architectures, focusing on the conserved αβββα core motif [7].
- Classification: Differentiate STAT-type from Src-type based on C-terminal elements (αB' helix versus βE-βF strands) [7] [2].
- Phylogenetic Analysis: Construct phylogenetic trees to establish evolutionary relationships among identified SH2 domains [12] [5].
Functional Characterization of STAT-type SH2 Domains
Protocol 2: DNA Binding and Transcriptional Function Analysis
Chromatin Immunoprecipitation (ChIP):
- Develop Dictyostelium to appropriate developmental stage (e.g., standing slug/first-finger stage) [11].
- Cross-link proteins to DNA with formaldehyde.
- Immunoprecipitate chromatin with anti-CudA antibody [11].
- Analyze precipitated DNA by PCR with promoter-specific primers (e.g., cotC promoter: 5'-CCCATACTACATTAAAATATTTG-3' and 5'-TCATATGCTTGTGTGTTGGG-3') [11].
DNA Affinity Chromatography:
- Prepare nuclear extracts from slug-stage cells by sonication in DB buffer (50 mM KPO4 pH 7.5, 10% glycerol, 0.5 mM EDTA, 0.1 mM ZnCl2, 0.1 mM MgCl2, 0.01% Brij 35) with 0.1 M NaCl and protease inhibitors [11].
- Concatemerize wild-type or mutated cotC 4×14-mer sequence element (5'-gatcTGAGAATTTTCTATTGAGAATTTTCTATTGAGAATTTTCTATTGAGAATTTTCTAT-3') and couple to CNBr-Sepharose 4B [11].
- Incubate nuclear extract with oligonucleotide-coupled Sepharose.
- Wash with DB buffer containing 0.1 M NaCl and elute with 0.4 M NaCl [11].
- Analyze bound protein by western blot with specific antibodies [11].
Band Shift Analysis:
- Express and purify recombinant STAT-type proteins (e.g., histidine-tagged ECudA in pET15b) [11].
- Label oligonucleotides with [α-32P]dATP or Cy5-dCTP.
- Incubate purified protein with labeled DNA probes.
- Separate protein-DNA complexes by native gel electrophoresis [11].
- Visualize using appropriate detection methods (phosphorimaging or infrared scanning) [11].
Structural Analysis of STAT-type SH2 Domains
Protocol 3: Structural Characterization Approaches
X-ray Crystallography:
- Express and purify recombinant STAT-type SH2 domains.
- Crystallize using vapor diffusion methods.
- Collect diffraction data and solve structures by molecular replacement using known SH2 domain structures as search models [10].
Analysis of Disease-Associated Mutations:
Visualization of Evolutionary Relationships and Domain Architectures
Diagram 1: Evolutionary Relationships of STAT-type SH2 Domains Across Eukaryotes
Diagram 2: Comparative Domain Architecture of STAT-type Proteins
Table 3: Key Research Reagents for STAT-type SH2 Domain Investigation
| Reagent/Resource | Function/Application | Example Implementation |
|---|---|---|
| Anti-CudA Antibody | Immunoprecipitation and chromatin immunoprecipitation | Dictyostelium nuclear protein detection and DNA binding studies [11] |
| cotC Promoter Probes | DNA binding assays | 4×14-mer sequence element for affinity chromatography and band shift analysis [11] |
| pET15b Expression Vector | Recombinant protein production | Histidine-tagged ECudA protein expression for in vitro studies [11] |
| CNBr-Sepharose 4B | DNA affinity chromatography | Immobilization of concatemerized DNA sequences for protein binding studies [11] |
| STAT SH2 Domain Crystallization Kits | Structural studies | Optimization of crystallization conditions for X-ray diffraction [10] |
| Phosphotyrosine Peptide Libraries | Binding specificity profiling | Screening SH2 domain binding preferences and specificity determinants [2] |
| Dictyostelium Knockout Strains | Functional analysis in vivo | cudA-null strains for phenotypic and gene expression studies [11] |
The cumulative evidence from plant and amoebozoan genomes establishes that STAT-type SH2 domains represent an ancient structural template that predates the divergence of major eukaryotic lineages. The conservation of domain architecture, DNA binding specificity, and dimerization mechanisms across evolutionary boundaries underscores the fundamental importance of this structural motif in eukaryotic signaling systems. These findings reposition STAT-type SH2 domains as primordial components of phosphotyrosine signaling rather than metazoan innovations.
From a drug discovery perspective, the ancient origin and structural conservation of STAT-type SH2 domains highlight their potential as therapeutic targets. The unique features of STAT-type domains, particularly their role in dimerization and DNA binding, offer opportunities for selective intervention in pathological signaling pathways. Understanding the evolutionary constraints on these domains may inform the development of targeted therapies with reduced off-target effects, particularly in oncology and immunology where STAT signaling is frequently dysregulated. Further exploration of STAT-type SH2 domains in diverse eukaryotic models will continue to reveal fundamental principles of signal transduction evolution and identify new avenues for therapeutic intervention.
The Src homology 2 (SH2) domain represents a fundamental modular unit in eukaryotic cellular signaling, specializing in phosphotyrosine (pTyr) recognition. While the canonical SH2 structure is well-characterized, recent evolutionary and structural analyses have revealed a distinct architectural subclass: the linker-SH2 domain of Signal Transducers and Activators of Transcription (STAT) proteins. This whitepaper delineates the unique structural blueprint of the STAT-type linker-SH2 domain, contrasting it with the canonical Src-type architecture. We frame these findings within the broader context of evolutionary conservation, demonstrating that the linker-SH2 domain predates the divergence of plants and animals and serves as a template for SH2 domain evolution. The analysis incorporates quantitative structural data, detailed experimental protocols for domain characterization, and discusses implications for targeted therapeutic development.
Src homology 2 (SH2) domains are approximately 100-amino-acid modular protein domains that mediate specific protein-protein interactions by recognizing and binding to phosphotyrosine (pTyr) containing motifs [4] [14]. These domains are fundamental components of intracellular signaling networks, defining specificity in phosphotyrosine signaling pathways that regulate critical cellular processes including growth, proliferation, differentiation, and immune responses [4] [15]. The human genome encodes approximately 111 SH2 domain-containing proteins, highlighting their extensive role in coordinating complex signaling networks [4].
Evolutionarily, SH2 domains expanded alongside protein-tyrosine kinases (PTKs) to coordinate cellular and organismal complexity throughout the evolution of the unikont branch of eukaryotes [4]. Examination of conserved PTK and SH2 domain protein families provides fiduciary marks that trace the developmental landscape for complex cellular systems in proto-metazoan and metazoan lineages. The evolutionary provenance of these families reveals how diversity is achieved through tissue-specific gene transcription, altered ligand binding, insertions of linear motifs, and domain gains or losses following gene duplication [4].
This review focuses on a specialized architectural variant: the linker-SH2 domain of STAT proteins. We provide a comprehensive structural and functional analysis of this unique domain architecture, situating it within evolutionary conservation research and highlighting its implications for targeted drug development.
Structural Anatomy of SH2 Domains: Canonical versus STAT-Type Architectures
The Canonical SH2 Domain Fold
The canonical SH2 domain structure consists of a central three-stranded antiparallel beta-sheet flanked by two alpha-helices, forming a characteristic "sandwich" structure [2]. The primary structural elements follow the pattern βA-αA-βB-βC-βD-αB, with most SH2 domains containing additional beta strands (βE, βF, βG) to form a total of seven core secondary structure elements [16] [2]. The N-terminal region is highly conserved and contains a deep pocket within the βB strand that binds the phosphate moiety of phosphotyrosine [2].
A defining feature of this pocket is the invariant arginine at position βB5 (the fifth residue of the βB strand), which forms part of the highly conserved "FLVR" or "FLVRES" motif [14] [2]. This arginine directly coordinates the phosphotyrosine residue through a salt bridge, contributing significantly to binding energy [14]. The C-terminal region of SH2 domains is more variable and contains determinants for specificity, recognizing residues C-terminal to the phosphotyrosine, typically at the +3 position [17] [14]. This creates the characteristic "two-pronged plug" interaction between the domain and its pTyr peptide ligand [14].
The Unique STAT-Type Linker-SH2 Architecture
In contrast to the canonical SH2 architecture, the STAT-type linker-SH2 domain exhibits distinct structural modifications essential for its specialized function in signal transduction and transcription. Comparative structural analysis reveals fundamental differences:
Table 1: Structural Comparison of Src-type and STAT-type SH2 Domains
| Structural Feature | Src-Type SH2 Domain | STAT-Type Linker-SH2 Domain |
|---|---|---|
| Core Secondary Structure | βA-αA-βB-βC-βD-αB with additional βE, βF, βG strands | βA-αA-βB-βC-βD-αB, lacks βE and βF strands |
| C-terminal Region | Contains βE-βF motif | Features αB' motif instead of βE-βF |
| αB Helix Configuration | Single continuous helix | Split into two helices |
| Dimerization Capability | Limited | Enhanced, facilitates STAT dimerization |
| Evolutionary Origin | Later development | Ancient, predates plant-animal divergence |
The STAT-type SH2 domain lacks the βE and βF strands present in Src-type domains and instead incorporates a unique αB' motif [7] [2]. This structural disparity represents an adaptation that facilitates STAT dimerization—a critical step in STAT-mediated transcriptional regulation [18] [2]. This architecture reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity, as organisms like Dictyostelium employ SH2 domain/phosphotyrosine signaling for transcriptional regulation [2].
The following diagram illustrates the key structural differences between these two SH2 domain architectures:
Evolutionary Conservation of the Linker-SH2 Architecture
Phylogenetic Evidence for Ancient Origin
The linker-SH2 domain of STAT proteins represents one of the most ancient and fully developed functional domains, serving as a template for the continuing evolution of the SH2 domain essential for phosphotyrosine signal transduction [7]. Research employing secondary structural alignment to characterize SH2 domains across eukaryotic model systems has revealed:
- Pre-divergence Origin: The linker-SH2 architecture existed prior to the divergence of plants and animals [7]. Two novel genes carrying the STAT-type linker-SH2 domain were cloned from Arabidopsis and designated as STAT-type linker-SH2 domain factors (STATL) [7].
- Broad Phylogenetic Distribution: These STATL factors are found in a wide array of vascular and nonvascular plants, indicating deep evolutionary conservation [7].
- Ancestral Template Role: The structural blueprint of the linker-SH2 domain appears to have served as an evolutionary template from which other SH2 domain variants diversified [7].
Sequence and Structural Conservation Patterns
Analysis of evolutionary conservation patterns across SH2 domains reveals critical conserved residues and structural motifs:
Table 2: Evolutionarily Conserved Features in SH2 Domains
| Feature | Conservation Pattern | Functional Significance |
|---|---|---|
| FLVR Motif (βB5 Arginine) | Near universal conservation; absent in only 3 of 120+ human SH2 domains [14] | Provides ~50% of binding free energy; specificity for pTyr over pSer/pThr [14] |
| pTyr Binding Pocket | High conservation of basic residues at positions αA2 and βD6 [14] | Coordinated phosphotyrosine recognition; defines Src-like (αA2 basic) vs. SAP-like (βD6 basic) classes [14] |
| Core β-sheet Structure | Conserved βA-βB-βC-βD arrangement across all SH2 domains [16] [2] | Maintains structural integrity of the phosphotyrosine binding pocket |
| Linker-αB' Region (STAT-type) | Conservation in STAT proteins across metazoans [7] [2] | Facilitates STAT dimerization and nuclear translocation |
The conservation of the FLVR arginine (βB5) is particularly remarkable, with mutation studies showing it can cause a 1,000-fold reduction in binding affinity [14]. This highlights the critical structural and functional constraints that have shaped SH2 domain evolution.
Experimental Approaches for Characterizing Linker-SH2 Architecture
Structural Determination and Analysis Protocols
X-ray Crystallography of SH2 Domain Complexes
- Purpose: Determine high-resolution structures of SH2 domains in complex with phosphotyrosine peptides
- Protocol:
- Express and purify recombinant SH2 domains (typically as GST-fusion proteins)
- Co-crystallize with synthetic phosphopeptides corresponding to known binding motifs
- Collect diffraction data and solve structures using molecular replacement
- Analyze binding interfaces focusing on pTyr pocket and specificity determinants
- Key Insights: Structures reveal the conserved "two-pronged plug" binding mechanism and structural variations between SH2 types [17] [14]
Secondary Structure Prediction and Alignment
- Purpose: Identify divergent SH2 domains through structural bioinformatics
- Protocol:
- Perform multiple sequence alignment of SH2 domain sequences
- Apply secondary structure prediction algorithms (e.g., Jpred, PSIPRED)
- Create two-dimensional structural alignments focusing on core elements
- Classify domains as Src-type or STAT-type based on presence/absence of βE-βF strands and αB' motif
- Key Insights: This approach enabled identification of STAT-type domains in Arabidopsis and expansion of putative SH2 domain genes in Dictyostelium [7]
Binding Affinity and Specificity Assays
Free Energy Calculations of SH2-Peptide Interactions
- Purpose: Quantitatively characterize binding specificity and affinity
- Protocol:
- Select SH2 domains representing different structural classes (Lck, Grb2, Cbl, p85αN, Stat1)
- Perform molecular dynamics simulations based on crystal structures
- Calculate absolute binding free energies using potential of mean force (PMF) methods
- Compare affinities of SH2 domains for different peptide motifs
- Key Insights: For three of five SH2 domains studied, computational results ranked native peptides as the most preferred binding motif [17]
Population Constraint Analysis with Missense Enrichment Score (MES)
- Purpose: Residue-level analysis of evolutionary and population constraint
- Protocol:
- Map population missense variants (gnomAD) to protein domain families (Pfam)
- Calculate Missense Enrichment Score (MES) quantifying constraint at each site
- Classify residues as missense-depleted (constrained), enriched, or neutral
- Correlate with evolutionary conservation and structural features
- Key Insights: Missense-depleted sites are enriched in buried residues and binding sites, revealing structural constraints [9]
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Linker-SH2 Domain Studies
| Reagent / Resource | Function / Application | Key Features / Examples |
|---|---|---|
| Recombinant SH2 Domains | Structural and biophysical studies; binding assays | GST-tagged domains for purification; point mutants (e.g., FLVR arginine mutants) [14] |
| Phosphotyrosine Peptide Libraries | Specificity profiling; binding motif identification | Diverse pY-containing peptides; positional scanning libraries [17] |
| Structural Biology Resources | SH2 domain structure determination | Crystallization screens; homology modeling templates (PDB: 1LKK, 1JYR, 1YVL) [17] |
| Computational Tools | Binding free energy calculations; structural analysis | Molecular dynamics simulations; implicit solvent models [17] |
| Population Variant Databases | Constraint analysis; pathogenicity assessment | gnomAD for missense variants; ClinVar for pathogenic mutations [9] |
| HMG-CoA Reductase-IN-1 | HMG-CoA Reductase-IN-1, MF:C27H29N3O7, MW:507.5 g/mol | Chemical Reagent |
| Val-Ala-PABC-Exatecan | Val-Ala-PABC-Exatecan, MF:C40H43FN6O8, MW:754.8 g/mol | Chemical Reagent |
Functional Implications in JAK/STAT Signaling and Therapeutic Targeting
Role in JAK/STAT Signaling Pathway
The unique linker-SH2 architecture of STAT proteins is essential for their function in the JAK/STAT signaling pathway, a critical pathway implicated in various diseases including cancer and autoimmune disorders [15]. The specialized structure enables:
- Receptor Association: SH2 domains mediate specific association between STATs and the cytoplasmic domains of cytokine receptors [18]
- Dimerization: Following phosphorylation by JAK kinases, STAT SH2 domains facilitate homo- and heterodimerization through reciprocal interactions with phosphotyrosine residues [18]
- Nuclear Translocation: STAT dimers translocate to the nucleus where they regulate transcription of target genes [15]
The following diagram illustrates the central role of the SH2 domain in JAK/STAT signaling:
Therapeutic Targeting Strategies
The critical role of STAT linker-SH2 domains in signaling pathways has made them attractive therapeutic targets. Several targeting strategies have emerged:
- Direct SH2 Domain Inhibitors: Small molecules designed to block phosphotyrosine binding pocket, preventing STAT dimerization and activation [2]
- Allosteric Modulators: Compounds targeting alternative surfaces of the SH2 domain to modulate function [2]
- Lipid-Binding Disruptors: Targeting cationic lipid-binding regions adjacent to pTyr-binding pockets in SH2 domains [2]
- Protein-Protein Interaction Inhibitors: Disrupting the multivalent interactions that drive phase separation in signaling condensates [2]
Recent research indicates that targeting lipid binding in SH2 domain-containing kinases may offer a promising avenue for developing small-molecule drugs, with successful development of nonlipidic inhibitors of Syk kinase demonstrating this approach [2].
The structural blueprint of the unique linker-SH2 architecture represents a fascinating example of evolutionary conservation coupled with functional specialization. STAT-type SH2 domains, with their distinctive lack of βE-βF strands and characteristic αB' motif, represent an ancient architectural variant that has been conserved from plants to humans. This conserved structure enables the specialized function of STAT proteins in signal transduction and transcriptional regulation through facilitated dimerization.
Understanding these structural nuances provides critical insights for therapeutic development, particularly for targeting the JAK/STAT pathway in cancer and autoimmune diseases. The experimental approaches outlined—from structural determination to binding analysis and population constraint studies—provide researchers with robust methodologies for further characterizing these important domains. As structural biology techniques advance and our understanding of allosteric mechanisms deepens, the unique linker-SH2 architecture will continue to offer valuable insights into the evolution of signaling systems and opportunities for targeted therapeutic intervention.
Co-evolution with Tyrosine Kinases and Phosphatases
Phosphotyrosine (pTyr) signaling is a cornerstone of cellular communication in multicellular organisms, governing critical processes such as cell proliferation, differentiation, and immune response [19] [4]. This sophisticated signaling system relies on a fundamental triad of components: protein tyrosine kinases (PTKs) that "write" the phosphorylation mark, protein tyrosine phosphatases (PTPs) that "erase" it, and Src homology 2 (SH2) domains that "read" the signal by binding to phosphorylated tyrosine residues [4] [20]. The co-evolution of these three components has been crucial for the development of metazoan complexity, facilitating the emergence of intricate cell communication networks necessary for tissue specialization and developmental programming [19] [5].
SH2 domains are protein interaction modules that specifically recognize pTyr-containing sequences, with the human genome encoding approximately 111 SH2 domain-containing proteins [5] [20]. The evolutionary expansion of SH2 domains alongside their catalytic counterparts represents a fascinating case of molecular co-evolution that mirrors increasing organismal complexity. This review examines the mechanistic basis and functional consequences of this co-evolutionary relationship, with particular emphasis on its implications for STAT-type SH2 domains and their role in health and disease.
Evolutionary Provenance of SH2 Domains and Their Catalytic Partners
Origin and Expansion of the pTyr Signaling System
The pTyr signaling system is a relatively recent evolutionary innovation compared to more primordial post-translational modifications such as Ser/Thr phosphorylation. Comprehensive genomic analyses across 21 eukaryotic species reveal that SH2 domains first emerged in the early Unikonta, with subsequent expansion occurring in the choanoflagellate and metazoan lineages [5].
Table 1: Evolutionary Expansion of pTyr Signaling Components Across Select Organisms
| Organism | SH2 Domain Proteins | Protein Tyrosine Kinases (PTKs) | Correlation Coefficient |
|---|---|---|---|
| H. sapiens (Human) | 111 | ~90 | 0.95 |
| M. musculus (Mouse) | 110 | ~88 | 0.95 |
| D. melanogaster (Fruit fly) | 43 | 32 | 0.95 |
| C. elegans (Roundworm) | 47 | 38 | 0.95 |
| M. brevicollis (Choanoflagellate) | 13 | 13 | 0.95 |
| S. cerevisiae (Yeast) | 1 | 0 | 0.95 |
The correlation between PTK and SH2 domain numbers across diverse organisms is striking (r = 0.95), indicating their coordinated expansion throughout evolution [5]. This parallel diversification suggests strong selective pressure to maintain balanced "writer-reader" relationships in pTyr signaling networks. The emergence of the complete pTyr signaling apparatus approximately 900 million years ago coincides with the transition from unicellular to multicellular life, underscoring its fundamental role in metazoan development [5] [4].
Evolutionary Trajectory of STAT-Type SH2 Domains
STAT (Signal Transducer and Activator of Transcription) proteins represent a crucial family of SH2 domain-containing transcription factors that directly link extracellular signals to gene expression programs. The evolutionary conservation of STAT SH2 domains is particularly remarkable, with orthologs identifiable from basal metazoans to mammals. These domains have maintained their core pTyr-binding function while acquiring specialized characteristics tailored to specific signaling pathways.
The conservation patterns in STAT SH2 domains reflect strong selective pressures preserving several key functionalities: (1) specific phosphopeptide recognition for receptor docking, (2) reciprocal SH2-pTyr interactions that mediate STAT dimerization upon phosphorylation, and (3) nuclear import mechanisms that enable transcriptional activity. Deep evolutionary conservation of these features highlights their fundamental importance to STAT function across metazoan signaling systems.
Molecular Mechanisms of Co-evolution
Structural and Dynamical Adaptations in SH2 Domains
Despite maintaining a conserved overall fold, SH2 domains have evolved considerable specificity in phosphopeptide recognition. Structural studies reveal that variations in surface loops, particularly the EF and BG loops, primarily dictate binding specificity by forming critical contacts with residues C-terminal to the phosphotyrosine [21]. These loops exhibit remarkable adaptability, with experimental evidence demonstrating that a single SH2 domain scaffold can be engineered to recognize distinct sequence motifs through combinatorial mutations in these flexible regions [21].
Table 2: Mechanisms Generating Diversity in SH2 Domain Specificity
| Mechanism | Molecular Basis | Functional Consequence |
|---|---|---|
| Loop Variation | Sequence diversity in EF and BG loops | Altered peptide binding specificity; enables recognition of different sequence motifs C-terminal to pTyr |
| Domain Shuffling | Gain or loss of protein domains in SH2-containing proteins | Creation of novel proteins with altered functions and regulatory mechanisms |
| Gene Duplication & Divergence | Duplication of SH2-encoding genes followed by functional specialization | Expansion of SH2 families with tissue-specific functions and binding preferences |
| Insertion of Linear Motifs | Acquisition of short sequence motifs that regulate interactions | Fine-tuning of binding properties and integration with other signaling networks |
Recent research has revealed that co-evolution extends beyond simple sequence conservation to encompass conserved conformational dynamics. In PTPs, residues distant from the active site undergo distinct intermediate timescale dynamics that correlate with catalytic activity, suggesting that conserved motions drive enzymatic function across enzyme families [22]. Similar dynamical properties likely operate in SH2 domains, where flexibility in critical loops enables functional adaptation while preserving structural integrity.
Co-evolutionary Networks and Constraint Analysis
Advanced computational analyses have begun mapping the complex co-evolutionary relationships within pTyr signaling networks. Covariation analysis of PTKs and SH2 domains reveals evolutionary couplings that reflect functional constraints and historical adaptations. These studies demonstrate that residues involved in protein-protein interactions and ligand binding show significant evolutionary constraint, with similar patterns observable in both deep evolutionary timescales and human population variants [9].
The integration of evolutionary conservation data with population constraint metrics (Missense Enrichment Score) provides a powerful framework for identifying functionally critical residues in SH2 domains [9]. This approach reveals that missense-depleted sites in SH2 domains are enriched in buried residues or those involved in small-molecule or protein binding, highlighting structural features under strongest selective pressure. For STAT SH2 domains, this combined analysis identifies both family-wide conserved sites critical for folding and function, as well as evolutionarily diverse functional residues that may determine pathway specificity.
Experimental Approaches for Studying SH2 Domain Co-evolution
Methodological Framework for Co-evolutionary Analysis
Understanding SH2 domain co-evolution requires integrated experimental approaches that bridge sequence analysis, structural biology, and functional assays. Below is a representative workflow for investigating co-evolutionary relationships in STAT-type SH2 domains.
Experimental Workflow for SH2 Domain Co-evolution Studies
Key Research Reagents and Methodologies
Table 3: Essential Research Reagents and Methods for Studying SH2 Co-evolution
| Reagent/Method | Specific Application | Technical Function |
|---|---|---|
| Coevolutionary Coupling Analysis | Identification of evolutionarily correlated residues | Statistical analysis of multiple sequence alignments to detect residue pairs that evolved in concert |
| Nuclear Magnetic Resonance (NMR) Spectroscopy | Characterization of protein dynamics and binding | Detection of conserved motions on microsecond timescales that correlate with function |
| Phage Display Libraries | Mapping SH2 domain specificity | Selection of SH2 variants with altered specificities through combinatorial mutagenesis of surface loops |
| Site-Directed Mutagenesis | Functional validation of co-evolving residues | Testing the impact of evolutionary coupled residues on folding, stability, and binding |
| Population Variant Analysis (MES) | Quantifying constraint in human populations | Missense Enrichment Score identifies residues under recent selective pressure in human populations |
Detailed Protocol: Coevolutionary Coupling Analysis of STAT SH2 Domains
Objective: Identify evolutionarily coupled residues in STAT SH2 domains that may underlie functional specificity.
Step 1: Sequence Compilation
- Collect STAT SH2 domain sequences from diverse vertebrate species using PFAM database (PF00017) and SMART database
- Include representative species from major evolutionary lineages (mammals, birds, reptiles, amphibians, fish)
- Curate sequences to ensure correct domain boundaries using CDD and InterPro
Step 2: Multiple Sequence Alignment
- Perform alignment using MAFFT or ClustalOmega with default parameters
- Manually inspect and refine alignment based on known secondary structure elements
- Trim alignment to include only unambiguously aligned positions
Step 3: Covariation Analysis
- Apply EVcouplings or plmDCA algorithms to detect statistically significant residue-residue couplings
- Use maximum entropy methods to distinguish direct from indirect correlations
- Apply empirical Bayesian shrinkage to regularize parameters and avoid overfitting
Step 4: Identification of Evolutionary Domains
- Partition the SH2 domain into evolutionary domains (EDs) using spectral clustering
- Validate EDs by comparison with known structural and functional data
- Map EDs onto three-dimensional structure using PyMOL
Step 5: Experimental Validation
- Select representative coupled residues for mutagenesis based on ED analysis
- Express and purify wild-type and mutant SH2 domains
- Assess phosphopeptide binding affinity using isothermal titration calorimetry (ITCAL) or surface plasmon resonance (SPR)
- Determine structural impacts using circular dichroism (CD) spectroscopy
This protocol successfully identified functionally important networks of co-evolving residues in PTP1B, including residues >20Ã… from the active site that undergo distinct dynamics correlated with catalytic activity [22]. Similar approaches can be applied to STAT SH2 domains to uncover allosteric networks governing their functional interactions.
Functional Consequences of Co-evolution
Evolution of Signaling Networks and Pathway Specificity
The co-expansion of SH2 domains with PTKs and PTPs facilitated the development of increasingly sophisticated signaling networks in higher organisms. Genomic analyses reveal that the innermost cores of domain co-occurrence networks gradually expand with increasing evolutionary complexity, from single-cellular eukaryotes to multicellular organisms [23]. These network cores are enriched with domains involved in cell-cell communication and signal transduction, reflecting their central role in metazoan biology.
For STAT proteins, co-evolution with specific JAK kinases and cytokine receptors has created highly specialized signaling pathways with precise cellular outcomes. The STAT SH2 domain has evolved to recognize specific phosphorylated motifs on cytokine receptors while maintaining conserved dimerization properties. This dual specialization-conservation paradigm enables pathway specificity while preserving core signaling mechanisms.
Structural and Dynamical Divergence Between Kinase Classes
Interesting evolutionary divergence is observed between tyrosine kinases and serine/threonine kinases in their conformational landscapes. Tyrosine kinases show stronger binding affinity for type-II inhibitors that target inactive "DFG-out" conformations, which appears to result from evolutionary adaptations that make the DFG-out state more accessible in TKs compared to STKs [24]. This divergence exemplifies how evolutionary pressures can shape conserved protein folds to exhibit distinct functional properties through modulation of conformational dynamics.
The conformational dynamics of SH2 domains themselves have likely undergone similar evolutionary optimization. While maintaining the conserved SH2 fold, different SH2 families have evolved distinct dynamic properties that facilitate their specific biological functions and regulatory mechanisms.
Implications for Disease and Therapeutic Development
Pathogenic Mutations and Evolutionary Constraint
The integration of evolutionary and population constraint data provides powerful insights into pathogenic mechanisms affecting SH2 domain function. Analysis of 2.4 million population variants mapped to 5,885 protein domain families demonstrates that missense-depleted sites in SH2 domains (under strong constraint) are enriched in buried residues and binding interfaces [9]. These constrained positions show significant overlap with known pathogenic mutations, highlighting the clinical relevance of evolutionary conservation patterns.
For STAT SH2 domains, this approach can distinguish between residues critical for structural stability versus those important for specific interactions. Mutations at evolutionarily conserved, structurally critical positions tend to cause complete loss-of-function, while mutations at more variable positions involved in specific binding interfaces may cause more subtle signaling defects.
Therapeutic Targeting of Co-evolved Networks
The co-evolutionary relationships between SH2 domains and their catalytic partners offer unique opportunities for therapeutic intervention. Several strategies have emerged for targeting these networks:
- Direct SH2 Domain Inhibition: Developing small molecules or peptidomimetics that block specific SH2-phosphopeptide interactions
- Allosteric Modulation: Targeting evolutionarily conserved dynamic networks rather than the binding pocket itself
- Multi-domain Targeting: Exploiting co-evolved domain combinations for enhanced specificity
The deep evolutionary conservation of PD-1/PD-L1 interactions with SHP-2 phosphatase, dating back to cartilaginous fish, underscores the fundamental importance of this immune checkpoint pathway and validates it as a therapeutic target [25]. Similarly, the ancient origin and conservation of STAT SH2 domains highlight their fundamental role in immunity and cell regulation, supporting their continued investigation as drug targets.
Understanding the co-evolutionary history of SH2 domains with their binding partners provides a framework for predicting resistance mechanisms, identifying synthetic lethal interactions, and developing context-specific therapeutic strategies that account for evolutionary constraints and adaptations.
The Expansion of SH2 Domains and the Rise of Metazoan Complexity
Src homology 2 (SH2) domains represent a fundamental protein interaction module that co-evolved with phosphotyrosine signaling to facilitate metazoan complexity. This review synthesizes current understanding of SH2 domain expansion across eukaryotic evolution, highlighting the crucial role of STAT-type SH2 domains in transcriptional regulation and immune function. Genomic analyses reveal that SH2 domains emerged in unicellular ancestors and underwent dramatic expansion at the unicellular-to-multicellular transition, correlating strongly with increases in organismal complexity. Structural and functional studies elucidate unique characteristics of STAT-type SH2 domains that enable their specialized role in JAK-STAT signaling. Emerging research further reveals non-canonical SH2 domain functions, including lipid binding and participation in liquid-liquid phase separation, providing novel insights into the mechanisms through which these domains contribute to sophisticated signaling networks. The therapeutic implications of targeting SH2 domains are discussed, with particular emphasis on STAT-type SH2 domains in disease contexts.
The evolution of complex multicellular organisms required sophisticated cell-cell communication systems capable of precise spatiotemporal regulation. Among these systems, phosphotyrosine-based signaling represents a relatively recent evolutionary innovation that emerged alongside metazoan development [5] [4]. At the heart of this signaling paradigm lies the Src homology 2 (SH2) domain, a protein interaction module that specifically recognizes and binds phosphorylated tyrosine residues, thereby directing the formation of transient signaling complexes [2]. The human genome encodes approximately 110-111 SH2 domain-containing proteins, which stand in stark contrast to their limited representation in unicellular eukaryotes [5] [4]. This dramatic expansion suggests a central role for SH2 domains in the development of metazoan complexity.
SH2 domains function as the primary "readers" of the phosphotyrosine code, working in concert with protein tyrosine kinases ("writers") and protein tyrosine phosphatases ("erasers") to establish dynamic signaling networks [4]. These approximately 100-amino-acid domains achieve specificity through recognition of both the phosphotyrosine residue and its surrounding amino acid sequence, enabling precise interaction with target proteins [2] [26]. While all SH2 domains share a conserved structural fold, they have diversified into two major classes: the Src-type and STAT-type SH2 domains, with the latter playing specialized roles in signal transduction and activator of transcription (STAT) proteins [2] [10].
This review examines the expansion of SH2 domains from an evolutionary perspective, focusing on their role in the emergence of metazoan complexity. Particular emphasis is placed on STAT-type SH2 domains, their structural and functional specialization, and their conservation across metazoans. We further discuss emerging non-canonical SH2 domain functions and experimental approaches for studying these critical signaling modules.
Evolutionary Expansion of SH2 Domains
Genomic Evidence for SH2 Domain Co-evolution with Metazoan Complexity
Comparative genomic analyses across 21 eukaryotic species reveal that SH2 domains first appeared in early Unikonta and expanded dramatically in the choanoflagellate and metazoan lineages [5]. This expansion paralleled the development of tyrosine kinases, creating an increasingly sophisticated phosphotyrosine signaling apparatus [5] [4]. The correlation between the percentage of protein tyrosine kinases (PTKs) and SH2 domains in various genomes is remarkably strong (correlation coefficient of 0.95), indicating their coordinated evolution [5].
Table 1: SH2 Domain Distribution Across Select Eukaryotes
| Organism | Classification | SH2 Domain-Containing Proteins | Protein Tyrosine Kinases |
|---|---|---|---|
| Saccharomyces cerevisiae (Yeast) | Unikont (Fungus) | 1 | 0 |
| Monosiga brevicollis (Choanoflagellate) | Unikont (Choanozoa) | 17 | 48 |
| Dictyostelium discoideum (Slime mold) | Unikont (Amoebozoa) | 6 | 0 |
| Caenorhabditis elegans (Roundworm) | Metazoa | 70 | 40 |
| Drosophila melanogaster (Fruit fly) | Metazoa | 42 | 32 |
| Homo sapiens (Human) | Metazoa | 111 | 90 |
The evolutionary trajectory of SH2 domains reveals their crucial role in metazoan development. The emergence of SH2 domain-containing proteins approximately 900 million years ago at the premetazoan boundary suggests that phosphotyrosine signaling may have facilitated the evolution of metazoans [5] [4]. This timeline corresponds with the development of specialized cell types and more elaborate body plans, highlighting the importance of selective intercellular communication in metazoan complexity [5].
The expansion of SH2 domains occurred primarily through gene duplication and domain shuffling events, which placed SH2 domains in novel protein contexts and enabled their participation in diverse cellular processes [5] [4]. This diversification allowed SH2 domains to integrate with existing signaling networks, positioning phosphotyrosine signaling as a crucial driver of robust cellular communication networks in metazoans [5].
STAT-Type SH2 Domains in Evolutionary Context
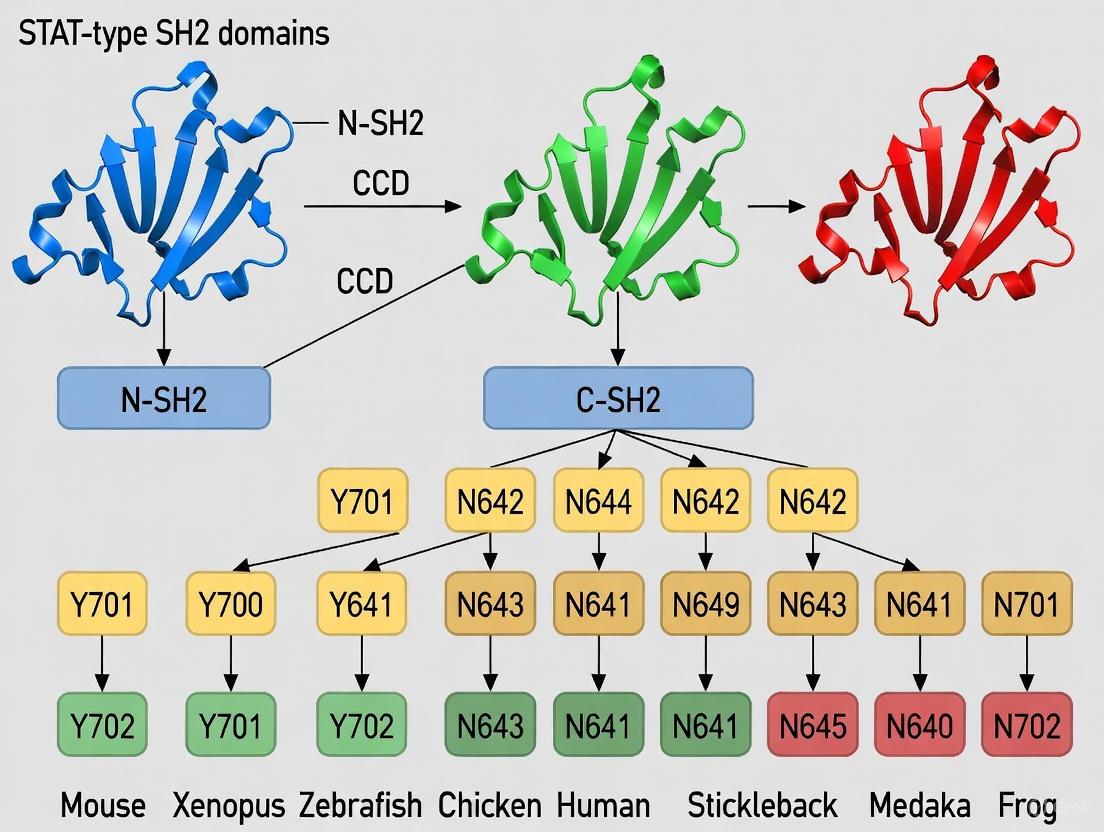
STAT-type SH2 domains represent a distinct evolutionary adaptation within the SH2 superfamily. Phylogenetic analysis has categorized SH2 domain-containing proteins into 38 different sub-families, with STAT SH2 domains forming a separate clade [10]. These domains lack the βE and βF strands found in Src-type SH2 domains and feature a split αB helix, structural adaptations that facilitate STAT dimerization—a critical step in STAT-mediated transcriptional regulation [2].
The evolutionary provenance of STAT-type SH2 domains can be traced to ancestral functions predating animal multicellularity, as observed in Dictyostelium, which employs SH2 domain/phosphotyrosine signaling for transcriptional regulation [2]. This conservation across deep evolutionary timescales underscores the fundamental importance of STAT-type SH2 domains in cellular signaling.
Table 2: Evolutionary Conservation of STAT Proteins Across Species
| STAT Gene | Mammalian Specialization | Fish Orthologs | Conserved Domains |
|---|---|---|---|
| STAT1 | Response to interferons, antiviral defense | stat1a, stat1b (duplicated) | NTD, CCD, DBD, Linker, SH2, TAD |
| STAT2 | Type I interferon signaling | stat2 | NTD, CCD, DBD, Linker, SH2, TAD |
| STAT3 | IL-6 family cytokine signaling, acute phase response | stat3 | NTD, CCD, DBD, Linker, SH2, TAD |
| STAT4 | IL-12 signaling, Th1 differentiation | stat4 | NTD, CCD, DBD, Linker, SH2, TAD |
| STAT5 | Prolactin, growth hormone signaling | stat5a, stat5b (separate chromosomes) | NTD, CCD, DBD, Linker, SH2, TAD |
| STAT6 | IL-4/IL-13 signaling, Th2 differentiation | stat6 | NTD, CCD, DBD, Linker, SH2, TAD |
In fish, including the lumpfish (Cyclopterus lumpus L.), the complete complement of STAT genes (stat1a, 2, 3, 4, 5a, 5b, and 6) is present and functionally conserved, demonstrating the deep evolutionary conservation of STAT proteins and their SH2 domains [27]. The presence of stat1a and stat1b duplicates in fish reflects a genome duplication event approximately 35 million years ago, with some fish species possessing up to five stat1 gene copies [27].
Structural and Functional Specialization of SH2 Domains
Conserved Architecture of SH2 Domains
All SH2 domains share a conserved structural fold despite significant sequence variation, suggesting this structure has evolved almost exclusively to bind phosphotyrosine-containing motifs [2]. The canonical SH2 domain structure consists of a three-stranded antiparallel beta-sheet flanked by two alpha helices in an αβββα configuration [2] [10]. The N-terminal region contains a deep pocket within the βB strand that binds the phosphate moiety, featuring an invariant arginine residue at position βB5 that directly interacts with the phosphotyrosine through a salt bridge [2].
The structural conservation across SH2 domains is remarkable, with family members sharing as little as ~15% pairwise sequence identity while maintaining nearly identical three-dimensional folds [2]. This conservation highlights the structural constraints required for phosphotyrosine recognition while allowing for diversification in sequence specificity.
Figure 1: SH2 Domain Structural Organization. All SH2 domains share a conserved αβββα fold with specialized binding pockets for phosphotyrosine recognition and sequence-specific interactions.
Unique Features of STAT-Type SH2 Domains
STAT-type SH2 domains possess distinct structural characteristics that differentiate them from Src-type SH2 domains and enable their specialized function in transcriptional regulation. Unlike Src-type domains, STAT-type SH2 domains lack the βE and βF strands and feature a split αB helix (designated αB and αB') [2] [10]. This structural adaptation facilitates STAT dimerization, a critical step in STAT-mediated transcriptional regulation [2].
The STAT-type SH2 domain contains several functionally critical regions:
- pY pocket: Binds the phosphotyrosine residue through conserved residues including the invariant arginine
- pY+3 pocket: Determines specificity by interacting with residues C-terminal to the phosphotyrosine
- Evolutionary active region (EAR): Contains additional α-helix (αB') in STAT-type domains
- Hydrophobic system: A cluster of non-polar residues that stabilizes the β-sheet conformation
- Dimerization interfaces: Surfaces on αB, αB', and BC* loop that mediate STAT dimerization [10]
These structural features allow STAT SH2 domains to participate in both receptor recognition and dimerization, two critical functions in JAK-STAT signaling. The flexibility of STAT SH2 domains, particularly in the pY pocket, presents both challenges and opportunities for drug discovery [10].
Molecular Recognition and Specificity Determinants
SH2 domain binding is characterized by a combination of high specificity toward cognate phosphotyrosine ligands with moderate binding affinity (Kd typically 0.1-10 μM) [2]. This affinity range allows for specific but transient interactions, a defining characteristic of dynamic cell signaling processes.
Specificity is determined by interactions between surface residues adjacent to the phosphotyrosine-binding pocket and amino acids C-terminal to the phosphotyrosine residue, particularly at the +1 to +5 positions [2] [26]. The EF loop (joining β-strands E and F) and BG loop (joining α-helix B and β-strand G) play crucial roles in determining binding selectivity by controlling access to ligand specificity pockets [2].
High-throughput profiling using bacterial peptide display has revealed that both tyrosine kinases and SH2 domains recognize specific sequence motifs surrounding their target tyrosine or phosphotyrosine residues [26]. This specificity profiling enables prediction of signaling pathways and identification of natural genetic variants that affect phosphosite recognition [26].
Non-Canonical SH2 Domain Functions and Regulatory Mechanisms
Lipid Binding by SH2 Domains
Recent research has revealed that SH2 domains possess non-canonical functions beyond phosphotyrosine recognition. Genome-wide screening demonstrates that approximately 75-90% of human SH2 domains bind plasma membrane lipids with high affinity and specificity [2] [28]. These interactions occur through surface cationic patches separate from phosphotyrosine-binding pockets, allowing simultaneous binding to lipids and phosphorylated proteins [28].
Table 3: Lipid-Binding SH2 Domain-Containing Proteins and Their Functions
| Protein Name | Lipid Specificity | Functional Role of Lipid Association |
|---|---|---|
| SYK | PIP3 | PIP3-dependent membrane binding required for SYK activation and noncatalytic activation of STAT3/5 |
| ZAP70 | PIP3 | Facilitates and sustains ZAP70 interactions with TCR-ζ in T cell signaling |
| LCK | PIP2, PIP3 | Modulates LCK interaction with binding partners in TCR signaling complex |
| ABL | PIP2 | Membrane recruitment and modulation of Abl activity |
| VAV2 | PIP2, PIP3 | Modulates VAV2 interaction with membrane receptors such as EphA2 |
| C1-Ten/Tensin2 | PIP3 | Regulation of Abl activity and IRS-1 phosphorylation in insulin signaling |
Lipid binding plays crucial regulatory roles in SH2 domain function. For example, phosphatidylinositol-3,4,5-trisphosphate (PIP3) binding to the SYK SH2 domain is required for SYK activation and its noncatalytic activation of STAT3/5 [2]. Similarly, lipid interactions with the ZAP70 SH2 domain facilitate and sustain its association with the T-cell receptor ζ chain [2] [28]. These findings reveal how lipids exert spatiotemporal control over SH2 domain-mediated protein-protein interactions and signaling activities [28].
SH2 Domains in Liquid-Liquid Phase Separation
Proteins with SH2 domains have increasingly been linked to the formation of intracellular condensates via protein phase separation [2]. Multivalent interactions involving SH2 domains and other modular domains (e.g., SH3 domains) drive condensate formation, creating membrane-less organelles that enhance signaling specificity and efficiency [2].
Notable examples include:
- GRB2, Gads, and LAT receptor: Contribute to liquid-liquid phase separation (LLPS) formation, enhancing T-cell receptor signaling [2]
- Adapter NCK in podocyte kidney cells: LLPS increases membrane dwell time of N-WASP and Arp2/3 complexes, promoting actin polymerization [2]
Post-translational modifications, including phosphorylation, modulate the assembly and disassembly of these condensates, providing a dynamic regulatory mechanism for controlling signal transduction [2]. This emerging role of SH2 domains in phase separation represents a novel mechanism for achieving signaling specificity and efficiency in complex metazoan cells.
SH2 Domain Mutations in Disease
The SH2 domain represents a mutational hotspot in disease, particularly for STAT proteins [10]. Sequencing analyses of patient samples have identified numerous point mutations within STAT3 and STAT5B SH2 domains that result in either hyperactivated or refractory STAT mutants [10].
Table 4: Disease-Associated Mutations in STAT3 and STAT5B SH2 Domains
| Mutation | Location | Pathology | Effect |
|---|---|---|---|
| STAT3 K591E/M | αA2 helix, pY pocket | AD-HIES (Germline) | Loss-of-function |
| STAT3 R609G | βB5 strand, pY pocket | AD-HIES (Germline) | Loss-of-function |
| STAT3 S614R | BC loop, pY pocket | T-LGLL, NK-LGLL (Somatic) | Gain-of-function |
| STAT3 E616K | BC loop, pY pocket | NKTL (Somatic) | Gain-of-function |
| STAT5B N642H/H→Y | SH2 domain | Multiple cancers | Gain-of-function |
The SH2 and transactivation domains (TAD) of STAT genes show higher mutation rates in the general population compared to other domains, with STAT SH2 domains exhibiting mutation rates of 24-34% across the STAT family [29]. This genetic volatility underscores the delicate evolutionary balance of wild-type STAT structural motifs in maintaining precise levels of cellular activity [10].
Mutations can have opposing effects depending on their specific location and nature. For instance, STAT3 S614R is a somatic gain-of-function mutation found in T-cell large granular lymphocytic leukemia, while STAT3 S614G is a germline loss-of-function mutation associated with autosomal-dominant hyper IgE syndrome [10]. This delicate balance highlights the evolutionary constraints on SH2 domain structure and function.
Experimental Approaches for SH2 Domain Research
High-Throughput Specificity Profiling
Understanding SH2 domain function requires comprehensive characterization of their binding specificities. Bacterial peptide display combined with deep sequencing represents a powerful platform for profiling sequence recognition by SH2 domains [26]. This method enables quantitative analysis of SH2 domain binding specificities across thousands of peptide sequences in a single experiment.
Figure 2: High-Throughput SH2 Domain Specificity Profiling. Bacterial peptide display enables comprehensive characterization of SH2 domain binding preferences using magnetic bead separation and deep sequencing.
The experimental workflow involves:
- Library construction: Creating genetically encoded peptide libraries displayed on the surface of E. coli as fusions to the eCPX surface display protein
- Binding selection: Incubating the peptide library with biotinylated SH2 domains followed by capture with avidin-functionalized magnetic beads
- Deep sequencing: Quantitatively analyzing selected peptides using high-throughput sequencing to determine binding preferences [26]
This approach can be adapted for various library types:
- X5-Y-X5 libraries: Contain 10â¶-10â· random 11-residue sequences with a central tyrosine for determining general specificity motifs
- pTyr-Var libraries: Include thousands of human tyrosine phosphorylation sites and their natural variants for assessing the impact of mutations on SH2 domain recognition
- Amber codon suppression: Enables incorporation of non-canonical or post-translationally modified amino acids to study their effects on binding [26]
Essential Research Reagents and Tools
Table 5: Research Reagent Solutions for SH2 Domain Studies
| Reagent/Tool | Function | Application Examples |
|---|---|---|
| Bacterial peptide display system (eCPX) | High-throughput specificity profiling | Determining SH2 domain binding motifs [26] |
| Oriented peptide libraries | In vitro binding specificity | Position-specific amino acid preferences [26] |
| Phosphotyrosine variant (pTyr-Var) library | Natural genetic variant analysis | Impact of disease-associated mutations on SH2 binding [26] |
| Amber codon suppression system | Non-canonical amino acid incorporation | Studying PTM effects on SH2 recognition [26] |
| Lipid binding assays | Lipid-protein interaction analysis | Characterizing membrane recruitment of SH2 domains [28] |
| Phase separation assays | LLPS formation analysis | SH2 domain role in biomolecular condensates [2] |
Therapeutic Targeting of SH2 Domains
The critical role of SH2 domains in signaling pathways, particularly in disease contexts, makes them attractive therapeutic targets. STAT-type SH2 domains have received particular attention due to their central role in JAK-STAT signaling and implication in numerous diseases, including cancer and immune disorders [10].
Several strategies have emerged for targeting SH2 domains:
- Small molecule inhibitors: Targeting the phosphotyrosine-binding pocket or adjacent specificity pockets to disrupt protein-protein interactions
- Nonlipidic inhibitors: Targeting lipid-binding sites to modulate membrane recruitment and activation, as demonstrated for Syk kinase [2]
- Stabilizers: Compounds that stabilize inactive conformations of SH2 domain-containing proteins
Targeting the SH2 domains of STAT proteins presents unique challenges due to their flexible nature and the shallow, dynamic characteristics of their binding surfaces [10]. The pY and pY+3 pockets represent the most targetable regions, with additional opportunities in the evolutionary active region (EAR) and hydrophobic system [10]. Understanding the structural dynamics of STAT SH2 domains is essential for rational drug design, as crystal structures do not always preserve targetable pockets in accessible states [10].
The high mutation rate observed in STAT SH2 domains in the general population [29] underscores the importance of personalized medicine approaches when developing SH2-targeted therapies, as genetic variation may significantly impact drug efficacy.
The expansion of SH2 domains represents a cornerstone in the evolution of metazoan complexity, enabling the sophisticated cell-cell communication required for multicellular life. The coordinated evolution of SH2 domains with protein tyrosine kinases and phosphatases created a dynamic signaling system capable of precise spatiotemporal regulation. STAT-type SH2 domains, with their unique structural adaptations for dimerization and transcriptional regulation, exemplify the functional specialization that accompanied this expansion.
Emerging research continues to reveal unexpected roles for SH2 domains beyond canonical phosphotyrosine recognition, including lipid binding and participation in liquid-liquid phase separation. These non-canonical functions expand our understanding of how SH2 domains contribute to the exquisite regulation of cellular signaling networks. The development of high-throughput profiling methods has accelerated our understanding of SH2 domain specificity and the functional consequences of natural genetic variation.
Future research directions include:
- Elucidating the structural dynamics of SH2 domains in full-length proteins and signaling complexes
- Investigating the crosstalk between phosphotyrosine, lipid, and phase separation functions of SH2 domains
- Developing innovative therapeutic strategies that target both canonical and non-canonical SH2 domain functions
- Exploring SH2 domain evolution in non-model organisms to understand adaptive changes in signaling networks
The deep evolutionary conservation of SH2 domains, particularly STAT-type SH2 domains, underscores their fundamental importance in metazoan biology. As we continue to unravel their diverse functions and regulatory mechanisms, we gain not only insights into the evolution of biological complexity but also opportunities for therapeutic intervention in human disease.
Techniques for Analyzing STAT-type SH2 Structure, Function, and Inhibition
Src homology 2 (SH2) domains are protein modules of approximately 100 amino acids that specifically recognize and bind to phosphorylated tyrosine (pY) residues, thereby facilitating critical protein-protein interactions in intracellular signaling networks [2] [30]. These domains are fundamental components of phosphotyrosine signaling, governing cellular processes including growth, differentiation, immune response, and cytoskeletal reorganization [4] [2]. In the human proteome, roughly 110 proteins contain SH2 domains, classifying them as enzymes, adaptors, docking proteins, and transcription factors [30]. From an evolutionary perspective, SH2 domains expanded alongside protein-tyrosine kinases (PTKs) and phosphatases (PTPs) to coordinate increasing cellular and organismal complexity in metazoans [4]. This review focuses on the application of two powerful structural biology tools—X-ray crystallography and AlphaFold—for analyzing SH2 domain structure and function, with particular emphasis on their utility for investigating the evolutionary conservation of STAT-type SH2 domains.
Structural Biology of SH2 Domains
Conserved Architecture and Specificity Determinants
All SH2 domains share a highly conserved three-dimensional fold despite significant sequence variation, with some family members sharing as little as ~15% pairwise identity [2] [30]. The canonical SH2 domain structure consists of a central three-stranded antiparallel beta-sheet flanked by two alpha helices in an αA-βB-βC-βD-αB arrangement [2] [30]. The N-terminal region contains a deep pocket within the βB strand that binds the phosphate moiety of phosphorylated tyrosine. This pocket invariably contains a critical arginine residue (at position βB5) that forms a salt bridge with the phosphorylated tyrosine residue of ligand peptides [2] [30].
SH2 domains recognize both the phosphotyrosine and specific residue sequences flanking it, primarily carboxy-terminal to the pY residue [4] [8]. This dual recognition provides specificity in signaling interactions, with binding affinities typically ranging from 0.1–10 μM [2]. The structural basis for specificity involves surface residues adjacent to the pY-binding pocket that interact with amino acids at positions C-terminal to the pY, creating a diverse recognition system capable of discriminating among different pY-containing motifs [4] [2].
Table 1: Key Structural Features of SH2 Domains
| Structural Element | Description | Functional Role |
|---|---|---|
| Central β-sheet | Three-stranded antiparallel β-sheet (βB-βC-βD) | Forms structural core of the domain |
| Flanking α-helices | Two α-helices (αA and αB) | Stabilize domain structure and contribute to binding surface |
| pY-binding pocket | Deep pocket within βB strand | Binds phosphotyrosine moiety via conserved arginine residue |
| Specificity pockets | Surface adjacent to pY-binding pocket | Recognizes residues C-terminal to pY, determining binding specificity |
| EF and BG loops | Variable loops connecting secondary structures | Control access to ligand specificity pockets |
STAT-type vs. Src-type SH2 Domains
SH2 domains are structurally classified into two major subgroups: Src-type and STAT-type, which have distinct structural and functional characteristics [2] [7]. STAT-type SH2 domains lack the βE and βF strands present in Src-type domains and feature a split αB helix [2]. This structural adaptation facilitates SH2 domain-mediated dimerization, which is critical for STAT protein activation and nuclear translocation [2]. Evolutionary studies suggest that the linker-SH2 domain of STAT transcription factors represents one of the most ancient and fully developed functional SH2 domains, serving as a template for continuing SH2 domain evolution [7]. This ancient origin makes STAT-type SH2 domains particularly interesting for evolutionary studies of phosphotyrosine signal transduction.
Table 2: Comparison of Src-type and STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core structure | αA-βB-βC-βD-αB with additional βE, βF, βG strands | αA-βB-βC-βD-αB' without βE and βF strands |
| αB helix | Single continuous helix | Split into two helices (αB and αB') |
| Primary function | Recruitment of signaling proteins to pY sites | Mediate dimerization and nuclear translocation |
| Evolutionary origin | More recent diversification | Ancient, predating plant-animal divergence |
| Representative proteins | Src, Grb2, PLCγ | STAT1, STAT3, STAT5 |
X-ray Crystallography for SH2 Domain Analysis
Methodology and Workflow
X-ray crystallography has been instrumental in elucidating SH2 domain structures and their interactions with phosphorylated ligands. To date, the structures of approximately 70 different SH2 domains have been experimentally determined using crystallography [2] [30]. The standard workflow involves:
Protein Expression and Purification: Recombinant SH2 domains or multi-domain constructs are expressed in systems like E. coli and purified using affinity chromatography [31].
Crystallization: Purified proteins are concentrated and subjected to crystallization trials using vapor diffusion or other methods. Obtaining high-quality crystals remains a critical and often challenging step.
Data Collection: X-ray diffraction data are collected at synchrotron facilities. Serial crystallography (SX) approaches, particularly at X-ray free-electron lasers (XFELs), have enabled studies of challenging proteins with limited sample availability [32].
Structure Determination: Diffraction patterns are processed to generate electron density maps, into which protein structures are built and refined.
Recent advances in serial crystallography have significantly reduced sample consumption, with specialized fixed-target devices and liquid injection methods enabling data collection from microcrystals [32]. These developments are particularly valuable for studying SH2 domain complexes with ligands or drugs.
Key Insights from Crystallographic Studies
X-ray crystallography has revealed fundamental aspects of SH2 domain structure and function:
Conserved Fold Architecture: Despite low sequence similarity, all SH2 domains maintain nearly identical tertiary structures optimized for pY recognition [2] [30].
Ligand Binding Mechanisms: Structures of SH2 domains complexed with phosphopeptides show how the conserved arginine in the FLVRES motif coordinates the phosphate group, while variable regions determine sequence specificity [2].
Multi-domain Organization: Crystallographic studies of tandem SH3-SH2 constructs revealed limited interdomain interactions in some proteins (Lck, Src) but more extensive interfaces in others (Abl) [31]. These arrangements may influence domain orientation and function in signaling regulation.
Regulatory Mechanisms: Structures of full-length Src-family kinases showed unanticipated interactions between SH2, SH3, and kinase domains that maintain the enzyme in an autoinhibited state [31].
AlphaFold for SH2 Domain Prediction and Analysis
Accuracy and Reliability Assessment
AlphaFold 2 has revolutionized structural biology by providing highly accurate protein structure predictions. For SH2 domain research, its predictions are particularly valuable for rapid structural analysis and hypothesis generation. Validation studies comparing AlphaFold predictions to experimental structures show:
The median root mean square deviation (RMSD) between AlphaFold models and experimental structures is approximately 1.0 Ã…, indicating excellent overall agreement [33].
In high-confidence regions, the median RMSD improves to 0.6 Ã…, matching the variation between different experimental structures of the same protein [33].
Approximately 93% of side chain conformations are roughly correct, with 80% showing perfect fit to experimental data [33].
Low-confidence regions (often corresponding to flexible loops or disordered regions) may show RMSD values exceeding 2.0 Ã… [33].
For multi-domain proteins containing SH2 domains, AlphaFold accurately predicts individual domain structures but may not reliably position domains relative to each other, especially when connected by flexible linkers [33]. This uncertainty is reflected in the predicted aligned error (PAE) output.
Applications in Evolutionary Studies of STAT-type SH2 Domains
AlphaFold enables large-scale evolutionary structural analyses that were previously impractical with experimental methods alone:
Conservation of Structural Folds: AlphaFold predictions confirm that STAT-type SH2 domains from diverse organisms maintain the characteristic split αB helix and absence of βE/F strands, despite sequence divergence [2] [7].
Ancestral Protein Reconstruction: Combined with evolutionary sequence analysis, AlphaFold can model structures of ancestral SH2 domains to trace structural adaptations throughout evolution.
Variant Impact Prediction: AlphaFold can model the structural consequences of natural variants, helping identify residues critical for maintaining structural integrity versus those tolerant to change.
Dimerization Interface Conservation: For STAT-type SH2 domains, AlphaFold predictions can assess conservation of dimerization interfaces across evolutionary lineages.
Table 3: AlphaFold Performance Characteristics for SH2 Domain Analysis
| Metric | Performance | Implications for SH2 Research |
|---|---|---|
| Overall RMSD | 1.0 Ã… (median) | High accuracy for general structural analysis |
| High-confidence regions | 0.6 Ã… (median) | Suitable for detailed mechanistic studies |
| Side chain accuracy | 80% perfect fit | Reliable for binding site analysis |
| Multi-domain proteins | Variable relative positioning | Limited utility for inter-domain arrangements |
| Low-confidence regions | >2.0 Ã… RMSD | Caution required for flexible regions |
Integrated Approaches for SH2 Domain Research
Complementary Use of Experimental and Computational Methods
The most powerful insights into SH2 domain structure and function emerge from integrating multiple approaches:
AlphaFold for Experimental Design: AlphaFold predictions can guide crystallography by identifying flexible regions that may require modification for crystallization and suggesting optimal construct boundaries [33].
Ligand Binding Studies: Computational predictions combined with high-throughput experimental profiling using bacterial peptide display and next-generation sequencing can generate accurate sequence-to-affinity models for SH2 domains [8].
Evolutionary Conservation Analysis: Population constraint metrics like the Missense Enrichment Score (MES) combined with evolutionary conservation patterns can identify structurally and functionally critical residues in SH2 domains [9].
Research Reagent Solutions for SH2 Domain Studies
Table 4: Essential Research Reagents and Resources for SH2 Domain Structural Biology
| Reagent/Resource | Specifications | Research Application |
|---|---|---|
| Recombinant SH2 Domains | 1-10 mg, >95% pure, isotopically labeled for NMR | Crystallization, binding assays, structural studies |
| Phosphopeptide Libraries | Diverse pY-containing peptides, random or proteome-derived | Specificity profiling, binding affinity measurements |
| Crystallization Screens | Commercial sparse matrix screens (e.g., Hampton Research) | Initial crystallization condition identification |
| Fixed-target Crystallography Chips | Silicon or polymer-based with microwells | Serial crystallography with minimal sample consumption |
| AlphaFold Database | Pre-computed structures for entire proteomes | Rapid access to SH2 domain predictions without computation |
| ProBound Software | Statistical learning method with free-energy regression | Building quantitative sequence-to-affinity models from NGS data |
X-ray crystallography and AlphaFold represent complementary and powerful approaches for elucidating the structure and function of SH2 domains. Crystallography continues to provide atomic-resolution insights into mechanistic aspects of SH2 domain function, particularly for ligand complexes and multi-domain architectures, while technological advances steadily reduce sample requirements. AlphaFold offers unprecedented capabilities for rapid structural prediction and large-scale evolutionary analyses, with particular strength in modeling individual domain structures accurately. For evolutionary studies of STAT-type SH2 domains, the integration of these tools with functional assays and evolutionary analysis enables researchers to trace the structural adaptations that underpin the conservation and diversification of phosphotyrosine signaling networks throughout eukaryotic evolution. This integrated structural biology approach continues to advance our understanding of how these modular domains have evolved to coordinate complex signaling processes essential for metazoan development and physiology.
In the field of protein bioinformatics, primary sequence alignment has long been the cornerstone of motif identification and evolutionary analysis. However, the limitations of this approach become particularly apparent when studying rapidly evolving or highly divergent protein domains such as the STAT-type Src homology 2 (SH2) domain. This technical review examines how secondary structural alignment overcomes these limitations by capturing conserved structural features that remain invisible to sequence-based methods. Within the context of evolutionary conservation research on STAT-type SH2 domains, we demonstrate how this approach has revealed the ancient origin of the linker-SH2 domain architecture, identified novel genes across eukaryotic species, and provided insights into phosphotyrosine signal transduction evolution. The integration of secondary structure prediction with proteomic-scale analysis represents a paradigm shift in our ability to trace domain evolution and identify functional motifs across distantly related species.
Protein domain identification and classification traditionally relies on primary sequence alignment, which operates under the assumption that conserved residues reflect conserved structures and functions. While effective for closely related sequences, this approach fails when sequence similarity drops below the "twilight zone" of alignment, typically around 20-30% identity. For protein motifs like the SH2 domain, which play crucial roles in phosphotyrosine-mediated signal transduction, primary structural alignment often cannot accurately identify the motif due to sequence divergence [7].
The Src homology 2 (SH2) domain exemplifies this challenge. Approximately 100 amino acids in length, SH2 domains are specialized modules that specifically bind phosphorylated tyrosine motifs, forming a crucial part of protein-protein interaction networks involved in cellular signaling, transcription, and metabolism [30]. Despite their functional conservation, SH2 domains exhibit significant sequence variation that complicates identification based solely on primary sequence.
Secondary structural alignment addresses this limitation by focusing on the conserved architectural blueprint of protein domains—their arrangement of α-helices and β-strands—which often persists even when sequences diverge beyond recognition by conventional methods. This approach has proven particularly valuable for studying the evolutionary conservation of STAT-type SH2 domains, revealing insights that have reshaped our understanding of phosphotyrosine signaling evolution.
Structural Fundamentals of SH2 Domains
Conserved Architecture of SH2 Domains
All SH2 domains share a conserved structural fold despite significant sequence variation. The fundamental architecture consists of a central three-stranded antiparallel beta-sheet flanked on both sides by two alpha helices, creating a characteristic "αβββα" structure [7] [30]. This core "sandwich" structure is maintained across diverse SH2 domain families and provides the structural framework for phosphotyrosine recognition.
The N-terminal region of the SH2 domain contains a deep pocket within the βB strand that specifically binds the phosphate moiety of phosphorylated tyrosine residues. This pocket contains an invariant arginine residue at position βB5, which is part of the FLVR motif found in most SH2 domains and directly interacts with the phosphotyrosine through salt bridge formation [30]. The structural conservation of this binding pocket underscores the functional conservation of phosphotyrosine recognition across diverse SH2 domains.
Classification of SH2 Domains: Src-type versus STAT-type
Secondary structural alignment has enabled the classification of SH2 domains into two distinct groups based on their structural features:
Src-type SH2 domains: Characterized by the basic "αβββα" structure with an additional extra β-strand (βE or βE-βF motif) [7]. These domains represent the canonical SH2 structure found in numerous signaling proteins.
STAT-type SH2 domains: Distinguished by the presence of a unique αB' motif and the conjugation of the SH2 domain with a linker domain [7]. This linker-SH2 architecture represents an evolutionarily distinct lineage within the SH2 superfamily.
Table 1: Structural Classification of SH2 Domain Types
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αβββα | αβββα |
| Additional Elements | Extra β-strand (βE or βE-βF motif) | αB' motif |
| Domain Architecture | Typically isolated SH2 domain | Linker-SH2 domain conjugation |
| Representative Proteins | SRC, ABL, FYN | STAT1, STAT3, STAT5A, STAT5B |
The differentiation between these two classes extends beyond structural features to encompass their evolutionary history and functional specialization. STAT-type SH2 domains represent one of the most ancient and fully developed functional domains, serving as a template for the continuing evolution of the SH2 domain essential for phosphotyrosine signal transduction [7].
Methodological Approaches to Secondary Structure Alignment
Workflow for Secondary Structure-Based Identification
The identification of SH2 domains through secondary structure alignment follows a systematic workflow that integrates bioinformatic prediction with experimental validation. The following diagram illustrates this process:
Key Algorithms and Tools
Implementation of secondary structure alignment requires specialized computational tools and algorithms:
A-Bruijn Alignment (ABA): A graph-based alignment method that represents alignments as directed graphs potentially containing cycles, providing more flexibility than traditional alignment matrices [34]. This approach is particularly valuable for proteins with shuffled or repeated domain structures.
Jalview: A cross-platform program for multiple sequence alignment editing, visualization, and analysis that provides integrated viewing of sequence and structural information [35]. The platform offers built-in DNA, RNA, and protein structure visualization capabilities.
CoDIAC (Comprehensive Domain Interface Analysis of Contacts): A Python-based package that extracts interaction interfaces from experimental and predicted structures, enabling domain-centric analysis of contact maps [36]. This tool facilitates the integration of structural data with post-translational modification and mutation information.
The application of these tools enables researchers to move beyond the limitations of primary sequence alignment and leverage the evolutionary conserved information embedded in protein secondary structures.
Experimental Validation Techniques
Computational predictions require experimental validation to confirm both structure and function:
X-ray Crystallography and NMR Spectroscopy: Provide high-resolution structural data for verifying predicted secondary structure elements and domain boundaries [36].
Genetically Encoded Biosensors: Tools like STATeLights enable real-time monitoring of STAT activation in live cells, providing functional validation of SH2 domain activity [37]. These biosensors typically employ FRET (Förster Resonance Energy Transfer) pairs to detect conformational changes associated with SH2 domain-mediated dimerization.
Contact Mapping: Systematic extraction of domain interfaces from structural data to understand binding specificity and interface conservation [36]. This approach verifies predicted interactions through experimental structural data.
Table 2: Research Reagent Solutions for SH2 Domain Studies
| Reagent/Tool | Type | Primary Function | Application Example |
|---|---|---|---|
| STATeLights | Genetically encoded biosensor | Real-time detection of STAT activation via FLIM-FRET | Monitoring STAT5 conformational changes in live cells [37] |
| CoDIAC | Python package | Comprehensive domain interface analysis from structures | Mapping SH2 domain interfaces with ligands and other domains [36] |
| Jalview | Alignment visualization software | Multiple sequence alignment editing and analysis | Integrating sequence and structural annotation [35] |
| A-Bruijn Aligner (ABA) | Alignment algorithm | Graph-based multiple sequence alignment | Aligning proteins with shuffled domain architectures [34] |
Evolutionary Insights into STAT-type SH2 Domains
Ancient Origin of the Linker-SH2 Domain
The application of secondary structural alignment to SH2 domain analysis has yielded profound insights into the evolutionary history of STAT-type domains. Research indicates that the linker-SH2 domain of the transcription factor STAT represents one of the most ancient and fully developed functional domains, serving as a template for the continuing evolution of the SH2 domain essential for phosphotyrosine signal transduction [7].
This conclusion is supported by the discovery of novel genes carrying the linker-SH2 domain in Arabidopsis, designated as STAT-type linker-SH2 domain factors (STATL). These genes are found in a wide array of vascular and nonvascular plants, suggesting that the linker-SH2 domain evolved prior to the divergence of plants and animals [7]. This finding fundamentally reshapes our understanding of phosphotyrosine signaling evolution, extending its origins deeper into eukaryotic history than previously recognized.
Conservation Patterns in SH2 Domains
Recent analysis of evolutionary and population constraints in protein domains has revealed distinctive conservation patterns in SH2 domains. Studies mapping 2.4 million population variants to 5,885 protein domain families have demonstrated that population constraint, as measured by Missense Enrichment Score (MES), strongly correlates with evolutionary conservation in SH2 domains [9].
Population-constrained sites in SH2 domains show significant enrichment in buried residues and binding interfaces, mirroring patterns observed in evolutionary conservation analysis. This dual constraint highlights the structural and functional importance of these regions and underscores how secondary structure dictates evolutionary trajectories [9].
The integration of population genetics with structural analysis provides a powerful framework for identifying functionally critical regions within SH2 domains and predicting the potential pathogenicity of mutations affecting these regions.
Applications in Drug Discovery and Development
Targeting SH2 Domains for Therapeutic Intervention
The structural insights gained from secondary structure alignment have direct applications in pharmaceutical development. SH2 domains represent attractive therapeutic targets due to their central role in signaling pathways associated with malignancy, autoimmunity, and immunodeficiency [30] [37]. STAT proteins in particular are valuable drug targets, with STAT5 playing a central role in signaling cascades triggered by cytokines, growth factors, and hormones [37].
Traditional approaches to measuring STAT activation rely on detecting phosphorylated tyrosine residues using specific antibodies, but this method requires cell fixation and permeabilization, preventing real-time monitoring in live cells [37]. Secondary structure-informed biosensor design has overcome this limitation, enabling continuous tracking of STAT activation and facilitating drug discovery efforts.
Structural Insights for Inhibitor Design
The detailed structural understanding of SH2 domains provided by secondary structure alignment has enabled more rational approaches to inhibitor design:
Lipid-binding pocket targeting: Recent research shows that nearly 75% of SH2 domains interact with lipid molecules in the membrane, with a tendency towards phosphatidylinositol-4,5-bisphosphate (PIP2) or phosphatidylinositol-3,4,5-trisphosphate (PIP3) [30]. Targeting these lipid-binding interfaces offers alternative approaches to modulating SH2 domain function.
Allosteric inhibition: Understanding the complete secondary structure architecture of SH2 domains has revealed potential allosteric sites distinct from the phosphotyrosine-binding pocket. These sites provide opportunities for developing more selective inhibitors with reduced off-target effects.
Liquid-liquid phase separation modulation: SH2 domain-containing proteins participate in intracellular condensate formation via liquid-liquid phase separation [30]. Small molecules that modulate these phase separation behaviors represent a novel approach to targeting SH2 domain-mediated signaling.
Table 3: SH2 Domain-Targeting Therapeutic Approaches
| Therapeutic Approach | Mechanism | Development Status |
|---|---|---|
| Phosphotyrosine Mimetics | Competitive inhibition of pY-binding pocket | Preclinical and clinical development [30] |
| Lipid-Binding Disruptors | Interference with membrane association | Early preclinical [30] |
| Allosteric Inhibitors | Modulation of SH2 domain conformation | Research phase |
| Phase Separation Modulators | Alteration of condensate formation | Emerging concept [30] |
The integration of secondary structural alignment with emerging technologies promises to further advance our understanding of STAT-type SH2 domains and their biological functions. The application of AlphaFold and other structure prediction tools to model full-length STAT proteins provides new insights into domain arrangements and conformational changes associated with activation [37] [36]. Meanwhile, comprehensive contact mapping approaches like CoDIAC enable systematic analysis of interaction interfaces across entire domain families [36].
Future research directions will likely focus on several key areas:
Integration of structural and population genetics data to better understand the pathogenicity of mutations affecting SH2 domains [9] [36].
Expansion of structural alignment approaches to include other domain types and protein families, creating a more comprehensive map of domain evolution.
Development of dynamic structural models that capture the conformational flexibility of SH2 domains and their role in allosteric regulation.
Application of secondary structure alignment to metagenomic data to discover novel SH2 domain variants and expand our understanding of phosphotyrosine signaling evolution.
In conclusion, secondary structure alignment represents a critical methodology that has dramatically advanced our understanding of STAT-type SH2 domain evolution, function, and therapeutic potential. By focusing on the evolutionarily conserved architectural blueprint of these domains, researchers have uncovered their ancient origin, identified novel family members across diverse species, and developed new approaches for therapeutic intervention in diseases driven by aberrant SH2 domain signaling. As structural bioinformatics continues to evolve, secondary structure alignment will remain an essential tool for deciphering the complex relationship between protein sequence, structure, function, and evolution.
Src Homology 2 (SH2) domains are protein-protein interaction modules that play an indispensable role in tyrosine phosphorylation-mediated signal transduction, a regulatory mechanism critical for fundamental cellular processes including proliferation, differentiation, and apoptosis [4] [38]. These domains, of which approximately 120 are encoded in the human genome, achieve signaling specificity by recognizing and binding to short peptide sequences containing phosphorylated tyrosine residues (pTyr) [39]. The high sequence conservation of SH2 domains across evolution underscores their fundamental role in metazoan cell communication systems, with their expansion coinciding with increasing organismal complexity [4]. This technical guide provides a comprehensive framework for characterizing the binding affinity and specificity of SH2 domain-phosphopeptide interactions, with particular emphasis on methodologies relevant to STAT-family SH2 domains and their conservation patterns. Accurate determination of dissociation constants (Kd) is paramount for understanding physiological signaling mechanisms, identifying pathological disruptions, and developing targeted therapeutic interventions [39].
Structural and Evolutionary Basis of SH2 Domain Specificity
Molecular Recognition Mechanisms
SH2 domains employ a conserved structural framework to achieve diverse binding specificities. The domain typically consists of 4-6 beta strands flanked by two alpha helices, forming a compact structure [40]. Recognition of phosphotyrosine-containing peptides occurs through two adjacent binding pockets: a highly conserved pTyr-binding pocket that interacts with the phosphorylated tyrosine side chain, and a specificity-determining pocket that recognizes residues C-terminal to the pTyr, typically with strong preference for the +3 position [39]. This dual-pocket architecture enables SH2 domains to bind pTyr motifs with nanomolar affinities while discriminating between different sequence contexts.
The structural constraints governing SH2 domain evolution manifest clearly in population-level genetic variation. Recent analyses of missense variants across human populations reveal that residues critical for phosphopeptide binding and structural integrity show significant depletion of variation, indicating strong selective constraint [9]. These evolutionarily conserved positions are predominantly buried within the protein core or participate directly in binding interactions, highlighting the relationship between structural functional constraints and evolutionary conservation patterns in SH2 domains [9].
Specificity Profiles of SH2 Domain Classes
Different SH2 domain families exhibit distinct recognition specificities, which can be quantified using peptide library approaches:
- SHP2 SH2 Domains: The N-SH2 domain of SHP2 displays broader specificity, while the C-SH2 domain shows more restricted binding preferences [41]. Both domains recognize Gab2 scaffolding protein through specific phosphotyrosine motifs, with the C-SH2 domain employing a highly conserved histidine residue for interaction with negative charges on the phosphotyrosine [40].
- SFK SH2 Domains: Src Family Kinase SH2 domains recognize motifs with the consensus pY-(I/V/L)-X-(I/V/L) [39]. Despite high sequence conservation, monobody technology has achieved unprecedented discrimination between SrcA (Yes, Src, Fyn, Fgr) and SrcB (Lck, Lyn, Blk, Hck) subgroup SH2 domains [39].
- Adaptor SH2 Domains: SH2 domains in proteins such as Grb2, CRK, and PIK3R1 exhibit distinct recognition patterns, with CRK preferring pY-X-X-(I/P) and PIK3R1 binding pY-(M/I/L/V/E)-X-M motifs [38].
Table 1: Representative SH2 Domain Specificity Profiles
| SH2 Domain | Representative Recognition Motif | Reported Kd Range | Biological Context |
|---|---|---|---|
| SHP2 N-SH2 | pY-(I/V/L)-X-(I/V/L) [38] [39] | Low nM [42] | Broad specificity; autoinhibition |
| SHP2 C-SH2 | Requires specific Gab2 sequence [40] [41] | -- | Orients ligand binding |
| SFK SH2 | pY-(I/V/L)-X-(I/V/L) [39] | -- | Kinase autoinhibition & substrate recruitment |
| CRK SH2 | pY-X-X-(I/P) [38] | -- | Adaptor protein signaling |
| PIK3R1 (p85) SH2 | pY-(M/I/L/V/E)-X-M [38] | Low nM [42] | PI3K signaling pathway |
Experimental Methodologies for Affinity and Specificity Determination
Binding Affinity Assays
Multiple biophysical techniques enable quantitative determination of SH2 domain-phosphopeptide interaction parameters:
Isothermal Titration Calorimetry (ITC) ITC provides direct measurement of binding affinity (Kd), stoichiometry (n), and thermodynamic parameters (ΔH, ΔS). For SH2 domain interactions, ITC has confirmed low nanomolar affinities for high-specificity interactions, with monobody-SH2 complexes exhibiting Kd values in this range [39]. The technique requires purified SH2 domains and phosphopeptides at concentrations typically above 10μM for detectable heat signals.
Surface-Based Binding Assays Biosensor-based methods (SPR, BLI) enable real-time monitoring of association and dissociation kinetics. These approaches have revealed complex binding mechanisms for SH2 tandems, where the N-SH2 and C-SH2 domains can exhibit cooperative interactions [41]. The immobilization strategy (domain vs peptide capture) significantly influences measured affinities and requires careful optimization.
Competition Binding Assays Quantitative competition assays demonstrate that closely related SH2 domains from proteins such as GAP and p85 bind to equivalent or overlapping sites on tyrosine-phosphorylated receptors [42]. These assays provide critical information about binding site occupancy and potential therapeutic competition even when absolute Kd values are similar.
Figure 1: Experimental Workflow for SH2 Binding Characterization
Quantifying Binding Kinetics and Energetics
The binding mechanism between SH2 domains and phosphopeptides involves complex kinetic pathways. Studies of the SHP2 C-SH2 domain binding to Gab2-derived peptides reveal that electrostatic interactions dominate the early recognition events, with a highly conserved histidine residue playing a critical role in phosphotyrosine coordination [40]. Folding and binding kinetic analyses using stopped-flow methodology demonstrate that SH2 domains can follow three-state folding mechanisms with high-energy metastable intermediates, and that pH significantly influences the folding landscape [40] [41].
For tandem SH2 domain proteins such as SHP2, the binding kinetics reveal a dynamic interplay between domains. When both SH2 domains in the tandem are engaged with their specific ligands, the microscopic association rate constant can be modulated compared to isolated domains [41]. This phenomenon highlights the importance of studying SH2 domains in their native supramodular contexts to fully understand their physiological binding mechanisms.
Research Reagent Solutions
Table 2: Essential Research Reagents for SH2 Domain Binding Studies
| Reagent Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Expression Systems | E. coli recombinant SH2 domains [39] | Production of purified SH2 domains for binding assays | Requires optimization for solubility and phosphorylation state |
| Binding Probes | Synthetic phosphopeptides [43]; Monobodies [39] | Target for affinity measurements; high-specificity inhibitors | Peptide purity critical; monobodies enable unprecedented selectivity |
| Enrichment Materials | IMAC; TiO2 beads [43] | Phosphopeptide enrichment from complex mixtures | IMAC recovery: ~38%; TiO2 recovery: ~58% [43] |
| Detection Reagents | Isotope-labeled peptides [43]; Fluorescence dyes | SRM/MS quantification; fluorescence polarization | Heavy isotope labels enable precise quantification |
| Stability Additives | DTT (1,4-dithiothreitol) [40] | Reduction of cysteine residues in SH2 domains | Typically used at 2mM concentration to maintain reduced state |
Evolutionary Conservation Informing Functional Characterization
The evolutionary provenance of SH2 domains provides critical insights for designing binding characterization experiments. Analysis of 2.4 million population variants mapped to protein domain families reveals that missense-depleted sites in SH2 domains—those under strong selective constraint—are significantly enriched in buried residues and binding interfaces [9]. This evolutionary constraint mapping can prioritize functional residues for mutational analysis and binding studies.
STAT-family SH2 domains exhibit characteristic conservation patterns that reflect their dual roles in phosphotyrosine recognition and dimerization. Evolutionary analysis indicates that SH2 domains expanded alongside protein-tyrosine kinases to coordinate cellular complexity in metazoan evolution [4]. This co-evolution has resulted in conservation patterns where the pTyr-binding pocket remains highly conserved, while specificity-determining regions show greater diversity, reflecting their adaptation to distinct signaling contexts.
Figure 2: Evolutionary Conservation Guides Functional Studies
Advanced Applications and Therapeutic Targeting
Targeting SH2 Domains with Engineered Proteins
The development of monobodies—synthetic binding proteins based on fibronectin type III domains—has enabled unprecedented selectivity in SH2 domain targeting [39]. These reagents can discriminate between highly homologous SFK SH2 domains, with crystal structures of monobody-SH2 complexes revealing distinct and only partially overlapping binding modes. Such engineered proteins serve both as mechanistic tools for dissecting SH2 domain functions and as potential therapeutic scaffolds for inhibiting aberrant SH2-mediated signaling in disease.
Quantitative Analysis of Signaling Networks
Targeted quantification of phosphorylation dynamics using enrichment methods coupled with selected reaction monitoring mass spectrometry (SRM-MS) enables precise measurement of pathway activation states [43]. For SH2 domain-mediated signaling, this approach can quantify the temporal dynamics of phosphorylation at specific tyrosine residues that serve as SH2 docking sites, providing systems-level understanding of SH2 domain function in physiological contexts.
Comprehensive characterization of SH2 domain-phosphopeptide binding affinity and specificity requires integration of multiple biochemical and biophysical approaches. The experimental frameworks outlined in this guide, informed by evolutionary conservation principles, provide a roadmap for elucidating the molecular determinants of SH2 domain specificity. As structural and population genetic data continue to expand, the ability to precisely quantify these interactions will remain fundamental to understanding tyrosine phosphorylation signaling networks and developing targeted interventions for pathological conditions driven by their dysregulation.
Liquid-liquid phase separation (LLPS) has emerged as a fundamental physicochemical process governing the spatial organization of cellular components, while Src homology 2 (SH2) domains serve as critical readers of phosphotyrosine signaling. The convergence of these paradigms—membrane lipid interactions and biomolecular condensation—represents a transformative frontier in understanding cellular signal transduction. LLPS refers to the process whereby biomacromolecules such as proteins and nucleic acids condense into structured aggregates at the nanoscale, separating into distinct liquid-like phases within cells [44]. These biomolecular condensates function as membraneless organelles that enable efficient regulation and dynamic cellular responses, playing critical roles in maintaining cellular functions and contributing to disease pathogenesis [44] [45].
Simultaneously, emerging research reveals that SH2 domains, previously characterized primarily as phosphotyrosine-binding modules, exhibit complex interactions with membrane lipids that profoundly influence their function and specificity [46]. This whitepaper examines the integrated mechanisms through which lipid-microenvironment organization and phase separation collaborate to regulate sophisticated signaling networks, with particular emphasis on the evolutionary conservation of STAT-type SH2 domains and implications for therapeutic intervention.
Fundamental Principles of Liquid-Liquid Phase Separation
Physical Mechanisms and Driving Forces
LLPS is driven by a balance between mixing entropy and energy interactions between polymers and solvents, as explained by the Flory-Huggins theory [44]. When attractive forces between biomolecules are sufficiently strong and their concentration exceeds a critical threshold, the system spontaneously undergoes phase separation to reduce overall free energy, forming a concentrated phase enriched with biomolecules and a dilute solution phase [44]. A key feature of LLPS is the existence of this concentration threshold, beyond which phase separation occurs spontaneously [44].
The process is primarily mediated by multivalent weak interactions between intrinsically disordered regions (IDRs) and low-complexity regions (LCRs) of proteins [44] [47]. These interactions include:
- π-π stacking between aromatic residues (phenylalanine, tyrosine, tryptophan)
- Cation-Ï€ interactions between positively charged and aromatic residues
- Electrostatic interactions between charged residues
- Hydrophobic contacts [44]
IDRs are enriched in specific amino acids that facilitate these interactions, including aromatic residues, charged residues, and hydrophilic residues [44]. The structural flexibility of IDRs makes them particularly conducive to forming the reversible, weak interactions that drive phase transitions [47].
Regulation and Material Properties of Biomolecular Condensates
The formation and dissolution of biomolecular condensates are regulated by multiple factors, including:
- Post-translational modifications (phosphorylation, acetylation, methylation, ubiquitination) that alter charge and interaction valency [47]
- RNA concentration and composition that can promote or dissolve condensates [47]
- Environmental conditions such as pH, temperature, ionic strength, and osmotic pressure [44]
- Macromolecular crowding that affects effective concentration and excluded volume [44]
The material properties of condensates range from liquid-like to gel-like states, with significant functional implications [45]. These properties can be assessed through techniques such as fluorescence recovery after photobleaching (FRAP), fluorescence loss in photobleaching (FLIP), and fluorescence correlation spectroscopy (FCS) [45].
SH2 Domains as Dual-Function Interaction Modules
Canonical Phosphotyrosine Recognition
SH2 domains are protein interaction domains that direct phosphotyrosine (pY) signaling pathways with an average length of approximately 100 amino acids [46]. They feature a conserved architecture comprising two α-helices flanking antiparallel β-strands [46]. These domains specifically recognize pY and a few residues immediately C-terminal to pY using a pY-binding pocket and a secondary binding site, respectively [46].
The human genome encodes 121 SH2 domains in 111 different proteins, including kinases, adaptors, phosphatases, and other signaling molecules that control the specificity of pY signaling [46]. Quantitative analyses have revealed that SH2 domains bind pY-containing peptides with variable affinity and a significant degree of promiscuity, suggesting that additional mechanisms must contribute to signaling specificity in cellular contexts [46].
Non-Canonical Lipid Binding Properties
Genome-wide screening of human SH2 domains has revealed that approximately 90% bind plasma membrane lipids, with many exhibiting high phosphoinositide specificity [46]. These lipid interactions occur through surface cationic patches distinct from pY-binding pockets, enabling SH2 domains to bind lipids and pY motifs independently [46].
Table 1: Lipid Binding Properties of Selected SH2 Domains
| SH2 Domain | Kd for PM-mimetic Vesicles (nM) | Lipid Binding Residues | Phosphoinositide Selectivity |
|---|---|---|---|
| STAT6-SH2 | 20 ± 10 | Not specified | Not specified |
| GRB7-SH2 | 70 ± 12 | Not specified | Low selectivity |
| FRK(PTK5)-SH2 | 80 ± 12 | Not specified | Not specified |
| YES1-SH2 | 110 ± 12 | R215, K216 | PI45P2 > PIP3 > others |
| BLNK-SH2 | 120 ± 19 | Not specified | PIP3 > PI45P2 ≫ others |
| ZAP70-cSH2 | 340 ± 35 | K176, K186, K206, K251 | PIP3 > PI45P2 > others |
| Lck-SH2 | Not specified | Surface-exposed basic, aromatic, and hydrophobic residues | Low specificity [48] |
Lipid binding occurs through two primary mechanisms: (1) grooves for specific lipid headgroup recognition, or (2) flat surfaces for non-specific membrane binding [46]. These interactions are functionally significant, as demonstrated in ZAP70, where multiple lipids bind its C-terminal SH2 domain in a spatiotemporally specific manner to control protein binding and signaling activities in T cells [46].
Evolutionary Conservation of STAT-Type SH2 Domains
Structural and Functional Conservation
STAT (Signal Transducer and Activator of Transcription) proteins represent a distinct class of SH2 domain-containing transcription factors that mediate cytokine and growth factor signaling [49]. STAT activation involves phosphorylation by receptor-associated Janus kinases, receptor tyrosine kinases, or cytoplasmic tyrosine kinases, leading to STAT dimerization through reciprocal SH2 domain-phosphotyrosine interactions [49]. These dimeric STATs then translocate to the nucleus, bind specific DNA sequences, and regulate target gene transcription [49].
Comparative structural analysis reveals that STAT-type SH2 domains represent one of the most ancient forms, serving as a template for SH2 domain evolution [7]. While conventional Src-type SH2 domains contain an basic "αβββα" structure with an extra β-strand (βE or βE-βF motif), STAT-type SH2 domains feature a characteristic linker domain-conjugated SH2 domain containing the αB' motif [7].
Table 2: Evolutionary Distribution of STAT-Type SH2 Domains
| Organism | STAT/SH2 Features | Evolutionary Significance |
|---|---|---|
| Mammals (Human/Mouse) | STAT1-SH2 with conserved residues [49] | Conventional STAT signaling |
| Zebrafish | STAT SH2 with high sequence conservation [49] | Early vertebrate conservation |
| Pooled snail (Hyriopsis schlegelii) | HsSTAT with STATint, STATalpha, STAT_bind, SH2 domains [50] | Functional conservation in invertebrates |
| Arabidopsis | STAT-type linker-SH2 domain factors (STATL) [7] | Pre-dates plant-animal divergence |
| Dictyostelium | Putative SH2 domain-bearing genes [7] | Ancient eukaryotic origin |
This evolutionary conservation is exemplified by the identification of STAT-type linker-SH2 domains in Arabidopsis, designated STATL (STAT-type linker-SH2 domain factors), which are found in diverse vascular and nonvascular plants [7]. This distribution indicates that the linker-SH2 domain evolved prior to the divergence of plants and animals, highlighting its fundamental role in cellular signaling [7].
Functional Implications of Ancient SH2 Architecture
The conservation of STAT-type SH2 domains across evolutionary timescales suggests preserved functional capabilities beyond canonical phosphotyrosine signaling. Research indicates that these ancient architectures facilitate:
- Nucleocytoplasmic shuttling of unphosphorylated STATs in resting cells [49]
- Tetramerization on promoters with tandem STAT binding sites through N-terminal domain interactions [49]
- Recruitment of transcriptional co-activators such as CREB binding protein (CBP)/p300 [49]
- Protein-protein interactions with regulatory partners including PIAS1, BRCA1, and mammalian target of rapamycin [49]
The structural conservation in diverse organisms such as the pooled snail (Hyriopsis schlegelii), where HsSTAT contains four classical conservative function domains (STATint, STATalpha, STAT_bind, and SH2), further supports the functional importance of this architecture in fundamental cellular processes [50].
Integrated Experimental Approaches
Methodologies for Investigating Lipid-SH2 Interactions
Surface Plasmon Resonance (SPR) for Lipid Binding Analysis: SPR provides quantitative measurements of lipid binding affinity and specificity for SH2 domains [46]. The experimental workflow involves:
- Vesicle Preparation: Generate vesicles with lipid composition recapitulating the cytofacial leaflet of the plasma membrane (PM-mimetic vesicles) [46]
- Protein Expression: Express SH2 domains as EGFP-fusion proteins to improve expression yield and stability [46]
- Binding Measurements: Immobilize lipid vesicles and measure SH2 domain binding kinetics
- Affinity Determination: Calculate dissociation constants (Kd) from binding curves [46]
This approach enabled the systematic characterization of 76 human SH2 domains, revealing that 74% have submicromolar affinity for PM-mimetic vesicles [46].
NMR and Mutational Analysis for Binding Site Mapping: Nuclear Magnetic Resonance (NMR) spectroscopy combined with mutational studies identifies specific lipid-binding residues:
- Electrostatic Potential Calculation: Predict membrane interaction surfaces
- NMR Chemical Shift Perturbation: Identify residues affected by lipid binding
- Site-Directed Mutagenesis: Validate functional contributions of specific residues [48]
Using this approach, researchers identified that the Lck SH2 domain lipid-binding site comprises surface-exposed basic, aromatic, and hydrophobic residues distinct from the phosphotyrosine-binding pocket [48].
Approaches for LLPS Characterization
In Vitro Reconstitution Assays: LLPS can be studied using purified components to determine specific phase separation conditions:
- Protein Purification: Express and purify target proteins from E. coli, yeast, or insect cells [45]
- Droplet Formation Assays: Monitor condensate formation under varying conditions (pH, temperature, salt concentration) [45]
- Component Titration: Assess the effects of RNA, partner proteins, or small molecules on phase behavior [47]
This approach allows systematic manipulation of factors known to influence LLPS, including RNA concentration, post-translational modifications, and ionic strength [45].
Imaging-Based Material Property Assessment: Advanced microscopy techniques characterize the physical properties of biomolecular condensates:
- Fluorescence Recovery After Photobleaching (FRAP): Measure fluidity and dynamics by monitoring fluorescence recovery post-bleaching [45]
- Super-Resolution Microscopy: Resolve fine structural details of condensates, such as core-shell architectures [45]
- Electron Microscopy: Visualize condensates in a label-free manner [45]
These techniques revealed that the material properties of condensates (liquid-like vs. gel-like) have functional consequences, as demonstrated with SARS-CoV-2 N protein condensates [45].
Optogenetic Manipulation in Living Cells: The optoDroplet system enables spatiotemporal control of LLPS in vivo:
- Domain Fusion: Fuse Cry2 (an Arabidopsis thaliana protein domain that oligomerizes with blue light) to IDRs of target proteins with fluorescent tags [45]
- Light Activation: Use blue light to induce condensate formation [45]
- Functional Assessment: Monitor biological consequences of controlled condensate assembly [45]
This system facilitates investigation of LLPS roles in promoting biological function or dysfunction in living cells [45].
Research Reagent Solutions Toolkit
Table 3: Essential Reagents and Tools for Lipid-LLPS Research
| Category | Specific Reagents/Tools | Function/Application | Example Use |
|---|---|---|---|
| Lipid Binding Assays | PM-mimetic lipid vesicles [46] | Recapitulate cytofacial leaflet of plasma membrane | SPR analysis of SH2 domain binding [46] |
| Phosphoinositide-containing vesicles [46] | Assess lipid specificity | Determine PIP2 vs. PIP3 preference [46] | |
| LLPS Reconstitution | Purified IDR-containing proteins [45] | In vitro droplet formation | Test phase separation conditions [45] |
| 1,6-hexanediol [45] | LLPS disruption agent | Confirm liquid-like properties of condensates [45] | |
| Imaging & Visualization | FRAP/FLIP/FCS [45] | Measure condensate dynamics | Assess material properties [45] |
| Super-resolution microscopy [45] | High-resolution condensate imaging | Reveal core-shell architectures [45] | |
| Electron microscopy [45] | Label-free condensate visualization | Ultrastructural analysis [45] | |
| In Vivo Manipulation | OptoDroplet system (Cry2-IDR fusions) [45] | Spatiotemporal control of LLPS | Light-inducible condensate formation [45] |
| Computational Tools | D2P2 database [45] | Predict disorder and binding sites | Identify potential LLPS-driving regions [45] |
| DrLLPS database [51] | Comprehensive LLPS-related genes | Screen for LLPS-associated factors [51] | |
| Nlrp3-IN-30 | Nlrp3-IN-30, MF:C19H17F3N4O2, MW:390.4 g/mol | Chemical Reagent | Bench Chemicals |
| Usp1-IN-7 | Usp1-IN-7, MF:C27H23F4N7O2, MW:553.5 g/mol | Chemical Reagent | Bench Chemicals |
Pathophysiological Implications and Therapeutic Opportunities
Dysregulation in Disease States
The integration of lipid interactions and LLPS has significant implications for human diseases, particularly cancer and chronic liver diseases. In cancer, dysregulated LLPS contributes to tumorigenesis through multiple mechanisms:
- Oncogenic Signaling Activation: LLPS of acetylation-mediated EZH2 causes sequestration of STAT3 within condensates, leading to sustained STAT3 activation and promotion of lung tumorigenesis [51]
- DNA Damage Response Modulation: In colorectal cancer, SUMOylated RNF168 undergoes LLPS, forming nuclear condensates that reduce DNA damage response efficiency [51]
- Transcriptional Dysregulation: Cancer cells exhibit altered transcriptional condensates that drive abnormal gene expression programs [45] [47]
In chronic liver diseases, LLPS dysregulation is linked to pathological progression of non-alcoholic fatty liver disease (NAFLD), liver fibrosis, and hepatocellular carcinoma (HCC) [44]. LLPS mediates these disease processes by regulating key mechanisms including lipid metabolism, inflammatory responses, and cell death [44].
Therapeutic Targeting Strategies
Several strategies have emerged for targeting pathological LLPS and lipid interactions:
- Small-Molecule Inhibitors: Compounds that specifically modulate phase separation [44]
- Protein Modification Regulators: Agents that control post-translational modifications (phosphorylation, acetylation) that influence LLPS [44] [47]
- RNA Interference: Approaches to restore LLPS homeostasis by targeting key condensate components [44]
These strategies hold potential for mitigating disease progression and preventing transitions to more severe pathological states, such as the transition from NAFLD to fibrosis and liver cancer [44].
The convergence of lipid interaction biology and liquid-liquid phase separation represents a paradigm shift in understanding cellular signal transduction and organization. The evolutionary conservation of STAT-type SH2 domains highlights the fundamental importance of these interaction modules across biological systems. Future research should focus on:
- Structural Integration: Determining how lipid interactions influence phase separation propensity and vice versa
- Dynamic Modeling: Developing computational models that incorporate both lipid binding and phase separation behaviors
- Therapeutic Exploitation: Leveraging these integrated mechanisms for targeted intervention in disease states
The emerging recognition that many signaling proteins, including those with SH2 domains, participate in both lipid-membrane interactions and biomolecular condensation suggests a sophisticated layering of organizational principles in cellular regulation. As research methodologies advance to better capture these dynamic processes in physiological contexts, our understanding of cellular signaling complexity will continue to evolve, revealing new therapeutic opportunities for manipulating these fundamental biological processes.
The Src Homology 2 (SH2) domain is a protein interaction module of approximately 100 amino acids that specifically recognizes and binds to phosphorylated tyrosine (pTyr) residues, thereby playing a fundamental role in orchestrating cellular signaling networks [2] [1]. Among the diverse families of SH2 domain-containing proteins, the Signal Transducer and Activator of Transcription (STAT) family, particularly its STAT-type SH2 domain, represents a critical class of transcription factors that transduce signals from cytokines and growth factors directly to the nucleus [49] [52]. The evolutionary conservation of the STAT-type SH2 domain is remarkable, with a characteristic structure distinct from Src-type SH2 domains, believed to be one of the most ancient and fully developed functional templates for phosphotyrosine signal transduction [7]. Its central role is to mediate the reciprocal pTyr-SH2 interaction that drives STAT dimerization—a key step for nuclear translocation, DNA binding, and the regulation of target genes involved in cell proliferation, survival, and immune responses [49] [2].
The dysregulation of STAT signaling, particularly through constitutive activation of STAT3 and STAT1 in cancers and inflammatory diseases, makes their SH2 domains a high-priority target for therapeutic intervention [52] [2]. Targeting the SH2 domain offers a strategic mechanism to block the pathogenic protein-protein interactions that drive oncogenic signaling, presenting an attractive alternative to traditional catalytic kinase inhibitors [53]. This technical guide outlines the process of discovering small-molecule inhibitors targeting the STAT-type SH2 domain, employing high-throughput virtual screening (HTVS) methodologies rooted in an understanding of its evolutionarily conserved structure and function. We frame this process within the context of a broader thesis on evolutionary conservation, which informs the strategic targeting of immutable, functionally critical regions of the protein.
Evolutionary and Structural Foundations of STAT-type SH2 Domains
Evolutionary Conservation and Functional Imperative
SH2 domains first emerged in the early Unikonta and expanded alongside protein tyrosine kinases (PTKs) and tyrosine phosphatases (PTPs), coupling phosphotyrosine signaling to downstream networks in multicellular organisms [5]. STAT proteins are a central part of this evolutionary story. The STAT-type SH2 domain is defined by a unique secondary structure that differentiates it from the Src-type SH2 domain. While Src-type domains possess extra β-strands (βE and βF), the STAT-type SH2 domain lacks these strands and features a split αB helix, an adaptation that facilitates its primary function: dimerization for transcriptional regulation [7] [2]. This domain architecture is highly conserved from social amoeba (e.g., Dictyostelium) to humans, underscoring its fundamental role in one of the most ancient phosphotyrosine signaling pathways [7] [5].
The critical functional regions of the SH2 domain exhibit significant sequence conservation across species. The core binding pocket, which engages the phosphotyrosine residue, is particularly immutable. The sequence alignment of the STAT1 SH2 domain illustrates this point, showing high conservation across diverse organisms, from humans and mice to zebrafish and zebra finches [49]. This deep evolutionary conservation is not merely structural; it signifies regions of the protein that are indispensable for function. From a drug discovery perspective, targeting these conserved, functionally critical surfaces increases the likelihood of identifying inhibitors that are effective and less prone to resistance through mutation.
Structural Mechanism of Phosphopeptide Recognition
All SH2 domains share a common structural fold: a central anti-parallel β-sheet flanked by two α-helices, forming a βαβββββαβ sandwich [2] [53]. The binding of phosphotyrosine-containing peptides is mediated by two key regions on the SH2 domain surface, as illustrated in the diagram below.
The STAT SH2 domain is essential for the canonical activation pathway. Upon phosphorylation by upstream kinases, two STAT monomers dimerize via a reciprocal phosphotyrosine-SH2 domain interaction, forming an active transcription factor. The following diagram illustrates this pathway and the strategic inhibition point.
Figure 2: STAT Signaling Pathway and SH2 Domain Inhibition. Small-molecule inhibitors block the critical phosphotyrosine-SH2 domain interaction, preventing dimerization and subsequent pro-oncogenic gene expression.
High-Throughput Virtual Screening (HTVS) Workflow
The discovery of small-molecule inhibitors targeting the STAT SH2 domain leverages computational high-throughput virtual screening (HTVS) to efficiently evaluate vast chemical libraries. This multi-tiered workflow is designed to prioritize molecules with a high probability of biological activity and favorable drug-like properties. A representative workflow, integrating a specific case study, is detailed below.
Figure 3: High-Throughput Virtual Screening Workflow. A funnel-based approach for identifying STAT SH2 domain inhibitors, from initial library screening to experimental validation.
Detailed Experimental Protocols
Protocol 1: Structure Preparation and Molecular Docking
- Protein Preparation: Obtain the 3D crystal structure of the STAT SH2 domain (e.g., PDB ID: 1BF5). Remove bound water and co-crystallized ligands. Add hydrogen atoms and assign protonation states using tools like Maestro's Protein Preparation Wizard. Perform energy minimization with a force field (e.g., OPLS4) to relieve steric clashes.
- Ligand Library Preparation: Download small-molecule libraries (e.g., ZINC, FDA-approved compounds, natural products). Prepare ligands using LigPrep, generating possible tautomers, stereoisomers, and protonation states at physiological pH (7.0 ± 0.5).
- Grid Generation and Docking: Define the binding site around the conserved pTyr pocket and specificity pocket. Generate a receptor grid. Perform high-throughput docking using Glide SP or HTVS mode. Post-process results by clustering poses and analyzing key interactions (e.g., hydrogen bonds with conserved Arg, hydrophobic contacts in specificity pocket).
Protocol 2: Binding Free Energy Calculation (MM-GBSA)
- System Setup: Select top-ranking docked complexes for further analysis. Solvate the protein-ligand complex in an orthorhombic water box (e.g., TIP3P water model) with a buffer of at least 10 Ã…. Add counterions to neutralize the system's charge.
- Energy Minimization and Equilibration: Conduct a multi-step minimization and equilibration protocol using a molecular dynamics (MD) engine (e.g., Desmond). This typically involves:
- Minimization with solute heavy atoms restrained (500 ps).
- Heating to 300 K under NVT conditions (100 ps).
- Equilibration under NPT conditions (100 ps).
- Free Energy Calculation: Use the MM-GBSA method (e.g., via the
hawkscript in Schrödinger) on a set of snapshots from a short production MD simulation. Calculate the binding free energy (ΔGbind) using the equation: ΔGbind = Gcomplex - (Gprotein + G_ligand).
Protocol 3: Molecular Dynamics Simulation
- System Preparation: Prepare the system as in Protocol 2, Step 1.
- Production Run: Run an unrestrained MD simulation for a minimum of 100 nanoseconds (ns). Use a force field (e.g., OPLS4) and a time step of 2 femtoseconds. Save trajectory frames every 100 picoseconds.
- Trajectory Analysis: Analyze the stability of the protein-ligand complex by calculating the root-mean-square deviation (RMSD) of the protein backbone and ligand heavy atoms. Evaluate binding persistence by measuring ligand-protein contacts over time and calculating the radius of gyration (Rg) to assess compactness.
Data Presentation and Analysis
The quantitative data generated from HTVS must be systematically organized to enable informed decision-making for lead candidate selection. The following tables summarize key metrics from a hypothetical screening campaign targeting the STAT3 SH2 domain, inspired by published methodologies [54] [52].
Table 1: Top Virtual Screening Hits Against the STAT3 SH2 Domain
| Compound ID | Chemical Class | Docking Score (kcal/mol) | MM-GBSA ΔG (kcal/mol) | Key Interactions |
|---|---|---|---|---|
| RH-01 | Flavonoid glycoside | -12.3 | -58.9 | H-bonds with Arg609, Ser611, Tyr640; π-cation with Arg609 |
| AH-02 | Aminoglycoside | -10.1 | -45.2 | Ionic with Arg609; H-bonds with Ser611, Glu638 |
| HL-03 | Flavonoid | -9.8 | -42.7 | H-bonds with Arg609, Ser611; hydrophobic with Leu637 |
| S3I-201 | Salicylic acid derivative | -8.5 | -35.1 | H-bond with Arg609; hydrophobic with Phe637 (Reference compound [52]) |
Table 2: Predicted ADME Properties of Top Screening Hits
| Compound ID | Molecular Weight (g/mol) | cLogP | H-Bond Donors | H-Bond Acceptors | TPSA (Ų) | Rule of 5 Violations | Predicted Solubility |
|---|---|---|---|---|---|---|---|
| RH-01 | 610.5 | -1.5 | 10 | 16 | 270 | 2 (MW, HBD) | Low |
| AH-02 | 585.6 | -7.2 | 13 | 19 | 389 | 2 (MW, HBD) | High |
| HL-03 | 302.2 | 2.1 | 4 | 6 | 107 | 0 | Moderate |
| S3I-201 | 340.4 | 3.5 | 2 | 4 | 66 | 0 | Low |
The Scientist's Toolkit: Research Reagent Solutions
Successful execution of the described HTVS pipeline requires a suite of specialized software, databases, and computational resources.
Table 3: Essential Research Reagents and Tools for SH2 Domain Inhibitor Screening
| Item Name | Provider / Example | Function in Workflow |
|---|---|---|
| Protein Data Bank (PDB) | RCSB PDB (e.g., PDB 1BF5) | Source of high-resolution 3D structures of the STAT SH2 domain for docking. |
| Small-Molecule Libraries | ZINC, ChEMBL, FDA-approved/Phase-I compounds | Collections of chemically diverse, purchasable molecules for virtual screening [54]. |
| Molecular Docking Suite | Schrödinger (Glide), AutoDock Vina | Software for predicting the binding pose and affinity of ligands to the SH2 domain. |
| Molecular Dynamics Engine | Desmond (Schrödinger), GROMACS, AMBER | Software for simulating the dynamic behavior and stability of protein-ligand complexes in a solvated environment. |
| Free Energy Calculation Tool | Schrödinger (Prime/MM-GBSA) | Module for calculating the binding free energy of protein-ligand complexes from MD trajectories. |
| ADMET Prediction Software | Schrödinger (QikProp), SwissADME | Tools for predicting the absorption, distribution, metabolism, excretion, and toxicity of hit compounds in silico. |
| T3SS-IN-4 | T3SS-IN-4|T3SS Inhibitor|For Research Use | T3SS-IN-4 is a potent type III secretion system (T3SS) inhibitor for anti-virulence research. This product is For Research Use Only. Not for human or veterinary use. |
| Hsd17B13-IN-15 | Hsd17B13-IN-15, MF:C21H17ClN2O4S, MW:428.9 g/mol | Chemical Reagent |
The journey from bench to bedside for small-molecule inhibitors targeting the evolutionarily conserved STAT-type SH2 domain is a rigorous process that begins with intelligently designed high-throughput virtual screening. By leveraging the deep evolutionary conservation of the SH2 domain's structure and function, screening strategies can be optimized to target the most critical and immutable interaction surfaces. The integrated computational workflow—encompassing docking, free energy calculations, ADME profiling, and molecular dynamics simulations—serves as a powerful funnel to identify promising lead compounds like rutin hydrate and 6-hydroxyluteolin, which have shown multi-target inhibitory potential in recent studies [54].
The subsequent translational pathway requires validating these computational hits through in vitro binding assays, cell-based models to confirm inhibition of STAT phosphorylation and dimerization, and ultimately, in vivo efficacy and toxicity studies in disease-relevant animal models. The continuous refinement of screening libraries and algorithms, coupled with a growing understanding of SH2 domain biology and its non-canonical roles (e.g., in liquid-liquid phase separation [2]), promises to enhance the efficiency and success of this pipeline. By grounding this discovery process in the principles of evolutionary conservation, researchers can develop more effective and specific immunotherapeutics for cancer and other human diseases driven by aberrant STAT signaling.
Navigating Research Challenges in STAT-type SH2 Domain Studies
Overcoming Low Sequence Identity in Divergent SH2 Domains
Src homology 2 (SH2) domains are modular protein domains of approximately 100 amino acids that specifically recognize and bind to phosphotyrosine (pY) motifs, thereby facilitating critical protein-protein interactions in cellular signaling networks [30]. While all SH2 domains share a conserved structural fold, certain lineages, particularly the STAT (Signal Transducers and Activators of Transcription) family, have undergone remarkable sequence divergence through evolution [55] [56]. This divergence presents significant challenges for researchers using standard sequence-based identification methods, which often fail to recognize these non-canonical SH2 domains [55]. Understanding and overcoming these challenges is not merely a bioinformatic exercise; it is essential for elucidating the full complexity of phosphotyrosine signaling across the evolutionary tree and for exploiting these domains as therapeutic targets.
The STAT-type SH2 domain represents one of the most ancient and fully developed functional SH2 domains, serving as an evolutionary template for the subsequent diversification of the SH2 domain superfamily [55]. Research indicates that the linker-SH2 domain of STAT predates the divergence of plants and animals, highlighting its deep evolutionary conservation despite its sequence variability [55]. In organisms like Dictyostelium, STAT proteins have been identified with SH2 domains containing a 15-amino acid insertion and substitutions at the arginine residue otherwise absolutely conserved in canonical SH2 domains for phosphotyrosine binding [56]. Despite these radical sequence changes, these proteins remain biologically functional, suggesting the existence of non-canonical activation mechanisms that operate independently of orthodox SH2 domain-phosphotyrosine interactions [56]. This technical guide provides a structured approach to identifying, characterizing, and studying these divergent SH2 domains, with a particular emphasis on STAT-type domains and their evolutionary context.
Structural and Evolutionary Basis of SH2 Domain Divergence
Core Structural Conservation Amidst Sequence Variation
Despite sometimes exhibiting sequence identity as low as ~15%, all SH2 domains share a highly conserved three-dimensional fold [30]. The core structure is a sandwich consisting of a central three-stranded antiparallel beta-sheet (βB, βC, βD) flanked by two alpha helices (αA and αB) on either side [30]. This structural unity is the foundation that enables the identification of divergent SH2 domains when sequence-based methods fail.
The primary functional site—the phosphotyrosine-binding pocket—is located in the βB strand and typically contains a highly conserved arginine residue (at position βB5) that forms a critical salt bridge with the phosphate moiety of the phosphotyrosine ligand [30]. It is in this very region that the most striking divergences occur. For example, the Dd-STATb protein in Dictyostelium has a leucine substitution at this conserved arginine position, yet remarkably retains its biological function, indicating a non-canonical mode of activation [56].
Classification: Src-type vs. STAT-type SH2 Domains
Comprehensive structural alignment has revealed a fundamental division of SH2 domains into two distinct groups:
- Src-type SH2 domains: These characteristic domains possess the basic "αββα" structure plus an extra beta-strand (βE or βE-βF motif) [55]. They represent the canonical SH2 domain architecture found in a wide array of signaling proteins.
- STAT-type SH2 domains: These domains are defined by the presence of an αB' motif connected to a linker region [55]. This structural variant is now recognized as one of the most ancient SH2 domain forms.
Table 1: Key Characteristics of Src-type and STAT-type SH2 Domains
| Feature | Src-type SH2 Domains | STAT-type SH2 Domains |
|---|---|---|
| Core Structure | αA-βB-βC-βD-αB with extra βE/βF strand | αA-βB-βC-βD-αB with αB' motif |
| Conserved Arg in βB5 | Almost universally present | Sometimes substituted (e.g., Leu in Dd-STATb) [56] |
| Insertions | Rare | Common (e.g., 15-aa insertion in Dd-STATb) [56] |
| Evolutionary Origin | Later divergence | Ancient, predating plant-animal divergence [55] |
The evolutionary trajectory of SH2 domains reveals a compelling narrative of expansion and diversification. SH2 domains first emerged in the early Unikonta, with their numbers expanding dramatically in the choanoflagellate and metazoan lineages alongside the development of tyrosine kinases [5]. The correlation between the percentage of protein tyrosine kinases (PTKs) and SH2 domains in genomes is remarkably high (correlation coefficient of 0.95), indicating their co-evolution [5]. This expansion facilitated the rapid elaboration of phosphotyrosine signaling in early multicellular animals, with STAT-type SH2 domains representing an ancient template from which other forms diversified [5] [55].
Diagram 1: Evolutionary pathway of SH2 domain diversification, highlighting the ancient origin of STAT-type domains.
Experimental and Computational Strategies for Identification
Secondary Structure Prediction and Alignment
Primary structural alignment often fails to identify divergent SH2 domains due to extensive sequence variation. The most effective solution involves combining secondary structure prediction with sequence alignment to identify the characteristic SH2 fold despite low sequence conservation [55].
Protocol: Two-Dimensional Structural Alignment
- Sequence Retrieval: Compile candidate sequences from genomic databases using relaxed BLAST parameters (E-value < 0.1) to cast a wide net.
- Secondary Structure Prediction: Process candidate sequences using algorithms such as PSIPRED or JPred to predict α-helices and β-strands.
- Motif Identification: Scan predictions for the characteristic "αββα" SH2 core pattern (αA-βB-βC-βD-αB).
- Variant Detection: Specifically look for:
- Insertions between βD and αB (indicative of STAT-type domains)
- Substitutions in the conserved FLVR motif, particularly Arg βB5
- Structural Modeling: Use tools like Phyre2 or SWISS-MODEL to generate three-dimensional models and verify conservation of the SH2 fold.
This approach successfully identified novel STAT-type linker-SH2 domain factors in Arabidopsis, proving its utility for discovering divergent SH2 domains in non-metazoan systems [55].
Advanced Bioinformatics and Machine Learning
For high-throughput identification and characterization, machine learning approaches offer significant advantages over traditional methods:
Permutation-Based Logistic Regression (PEBL) Classifier This method was specifically developed to address the limitations of traditional algorithms in predicting interactions with biologically derived peptide sequences that often deviate from optimal binding motifs [57].
Table 2: Comparison of SH2 Domain Prediction Algorithms
| Algorithm | Principle | Strength | Weakness |
|---|---|---|---|
| Traditional Motif-Based | Position-specific scoring matrices from oriented peptide libraries | Excellent for optimal motifs | Poor performance on biological peptides [57] |
| SMALI | Structural modeling and affinity calculation | Good for well-characterized domains | Fails with divergent sequences |
| PEBL Classifier | Logistic regression on permuted biological peptide data | Superior for biological contexts; handles low-affinity interactions [57] | Requires substantial training data |
Implementation Protocol:
- Data Collection: Assemble a dataset of known interactions between SH2 domains and biologically derived phosphopeptides using fluorescence polarization or similar quantitative methods.
- Permutation Analysis: For each SH2 domain, randomly sample peptide sequences multiple times (e.g., 100 permutations) to establish a null distribution for amino acid frequencies at each position.
- Feature Calculation: For each amino acid at each position relative to phosphotyrosine, calculate enrichment or depletion p-values compared to the permuted background.
- Model Training: Build a classifier that sums transformed p-values for each amino acid in a query peptide to generate an interaction prediction score.
- Validation: Test classifier performance using independent datasets, such as SPOT array measurements [57].
This PEBL classifier has demonstrated significantly improved performance in predicting the interaction potential of SH2 domains with physiologically relevant peptide sequences compared to motif-based approaches [57].
Functional Characterization of Divergent SH2 Domains
High-Throughput Specificity Profiling
Once identified, determining the binding specificity of divergent SH2 domains is essential for understanding their biological roles. Bacterial peptide display provides a powerful platform for high-throughput specificity profiling [26] [58].
Protocol: Bacterial Peptide Display with Deep Sequencing
- Library Construction:
- Random Library: Create an Xâ‚…-Y-Xâ‚… library with 10â¶-10â· random 11-residue sequences with a central tyrosine for de novo motif discovery.
- Proteomic Library: Generate a library containing thousands of human proteome-derived phosphosites and their natural variants (e.g., pTyr-Var library) [26].
- Bacterial Display: Clone library into eCPX surface display vector and express in E. coli.
- Selection:
- For kinase specificity: Incubate cells with purified tyrosine kinase, then label with pan-phosphotyrosine antibody.
- For SH2 specificity: Use pre-phosphorylated libraries and incubate with biotinylated SH2 domains.
- Magnetic Separation: Isolate bound cells using avidin-functionalized magnetic beads (more efficient than FACS for large libraries).
- Deep Sequencing: Amplify and sequence DNA from selected cells using Illumina platforms.
- Data Analysis: Calculate enrichment scores for each peptide by comparing frequency before and after selection [26] [58].
Diagram 2: Workflow for high-throughput specificity profiling of SH2 domains using bacterial peptide display.
This method has been successfully used to profile sequence recognition by tyrosine kinases and SH2 domains, revealing hundreds of phosphosite-proximal mutations that impact phosphosite recognition and enabling the design of high-activity sequences [26].
Quantitative Binding Affinity Measurements
Understanding the functional consequences of sequence divergence requires quantitative assessment of binding affinity. Fluorescence polarization (FP) provides a robust solution for high-throughput determination of dissociation constants (K_D) [57].
Protocol: Fluorescence Polarization Saturation Binding Assay
- Protein Purification: Express and purify recombinant SH2 domains as monomeric proteins (≥50% monomeric by size exclusion chromatography).
- Peptide Synthesis: Synthesize target phosphopeptides with an N-terminal fluorescent tag (e.g., FITC).
- Titration Series: Incubate a fixed concentration of fluorescent peptide with increasing concentrations of SH2 domain protein.
- Polarization Measurement: Measure fluorescence polarization in millipolarization units.
- K_D Calculation: Fit data to a binding isotherm using nonlinear regression to determine dissociation constants [57].
This approach has been scaled to analyze 93 human SH2 domains against hundreds of phosphopeptides, generating over 1,000 novel peptide-protein interactions and providing quantitative data on binding specificities [57].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Divergent SH2 Domains
| Reagent/Tool | Function | Application Example |
|---|---|---|
| eCPX Display Vector | Bacterial surface display of peptide libraries | High-throughput specificity profiling [26] |
| pTyr-Var Library | Defined sequences of human phosphosites with variants | Assessing impact of natural mutations on recognition [26] |
| Xâ‚…-Y-Xâ‚… Random Library | 10â¶-10â· random 11-residue sequences | De novo motif discovery for divergent domains [26] |
| Fluorescently Labeled Peptides | FITC-conjugated phosphopeptides | Quantitative FP binding assays [57] |
| Recombinant SH2 Domains | Purified monomeric SH2 domain proteins | Structural and functional studies [57] |
| Pan-phosphotyrosine Antibodies | Recognize phosphorylated tyrosine residues | Detection in far-Western blotting and display systems [59] |
| PEBL Classifier | Machine learning prediction algorithm | Predicting interactions of divergent SH2 domains [57] |
| Dyrk2-IN-1 | Dyrk2-IN-1, MF:C29H31FN8O2S, MW:574.7 g/mol | Chemical Reagent |
| Gly-Phe-Gly-Aldehyde semicarbazone | Gly-Phe-Gly-Aldehyde semicarbazone, MF:C14H20N6O3, MW:320.35 g/mol | Chemical Reagent |
Case Study: Dd-STATb - A Paradigm of Functional Divergence
The Dictyostelium STAT protein Dd-STATb exemplifies the challenges and opportunities in studying divergent SH2 domains. Despite containing a highly aberrant SH2 domain with a 15-amino acid insertion and a leucine substitution at the conserved arginine residue (βB5) critical for phosphotyrosine binding, Dd-STATb remains biologically functional [56]. This protein plays a role in growth regulation and gene expression during early development, with null cells showing discoidin 1 overexpression [56].
Remarkably, Dd-STATb sediments as a homodimer and shows constitutive nuclear localization, even when its predicted tyrosine phosphorylation site is mutated to phenylalanine [56]. This suggests a completely non-canonical mode of activation that does not rely on orthodox SH2 domain-phosphotyrosine interactions. Studying such extreme examples of divergence provides invaluable insights into the structural plasticity of the SH2 fold and alternative mechanisms of signal transduction in evolutionary distant organisms.
Overcoming the challenges posed by low sequence identity in divergent SH2 domains requires a multidisciplinary approach that combines evolutionary biology, structural prediction, and high-throughput experimental characterization. The STAT-type SH2 domains, as ancient representatives of this protein family, offer a unique window into the evolutionary plasticity of phosphotyrosine signaling. By employing the strategies outlined in this guide—secondary structure alignment, machine learning prediction, quantitative binding assays, and functional screening—researchers can decipher the structure-function relationships of these non-canonical domains. This knowledge not only expands our understanding of signaling evolution but also opens new avenues for therapeutic intervention by revealing alternative signaling mechanisms in pathogenic organisms or disease states.
Src homology 2 (SH2) domains represent a crucial family of protein interaction modules that specifically recognize phosphotyrosine (pTyr) motifs, thereby enabling the assembly of specific signaling complexes in tyrosine kinase pathways [30] [53]. Within the human proteome, approximately 110 proteins contain SH2 domains, which have undergone significant evolutionary expansion alongside protein tyrosine kinases to coordinate complex cellular communication systems in metazoans [4] [1]. From an evolutionary perspective, SH2 domains exhibit a remarkable conservation of three-dimensional structure despite considerable sequence divergence, with some family members sharing as little as 15% pairwise sequence identity while maintaining nearly identical folds [30]. Research into the evolutionary provenance of SH2 domains reveals that they can be broadly classified into two distinct groups based on structural characteristics: the STAT-type and SRC-type SH2 domains [7]. This classification provides critical insights into the molecular evolution of phosphotyrosine signaling networks, with evidence suggesting that the STAT-type SH2 domain represents one of the most ancient and fully developed functional domains that served as a template for continuing SH2 domain evolution [7]. Understanding the structural and functional distinctions between these two SH2 domain types is essential for researchers investigating signal transduction mechanisms and developing targeted therapeutic interventions.
Structural Foundations: Comparative Architecture of SH2 Domains
Core Structural Motifs and Variations
All SH2 domains share a conserved structural core consisting of a three-stranded antiparallel beta-sheet flanked by two alpha helices, arranged in a characteristic βαβββββαβ fold [30] [53]. This fundamental "sandwich" structure—denoted as αA-βB-βC-βD-αB—provides the scaffold for phosphotyrosine recognition and binding. Despite this conserved framework, significant structural variations distinguish STAT-type and SRC-type SH2 domains, particularly in their secondary structural elements and terminal regions.
The N-terminal region of SH2 domains is highly conserved across both types and contains a deep binding pocket within the βB strand that specifically recognizes the phosphate moiety of phosphotyrosine [30]. This pocket invariably contains a critical arginine residue at position βB5 (with rare exceptions), which forms part of the conserved FLVR motif and directly engages the phosphotyrosine through salt bridge interactions [30] [14]. In contrast, the C-terminal region exhibits considerable structural variation between STAT-type and SRC-type SH2 domains, contributing to their functional specialization.
Table 1: Core Structural Features of STAT-type and SRC-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | SRC-type SH2 Domains |
|---|---|---|
| Basic Fold | Central β-sheet flanked by two α-helices | Central β-sheet flanked by two α-helices |
| Characteristic Motif | Contains αB' motif | Contains extra β-strand (βE or βE-βF motif) |
| N-terminal Region | Highly conserved with phosphotyrosine pocket | Highly conserved with phosphotyrosine pocket |
| C-terminal Region | Variable with linker domain conjugation | Variable with additional β-strands E, F, G |
| FLVR Motif | Conserved arginine at βB5 position | Conserved arginine at βB5 position |
| Representative Proteins | STAT family transcription factors | SRC, ABL, LCK tyrosine kinases |
Specialized Structural Elements and Domain Arrangements
STAT-type SH2 domains are characterized by their conjugation with a linker domain (forming the linker-SH2 domain) and the presence of an αB' structural motif [7]. This distinctive architectural arrangement appears evolutionarily ancient, with bioinformatic analyses identifying STAT-type linker-SH2 domains in diverse eukaryotic model systems including Arabidopsis, Dictyostelium, and Saccharomyces [7]. The discovery of genes encoding STAT-type linker-SH2 domains in a wide array of vascular and nonvascular plants suggests that this structural paradigm evolved prior to the divergence of plants and animals [7].
In contrast, SRC-type SH2 domains typically contain additional C-terminal beta strands (βE, βF, and βG) that are absent in STAT-type domains [7]. The presence of these extra structural elements in SRC-type domains correlates with their emergence later in evolutionary history and their specialization for specific aspects of tyrosine kinase signaling. The intervening loops between secondary structural elements also contribute to functional diversity, with SH2 domains of enzymatic proteins typically possessing longer loops compared to non-enzymatic proteins such as STATs [30]. These structural variations directly influence phosphopeptide binding specificity and affinity, enabling the functional diversification of SH2 domains across signaling networks.
Functional Implications of Structural Differences
Mechanisms of Phosphopeptide Recognition and Binding
The structural distinctions between STAT-type and SRC-type SH2 domains directly influence their mechanisms of phosphopeptide recognition and binding. While both domain types maintain the fundamental requirement for phosphotyrosine engagement, they employ different strategies for achieving binding specificity and regulating downstream signaling events.
SRC-type SH2 domains typically recognize phosphotyrosine-containing peptides through a canonical "two-pronged plug" binding mechanism, where the phosphotyrosine inserts deeply into a conserved binding pocket while residues C-terminal to the phosphotyrosine (particularly the +3 position) engage a specificity-determining region [53] [14]. This binding mode positions the peptide backbone in an extended conformation, allowing optimal contact with the SH2 domain surface. The binding energy for this interaction is dominated by the phosphotyrosine engagement, which accounts for approximately half of the free energy of binding through interactions with the invariant arginine at the βB5 position of the FLVR motif [14].
STAT-type SH2 domains employ variations on this binding theme, with their unique structural features enabling distinct regulatory mechanisms. The conjugation of the SH2 domain with a linker region in STAT proteins facilitates specific conformational changes upon phosphorylation that are essential for STAT dimerization, nuclear translocation, and DNA binding activity [7]. This integrated structural arrangement allows STAT-type SH2 domains to participate in both signal reception and transcriptional activation, representing a functional adaptation of the core SH2 fold for nuclear signaling.
Roles in Cellular Signaling and Disease
The structural and functional differences between STAT-type and SRC-type SH2 domains underpin their specialized roles in cellular signaling pathways and their differential involvement in human diseases. SRC-type SH2 domains are frequently found in cytoplasmic signaling proteins including adaptors, kinases, and phosphatases, where they facilitate the assembly of signaling complexes in response to tyrosine phosphorylation [30] [53]. These domains typically exhibit moderate binding specificity, allowing them to participate in overlapping signaling networks while maintaining preference for specific sequence contexts C-terminal to the phosphotyrosine residue.
STAT-type SH2 domains function primarily in the JAK-STAT signaling pathway, where they mediate the recruitment of STAT transcription factors to activated cytokine receptors [30]. Following phosphorylation by JAK kinases, STAT proteins undergo SH2 domain-mediated homodimerization or heterodimerization, leading to their nuclear translocation and regulation of target gene expression. The specialized structure of STAT-type SH2 domains enables this dual functionality—both receptor engagement and protein dimerization—within a single domain architecture.
Table 2: Functional Roles of STAT-type and SRC-type SH2 Domains in Cellular Signaling
| Functional Aspect | STAT-type SH2 Domains | SRC-type SH2 Domains |
|---|---|---|
| Primary Signaling Role | JAK-STAT pathway; transcription factor regulation | Tyrosine kinase signaling; adaptor functions |
| Cellular Localization | Cytoplasmic and nuclear | Predominantly cytoplasmic |
| Dimerization Capacity | Homodimerization and heterodimerization | Typically monomeric or heterodimeric |
| Disease Associations | Cancer, immune disorders | Cancer, immunodeficiencies, bone disorders |
| Therapeutic Targeting | STAT3 inhibitors in clinical development | Src, Grb2 inhibitors extensively studied |
Mutations in both STAT-type and SRC-type SH2 domains have been implicated in human diseases, particularly cancers and immunodeficiencies [1] [60]. For example, gain-of-function mutations in STAT3 SH2 domain are associated with various malignancies, while loss-of-function mutations in SRC-type SH2 domains of BTK and ZAP70 can cause immunodeficiencies such as X-linked agammaglobulinemia and severe combined immunodeficiency [1]. Understanding the structure-function relationships of these distinct SH2 domain types provides critical insights for developing targeted therapies that specifically disrupt pathogenic signaling interactions.
Experimental Approaches for SH2 Domain Characterization
Structural Biology Methodologies
The elucidation of structural differences between STAT-type and SRC-type SH2 domains relies on a combination of experimental techniques that provide high-resolution information about domain architecture and ligand interactions. X-ray crystallography has been instrumental in determining the three-dimensional structures of numerous SH2 domains, with over 70 SH2 domain structures experimentally solved to date [30]. This technique enables precise mapping of the binding interfaces and conformational changes associated with phosphopeptide engagement.
For dynamic studies of SH2 domain behavior, nuclear magnetic resonance (NMR) spectroscopy provides valuable insights into domain flexibility, binding kinetics, and transient interactions. NMR has been particularly useful for characterizing the structural transitions that occur upon ligand binding and for identifying allosteric regulatory mechanisms. More recently, cryo-electron microscopy (cryo-EM) has emerged as a powerful tool for studying larger SH2-containing complexes and membrane-proximal signaling assemblies that have proven challenging for traditional crystallographic approaches.
Biochemical and Biophysical Assessment Techniques
Comprehensive characterization of SH2 domain function requires quantitative assessment of binding affinity and specificity. Isothermal titration calorimetry (ITC) provides direct measurements of binding thermodynamics, enabling determination of dissociation constants (Kd), stoichiometry (n), and thermodynamic parameters (ΔH, ΔS) for SH2-phosphopeptide interactions [60]. Surface plasmon resonance (SPR) offers complementary information about binding kinetics, including association (ka) and dissociation (kd) rate constants, through real-time monitoring of molecular interactions.
Phage display and combinatorial peptide library screening represent powerful approaches for defining the sequence specificity of SH2 domains [53]. These techniques have revealed that while SRC-type SH2 domains typically recognize specific motifs C-terminal to the phosphotyrosine, STAT-type SH2 domains may exhibit distinct specificity profiles influenced by their linker regions and dimerization properties. Fluorescence polarization assays provide a high-throughput alternative for validating binding specificities and screening potential inhibitors of SH2 domain interactions.
Research Workflow for SH2 Domain Characterization
Evolutionary Conservation and Diversification Patterns
Phylogenetic Distribution and Conservation
The evolutionary history of SH2 domains reveals distinct patterns of conservation and diversification between STAT-type and SRC-type domains. STAT-type SH2 domains represent evolutionarily ancient forms, with homologs identified in diverse eukaryotic lineages including plants, social amoebae, and yeast [7]. The presence of STAT-type linker-SH2 domains in Arabidopsis and other plant species indicates that this architectural paradigm predates the divergence of plant and animal lineages, suggesting its fundamental role in early eukaryotic signaling.
In contrast, SRC-type SH2 domains exhibit a more restricted phylogenetic distribution, emerging later in evolutionary history and undergoing substantial expansion in metazoans [4] [7]. The co-evolution of SRC-type SH2 domains with tyrosine kinases correlates with increasing multicellular complexity and the development of specialized cell communication systems in animals. This differential evolutionary history has profound implications for understanding the structural constraints and functional adaptations of these two SH2 domain classes.
Mechanisms of Functional Diversification
The diversification of STAT-type and SRC-type SH2 domains has occurred through several evolutionary mechanisms, including gene duplication, domain shuffling, and selective modification of binding specificities. Gene duplication events have enabled the functional specialization of SH2 domains, allowing copies to acquire new specificities while preserving essential functions in ancestral copies [4]. Domain shuffling has created novel combinatorial arrangements, with SH2 domains appearing in conjunction with diverse catalytic and protein-interaction modules including kinase domains, phosphatase domains, SH3 domains, and DNA-binding domains [30] [4].
Modifications in binding specificity have been achieved through mutations in key residues lining the phosphotyrosine pocket and specificity-determining regions. For instance, point mutations in the EF loop region can dramatically alter peptide binding preferences, as demonstrated by the conversion of Src SH2 domain specificity to Grb2-like preference through a single Thr to Trp substitution [53]. Such evolutionary tinkering with binding specificity has enabled the functional diversification of SH2 domains while maintaining the core structural scaffold and phosphotyrosine dependence.
Research Toolkit: Essential Reagents and Methodologies
Table 3: Research Reagent Solutions for SH2 Domain Studies
| Reagent/Method | Function/Application | Technical Considerations |
|---|---|---|
| Recombinant SH2 Domains | Structural and biophysical studies; binding assays | Define domain boundaries carefully; often require phosphopeptide for stability |
| Phosphotyrosine Peptide Libraries | Specificity profiling; binding motif identification | Include diverse flanking sequences; proper phosphorylation critical |
| ITC & SPR Instrumentation | Quantitative binding affinity and kinetics | Requires purified components; controls for non-specific binding |
| X-ray Crystallography | High-resolution structure determination | May require engineered constructs; co-crystallization with peptides often needed |
| NMR Spectroscopy | Solution studies; dynamics and folding | Isotope labeling required; size limitations for larger domains |
| Phage Display Systems | Rapid specificity profiling; engineered binders | Library diversity critical; panning conditions affect outcomes |
| Cellular Signaling Assays | Validation of physiological relevance | Context-dependent results; redundancy considerations important |
Therapeutic Targeting and Future Directions
Current Targeting Strategies
The structural and functional differences between STAT-type and SRC-type SH2 domains have important implications for therapeutic development. SRC-type SH2 domains have been extensively targeted for drug development, with inhibitors of Grb2 and Src SH2 domains representing advanced candidates for targeting Ras pathway activation and osteoclastic bone resorption, respectively [53]. The well-defined binding pockets and characterized specificity determinants of SRC-type SH2 domains facilitate structure-based drug design approaches.
STAT-type SH2 domains present more challenging targets due to their dual functionality in receptor engagement and dimerization. However, significant progress has been made in developing inhibitors targeting the STAT3 SH2 domain, with several candidates reaching clinical development [30] [61]. These inhibitors typically block STAT3 phosphorylation, dimerization, or nuclear translocation by competing with native binding partners for SH2 domain engagement. The unique structural features of STAT-type SH2 domains, particularly their linker interactions and dimerization interfaces, provide opportunities for developing highly specific inhibitors with reduced off-target effects.
Emerging Research Frontiers
Several emerging research areas are advancing our understanding of STAT-type and SRC-type SH2 domain biology. The role of SH2 domains in liquid-liquid phase separation (LLPS) represents a frontier in signal transduction research, with evidence that multivalent interactions involving SH2 and SH3 domains drive the formation of membrane-free signaling condensates [30]. For example, interactions among GRB2, Gads, and the LAT receptor contribute to phase-separated condensate formation that enhances T-cell receptor signaling [30].
Another emerging area involves the non-canonical functions of SH2 domains, including their interactions with membrane lipids. Recent research indicates that nearly 75% of SH2 domains interact with lipid molecules, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [30]. These interactions modulate cellular signaling by influencing membrane recruitment and enzymatic activity of SH2-containing proteins, with disease-causing mutations often localized within lipid-binding pockets [30]. Understanding these non-canonical functions provides new insights into the functional diversification of STAT-type and SRC-type SH2 domains and suggests novel therapeutic targeting strategies.
Structural and Functional Distinctions Between SH2 Domain Types
The structural and functional distinctions between STAT-type and SRC-type SH2 domains reflect their divergent evolutionary histories and specialized roles in cellular signaling. STAT-type SH2 domains, with their characteristic linker conjugation and αB' structural motif, represent evolutionarily ancient forms adapted for nuclear signaling and transcription factor regulation. In contrast, SRC-type SH2 domains, distinguished by additional beta strands and classical two-pronged binding mechanisms, emerged later in evolution to support complex tyrosine kinase signaling networks in metazoans. These fundamental differences inform research methodologies and therapeutic targeting strategies, with implications for understanding signal transduction mechanisms and developing treatments for cancer, immunodeficiencies, and other diseases linked to SH2 domain dysfunction. As research continues to unveil novel aspects of SH2 domain biology—including their roles in phase-separated condensates and non-canonical interactions with membrane lipids—the distinction between STAT-type and SRC-type domains provides an essential framework for advancing our understanding of cellular communication systems.
Addressing the Complexity of Redundancy and Specificity in Signaling Networks
Src homology 2 (SH2) domains represent a fundamental paradigm for understanding how specificity emerges within complex tyrosine kinase signaling networks. These approximately 100-amino acid modules specifically recognize phosphorylated tyrosine (pY) residues, directing the formation of transient protein complexes that underlie cellular communication. This technical guide examines the molecular mechanisms that enable STAT-type SH2 domains and their paralogs to achieve binding specificity despite structural conservation, focusing on both canonical pY recognition and emerging non-canonical functions. We integrate structural biology, high-throughput specificity profiling, and evolutionary analysis to provide a framework for understanding how functional redundancy and specificity coexist in phosphotyrosine signaling. The implications for targeted therapeutic development, particularly for STAT3-dependent pathologies, are discussed throughout.
SH2 domains constitute the largest class of pTyr recognition domains in the human proteome, with approximately 120 domains across 110 proteins [62]. They function as modular regulators within multidomain proteins, including enzymes, adaptors, docking proteins, and transcription factors like the STAT family [30]. Their primary function involves coupling activated protein tyrosine kinases (PTKs) to intracellular signaling pathways by recognizing specific pY-containing motifs, thereby establishing signaling networks essential for development, homeostasis, and immune responses [30] [63].
The evolutionary conservation of SH2 domains presents a fascinating paradox: despite maintaining a highly conserved structural fold, they have evolved distinct recognition specificities that enable precise signal transduction. This guide examines the molecular principles underlying this paradox, with particular emphasis on STAT-type SH2 domains as a model system for understanding how specificity is achieved within conserved architectural frameworks.
Structural Basis of SH2 Domain Specificity
Conserved Structural Architecture
All SH2 domains adopt a conserved "sandwich" fold consisting of a three-stranded antiparallel beta-sheet flanked on each side by an alpha helix, typically arranged as αA-βB-βC-βD-αB [30]. The N-terminal region contains a deep pocket within the βB strand that binds the phosphate moiety of phosphotyrosine. This pocket features an invariable arginine residue at position βB5 (part of the FLVR motif) that directly coordinates the phosphate group through a salt bridge [30]. The C-terminal region is more variable and contains additional structural elements that contribute to specificity.
Molecular Determinants of Ligand Recognition
SH2 domains recognize their ligands through two primary binding surfaces:
- The pY-binding pocket: A highly conserved pocket that engages the phosphotyrosine residue
- Specificity-determining regions: Surrounding surfaces that interact with residues C-terminal to the pY, typically at positions +1 to +5 [63]
The structural basis for specificity extends beyond simple permissive interactions that enhance binding to include non-permissive residues that actively oppose binding through steric clash or charge repulsion [63]. This complex integration of positive and negative determinants enables SH2 domains to distinguish subtle differences in peptide ligands, substantially increasing the accessible information content embedded in short linear motifs.
Table 1: Key Structural Features of SH2 Domains
| Structural Feature | Location | Functional Role | Conservation |
|---|---|---|---|
| βB5 Arginine | βB strand | Forms salt bridge with phosphate moiety | Nearly invariant |
| FLVR Motif | N-terminal | pY coordination and stabilization | Highly conserved |
| Specificity Pocket | C-terminal | Binds residues C-terminal to pY | Variable |
| BC Loop | Between βB and βC | Contacts peptide ligands | Variable |
| Lipid-binding site | Near pY pocket | Membrane association | Present in ~75% of SH2 domains |
Experimental Methodologies for Profiling SH2 Domain Specificity
High-Throughput Specificity Profiling Techniques
Peptide Microarray Technology
Advanced peptide microarray technologies enable comprehensive profiling of SH2 domain binding specificities. The tyrosine phosphopeptide chip (pTyr-chip) represents a nearly complete complement of the human phosphotyrosine proteome, containing up to 6,202 phosphopeptides (13 residues long with pTyr in the middle position) printed in triplicates with appropriate controls [62]. The experimental workflow involves:
- Membrane-based peptide synthesis using SPOT synthesis approach
- Punch-pressing peptide spots into microtiter plates
- Peptide release from cellulose discs
- Printing onto aldehyde-modified glass surfaces to create high-density chips
- Profiling with GST-tagged SH2 domains detected by fluorescent anti-tag antibodies
This approach demonstrates excellent reproducibility, with Pearson correlation coefficients of 0.7-0.99 for intra-chip comparisons and approximately 0.95 for inter-experimental replicates [62].
Quantitative Bacterial Peptide Display
Recent advances combine bacterial display of genetically-encoded peptide libraries with enzymatic phosphorylation and next-generation sequencing (NGS) to quantify binding affinities [8]. The ProBound computational framework enables transformation of selection data into quantitative sequence-to-affinity models that predict binding free energy across the full theoretical ligand sequence space. This approach provides:
- Quantitative affinity predictions in biophysically meaningful units (ΔΔG)
- Coverage of complete sequence space through degenerate libraries (10â¶-10â· sequences)
- Joint analysis of multi-round selection experiments
Figure 1: Bacterial Peptide Display Workflow for SH2 Specificity Profiling
Data Analysis and Specificity Classification
Binding data from high-throughput experiments are analyzed using computational approaches including:
- Sequence logo generation from aligned binding peptides
- Hierarchical clustering to group domains by specificity preferences
- Artificial neural networks (ANN) to predict binding for uncharacterized peptides
- Position-specific scoring matrices (PSSM) for binding site prediction
For 70 profiled SH2 domains, ANN predictors (NetSH2) demonstrated an average Pearson correlation coefficient of 0.4 between predicted and experimental binding [62]. These computational tools enable researchers to rapidly scan protein sequences for potential SH2 binding sites and predict the impact of phosphosite variants on binding affinity.
Quantitative Analysis of SH2 Domain Binding
Affinity Ranges and Specificity Classes
Quantitative binding measurements reveal that SH2 domains exhibit nanomolar to micromolar affinities for their physiological ligands, with significant variation between domains. Studies profiling 50 SH2 domains against 192 physiological phosphopeptides from FGF, insulin, and IGF-1 receptor pathways demonstrate that individual SH2 domains possess distinct recognition properties beyond previously described binding motifs [63].
Table 2: SH2 Domain Specificity Classes and Representative Members
| Specificity Class | Representative Members | Preferred Motif | Affinity Range (K_d) |
|---|---|---|---|
| Class I | Src, Fyn | pYEEI | 0.1-1 μM |
| Class II | PLCγ1 C-SH2 | pYVPV | nM range |
| Class III | PI3K p85 N-SH2 | pYMXM | 50-500 nM |
| STAT-type | STAT1, STAT3, STAT5 | pYXPQ | Varies by STAT |
| SHP2-type | PTPN11 N-SH2 | pYIXL | nM range |
Analysis of 99 human SH2 domains identified 17 distinct specificity classes based on their preference for phosphotyrosine sequence context [62]. Notably, the correlation between overall domain sequence homology and peptide recognition specificity is surprisingly poor (PCC=0.30), indicating that subtle sequence variations can significantly alter binding preferences [62].
Contextual Sequence Recognition
A fundamental insight from quantitative studies is that SH2 domains exhibit context-dependent recognition where neighboring positions affect one another, creating a complex "linguistics" of binding specificity [63]. This contextual dependence allows SH2 domains to integrate various permissive and non-permissive factors to produce sophisticated recognition profiles.
Experimental evidence demonstrates that non-permissive residues can inhibit binding through:
- Steric hindrance from bulky side chains
- Charge repulsion from acidic residues
- Structural incompatibility with binding pocket geometry
This complex recognition mechanism substantially increases the information content accessible to SH2 domains, enabling them to distinguish subtle differences in peptide ligands that would appear identical to simpler recognition models.
Evolutionary Conservation and Population Constraint in SH2 Domains
Evolutionary Conservation Patterns
Analysis of evolutionary conservation across SH2 domains reveals characteristic patterns constrained by structure and function. A unified analysis of evolutionary and population constraint mapped 2.4 million missense variants to 5,885 protein domain families, quantifying residue-level constraint with a Missense Enrichment Score (MES) [9].
Key findings for SH2 domains include:
- Buried residues show strong evolutionary conservation and missense depletion
- Binding interfaces for proteins and ligands exhibit significant constraint
- Surface-exposed residues display greater evolutionary diversity and missense enrichment
The correlation between evolutionary conservation and population constraint is remarkably strong, with 85% of protein families showing significant positive association when sufficient human paralogs exist for analysis [9].
STAT-Type SH2 Domain Conservation
STAT-type SH2 domains exhibit distinctive conservation patterns that reflect their specialized functions in signal transduction and gene regulation. These domains must maintain dual functionalities: specific phosphopeptide recognition and participation in receptor-mediated dimerization.
Analysis of evolutionary rates across STAT family SH2 domains reveals:
- Strong conservation of pY-binding pocket residues
- Variable conservation in specificity-determining regions
- Family-specific conservation patterns corresponding to functional specialization
The combination of evolutionary conservation analysis with population constraint metrics enables identification of residues critical for structural stability versus those involved in functional specificity, providing insights into potential mutational vulnerabilities.
Research Reagent Solutions
Table 3: Essential Research Reagents for SH2 Domain Studies
| Reagent/Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Expression Vectors | pGEX-2TK | Bacterial expression of GST-tagged SH2 domains | GST tag enables purification and detection |
| Peptide Array Platforms | SPOT synthesis on cellulose membranes | Semiquantitative binding specificity profiling | Addressable synthesis of 1000+ peptides |
| High-Density Peptide Chips | pTyr-chip with 6202 peptides | Comprehensive specificity profiling | Nearly complete human pY proteome coverage |
| Peptide Libraries | Oriented peptide libraries; degenerate libraries | Specificity profiling and affinity selection | 18-20 amino acid diversity at selected positions |
| Display Technologies | Bacterial peptide display | Quantitative affinity measurements | Genetically-encoded libraries with NGS readout |
| Computational Tools | ProBound; NetSH2 ANN predictors | Binding affinity prediction and data analysis | Quantitative sequence-to-affinity modeling |
Therapeutic Targeting of SH2 Domains
SH2 Domains as Drug Targets
The critical role of SH2 domains in signaling pathways, particularly in oncogenic processes, makes them attractive therapeutic targets. STAT3, in particular, has been extensively pursued due to its involvement in numerous cancers and inflammatory diseases [30]. Several targeting strategies have emerged:
- Competitive inhibitors that disrupt pY-peptide binding
- Allosteric modulators that stabilize inactive conformations
- Bifunctional compounds that target both SH2 and adjacent domains
Recent research has also revealed that approximately 75% of SH2 domains interact with lipid molecules, predominantly phosphatidylinositol-4,5-bisphosphate (PIP₂) or phosphatidylinositol-3,4,5-trisphosphate (PIP₃) [30]. These lipid-binding sites represent novel targeting opportunities, as demonstrated by the development of nonlipidic inhibitors of Syk kinase that disrupt both lipid and protein interactions [30].
Emerging Paradigms: Phase Separation and Signaling Condensates
SH2 domain-containing proteins increasingly are recognized as contributors to intracellular condensate formation through protein phase separation (PPS) [30]. Multivalent interactions mediated by SH2 and other modular domains drive condensate formation, creating specialized signaling compartments that enhance pathway specificity and efficiency.
Examples include:
- LAT-GRB2-SOS1 condensates in T-cell receptor signaling
- FGFR2:SHP2:PLCγ1 complexes in RTK signaling
- NCK-N-WASP assemblies promoting actin polymerization
This phase separation paradigm represents a new frontier for therapeutic intervention, potentially offering strategies to modulate signaling amplitude without completely abrogating pathway function.
The complexity of redundancy and specificity in SH2 domain-mediated signaling networks reflects sophisticated evolutionary optimization. STAT-type SH2 domains exemplify how conserved structural frameworks can yield highly specific functionalities through subtle variations in sequence and recognition mechanisms. The integration of high-throughput experimental profiling, quantitative computational modeling, and evolutionary analysis provides researchers with powerful tools to decipher this complexity and develop targeted therapeutic interventions. Future research will likely focus on understanding the dynamic regulation of SH2-mediated interactions in space and time, including their roles in biomolecular condensates and non-canonical signaling functions.
The Src homology 2 (SH2) domain has long been recognized as a central module in phosphotyrosine (pTyr) signaling, classically mediating specific protein-protein interactions by recognizing phosphorylated tyrosine motifs [4] [30]. However, emerging research has revealed that SH2 domains possess non-canonical functions that extend far beyond this established role, including specific lipid binding and participation in biomolecular condensate formation via liquid-liquid phase separation (LLPS) [64] [65] [2]. These findings necessitate a re-evaluation of SH2 domain functionality and the experimental approaches used to study them. Furthermore, these non-canonical functions must be understood within an evolutionary framework that recognizes the STAT-type SH2 domain as one of the most ancient and fully developed functional templates, predating the divergence of plants and animals [7]. This technical guide provides researchers with advanced methodologies for investigating these non-canonical functions, places these functions in the context of SH2 domain evolution, and offers standardized assays for quantifying lipid binding and condensate formation, thereby enabling more comprehensive analysis of SH2 domain biology in health and disease.
Evolutionary Context: The Ancient Origin of STAT-Type SH2 Domains
Understanding the non-canonical functions of SH2 domains requires appreciation of their evolutionary trajectory. Comparative genomic analyses reveal that the linker-SH2 domain of the transcription factor STAT represents one of the most ancient and fully developed functional domains, serving as an evolutionary template for SH2 domain diversification [7]. STAT-type SH2 domains are structurally distinct from Src-type domains; they lack the βE and βF strands as well as the C-terminal adjoining loop, and feature a split αB helix [2]. This structural disparity is likely an adaptation that facilitates STAT dimerization, a critical step in transcriptional regulation, and reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity [7] [2]. The discovery of STAT-type linker-SH2 domain factors (STATL) in a wide array of vascular and non-vascular plants confirms that this domain architecture evolved prior to the divergence of plants and animals [7]. This deep evolutionary conservation suggests that the fundamental structural properties of STAT-type SH2 domains have been maintained across billion years of evolution, possibly due to their optimal structural features for dimerization and their potential involvement in primordial non-canonical functions.
Lipid Binding Assays: Methodologies and Applications
Quantitative Lipid Binding Profiling
The discovery that approximately 75% of human SH2 domains interact with plasma membrane lipids represents a paradigm shift in understanding SH2 domain function [46]. These interactions occur through surface cationic patches separate from pTyr-binding pockets, enabling simultaneous binding to lipids and pTyr motifs [46]. To systematically investigate these interactions, researchers should employ the following quantitative approaches:
Surface Plasmon Resonance (SPR) Methodology:
- Liposome Preparation: Create plasma membrane-mimetic vesicles with lipid composition recapitulating the cytofacial leaflet (typically 36% phosphatidylcholine, 21% phosphatidylethanolamine, 11% phosphatidylserine, 9% phosphatidylinositol, 9% sphingomyelin, 8% cholesterol, 4% phosphatidic acid, and 2% PIP2) [46].
- Sensor Chip Functionalization: Immobilize liposomes on L1 sensor chips in running buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT, 0.005% P20 surfactant).
- Binding Measurements: Inject purified SH2 domains (as EGFP-fusions to improve expression yield) over functionalized surfaces at concentrations ranging from 1 nM to 5 μM.
- Data Analysis: Determine equilibrium dissociation constants (Kd) by fitting sensorgrams to a 1:1 Langmuir binding model. Include reference surface subtraction to correct for bulk refractive index changes.
This approach revealed that 74% of human SH2 domains have submicromolar affinity for plasma membrane-mimetic vesicles, with only approximately 10% showing no detectable binding [46]. The table below summarizes representative lipid binding affinities for selected SH2 domains:
Table 1: Lipid Binding Affinities of Selected SH2 Domains
| SH2 Domain | Kd for PM-mimetic Vesicles | Phosphoinositide Selectivity | Key Lipid-Binding Residues |
|---|---|---|---|
| STAT6-SH2 | 20 ± 10 nM | Not determined | Not determined |
| GRB7-SH2 | 70 ± 12 nM | Low selectivity | Not determined |
| FRK-SH2 | 80 ± 12 nM | Not determined | Not determined |
| YES1-SH2 | 110 ± 12 nM | PI(4,5)P2 > PIP3 > others | R215, K216 |
| BLNK-SH2 | 120 ± 19 nM | PIP3 > PI(4,5)P2 ≫ others | Not determined |
| ZAP70-cSH2 | 340 ± 35 nM | PIP3 > PI(4,5)P2 > others | K176, K186, K206, K251 |
| GRB2-SH2 | 520 ± 15 nM | Not determined | Not determined |
Cellular Validation Assays
To confirm physiological relevance of lipid binding interactions:
- Live-Cell Imaging: Express mCherry-tagged SH2 domains in appropriate cell lines and monitor plasma membrane localization before and after phosphoinositide depletion via rapamycin-induced recruitment of pseudojanin or iFYVE domains [46].
- Mutational Analysis: Introduce point mutations in cationic lipid-binding patches (e.g., lysine to glutamate) and compare membrane localization with wild-type domains.
- FRET-Based Sensors: Develop biosensors that detect proximity between SH2 domains and membrane compartments using lipid-anchored FRET acceptors.
The experimental workflow for comprehensive lipid binding analysis is illustrated below:
Diagram 1: Lipid Binding Assay Workflow
Condensate Formation Assays: Investigating Phase Separation
In Vitro Reconstitution Assays
Biomolecular condensates formed through liquid-liquid phase separation (LLPS) represent a crucial non-canonical function of multivalent SH2 domain-containing proteins [30] [65] [2]. The following methodology outlines a minimal-component system for studying SH2-mediated condensate formation:
Reconstitution Protocol:
- Component Purification: Express and purify full-length proteins or minimal multivalent constructs containing SH2 domains (e.g., Nephrin, Nck, NWASP for actin nucleation studies).
- Buffer Conditions: Use physiological buffer conditions (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT) with optional molecular crowding agents (5% PEG-8000 or Ficoll PM-70) to mimic intracellular conditions.
- Phase Separation Induction: Combine proteins at physiological concentrations (50-500 nM for scaffold proteins, 1-5 μM for client proteins) in 8-well chambered coverslips.
- Imaging and Analysis: Acquire images using differential interference contrast (DIC) or fluorescence microscopy every 30 seconds for 30-60 minutes to monitor condensate formation.
Table 2: Key Proteins in SH2 Domain-Mediated Condensates
| Condensate Complex | SH2-Containing Proteins | Biological Role | Reference |
|---|---|---|---|
| FGFR2:SHP2:PLCγ1 | SHP2, PLCγ1 | RTK Signaling | [30] |
| LAT-GRB2-SOS1 | GRB2, ZAP70, LCK, PLCγ1 | T-cell Activation | [30] |
| N-WASP–NCK | NCK | T-cell Signaling | [30] |
| SLP65, CIN85 | SLP65 | B-cell Signaling | [30] |
Computational Modeling Approaches
Complement experimental studies with computational approaches to understand condensate dynamics:
- Langevin Dynamics Simulations: Utilize bead-spring models of multivalent components with specific interaction energies between complementary stickers (SH2-pTyr, SH3-PRM) and weak non-specific background interactions [65].
- Reaction-Diffusion Modeling: Couple condensate geometry with protein diffusion and activation kinetics to model downstream signaling output.
- Parameter Scanning: Systematically vary interaction strengths (Es for specific, Ens for non-specific) to map phase diagrams and identify conditions supporting multi-condensate states.
The molecular interactions driving condensate formation are illustrated below:
Diagram 2: Condensate Assembly Mechanism
Functional Validation in Cellular Systems
To assess the functional consequences of SH2-mediated condensate formation:
- Actin Polymerization Assay: In the Nephrin-Nck-NWASP-Arp2/3 system, monitor F-actin formation using fluorescently-labeled actin or phalloidin staining after condensate induction [65].
- Signaling Output Quantification: Measure phosphorylation levels of downstream targets (e.g., STAT proteins) via Western blotting or FRET-based biosensors.
- Condensate Disruption Experiments: Introduce 1,6-hexanediol (5-10%) to disrupt hydrophobic interactions in condensates or use dominant-negative SH2 mutants to test functional necessity.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Non-Canonical SH2 Domain Studies
| Reagent/Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Lipid Binding Assays | PI(4,5)P2, PIP3 vesicles | SH2 domain membrane recruitment studies | Use natural lipid composition with 2-5% phosphoinositides |
| SPR Consumables | L1 sensor chips | Liposome immobilization for binding studies | Maintain lipid integrity with 1 mM DTT in running buffer |
| Fluorescent Tags | mCherry, EGFP | Protein localization and dynamics | N-terminal fusions improve expression yield for problematic SH2 domains [46] |
| Phase Separation Inducers | PEG-8000, Ficoll PM-70 | Molecular crowding to mimic intracellular environment | Optimize concentration (2-10%) for specific SH2 domain systems |
| Computational Models | Bead-spring models | Simulating multivalent interactions in condensates | Parameterize with Es=3-5kT for specific, Ens=0.4-0.5kT for non-specific interactions [65] |
| Cellular Perturbation Reagents | 1,6-hexanediol, Rapamycin | Condensate disruption, Acute phosphoinositide depletion | Titrate concentration to avoid nonspecific effects (5-10% for 1,6-hexanediol) |
The experimental frameworks outlined in this guide provide standardized methodologies for investigating the non-canonical functions of SH2 domains, particularly lipid binding and condensate formation. When applying these approaches, it is essential to consider the evolutionary context of the specific SH2 domain under investigation, particularly whether it belongs to the ancient STAT-type or more derived Src-type structural categories [7] [2]. The growing understanding of these non-canonical functions not only expands our fundamental knowledge of SH2 domain biology but also opens new therapeutic avenues. Targeting lipid-binding interfaces or specifically disrupting pathogenic condensates offers promising strategies for modulating SH2 domain function in cancer, immunodeficiencies, and other diseases [30] [46]. By employing the comprehensive assay systems described herein, researchers can systematically characterize these non-canonical functions across the diverse family of SH2 domains, ultimately leading to a more complete understanding of their roles in health and disease.
Strategies for Targeting Challenging Protein-Protein Interactions with Drugs
Protein-protein interactions (PPIs) are fundamental to cellular signaling and transduction, making them attractive therapeutic targets. However, a significant portion of the proteome has been historically classified as "undruggable" due to several inherent challenges. It is estimated that only 15% of drug targets (including enzymes, ion channels, and receptors) are considered druggable, while the remaining 85% fall into the undruggable category [66]. These challenging targets typically exhibit one or more of the following characteristics: lack of deep hydrophobic pockets suitable for small-molecule binding, function through extensive protein-protein interfaces, highly conserved active sites among protein family members, and intrinsically disordered regions or unknown tertiary structures [66].
Among the most challenging PPI classes are those mediated by Src Homology 2 (SH2) domains, which specifically recognize and bind phosphotyrosine (pY) motifs. SH2 domains are approximately 100 amino acids long and are crucial for phosphotyrosine-mediated signaling networks, inducing proximity of protein tyrosine kinases and phosphatases to specific substrates and signaling effectors [2]. The human proteome contains roughly 110 SH2 domain-containing proteins, which function as modular regulators in diverse multidomain proteins including enzymes, signaling regulators, adapter proteins, docking proteins, transcription factors, and cytoskeleton proteins [2]. This review will explore innovative strategies to overcome these challenges, with particular focus on the evolutionary conservation of STAT-type SH2 domains as a case study in targeting difficult PPIs.
SH2 Domain Structure and Function: A Focus on STAT-Type Domains
Structural Fundamentals of SH2 Domains
All SH2 domains share a conserved structural fold despite varying sequence identity, which can be as low as 15% among family members. The canonical structure consists of a three-stranded antiparallel beta-sheet flanked on each side by an alpha helix, forming an αA-βB-βC-βD-αB "sandwich" [2]. The N-terminal region contains a deep pocket within the βB strand that binds the phosphate moiety of phosphotyrosine, featuring an invariable arginine at position βB5 (part of the FLVR motif) that directly engages the pY residue through a salt bridge [2].
STAT-type SH2 domains exhibit distinct structural adaptations that differentiate them from SRC-type domains. Notably, STAT-type domains lack the βE and βF strands and the C-terminal adjoining loop present in other SH2 domains. Additionally, their αB helix is split into two separate helices [2]. These structural modifications represent evolutionary adaptations that facilitate the dimerization function critical for STAT-mediated transcriptional regulation, reflecting ancestral functions that predate animal multicellularity, as observed in Dictyostelium which employs SH2 domain/phosphotyrosine signaling for transcriptional regulation [2].
Binding Characteristics and Specificity Determinants
SH2 domain binding is characterized by a combination of high specificity toward cognate pY ligands with moderate binding affinity (Kd typically 0.1–10 µM) [2]. This balanced affinity allows for specific yet transient interactions suitable for dynamic cellular signaling. Specificity is achieved through interactions with residues C-terminal to the phosphotyrosine, particularly the +1 to +3 positions, which engage in complementary interactions with specificity-determining regions of the SH2 domain, primarily the EF loop (joining β-strands E and F) and the BG loop (joining α-helix B and β-strand G) [2].
Table 1: Key Structural Features of STAT-type vs. SRC-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | SRC-type SH2 Domains |
|---|---|---|
| Beta strands | Lacks βE and βF strands | Contains βE and βF strands |
| αB helix | Split into two helices | Single continuous helix |
| C-terminal loop | Lacks adjoining loop | Contains adjoining loop |
| Primary function | Facilitates dimerization for transcription | Signal transduction scaffolding |
| Evolutionary origin | Predates animal multicellularity | Metazoan signaling adaptation |
Emerging Strategies for Targeting SH2 Domain-Mediated PPIs
Direct Small-Molecule Inhibition
Direct targeting of SH2 domains with small molecules represents a promising therapeutic strategy. The pY binding pocket of SH2 domains is typically divided into three sub-pockets: pY+X (hydrophobic side), pY+0 (binds pY705), and pY+1 (binds L706 in STAT3) [67]. Successful targeting requires compounds that can effectively compete with endogenous pY-containing peptides while achieving sufficient selectivity among closely related SH2 domains.
Recent advances in targeting the STAT3 SH2 domain demonstrate this approach's potential. Computational screening of 182,455 natural compounds identified several promising inhibitors with high binding affinity to the SH2 domain [67]. The top candidates, including ZINC255200449, ZINC299817570, ZINC31167114, and ZINC67910988, exhibited docking scores ranging from -10.5 to -12.3 kcal/mol and favorable pharmacokinetic properties [67]. Molecular dynamics simulations confirmed the stability of these complexes, with ZINC67910988 showing particular promise for further development.
Allosteric and Alternative Modulation Strategies
Beyond direct inhibition, several innovative approaches are emerging for targeting SH2 domain-mediated PPIs:
PROTAC-Based Degradation: Proteolysis Targeting Chimeras (PROTACs) represent a novel strategy that moves beyond traditional occupancy-driven pharmacology. These bifunctional molecules simultaneously bind the target protein and an E3 ubiquitin ligase, leading to ubiquitination and proteasomal degradation of the target [66]. This approach has shown promise for targets traditionally considered undruggable, including KRAS mutants [66].
Stabilization of Inactive States: Some successful strategies involve stabilizing inactive conformations of SH2 domain-containing proteins. For example, BTK SH2 domain inhibitors developed by Recludix Pharma employ a prodrug approach that achieves sustained intracellular concentrations and prolonged target engagement [68] [69]. This strategy demonstrates superior selectivity compared to kinase domain-targeting inhibitors, avoiding off-target effects on TEC kinase and associated platelet dysfunction [68].
Targeting Non-Canonical Functions: Emerging research reveals that approximately 75% of SH2 domains interact with membrane lipids, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [2]. These interactions involve cationic regions near the pY-binding pocket, flanked by aromatic or hydrophobic residues. Targeting these lipid-binding interfaces represents an alternative strategy for modulating SH2 domain function, as demonstrated by the development of nonlipidic inhibitors of Syk kinase that block its lipid protein interactions [2].
Experimental Protocols for SH2 Domain-Targeted Drug Discovery
Computational Screening and Validation
Computational approaches provide efficient initial screening for SH2 domain inhibitors. The following protocol outlines a comprehensive in silico screening methodology:
Protein Preparation: Retrieve the SH2 domain crystal structure from the Protein Data Bank (e.g., STAT3 SH2 domain PDB: 6NJS). Process the structure using protein preparation software to add hydrogen atoms, fill missing side chains, assign bond orders, and optimize hydrogen bonding networks. Employ the OPLS3e force field for energy minimization to achieve a low-energy protein structure [67].
Ligand Library Preparation: Obtain natural compound libraries from databases such as ZINC15. Prepare ligands using LigPrep or similar tools to generate three-dimensional structures with optimized ionization states at physiological pH (7.4 ± 0.5). Generate stereoisomers and confirm chirality [67].
Molecular Docking: Establish a grid box centered on the known ligand-binding site (e.g., coordinates X:13.22, Y:56.39, Z:0.27 for STAT3 SH2 domain). Perform sequential docking using high-throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP) protocols. Validate the docking protocol by redocking the cognate ligand and calculating root-mean-square deviation (RMSD) between docked and crystallographic poses [67].
Binding Affinity Assessment: Perform Molecular Mechanics Generalized Born Surface Area (MM-GBSA) calculations to estimate binding free energies using the equation: ΔGBinding = ΔGComplex - (ΔGReceptor + ΔGLigand). More negative values indicate stronger binding potential. Utilize the OPLS3e force field and VSGB solvation model for these calculations [67].
Molecular Dynamics Simulations: Conduct simulations using Desmond or similar software with an OPLS3e force field. Solvate the protein-ligand complex in an orthorhombic water box with SPC water molecules and neutralize the system with appropriate ions. Run simulations for至少 100 ns while monitoring root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) to assess complex stability [67].
Machine Learning Approaches for PPI Prediction
Machine learning methods are increasingly valuable for predicting PPIs and identifying potential intervention points:
Feature Extraction: Convert protein sequences into Position-Specific Scoring Matrices (PSSM) using PSI-BLAST with an e-value threshold of 0.001 and three iterations. Transform PSSM matrices into uniform 20×20 matrices by calculating P̂PSSM = PPSSMT × PPSSM to handle variable sequence lengths [70].
Model Architecture: Implement a Deep Denoising Autoencoder (DAE) to extract robust feature representations. The encoder compresses input features into latent space through the function h = f(Wx + b), where f is a non-linear activation function, W is the encoder weight, and b is the encoder bias. The decoder then reconstructs the input from latent features using x̂ = f(Ŵh + b̂), where Ŵ is the decoder weight and b̂ is the decoder bias [70].
Model Training and Validation: Train the model using the CatBoost gradient boosting framework, particularly effective for datasets containing both categorical and continuous features. Validate model performance using yeast and human PPI datasets, with typical accuracy benchmarks of 97.85% and 98.49% respectively [70] [71].
Diagram 1: Machine Learning Workflow for PPI Prediction. This diagram illustrates the computational pipeline for predicting protein-protein interactions using sequence data and machine learning.
Case Study: Targeting STAT3 SH2 Domain for Cancer Therapy
STAT3 Signaling and Dimerization Mechanism
STAT3 is a key transcription factor regulating cell growth, survival, and differentiation, with constitutive activation observed in numerous cancers including breast, prostate, lung, and hematological malignancies [67]. Activation occurs through phosphorylation at tyrosine 705 (Y705), primarily driven by sustained cytokine signaling (e.g., IL-6) or growth factors (VEGF, EGF, PDGF) [67]. The SH2 domain mediates STAT3 dimerization by binding to the phosphorylated Y705 of another STAT3 molecule, forming an active dimer that translocates to the nucleus and promotes expression of proliferation and survival genes [67].
Key residues in the STAT3 SH2 domain that facilitate this interaction include Arg609, Glu594, Lys591, Ser636, Ser611, Val637, Tyr657, Gln644, Thr640, Glu638, and Trp623, which establish direct or indirect interactions with the phosphotyrosine motif [67]. Disruption of these interactions prevents dimerization and subsequent nuclear translocation, effectively inhibiting STAT3's oncogenic functions.
Successful Targeting Approaches
Several strategies have demonstrated success in targeting the STAT3 SH2 domain:
Small-Molecule Inhibitors: Compounds such as Stattic and SD36 represent well-characterized small molecules designed to target the STAT3 SH2 domain [67]. Recent computational screening has identified natural compounds with potentially superior binding characteristics. ZINC67910988 demonstrated exceptional stability in molecular dynamics simulations and favorable binding free energies (-68.23 kcal/mol) in MM-GBSA calculations [67].
Network Pharmacology: Integrating compound-target networks reveals multitarget potential and helps minimize off-target effects. This approach maps interactions within biological networks, identifying key nodes where intervention may yield maximal therapeutic benefit with reduced toxicity [67].
Combination with Predictive Modeling: Machine learning approaches that predict thermodynamic stability changes upon tyrosine phosphorylation can identify vulnerable nodes in signaling networks. One such method based on computational biophysics-informed machine learning accurately predicts destabilizing phosphorylations in both oncogenes and tumor suppressors, with ΔΔG values and local protein circuit topology features distinguishing phosphoproteins dysregulated in cancer [71].
Table 2: Experimental Results for STAT3 SH2 Domain Inhibitors
| Compound ID | Docking Score (kcal/mol) | Binding Free Energy (MM-GBSA) | Key Interactions | Stability in MD Simulation |
|---|---|---|---|---|
| ZINC255200449 | -11.2 | -64.55 kcal/mol | Arg609, Ser611, Ser636 | Stable (RMSD < 2.0 Ã…) |
| ZINC299817570 | -10.5 | -59.82 kcal/mol | Glu594, Lys591, Tyr657 | Moderate stability |
| ZINC31167114 | -11.8 | -66.74 kcal/mol | Arg609, Glu638, Trp623 | Stable (RMSD < 2.2 Ã…) |
| ZINC67910988 | -12.3 | -68.23 kcal/mol | Multiple hydrophobic and polar contacts | High stability (RMSD < 1.8 Ã…) |
The Scientist's Toolkit: Essential Research Reagents and Methods
Table 3: Key Research Reagent Solutions for SH2 Domain-Targeted Drug Discovery
| Reagent/Method | Function/Application | Key Features | Representative Examples |
|---|---|---|---|
| DNA-Encoded Libraries (DELs) | Generation of diverse compound libraries for SH2 domain screening | Custom-designed libraries targeting specific domain features | Recludix Pharma SH2 platform [68] |
| Position-Specific Scoring Matrix (PSSM) | Encoding evolutionary information from protein sequences | L×20 matrix representing conservation patterns; input for ML models | PSI-BLAST with e-value 0.001 [70] |
| Molecular Dynamics Software | Simulating protein-ligand interactions and stability | OPLS3e force field; explicit solvation models | Desmond, GROMACS [67] |
| Deep Denoising Autoencoders | Feature extraction from protein sequence data | Robust representation learning from corrupted inputs | DAE with CatBoost integration [70] |
| MM-GBSA Calculations | Binding free energy estimation | Combines molecular mechanics and solvation models | Prime MM-GBSA with VSGB solvation [67] |
| SH2-Targeted Crystallography | Structural characterization of inhibitor complexes | High-resolution mapping of binding interactions | STAT3 SH2 domain with inhibitors [67] [2] |
Future Perspectives and Concluding Remarks
The field of targeting challenging PPIs, particularly SH2 domain-mediated interactions, is rapidly evolving with several promising directions:
Integration of Artificial Intelligence: Machine learning and deep learning approaches are revolutionizing PPI prediction and drug discovery. Methods like Deep Denoising Autoencoders (DAEPPI) achieve impressive accuracy (97.85-98.49%) in predicting PPIs from sequence information alone [70]. These approaches will increasingly incorporate evolutionary conservation data to identify targetable interfaces conserved in pathogenicity but dispensable for normal function.
Expanding Therapeutic Modalities: Beyond small molecules, emerging modalities including proteolysis-targeting chimeras (PROTACs), molecular glues, and stabilized peptides offer new avenues for targeting challenging PPIs [66] [72]. The success of BTK SH2 domain inhibitors demonstrates that alternative targeting strategies can overcome limitations of traditional approaches [68] [69].
Structural Biology Advances: Improvements in cryo-electron microscopy and computational structure prediction (AlphaFold, RosettaFold) are providing unprecedented insights into PPI interfaces [72] [2]. These advances enable structure-based drug design for targets previously considered intractable.
Network Pharmacology and Polypharmacology: Understanding PPIs within broader biological networks will facilitate the design of multitarget strategies that achieve efficacy through modest modulation of multiple nodes rather than potent inhibition of single targets [67] [73]. This approach may improve therapeutic outcomes while reducing toxicity.
Targeting challenging PPIs, particularly those mediated by evolutionarily conserved domains like STAT-type SH2 domains, requires integrated approaches combining computational prediction, structural biology, and mechanistic biology. As these strategies mature, they will transform our ability to drug the undruggable, opening new therapeutic avenues for cancer, inflammatory diseases, and other conditions driven by dysregulated PPIs.
Diagram 2: Strategic Framework for Targeting Challenging PPIs. This diagram outlines the relationship between challenging PPI characteristics and corresponding targeting approaches enabled by modern technologies.
Validating Critical Roles: From Genetic Constraint to Clinical Pipelines
This whitepaper provides a comprehensive comparative analysis of two major classes of Src homology 2 (SH2) domains: the STAT-type and SRC-type. SH2 domains are protein interaction modules that specifically recognize phosphorylated tyrosine residues, playing crucial roles in cellular signal transduction. Through evolutionary, structural, and functional examination, we demonstrate that STAT-type SH2 domains represent an ancient architectural lineage with distinctive features compared to the canonical SRC-type domains. This analysis reveals significant implications for understanding phosphotyrosine signaling evolution and developing targeted therapeutic interventions.
Src homology 2 domains are approximately 100-amino-acid protein modules that specifically recognize and bind to phosphorylated tyrosine residues, thereby facilitating protein-protein interactions in cellular signaling pathways [30] [1]. First identified in the Src oncoprotein, SH2 domains have since been documented in over 110 human proteins [30] [1]. While these domains share a conserved structural fold, recent research has revealed substantial diversity in their architecture and binding mechanisms [14].
The STAT-type and SRC-type SH2 domains represent two evolutionarily and structurally distinct classes within the SH2 superfamily [7]. STAT (Signal Transducer and Activator of Transcription) proteins are transcription factors that contain SH2 domains critical for their dimerization and nuclear translocation [74]. Secondary structural analysis has revealed that the linker-SH2 domain of STAT represents one of the most ancient and fully developed functional domains, serving as an evolutionary template for SH2 domain development [7].
This technical guide provides an in-depth comparative analysis of these two SH2 domain classes, focusing on their structural characteristics, evolutionary conservation, and functional implications within cellular signaling networks, with particular relevance to drug discovery efforts targeting specific SH2 domain interactions.
Evolutionary Origins and Conservation
Evolutionary Trajectory
SH2 domains emerged early in eukaryotic evolution, with an ancestral form identified in SPT6, a transcription elongation factor present from yeast to humans [14]. This ancestral SH2 domain maintains the overall SH2 fold but binds to phosphoserine and phosphothreonine rather than phosphotyrosine, representing an evolutionary stepping stone to pTyr recognition [14]. The linker-SH2 domain of STAT is considered one of the most ancient and fully developed functional domains, serving as a template for the continuing evolution of the SH2 domain essential for phosphotyrosine signal transduction [7].
Comparative genomic analyses have identified SH2 domains in various eukaryotic model systems, including Arabidopsis, Dictyostelium, and Saccharomyces [7]. The discovery of STAT-type linker-SH2 domain factors in a wide array of vascular and nonvascular plants suggests that this domain architecture evolved prior to the divergence of plants and animals [7].
Conservation Patterns
Evolutionary conservation analysis reveals that SH2 domains are constrained by structure and function, creating patterns in residue conservation that can be exploited to predict structural features [9]. Population constraint studies mapping 2.4 million missense variants to protein domains show that missense-depleted sites in SH2 domains are enriched in buried residues or those involved in small-molecule or protein binding [9]. These constrained sites align closely with functional regions critical for maintaining SH2 domain structure and ligand recognition capabilities.
Table 1: Evolutionary Distribution of SH2 Domain Types
| Organism Category | STAT-type SH2 Presence | SRC-type SH2 Presence | Key Evolutionary Notes |
|---|---|---|---|
| Mammals | Yes (8 STAT members) | Yes (>100 proteins) | Full diversification |
| Teleost Fish | Yes (6 core subtypes) | Yes | Lineage-specific duplication |
| Plants | Yes (STATL genes) | Limited | Ancient origin predating plant-animal divergence |
| Social Amoeba | Yes | Yes | Early eukaryotic expansion |
| Yeast | Limited (SPT6) | No | Ancestral forms binding pSer/pThr |
Structural Characteristics
Canonical SH2 Domain Architecture
The fundamental SH2 domain structure consists of a central antiparallel β-sheet flanked by two α-helices, forming a compact αββα "sandwich" structure [30] [14]. This architecture creates two primary binding sites: a deep phosphotyrosine binding pocket and a specificity pocket that recognizes residues C-terminal to the phosphotyrosine [14]. The binding interaction has been described as a "two-pronged plug" mechanism where the phosphorylated peptide binds perpendicularly to the β-sheet [14].
A highly conserved arginine residue at position βB5 (part of the FLVR motif) is critical for phosphotyrosine recognition, forming a salt bridge with the phosphate moiety and contributing significantly to binding energy [30] [14]. This residue is conserved in all but three of the 120+ human SH2 domains [14].
SRC-type SH2 Domain Features
SRC-type SH2 domains represent the canonical SH2 architecture with the characteristic "αβββα" structure supplemented by an extra β-strand (βE or βE-βF motif) [7]. These domains typically recognize phosphotyrosine residues followed by a hydrophobic residue at the +3 position [14]. The pTyr binding pocket in SRC-type domains often contains a basic residue at position αA2 (Src-like) rather than at βD6 (SAP-like) [14].
STAT-type SH2 Domain Features
STAT-type SH2 domains exhibit distinct structural characteristics, most notably the presence of a linker domain-conjugated SH2 domain containing the αB' motif instead of the extra β-strand found in SRC-type domains [7]. This linker region connects the DNA-binding domain to the SH2 domain and plays a critical role in STAT dimerization and nuclear translocation [74]. STAT SH2 domains are exceptional in that they must recognize specific phosphotyrosine motifs on cytokine receptors while also participating in reciprocal phosphotyrosine-SH2 interactions between STAT monomers during dimerization [74].
Table 2: Comparative Structural Features of STAT-type vs. SRC-type SH2 Domains
| Structural Feature | STAT-type SH2 Domains | SRC-type SH2 Domains |
|---|---|---|
| Core Structure | αβββα + αB' motif | αβββα + βE/βE-βF motif |
| Linker Region | Conjugated linker domain | Typically isolated domain |
| Conserved Binding Motif | FLVR (with exceptions) | FLVRES |
| pTyr Coordination | Often βD6 basic residue | Often αA2 basic residue |
| Dimerization Capability | Reciprocal pTyr-SH2 binding | Typically monomeric |
| Biological Function | Transcription factor activation | Signal transduction adaptor |
Figure 1: Evolutionary trajectory and structural diversification of STAT-type and SRC-type SH2 domains from a common ancestral form, highlighting their distinct structural features and therapeutic applications.
Ligand Recognition and Specificity
Binding Mechanisms
SH2 domains recognize phosphorylated tyrosine residues within specific sequence contexts, with residues C-terminal to the phosphotyrosine contributing significantly to binding specificity [63] [75]. The recognition process involves both permissive residues that enhance binding and non-permissive residues that oppose binding through steric clash or charge repulsion [63]. This complex linguistics allows SH2 domains to distinguish subtle differences in peptide ligands, substantially increasing the accessible information content embedded in peptide ligands [63].
STAT SH2 domains exhibit particularly stringent specificity requirements as they must recognize specific phosphotyrosine motifs on cytokine receptors while also engaging in reciprocal interactions during STAT dimerization [74]. This dual recognition capability distinguishes them from many SRC-type SH2 domains that primarily function as adaptor modules.
Specificity Profiling
Large-scale specificity profiling of 76 human SH2 domains against oriented peptide array libraries has revealed distinct selectivity patterns between different SH2 domain classes [75]. The development of scoring matrix-assisted ligand identification has enabled prediction of binding partners for SH2-containing proteins based on these specificity profiles [75].
For STAT SH2 domains, this approach has identified key interactions in regulatory networks, while for SRC-type domains like BRDG1, novel binding motifs have been discovered, including selection for a bulky, hydrophobic residue at the P+4 position relative to the phosphotyrosine [75].
Experimental Approaches for SH2 Domain Analysis
Structural Determination Methods
The structures of approximately 70 SH2 domains have been experimentally solved to date using X-ray crystallography and NMR spectroscopy [30]. These approaches have revealed that despite sometimes having as little as 15% pairwise sequence identity, all SH2 domains assume nearly identical folds [30]. Comparison of STAT-type and SRC-type structures has been instrumental in identifying their distinguishing characteristics.
Figure 2: Experimental workflow for comparative analysis of SH2 domains, integrating structural, biophysical, and specificity profiling approaches to elucidate differences between STAT-type and SRC-type domains.
Binding Affinity Measurements
Fluorescence polarization measurements of interactions with soluble peptides and solid-phase peptide arrays (SPOT method) provide semiquantitative approaches for studying SH2 domain interactions [63]. These methods have been particularly valuable for examining the role of non-permissive residues and contextual information in determining SH2 domain binding selectivity [63].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for SH2 Domain Studies
| Reagent / Method | Application | Key Features |
|---|---|---|
| GST-SH2 Fusion Proteins | Binding assays, pull-down experiments | Recombinant expression in E. coli, purification via glutathione-Sepharose |
| Oriented Peptide Array Libraries | Specificity profiling | 192 physiological phosphotyrosine peptides, SPOT synthesis method |
| Fluorescence Polarization | Binding affinity quantification | Solution-based measurements, quantitative Kd determination |
| Phosphotyrosine Peptide Libraries | Specificity determinant mapping | Degenerate peptides, position-specific scoring matrices |
| Structural Biology Tools | 3D structure determination | X-ray crystallography, NMR spectroscopy |
Functional Implications in Signaling and Disease
Cellular Signaling Networks
STAT-type SH2 domains function primarily in the JAK-STAT signaling pathway, transducing signals from cytokine receptors directly to the nucleus to regulate gene expression [74]. Following receptor activation and STAT phosphorylation, STAT SH2 domains mediate reciprocal interactions between STAT monomers, forming dimers that translocate to the nucleus [74].
SRC-type SH2 domains participate in diverse signaling pathways, including growth factor signaling, immune receptor signaling, and cytoskeletal reorganization [30]. These domains typically function as adaptors or regulators rather than as direct transcriptional activators.
Disease Associations
Mutations disrupting SH2 domain structure or phosphotyrosine peptide binding are implicated in various diseases [1]. For STAT SH2 domains, dysregulation contributes to immune disorders and cancers through altered JAK-STAT signaling [74]. SRC-type SH2 domain mutations are associated with X-linked agammaglobulinemia and severe combined immunodeficiency [1].
The distinct functions of STAT-type and SRC-type SH2 domains necessitate different therapeutic targeting strategies. STAT SH2 domains are attractive targets for disrupting aberrant transcriptional programs in cancer and autoimmune diseases, while SRC-type SH2 domains are often targeted to modulate kinase signaling pathways.
This comparative analysis demonstrates that STAT-type and SRC-type SH2 domains represent evolutionarily and structurally distinct lineages within the SH2 superfamily. STAT-type SH2 domains, with their conjugated linker domains and αB' motifs, represent an ancient architectural class specialized for dual recognition roles in transcription factor activation. SRC-type domains exhibit the canonical SH2 fold supplemented by additional β-strands and function primarily as adaptor modules in signal transduction cascades.
Understanding these structural and functional differences has significant implications for drug discovery efforts targeting SH2 domain interactions. The distinctive features of STAT-type SH2 domains, particularly their role in STAT dimerization, offer unique opportunities for therapeutic intervention in diseases characterized by dysregulated JAK-STAT signaling. Future research leveraging emerging structural and proteomic approaches will continue to elucidate the nuanced functional specialization of these critical signaling domains within cellular networks.
The Src Homology 2 (SH2) domain is a critical protein module of approximately 100 amino acids that specifically recognizes and binds to phosphorylated tyrosine (pY) motifs, thereby facilitating key signaling events in multicellular organisms [2]. Within the human proteome, SH2 domains are found in roughly 110 functionally diverse proteins, including enzymes, adapters, and transcription factors, playing indispensable roles in development, homeostasis, and immune responses [2]. The STAT-type SH2 domain, found in Signal Transducer and Activator of Transcription (STAT) proteins, exhibits distinct structural characteristics that set it apart from Src-type SH2 domains. Notably, STAT-type domains lack the βE and βF strands present in Src-type domains and feature a split αB helix, an adaptation believed to facilitate the dimerization essential for STAT-mediated transcriptional regulation [2] [10].
The advent of large-scale sequencing has revealed the SH2 domain as a hotspot for mutations in STAT proteins, particularly STAT3 and STAT5B, with profound implications for human disease [10]. These mutations can disrupt the delicate evolutionary balance of wild-type STAT structural motifs, leading to either hyperactivated or refractory STAT mutants. The accurate interpretation of these variants, facilitated by resources like ClinVar and an understanding of molecular evolutionary signatures (MES) or population constraint, provides the foundation for novel therapeutic interventions and a deeper understanding of disease mechanisms [10] [76]. This technical guide synthesizes current structural insights, mutational landscapes, and experimental methodologies for mapping pathogenic mutations within the evolutionarily conserved framework of STAT-type SH2 domains.
Structural and Functional Mechanisms of STAT-type SH2 Domains
Canonical Structure and Phosphopeptide Binding
All SH2 domains share a conserved structural core: a central sandwich of a three-stranded antiparallel beta-sheet (βB-βC-βD) flanked by two alpha-helices (αA and αB) [2] [10]. This architecture forms two primary ligand-binding subpockets:
- The pY pocket: Binds the phosphate moiety of the phosphorylated tyrosine. This pocket contains a nearly invariant arginine residue (at position βB5) that forms a salt bridge with the phosphate group [2].
- The pY+3 pocket: Determines binding specificity by interacting with residues C-terminal to the phosphotyrosine, particularly the amino acid at the pY+3 position [10].
In STAT proteins, SH2 domain-mediated interactions are fundamental to canonical activation. Cytokine or growth factor stimulation triggers the SH2 domain-mediated recruitment of monomeric STATs to phosphorylated receptor cytoplasmic domains. Following phosphorylation, STAT proteins form parallel homodimers or heterodimers via reciprocal SH2-pY interactions, enabling nuclear translocation and DNA binding [10] [37]. The structural integrity of the SH2 domain is therefore paramount for proper STAT function, with mutations potentially altering phosphopeptide binding affinity, dimerization stability, or DNA binding capacity.
Non-Canonical Functions and Structural Dynamics
Beyond phosphotyrosine binding, SH2 domains can engage in non-canonical functions. Nearly 75% of SH2 domains interact with membrane lipids, particularly phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3) [2]. These interactions, often mediated by cationic regions near the pY-binding pocket, facilitate membrane recruitment and modulate the signaling activity of SH2-containing proteins like SYK, ZAP70, and ABL [2].
Furthermore, SH2 domain-containing proteins are increasingly implicated in driving the formation of intracellular condensates via liquid-liquid phase separation (LLPS) [2]. Multivalent interactions among proteins like GRB2, Gads, and the LAT receptor contribute to LLPS formation, enhancing T-cell receptor signaling amplitude and specificity [2]. In kidney podocytes, phase separation of the adapter NCK increases membrane dwell time of actin polymerization complexes, promoting efficient actin assembly [2]. These non-canonical roles expand the functional repertoire of SH2 domains and present additional mechanisms through which mutations can dysregulate cellular signaling.
Mutational Landscape of STAT SH2 Domains in Human Disease
STAT3 SH2 Domain Mutations
The STAT3 SH2 domain is a mutational hotspot in numerous human pathologies. Loss-of-function (LOF) mutations are frequently identified in patients with autosomal-dominant Hyper IgE Syndrome (AD-HIES), an immunological disorder characterized by recurrent staphylococcal infections, eczema, and eosinophilia [10]. These mutations disrupt STAT3-mediated Th17 T-cell differentiation, impairing immune responses. Conversely, gain-of-function (GOF) mutations are found in various hematologic malignancies, including T-cell large granular lymphocytic leukemia (T-LGLL) and natural killer cell LGL leukemia (NK-LGLL) [10].
Table 1: Pathogenic Mutations in the STAT3 SH2 Domain
| Mutation | Location | Pathology | Type | Functional Effect |
|---|---|---|---|---|
| K591E/M | αA2 helix, pY pocket | AD-HIES | Germline | Loss-of-function [10] |
| R609G | βB5 strand, pY pocket | AD-HIES | Germline | Loss-of-function [10] |
| S611N/G/I | βB7 strand, pY pocket | AD-HIES | Germline | Loss-of-function [10] |
| S614R | BC loop, pY pocket | T-LGLL, NK-LGLL, ALCL* | Somatic | Gain-of-function [10] |
| E616K/G | BC loop, pY pocket | DLBCL, NKTL | Somatic | Gain-of-function [10] |
| G617E/R/V | BC loop, pY pocket | AD-HIES | Germline | Loss-of-function [10] |
ALCL: Anaplastic Large Cell Lymphoma; DLBCL: Diffuse Large B-Cell Lymphoma; NKTL: Natural Killer T-cell Lymphoma.
STAT5B SH2 Domain Mutations
STAT5B SH2 domain mutations similarly drive both malignant and non-malignant disorders. LOF mutations are associated with growth hormone insensitivity (Laron syndrome) and immune pathology, while GOF mutations are linked to T-cell leukemias [10] [77]. The residue Y665 exemplifies this delicate balance, where different substitutions can produce opposing functional consequences.
Table 2: Pathogenic Mutations in the STAT5B SH2 Domain
| Mutation | Location | Pathology | Type | Functional Effect |
|---|---|---|---|---|
| Y665F | pY+3 pocket / Dimer interface | T-LGLL, T-PLL* | Somatic | Gain-of-function [77] |
| Y665H | pY+3 pocket / Dimer interface | T-PLL (single case) | Somatic | Loss-of-function [77] |
| N642H | SH2 domain | T-LGLL | Somatic | Gain-of-function [77] |
T-PLL: T-cell Prolymphocytic Leukemia.
The Y665F mutation introduces a phenylalanine, which is predicted to stabilize the active parallel dimer through intramolecular aromatic stacking interactions with F711 [77]. In contrast, the Y665H mutation introduces a histidine imidazole group, destabilizing the C-terminal tail binding and SH2 domain structure, resulting in a LOF phenotype [77]. This illustrates how single nucleotide variants at the same codon can push the immune system and hematopoiesis in opposing directions, fine-tuning systems either up or down [77].
Experimental and Computational Methodologies for Mutation Analysis
ClinVar and Population Genomics for Variant Interpretation
ClinVar is a critical, publicly accessible database maintained by the NIH that aggregates information about genomic variation and its relationship to human health [78] [76]. ClinVar employs a systematic classification system for variants:
- Pathogenic (P)/Likely Pathogenic (LP): Variants considered disease-causing.
- Benign (B)/Likely Benign (LB): Variants not considered disease-causing.
- Uncertain Significance (VUS): Variants with insufficient evidence for classification.
The accuracy of ClinVar has improved over time, facilitated by the implementation of the ACMG/AMP guidelines, growing allele frequency databases (e.g., gnomAD), and increasing submission from multiple independent clinical laboratories [76]. For example, the STAT5B Y665F variant is cataloged in ClinVar with supporting evidence from multiple submitters. The review status of a variant (e.g., multiple submitters, no conflicts) is a key indicator of its reliability [76].
Population constraint metrics, such as Molecular Evolutionary Signatures (MES) derived from comparative genomics, help identify genomic regions intolerant to variation. Residues under strong negative selection are likely to be functionally critical, and mutations at these positions are more likely to be pathogenic. Tyrosine 665 in STAT5B, for instance, is highly conserved across vertebrate species, underscoring its functional importance [77].
Biosensors for Real-Time Functional Analysis
Genetically encoded biosensors represent a breakthrough for monitoring STAT activation dynamics in live cells. STATeLights are a class of highly sensitive FRET-based biosensors that allow direct, continuous detection of STAT activity with high spatiotemporal resolution [37].
The optimal STATeLight design for STAT5A involves C-terminal fusion of the fluorophores mNeonGreen (donor) and mScarlet-I (acceptor) to a truncated STAT5A containing the core fragment (CCD, DBD, LD, SH2) [37]. Upon cytokine-induced activation and transition to the parallel dimer conformation, the close proximity of the SH2 domains (< 50 Å) brings the fused fluorophores into close proximity, resulting in a measurable Förster Resonance Energy Transfer (FRET) signal detectable by Fluorescence Lifetime Imaging Microscopy (FLIM) [37].
Protocol: Using STATeLight5A Biosensor
- Cell Preparation: Transfect IL-2-sensitive cells (e.g., HEK-Blue IL-2) with the STATeLight5A construct.
- Stimulation: Treat cells with IL-2 cytokine to activate the JAK-STAT5 pathway.
- Image Acquisition: Perform FLIM measurements to record the fluorescence lifetime of the donor fluorophore (mNeonGreen).
- Data Analysis: Calculate FRET efficiency, which is inversely correlated with donor fluorescence lifetime. A decrease in lifetime indicates STAT5 activation and dimerization.
- Application: Compare activation kinetics of wild-type STAT5 versus disease-associated mutants (e.g., Y665F vs. Y665H) or screen compounds for JAK-STAT5 inhibitory activity [37].
This methodology provides a specific readout of conformational rearrangement to the active dimer state, making it less susceptible to spurious signals from inactive phosphorylated monomers than traditional phospho-specific antibody staining [37].
Computational Prediction of Mutation Impact
In silico tools are indispensable for predicting the functional and structural consequences of SH2 domain mutations:
- AlphaFold3 and COORDinator: Predict protein structures and quantify the energetic contributions of specific residues to stability and dimerization. These tools can distinguish whether a mutation primarily affects domain stability or specific protein-protein interactions [77].
- Pathogenicity Predictors:
- AlphaMissense: Categorizes variants as benign or pathogenic based on a trained model.
- CADD (Combined Annotation Dependent Depletion): PHRED-scaled scores >20 suggest deleteriousness.
- REVEL (Rare Exome Variant Ensemble Learner): Scores >0.5 indicate a higher probability of pathogenicity.
For STAT5B Y665, computational predictions reveal a complex picture: AlphaMissense predicts mild impact for both Y665F and Y665H, while CADD scores (24.3 and 23.1, respectively) suggest potential deleterious effects. REVEL scores (0.535 for Y665F vs. 0.304 for Y665H) indicate a higher probability of pathogenicity for the Y665F variant, consistent with its GOF behavior in functional assays [77].
Research Toolkit for STAT SH2 Domain Investigation
Table 3: Essential Research Reagents and Resources
| Resource Category | Specific Example | Function and Application |
|---|---|---|
| Databases | ClinVar [78] | Archive of genomic variants and clinical interpretations |
| COSMIC [79] | Catalog of somatic mutations in cancer | |
| gnomAD [76] | Population genome variant frequency database | |
| Computational Tools | AlphaFold3 [77] | Protein structure prediction |
| CADD/REVEL [77] | In silico pathogenicity prediction | |
| COORDinator [77] | Predicts energetic impact of mutations | |
| Experimental Reagents | STATeLight Biosensors [37] | Live-cell, real-time monitoring of STAT activation via FLIM-FRET |
| SH2 Domain Profiling Arrays [80] | High-throughput profiling of SH2-phosphopeptide interactions | |
| Cell-Based Assays | Primary T-cell Cultures [77] | Functional validation of immune cell phenotypes |
| Reporter Cell Lines [37] | Measure STAT transcriptional activity |
Visualization of Signaling and Mutation Analysis Workflows
The following diagrams illustrate the core signaling pathway and the integrated workflow for mutation analysis described in this guide.
STAT Canonical Activation Pathway: Cytokine binding triggers receptor-associated JAK kinase activity, leading to STAT phosphorylation, conformational change to parallel dimers, nuclear translocation, and target gene transcription.
Mutation Analysis Workflow: Integrated pipeline from variant identification through computational prediction and experimental validation to therapeutic application.
The integration of population constraint data from resources like ClinVar with advanced experimental and computational methodologies provides a powerful framework for mapping pathogenic mutations in STAT-type SH2 domains. The structural and functional insights gained from these integrated approaches are driving the development of targeted therapeutic strategies, with the STAT SH2 domain itself representing an attractive drug target for cancers and immune disorders [2] [10]. As variant classification continues to improve and novel biosensor technologies enable real-time monitoring of STAT dynamics in live cells, researchers are better equipped than ever to decipher the complex genotype-phenotype relationships governing SH2 domain biology and pathology [76] [37].
The evolutionary conservation of protein domains is a cornerstone of cellular signaling, yet functional divergence of these domains across organisms reveals the adaptive landscape of molecular pathways. This whitepaper examines the evolutionary trajectory of STAT-type Src Homology 2 (SH2) domains, from their origins in early eukaryotes to their specialized functions in modern metazoans, providing a framework for understanding domain-centric evolution and its implications for therapeutic development. SH2 domains, approximately 100 amino acids in length, function as critical mediators of phosphotyrosine (pTyr) signaling networks by recognizing phosphorylated tyrosine motifs and facilitating protein-protein interactions essential for cellular communication [2]. The STAT-type SH2 represents one of the most ancient and fully developed functional domains, serving as an evolutionary template for the continuing development of phosphotyrosine signal transduction [7].
Research spanning diverse organisms from Dictyostelium to humans reveals that while the core structure of SH2 domains remains remarkably conserved, their sequences, binding specificities, and biological functions have undergone substantial divergence. This evolutionary perspective provides unique insights for drug development professionals seeking to target SH2 domain-mediated interactions in human disease, particularly in cancer and immune disorders where STAT signaling is frequently dysregulated.
Evolutionary Origins and Diversity of SH2 Domains
Phylogenetic Distribution and Expansion
SH2 domains first emerged in early Unikonta, with subsequent expansion correlating with metazoan complexity. Genomic analyses across 21 eukaryotic species reveal that SH2 domains co-evolved with protein tyrosine kinases (PTKs) and tyrosine phosphatases, creating sophisticated phosphotyrosine signaling networks [5]. The number of SH2 domain-containing genes expanded dramatically at the unicellular-to-multicellular transition, with humans possessing approximately 111 SH2 domain-containing proteins compared to just a single SH2 protein in Saccharomyces cerevisiae [5].
Table 1: Evolutionary Expansion of SH2 Domains and Tyrosine Kinases
| Organism | SH2 Domain-Containing Proteins | Protein Tyrosine Kinases (PTKs) | Key Evolutionary Position |
|---|---|---|---|
| S. cerevisiae (Yeast) | 1 | 0 | Unicellular opisthokont |
| M. brevicollis (Choanoflagellate) | 17 | 128 | Unicellular ancestor of metazoa |
| D. discoideum (Slime mold) | 6 | 0 | Social amoebozoa |
| C. elegans (Roundworm) | 70 | 90 | Simple metazoan |
| D. melanogaster (Fruit fly) | 42 | 32 | Protostome invertebrate |
| D. rerio (Zebrafish) | 75 | 112 | Vertebrate model |
| H. sapiens (Human) | 111 | 142 | Complex metazoan |
This expansion occurred primarily through gene duplication combined with domain gain or loss, producing novel SH2-containing proteins that function within phosphotyrosine signaling networks [5]. The correlation between the percentage of PTKs and SH2 domains across genomes is striking (r = 0.95), indicating their coordinated evolution [5].
Structural Classification: STAT-type vs. Src-type SH2 Domains
SH2 domains are structurally classified into two major subgroups: STAT-type and Src-type. All SH2 domains share a common "αβββα" sandwich structure with a three-stranded antiparallel beta-sheet flanked by alpha helices, but STAT-type SH2 domains are distinct in that they lack the βE and βF strands as well as the C-terminal adjoining loop [2]. The αB helix in STAT-type domains is split into two helices, an adaptation that facilitates dimerization—a critical step in STAT-mediated transcriptional regulation [2].
This structural disparity reflects the ancestral function of SH2 domain-containing proteins that predate animal multicellularity. The linker domain-conjugated SH2 domain in STAT contains the αB' motif, making it one of the most ancient and fully developed functional domains [7]. STAT-type SH2 domains have been identified in a wide array of vascular and non-vascular plants, suggesting they evolved prior to the divergence of plants and animals [7].
Figure 1. Evolutionary Pathway of SH2 Domains. This diagram traces the structural divergence of SH2 domains from their origins in early eukaryotes to their expansion in metazoans, highlighting the emergence of distinct STAT-type and Src-type variants.
Case Study: Divergent SH2 Domains in Dictyostelium
Dd-STATb: A STAT Protein with Non-Canonical Activation
The social amoeba Dictyostelium discoideum provides a fascinating model for studying ancestral STAT proteins. Dd-STATb possesses a remarkably divergent SH2 domain containing a 15-amino acid insertion and a critical substitution: the arginine residue conserved in all other known SH2 domains, which interacts with phosphotyrosine, is replaced by leucine [56]. Despite these structural abnormalities, Dd-STATb remains biologically functional with a subtle role in growth—Dd-STATb-null cells are gradually lost from populations when co-cultured with parental cells [56].
Microarray analysis identified several genes that are either underexpressed or overexpressed in Dd-STATb null strains. The best characterized of these, discoidin 1, is a marker of the growth-development transition and is overexpressed during growth and early development of Dd-STATb null cells [56]. Surprisingly, Dd-STATb sediments at the size expected for a homodimer and is constitutively enriched in the nucleus, even when the predicted site of tyrosine phosphorylation is substituted by phenylalanine [56]. This suggests a non-canonical mode of activation that does not rely on orthodox SH2 domain:phosphotyrosine interactions, representing a significant functional divergence from mammalian STAT proteins.
Experimental Analysis of Dd-STATb
Protocol 1: Characterizing Divergent SH2 Domain Function
- Gene Disruption: Generate Dd-STATb-null cells through homologous recombination to create knockout strains [56].
- Competitive Growth Assay: Co-culture Dd-STATb-null cells with parental cells in a 1:1 ratio under standard growth conditions. Monitor population composition over multiple generations using flow cytometry or fluorescent markers to quantify the relative fitness [56].
- Transcriptome Profiling: Isolate RNA from wild-type and Dd-STATb-null cells during growth and early development phases. Perform microarray analysis or RNA sequencing to identify differentially expressed genes, with particular attention to discoidin I expression as a marker [56].
- Sedimentation Analysis: Use ultracentrifugation through sucrose density gradients to determine the oligomeric state of Dd-STATb. Compare sedimentation coefficients to protein standards to confirm dimerization [56].
- Subcellular Localization: Employ immunofluorescence microscopy or GFP-tagged Dd-STATb constructs to visualize nuclear localization under various conditions, including after tyrosine phosphorylation site mutagenesis [56].
STAT SH2 Domains in Vertebrate Evolution
Gene Duplication and Functional Divergence in Teleost Fish
Teleost fish, which underwent a specific whole-genome duplication (WGD) event approximately 305-450 million years ago, provide exceptional models for studying STAT gene evolution. Lumpfish (Cyclopterus lumpus), belonging to the order Perciformes, possess stat1a, stat2, stat3, stat4, stat5a, stat5b, and stat6 genes, with most components of the JAK-STAT pathway present in their transcriptome [27]. Research shows that gene duplicates often evolve at different rates, with evolutionary rate asymmetry in overall proteins largely explained by asymmetric evolution within specific protein domains [81].
Domain-centric analysis of asymmetric evolution in teleost fish duplicates reveals that approximately 32% of domains tested were evolving asymmetrically, with certain protein domains like Tyrosine and Ser/Thr Kinase domains having a much greater prevalence of asymmetric evolution [81]. In cases of asymmetrically evolving domains, non-synonymous substitutions often cluster within fast-evolving domains, with rare substitutions preferred within these domains—a pattern suggestive of functional divergence [81].
Table 2: Functional Divergence of STAT Genes in Lumpfish Immune Responses
| STAT Gene | Expression Pattern | Proposed Function in Lumpfish | Activating Stimuli |
|---|---|---|---|
| stat1 | Upregulated 24 hpe against poly(I:C) | Antiviral defense, IFN signaling | Viral mimic (poly(I:C)) |
| stat2 | Upregulated 24 hpe against poly(I:C) | Antiviral defense, IFN signaling | Viral mimic (poly(I:C)) |
| stat3 | Upregulated 6 hpe against bacteria | Antibacterial response, IL-6/IL-10/IL-21 signaling | Bacterial (V. anguillarum) |
| stat4 | Not differentially regulated | T-cell differentiation, potentially conserved | Not determined in study |
| stat5a/5b | Not differentially regulated | Growth hormone signaling, potentially conserved | Not determined in study |
| stat6 | Not differentially regulated | IL-4/IL-13 signaling, Th2 response | Not determined in study |
hpe = hours post-exposure
Experimental Analysis of Teleost STAT Function
Protocol 2: Transcriptome-Wide Analysis of JAK-STAT Pathway
- Immune Challenge: Administer bacterial (Vibrio anguillarum) or viral mimic (poly(I:C)) stimuli to lumpfish via intraperitoneal injection or immersion bath [27].
- Leukocyte Isolation: Aseptically dissect head kidney (the major hematopoietic tissue in fish) and isolate leukocytes using discontinuous Percoll gradient centrifugation at 4°C [27].
- RNA Extraction and Sequencing: Extract high-quality total RNA from leukocytes at multiple time points post-exposure (e.g., 6, 24 hours). Prepare cDNA libraries and perform RNA sequencing on an appropriate platform [27].
- Bioinformatic Analysis: Map sequencing reads to the reference genome, then perform differential gene expression analysis. Conduct KEGG pathway enrichment analysis to identify activated signaling pathways, particularly components of the JAK-STAT system [27].
- qPCR Validation: Design gene-specific primers for identified STAT genes and key cytokines (IL-6, IL-10, IL-21, type I IFNs). Perform quantitative PCR to validate expression patterns from RNA-seq data [27].
Structural and Functional Constraint in SH2 Domains
Evolutionary and Population Constraint Analyses
Recent research combining evolutionary conservation patterns with human population variant data reveals structural constraints on SH2 domains. A unified analysis mapping 2.4 million population variants to 5,885 protein families quantified residue-level constraint using a Missense Enrichment Score (MES), demonstrating that population-constrained sites are enriched in buried residues and binding sites [9]. This pattern aligns closely with observations at evolutionarily conserved sites, suggesting that constraint captured by MES could be useful for predicting structural and functional features.
In SH2 domains specifically, evolutionary conservation and population constraint both indicate structural constraints observable in protein structures, including inter-domain interaction sites on the SH2 surface [9]. The strong correlation between population missense variants and evolutionary conservation suggests that population variants are broadly constrained by the same features that constrain evolutionary substitutions [9].
Figure 2. Workflow for Analyzing Evolutionary Constraint. This diagram illustrates the pipeline for mapping human population variants to protein domains to classify structural and functional constraints on residues.
SH2 Domain Binding Specificity and Affinity Modeling
Advanced experimental-computational approaches now enable accurate modeling of SH2 domain binding affinities across theoretical ligand sequence space. Integrated strategies combining bacterial peptide display, enzymatic phosphorylation of displayed peptides, affinity-based selection, and next-generation sequencing allow researchers to build quantitative sequence-to-affinity models for SH2 domains [8]. The ProBound statistical learning method can infer these models from multi-round selection data generated using fully random peptide libraries, generating predictions valid over multiple orders of magnitude of affinity/activity [8].
These approaches reveal that SH2 domain binding is characterized by a combination of high specificity toward cognate pY ligands with moderate binding affinity (Kd 0.1-10 μM) [2]. This affinity range allows for specific but short-lived interactions, a defining characteristic of most cell signaling mediator interactions [2].
Research Reagent Solutions
Table 3: Essential Research Tools for SH2 Domain Investigation
| Reagent/Method | Function/Application | Key Features | Research Context |
|---|---|---|---|
| Discontinuous Percoll Gradient | Leukocyte isolation from tissues | Maintains cell viability and function | Isolation of head kidney leukocytes from fish [27] |
| Poly(I:C) | Viral immune challenge mimic | Synthetic double-stranded RNA analog | Stimulation of antiviral STAT1/STAT2 pathways [27] |
| Bacterial Display + NGS | SH2 binding specificity profiling | High-throughput affinity characterization | Mapping SH2 domain binding specificities [8] |
| ProBound Algorithm | Sequence-to-affinity modeling | Quantitative binding free energy prediction | Building accurate SH2 affinity models [8] |
| Sedimentation Analysis | Protein oligomerization state determination | Measures hydrodynamic properties | Confirming STAT dimerization [56] |
| Missense Enrichment Score (MES) | Population constraint quantification | Residue-level constraint mapping | Identifying functionally constrained SH2 residues [9] |
Therapeutic Implications and Targeting Strategies
The functional divergence of STAT-type SH2 domains across organisms presents unique opportunities for therapeutic intervention. SH2 domains are increasingly recognized as potential drug targets due to their central role in signal transduction networks dysregulated in cancer, immune disorders, and other diseases [2]. Several targeting strategies have emerged:
Small Molecule Inhibitors: Traditional approaches focus on developing competitive inhibitors that target the pY-binding pocket. Recent advances include nonlipidic small molecules that specifically and potently inhibit lipid-protein interactions, as demonstrated with Syk kinase inhibitors [2].
Lipid-Binding Disruption: Nearly 75% of SH2 domains interact with lipid molecules in the membrane, with a tendency towards phosphatidylinositol-4,5-bisphosphate (PIP2) or phosphatidylinositol-3,4,5-trisphosphate (PIP3) [2]. Targeting lipid-binding interfaces offers an alternative to conventional pY-pocket inhibition and may produce more selective therapeutic agents.
Phase Separation Modulation: SH2 domain-containing proteins increasingly link to intracellular condensate formation via protein phase separation. Multivalent interactions involving SH2 and SH3 domains drive condensate formation, with phosphorylation modulating their assembly and disassembly [2]. This emerging mechanism represents a new frontier for therapeutic manipulation.
Understanding the evolutionary divergence of STAT-type SH2 domains from Dictyostelium to humans provides valuable insights for drug development professionals targeting these critical signaling modules. The conservation of core structural features alongside species-specific adaptations informs both target selection and species translation in preclinical development.
The Signal Transducer and Activator of Transcription (STAT) family of proteins represents a critical node in cellular signaling, translating extracellular cues from cytokines and growth factors into transcriptional programs within the nucleus [82]. The "canonical" signaling paradigm involves tyrosine phosphorylation of latent cytoplasmic STATs by upstream kinases like JAKs, prompting STAT dimerization via reciprocal SH2 domain-phosphotyrosine interactions, nuclear translocation, and DNA binding to regulate genes controlling proliferation, survival, differentiation, and immune responses [83]. Among the seven STAT family members (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, STAT6), STAT3 and STAT5 are frequently aberrantly activated in cancers and inflammatory disorders, driving pathological processes like tumor growth, immune evasion, and chronic inflammation [84] [82]. This established them as high-priority therapeutic targets, spurring the development of STAT inhibitors.
Growing understanding of STAT biology reveals significant complexity beyond the canonical model, including "non-canonical" functions involving unphosphorylated STATs in gene regulation and roles outside the nucleus [83]. This functional diversity is rooted in the evolutionarily conserved protein architecture of STATs, which features six key domains. The SH2 domain is particularly crucial, as it mediates both the recruitment to phosphorylated receptor complexes and the subsequent dimerization of STAT proteins [83]. This foundational role makes the SH2 domain a prime target for therapeutic intervention. The following pipeline analysis details the current clinical landscape of inhibitors designed to disrupt this critical pathway.
The Current STAT Inhibitor Clinical Pipeline
The STAT inhibitor pipeline is dynamic, characterized by a diverse array of drug candidates from over 18 companies utilizing various mechanisms to inhibit STAT signaling [84] [85]. The current pipeline encompasses 22 drugs across phases of clinical development, from discovery to Phase III. The following table provides a quantitative summary of the pipeline, categorized by developmental stage and key characteristics.
Table 1: STAT Inhibitor Pipeline Overview by Clinical Stage
| Drug Name | Company | Therapeutic Target | Mechanism of Action | Key Indications in Development | Development Stage |
|---|---|---|---|---|---|
| TTI-101 | Tvardi Therapeutics | STAT3 | Small molecule, SH2 domain binder [82] | Breast cancer, Idiopathic Pulmonary Fibrosis, Liver cancer [84] | Phase II [84] |
| KT-621 | Kymera Therapeutics | STAT6 | Oral STAT6 degrader [85] | Atopic Dermatitis [85] | Phase I [85] |
| VVD-850 | Vividion Therapeutics | STAT3 | Small molecule, allosteric DNA-binding inhibitor [82] | Solid & hematologic tumors [82] | Phase I [82] |
| BAY 3630914 | Bayer | STAT | Not Specified in Search Results | Not Specified in Search Results | Phase I (Inferred) |
| Danvatirsen | AstraZeneca | STAT3 | Antisense Oligonucleotide | Not Specified in Search Results | Phase I (Inferred) |
| WP1066 | Moleculin | STAT3 | Small molecule inhibitor | Not Specified in Search Results | Preclinical/Discovery |
| NT-219 | Purple Biotech | STAT3 | Dual inhibitor (IRS1/2 & STAT3) | Not Specified in Search Results | Preclinical/Discovery |
| Pipeline Candidates | 18+ Companies | STAT3/STAT5/STAT6 | Small Molecules, Degraders, Biologics | Cancers, Inflammatory Disorders | Preclinical & Discovery Stages |
The pipeline is dominated by efforts to target STAT3, reflecting its central role in oncogenesis [82]. Therapies in later development stages (Phase II and Phase I) include TTI-101, KT-621, and VVD-850, which employ distinct mechanisms from traditional SH2 domain blockade to targeted protein degradation [84] [85] [82]. The high number of candidates in preclinical and discovery phases indicates robust and ongoing research, with key players including Tvardi Therapeutics, Kymera Therapeutics, Vividion Therapeutics, Bayer, and AstraZeneca, among others [84] [82].
Evolutionary Context: The Conservation of STAT-type SH2 Domains
To fully appreciate the therapeutic strategy of targeting the STAT SH2 domain, one must view it through an evolutionary lens. SH2 domains are modular protein interaction domains that specifically bind to phosphotyrosine (pTyr)-containing motifs, forming a core component of metazoan cell signaling networks [4] [5]. The human genome encodes roughly 110 proteins containing SH2 domains, which mediate a vast array of protein-protein interactions in pTyr signaling pathways [2].
Evolutionary bioinformatics reveals that SH2 domains and phosphotyrosine signaling first emerged in the early Unikonta and expanded alongside protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) in the metazoan lineage [5]. This co-evolution facilitated the development of complex, robust cellular communication systems necessary for multicellularity [4] [5]. Phylogenetic analyses classify SH2 domains into two major structural subgroups: the SRC-type and the STAT-type [2].
The STAT-type SH2 domain is one of the most ancient forms. Research indicates that the linker-SH2 domain of STAT serves as the evolutionary origin for the SH2 domain itself, a template from which other SH2 domains continued to evolve [7]. STAT-type SH2 domains possess a distinctive "αβββα" core structure but lack the extra β-strands (βE-βF motif) found in SRC-type SH2 domains. Instead, they feature a characteristic αB' motif within the linker domain [7] [2]. This ancient, conserved structure is dedicated to the critical function of facilitating STAT dimerization, a process fundamental to its canonical role as a transcription factor.
Diagram: Evolutionary and Structural Classification of SH2 Domains
This profound evolutionary conservation underscores the functional importance of the STAT SH2 domain and validates it as a therapeutic target. Inhibiting this ancient, structurally unique module represents a direct strategy for disrupting pathogenic STAT signaling at its root.
Mechanisms of Action: A Technical Breakdown of Therapeutic Inhibition
STAT inhibitors in development employ a sophisticated range of mechanisms to achieve target disruption. The primary strategies can be categorized as follows:
Direct SH2 Domain Binding: This canonical approach involves small molecules that competitively occupy the phosphotyrosine-binding pocket of the SH2 domain. TTI-101 is a prime example; it is an oral small molecule that binds tightly to the SH2 domain of STAT3, preventing its recruitment to activated receptor complexes and subsequent phosphorylation at tyrosine 705. This blockade inhibits STAT3 dimerization and nuclear translocation, thereby suppressing its canonical transcriptional activity [82].
Targeted Protein Degradation: This novel modality uses heterobifunctional small molecules (PROTACs) to recruit the cell's own protein degradation machinery. KT-621 is an oral STAT6 degrader that binds to both STAT6 and an E3 ubiquitin ligase, leading to STAT6 ubiquitination and proteasomal degradation. This approach offers the potential for sustained pathway suppression and efficacy against traditional "undruggable" targets [85].
Allosteric Inhibition and DNA Binding Blockade: Some inhibitors bypass the SH2 domain entirely. VVD-850, for instance, is an orally bioavailable, highly selective small molecule that allosterically inhibits STAT3, preventing it from binding to DNA and driving downstream gene expression [82].
Antisense Oligonucleotides (ASOs): This strategy, exemplified by Danvatirsen, involves short nucleic acid sequences designed to bind to STAT3 mRNA, prompting its degradation by cellular enzymes and thereby reducing the total levels of STAT3 protein available for signaling [82].
Diagram: Molecular Mechanisms of STAT Inhibitors
Table 2: Experimental Models and Reagents for STAT Inhibitor Development
| Research Tool / Reagent | Type | Key Function in R&D | Experimental Application Example |
|---|---|---|---|
| Phospho-STAT Specific Antibodies | Antibody | Detects activated (tyrosine-phosphorylated) STAT proteins [83] | Western blot, IHC to measure pathway inhibition in cell/tissue lysates [83] |
| SH2 Domain Phosphopeptide Libraries | Peptide Library | Profiling SH2 domain binding specificity and selectivity [2] | Screen inhibitor candidates for competitive binding in FP or SPR assays [2] |
| Reporter Gene Assays (e.g., GAS-Luciferase) | Cell-based Assay | Measures STAT-dependent transcriptional activity [83] | High-throughput screening of compound libraries for functional activity [83] |
| Surface Plasmon Resonance (SPR) | Biophysical Instrument | Quantifies binding affinity (Kd) and kinetics of inhibitor-SH2 domain interaction [2] | Characterize direct binding of TTI-101 to recombinant STAT3 SH2 domain [2] |
| Recombinant SH2 Domain Proteins | Protein | Provides purified target for structural and binding studies [2] | X-ray crystallography to determine inhibitor co-structure [2] |
Discussion: Future Perspectives and Challenges
The development of STAT inhibitors faces several scientific and clinical hurdles. A primary challenge is achieving selectivity among highly conserved STAT family members to minimize off-target effects, a task complicated by the shared and ancient nature of the SH2 domain [2]. Furthermore, the integration of STAT inhibitors into combination therapies, particularly with established modalities like immunotherapy or chemotherapy, requires careful empirical evaluation to maximize efficacy and manage potential toxicities [84]. The field would also benefit significantly from the identification and validation of * predictive biomarkers* to select patient populations most likely to respond to therapy [84].
Despite these challenges, the future is promising. The pipeline is rich with innovative modalities, and the first drug candidates are advancing through clinical trials. The profound evolutionary conservation of the STAT-type SH2 domain underscores its fundamental biological role and provides a strong rationale for its continued investigation as a therapeutic target. As our understanding of both canonical and non-canonical STAT functions deepens, it will undoubtedly inform the next generation of targeted therapies, offering new hope for patients with cancers and other STAT-driven diseases.
STAT-type SH2 Domains as Biomarkers for Disease Progression and Treatment Response
Signal Transducer and Activator of Transcription (STAT) proteins are critical mediators of cytokine signaling with central roles in immunity, cell proliferation, and cancer progression. Their Src Homology 2 (SH2) domains facilitate phosphotyrosine-dependent dimerization and nuclear translocation, making them essential for transcriptional activity. Recent evidence identifies STAT-type SH2 domains as mutational hotspots in various pathologies, offering significant potential as diagnostic and prognostic biomarkers. This whitepaper examines the structural, evolutionary, and functional basis for utilizing STAT-type SH2 domains as biomarkers, detailing experimental methodologies for their assessment and discussing their emerging role in therapeutic development. The conservation of these domains across metazoans underscores their fundamental role in signaling networks, while their genetic volatility in disease states highlights their clinical relevance.
STAT proteins are intracellular transcription factors that transduce signals from cytokines and growth factors directly to the nucleus, regulating genes involved in proliferation, survival, and immune responses [10] [86]. The seven STAT family members (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) share a conserved domain architecture consisting of an N-terminal domain, coiled-coil domain, DNA-binding domain, linker domain, SH2 domain, and C-terminal transactivation domain [86]. Among these, the SH2 domain is the critical mediator of STAT activation through its reciprocal phosphotyrosine-binding function, enabling receptor recruitment and STAT dimerization [2] [10].
STAT-type SH2 domains represent a distinct structural subclass characterized by an α-helical C-terminal region (αB') rather than the β-sheet structure found in Src-type SH2 domains [10] [7]. This unique architecture facilitates the specific dimerization interface necessary for STAT transcriptional function. The central hypothesis driving biomarker development is that pathogenic mutations within STAT-type SH2 domains disrupt normal phosphotyrosine signaling, leading to constitutive activation or loss-of-function across diverse pathologies, particularly in cancer and immunodeficiencies [10]. The SH2 domain consequently serves as a mutational hotspot, with specific residues exhibiting significant clinical volatility that correlates with disease progression and treatment outcomes.
Evolutionary Conservation of STAT-type SH2 Domains
Origin and Expansion in Metazoan Lineages
SH2 domains first emerged in unicellular eukaryotes approximately 900 million years ago, coinciding with the development of multicellularity in metazoans [5]. Comparative genomic analyses across 21 eukaryotic species reveal that SH2 domains and phosphotyrosine signaling components expanded rapidly alongside tyrosine kinases and phosphatases in the choanoflagellate and metazoan lineages [5]. This co-evolutionary pattern suggests that SH2 domain-mediated signaling was crucial for the development of intercellular communication networks necessary for complex multicellular organisms.
STAT-type SH2 domains represent one of the most ancient functional templates, predating the divergence of plants and animals [7]. Research has identified STAT-type linker-SH2 domain factors (STATL) in Arabidopsis and other vascular plants, indicating this domain architecture evolved prior to plant-animal divergence [7]. The deep evolutionary conservation of the STAT-type SH2 domain underscores its fundamental role in transcriptional regulation across diverse eukaryotic organisms.
Sequence and Structural Conservation
STAT-type SH2 domains maintain remarkable structural fidelity despite sequence divergence. All SH2 domains assume a conserved αβββα fold with a central anti-parallel β-sheet flanked by two α-helices [2] [10]. The STAT-type SH2 domain is distinguished by:
- Presence of an additional α-helix (αB') at the C-terminus instead of the βE and βF strands found in Src-type SH2 domains [10] [7]
- A split αB helix that facilitates STAT dimerization [2]
- Conservation of critical binding residues despite overall sequence variability
The functional constraint on STAT-type SH2 domains is evident in residue-level conservation patterns. Analysis of missense variant distribution reveals strong evolutionary pressure on buried residues and binding interfaces, highlighting structural features essential for maintaining SH2 domain function [9].
Table 1: Evolutionary Features of STAT-type SH2 Domains
| Feature | Description | Functional Significance |
|---|---|---|
| Structural Fold | αβββα core with C-terminal αB' helix | Distinct from Src-type SH2 domains; facilitates STAT dimerization |
| Origin Timeline | ~900 million years ago | Coincides with emergence of multicellularity |
| Conservation Pattern | High structural conservation despite sequence divergence | Maintains phosphotyrosine binding and dimerization functions |
| Domain Architecture | Linker-SH2 conjunction in STAT proteins | Ancient configuration predating plant-animal divergence |
| Expansion Pattern | Co-evolved with tyrosine kinases | Correlated with increasing metazoan complexity |
Structural and Functional Basis for Biomarker Potential
Molecular Architecture of STAT-type SH2 Domains
The STAT-type SH2 domain contains several structurally and functionally distinct subpockets that determine its biomarker potential:
pY (Phosphate-Binding) Pocket: Formed by the αA helix, BC loop, and one face of the central β-sheet, this pocket contains an invariant arginine residue (βB5) that directly engages phosphotyrosine through a salt bridge [2] [10]. Mutations in this pocket frequently disrupt phosphopeptide binding and STAT activation.
pY+3 (Specificity) Pocket: Created by the opposite face of the β-sheet along with residues from the αB helix and CD/BC* loops, this pocket determines binding specificity by accommodating residues C-terminal to the phosphotyrosine [10]. The evolutionary active region (EAR) within this pocket exhibits significant genetic volatility in disease states.
Hydrophobic System: A cluster of non-polar residues at the base of the pY+3 pocket stabilizes the β-sheet conformation and maintains overall SH2 domain integrity [10]. This system represents a critical structural constraint with biomarker implications.
The structural flexibility of STAT SH2 domains, particularly in the pY pocket, presents both challenges and opportunities for biomarker development. Molecular dynamics simulations reveal substantial conformational heterogeneity even on sub-microsecond timescales, suggesting that dynamic behavior rather than static structure may correlate with pathological states [10].
Pathogenic Mutations in STAT SH2 Domains
Sequencing analyses of patient samples have identified the SH2 domain as a mutational hotspot in STAT proteins. The following table catalogues disease-associated mutations in STAT3 and STAT5B SH2 domains, illustrating their distribution and clinical correlates:
Table 2: Disease-Associated Mutations in STAT3 and STAT5B SH2 Domains
| Mutation | Location | Pathology | Mutation Type | Functional Effect |
|---|---|---|---|---|
| STAT3 K591E/M | αA2 helix | AD-HIES | Germline | Loss-of-function |
| STAT3 S611N | βB7 strand | AD-HIES | Germline | Loss-of-function |
| STAT3 S614R | BC loop | T-LGLL, NK-LGLL, ALK-ALCL | Somatic | Gain-of-function |
| STAT3 E616K | BC loop | NKTL | Somatic | Gain-of-function |
| STAT5B H683Y | βD4 strand | T-PLL, T-LGLL | Somatic | Gain-of-function |
| STAT5B N642H | βC2 strand | Growth hormone insensitivity | Germline | Loss-of-function |
The genetic volatility of specific SH2 domain residues creates a molecular signature of disease progression. Remarkably, identical residues can harbor either activating or deactivating mutations depending on the specific amino acid substitution, underscoring the delicate structural balance in STAT signaling [10]. For instance, the STAT3 S614 residue demonstrates this context-dependent volatility, with S614R mutations driving oncogenesis while other substitutions at this position cause immunodeficiencies.
Experimental Methodologies for Biomarker Assessment
Structural Analysis Techniques
X-ray Crystallography and Cryo-Electron Microscopy Protocol: For structural characterization of STAT-type SH2 domains, express recombinant proteins in mammalian or insect cell systems to ensure proper post-translational modifications. Purify using affinity chromatography followed by size-exclusion chromatography. Crystallize using vapor diffusion methods with optimized cryoprotection. For cryo-EM, grid preparation requires ultra-thin ice with optimal protein distribution. Data collection at resolutions better than 3.0 Ã… enables identification of pathogenic mutation effects on domain architecture and binding pocket conformation [2] [10].
Nuclear Magnetic Resonance (NMR) Spectroscopy Protocol: Prepare isotopically labeled (^15^N, ^13^C) STAT SH2 domains using bacterial or eukaryotic expression systems. Conduct titration experiments with phosphopeptide ligands to monitor chemical shift perturbations. Analyze backbone dynamics through ^15^N relaxation measurements to identify regions with altered flexibility in disease-associated variants. This approach effectively captures the dynamic features of SH2 domains that correlate with pathological activation states [10].
Functional Assays for Biomarker Validation
Surface Plasmon Resonance (SPR) Protocol: Immobilize phosphopeptide ligands corresponding to canonical STAT binding motifs on CMS sensor chips via amine coupling. Inject purified wild-type and mutant STAT SH2 domains at concentrations ranging from 10 nM to 100 μM in HBS-EP buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4). Monitor association (10 minutes) and dissociation (15 minutes) phases at 25°C. Calculate kinetic parameters (K~D~, k~on~, k~off~) using a 1:1 Langmuir binding model. This quantitatively assesses how pathogenic mutations alter binding affinity and kinetics [2].
Cellular Signaling and Transcriptional Reporter Assays Protocol: Transfect STAT-deficient cells with plasmids encoding wild-type or mutant STAT proteins along with luciferase reporters under control of STAT-responsive promoters (e.g., M67/SIE for STAT3). Stimulate with appropriate cytokines (IL-6 for STAT3, IL-4 for STAT6) for 24 hours. Measure luciferase activity normalized to co-transfected Renilla luciferase. Parallel samples should assess STAT phosphorylation (tyrosine and serine) and nuclear translocation via immunoblotting and immunofluorescence. This comprehensive approach validates the functional impact of SH2 domain mutations on signaling output [10].
The following diagram illustrates the key experimental workflow for comprehensive STAT-type SH2 domain biomarker validation:
Research Reagent Solutions for SH2 Domain Studies
Table 3: Essential Research Tools for STAT-type SH2 Domain Biomarker Investigation
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Recombinant Proteins | Purified STAT SH2 domains (wild-type and mutant) | Structural studies, binding assays | Ensure proper folding; eukaryotic expression preferred |
| Phosphopeptide Ligands | pY-containing peptides from receptors (e.g., gp130) | SPR, competitive binding assays | Include +1 to +5 residues C-terminal to pY for specificity |
| Cell Line Models | STAT-deficient cells (e.g., STAT1-/- fibroblasts) | Functional complementation assays | Verify STAT deficiency; control for compensatory signaling |
| Antibodies | Phospho-STAT specific antibodies (pY705 for STAT3) | Immunofluorescence, Western blotting | Validate specificity with phosphorylation-deficient mutants |
| Reporter Constructs | Luciferase under STAT-responsive promoters | Transcriptional activity measurement | Use minimal promoters with multimerized response elements |
| Crystallography Reagents | Crystallization screens (commercial sparse matrix) | Structure determination | Optimize for SH2 domain-specific conditions (PEG-based) |
Signaling Pathways and Pathological Mechanisms
STAT activation follows a conserved pathway initiated by extracellular signals and mediated through SH2 domain functionality. The following diagram illustrates the canonical STAT activation pathway and points of dysregulation by SH2 domain mutations:
Pathological mechanisms of SH2 domain mutations include:
- Constitutive Dimerization: Mutations like STAT3 S614R stabilize the phosphorylated dimer independent of upstream activation, leading to persistent signaling in malignancies [10].
- Signaling Hyper-sensitivity: Some SH2 domain variants lower the activation threshold, enabling response to sub-physiological cytokine levels.
- Altered Specificity: Mutations in the pY+3 pocket can rewire signaling networks by enabling recognition of non-cognate phosphopeptides.
- Differential Immune Effects: The same SH2 domain can harbor distinct mutations causing either immunodeficiencies (AD-HIES) or autoimmune proliferation (T-LGLL), highlighting the context-dependent nature of these biomarkers [10].
Therapeutic Targeting and Clinical Applications
STAT Inhibitors in Clinical Development
The recognition of STAT-type SH2 domains as biomarkers has accelerated therapeutic development targeting these domains:
Table 4: STAT Inhibitors in Clinical Development Targeting SH2-Mediated Signaling
| Therapeutic Agent | Developer | Stage | Molecular Target | Primary Indications |
|---|---|---|---|---|
| TTI-101 | Tvardi Therapeutics | Phase II | STAT3 inhibitor | Hepatocellular carcinoma, breast cancer, IPF |
| KT-621 | Kymera Therapeutics | Phase I | STAT6 degrader | Atopic dermatitis |
| VVD-850 | Vividion Therapeutics | Phase I | STAT3 inhibitor | Advanced tumors |
| Undisclosed Compounds | Multiple companies | Preclinical | STAT SH2 domains | Oncology, inflammation |
The biomarker potential of STAT-type SH2 domains extends to predicting response to these targeted therapies. Specific mutation profiles may indicate susceptibility to SH2 domain-targeting compounds, enabling patient stratification for precision medicine approaches.
Biomarker Applications in Clinical Practice
STAT-type SH2 domains serve multiple biomarker functions in clinical settings:
- Diagnostic Biomarkers: Specific SH2 domain mutations (e.g., STAT3 dominant-negative mutations) provide molecular confirmation of immunodeficiencies like AD-HIES [10].
- Prognostic Indicators: Mutation burden in STAT SH2 domains correlates with disease progression in hematologic malignancies and solid tumors.
- Predictive Biomarkers: SH2 domain mutation profiles may forecast response to JAK inhibitors, immunotherapies, and targeted STAT inhibitors.
- Pharmacodynamic Markers: SH2 domain phosphorylation status or conformational changes can monitor therapeutic target engagement during treatment.
The regulatory considerations for implementing STAT SH2 domains as clinical biomarkers require standardized detection methodologies, validated cutoff values for mutation significance, and demonstrated clinical utility in controlled trials.
STAT-type SH2 domains represent promising biomarkers based on their essential signaling functions, evolutionary conservation, and high mutational frequency in disease states. Future research directions should focus on:
- Comprehensive Mutational Mapping: Systematically characterizing all possible SH2 domain variants and their functional consequences using deep mutational scanning approaches.
- Structural Dynamics Correlation: Establishing relationships between SH2 domain flexibility and pathological signaling using advanced biophysical methods.
- Single-Cell Profiling: Developing techniques to assess STAT SH2 domain status at single-cell resolution within heterogeneous tumor microenvironments.
- Therapeutic Biomarker Validation: Prospectively validating SH2 domain mutations as predictive biomarkers in clinical trials of STAT pathway inhibitors.
The clinical translation of STAT-type SH2 domain biomarkers will require developing accessible detection platforms, establishing standardized interpretation guidelines, and demonstrating utility in guiding therapeutic decisions. As STAT-targeted therapies advance through clinical development, these biomarkers will become increasingly important for optimizing patient selection and treatment outcomes.
The evolutionary conservation of STAT-type SH2 domains underscores their fundamental role in metazoan signaling, while their mutational volatility in diseases highlights their clinical significance as biomarkers. Integrating structural, functional, and clinical assessment of these domains provides a powerful framework for advancing precision medicine in oncology, immunology, and beyond.
Conclusion
The evolutionary journey of the STAT-type SH2 domain underscores its fundamental role as an ancient and conserved orchestrator of phosphotyrosine signaling. Its deep evolutionary conservation, validated by modern genetic constraint analyses, highlights its non-redundant biological importance. The unique structural features that distinguish it from other SH2 families not only trace back to the earliest multicellular life but also present unique vulnerabilities that can be therapeutically exploited. The active and growing pipeline of STAT inhibitors targeting these domains in cancer and inflammatory diseases confirms their clinical translatability. Future research must focus on deciphering the full spectrum of their non-canonical functions, such as in liquid-liquid phase separation, and leveraging advanced structural insights to develop next-generation, high-specificity therapeutics that can disrupt pathogenic signaling networks with greater precision and fewer off-target effects.
References
- 1. SH2 domain [https://en.wikipedia.org/wiki/SH2_domain]
- 2. Update on Structure and Function of SH2 Domains [https://www.mdpi.com/1422-0067/26/18/9060]
- 3. Phosphotyrosine recognition domains: the typical, the atypical ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-10-32]
- 4. Evolution of SH2 domains and phosphotyrosine signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3415846/]
- 5. The SH2 Domain–Containing Proteins in 21 Species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4255630/]
- 6. Identification and characterization of a large family of ... [https://www.nature.com/articles/s41467-018-06943-2]
- 7. Identification of the Linker-SH2 Domain of STAT as ... [https://www.sciencedirect.com/science/article/pii/S1535947620338639]
- 8. Accurate affinity models for SH2 domains from peptide binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12521615/]
- 9. A unified analysis of evolutionary and population constraint ... [https://www.nature.com/articles/s42003-024-06117-5]
- 10. Structural Implications of STAT3 and STAT5 SH2 Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6895964/]
- 11. A new family of transcription factors - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3586674/]
- 12. Acanthamoeba castellanii STAT protein - PubMed [https://pubmed.ncbi.nlm.nih.gov/25338074/]
- 13. Comparative genomics of the social amoebae Dictyostelium ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2011-12-2-r20]
- 14. SH2 Domain Binding: Diverse FLVRs of Partnership [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2020.575220/full]
- 15. The JAK/STAT signaling pathway: from bench to clinic [https://www.nature.com/articles/s41392-021-00791-1]
- 16. Conservation analysis and structure prediction of the SH2 ... [https://pubmed.ncbi.nlm.nih.gov/1377638/]
- 17. Binding Specificity of SH2 Domains: Insight from Free ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2635922/]
- 18. The SH2 domains of Stat1 and Stat2 mediate multiple ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC450005/]
- 19. Evolution of SH2 domains and phosphotyrosine signalling networks - PubMed [https://pubmed.ncbi.nlm.nih.gov/22889907/]
- 20. SH2 Domains: Folding, Binding and Therapeutical ... [https://www.mdpi.com/1422-0067/23/24/15944]
- 21. Surface Loops in a Single SH2 Domain Are Capable of ... [https://www.sciencedirect.com/science/article/pii/S1535947620318776]
- 22. Conserved conformational dynamics determine enzyme activity [https://pmc.ncbi.nlm.nih.gov/articles/PMC9348788/]
- 23. Evolutionary cores of domain co-occurrence networks [https://bmcecolevol.biomedcentral.com/articles/10.1186/1471-2148-5-24]
- 24. Evolutionary divergence in the conformational landscapes ... [https://elifesciences.org/articles/83368]
- 25. PD-1 is conserved from sharks to humans: new insights ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1573492/full]
- 26. High-throughput profiling of sequence recognition by ... [https://elifesciences.org/articles/82345]
- 27. Evolutionary, comparative, and functional analyses of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10556531/]
- 28. SH2 Domains Serve as Lipid-Binding Modules for pTyr- ... [https://www.sciencedirect.com/science/article/pii/S109727651600054X]
- 29. Spotlight on amino acid changing mutations in the JAK ... [https://www.nature.com/articles/s41598-025-90788-5]
- 30. Update on Structure and Function of SH2 Domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC12470777/]
- 31. Comparison of SH3 and SH2 domain dynamics when ... [https://www.sciencedirect.com/science/article/pii/S0022283699926190]
- 32. Sample delivery methods for protein X-ray crystallography ... [https://pubmed.ncbi.nlm.nih.gov/41203635/]
- 33. How accurate are AlphaFold 2 structure predictions? [https://www.ebi.ac.uk/training/online/courses/alphafold/validation-and-impact/how-accurate-are-alphafold-structure-predictions/]
- 34. A novel method for multiple alignment of sequences with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC525693/]
- 35. Jalview Home Page - Jalview [https://www.jalview.org/]
- 36. CoDIAC: A comprehensive approach for interaction analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11291013/]
- 37. Real-time visualization of STAT activation in live cells using ... [https://www.nature.com/articles/s41589-025-02012-0]
- 38. PepCyber:P∼PEP: a database of human protein–protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2238930/]
- 39. Selective Targeting of SH2 Domain–Phosphotyrosine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5417323/]
- 40. Determining folding and binding properties of the C†... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8605372/]
- 41. Folding and Binding Kinetics of the Tandem of SH2 ... [https://www.mdpi.com/1422-0067/25/12/6566]
- 42. Quantitative analysis of SH2 domain binding. Evidence for ... [https://www.sciencedirect.com/science/article/pii/S0021925819496890]
- 43. Targeted Quantification of Phosphorylation Dynamics in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5912210/]
- 44. Molecular mechanisms mediated by liquid-liquid phase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12637247/]
- 45. Liquid–liquid phase separation in human health and ... [https://www.nature.com/articles/s41392-021-00678-1]
- 46. SH2 domains serve as lipid binding modules for pTyr ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4826312/]
- 47. Liquid-liquid phase separation in cell physiology and ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1540427/full]
- 48. Lipids Regulate Lck Protein Activity through Their ... [https://www.sciencedirect.com/science/article/pii/S0021925820334050]
- 49. CDD Conserved Protein Domain Family: SH2_STAT1 [https://www.ncbi.nlm.nih.gov/Structure/cdd/cd10372]
- 50. æ± è¶èšŒSTATåŸºå› çš„å…‹éš†åŠåŠŸèƒ½åˆ†æž - 水生生物å¦æŠ¥ [http://ssswxb.ihb.ac.cn/cn/article/id/5d545ac0-fdee-4b45-8355-dbc8882f1890]
- 51. Investigating Liquid–Liquid Phase Separation in Lung ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12372984/]
- 52. A Cell-permeable Stat3 SH2 Domain Mimetic Inhibits Stat3 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2975209/]
- 53. Review The SH2 domain: versatile signaling module and ... [https://www.sciencedirect.com/science/article/abs/pii/S1570963904002882]
- 54. High-throughput virtual screening of small-molecule ... [https://pubmed.ncbi.nlm.nih.gov/35633442/]
- 55. Identification of the linker-SH2 domain of STAT as ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/15073273/]
- 56. Dd-STATb, a Dictyostelium STAT protein with a highly ... [https://pubmed.ncbi.nlm.nih.gov/14701681/]
- 57. Enhanced Prediction of Src Homology 2 (SH2) Domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4083110/]
- 58. High-throughput profiling of sequence recognition by tyrosine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10065799/]
- 59. Time-resolved multimodal analysis of Src Homology 2 (SH2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4841779/]
- 60. Structural and functional analysis of target recognition by ... [https://www.nature.com/articles/s41467-021-26394-6]
- 61. Update on Structure and Function of SH2 Domains [https://pubmed.ncbi.nlm.nih.gov/41009621/]
- 62. The SH2 domain interaction landscape - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4110347/]
- 63. SH2 Domains Recognize Contextual Peptide Sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2984226/]
- 64. Novel Roles of SH2 and SH3 Domains in Lipid Binding [https://www.mdpi.com/2073-4409/10/5/1191]
- 65. Multi-condensate state as a functional strategy to optimize ... [https://www.nature.com/articles/s41467-024-50489-5]
- 66. Emerging Pharmacotherapeutic Strategies to Overcome Undruggable... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10367563/]
- 67. Computational screening and molecular dynamics of natural... [https://link.springer.com/article/10.1007/s11030-024-11075-5]
- 68. Bruton's tyrosine kinase (BTK) SH2 inhibitor reduced skin ... [https://recludixpharma.com/recludix-pharma-presents-preclinical-data-on-first-in-class-btk-sh2-domain-inhibitor-demonstrating-powerful-btk-inhibition-exceptional-selectivity-and-encouraging-efficacy-in-a-model-of-chronic-spon/]
- 69. Recludix Unveils First BTK SH2 Domain Inhibitor [https://www.dermatologytimes.com/view/recludix-unveils-first-btk-sh2-domain-inhibitor]
- 70. Predicting protein–protein interactions in microbes ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1565860/full]
- 71. A structural machine learning approach for rapid prediction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539237/]
- 72. New insights into protein – protein modulators in interaction ... drug [https://www.nature.com/articles/s41392-024-02036-3?error=cookies_not_supported&code=fb39c5a0-724a-41d1-ab63-7ae8af18bc3c]
- 73. Protein interactions, network pharmacology, and machine ... [https://www.nature.com/articles/s41598-025-97534-x]
- 74. Pathogen-Driven Modulation of Eight STATs in ... [https://www.sciencedirect.com/science/article/abs/pii/S1050464825008964]
- 75. Defining the Specificity Space of the Human Src Homology ... [https://www.sciencedirect.com/science/article/pii/S1535947620312007]
- 76. ClinVar and HGMD genomic variant classification accuracy ... [https://www.medrxiv.org/content/10.1101/2022.10.26.22281567v1.full]
- 77. STAT5B leukemic mutations, altering SH2 tyrosine 665, have ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11999048/]
- 78. ClinVar Genomic variation as it relates to human health - NCBI [https://www.ncbi.nlm.nih.gov/clinvar/variation/344]
- 79. Recognizing Pattern and Rule of Mutation Signatures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8427289/]
- 80. High-Throughput Phosphotyrosine Profiling Using SH2 ... [https://www.sciencedirect.com/science/article/pii/S1097276507003346]
- 81. Functional divergence of gene duplicates – a domain-centric ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/1471-2148-12-126]
- 82. STAT Inhibitors - Pipeline Insight, 2025 [https://www.researchandmarkets.com/report/stat-inhibitor-market?srsltid=AfmBOooehs2acWjlbheNBQdyLgqKtUmTklqmn2TD6-bFm1fYy4rVvaLu]
- 83. STAT proteins: a kaleidoscope of canonical and non ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-021-01214-y]
- 84. STAT Inhibitors Pipeline Market Landscape Report 2025 [https://www.globenewswire.com/news-release/2025/11/14/3188124/0/en/STAT-Inhibitors-Pipeline-Market-Landscape-Report-2025-Over-18-Companies-and-22-Drugs-in-Various-Stages-of-Development.html]
- 85. STAT Inhibitors Pipeline Market Insights Report 2025 [https://www.businesswire.com/news/home/20250901475308/en/STAT-Inhibitors-Pipeline-Market-Insights-Report-2025-18-Companies-and-22-Pipeline-Drugs-in-Stages-Ranging-from-Clinical-to-Nonclinical---ResearchAndMarkets.com]
- 86. Genome-Wide Identification and Expression Analysis of the ... [https://link.springer.com/article/10.1007/s10528-024-10820-7]
- 87. STAT Inhibitors Pipeline Market Landscape Report 2025 [https://finance.yahoo.com/news/stat-inhibitors-pipeline-market-landscape-100100382.html]